 Hannes Zwickl
Hannes Zwickl Elisabeth Zwickl-Traxler
Elisabeth Zwickl-Traxler- Department of Internal Medicine 2, University Hospital Krems, Karl Landsteiner Private University of Health Sciences, Krems, Austria
In this paper, we present evidence in support of our hypothesis that the neuronal histaminergic system might be involved in cancer cachexia1. To build our premise, we present the research and the reasonable inferences that can be drawn from it in a section by section approach starting from one of the key issues related to cachexia, increased resting energy expenditure (REE), and progressing to the other, anorexia. Based on an extensive survey of the literature and our own deliberations on the abovementioned topics, we investigate whether histamine signaling might be the mechanism used by a tumor to hijack the body's thermogenic machinery. Our hypothesis in short is that hypothalamic histaminergic neurons are stimulated by inputs from the parasympathetic nervous system (PSNS), which senses tumor traits early in cancer development. Histamine release in the preoptic area of the hypothalamus primarily activates brown adipose tissue (BAT), triggering a highly energy demanding mechanism. Chronic activation of BAT, which, in this context, refers to intermittent and/or low grade activation by the sympathetic nervous system, leads to browning of white adipose tissue and further enhances thermogenic potential. Aberrant histamine signaling not only triggers energy-consuming processes, but also anorexia. Moreover, since functions such as taste, smell, and sleep are governed by discrete structures of the brain, which are targeted by distinct histaminergic neuron populations even relatively minor symptoms of cachexia, such as sleep disturbances and taste and smell distortions, also might be ascribed to aberrant histamine signaling. In late stage cachexia, the sympathetic tone in skeletal muscle breaks down, which we hypothesize might be caused by a reduction in histamine signaling or by the interference of other cachexia related mechanisms. Histamine signaling thus might delineate distinct stages of cachexia progression, with the early phase marked by a PSNS-mediated increase in histamine signaling, increased sympathetic tone and symptomatic adipose tissue depletion, and the late phase characterized by reduced histamine signaling, decreased sympathetic tone and symptomatic muscle wasting. To support our hypothesis, we review the literature from across disciplines and highlight the many commonalities between the mechanisms underlying cancer cachexia and current research findings on the regulation of energy homeostasis (particularly as it relates to hypothalamic histamine signaling). Extrapolating from the current body of knowledge, we develop our hypothetical framework (based on experimentally falsifiable assumptions) about the role of a distinct neuron population in the pathophysiology of cancer cachexia. Our hope is that presenting our ideas will spark discussion about the pathophysiology of cachexia, cancer's devastating and intractable syndrome.
Introduction
Affecting patients' quality of life, prognosis, and response to and tolerance of therapies (1–3), cachexia is a major issue in cancer management, all the more so as effective treatment options to halt or reverse wasting are still lacking. Cancer-associated body weight loss reflects the profound dysregulation of energy homeostasis, which manifests in increased resting energy expenditure (REE) and in anorexia (1, 2, 4, 5). Moreover, cancer cachexia is associated with perturbations of lipid, glucose, and protein metabolism, and involves most, if not all, organs (6–8).
Systemic inflammation has been identified as a major driver of cancer cachexia (9–11), with cancer-associated anorexia having been attributed to neuroinflammation resulting from the effects of dysregulated cytokine networks on neurons, microglia, and endothelial cells (12–14). Nevertheless, there is consensus that treatment of cancer cachexia requires a multimodal approach and that controlling inflammation alone is a downstream solution targeting symptoms, which is not sufficient to obtain optimal therapy results (15). This indicates that cancer cachexia involves a pathophysiological mechanism triggered by the tumor (16), but not necessarily mediated by tumor-associated systemic inflammation.
A mechanism that meets this criterion recently has been identified in research on the neuronal control of energy homeostasis2. Enhanced histamine signaling ameliorates obesity by concomitantly increasing REE and inducing anorexia, the major characteristics of cancer cachexia (17, 18). The central histaminergic system comprises brain mast cells and histaminergic neurons (19), of which the latter are involved in controlling sleep and wakefulness, biological rhythms, mood, and cognition (2, 19–21), all of which may be disrupted in cancer cachexia (2). Indeed, the degree of overlap between the spectrum of physiological functions responsive to neuronal histamine signaling and the clinical manifestations of cancer cachexia is quite remarkable. It appears that in cachexia, neuronal histamine signaling takes on a more prominent role than its mediating function in normal physiology, disconnecting from the biological context normally shaped by other neurotransmitters to the extent that it subverts the action of upstream neurotransmitters and hormones. To wit, neuronal histamine signaling is presumed to work in parallel and largely independent of factors having been implicated in cancer cachexia so far.
We will begin our exploration by taking a closer look at the mechanisms that may play a role in increased REE.
Increased REE
Increased REE is the main cause of wasting (22). Several organs contribute to REE, including skeletal and cardiac muscle, liver, and both brown and white adipose tissue (BAT and WAT, respectively) (6, 23).
Activation of BAT
BAT activation has been found in cancer patients (24–27) and its correlation with weight loss demonstrated in rodent models of cancer cachexia (23, 28, 29). However, the significance of activated BAT for cancer cachexia in humans is still a matter of debate. This is mainly due to the fact that 18F-fluorodeoxyglucose positron emission tomography/computer tomography (18F-FDG-PET-CT), which is used to detect metabolically active BAT in humans, has yielded discrepant results (26, 27). Further studies are required, with appropriate consideration given to variables affecting BAT activity (27). One such potentially significant, but so far neglected, variable is circadian rhythmicity of BAT activation, which has been found in rodent models of cancer cachexia. If the findings in rodents apply to humans, this would suggest that BAT is activated mainly during the night in human cancer cachexia (23, 30). A recent retrospective study on variables influencing BAT activation based on retrospective re-examinations of 18F-FDG-PET-CT images from over 15,000 people demonstrated that its incidence changes with time of day (31). Furthermore, since BAT-mediated thermogenesis is a highly energy-demanding process, a slight but persistent increase in BAT activity is sufficient to induce pronounced weight loss (32). One drawback of 18F-FDG-PET-CT is that it is not sensitive enough to reliably detect BAT3 (33).
Hypothesis: Both the time of day of measurement and the lack of sensitivity of 18F-FDG-PET-CT might account for the inconsistency of data on BAT activation in cancer cachexia. Data inconsistency also might reflect peculiarities of the underlying mechanism, i.e., circadian rhythmicity and/or low-grade BAT activation. Given that BAT activation data appears to track with circadian rhythms, long-term continuous monitoring of body temperature is needed employing an appropriate measurement technique.
Browning of WAT
The emergence of beige adipocytes in WAT is associated with cancer cachexia in animal models and humans (24, 25, 34–36). Like brown adipocytes, beige adipocytes express uncoupling protein 1, which uncouples mitochondrial respiration from ATP synthesis in favor of thermogenesis and thereby increases REE (37). However, the extent of its contribution to REE in this context cannot be assessed by 18F-FDG-PET-CT due to its limited sensitivity (see above), thus is prone to being underestimated (26). Several humoral factors, the tumorkine parathyroid hormone-related protein (PTHrP) and the cytokine interleukin 6 (IL-6), recently have been suggested as potential causes of WAT browning. Kir et al. identified tumor-derived PTHrP as an inducer of WAT browning in a rodent cancer model and reported that approximately one third of patients with metastatic non-small-cell lung cancer (NSCLC) or colorectal cancer had detectable serum PTHrP, together with higher REE and lower lean body mass, compared to PTHrP-negative patients (34, 38–40). In addition, Hong et al. showed that increased serum PTHrP correlated with increased probability of weight loss in cancer patients, but that weight loss also occurred in the absence of PTHrP. This suggests that PTHrP, rather than being a causative factor, catalyzes already progressive cancer cachexia (41). IL-6, an immanent factor of cachexia-associated systemic inflammation, promotes browning, increases REE, and drives weight loss in cancer cachexia (35). However, though anti-IL-6 therapy alleviates cachectic symptoms, it is most effective in the treatment of cancer cachexia when used as part of a multimodal approach (42).
Hypothesis: These research findings indicate that there might be an underlying pathophysiological mechanism, which current treatments for cachectic cancer patients do not yet effectively target. This suggests that tumorkine-mediated mechanisms and systemic inflammation can be active early on as secondary mechanisms contributing to REE, but gain in significance during tumor progression, amplifying tumor-triggered REE and exacerbating systemic wasting in proportion to the rate of tumorkine production and cytokine levels in individual patients. This is consistent with the heterogeneity of cachexia across cancer patients, which manifests as symptoms that vary in type and severity (2).
Non-shivering Thermogenesis (NST)
In normal physiology, BAT is an effector organ of non-shivering thermogenesis (NST), which is activated by cold stress to maintain body temperature (43). Chronic cold stress promotes the proliferation of brown adipocytes and browning of WAT (37, 44) in rodents (45–47) and humans (48, 49).
Hypothesis: Cancer cachexia sharing characteristic features of the body's response to chronic cold stress suggests that a tumor might act as a chronic stressor, triggering the homeostatic response in a manner similar to cold exposure, the physiological stressor. Could NST serve as a blueprint for adipose tissue-driven energy expenditure in cancer cachexia?
NST primarily activates BAT, inducing WAT browning only if cold stress persists (44, 49). This implies that BAT activation precedes WAT browning in normal physiology or, conversely, that browning should not occur without chronically increased BAT activation. However, as discussed earlier, whereas WAT browning has been shown to occur in cancer cachexia (35), BAT activation is still controversial. In humans, intermittent, moderate cold stress4—as opposed to continuous triggering of NST or overly pronounced BAT activation—suffices to significantly increase the thermogenic capacity of WAT due to browning (37, 48, 50), with the increase in thermogenic capacity being a function of both duration and extent of the thermogenic trigger (26). In this view, WAT browning, as well as presumed BAT activation, in cachectic cancer patients mirrors the normal function of NST in response to chronic cold stress and aligns with the abovementioned findings re circadian rhythmicity and low-grade activation of BAT.
Increased Lipolysis
Chronic activation of BAT requires a supply of energy-rich compounds, either lipids from WAT or glucose from the liver (51). NST so effectively removes glucose and free fatty acids (FFA) from systemic circulation (52, 53), and reduces hyperglycemia and hyperlipidemia (54), that its intentional activation already has been attempted to treat obesity and metabolic syndrome (37, 52). The therapeutic application of a moderate cold exposure regimen has been shown to reduce body weight and increase insulin sensitivity in humans (52).
In normal physiology, the high rate of lipid turnover is due to a dynamic equilibrium of lipogenesis and lipolysis in white adipocytes (55). In cancer cachexia, the loss of adipose tissue reflects the depletion of cellular lipid stores (56) and an increase in lipid turnover primarily mediated by accelerated WAT lipolysis (57). Cancer cachexia has been linked to the increased expression of adipose triglyceride lipase and hormone-sensitive lipase, which, once stimulated, increase the lipolytic capacity of white adipocytes (58), while basal lipolysis remains unaltered or is reduced (58). This suggests that pro-lipolytic stimuli actively drive lipid mobilization in cancer cachexia. For instance, zinc-alpha2-glycoprotein, a lipid-mobilizing factor, has been associated with cancer cachexia in rodents and humans (59, 60). Insulin is a potent inhibitor of lipolysis, and resistance to its inhibiting effect contributes to increased lipolysis in cancer cachexia (58).
The inverse correlation of WAT-associated triacylglycerol hydrolase activity, the rate-limiting step of lipolysis, with the body mass index of cachectic cancer patients might reflect an adaptation to chronic activation of NST (61). Moreover, the increased FFA oxidation (62) and alterations at the transcriptome level of WAT observed in cachectic cancer patients indicates an increased capacity by white adipocytes to utilize fatty acids and suggests another potential link to browning and concomitant chronicity (63).
In humans, increased lipolysis is mediated mainly by catecholamines and natriuretic peptides (58, 64). In particular, the sympathetic nervous system (SNS) and related noradrenergic signaling are the most important mechanisms for conveying the homeostatic response to cold stress (65, 66). Increased sympathetic tone is sufficient to induce activation of BAT, browning of WAT, and lipolysis (37, 49). In normal physiology, SNS-associated noradrenergic signaling mediates the body's acute stress response5. If this stress response were to become chronic, its normal physiological role would be perverted and the mechanisms it triggers—now chronic—would increase REE and cause the loss of body weight, exactly as happens in cachexia.
Hypothesis: Consistent with increased lipolysis, plasma levels of triglycerides, FFA, glycerol, and cholesterol have been reported to increase in cancer patients who are losing weight (7, 56). However, other studies found lower serum cholesterol levels in newly diagnosed cancer patients and lung cancer patients (68, 69) and either the same or lower triglyceride levels in lung cancer patients (69) in comparison to non-malignant cohorts. NST is a highly energy-demanding process, thus serum lipid levels might change inversely with NST activation. The reduction in cholesterol levels of cancer patients after an overnight fast, as observed by us (publication in preparation) and others, is consistent with increased NST during the night and correlates with the circadian rhythmicity of BAT activation (68). As a consequence, analogous to BAT activation (see above), the time of day of measurement might be a crucial determinant of plasma cholesterol level.
It has long since been recognized that cholesterol levels begin to decline years before cancer diagnosis. This finding led to the hypothesis that the change in cholesterol reflects metabolic alterations attributable to subclinical disease (68)6; thus, inexplicable decreases in cholesterol levels which do not result from lifestyle changes might be an indicator of metabolic dysregulation induced by early malignancy. A recent retrospective study examining abnormal radiotracer uptake in PET-CTs revealed that tumor activity and volume correlated positively with BAT activation in non-cachectic cancer patients (70), indicating that NST—as a presumed mechanism of cachexia—is activated in the early stages of tumor progression, but appears as unintentional weight loss only once the increased energy expenditure can no longer be compensated.
The research findings on the SNS and noradrenergic signaling lead us to infer that a tumor uses the neuroanatomical structures mediating NST, including sympathetic efferents, to render this acute stress mechanism chronic. Moreover, as a mechanism of both tumor-induced changes to bodily homeostasis and tumor-induced immunosuppression, the SNS provides a link between cachexia and cancer biology. Pursuing this line of reasoning implies that the tumor might interfere with the central neural pathways of thermoregulation.
Central Neural Pathways of Thermoregulation
Under normal conditions, thermal inputs from various body sites are transmitted to central thermoregulatory neuronal circuits located in the preoptic area (POA) of the anterior hypothalamus (43). Thermogenic activation occurs when cold stress activates neurons in the median POA (MnPOA), which in turn decrease the activity of warm-sensitive neurons in the medial POA (MPOA), thereby inhibiting their tonic suppression of sympathetic premotor neurons in the rostral raphe pallidus nucleus.
Which of the thermoregulatory effector mechanisms is activated depends on the duration of cold stress and specific body temperature thresholds. Whereas, NST and tachycardia are rapidly provoked by skin cooling, shivering thermogenesis (ST) is activated only when core body temperature is at stake. This finding has led to the suggestion that the different cold defense mechanisms are mediated by distinct neuron populations (43).
Hypothesis: Mediation of cold defense mechanisms by distinct neuron populations provides a rationale for why NST—but not other cold defense mechanisms, such as cutaneous vasoconstriction or piloerection—has been observed to be activated in cancer cachexia. In normal physiology, sympathetically mediated tachycardia is always co-activated as a cardiac thermogenic mechanism along with both NST and ST (43). Assuming that a tumor co-opts the neurocircuits that induce NST in response to cold stress, one might expect to see tachycardia in cancer patients as well. Although the co-occurrence of NST and tachycardia has not been directly studied, an increased resting heart rate has indeed been described in patients with advanced colorectal, pancreatic cancer as well as NSCLC and suggested as an independent predictor of a poor chance of survival (71).
Might the tumor activate hypothalamic neurocircuits, which, by means of sympathetic efferents, trigger BAT activation, WAT browning, and increased lipid mobilization? Although the findings on cancer cachexia inducing low-grade BAT activation that fluctuates to circadian rhythms are still ambiguous, the chronic effects of cachexia on WAT (browning and increased lipolysis) have emerged more clearly. In answer as to how that might be implemented at the neurophysiological level, we refer to what is known about the normal mechanisms involved in thermoregulation and explore these intersections next.
Histamine Signaling
Histamine signaling plays a role in POA-mediated thermoregulation (72), which depends on the site and receptor type, as shown in rodent models (17). Hyperthermia can be induced by experimentally raising histamine levels in the MPOA and MnPOA (73), which involves H1R or H3R (74) and H2R signaling (75), respectively. Moreover, it has been suggested that H2R and H3R histamine signaling maintains hyperthermic tone (76, 77). However, histamine signaling also has been implicated in hypothermic effects, although only in rather harsh experimental settings, such as exposure to ionizing radiation (78) or in association with anaphylaxis (79). Notably, hypothermic effects do not involve the POA (17). Histamine signaling in the POA reduces the respiratory quotient, which is an indicator of increased utilization of lipids as an energy source, and increases the expression of uncoupling protein (UCP) in thermogenic tissues, in particular UCP1 in BAT, as well as UCP2 in WAT (73, 80, 81).
Hypothesis: WAT browning, and possibly BAT activation, are associated with cancer cachexia. As necessary and sufficient to induce hyperthermia, as well as produce the effects of chronic NST activation, histamine signaling in the POA provides a mechanism by which the tumor might gain control of the thermogenic machinery. Indeed, cancer patients frequently exhibit increased body temperature and night sweats, the latter again pointing to circadian rhythmicity in NST (82).
Circadian Rhythmicity and Night Sweats
Determining metabolic cycles (83, 84) and body temperature (30), circadian rhythmicity is highly conserved in mammals. In rats7, experimentally increasing histamine levels in the MPOA and MnPOA induces significant hyperthermia in their diurnal (inactive) phase, which diminishes in their nocturnal (active) phase (73). This effect likely is mediated by H1R activation in the MnPOA, which triggers NST and concomitantly reduces active phase motor activity, thereby relegating maintenance of body temperature toward the NST in histamine-dosed animals vs. toward motor activity in control animals.
Hypothesis: If this mechanism were to be active in (human) cachectic cancer patients, it would manifest as a body temperature higher than would otherwise occur with the normal nighttime circadian lowering of temperature. Since the extent of NST activation depends on the degree of histamine receptor activation, it can range from low (e.g., by only compensating the normal drop of body temperature at night, which is prone to remain unnoticed) to the fever described in lung and pancreatic cancer, leukemia/lymphoma and pediatric malignancies, among others (85–88).
Sweating represents a thermoregulatory mechanism which is activated in response to a rise in body temperature. In cancer cachexia, increased histamine signaling might promote thermogenesis while the body's temperature set point remains unchanged. Might cancer patients' night sweats reflect the body's attempt to reduce the differential between the actual—histamine-induced—elevated body temperature and the normal set point temperature, similar to what occurs after anti-pyrogenic drugs are administered to reduce fever? By contrast, inflammation-triggered and prostaglandin E2-mediated fever corresponds to the adaptation of the actual body temperature to a set point increased by inflammation. Thus, different neurophysiological mechanisms would underlie cancer cachexia-associated thermogenesis and pyrogenic fever, which is why it is possible to observe simultaneous fever and sweating in cachectic cancer patients.
The Histaminergic System
The two principal sources of histamine in the brain are mast cells and histaminergic neurons; together they comprise the central histaminergic system (19). Notably, histamine from peripheral sources such as the tumor itself (89, 90) do not affect brain physiology, since histamine cannot pass the blood-brain barrier, but is produced de novo from histidine (21). However, the expression of histamine by tumors underlines its dual role in malignancies (91).
Brain mast cells are important mediators of neuroinflammation (92), which has been implicated in cancer cachexia (13, 14). Recent research indicates that activation of brain mast cells induces their degranulation, which in turn activates microglia, and thereby mediates neuroinflammation, and that suppressing mast cells' degranulation prevents neuroinflammation (92). Therefore, the involvement of brain mast cells and thus, histamine signaling, is consistent with neuroinflammation. Moreover, there is evidence for an association between systemic and neuroinflammation (13).
Histaminergic neurons (19–21, 93) are located in the tuberomamillary nucleus (TMN) of the posterior hypothalamus (19–21, 93). They act as pacemakers, autonomously firing at a low rate, and are excitable by various neurotransmitters including noradrenaline, ATP, serotonin, and orexin. Histaminergic neurons are integral to POA-mediated thermogenesis and the regulation of food intake (17). Indeed, stimulating hypothalamic histamine signaling in distinct nuclei of the hypothalamus has been shown to promote NST and anorexia, both major symptoms of cancer cachexia (17, 18).
Hypothesis: Systemic inflammation and neuroinflammation may play different roles in cachexia than previously thought. Immunoediting, reflecting the interplay of immune system and tumor, is complex and has been divided into three distinct phases (elimination, equilibrium, and escape), whereby many crucial events occur in the local tumor environment, which in turn affect the immune response at the systemic level (94). Beginning in the early stages of cancer, there are physiological changes in plasma cholesterol levels and resting heart rate that are consistent with NST activation. We hypothesize that these effects occur before systemic inflammation and thus also before neuroinflammation. Physiological effects preceding cancer-associated systemic inflammation (but not necessarily local inflammation) point to an inflammation-independent mechanism as the primary central nervous system (CNS) mechanism in cancer cachexia. Hence, neuroinflammation might exacerbate wasting, rather than being one of its primary causes (see above mentioned discussion on PTHrP).
Cancer Cachexia-Associated Anorexia
Food intake is regulated at multiple sites in the body, including the cortical areas, hypothalamus, and brainstem. Research on the neurobiological aspects of cancer cachexia has been focused mainly on the hypothalamic melanocortin system (13, 14), which integrates signals about nutritional status and the perception of food (95, 96) and consists of two distinct subpopulations of neurons in the arcuate nucleus (ARC): the agouti-related peptide (AgRP) and pro-opiomelanocortin (POMC)-expressing neurons (95), whose function is to project to and modulate the activity of neurons in the paraventricular nucleus (PVN) by means of the melanocortin 4 receptor. A proteolytic cleavage product of POMC, α-melanocyte-stimulating hormone stimulates, while AgRP inhibits, these neurons; in combination, they determine all aspects of food intake (97). The activity of AgRP and POMC neurons is determined by humoral inputs, including anorexigenic leptin and insulin, orexigenic ghrelin, and satiety-mediating hormones, such as cholecystokinin.
Neuronal histamine signaling is integral to the regulation of food intake (14, 18). The active sites of histamine in this context are the hypothalamic ventromedial nucleus (VMN) and the PVN, but not the lateral hypothalamus, dorsomedial nucleus, or POA. Indeed, the ARC, VMN, and PVN receive histaminergic fibers and markedly express H1R (19). H1R is expressed in hypothalamic satiety centers and mediates hypothalamic AMP-activated protein kinase (AMPK) activation, while the inhibitory H3R inhibits hypothalamic hunger centers (98, 99). The spatial specificity of histaminergic neuron signaling with respect to their biological functions neuroanatomically mirrors the different ways that cancer cachexia manifests, with some patients developing taste or smell distortions, many patients developing anorexia, and some not experiencing these symptoms.
Increased Sensitivity to Leptin, Decreased Sensitivity to Ghrelin, and the Beneficial Effect of Anamorelin in Cancer Cachexia
One of the functions of neuronal histamine signaling is mediating factors that regulate food intake, such as leptin, orexin, and glucagon-like peptide 1 (19, 100). Histamine signaling being downstream of the anorexigenic hormones leptin and amylin suggests a mechanism of action in cancer-associated anorexia (101–103). We surmise that excessive histamine signaling can overrule humoral signals regulating food intake, thereby inducing and perpetuating an appetite-suppressed state. Plasma leptin correlates with fat mass and cell size and is considered to be an adiposity sensor (104). Cachectic cancer patients present with hypoleptinemia, which, contrary to what one would expect, fails to promote food intake (104). Tonic histamine signaling might account for the apparent increase of sensitivity to anorexigenic factors, above all leptin, associated with cachexia (6) (Figure 1).
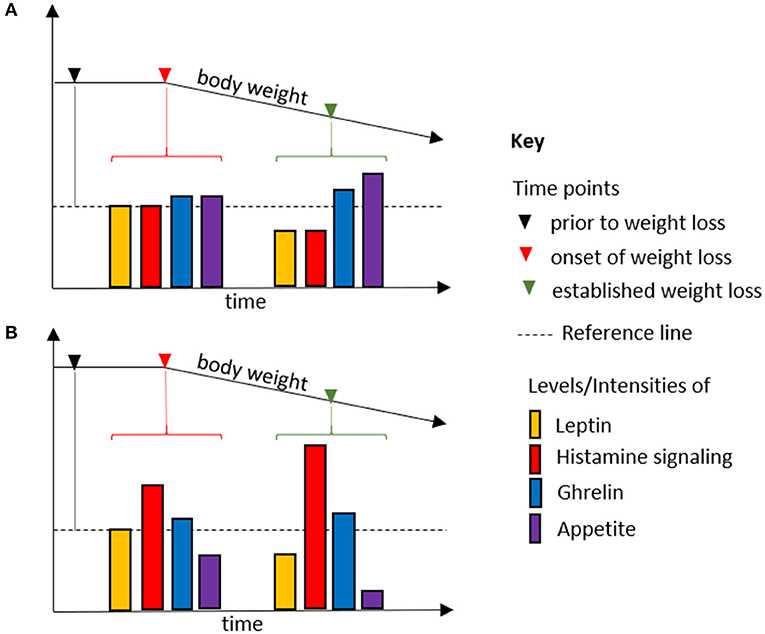
Figure 1. Body weight loss due to fasting (A) and in cancer cachexia (B). Histamine signaling is downstream and an integral part of the anorexigenic effect of leptin. As an adiposity sensor, leptin plasma levels, and thus histamine signaling, mirror the course of weight loss during fasting and in cancer cachexia. Analogously, average ghrelin levels are increased in (A,B). However, whereas histamine signaling is determined by leptin levels in fasting, it is uncoupled from it in cancer cachexia. Thus, relative levels of leptin and ghrelin promote appetite in fasting, but not in cancer cachexia. Increased histamine signaling overrules the hormonal regulatory mechanisms in food intake, allowing anorexia to be established as the default state.
Cancer cachexia also has been associated with a decreased sensitivity to orexigenic factors. Since food intake is determined by the balance of orexigenic and anorexigenic factors, a shift toward its tonic suppression would manifest in an apparent loss of sensitivity to orexigenic stimuli. Indeed, plasma levels of ghrelin are increased in cancer patients, but fail to promote food intake, a condition referred to as ghrelin resistance (6). Ghrelin is not directly involved in histamine signaling (105), which might explain why the ghrelin analog anamorelin has proved promising in the treatment of cancer cachexia in advanced clinical studies (106). If ghrelin's orexigenic effect were to require reduced neuronal histamine signaling, this therapeutic approach would not work.
In summary, histamine signaling can induce the main symptoms of cancer cachexia, increased REE and anorexia. In normal physiology, histamine is both a neuromodulator and a neuroinflammation mediator. Considering that inflammation is local rather than systemic in the early stages of cancer and that anti-inflammatory intervention cannot completely abolish wasting once systemic inflammation has been established in advanced cancer progression, our considerations so far support the view that neuronal histaminergic system is involved in the development of cachexia in cancer patients. This mechanism can provide a basic tonus toward wasting, which can be further exacerbated by tumorkines and (systemic and central) inflammation during cancer progression, with this combination of variables contributing to the different manifestations of cachexia in cancer patients. Moreover, the neuronal histaminergic system by itself provides a physiological basis for symptomatic heterogeneity, particularly with regard to relatively minor symptoms.
Minor Symptoms of Cancer Cachexia
Diversification of Histaminergic Neurons and Symptomatic Heterogeneity in Cancer Cachexia
Histaminergic neurons are located within the TMN, which is interconnected with almost every part of the brain and the spinal cord. Specifically, the anterior part of the hypothalamus, including the POA, is particularly rich in histaminergic fibers (17). Histaminergic neurons also project to brain areas regulating food intake. Moreover, TMN is innervated by neurons of the brainstem including serotoninergic cell groups, which have already been associated with cachexia-associated anorexia (12, 13, 107). Histaminergic neurons are organized into distinct pathways in terms of origin, terminal projections, and function (108).
The occurrence and severity of minor symptoms vary between individual cancer patients (2). Moreover, the sheer range and heterogeneity of disorders associated with aberrant histaminergic neuron signaling—including hypersomnia, cognitive dysfunction, and Parkinson's disease—suggests the involvement of distinct histaminergic neuronal populations (19, 21). Furthermore, histaminergic neurons can act as stress sensors integrating and responding to a variety of different inputs, including from the PSNS. Recent studies have demonstrated the existence of distinct subpopulations of histaminergic neurons, which specifically target distinct regions of the brain for histamine release (20, 109), and differ in their responsivity to stressors such as restraint, metabolic, hypercapnic, and food deprivation stress, as well as in their sensitivity to neurotransmitters and cannabinoid receptor 1 agonists (110–116). For instance, histaminergic neurons can be divided into two subpopulations with respect to their sensitivity to GABAergic inputs from the ventrolateral POA (VLP) (108), which largely determines sleep and wakefulness (21, 117).
Notably, and concordant with our hypothesis, histaminergic neurons provide the neurobiological prerequisites to induce various clinical manifestations of cancer cachexia.
Change in the Perception of Taste and Smell
Cancer patients commonly report changes in the perception of taste and smell; the severity of these symptoms correlates with weight loss, reduced food intake, and lower quality of life scores (118, 119). These changes in perception not only are a side effect of chemotherapy and radiotherapy, but also occur in a significant proportion of treatment-naive patients with solid tumors, where they are significantly associated with fatigue and early satiety (120–122). Hypothalamic histamine release is increased by adverse taste stimuli such as saltiness or bitterness, and decreased by stimuli mediating sweetness (123, 124). Normally, taste perception is mediated by the chorda tympani nerve, which is excited by perception of the physical substance (123). It appears that aberrant histamine signaling can invert this mechanism by rendering the default state of food perception to be unpleasant. Indeed, cancer patients complain about a bad taste in the mouth and report taste distortions and occasionally an increased sensitivity to odors (119). There is scant evidence linking smell perception and histamine signaling, but some research findings show that the scent of grapefruit and lavender oil affects autonomic neurotransmission and blood pressure by means of histaminergic neurons (125). It remains to be proven, but would not be unreasonable to presume that enhanced histamine signaling might also influence the perception of smell, not just taste.
Sleep Disturbances
Cancer patients occasionally experience problems sleeping (2). Neuronal histamine signaling is crucially involved in the regulation of sleep and wakefulness (20). Histaminergic neurons are part of an extensive neuronal network; they include orexinergic neurons, which are located in the perifornical area in the posterior lateral hypothalamus, locally and functionally closely associated with the TMN. Hypothalamic orexins are involved not only in leptin signaling, but also in regulating sleeping and waking (20, 126). The lack of orexinergic neurons induces narcolepsy. Histaminergic neurons are highly active during waking and sluggish or dormant during sleep. Their rate of firing, and thus the extent of histamine release, is regulated by GABAergic neurons of the VLP, which project to the TMN and are complementarily active during slow-wave sleep, the deepest phase of sleep.
The physiological role of histamine as an integral part of orexin signaling points to its possible role in the sleep disturbances of cancer patients and links it to leptin signaling, and thus anorexia and the circadian rhythmicity of NST in cancer cachexia.
Implications of the Role of the Neuronal Histaminergic System as Tumor Sensor
The physiological function of neuronal histamine signaling and the correlation of tumor activity and BAT activation are consistent with the idea that neuronal histamine signaling could be tumor-triggered, gradually increasing as cancer progresses from the early or subclinical to the advanced stages. The three most significant features of the neuronal histaminergic system which would imply that histaminergic neurons in the TMN might be critically involved in sensing the tumor and thus link tumor growth to cachexia progression are: (1) histaminergic neurons are stress sensors, which (2) differ in their sensitivity to stressors or stress intensities, and (3) provide functional specificity.
The neuronal histaminergic system represents an alternative, inflammation-independent mechanism of how tumors can be sensed and how they could induce changes at the systemic level (13) (Figure 2). This mechanism is presumed to be based on tumor-to-brain signaling mediated by the parasympathetic nervous system (PSNS), as has been postulated for serotoninergic signaling (12). The vagus nerve conveys information from visceral regions to the brain (127), including, as we and others suspect, from subclinical tumors (12, 128). An analogous signaling path has been implicated in heart failure, whereby vagal afferent signaling is induced by pressure-sensitive atrial volume receptors in the wall of the right atrium. This signal is then conveyed to the PVN, which responds by increasing sympathetic tone, resulting in heart failure (129).
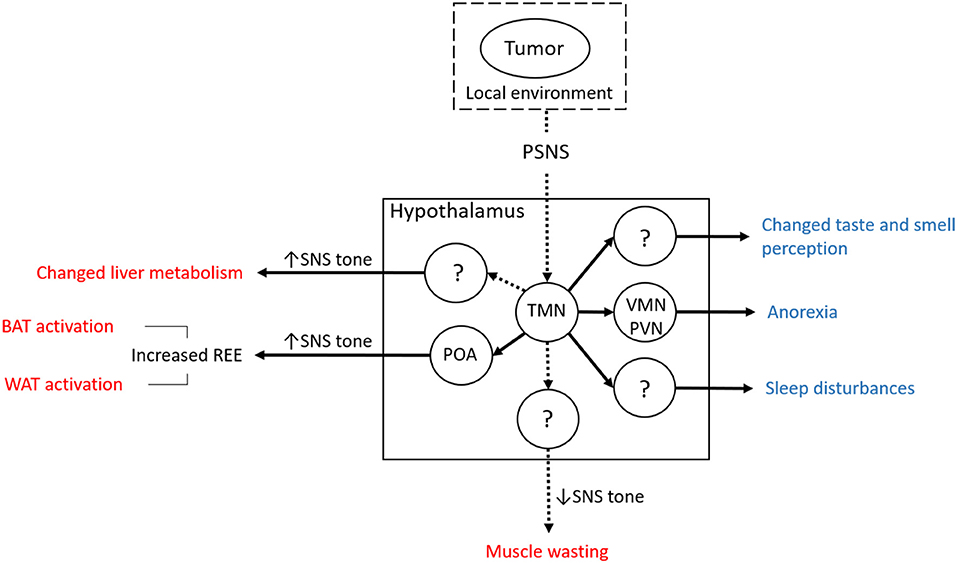
Figure 2. Overview of the hypothesis concerning the involvement of the neuronal part of the histaminergic system in cancer cachexia. Muscle wasting is suggested to involve the loss of SNS tone. The mechanism and the time of onset in the course of cancer cachexia is still not clear, although the switch from increased to reduced neuronal histamine signaling might delineate the late phase (muscle wasting). The notion of histaminergic neurons in the TMN mediating cancer cachexia is consistent with a mechanism involving the sensing of tumor-derived signals (including those reflecting changes in its local tissue environment via the PSNS). Since this mechanism does not rely on systemic inflammation as a trigger of neuroinflammation, it represents an alternative route for tumors to affect systemic metabolism and cause cancer cachexia (BAT, brown adipose tissue; POA, preoptic area; PSNS, parasympathetic nervous system; PVN, paraventricular nucleus; REE, resting energy expenditure; SNS, sympathetic nervous system; TMN, tuberomamillary nucleus; VMN, ventromedial nucleus; WAT, white adipose tissue). Solid lines originating from the TMN indicate that connections have already been shown although in other contexts than cancer cachexia. Dashed lines indicate that while there is no direct evidence, it can be inferred indirectly. Solid lines between circles (hypothalamic nuclei and the periphery, BAT and WAT) indicate that increased SNS tone has been shown to be decisive in the respective physiological context. Central effects of tumor-induced changes of neuronal histamine signaling are shown in blue font, peripheral effects in red.
Histaminergic neurons are responsive to a variety of peripheral signals. For instance, oleoylethanolamide (OEA) is released by enterocytes in response to fat intake and mediates satiety via hypothalamic mechanisms (130, 131). OEA activates sensory fibers of the vagus nerve, which projects to the nucleus tractus solitarii (NTS), where the signal is relayed toward the PVN. Noradrenergic NTS-PVN projections activate the hypothalamic oxytocin system, which mediates the anorectic effect of OEA (132). Moreover, OEA induces expression of c-Fos, a marker for neuronal activation, in a subgroup of TMN neurons, thus betokening the concept of neuronal diversification. Current research supports the idea that OEA might increase histamine levels in the PVN, which in turn stimulates oxytocin release through H1R (133).
We posit that the neuronal histaminergic system might be crucially involved in PSNS-mediated tumor sensing and in mediating cancer cachexia, in that it responds to afferent PSNS signals, which in turn complement efferent neuronal mechanisms influencing tumor biology (134). Based on the evidence, the vagus nerve might convey the physical, biochemical, and/or inflammatory traits of the tumor and/or its environment (134) to a subpopulation of histaminergic neurons, which primarily activate NST via the SNS. In the course of cancer progression, more and more functionally distinct histaminergic neurons, specifically those projecting to the PVN and mediating anorexia, become activated, as well as secondary mechanisms involved in modulating cancer cachexia. Following this line of reasoning would suggest that the varying prevalence of cancer cachexia with respect to tumor entities reflects organ-specific differences in parasympathetic innervation (135).
The Liver and Central Histamine Signaling
Evidence for the involvement of central histamine signaling in the dysregulation of the hepatic metabolism in cancer cachexia is still lacking. Nevertheless, it might be inferred for several reasons:
1) Cancer cachexia is associated with the dysregulation of the hepatic glucose and lipid metabolism (6, 8), which is under the control of the autonomic nervous system, including the SNS under normal conditions (136, 137);
2) Metabolic and humoral signals from the periphery converge with brain-derived inputs in cachexia-relevant brain areas such as the NTS and PVN, where histamine exerts its normal function;
3) Together, BAT, WAT, and the liver constitute a functional unit in chronic NST under normal conditions (65, 138–140).
Point 3 is especially persuasive in support of the histaminergic neuron signaling playing a role in increasing the sympathetic tone in the liver.
When Sympathetic Tone Breaks Down—Muscle Wasting in Cancer Cachexia
The impact of histamine signaling on muscle physiology and muscle wasting in cancer cachexia has not been studied yet. However, skeletal muscle is the effector organ of ST, which is activated when NST is insufficient to maintain body temperature (43, 141). Both ST and NST mechanisms are triggered by neurons of the POA and involve SNS activation, which provides the link between hypothalamic histamine signaling and noradrenergic signaling at the periphery. Sarcopenic obesity being associated with both reduced neuronal sensitivity to histamine and muscle atrophy underscores the importance of sympathetic tone, i.e., noradrenergic signaling, for skeletal muscle integrity and implicates histamine signaling (142).
The CNS-SNS skeletal muscle axis plays an important role in muscle wasting in cancer cachexia. ACT-ONE, a recent multicenter phase II study of cachectic patients with colorectal cancer and NSCLC, proved the impact of catecholamine-mediated signaling on muscle wasting and loss of lean body mass (143, 144). Under administration of different doses of espindolol8, study participants' showed significant increases in body weight and, at the higher dose tested, lean body mass with no effect on fat mass (143).
Espindolol and similar β2-adrenoceptor agonists have demonstrated beneficial effects on muscle wasting (145). In an animal model of age-related muscle wasting, espindolol increased muscle mass, and decreased fat mass (146). In another study, espindolol increased grip strength, a measure of skeletal muscle function, in cachectic cancer patients (143). Non-specific beta blockers have been shown to improve the survival rates of ovarian cancer patients (147) and to prevent wasting of skeletal and heart muscles in animal models (148, 149). In sum, both pharmacologic stimulation of β2-adrenoceptor and exercise have proven to be beneficial as part of a multimodal approach for the treatment of cancer cachexia (42).
Khan et al. recently described a mechanism which links β2-adrenoceptor-mediated signaling to muscle function (150), which underscores the likelihood of its involvement in the muscle wasting experienced by cachectic cancer patients. Their research showed that the integrity of neuromuscular junctions depends on their innervation by sympathetic neurons (151, 152). Indeed, signaling via cAMP/PKA, components of the signaling pathway of noradrenaline, have been found to be important for synapse maintenance (153–157). Sympathetic nerve stimulation has been shown to promote the importation of PPARGC1A, an activator of mitochondrial biogenesis, into myonuclei (158, 159), which underlies the positive effect of muscle training on health promotion and disease mitigation (160, 161).
Increased sympathetic tone mediates cachexia-related alterations of adipose tissue and liver metabolism, whereas decreased sympathetic tone is associated with muscle wasting. Changes in sympathetic tone might accompany distinct stages of cancer cachexia progression, with the first stage characterized by increased sympathetic tone inducing NST, WAT browning, and increased lipolysis and causing adipose tissue depletion; and the second stage characterized by loss of sympathetic tone causing muscle wasting. This would signify (1) that adipose tissue depletion precedes muscle wasting, which has indeed been found in cachectic cancer patients (162, 163), and (2) that histamine signaling, be it of neuronal or mast cell origin, is abolished in the second stage of cancer cachexia, which is supported by research showing that reduced neuronal histamine sensitivity has been implicated in obesity, which in turn is associated with sarcopenia (i.e., obese sarcopenia) (17, 142). The mechanism could be that reduced histamine signaling causes reduced SNS tone in muscles, which could engender the disintegration of neuromuscular junctions, thereby the loss of motor-neuronal inputs, leading to further functional and structural muscle loss.
Do Histaminergic and Serotoninergic Neurons Team up in Cancer Cachexia?
Serotonin is associated with cancer-associated anorexia (12, 13, 107). Tumor resection indeed brings about the normalization of serotonin signaling with respect to serotonin levels and receptor abundance, while intra-hypothalamic injection of a serotonin antagonist exerts an anti-anorexigenic effect in tumor-bearing rats (13). Histaminergic neurons in the TMN and serotonergic neurons in the raphe nuclei of the brain stem are intensely interconnected (21), with serotonergic neurons directly excited by histamine via H1R activation and suppressed via H2R (21). In turn, the TMN receives projections from aminergic nuclei, among others, and serotonin excites histaminergic neurons via 5-hydroxytryptamine receptor 2C (21). Serotonin and histamine appear to exert complementary effects on food intake, with serotonin mediating satiety and histamine reducing appetite (13, 17). By establishing a positive feedback loop between aminergic neuron populations, the feature of mutual excitation points to a possible mechanism of dysregulation in cancer cachexia, whereby the activation of histaminergic neurons might trigger the activation of serotoninergic neurons. This sequence of events seems all the more plausible when considering that histaminergic neurons can act as stress sensors, manifesting many clinical symptoms of cancer cachexia, in contrast to serotonin, which primarily affects food intake.
Caveat: Pharmacologically Targeting the Central Histaminergic System to Treat Cancer Cachexia?
Histamine signaling has been implicated in tumor biology and antihistaminic treatment9 is now viewed as an auspicious approach in cancer therapy (14, 164, 165). Still, this promising new treatment must be approached with caution, as pharmacologically interfering with brain histamine signaling may lead to undesirable side effects due to histamine's physiological role as a neuromodulator of a variety of neurotransmitters (19–21, 93). Nonetheless, in a positive development, mirtazapine—an anti-depressant which not only targets noradrenergic and serotonergic signaling but also interferes with histamine signaling—counteracted chemically induced cachexia in mice and, in combination with olanzapine, an antagonist of several neurotransmitter receptors including H1R, yielded good anti-nausea results in a phase II clinical trial (166–168). Cyproheptadine, an antagonist of both serotonin and histamine signaling, in combination with megestrol acetate to counteract skeletal muscle wasting, demonstrated an appetite-enhancing effect in pediatric cancer patients (169), but failed to abate progressive weight loss in advanced cancer patients (170–172). In terms of isolating the precise mechanism of action of each individual drug, however, it is not possible to infer the specific role of histamine signaling in this context.
Discussion
For this work, we scoured publications to gather evidence to support (or disprove) our hypothesis that the neuronal histaminergic system might be involved in cancer cachexia.
Although the data skew positively in support of our hypothesis, we want to anticipate two presumably major objections:
1) It is likely that one or the other line of our argumentation is not commonly accepted. This is certainly true for BAT activation as a mechanism of REE. In this particular case, the controversy on that topic is part of our argumentation and even supports our hypothesis.
2) The issues are not covered appropriately in terms of depth and/or detail and some aspects, especially empirical evidence, are missing. What we aimed to do in this paper is outline the conceptual framework around our hypothesis and its implications, and we hope we have accomplished this in a manner to engage the reader and spark discussion as to the merits of our line of reasoning.
The topic requires at least a high-level understanding of a variety of different disciplines, including oncology, endocrinology, and neurophysiology, which makes it a Herculean task to survey every possibly relevant finding. Fortunately, this was not necessary to draft a useful conceptual framework based on testable assumptions, since the selection of issues was mainly dictated by the line of inquiry. The depth of coverage of each topic, in particular the basic information, was discretionary and reflects the authors' personal choice.
Conclusion
We would like to conclude by summarizing the cornerstones of our hypothesis. We hypothesize that hypothalamic histaminergic neurons are stimulated early in cancer development via the PSNS sensing tumor traits. Histamine release in the POA primarily activates BAT, which is a highly energy-demanding mechanism. Chronic activation leads to browning of WAT, which further enhances thermogenic potential. In this view, the tumor hijacks the neuronal circuit and parts of the effector system of adaptive thermogenesis. In this context, chronicity refers to intermittent and/or low-grade activation by the SNS. Anorexia, as well as minor symptoms of cachexia, such as taste and smell distortions and sleep disturbances, can be ascribed to aberrant histamine signaling. These functions are exerted by different structures of the brain, which are targeted by distinct histaminergic neuron populations.
As cancer advances, further mechanisms such as systemic inflammation10, neuroinflammation, or tumorkines might gain importance and promote cachexia progression. Histaminergic neurons remain active as long as the tumor is present, although their significance for the manifestation of cachexia might lessen. Thus, patients with unresectable cachexia-inducing tumors lose weight even when receiving the optimal multimodal therapy. We hypothesize that in late-stage cachexia, the sympathetic tone in skeletal muscle breaks down, which might be caused by a reduction in histamine signaling or by the interference of other mechanisms in cancer cachexia. Histamine signaling thus might delineate distinct stages of cachexia progression, with the early phase marked by increased histamine signaling, increased sympathetic tone and adipose tissue depletion, and the late phase characterized by reduced histamine signaling, decreased sympathetic tone, and muscle wasting. A possible mechanism of muscle wasting in cancer cachexia mimics that of sarcopenia in obesity, whereby the sympathetic tone in muscle tissue is reduced, leading to the disintegration of neuromuscular junctions. The loss of histamine sensitivity of distinct neurons in obesity might be another candidate for a mechanism behind muscle wasting in cachexia. Pharmacologically targeting neuronal histamine signaling might result in intolerable side effects due to its physiological role as a neuromodulator of a variety of fast-acting neurotransmitters.
Our hypothesis provides a framework for the mechanisms that might be driving cachexia. There is no pharmacological treatment for the histaminergic system currently available, so for now this represents an attempt to better understand this seemingly intractable side effect of cancer. However, if it should prove to be true that energy-demanding mechanisms are activated early in cancer development, it might be possible to exploit the resultant effects, such as a decrease in cholesterol in the absence of substantiating lifestyle changes, for the detection of early malignancy and possible implementation as a screening tool.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Author Contributions
HZ, EZ-T, and MP equally contributed to the conceptualization of the hypothesis presented here as well as its implications for cancer diagnosis. HZ wrote the manuscript, which was discussed with and proof-read by EZ-T and MP.
Funding
All sources of funding for this work have been submitted. The funder is NÖ Forschungs- und Bildungsges.mb.H (Grant number LSC14-021), an official institution dedicated to fund research in Lower Austria.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AgRP, agouti-related peptide; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; α-MSH, α-melanocyte-stimulating hormone; ANS, autonomous nervous system; ARC, arcuate nucleus; ATGL, adipose triglyceride lipase; ATP, adenosine triphosphate; BAT, brown adipose tissue; BMI, body mass index; cAMP, cyclic AMP; CCK, cholecystokinin; c-Fos, Fos proto-oncogene; CNS, central nervous system; DMN, dorsomedial nucleus; 18F-FDG-PET-CT, 18F-fluorodeoxyglucose positron emission tomography/computer tomography; FFA, free fatty acids; GABA, gamma-aminobutyric acid; GLP-1, glucagon-like peptide 1; H1R, H2R, H3R, histamine receptor H1, histamine receptor H2, histamine receptor H3; HSL, hormone-sensitive lipase; 5-HT2CR, 5-hydroxytryptamine receptor 2C; IL-6, Interleukin 6; LH, lateral hypothalamus; MC4R, melanocortin 4 receptor; MnPOA, median POA; MPOA, medial POA; NMJ, neuromuscular junction; NSCLC, non-small-cell lung cancer; NST, non-shivering thermogenesis; NTS, nucleus tractus solitarii; OEA, oleoylethanolamide; PTHrP, parathyroid hormone-related protein; PGE2, prostaglandin E2; PKA, protein kinase A; POA, preoptic area; POMC, pro-opiomelanocortin; PSNS, parasympathetic nervous system; PVN, paraventricular nucleus; REE, resting energy expenditure; rRPA, rostral raphe pallidus; ST, shivering thermogenesis; SNS, sympathetic nervous system; TAG, triacylglycerol; TMN, tuberomamillary nucleus; UCP, uncoupling protein; VLP, ventrolateral POA; VMN, ventromedial nucleus; WAT, white adipose tissue; ZAG, zinc-alpha2-glycoprotein.
Footnotes
1. ^This hypothesis grew out of research on the role of histamine in energy homeostasis done by Dr. Iustin V. Tabarean. In the abstract of his paper “Histamine receptor signaling in energy homeostasis”, Neuropharmacology, 2016 Jul; 106: 13–19, Tabarean wrote: “Histamine modulates several aspects of energy homeostasis. By activating histamine receptors in the hypothalamus the bioamine influences thermoregulation, its circadian rhythm, energy expenditure and feeding. These actions are brought about by activation of different histamine receptors and/or the recruitment of distinct neural pathways.” Wondering if this could also be a description of cancer cachexia in neurobiological terms, Zwickl began scouring the literature, pondering possible mechanisms involved, checking plausibility, looking at phenomena where distinct mechanisms have already been shown, and eventually coming up with what is being presented in this paper.
2. ^There is increasing evidence that histamine signaling in the hypothalamus is involved in the development of obesity (17).
3. ^BAT is detected via its uptake of labeled glucose. If this occurs at low levels, BAT will not be detected by 18F-FDG-PET-CT.
4. ^Either 2 h/day for 6 weeks at 17°C; or up to 6 h/day at 15–16°C for 10 days.
5. ^SNS-associated noradrenergic signaling also has been implicated in cancer-associated immunosuppression (67).
6. ^And citations therein.
7. ^Rats are nocturnal, so their circadian rhythmicity will be the opposite of humans'.
8. ^Espindolol is a non-specific beta blocker with pro-anabolic, anti-catabolic, central appetite-stimulating, and thermogenesis-reducing effects via its β2-adrenoceptor-stimulating and β1-adrenoceptor- and 5-HT1α-receptor-blocking activity (143, 144).
9. ^Peripheral anti-histaminic treatment, not CNS anti-histaminic treatment.
10. ^Please note: We are not implying that inflammation is not present in the subclinical stage. It may very well be present at a low-grade level. As cancer progresses, it takes on an increasingly prominent role.
References
1. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. (2011) 12:489–95. doi: 10.1016/S1470-2045(10)70218-7
2. Argiles JM, Lopez-Soriano FJ, Toledo M, Betancourt A, Serpe R, Busquets S. The cachexia score (CASCO): a new tool for staging cachectic cancer patients. J Cachexia Sarcopenia Muscle. (2011) 2:87–93. doi: 10.1007/s13539-011-0027-5
3. Kalantar-Zadeh K, Rhee C, Sim JJ, Stenvinkel P, Anker SD, Kovesdy CP. Why cachexia kills: examining the causality of poor outcomes in wasting conditions. J Cachexia Sarcopenia Muscle. (2013) 4:89–94. doi: 10.1007/s13539-013-0111-0
4. Bosaeus I, Daneryd P, Svanberg E, Lundholm K. Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients. Int J Cancer. (2001) 93:380–3. doi: 10.1002/ijc.1332
5. Laviano A, Meguid MM, Inui A, Muscaritoli M, Rossi-Fanelli F. Therapy insight: Cancer anorexia-cachexia syndrome–when all you can eat is yourself. Nat Clin Pract Oncol. (2005) 2:158–65. doi: 10.1038/ncponc0112
6. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. (2016) 5:e200. doi: 10.1038/oncsis.2016.3
7. Tisdale MJ Mechanisms of cancer cachexia. Physiol Rev. (2009) 89:381–410. doi: 10.1152/physrev.00016.2008
8. Petruzzelli M, Wagner EF. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. (2016) 30:489–501. doi: 10.1101/gad.276733.115
9. Fearon KC, Voss AC, Hustead DS, G. Cancer Cachexia Study, Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr. (2006) 83:1345–50. doi: 10.1093/ajcn/83.6.1345
10. Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J Cachexia Sarcopenia Muscle. (2015) 6:317–24. doi: 10.1002/jcsm.12033
11. Pfitzenmaier J, Vessella R, Higano CS, Noteboom JL, Wallace D Jr, Corey E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer. (2003) 97:1211–6. doi: 10.1002/cncr.11178
12. Laviano A, Inui A, Marks DL, Meguid MM, Pichard C, Rossi Fanelli F, et al. Neural control of the anorexia-cachexia syndrome. Am J Physiol Endocrinol Metab. (2008) 295:E1000–8. doi: 10.1152/ajpendo.90252.2008
13. Molfino A, Gioia G, Rossi Fanelli F, Laviano A. Contribution of neuroinflammation to the pathogenesis of cancer cachexia. Mediators Inflamm. (2015) 2015:801685. doi: 10.1155/2015/801685
14. Grossberg AJ, Scarlett JM, Marks DL. Hypothalamic mechanisms in cachexia. Physiol Behav. (2010) 100:478–89. doi: 10.1016/j.physbeh.2010.03.011
15. Fearon K Cachexia: treat wasting illness on multiple fronts. Nature. (2016) 529:156. doi: 10.1038/529156b
16. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol. (2013) 10:90–9. doi: 10.1038/nrclinonc.2012.209
17. Tabarean IV. Histamine receptor signaling in energy homeostasis. Neuropharmacology. (2016) 106:13–9. doi: 10.1016/j.neuropharm.2015.04.011
18. Provensi G, Blandina P, Passani MB The histaminergic system as a target for the prevention of obesity and metabolic syndrome. Neuropharmacology. (2016) 106:3–12. doi: 10.1016/j.neuropharm.2015.07.002
19. Panula P, Nuutinen S. The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci. (2013) 14:472–87. doi: 10.1038/nrn3526
20. Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. (2003) 4:121–30. doi: 10.1038/nrn1034
21. Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. (2008) 88:1183–241. doi: 10.1152/physrev.00043.2007
22. Argiles JM, Fontes-Oliveira CC, Toledo M, Lopez-Soriano FJ, Busquets S. Cachexia: a problem of energetic inefficiency. J Cachexia Sarcopenia Muscle. (2014) 5:279–86. doi: 10.1007/s13539-014-0154-x
23. Tsoli M, Moore M, Burg D, Painter A, Taylor R, Lockie SH, et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. (2012) 72:4372–82. doi: 10.1158/0008-5472.CAN-11-3536
24. Bianchi A, Bruce J, Cooper AL, Childs C, Kohli M, Morris ID, et al. Increased brown adipose tissue activity in children with malignant disease. Horm Metab Res. (1989) 21:640–1. doi: 10.1055/s-2007-1009308
25. Shellock FG, Riedinger MS, Fishbein MC Brown adipose tissue in cancer patients: possible cause of cancer-induced cachexia. J Cancer Res Clin Oncol. (1986) 111:82–5. doi: 10.1007/BF00402783
26. Beijer E, Schoenmakers J, Vijgen G, Kessels F, Dingemans AM, Schrauwen P, et al. A role of active brown adipose tissue in cancer cachexia? Oncol Rev. (2012) 6:e11. doi: 10.4081/oncol.2012.e11
27. Dong M, Lin J, Lim W, Jin W, Lee HJ. Role of brown adipose tissue in metabolic syndrome, aging, and cancer cachexia. Front Med. (2018) 12:130–8. doi: 10.1007/s11684-017-0555-2
28. Oudart H, Calgari C, Andriamampandry M, Le Maho Y, Malan A. Stimulation of brown adipose tissue activity in tumor-bearing rats. Can J Physiol Pharmacol. (1995) 73:1625–31. doi: 10.1139/y95-724
29. Roe S, Cooper AL, Morris ID, Rothwell NJ. Mechanisms of cachexia induced by T-cell leukemia in the rat. Metabolism. (1996) 45:645–51. doi: 10.1016/S0026-0495(96)90037-2
30. Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. (2008) 9:764–75. doi: 10.1038/nrg2430
31. Steinberg JD, Vogel W, Vegt E. Factors influencing brown fat activation in FDG PET/CT: a retrospective analysis of 15,000+ cases. Br J Radiol. (2017) 90:20170093. doi: 10.1259/bjr.20170093
32. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. (2009) 360:1518–25. doi: 10.1056/NEJMoa0808949
33. Lee P, Greenfield JR, Ho KK, Fulham MJ. A critical appraisal of the prevalence and metabolic significance of brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. (2010) 299:E601–6. doi: 10.1152/ajpendo.00298.2010
34. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. (2014) 513:100–4. doi: 10.1038/nature13528
35. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. (2014) 20:433–47. doi: 10.1016/j.cmet.2014.06.011
36. Kir S, Spiegelman BM. Cachexia & brown fat: a burning issue in cancer. Trends Cancer. (2016) 2:461–3. doi: 10.1016/j.trecan.2016.07.005
37. Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature. (2014) 510:76–83. doi: 10.1038/nature13477
38. Mourtzakis M, Prado CM, Lieffers JR, Reiman T, McCargar LJ, Baracos VE. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl Physiol Nutr Metab. (2008) 33:997–1006. doi: 10.1139/H08-075
39. Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, Baracos VE. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole-body energy demands. Am J Clin Nutr. (2009) 89:1173–9. doi: 10.3945/ajcn.2008.27273
40. Prado CM, Lieffers JR, Bergsten G, Mourtzakis M, Baracos VE, Reiman T, et al. Dietary patterns of patients with advanced lung or colorectal cancer. Can J Diet Pract Res. (2012) 73:e298–303. doi: 10.3148/73.4.2012.e298
41. Hong N, Yoon HJ, Lee YH, Kim HR, Lee BW, Rhee Y, et al. Serum PTHrP predicts weight loss in cancer patients independent of hypercalcemia, inflammation, and tumor burden. J Clin Endocrinol Metab. (2016) 101:1207–14. doi: 10.1210/jc.2015-3785
42. Argiles JM, Lopez-Soriano FJ, Stemmler B, Busquets S. Novel targeted therapies for cancer cachexia. Biochem J. (2017) 474:2663–78. doi: 10.1042/BCJ20170032
43. Morrison SF, Nakamura K. Central neural pathways for thermoregulation. Front Biosci. (2011) 16:74–104. doi: 10.2741/3677
45. Walden TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am J Physiol Endocrinol Metab. (2012) 302:E19–31. doi: 10.1152/ajpendo.00249.2011
46. Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol. (2013) 15:659–67. doi: 10.1038/ncb2740
47. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. (2013) 19:1338–44. doi: 10.1038/nm.3324
48. van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. (2013) 123:3395–403. doi: 10.1172/JCI68993
49. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol. (2014) 10:24–36. doi: 10.1038/nrendo.2013.204
50. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. (2013) 123:3404–8. doi: 10.1172/JCI67803
51. Scheja L, Heeren J. Metabolic interplay between white, beige, brown adipocytes and the liver. J Hepatol. (2016) 64:1176–86. doi: 10.1016/j.jhep.2016.01.025
52. Cohen P, Spiegelman BM. Brown and beige fat: molecular parts of a thermogenic machine. Diabetes. (2015) 64:2346–51. doi: 10.2337/db15-0318
53. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. (2011) 17:200–5. doi: 10.1038/nm.2297
54. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. (2009) 58:1526–31. doi: 10.2337/db09-0530
55. Arner P, Bernard S, Salehpour M, Possnert G, Liebl J, Steier P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. (2011) 478:110–3. doi: 10.1038/nature10426
56. Das SK, Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med. (2013) 19:292–301. doi: 10.1016/j.molmed.2013.02.006
57. Ryden M, Agustsson T, Laurencikiene J, Britton T, Sjolin E, Isaksson B, et al. Lipolysis–not inflammation, cell death, or lipogenesis–is involved in adipose tissue loss in cancer cachexia. Cancer. (2008) 113:1695–704. doi: 10.1002/cncr.23802
58. Arner P, Langin D. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol Metab. (2014) 25:255–62. doi: 10.1016/j.tem.2014.03.002
59. Bing C, Bao Y, Jenkins J, Sanders P, Manieri M, Cinti S, et al. Zinc-alpha2-glycoprotein, a lipid mobilizing factor, is expressed in adipocytes and is up-regulated in mice with cancer cachexia. Proc Natl Acad Sci USA. (2004) 101:2500–5. doi: 10.1073/pnas.0308647100
60. Taylor DD, Gercel-Taylor C, Jenis LG, Devereux DF. Identification of a human tumor-derived lipolysis-promoting factor. Cancer Res. (1992) 52:829–34.
61. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. (2011) 333:233–8. doi: 10.1126/science.1198973
62. Legaspi A, Jeevanandam M, Starnes HF Jr, Brennan MF. Whole body lipid and energy metabolism in the cancer patient. Metabolism. (1987) 36:958–63. doi: 10.1016/0026-0495(87)90132-6
63. Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, et al. Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer. (2010) 102:1541–8. doi: 10.1038/sj.bjc.6605665
64. Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol Metab. (2008) 19:130–7. doi: 10.1016/j.tem.2007.11.006
65. Bartness TJ, Song CK Thematic review series: adipocyte biology. Sympathetic and sensory innervation of white adipose tissue. J Lipid Res. (2007) 48:1655–72. doi: 10.1194/jlr.R700006-JLR200
66. Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes. (2010) 34(Suppl 1):S36–42. doi: 10.1038/ijo.2010.182
67. Repasky EA, Eng J, Hylander BL. Stress, metabolism and cancer: integrated pathways contributing to immune suppression. Cancer J. (2015) 21:97–103. doi: 10.1097/PPO.0000000000000107
68. Fiorenza AM, Branchi A, Cardena A, Molgora M, Rovellini A, Sommariva D. Serum cholesterol levels in patients with cancer. Relationship with nutritional status. Int J Clin Lab Res. (1996) 26:37–42. doi: 10.1007/BF02644772
69. Siemianowicz K, Gminski J, Stajszczyk M, Wojakowski W, Goss M, Machalski M, et al. Serum total cholesterol and triglycerides levels in patients with lung cancer. Int J Mol Med. (2000) 5:201–5. doi: 10.3892/ijmm.5.2.201
70. Bos SA, Gill CM, Martinez-Salazar EL, Torriani M, Bredella MA Preliminary investigation of brown adipose tissue assessed by PET/CT and cancer activity. Skeletal Radiol. (2019) 48:413–9. doi: 10.1007/s00256-018-3046-x
71. Anker MS, Ebner N, Hildebrandt B, Springer J, Sinn M, Riess H, et al. Resting heart rate is an independent predictor of death in patients with colorectal, pancreatic, and non-small cell lung cancer: results of a prospective cardiovascular long-term study. Eur J Heart Fail. (2016) 18:1524–34. doi: 10.1002/ejhf.670
72. Colboc O, Protais P, Costentin J Histamine-induced rise in core temperature of chloral-anaesthetized rats: mediation by H2-receptors located in the preopticus area of hypothalamus. Neuropharmacology. (1982) 21:45–50. doi: 10.1016/0028-3908(82)90209-X
73. Sethi J, Sanchez-Alavez M, Tabarean IV. Loss of histaminergic modulation of thermoregulation and energy homeostasis in obese mice. Neuroscience. (2012) 217:84–95. doi: 10.1016/j.neuroscience.2012.04.068
74. Lundius EG, Sanchez-Alavez M, Ghochani Y, Klaus J, Tabarean IV. Histamine influences body temperature by acting at H1 and H3 receptors on distinct populations of preoptic neurons. J Neurosci. (2010) 30:4369–81. doi: 10.1523/JNEUROSCI.0378-10.2010
75. Tabarean IV, Sanchez-Alavez M, Sethi J. Mechanism of H(2) histamine receptor dependent modulation of body temperature and neuronal activity in the medial preoptic nucleus. Neuropharmacology. (2012) 63:171–80. doi: 10.1016/j.neuropharm.2012.02.006
76. Hirose M, Hara Y, Matsusaki M. Premedication with famotidine augments core hypothermia during general anesthesia. Anesthesiology. (1995) 83:1179–83. doi: 10.1097/00000542-199512000-00008
77. Toyota H, Dugovic C, Koehl M, Laposky AD, Weber C, Ngo K, et al. Behavioral characterization of mice lacking histamine H(3) receptors. Mol Pharmacol. (2002) 62:389–97. doi: 10.1124/mol.62.2.389
78. Kandasamy SB, Hunt WA. Involvement of histamine H1 and H2 receptors in hypothermia induced by ionizing radiation in guinea pigs. Life Sci. (1988) 42:555–63. doi: 10.1016/0024-3205(88)90097-5
79. Kang M, Yoshimatsu H, Kurokawa M, Ogawa R, Sakata T. Prostaglandin E2 mediates activation of hypothalamic histamine by interleukin-1beta in rats. Proc Soc Exp Biol Med. (1999) 220:88–93. doi: 10.3181/00379727-220-44349
80. Tsuda K, Yoshimatsu H, Niijima A, Chiba S, Okeda T, Sakata T. Hypothalamic histamine neurons activate lipolysis in rat adipose tissue. Exp Biol Med. (2002) 227:208–13. doi: 10.1177/153537020222700309
81. Malmlof K, Zaragoza F, Golozoubova V, Refsgaard HH, Cremers T, Raun K, et al. Influence of a selective histamine H3 receptor antagonist on hypothalamic neural activity, food intake and body weight. Int J Obes. (2005) 29:1402–12. doi: 10.1038/sj.ijo.0803036
82. Tsai JS, Wu CH, Chiu TY, Hu WY, Chen CY. Symptom patterns of advanced cancer patients in a palliative care unit. Palliat Med. (2006) 20:617–22. doi: 10.1177/0269216306071065
83. Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. (2011) 13:125–37. doi: 10.1016/j.cmet.2011.01.006
84. Tu BP, McKnight SL. Metabolic cycles as an underlying basis of biological oscillations. Nat Rev Mol Cell Biol. (2006) 7:696–701. doi: 10.1038/nrm1980
85. Sharma R, Cunningham D, Smith P, Robertson G, Dent O, Clarke SJ. Inflammatory (B) symptoms are independent predictors of myelosuppression from chemotherapy in Non-Hodgkin Lymphoma (NHL) patients–analysis of data from a British National Lymphoma Investigation phase III trial comparing CHOP to PMitCEBO. BMC Cancer. (2009) 9:153. doi: 10.1186/1471-2407-9-153
86. Falconer JS, Fearon KC, Plester CE, Ross JA, Carter DC Cytokines, the acute-phase response, and resting energy expenditure in cachectic patients with pancreatic cancer. Ann Surg. (1994) 219:325–31. doi: 10.1097/00000658-199404000-00001
87. Simons JP, Schols AM, Buurman WA, Wouters EF Weight loss and low body cell mass in males with lung cancer: relationship with systemic inflammation, acute-phase response, resting energy expenditure, and catabolic and anabolic hormones. Clin Sci. (1999) 97:215–23. doi: 10.1042/cs0970215
88. Andrassy RJ, Chwals WJ. Nutritional support of the pediatric oncology patient. Nutrition. (1998) 14:124–9. doi: 10.1016/S0899-9007(97)00225-6
89. Kennedy L, Hodges K, Meng F, Alpini G, Francis H. Histamine and histamine receptor regulation of gastrointestinal cancers. Transl Gastrointest Cancer. (2012) 1:215–227. doi: 10.3978/j.issn.2224-4778.2012.07.02
90. Massari NA, Nicoud MB, Medina VA. Histamine receptors and cancer pharmacology: an update. Br J Pharmacol. (2018). doi: 10.1111/bph.14535
91. Vila-Leahey A, Oldford SA, Marignani PA, Wang J, Haidl ID, Marshall JS. Ranitidine modifies myeloid cell populations and inhibits breast tumor development and spread in mice. Oncoimmunology. (2016) 5:e1151591. doi: 10.1080/2162402X.2016.1151591
92. Dong H, Zhang X, Wang Y, Zhou X, Qian Y, Zhang S. Suppression of brain mast cells degranulation inhibits microglial activation and central nervous system inflammation. Mol Neurobiol. (2017) 54:997–1007. doi: 10.1007/s12035-016-9720-x
93. Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. (2001) 63:637–72. doi: 10.1016/S0301-0082(00)00039-3
94. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
95. Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. (2014) 15:367–78. doi: 10.1038/nrn3745
96. Benarroch EE. Neural control of feeding behavior: overview and clinical correlations. Neurology. (2010) 74:1643–50. doi: 10.1212/WNL.0b013e3181df0a3f
97. Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. (1999) 24:155–63. doi: 10.1016/S0896-6273(00)80829-6
98. Siafis S, Tzachanis D, Samara M, Papazisis G. Antipsychotic drugs: from receptor-binding profiles to metabolic side effects. Curr Neuropharmacol. (2018) 16:1210–23. doi: 10.2174/1570159X15666170630163616
99. Singh R, Bansal Y, Medhi B, Kuhad A. Antipsychotics-induced metabolic alterations: recounting the mechanistic insights, therapeutic targets and pharmacological alternatives. Eur J Pharmacol. (2019) 844:231–40. doi: 10.1016/j.ejphar.2018.12.003
100. Passani MB, Blandina P, Torrealba F. The histamine H3 receptor and eating behavior. J Pharmacol Exp Ther. (2011) 336:24–9. doi: 10.1124/jpet.110.171306
101. Masaki T, Yoshimatsu H, Chiba S, Watanabe T, Sakata T. Central infusion of histamine reduces fat accumulation and upregulates UCP family in leptin-resistant obese mice. Diabetes. (2001) 50:376–84. doi: 10.2337/diabetes.50.2.376
102. Seth R, Terry DE, Parrish B, Bhatt R, Overton JM. Amylin-leptin coadministration stimulates central histaminergic signaling in rats. Brain Res. (2012) 1442:15–24. doi: 10.1016/j.brainres.2011.12.022
103. Davidowa H. Histamine H1-receptors differentially mediate the action of amylin on hypothalamic neurons in control and in overweight rats. Behav Brain Res. (2007) 182:28–35. doi: 10.1016/j.bbr.2007.05.001
104. Ebadi M, Mazurak VC. Potential biomarkers of fat loss as a feature of cancer cachexia. Mediators Inflamm. (2015) 2015:820934. doi: 10.1155/2015/820934
105. Ishizuka T, Nomura S, Hosoda H, Kangawa K, Watanabe T, Yamatodani A. A role of the histaminergic system for the control of feeding by orexigenic peptides. Physiol Behav. (2006) 89:295–300. doi: 10.1016/j.physbeh.2006.05.049
106. Prommer E. Oncology update: anamorelin. Palliat Care. (2017) 10:1178224217726336. doi: 10.1177/1178224217726336
107. Laviano A, Russo M, Freda F, Rossi-Fanelli F. Neurochemical mechanisms for cancer anorexia. Nutrition. (2002) 18:100–5. doi: 10.1016/S0899-9007(01)00727-4
108. Blandina P, Munari L, Provensi G, Passani MB. Histamine neurons in the tuberomamillary nucleus: a whole center or distinct subpopulations? Front Syst Neurosci. (2012) 6:33. doi: 10.3389/fnsys.2012.00033
109. Inagaki N, Yamatodani A, Shinoda K, Shiotani Y, Tohyama M, Watanabe T, et al. The histaminergic innervation of the mesencephalic nucleus of the trigeminal nerve in rat brain: a light and electron microscopical study. Brain Res. (1987) 418:388–91. doi: 10.1016/0006-8993(87)90109-0
110. Miklos IH, Kovacs KJ. Functional heterogeneity of the responses of histaminergic neuron subpopulations to various stress challenges. Eur J Neurosci. (2003) 18:3069–79. doi: 10.1111/j.1460-9568.2003.03033.x
111. Passani MB, Blandina P. Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol Sci. (2011) 32:242–9. doi: 10.1016/j.tips.2011.01.003
112. Yanovsky Y, Zigman JM, Kernder A, Bein A, Sakata I, Osborne-Lawrence S, et al. Proton- and ammonium-sensing by histaminergic neurons controlling wakefulness. Front Syst Neurosci. (2012) 6:23. doi: 10.3389/fnsys.2012.00023
113. Umehara H, Mizuguchi H, Mizukawa N, Matsumoto M, Takeda N, Senba E, et al. Deprivation of anticipated food under scheduled feeding induces c-Fos expression in the caudal part of the arcuate nucleus of hypothalamus through histamine H(1) receptors in rats: potential involvement of E3 subgroup of histaminergic neurons in tuberomammillary nucleus. Brain Res. (2011) 1387:61–70. doi: 10.1016/j.brainres.2011.02.018
114. Sergeeva OA, Eriksson KS, Sharonova IN, Vorobjev VS, Haas HL. GABA(A) receptor heterogeneity in histaminergic neurons. Eur J Neurosci. (2002) 16:1472–82. doi: 10.1046/j.1460-9568.2002.02221.x
115. Sergeeva OA, Eriksson KS, Haas HL. Glycine receptor mediated responses in rat histaminergic neurons. Neurosci Lett. (2001) 300:5–8. doi: 10.1016/S0304-3940(01)01509-9
116. Cenni G, Blandina P, Mackie K, Nosi D, Formigli L, Giannoni P, et al. Differential effect of cannabinoid agonists and endocannabinoids on histamine release from distinct regions of the rat brain. Eur J Neurosci. (2006) 24:1633–44. doi: 10.1111/j.1460-9568.2006.05046.x
117. McKinley MJ, Yao ST, Uschakov A, McAllen RM, Rundgren M, Martelli D. The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiol. (2015) 214:8–32. doi: 10.1111/apha.12487
118. McGreevy J, Orrevall Y, Belqaid K, Wismer W, Tishelman C, Bernhardson BM. Characteristics of taste and smell alterations reported by patients after starting treatment for lung cancer. Support Care Cancer. (2014) 22:2635–44. doi: 10.1007/s00520-014-2215-2
119. Hutton JL, Baracos VE, Wismer WV. Chemosensory dysfunction is a primary factor in the evolution of declining nutritional status and quality of life in patients with advanced cancer. J Pain Symptom Manage. (2007) 33:156–65. doi: 10.1016/j.jpainsymman.2006.07.017
120. Wagland R, Richardson A, Ewings S, Armes J, Lennan E, Hankins M, et al. Prevalence of cancer chemotherapy-related problems, their relation to health-related quality of life and associated supportive care: a cross-sectional survey. Support Care Cancer. (2016) 24:4901–4911. doi: 10.1007/s00520-016-3346-4
121. Spotten LE, Corish CA, Lorton CM, Ui Dhuibhir PM, O'Donoghue NC, O'Connor B, et al. Subjective and objective taste and smell changes in cancer. Ann Oncol. (2017) 28:969–84. doi: 10.1093/annonc/mdx018
122. Spotten L, Corish C, Lorton C, Dhuibhir PU, O'Donoghue N, O'Connor B, et al. Subjective taste and smell changes in treatment-naive people with solid tumours. Support Care Cancer. (2016) 24:3201–8. doi: 10.1007/s00520-016-3133-2
123. Smith KR, Treesukosol Y, Paedae AB, Contreras RJ, Spector AC. Contribution of the TRPV1 channel to salt taste quality in mice as assessed by conditioned taste aversion generalization and chorda tympani nerve responses. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R1195–205. doi: 10.1152/ajpregu.00154.2012
124. Treesukosol Y, Ishizuka T, Yamamoto C, Senda K, Tsutsumi S, Yamatodani A, et al. Hypothalamic histamine release by taste stimuli in freely moving rats: possible implication of palatability. Behav Brain Res. (2005) 164:67–72. doi: 10.1016/j.bbr.2005.06.004
125. Tanida M, Niijima A, Shen J, Nakamura T, Nagai K. Olfactory stimulation with scent of essential oil of grapefruit affects autonomic neurotransmission and blood pressure. Brain Res. (2005) 1058:44–55. doi: 10.1016/j.brainres.2005.07.048
126. Inutsuka A, Yamanaka A. The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front Endocrinol. (2013) 4:18. doi: 10.3389/fendo.2013.00018
127. Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci. (2000) 85:1–17. doi: 10.1016/S1566-0702(00)00215-0
128. Gidron Y, Perry H, Glennie M. Does the vagus nerve inform the brain about preclinical tumours and modulate them? Lancet Oncol. (2005) 6:245–8. doi: 10.1016/S1470-2045(05)70096-6
129. Shenton FC, Pyner S. Vagal afferents, sympathetic efferents and the role of the PVN in heart failure. Auton Neurosci. (2016) 199:38–47. doi: 10.1016/j.autneu.2016.08.009
130. Provensi G, Coccurello R, Umehara H, Munari L, Giacovazzo G, Galeotti N, et al. Satiety factor oleoylethanolamide recruits the brain histaminergic system to inhibit food intake. Proc Natl Acad Sci USA. (2014) 111:11527–32. doi: 10.1073/pnas.1322016111
131. Romano A, Cassano T, Tempesta B, Cianci S, Dipasquale P, Coccurello R, et al. The satiety signal oleoylethanolamide stimulates oxytocin neurosecretion from rat hypothalamic neurons. Peptides. (2013) 49:21–6. doi: 10.1016/j.peptides.2013.08.006
132. Gaetani S, Fu J, Cassano T, Dipasquale P, Romano A, Righetti L, et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J Neurosci. (2010) 30:8096–101. doi: 10.1523/JNEUROSCI.0036-10.2010
133. Bealer SL, Crowley WR. Histaminergic control of oxytocin release in the paraventricular nucleus during lactation in rats. Exp Neurol. (2001) 171:317–22. doi: 10.1006/exnr.2001.7770
134. Ondicova K, Mravec B. Role of nervous system in cancer aetiopathogenesis. Lancet Oncol. (2010) 11:596–601. doi: 10.1016/S1470-2045(09)70337-7
135. Sun L, Quan XQ, Yu S. An epidemiological survey of cachexia in advanced cancer patients and analysis on its diagnostic and treatment status. Nutr Cancer. (2015) 67:1056–62. doi: 10.1080/01635581.2015.1073753
136. Yi CX, la Fleur SE, Fliers E, Kalsbeek A. The role of the autonomic nervous liver innervation in the control of energy metabolism. Biochim Biophys Acta. (2010) 1802:416–31. doi: 10.1016/j.bbadis.2010.01.006
137. Mizuno K, Ueno Y. Autonomic nervous system and the liver. Hepatol Res. (2016) 47:160–5. doi: 10.1111/hepr.12760
138. Zhang W, Bi S. Hypothalamic regulation of brown adipose tissue thermogenesis and energy homeostasis. Front Endocrinol. (2015) 6:136. doi: 10.3389/fendo.2015.00136
139. Lowell BB, Bachman ES. Beta-Adrenergic receptors, diet-induced thermogenesis, and obesity. J Biol Chem. (2003) 278:29385–8. doi: 10.1074/jbc.R300011200
140. Zeng W, Pirzgalska RM, Pereira MM, Kubasova N, Barateiro A, Seixas E, et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell. (2015) 163:84–94. doi: 10.1016/j.cell.2015.08.055
141. Morrison SF, Madden CJ, Tupone D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab. (2014) 19:741–56. doi: 10.1016/j.cmet.2014.02.007
142. Kalinkovich A, Livshits G Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res Rev. (2017) 35:200–21. doi: 10.1016/j.arr.2016.09.008
143. Stewart Coats AJ, Ho GF, Prabhash K, von Haehling S, Tilson J, Brown R, et al. Espindolol for the treatment and prevention of cachexia in patients with stage III/IV non-small cell lung cancer or colorectal cancer: a randomized, double-blind, placebo-controlled, international multicentre phase II study (the ACT-ONE trial). J Cachexia Sarcopenia Muscle. (2016) 7:355–65. doi: 10.1002/jcsm.12126
144. Lainscak M, Laviano A, ACT-ONE - ACTION at last on cancer cachexia by adapting a novel action beta-blocker. J Cachexia Sarcopenia Muscle. (2016) 7:400–2. doi: 10.1002/jcsm.12136
145. Joassard OR, Durieux AC, Freyssenet DG. beta2-Adrenergic agonists and the treatment of skeletal muscle wasting disorders. Int J Biochem Cell Biol. (2013) 45:2309–21. doi: 10.1016/j.biocel.2013.06.025
146. Potsch MS, Tschirner A, Palus S, von Haehling S, Doehner W, Beadle J, et al. The anabolic catabolic transforming agent (ACTA) espindolol increases muscle mass and decreases fat mass in old rats. J Cachexia Sarcopenia Muscle. (2014) 5:149–58. doi: 10.1007/s13539-013-0125-7
147. Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer. (2015) 121:3444–51. doi: 10.1002/cncr.29392
148. Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J. (2014) 35:932–41. doi: 10.1093/eurheartj/eht302
149. Toledo M, Penna F, Oliva F, Luque M, Betancourt A, Marmonti E, et al. A multifactorial anti-cachectic approach for cancer cachexia in a rat model undergoing chemotherapy. J Cachexia Sarcopenia Muscle. (2016) 7:48–59. doi: 10.1002/jcsm.12035
150. Khan MM, Lustrino D, Silveira WA, Wild F, Straka T, Issop Y, et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci USA. (2016) 113:746–50. doi: 10.1073/pnas.1524272113
151. Kim YS, Sainz RD, Molenaar P, Summers RJ. Characterization of beta 1- and beta 2-adrenoceptors in rat skeletal muscles. Biochem Pharmacol. (1991) 42:1783–9. doi: 10.1016/0006-2952(91)90516-8
152. Williams RS, Caron MG, Daniel K. Skeletal muscle beta-adrenergic receptors: variations due to fiber type and training. Am J Physiol. (1984) 246(2 Pt 1): p. E160-7. doi: 10.1152/ajpendo.1984.246.2.E160
153. Roder IV, Choi KR, Reischl M, Petersen Y, Diefenbacher ME, Zaccolo M, et al. Myosin Va cooperates with PKA RIalpha to mediate maintenance of the endplate in vivo. Proc Natl Acad Sci USA. (2010) 107:2031–6. doi: 10.1073/pnas.0914087107
154. Choi KR, Berrera M, Reischl M, Strack S, Albrizio M, Roder IV, et al. Rapsyn mediates subsynaptic anchoring of PKA type I and stabilisation of acetylcholine receptor in vivo. J Cell Sci. (2012) 125(Pt 3):714–23. doi: 10.1242/jcs.092361
155. Roder IV, Strack S, Reischl M, Dahley O, Khan MM, Kassel O, et al. Participation of myosin Va and Pka type I in the regeneration of neuromuscular junctions. PLoS ONE. (2012) 7:e40860. doi: 10.1371/journal.pone.0040860
156. Nelson PG, Lanuza MA, Jia M, Li MX, Tomas J. Phosphorylation reactions in activity-dependent synapse modification at the neuromuscular junction during development. J Neurocytol. (2003) 32:803–16. doi: 10.1023/B:NEUR.0000020625.70284.a6
157. Martinez-Pena y Valenzuela I, Pires-Oliveira M, Akaaboune M PKC and PKA regulate AChR dynamics at the neuromuscular junction of living mice. PLoS ONE. (2013) 8:e81311. doi: 10.1371/journal.pone.0081311
158. Miura S, Kawanaka K, Kai Y, Tamura M, Goto M, Shiuchi T, et al. An increase in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to exercise is mediated by beta-adrenergic receptor activation. Endocrinology. (2007) 148:3441–8. doi: 10.1210/en.2006-1646
159. Bruno NE, Kelly KA, Hawkins R, Bramah-Lawani M, Amelio AL, Nwachukwu JC, et al. Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J. (2014) 33:1027–43. doi: 10.1002/embj.201386145
160. Irrcher I, Adhihetty PJ, Joseph AM, Ljubicic V, Hood DA Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med. (2003) 33:783–93. doi: 10.2165/00007256-200333110-00001
161. Balakrishnan VS, Rao M, Menon V, Gordon PL, Pilichowska M, Castaneda F, et al. Resistance training increases muscle mitochondrial biogenesis in patients with chronic kidney disease. Clin J Am Soc Nephrol. (2010) 5:996–1002. doi: 10.2215/CJN.09141209
162. Bing C, Trayhurn P. Regulation of adipose tissue metabolism in cancer cachexia. Curr Opin Clin Nutr Metab Care. (2008) 11:201–7. doi: 10.1097/MCO.0b013e3282f948e2
163. Agustsson T, Wikrantz P, Ryden M, Brismar T, Isaksson B Adipose tissue volume is decreased in recently diagnosed cancer patients with cachexia. Nutrition. (2012) 28:851–5. doi: 10.1016/j.nut.2011.11.026
164. Faustino-Rocha AI, Ferreira R, Gama A, Oliveira PA, Ginja M. Antihistamines as promising drugs in cancer therapy. Life Sci. (2017) 172:27–41. doi: 10.1016/j.lfs.2016.12.008
165. Varga B, Csonka A, Csonka A, Molnar J, Amaral L, Spengler G. Possible biological and clinical applications of phenothiazines. Anticancer Res. (2017) 37:5983–93. doi: 10.21873/anticanres.12045
166. Jiang SM, Wu JH, Jia L Intervention of mirtazapine on gemcitabine-induced mild cachexia in nude mice with pancreatic carcinoma xenografts. World J Gastroenterol. (2012) 18:2867–71. doi: 10.3748/wjg.v18.i22.2867
167. Riechelmann RP, Burman D, Tannock IF, Rodin G, Zimmermann C. Phase II trial of mirtazapine for cancer-related cachexia and anorexia. Am J Hosp Palliat Care. (2010) 27:106–10. doi: 10.1177/1049909109345685
168. Kast RE, Foley KF. Cancer chemotherapy and cachexia: mirtazapine and olanzapine are 5-HT3 antagonists with good antinausea effects. Eur J Cancer Care. (2007) 16:351–4. doi: 10.1111/j.1365-2354.2006.00760.x
169. Couluris M, Mayer JL, Freyer DR, Sandler E, Xu P, Krischer JP. The effect of cyproheptadine hydrochloride (periactin) and megestrol acetate (megace) on weight in children with cancer/treatment-related cachexia. J Pediatr Hematol Oncol. (2008) 30:791–7. doi: 10.1097/MPH.0b013e3181864a5e
170. Kardinal CG, Loprinzi CL, Schaid DJ, Hass AC, Dose AM, Athmann LM, et al. A controlled trial of cyproheptadine in cancer patients with anorexia and/or cachexia. Cancer. (1990) 65:2657–62.
171. Tazi E, Errihani H Treatment of cachexia in oncology. Indian J Palliat Care. (2010) 16:129–37. doi: 10.4103/0973-1075.73644
Keywords: cancer cachexia, central histaminergic system, histaminergic neurons, histamine signaling, food intake, energy expenditure, parasympathetic nervous system, anorexia
Citation: Zwickl H, Zwickl-Traxler E and Pecherstorfer M (2019) Is Neuronal Histamine Signaling Involved in Cancer Cachexia? Implications and Perspectives. Front. Oncol. 9:1409. doi: 10.3389/fonc.2019.01409
Received: 09 July 2019; Accepted: 28 November 2019;
Published: 20 December 2019.
Edited by:
Maria Ida Amabile, Sapienza University of Rome, ItalyReviewed by:
Stephen John Ralph, Griffith University, AustraliaPaul Dent, Virginia Commonwealth University, United States
Fabio Penna, University of Turin, Italy
Copyright © 2019 Zwickl, Zwickl-Traxler and Pecherstorfer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannes Zwickl, aGFubmVzLnp3aWNrbEBrbC5hYy5hdA==