 Wei Mu
Wei Mu Zhe Wang
Zhe Wang Margot Zöller
Margot Zöller- 1School of Public Health, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Oncology, The First Affiliated Hospital of Guangdong, Pharmaceutical University, Guangzhou, China
Metastasis is the main cause of high pancreatic cancer (PaCa) mortality and trials dampening PaCa mortality rates are not satisfying. Tumor progression is driven by the crosstalk between tumor cells, predominantly cancer-initiating cells (CIC), and surrounding cells and tissues as well as distant organs, where tumor-derived extracellular vesicles (TEX) are of major importance. A strong stroma reaction, recruitment of immunosuppressive leukocytes, perineural invasion, and early spread toward the peritoneal cavity, liver, and lung are shared with several epithelial cell-derived cancer, but are most prominent in PaCa. Here, we report on the state of knowledge on the PaCIC markers Tspan8, alpha6beta4, CD44v6, CXCR4, LRP5/6, LRG5, claudin7, EpCAM, and CD133, which all, but at different steps, are engaged in the metastatic cascade, frequently via PaCIC-TEX. This includes the contribution of PaCIC markers to TEX biogenesis, targeting, and uptake. We then discuss PaCa-selective features, where feedback loops between stromal elements and tumor cells, including distorted transcription, signal transduction, and metabolic shifts, establish vicious circles. For the latter particularly pancreatic stellate cells (PSC) are responsible, furnishing PaCa to cope with poor angiogenesis-promoted hypoxia by metabolic shifts and direct nutrient transfer via vesicles. Furthermore, nerves including Schwann cells deliver a large range of tumor cell attracting factors and Schwann cells additionally support PaCa cell survival by signaling receptor binding. PSC, tumor-associated macrophages, and components of the dysplastic stroma contribute to perineural invasion with signaling pathway activation including the cholinergic system. Last, PaCa aggressiveness is strongly assisted by the immune system. Although rich in immune cells, only immunosuppressive cells and factors are recovered in proximity to tumor cells and hamper effector immune cells entering the tumor stroma. Besides a paucity of immunostimulatory factors and receptors, immunosuppressive cytokines, myeloid-derived suppressor cells, regulatory T-cells, and M2 macrophages as well as PSC actively inhibit effector cell activation. This accounts for NK cells of the non-adaptive and cytotoxic T-cells of the adaptive immune system. We anticipate further deciphering the molecular background of these recently unraveled intermingled phenomena may turn most lethal PaCa into a curatively treatable disease.
Introduction
The Metastatic Cascade and Tumor Cell Dissemination
More than 90% of cancer mortality is related to metastasis (1), which in carcinoma requires completion of the metastatic cascade starting with local invasion of the surrounding extracellular matrix (ECM) and cells and processing through intravasation, surviving transport in vessels, arrest at distant organs, extravasation, surviving in the foreign environment and reinitiating tumor growth (2). These complex biological events are orchestrated by cell autonomous and non-autonomous signaling cascades. Local invasion requires breaching the basal membrane (BM) promoted by tumor-derived proteases and leading to liberation of growth factors and integrin activation affecting cell polarity and survival (3). Alternatively, tumor cells may use a protease- and integrin-independent, Rho1/ROCK1-dependent amoeboid invasion program (4). For local invasion of individual cells, tumor cells adopt a developmental epithelial-mesenchymal transition (EMT) program, which orchestrates activation of sets of transcription factors (Tf) that repress cell-cell adhesion molecules and induce expression of mesenchymal markers (5). Having passed the BM, tumor cells encounter the tumor stroma, which consists of endothelial cells (EC), pericytes, adipocytes, fibroblasts (FB), and bone marrow mesenchymal cells. Tumor cells push the reactive stroma toward pro-tumorigenic factor secretion and pro-tumorigenic cell recruitment. Thus, contact with the surrounding stroma is the first step where tumor cells receive a self-amplifying feedback (6, 7). The following step of invasion is strongly promoted by tumor-induced angiogenesis/lymphangiogenesis, the newly formed vessels being tortuous, leaky and continuously reconfiguring themselves, weak interactions between adjacent EC and the incomplete pericyte coverage facilitate tumor cell intravasation. EC wall passage is assisted by TGFβ1 and tumor-associated macrophages (TAM), providing CFS1/MCSF1 and EGF1. In addition, metabolic adaptations of growing and sprouting EC support (lymph)angiogenesis (8–10). In the vasculature, tumor cells are exposed to a variety of stresses. In the absence of cell-cell or cell-matrix adhesion, epithelial cell would undergo apoptosis/anoikis, which is circumvented by metabolic shifts toward the pentosephosphate pathway and anaerobic glycolysis. Matrix detachment-forced reduced glucose uptake assists LKB11 activation,1 which increases protein kinase AMP1 catalytic subunit PRKAA1 activity. This inhibits acetyl-CoA carboxylases ACACA/B1, lowers NADPH1 consumption in fatty acid (FA) synthesis, but increases NADPH generation through an alternative pathway. This process reduces reactive oxygen species (ROS), essential for precluding detached cancer cell anoikis (10–13). Shear stress and the attack by the innate immune system are circumvented by tumor cell tissue factor (TF1) and selectins binding to platelets to form microemboli, which act as protective shields for the tumor cells (14, 15). Tumor cells mostly extravasate between adjacent EC. Adhesion to EC is facilitated by selectins, cadherins, integrins, CD44, Ig superfamily members, CD146/MUC181, and by homophilic interactions between JAM1. Interactions between tumor cell-provided factors such as ANGPTL41 and α5β1, CDH5/CD1441, CLDN51, EREG1, COX21, and MMP1 support extravasation. Actin remodeling, opening of junctions, necroptosis and APP1-DR61-assisted EC death are discussed as underlying mechanisms. Platelet-, neutrophil- and monocyte-provided cytokines and chemokines also assist extravasation (16, 17).
Metastatic Growth
There is ample evidence that migrating cancer cells leave the circulation for well-prepared soil, known as premetastatic niche. It is arranged in advance of cancer cell arrival by receiving information via tumor exosomes (TEX). Integrins, tetraspanins, receptor tyrosine kinases (RTK) and G-protein coupled receptors (GPCR) are important for message transfer (18–21). Established micrometastases may persist for weeks to years in a state of long-term dormancy. This dormancy relies on resting state persistence or failure to initiate angiogenesis, or on apoptosis-promoting host cells. Macroscopic metastatic outgrowth requires a multitude of adaptive programs that vary depending on the organ site of the metastasis and the original tumor. No metastasis-specific genetic changes being observed, outgrowth is supposed relying on epigenetic changes, like aberrant DNA methylation, altered chromatin structure, and activation of transcriptional programs that can be facilitated/guided by long non-coding (lnc)RNA. Two prerequisites must be fulfilled. One is the presence of cancer-initiating cells (CIC) with the capacity for self-renewal that in part is promoted by EMT-related Tf. The other is the establishment of adaptive programs enabling growth in the foreign environment. This includes some common traits such as metabolic adaptation and survival pathway activation. Other adaptive programs vary with the site of metastasis. Thus, similar to primary tumor growth, metastatic outgrowth is supported by the surrounding stroma including TGFβ1 and periostin, pro-inflammatory cells, local fibroblasts, and supportive ECM components (22–24). There remains a last query. CIC-derived metastases frequently reflect the mixed phenotype of the primary tumor. This may be due to the reversibility of EMT, called mesenchymal-epithelial transition (MET). However, further studies are required to elucidate tumor-inherent and surrounding-supported MET reprogramming (25–27).
Twenty-five years ago, the metastatic cascade was described as sequential processes in microecosystems (28). This still holds true, where striking progress in molecular characterization, important insights into stem cell (SC)/CIC plasticity, signaling pathways, networking connectivity and the modes of epigenetic regulation allowed deciphering the paths toward tumor progression.
After briefly introducing the clinical features of PaCa and exosome composition, we discuss current theories on the molecular mechanisms underlying the steps of the metastatic cascade particularly in PaCa.
Clinical Features of Pancreatic Cancer Growth and Metastasis
Pancreatic cancer (PaCa) is the most lethal cancer, with a mortality rate close to the incidence rate. The overall 5-year survival rate is ~5% (29) and does not exceed 15–20% after surgery, the only curative treatment option, owing to local recurrence and metastatic spread. Furthermore, 80% of patients are inoperable at diagnosis (30). Though mortality rates for several common cancers decreased over the last decades (29), mortality rates increased for PaCa. Ductal PaCa, the most frequent subtype, is expected to be the second cancer-related cause of death after lung cancer by 2030 (31). The high mortality, due to early spread and radio- and chemotherapy resistance (32), is caused by a small population of CIC (33). Three additional contributing features are abundant stroma reactions, preferential dissemination along intrapancreatic nerves and pronounced immune deviation.
Unlike most tumors, PaCa cells may form only small islands within an abundant tumor stroma. The main cellular components are cancer-associated fibroblasts (CAF), predominantly deriving from pancreatic stellate cells (PSC) and inflammatory cells. The ECM consists of collagens, laminin (LN), fibronectin (FN), proteoglycans, and glycosaminoglycans and harbors soluble factors affecting tumor and host cells (34, 35). The PaCa stroma reaction, primarily promoting tumor growth, may hamper tumor progression in certain circumstances, indicating the need for further studies on composition and activities (36).
Perineural invasion (PNI) is most common in PaCa and an indicator of aggressive tumors and short survival (37). The pancreatic nerve fibers from the splanchnic nerves, dorsal root ganglion and the vagus become hyperinnervated and hypertrophic. The nervous system participates in all stages of PaCa development with neurotrophic factors and axon guidance genes overrepresentation or mutation. CAF and intrapancreatic immune cells also affect the intrapancreatic neurons (38), but intrapancreatic neurons and Schwann cells also signal toward the tumor cells (39, 40).
Finally, the PaCa stroma is replete with immune cells (41) that are almost exclusively immunosuppressive (42).
The steeply increasing incidence of most malignant PaCa demands intensifying efforts to clarify the underlying mechanisms. PaCa shares the consecutive steps of the metastatic cascade with most epithelial carcinoma, but also displays several peculiarities. Extensive stroma dysplasia, preferred routing of migrating tumor cells along intrapancreatic nerves and striking deviations toward immunosuppressive cells and factors account for the early spread. We will discuss those features, which quantitatively differentiate PaCa from the majority of epithelial cancer. Exosomes and PaCIC markers, both essentially contributing to the selective features, are introduced in advance.
The Importance of Exosomes in Tumor Progression
Contact between single tumor cells detaching from the tumor mass and distinct non-transformed tissues and cells is an essential prerequisite for tumor progression. The crosstalk between metastasizing and non-metastasizing tumor cells and non-transformed cells mostly relies on message delivery by TEX and stroma cell-derived Exo.
Exo, small 40–100 nm vesicles delivered by live cells (43), disperse throughout the body, which allows for short and long-range communication (44). Exo expressing donor cell-derived components allows differentiating non-transformed cell-derived Exo from TEX (45). Exo components are function-competent (46) and highly effective intercellular communicators (47). Delivered messages modulate the ECM, non-metastasizing tumor cells (Non-CIC), and non-transformed cells including hematopoietic cells, EC, FB, nerves, and epithelial cells (48–51).
Exo biogenesis starts with early endosome (EE) formation. EE derive from the trans-Golgi network or internalized membrane microdomains (52). Distinct transport machineries guide EE toward multivesicular bodies (MVB) (53). Exo collect their cargo during inward budding of endosomes, called intraluminal vesicles (ILV), into MVB (54–56). LPAR11, Alix/PDCD6IP1, and HSP701 spur inward budding and SGPP11 and diaglycerol1 are engaged in cargo sorting (57, 58). Loading are nonrandom processes. Protein loading is facilitated by mono-ubiquitination, acylation, myristoylation, higher order oligomerization, or sphingolipids forming ceramide (59–61). Annexin-II supports RNA sorting (62). Optionally, RNA becomes incorporated by affinity for the outer (cytoplasmic) raft-like MVB membrane (63). MiRNA loading is guided by a zip code in the 3′-UTR and coupling of RISC (RNA induced silencing complex) to specific EXO motifs binding to HNRNP (heterogeneous ribonucleoprotein) (55, 64). Selective lncRNA recruitment requires clarification (65, 66). ILV are guided toward the proteasome for degradation or toward the plasma membrane, supported by microtubules and Rab1 proteins (53, 67). SNARE1 and synaptogamins assist fusion with the plasma membrane (52, 53, 67). Released vesicles are called exosomes.
Exosome Composition
The Exo membrane lipid bilayer contains integrated membrane proteins and lipid- or membrane protein-attached cytoskeletal and cytosolic signaling molecules. The Exo lipid envelop is composed of phosphatidylcholine, -ethanolamine, -inositol, prostaglandins, lysobisphosphatidic acid, sphingomyelin, cholesterol, GM31/GRM61, and PS1 (phosphatidylserine) (68), high PS levels differentiating Exo from microvesicles (69). Lipids are organized along with lipid carriers such as lipid-transporting FABP1. Lipid second messengers are involved in biogenesis, some requiring a link to lipids during ILV invagination, e.g., HSPA8 needs battenin (CLN31) (70), formed by PLD21 (71, 72). Ceramide triggers an ESCRT (endosomal sorting complex required for transport)-independent pathway of Exo biogenesis (73). Cholesterol enhances flotillin-2 positive Exo secretion (74). Lipid transporters such as ABCA31 are also involved in Exo production (75). Thus, Exo carry bioactive lipids, related enzymes, fatty acid transporters, and lipid-related enzyme carriers and use lipids to fuse with target cells (76–78).
Exo protein characterization profited from improved mass spectrometry (MS) (79) to be followed by the exocharta database [http://exocarta.org/exosome_markers]. Exo also contain proteins engaged in biogenesis and vesicle transport and proteins actively recruited during ILV invagination. Tetraspanins are most strongly enriched constitutive Exo component (80–82). Other abundant proteins include adhesion molecules, proteases, major histocompatibility complex (MHC) molecules, HSP, TSG1011, ALIX, annexins, cytoskeleton proteins, metabolic enzymes, cytosolic signal transduction molecules, and ribosomal proteins (82, 83). Finally, PaCIC biomarkers are enriched in TEX (84–86). This is important as CIC drive the metastatic process (87–90), where Tspan8 (86, 91) and associated α6β4 (92–94), CD44v6 (95, 96), and linked cMET1 (96, 97), CD184/CXCR41 that can associate with Tspan8 and CD44v6 (98–100), cldn7 (84, 101, 102), and associated EpCAM1 (84, 103, 104), LGR5/GPR491 (105, 106) and CD133/PROM11 (107, 108) are engaged in distinct steps of tumor progression.
Exo also contain mRNA. mRNA is produced and processed in the nucleus, transported to the cytoplasm and translated. These processes are controlled by proteins, mostly RNA binding proteins (RBP), which interact with mRNA (109) and together with additional regulatory RNA constitute the mRNA binding protein code (110–113). Notably, the activity of RBP varies depending on the cell's activation state. Thus, GAPDH1 binds the 3′UTR of IFNγ1 and represses translation in inactive, but not activated T-cells (114). RBP also account for localization and trafficking of RNA-protein complexes in cells (115, 116). Finally, the mRNA profile of Exo differs from that of cells (117), metabolic enzymes and proteins engaged in cell-cell and cell-matrix adhesion being frequently overrepresented (118–120), and possibly translated in Exo (121, 122).
Exo contain a large range of non-coding (nc)RNA. Most abundant are microRNA (miRNA) and lncRNA. miRNA host genes are transcribed by RNA polymerase II to form primary (pri)-miRNA. The Drosha1 endonuclease associates with the RBP DGCR81 releasing the stemloop precursor from the flanking pri-miRNA transcript sequence. After export from the nucleus by exportin-5, Dicer in association with TRBP1 cleaves the precursor loop releasing the mature miRNA (123). One strand of this duplex RNA is integrated into the RISC complex, which contains argonaute linking the miRNA to target mRNA (124, 125). Importantly, miRNA with sequence motifs for sorting into ILV are efficiently transferred into Exo, some miRNA becoming undetectable in the donor cell (126, 127). Most miRNA bind to a large number of mRNA and most mRNA are targeted by more than one miRNA, providing hurtles for their potential therapeutic use, aggravated by the discussed mode whereby miRNA affect target cells (117, 128).
LncRNA, defined by a length of >200 bp, are abundantly recovered from Exo (129). LncRNA are involved in a large range of activities, including chromatin organization, gene transcription, mRNA turnover, protein translation, and macromolecular complex assembly (130–132). LncRNA can also be grouped according to functioning as signal, decoy, scaffold, guide, enhancer RNA, and short peptides (133). Signaling lncRNA regulate transcription (134). Decoy lncRNA sequesters regulatory factors including Tf, catalytic proteins, subunits of larger chromatin modifying complexes and miRNA (135). Scaffold lncRNA provide platforms for assembly of multiple-component complexes, e.g., ribonucleoprotein (RNP) complexes (136). Guide lncRNA drive RNP to specific target genes (137). Enhancer lncRNA (eRNA) influence the 3-dimensional organization of DNA, which may result from lncRNA being not released and tethering interacting proteins to enhancer regions (138). Finally, lncRNA can encode function-competent short peptides (139). Evidence for selective recruitment into Exo derives from enrichment of some lncRNA harboring seed regions for miRNA in Exo (140, 141). LncRNA recovery in Exo only recently receiving attention, important information on the multiple functions of lncRNA can be expected in the near future.
Exo contain mitochondrial, genomic, or retrotransposon double and single stranded DNA (142, 143). Without hints toward sorting and disputed functionality, a possible contribution of Exo DNA to tumor progression remains to be elaborated.
Taken together, TEX are optimally furnished to drive all steps of the metastatic cascade using their lipid, protein and RNA armament, where PaCIC markers contribute to biogenesis (Tspan8), miRNA loading (CD44v6), and lipid transport (cldn7) (144, 145) (Figures 1A–C).
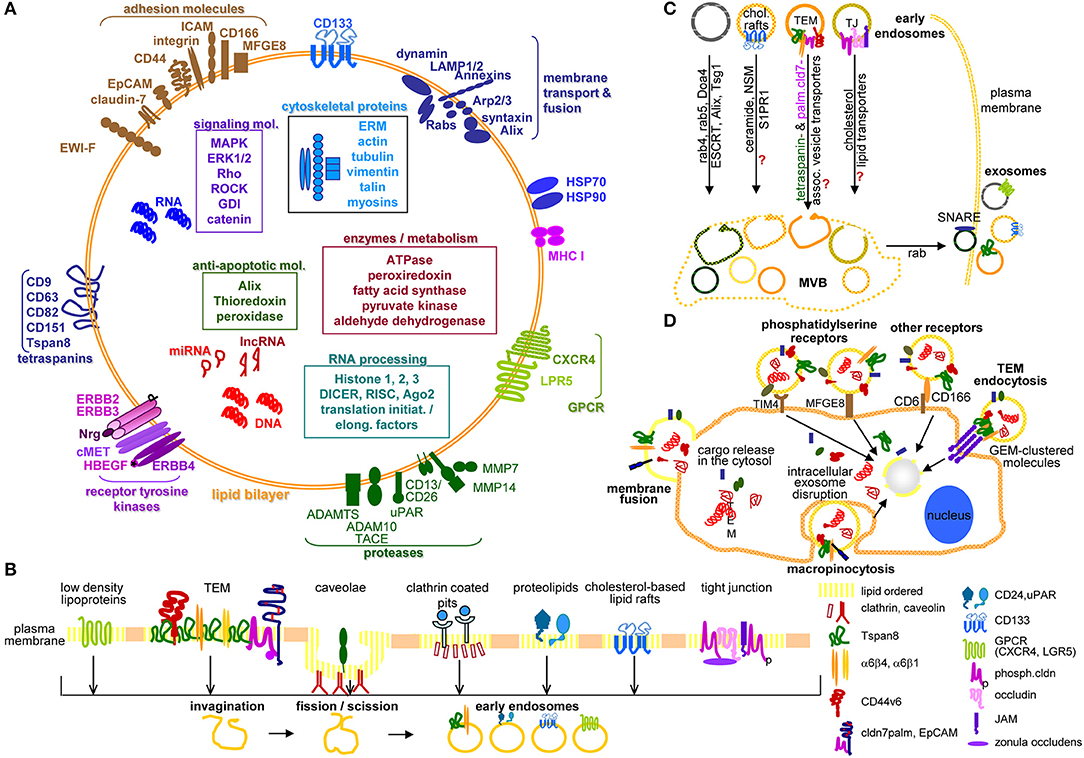
Figure 1. Exosome characterization, biogenesis, and targeting. (A) Exosomes are composed of a lipid bilayer, transmembrane protein and the cytoplasm containing proteins, mRNA, non-coding RNA like miRNA and lncRNA and DNA, where PaCIC-TEX, express the CIC markers Tspan8, integrin α6β1/α6β4, CD44v6, CD133, CXCR4, LRP5, EpCAM, and cldn7. Other transmembrane proteins are linked to Exo biogenesis. (B) Exo biogenesis starts with the invagination of membrane microdomains that are characterized by ordered lipids, like low-density lipoprotein, caveolae, clathrin-coated pits, cholesterol-based lipid rafts, and others. (C) After fission and scission of invaginated membrane domains, the EE are guided toward MVB, the traffic differs between the origins from distinct lipid-enriched domains. Most abundant is rab4, rab5, Doa4 promoted migration and invagination into MVB via the ESCRT system. Components of cholesterol-based lipid raft-, TJ-, or TEM-derived EE are not completely explored. Guidance from MVB to the plasma membrane involves rab proteins, phospholipase D, and SNARE. (D) The contact between Exo and target cells can proceed via fusion of the Exo membrane with the cell membrane, by macropinocytosis, receptor ligand binding such as phosphatidylserine binding to TIM4 or MFGE8 or CD166 binding to CD6 or may be facilitated by Exo membrane protein complexes binding to invagination-prone complexes as described for TEM binding to the TCR complex. Exo also bind to the ECM or matricellular proteins, CD44 and integrins being most frequently involved. Full name of proteins are listed in Table S1. In brief, cells use a variety of pathways for the generation of EE, the traffic toward MVB, the loading of ILV with proteins, coding and non-coding RNA and DNA. Exo may preferentially bind to and be taken up by receptor-ligand binding, uptake being facilitated by the engagement of protein complexes at both the Exo and the target cell.
Exosome Targeting and Uptake
Exosomes bind to the ECM and cells, using for both a similar appurtenance.
Exo binding mostly relies on surface receptor and adhesion molecules, such as tetraspanins, integrins, proteoglycans, and lectins, docking to appropriate ligands on the ECM and cells (146). Tetraspanin-associated adhesion molecules account for target-selective binding. Thus, Tspan8-α4 preferentially binds EC, whereas Tspan8-α6β4 preferentially binds FB (147, 148). Integrins, receptors for ECM proteins, also are involved in Tspan8-independent Exo binding (149), e.g., preventing α5β1-FN binding inhibits anchorage independent growth (150). ECM-binding proteins also guide Exo docking and uptake by recipient cells, demonstrated for β1, αv, β3, and αL integrin chains and ICAM11 (151). Recipient cell integrins contribute to Exo binding. PaCa-TEX preferentially bind ADGRE11 and CD11b1 on Kupffer cells (152). Premetastatic niche formation relies on an integrin-dependent TEX tropism. (Tspan8)/β4 preferentially binds to lung (148, 153), αvβ5 preferentially to liver cells (153).
A second Exo docking system also is highly relevant (154). Exo proteoglycans bind to their receptors such as galectins, CD62E1, CD169/SIGLEC11 (155, 156), and CD44 binds to hyaluronan (HA1) (157). Blocking Exo heparan sulfate proteoglycan (HSPG), the proteoglycan CD44 or the target cell ligands interferes with Exo binding in vitro and in vivo (157–160). PS binding TIM41, TIM11, TIM31, GAS61, MFGE81, Stabilin1, ADGRB11, and RAGE/AGER1 also contributes to Exo docking (146, 154, 161). Furthermore, we want to stress that protein complexes rather than individual molecules, many of which are abundantly expressed, likely account for the selectivity of Exo binding. This is well-demonstrated for tetraspanin complexes in glycolipid-enriched membrane domains (TEM), the multiple interactions between clustered proteins and target ligands strengthening and stabilizing docking (162). Finally, in view of the ongoing discussion on rapid Exo clearance in vivo, which could interfere with their therapeutic efficacy, an excellent report on CD47 binding to SIRPα1 preventing Exo clearance should be mentioned. Particularly in PaCa, oncogenic KRAS contributes to Exo uptake by yet undefined mechanisms such that long-term persisting Exo manipulated to target oncogenic KRAS is currently the most efficient therapeutics (163).
Exo uptake proceeds by Exo fusion (164, 165) or preferentially endocytosis, a process requiring actin modulation (166). Endocytosis occurs via phagocytosis, macropinocytosis, or clathrin-dependent lipid raft/caveolae endocytosis (167). Phagocytosis, facilitated by LAMP11 and TIM4 proceeds by forming cup-like extensions, the tips fusing and becoming internalized (168, 169). Macropinocytosis relies on lamellipodia folding back and fusing with the plasma membrane. Dynamin, Na+H+ exchange, RAC11, EGF, and SDF11 are also engaged in uptake (170). Endocytosis via clathrin-coated pits, rafts, TEM or caveolae are most frequent (171, 172). In clathrin-dependent endocytosis, the membrane invagination becomes coated with clathrin. Clathrin-coated pits are released after scission by dynamin, dominant-negative forms of clathrin reducing Exo uptake (146). Ligand clustering in TEM also supports Exo uptake (162, 171) and a caveolin knockdown (kd) reduces exosome uptake (173, 174). Uptaken Exo are targeted to lysosomes for degradation. Exo content can directly modulate target cells or stimulate target cells' signaling cascades, transcription and silencing processes (175–177) (Figure 1D).
Exo/TEX binding and uptake drastically influence targets. In PaCa, TEX, but also PSC/CAF, immune cell and nerve Exo contribute to PaCa progression.
Pancreatic Cancer-Initiating Cell Markers and the Metastatic Cascade
Metastasis depends on CIC. Stem cells are a rare cell population with the capacity for self-renewal and differentiation, which relies mostly on Tf activation, the nuclear equivalent remaining unaltered (178–180). This also accounts for CIC (181, 182), characterized by infrequent division (183, 184), longevity (185), drug and radiation resistance (186–192), and migratory activity (193–196). Since CIC depend on crosstalk with surrounding tissues (197, 198), we wondered whether the PaCIC biomarkers CD44v6 (Figure 2A), Tspan8 (Figure 2B) and associated α6β4 (Figure 2C), LGR5/GPR49 (Figure 2D), CXCR4/CD184 that associates with Tspan8 and CD44v6 (Figure 2E), cldn7 (Figure 2F), EpCAM and cld7-associated EpCAM (Figure 2G), and CD133 (Figure 2H) might provide hints toward feedback communications between PaCIC and the stroma.
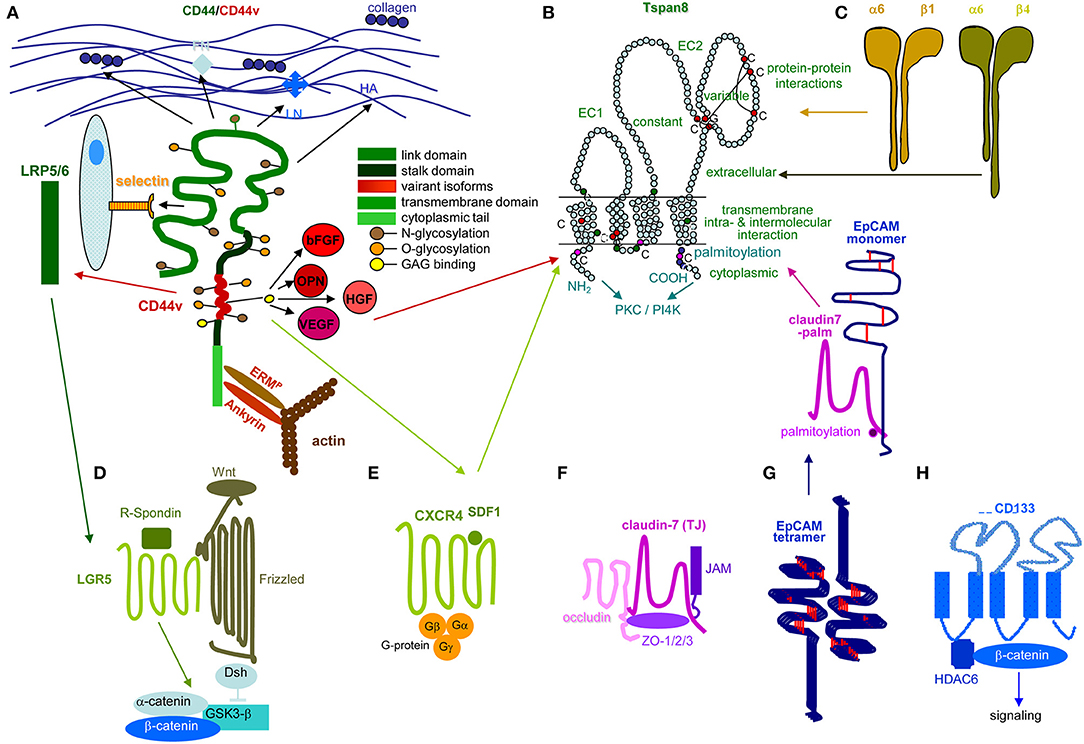
Figure 2. Prominent PaCIC markers. (A) The lead PaCIC marker is CD44v6, a type I transmembrane protein that contributes to the crosstalk with the ECM via its link domain and the HA binding site. It has binding sites for selectins and LRP5/6. The v6 exon product carries binding sites for several growth factors. The cytoplasmic tail has binding sites for ankyrin and ERM proteins including merlin, which promote cytoskeleton association and downstream signaling. (B) Tspan8 is a tetraspanin with a small and a large extracellular loop, the latter mostly accounts for protein-protein interactions. The four transmembrane regions account for intramolecular and intermolecular interactions. The cytoplasmic tail binds PKC and PI4K. Main activities rely on the association with a large range of proteins. Dominant are integrins, but also CD44v6 and an EpCAM-cldn7 complex. (C) Particularly α6β4 is known as a PaCIC marker. Similar to other integrins, it binds matrix proteins, particularly LN. It is a major component of hemidesmosomes anchoring epithelial cells in the basal membrane. Upon activation, it leaves the desmosome complex and associates preferentially with Tspan8. It differs from other integrins by a long cytoplasmic domain of the β4 chain, which promotes multiple signaling pathways. (D) LGR5 is a seven transmembrane protein located close to frizzled. Upon R-spondin binding, it contributes via Wnt activation to ß-catenin liberation. LGR5 activity is supported by CD44v6-associated LRP5/6. (E) CXCR4 is another seven transmembrane protein. This GPCR becomes activated by SDF1 binding. It predominantly signals via trimeric G-proteins. CD44 crosslinking via HA promotes CXCR4 recruitment and strengthens activation of downstream signaling cascades. Activated CXCR4 also associates with Tspan8 (F) Claudin7 is a 4 transmembrane protein, which can be integrated in TJ, where it associates with other claudins, JAM, and occludin and the cytoplasmic zonula occludens proteins. Cldn7 is also recovered outside of TJ. Upon palmitoylation, it associates via a direct protein-protein interaction in the transmembrane region with monomeric EpCAM. The cldn7-EpCAM complex is recruited into TEM and associates with Tspan8. (G) EpCAM is a type I transmembrane protein of many epithelial cells. It forms tetramers, which promote homophilic binding to EpCAM on neighboring cells. It is engaged in signal transduction, predominantly via the liberated cytoplasmic tail that acts as cotranscription factor. (H) CD133 is a five transmembrane protein located in cholesterol-rich membrane domains. It is associated with HDAC6 that stabilizes a ternary CD133-HDAC6-β-catenin complex and β-catenin target activation, which present one of the signaling cascades initiated via CD133. The seven most prominent PaCIC markers belong to distinct protein families and exert non-related functions. Five of these molecules can become recruited into TEM, where they associate via weak, non-protein-protein interactions with Tspan8. This significantly expands the range of activities of TEM and TEM-derived Exo. Of note, all seven CIC markers contribute via different routes to maintain stem cell features.
Tspan8 and the α6β4 Integrin
Tetraspanins are highly conserved 4-transmembrane proteins with a small and a large extracellular loop (199). The latter accounts for dimerization and association with non-tetraspanin molecules (200, 201). Prominent partners are integrins, proteases, cytoskeleton, and cytosolic signal transduction molecules (202–205). Intracellular, juxtamembrane cysteine palmitoylation supports tetraspanin-tetraspanin web formation, protects tetraspanins from lysosomal degradation and provides a link to cholesterol and gangliosides, tetraspanins mostly acting as molecular facilitators for associated molecules (206–209). As mentioned, Tspan8 contributes to Exo biogenesis (210) and is upregulated in PaCIC and -TEX (211–214).
Tspan8-promoted PaCa migration, invasion, and progression (215–220) relies on the recruitment of additional CIC markers. Tspan8 associates with CD44v6 (213), which recruits cMET and VEGFR21 via CD44v6-bound HGF1 and VEGF1 (216, 221, 222), α6β1 and α6β4 (213, 223, 224), cldn7 and EpCAM (225–227). Some associations depend on the cells' activation state in particular α6β4 (228), a major hemidesmosome component in non-activated cells (229, 230). Upon association with Tspan8, integrins become activated and initiate downstream signaling (231, 232). The tight junction (TJ) component cldn7 (233, 234) only associates upon palmitoylation (234) and recruits EpCAM (235–238). Tspan8 also cooperates with proteases (239–241).
Tspan8/Tspan8-TEX engage in crosstalk with the tumor stroma and premetastatic niche tissue (210) and promote EC progenitor maturation and activation (147, 148, 242). The interaction with the ECM is initiated by Tspan8-associated integrins. Collagen crosslinking assists associated protease activation, which degrade collagen and LN (243). Tspan8-associated α6β4 binding to the LN3321-rich BM promotes tumor cell migration. Liberation of growth factors, chemokines and proteases deposited in the ECM supports tumor cell migration and distant organ settlement (157). TEX Tspan8-integrin and -protease complexes distinctly affect gene expression in different target cells. Tumor cells respond with vimentin, Snail1, and Slug1 expression. In FB proteases (ADAM171, MMP14, TIMP1, and 21) are mainly upregulated (240). Bone marrow cells (BMC) respond with TNFα1 upregulation and STAT41 activation. Lymph node cells (LNC) upregulate TNFα, TGFβ, and FoxP31 expression (240). TEX Tspan8-α4β1/α5β1 (147, 148) targeting EC/EC progenitors induce CXCL51, MIF1, vWF1, and CCR11 mRNA translation. The increase in mRNA after 1d−5d indicates induction of transcription (147). In vivo, EC/lymphatic EC respond with FGF21, VEGFR1, VEGFR2, and VEGFR3 upregulation (244).
In brief, Tspan8 contributes to tumor progression at different levels of the metastatic cascade. Tspan8 is engaged in TEX biogenesis and binding/uptake and acts by clustering integrins, RTK, and proteases, which facilitate downstream signaling (Figure 3).
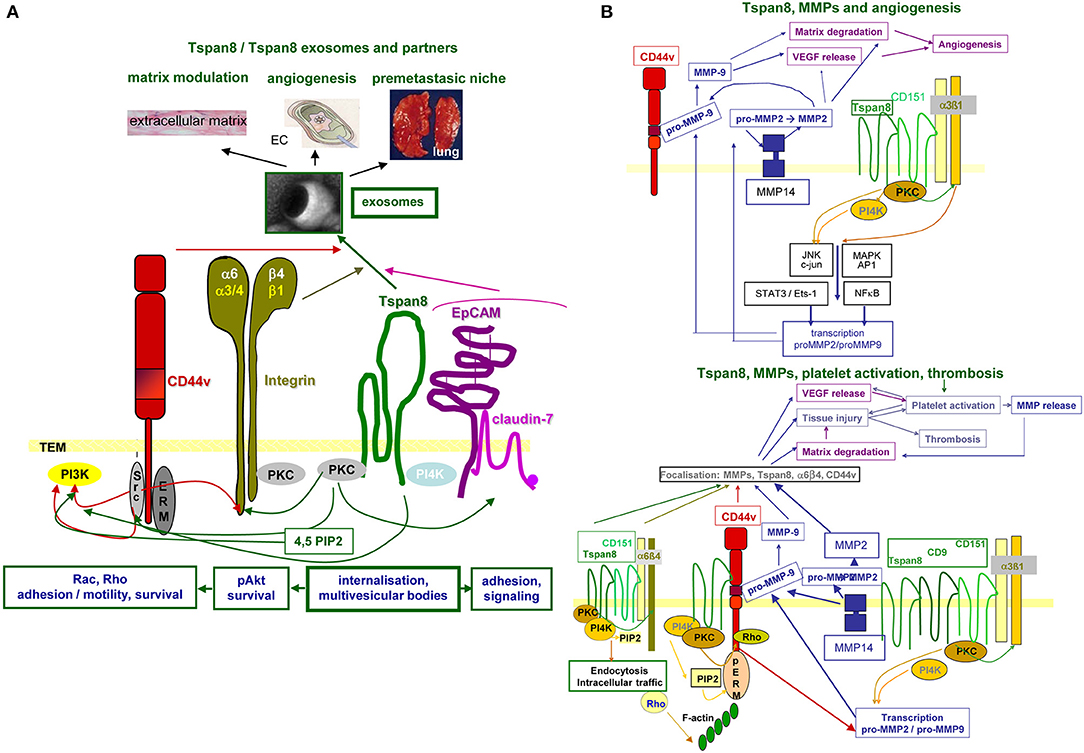
Figure 3. Tspan8 promoted tumor progression. (A) Tspan8 acts as a facilitator. This accounts for membrane bound Tspan8, where it strengthens CD44v6, integrin, and cldn7palm/EpCAM complex signaling activity via its association with PKC and PI4K. This also holds true for the Exo-recruited TEM complex described to modulate the ECM, to promote or inhibit angiogenesis and to contribute actively to premetastatic niche formation. (B) Tspan8 is associated with MMP14 and the association of Tspan8 with α6β1 promotes, besides other the transcription of MMP2 and MMP9. Upon proform activation, also assisted by the proximity to CD44v6, matrix proteins become degraded and VEGF is released. VEGF, in collaboration with collagen degradation products, promotes angiogenesis. In addition, a complex between Tspan8, CD44v6, α6β4, and MMP is found in focal contact. The matrix degradation promoted tissue injury contributes to platelet activation and thrombosis, where together with the release of VEGF a positive feedback loop is created further pushing platelet activation and thrombus formation. Full name of proteins are listed in Table S1. With the multitude of Tspan8 associating molecules, we only present one example building on the association with MMP, which strengthens angiogenesis and thrombus formation. However, it should at least be mentioned that Tspan8 also associates with TACE, which strongly affect e.g., the delivery of the NOTCH and the EpCAM ICD, both acting as cotranscription factors.
The α6β4 integrin was one of the first genes described to be metastasis-associated (245, 246). It is expressed in several normal epithelia, Schwann cells and EC, the β4 chain being characterized by a long cytoplasmic tail (245). A6β4 binds to LN in the BM facilitating adhesion through the formation of hemidesmosomes, nucleating the connection between LN and cytokeratin intermediate filaments (247). Upon stimulation, hemidesmosomes are dismantled allowing leading edge migration (248, 249). Hemidesmosome disassembly is accompanied by α6β4 forming a complex with MST1R/RON1, which interrupts its association with plectin (250). β4-linked activated ERBB21 associates with src1, which initiates phosphorylation of the three components and signaling toward STAT3, which accounts for the breakdown of cell-cell junctions and initiation of invasion (251). Motility involves PI3K1 catalytic subunit beta activation, proceeding via α6β4 promoted IRS1 and−21 phosphorylation (252), PI3K localization into lipid rafts or TEM (253, 254), or ERBB2/ERBB3 activation (255, 256). RAC1 activation strengthens the formation of F-actin-rich motility structures by the cooperation of α6β4 with RTK (257). α6β4-increased cAMP-specific phosphodiesterase activity decreases cAMP and activates RhoA (258). FAK1 regulates β4 tyrosine phosphorylation, which further promotes migration (259). Intravasation and extravasation are assisted by β4 cytoplasmic domain-dependent upregulation of VEGF enhancing transendothelial permeability (260). TEX Tspan8-α6β4 supports premetastatic niche preparations in the lung (92, 261).
β4 contributes to apoptosis resistance via tyrosine phosphorylation of the C-terminal segment of β4 by src family kinases downstream of RTK, but also by syndecan, which directly binds to the β4 cytoplasmic domain (262). Regardless of the initial signals, apoptosis resistance progresses by antiapoptotic PI3K pathway activation (263). TEX β4-vinculin complexes also cope with resistance toward a complex diterpene alkaloid, likely via plectin transfer by TEX (264).
Finally, α6β4 regulates transcription of invasion/metastasis-associated molecules by controlling promoter DNA demethylation. This was demonstrated for NFAT11 (265), which assists autotoxin expression, a motility factor stimulating lipoproteinA production (266). Metastasin1/S100A41 (267) spurs membrane ruffling via rhotekin (268), regulated through NFAT5 in conjunction with S100A4 promoter demethylation (269). S100A4 is also engaged in ERBB2 translation (270).
A6β4 is expressed on mature EC, a contribution to angiogenesis being disputed (271). Although reported to inhibit angiogenesis (148, 272, 273), α6β4 may be engaged in an early stage of angiogenesis (274) via stimulating VEGF translation and signaling (275). The β4 C-terminal domain is important for responding to FGF2 and VEGF (276) and arteriolar remodeling is defective in β4 knockout (ko) cells due to altered TGFβ signaling (271).
Long-known as metastasis-associated, molecular pathways of α6β4 are not fully unraveled. Central are the signaling domain of the β4 tail and the dislodgement from hemidesmosomes. In PaCIC/-TEX, we consider the linkage to Tspan8 as a central coordinator (Figure 4).
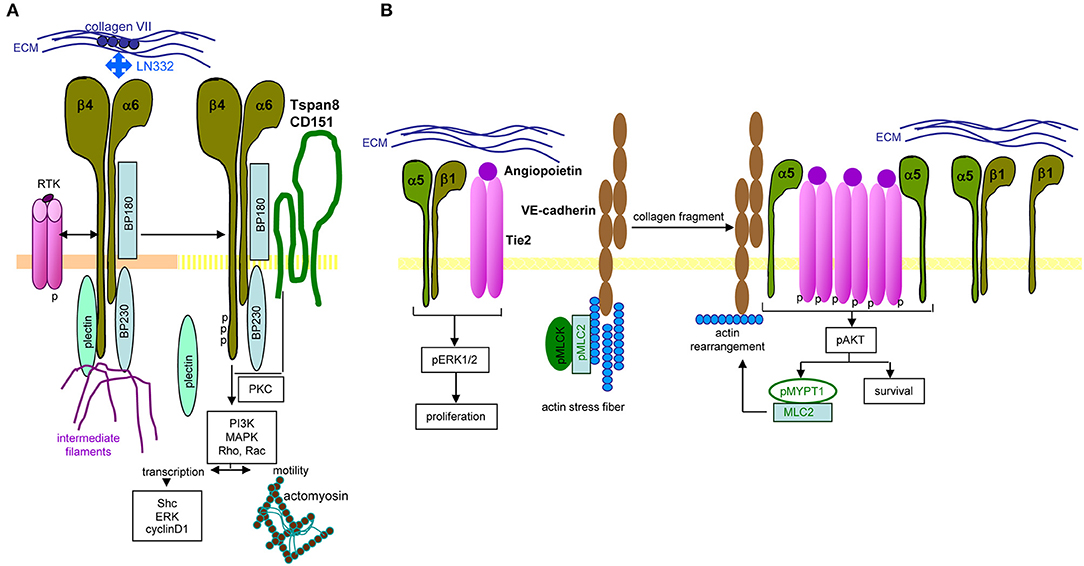
Figure 4. Distinct integrin signaling in PaCIC. (A) Hemidesomosome-integrated α6β4 is associated with BP160/320 and plectin, the complex being linked to intermediate filament. Upon contact with RTK, the β4 cytoplasmic tail becomes phosphorylated, plectin is released from the complex and phosphorylated β4, supported by Tspan8-associated PKC promotes PI3K, MAPK, Rho, and RAC activation. Besides initiating transcription, the complex assists the association with actomyosin and motility. (B) Instead, when α5β1 associates with angiopoietin-activated Tie2, proliferation is initiated via ERK phosphorylation. In the presence of VE-cadherin, linked to actin stress fibers, pMLCK, and pMLC2 collagen fragments initiate actin rearrangement that promotes dissociation of the α5 from the β1 chain, which enclose phosphorylated Tie2. The phosphorylated Tie2 promotes Akt phosphorylation, which supports MYPT1 phosphorylation and MLC2 association that evoke actin rearrangement. Full name of proteins are listed in Table S1. In brief, only parts of integrin-mediated activities are affected by the association with Tspan8. Notably, the same stimulus distinctly affects integrin activation depending on the α or β chain of the integrin.
CD44v6 and CD44v6-Associated Receptor Tyrosine Kinases
CD44v6, the alternatively spliced isoform of the adhesion molecule CD44 is a PaCIC marker involved in several steps of the metastatic cascade (277, 278). CD44, a type I transmembrane glycoprotein, varies in size by N- and O-glycosylation and insertion of alternatively spliced exon products between exons 5 and 6 of the CD44 standard isoform (CD44s) (279–281). CD44 belongs to the cartilage link protein family (282), the globular structure being stabilized by conserved cysteines (283). After the globular domain a heavily glycosylated stalk-like structure has putative proteolytic cleavage sites (284) and contains the variable exon products (285). The transmembrane region facilitates oligomerisation and recruitment into TEM, important for the interaction between CD44 and extracellular ligands and other transmembrane and cytoplasmic molecules (286). The cytoplasmic tail binds signaling and cytoskeletal linker proteins (287, 288). Most CD44s activities are maintained by CD44v.
CD44 has multiple ligands, which contribute to tumor progression. The link domain binds collagen, LN, FN, E-, and L-selectin (289, 290). CD44 has binding sites for glycosaminoglycans (GAG) and is the major HA receptor that binds to a basic motif outside the link domain (291–293). CD44v6 binds HGF, VEGF, and osteopontin (294–296). These associations are of central importance for its lateral associations with RTK. HGF binding brings CD44v6 into proximity with cMET and expedites cMET activation, which requires interaction between the CD44 cytoplasmic tail and ERM (ezrin, radixin, moesin) proteins for Ras1-MAPK1 pathway activation (297). CD44v6-ECM binding also contributes to cMET transcription (298). Lateral association-initiated signal transduction also accounts for IGFR11 and PDGFR1 (299). The HA crosslinking-initiated CD44 association with CXCR4 promotes SDF1 binding (300). The association with the low-density lipoprotein (LDL1) receptor-related LRP61 strengthened activation of the EMT-related Wnt1 signaling pathway (301). Cytoplasmic tail-bound ankyrin contacts with spectrin support HA-dependent adhesion and motility (287). ERM proteins regulate migration, cell shape, and protein resorting (302, 303). The N-terminus of activated ERM proteins binds CD44, the C-terminus F-actin (304). Cytoskeletal linker protein binding expands the range of CD44-mediated downstream signaling pathways (303, 305), which can also proceed directly from TEM-located CD44v (306–308) or associated non-RTK (309, 310). The CD44/CD44v6-associated membrane-bound proteases MMP14 and Hyal21 (311) support tumor cell migration through matrix degradation and remodeling (312). CD44 contributes to drug resistance (313) by associating with ABC1 transporters (314, 315) and additional antiapoptotic proteins (316, 317). Last, not least, the CD44 cytoplasmic tail (CD44ICD) moves toward the nucleus functioning as a cotranscription factor (318). Alternatively, the CD44v6 cytoplasmic tail can affect transcription by activation of signal-transducing complexes. With regard to the metastatic cascade, CD44v6 was described to directly or indirectly activate Tspan8, MMP9, MDR11, and NOTCH11 transcription (221, 319–321). Finally, CD44v6, but not CD44s, is engaged in loading ILV with miRNA (159, 322), which might rely on its association with Dicer (322) and contributes to tumor progression (323).
In brief, CD44v6 engages in EMT induction by supporting Wnt signaling and Nanog and Notch activation (324–326). It contributes to intravasation through binding and degradation via associated proteases. It supports extravasation by selectin binding to EC, allowing crawling toward EC-EC gaps. It assists tumor stroma formation and premetastatic niche preparation by HA, matrix-remodeling enzyme, cytokine, and chemokine provision (91, 327). Recruiting miRNA into ILV expands the range of TEX activities (322). A few of the many CD44v6 activities in tumor progression are shown in the accompanying figure (Figure 5).
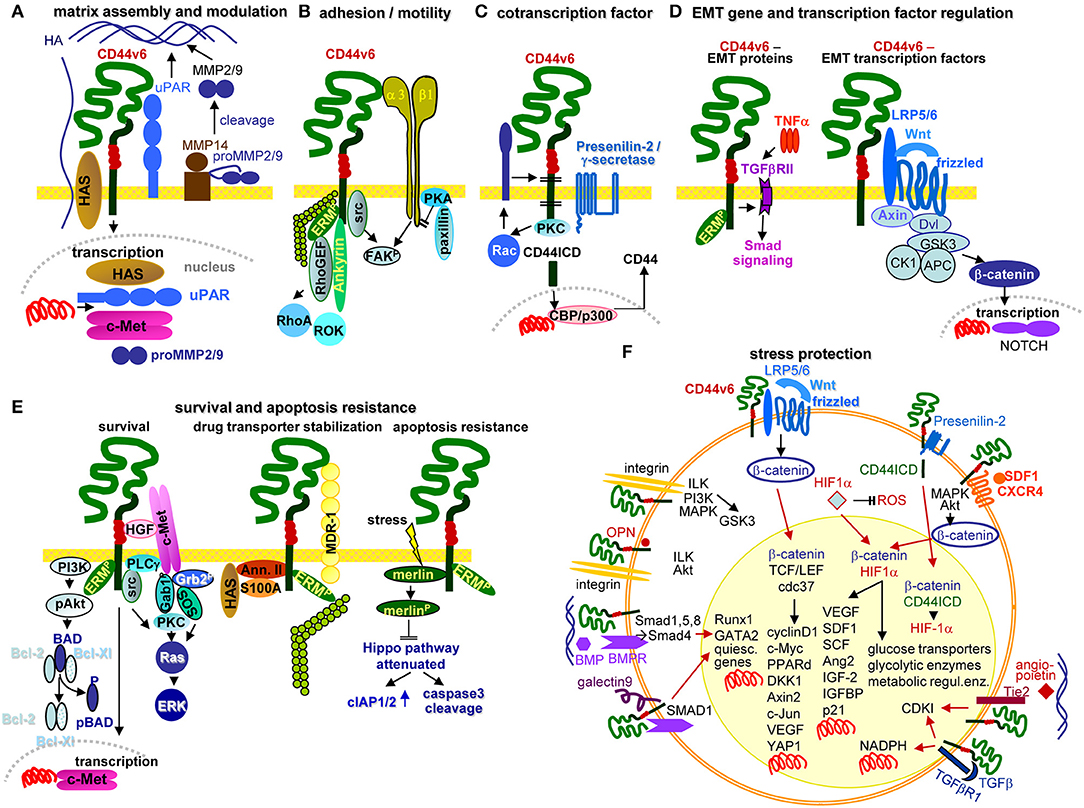
Figure 5. Multifaceted activities of CD44v6 in PaCIC. (A) Upon HA crosslinking, CD44v6 initiates HAS, uPAR, MMP2, and MMP9 transcriptions, which promote HA assembly and matrix remodeling, where MMP14 contributes to proMMP2 and MMP9 cleavages. (B) CD44v6 can associate with α3β1 such that both molecules jointly contribute to FAK activation and motility. (C) CD44v6 can be cleaved by TACE and subsequently by the presenilin2 complex. The CD44ICD acts as a cotranscription factor, which together with CBP/p300 promotes CD44 transcription (D) By TNFα associating with TGFβRII, EMT protein expression is supported via Smad signaling. The association of CD44v6 with LRP5/6 supports Wnt/frizzled activation such that β-catenin leaves the suppressive complex and acts as cotranscription factor in NOTCH transcription. (E) There are several pathways whereby CD44v6 strengthens PaCIC survival and apoptosis resistance. cMET comes into proximity of CD44v6 via CD44v6-bound HGF. This initiates activation of the PI3K/Akt anti-apoptotic and of the Ras-ERK pathways. In addition, CD44v6 supports cMET transcription. A complex of CD44v6 with HAS, Annexin II, S100A, and activated ERM stabilizes MDR1 expression, which contributes to drug efflux. Finally, stress induces the association with and dephosphorylation of merlin, which attenuates the HIPPO pathway with upregulation of cIAP1/2 and caspace3 cleavage. (F) Some of the multiple activities of CD44v6 in stress protection via affecting the cells metabolism are summarized indicating whether altered metabolism is promoted by signaling cascades in the cytosol or depends on transcriptional activation (red arrows). The latter accounts particularly for β-catenin-TCF/LEF, β-catenin-HIF1α, and β-catenin-CD44ICD complexes, but also for the cooperation of CD44v6 with Tie2, TGFβR1, galectin 9, and BMPR, which affect transcription of a large range of distinct genes. Full name of proteins are listed in Table S1. CD44v6 is engaged in most steps of the metastatic cascade. The strongest impacts are seen in terms of survival, EMT induction and metabolic changes that guarantee unimpaired survival under hypoxic and poor nutrient conditions.
CXCR4 and Its Association With Tspan8 and CD44v6
CXCR4 has been tied to tumor progression and poor prognosis (328, 329) and expression of its ligand SDF1 correlates with poor survival (97).
CXCR4 is expressed in BMC/-precursors, lymphocytes, resident macrophages (Mϕ), EC precursors, FB, and CIC. CXCR4 is a seven transmembrane GPCR (330), transcription increasing in response to several signaling molecules such as cyclic AMP, some cytokines including TGFβ and the growth factors FGF2 and VEGF (331). Upon ligand binding, CXCR4 undergoes a conformational change activating the intracellular trimeric G protein leading to the Gαi dissociation, which stimulates src, Ras/Raf1/MAPK (332) and PI3K pathways (331, 333). Gβγ triggers PLC, which catalyzes PIP2 into IP3 and DAG leading to Ca++ mobilization and PKC1 and MAPK activation (334). CXCR4 also triggers a G-protein-independent pathway (335) promoting recruitment of GRK21 that phosphorylates the C-terminus resulting in β-arrestin association. CXCR4 thereby uncouples from G proteins and becomes internalized (336, 337). GRK2 is supported by PKC, PKA, and src (338). β-arrestin serves as a scaffold for downstream signaling promoting ERK/MAPK1 and p38/MAPK14 activation (339). Proper folding depends on HSP90, a chaperone for members of the CXCR4 phosphorylation cascade (340). Colocalization of these complexes in cholesterol-enriched lipid rafts (341) facilitates signal transfer (342).
CXCR4 contributes to tumor progression at multiple levels. CXCR4 sustains proliferation through direct activation of MAPK, PI3K, Wnt, and HH1 signaling (343), where HH additionally induces SDF1 expression in the tumor surrounding (344) and activation of the intrinsic anti-apoptotic pathway via ERK and Akt1 (344, 345). CXCR4 assists invasion, HH signaling being associated with EMT and loss of adhesion (344). SDF1 increases MMP2, MMP9, and urokinase expression (346, 347). Particularly in PaCa, CXCR4 expression is linked to a subpopulation of migrating, metastasis-promoting PaCIC (348) that is highly chemotherapy resistant (349–351).
The involvement of CXCR4 in tumor progression is not restricted to tumor cells. EC respond to HIF1α1 with CXCR4 upregulation (352). The SDF1-CXCR4 axis enhances VEGF and MMP production through ERK and Akt signaling (353), which promotes EC migration and capillary tube formation (354). Activated PSC (aPSC) promote SDF1 secretion, which binds to EC CXCR4 and is supported by PAUF1. SDF1 together with VEGFC also attracts lymphatic EC (354). Furthermore, tumor stroma cell-secreted SDF1 assists CXCR4 activation in tumor cells and CXCR4-induced HH expression stimulates CAF recruitment (344). By stimulating IL61 production, CXCR4 assists TAM recruitment (343) and mast cell recruitment and activation. Mast cells release IL13, which activates PSC, further promoting tumor growth (355). Other CXCR4-recruited immune cells force CXCR4 expression via IFNγ creating a positive feedback loop (356). The link between high CXCR4 expression and bone metastases relies on circulating tumor cells passing through the bone vessels, hematopoietic and mesenchymal progenitors highly expressing SDF1 (357). Another CXCR4 ligand is SDF1-associated HMGB11, which is also a ligand for AGER (358) and TLR2, 4, and 91 (359, 360). SDF1/HMGB1 complex binding to CXCR4 promotes inflammatory cell recruitment (361) (Figure 6).
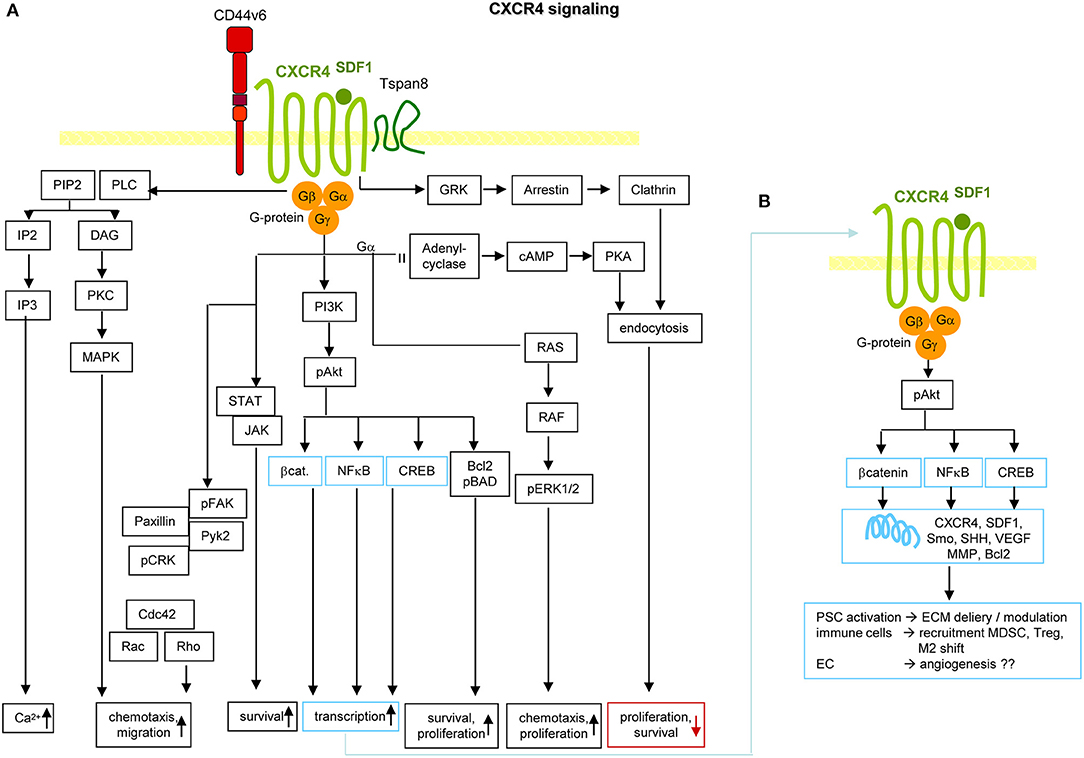
Figure 6. CXCR4 and PaCIC survival and motility. (A) CXCR4 is a G-protein coupled receptor (GPCR) that in PaCa is mostly recovered in association with CD44v6 and/or Tspan8. Activation is initiated by binding of its ligand SDF1. Signals are transferred via the G protein subunits, which promote Ca2+ influx, and either via MAPK or Rho chemotaxis and migration. Chemotaxis and proliferation can also proceed via the Gα, Ras, Raf, pERK1,2 activation route. Activation of PI3K/Akt, Bcl2/pBAD promotes proliferation and survival. The latter is also supported by activation of the STAT-Jak pathway. PI3K/Akt can also initiate activation of transcription factors. Independent of the trimeric G-protein complex, CXCR4 associates with GRK, arrestin and clathrin. The complex becomes internalized, which is accompanied by reduced proliferation and survival. (B) Activation of β-catenin, NFκB and CREB supports transcription of CXCR4, SDF1, Smo, SHH, VEGF, MMP, and Bcl2. These genes are important in PSC activation, recruitment of immunosuppressive MDSC and Treg and the shift of M1 to M2 and in supporting angiogenesis, which may not be dominating in PaCa. Full name of proteins are listed in Table S1. It should be noted that the dominant activity of CXCR4 in promoting chemotaxis and motility covers only one, not essentially dominating feature in PaCIC.
In 2007, a first series of reviews pointed out the special role of CXCR4 in subpopulations of migrating/metastasizing CIC (348, 362, 363). Great progress over the last decade extended original findings toward the involvement of tumor stroma and EC. Although the extent of CXCR4 heterocomplex engagement in leukocyte recruitment awaits further exploration (364), based on promising results, great efforts are taken toward therapeutic translation (100, 365, 366).
Claudin7 and EpCAM
Claudins, including cldn7, are a four-pass proteins, which are the central TJ components (232, 367). TJ are found in epithelial and endothelial cells, cldn7 expression being particularly high in the gastrointestinal tract and lymphatic vessels (368). TJ, composed of the transmembrane proteins occludin, JAM and cldn, linked to zonula occludens proteins (ZO1), are located in the most apical lateral region of cell-cell contact sites (367). The transmembrane proteins are laterally linked via claudins, and tightly associate with TJ on opposing cells (369). TJ seal the organism from the outside and are involved in paracellular transport (370). The latter is determined by the polarity of the β-sheet of the extracellular loops of cldn, which differs between individual cldn and is adjusted to selective organs' demands (371). Both barrier and channel functions of TJ-integrated cldn are vital. Cldn7ko mice die within 10 days after birth due to gut destruction that might rely on a missing association with integrins and strong MMP3 upregulation or on enhanced paracellular influx of colonic inflammation-inducing bacterial products (372, 373). Apart from sealing and paracellular transport (370, 371, 374–376), few reports explore cldn-Exo/TEX activities. It was recently realized that a comparably large amount of continuously remodeled TJ components is recovered insight the cell and at distinct membrane locations (377–379). TJ remodeling rests on claudins being PKA, PKC, and MLCK1 targets, cldn phosphorylation prohibiting TJ integration (380–385). Furthermore, TJ formation depends on sphingomyelin with long-chain fatty acids and cholesterol enrichment in membrane subdomains, cholesterol depletion affecting cldn integration and abolishing TJ formation (386). Finally, cldn7 is also located in the plasma membrane outside of TJ. Cldn7 palmitoylation is a precondition for partitioning into TEM, where palmitoylated cldn7 associates with EpCAM and tetraspanins (234, 387).
Internalized, TJ-derived cldn can be degraded, recycle or integrate into EE and, after passage through MVB, into Exo. In fact, TEM-located, palmitoylated and EpCAM-associated cldn7 is exclusively recovered from apical plasma membrane derived TEX (388, 389). In organoids, a second population of cldn7+/EpCAM- TEX is delivered at the basal membrane (389), which might derive from internalized TJ, facilitated by the high cholesterol content. Intracellular vesicle traffic remains to be defined (378). Alternatively, Exo-recruitment during biogenesis is not excluded (390) and would be consistent with pronounced coimmunoprecipitation of cldn7 with Golgi-endoplasmic reticulum (ER) transporters (391).
Pinpointing the activity of cldn7 in the metastatic cascade is difficult. Palmitoylated, EpCAM-associated cldn7 might favor signaling by supporting EpCAM cleavage and EPICD cotranscription factor activity in EMT. However, it is hard to demarcate from support by other TEM-located CIC markers. TJ-integrated and phosphorylated cldn7 is associated with a wide range of transporters, which likely impacts altered metabolism and signal transduction of CIC (Figures 7A,C). These options await untangling exploration.
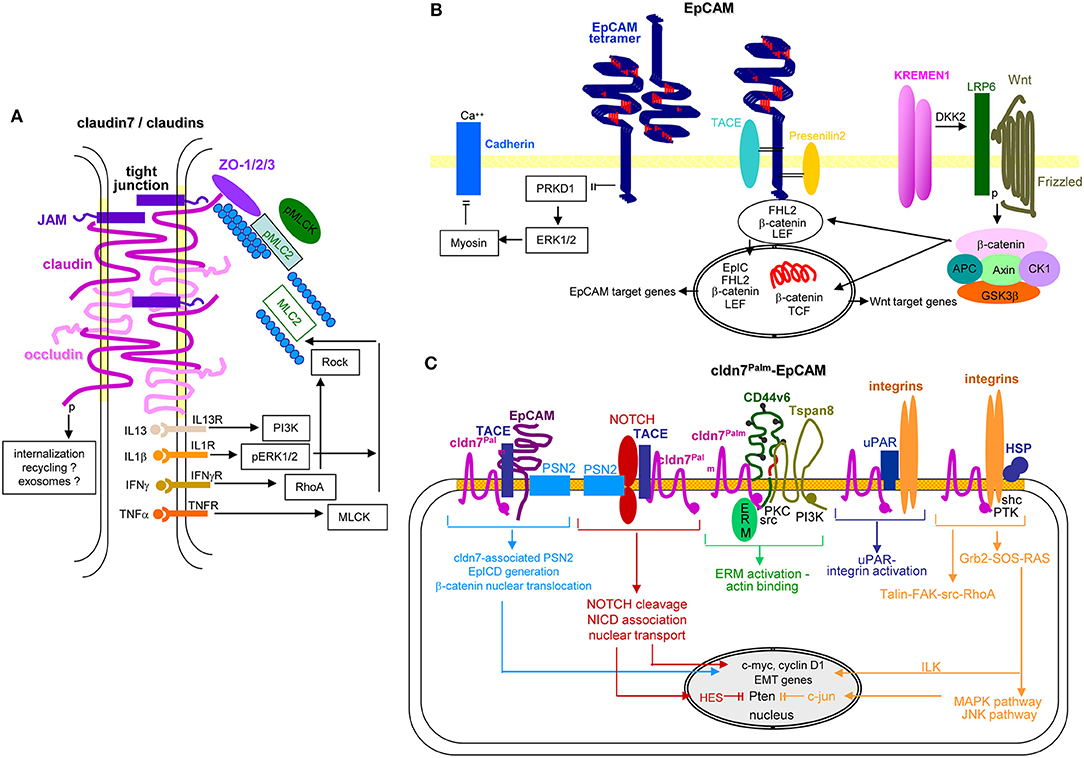
Figure 7. EpCAM, claudin7 and their cooperation in PaCa. (A) In tight junctions, cldn7 is associated with additional cldns, occludin, JAM, and ZO1, 2, and 3, the latter being associated with pMIC2 and the cytoskeleton. Upon stimulation by several cytokine receptors, PI3K, ERK, RhoA/Rock, and MLCK promote MLC2 dephosphorylation, which promotes reorganization of the cytoskeleton with consequences on the cldn associated transporter activity. A stress response provokes internalization of the TJ complex. It is suggested that the internalized complex may be partly digested, recycle and become integrated into Exo. (B) EpCAM form tetramers, which with low affinity bind to EpCAM tetramers on neighboring cells and concomitantly prevent PRKD1 activation that leads to ERK1/2 and myosin activation, which inhibits Ca++-dependent Cadherin adhesion. Alternatively, EpCAM becomes cleaved by TACE and subsequently by PSN2. The cotranscription factor EpICD becomes supported by LEF and ß-catenin that might derive from Kremen1-DKK2-LRP6 promoted Wnt-Frizzled activation. This transcription factor complex mostly supports EpCAM and Wnt target gene expression. (C) Palmitoylated cld7 associates with monomeric EpCAM. As cldn7 is associated with PSN2, EpICD generation is augmented. Palmitoylated cldn7 may also contribute to NICD generation that acts as cotranscription factor. In association with CD44v6 and Tspan8, cldn7palm associates with ERM and contributes to ERM activation and actin binding. In association with uPAR and integrins it promotes both uPAR and integrin activation. Finally, a cldn7Palm-integrin-HSP complex assists talin-FAK-src-RhoA activation and by activation of the Grb2-SOS-RAS pathway ILK and the MAPK-JNK pathway. EpICD, NICD, and ILK contribute to c-myc, cyclinD1 and EMT gene transcription; NICD via HES and c-jun interfere with Pten transcription. Full name of proteins are listed in Table S1. Thus, TJ cldn7 is important particularly in lipid transport and cytoskeleton organization, EpCAM by promoting oncogenes and EMT genes, which also accounts for cldn7Palm-associated EpCAM. Cldn7Palm additionally contributes to Pten silencing.
The epithelial cell adhesion molecule EpCAM, overexpressed in many epithelial cancer, serves as diagnostic and therapeutic target (392). EpCAM mediates homophilic cell-cell adhesion (393, 394) and fulfills condition-dependent distinct functions (395). An initial, straight-forward explanation that oncogenic and tumor progression supporting EpCAM activities rest on interfering with E-cadherin-mediated adhesion required revisiting, when it was realized that EpCAM can be cleaved by TACE and subsequently presenilin1, which generates EpICD (396). EpICD functions together with TCF/LEF1 as a cotranscription factor for MYC, cyclinA/E, Oct41, and Nanog amongst others (397, 398). EpICD is also engaged in hypermethylation and activation of BMP1 promoters (399) and can promote EMT through increased Slug and PTEN/Akt/mTOR1 signaling pathway activation (400) and engagement in Wnt signaling. PKC downregulation and MMP7 upregulation backs EpCAM-promoted motility (401–406). Indicating its regulatory effect on another major pathway, EpCAM silencing reduces Ras/Raf/ERK signaling (407). However, EpCAM expression is transiently downregulated during EMT (401, 408, 409), which could argue for EpCAM prohibiting tumor cell dissemination (410, 411). Nonetheless, strong overexpression on embryonic SC endorses a contribution to pluripotency maintenance (412, 413).
EpCAM expression is epigenetically regulated. High EpCAM expression correlates with hypomethylation of a fragment of exon 1 and the proximal promoter, lack of EpCAM expression correlates with methylation at a proposed Sp1 binding site (414, 415). Furthermore, activating histone modifications acH41, acH31, and H3K4me31 correlate and repressive histone modifications H3K9me31 and H3K27me31 inversely correlate with EpCAM expression (413, 416, 417). Additionally, EpCAM regulation by ncRNA might be relevant to the crosstalk between TEX and targets. LncRNA LINC00152 activates mTOR through binding to the EpCAM promoter region (418). Furthermore, miR-150, miR-155, miR-181, and miR-223 expression is increased in EpCAM+ hepatocellular carcinoma (HCC). MiR-155 contributes to EpCAM-promoted migration and invasion (419) and miR-29b to proliferation and inhibition of liver progenitor cell differentiation (418). Since miR-16-5p, miR-23a-3p, miR-23b-3p, miR-27a-3p, miR-27b-3p, miR-30b-5p, miR-30c-5p, and miR-222-3p are high in EpCAM+ colorectal cancer (CoCa) TEX, an EpCAM-dependent recruitment is discussed (420).
In brief, possibly due to abundant expression in most epithelial cells and upregulated expression in many primary tumors, the CIC features of EpCAM are more difficult to define than originally expected. Notwithstanding, EpICD contributes to the metastatic process by acting as a cotranscription factor. We personally interpret the transient downregulation during EMT as evidence for EpCAM not contributing to intravasation, intravascular traffic or extravasation. Expression during settlement of migrating tumor cells in distant organs could indicate a share in premetastatic niche preparation (Figures 7B,C). An interpretation of EpCAM regulation by lncRNA and miRNA might be premature.
LGR5
The leucin-rich repeat containing GPCR-5 (LGR51) is a Rhodopsin GPCR, expressed in adult SC and best explored in intestinal SC and CIC (421). Secreted Wnt proteins interact with the Wnt receptor complex consisting of Frizzled and LPR5/6. Wnt binding sustains dissolving the downstream destruction complex and liberated β-catenin acts together with TCF/LEF as Tf (422). LGR5 is one of the targets of TCF41 (423), which regulates Wnt signaling. In the absence of Wnt, Frizzled, LPR5/6 and the RING-type E3 ubiquitin ligases RNF431 and ZNRF31 form a complex, which promotes Frizzled ubiquitination and degradation. Upon soluble R-spondin binding to LGR5, RNF43 becomes phosphorylated and sequestered generating a more stable complex between R-spondin, LRP5/6, and Wnt-Frizzled, which promotes β-catenin liberation (424, 425). This suggests LGR5 elimination hampering tumor progression. LGR5 elimination transiently retarded local tumor growth, possibly reflecting CIC plasticity, where differentiated cells can revert to LGR5+ CIC. Instead, metastatic growth was enduringly inhibited (426, 427).
Briefly, by regulating Wnt signaling, LGR5 is important for CIC maintenance and thereby tumor progression (Figure 8).
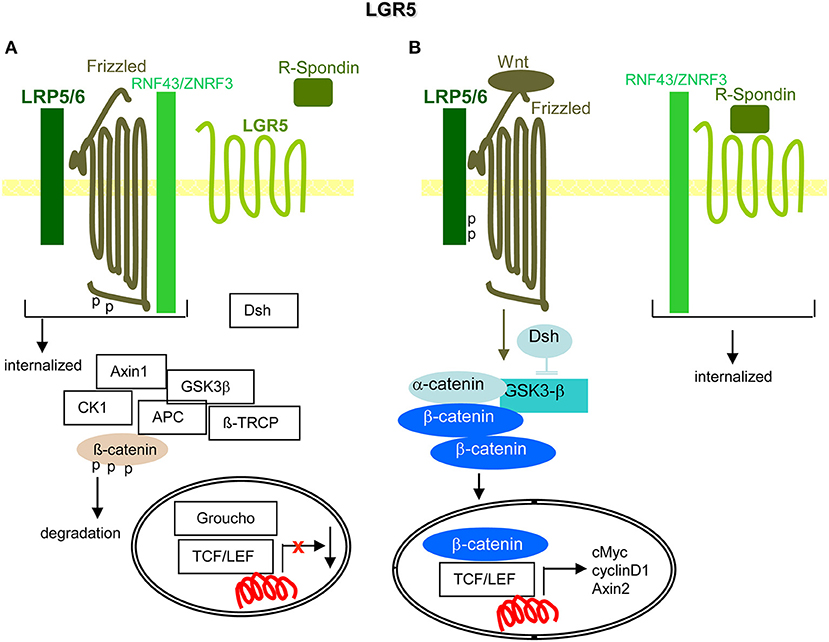
Figure 8. LGR5 and the contribution to PaCIC maintenance. (A) The leucine-rich repeat-containing GPCR is engaged in Wnt signaling. Its ligand is R-Spondin. In the absence of R-Spondin, the transmembrane E ligases RNF43/ZNRF3 associate with Frizzled and LRP5/6 that leads to Frizzled phosphorylation and internalization of the complex. (B) However, in the presence of R-Spondin, RNF43/ZNRF3 is recruited toward LGR5 such that Wnt can bind to Frizzled and LRP5/6 becomes phosphorylated. Dsh blocks GSK3-β and β-catenin is liberated to move to the nucleus, where it together with TCF/LEF promotes cMyc, cyclinD1, and Axin1 transcription. Full name of proteins are listed in Table S1. The upregulated expression of LGR5 in PaCIC suggests its engagement in PaCIC maintenance.
CD133
CD133 (Prominin1) is a hematopoietic SC and CIC marker in many malignancies (428, 429), high expression being associated with poor prognosis (430). CD133 is a 5-transmembrane molecule in protruding membrane subdomains, where it interacts with cholesterol-based lipid rafts (428, 431). CD133 contributes to cell polarity, cell-cell and cell-ECM interactions (432) and signaling cascade activation (433). Expression is enhanced by binding to HDAC61 that stabilizes β-catenin in a ternary CD133-HDAC6-β-catenin complex promoting β-catenin target activation. Loss of CD133 is accompanied by reduced SLUG, LAMC11, and MMP7 expression and a shift toward MET (434). CD133 activity is regulated by the tyrosine phosphatase receptor PTPRK1, which dephosphorylates tyrosines 828 and 852. Low PTPRK expression in patients with cancer is associated with pronounced AKT activation and poor prognosis (435).
CD133 interferes with CIC differentiation by suppressing NTRK2 via p38MAPK and PI3K signaling (436). A CD133kd is also accompanied by a strong decrease in invasion and TIMP2 expression, the pathway remaining to be explored (437). CD133 affects migration via Akt or src activation and FAK phosphorylation (438, 439). A suggested engagement in drug resistance might proceed via CD133 directly interacting with PI3K-p85, resulting in multidrug resistance (440). Finally, neighboring cells support CD133 activities, e.g., EC secrete a soluble form of Jagged11 promoting Notch activation (441).
According to the location in internalization-prone rafts, CD133 is recovered in Exo/TEX (442–444). CD133 intracellular traffic follows an ESCRT-independent pathway and requires ceramide, neutral sphingomyelinases and the sphingosine-1 phosphate receptor S1PR11, confirmed by reduced MVB formation upon expulsion of S1PR1 by α-synuclein1 (445, 446). The expected CD133-TEX contribution to intercellular communication requires exploration (107). However, endosomal CD133 at the pericentrosomal region captures GABARAP1, an initiator of autophagy. This prevents GABARAP from mediating ULK11 activation and autophagy, whereby pericentrosomal CD133 sustains CIC undifferentiated state maintenance (447).
CD133 shares with several metastasis-associated markers the recovery in SC and CIC. It is engaged in CIC maintenance, Wnt/β-catenin signaling and contributes to migration and invasion, molecular mechanisms being not fully elucidated.
CIC Markers, Stemness, and EMT
Before summarizing the importance of PaCIC markers in tumor progression, we need commending on the linkage between CIC and EMT. Partial activation of the embryonic EMT program was considered a central feature of CIC and a prerequisite for metastasis formation (5). This was recently questioned for PaCa, where the EMT-related Tf Snail and Twist do not contribute to PaCa metastasis, but promote proliferation (448). On the other hand Notch2 and its ligand Jagged-1 are highly upregulated in drug-resistant PaCa cells and a NOTCH2 kd is associated with a partial reversion of the EMT phenotype with decreased vimentin, ZEB1, Slug, Snail, and NFκB expression (449). A more recent publication, describing ZEB1 being essential for PaCa progression, offers a plausible explanation, proposing context-dependent complementary subfunctions of distinct EMT-related Tf (450). Thus, the suggestions of CIC stemness and (partial) EMT requirement in supporting tumor progression, are not yet unambiguously answered (5). Taking the frequently unimpaired growth of the primary tumor mass and of established metastases after therapeutic trials to deplete CIC markers and/or selected Tf, we expect that both stemness markers and partial EMT greatly facilitate tumor progression.
Despite remaining open questions, we want to close this chapter with a personal experience, dating back to 1978, where a local tumor and ascites of a spontaneously arising PaCa were isolated from a rat (451). After subcutaneous transfer, rats receiving local tumor tissue developed local tumors, but not metastases. Rats receiving ascites did not develop a local tumor, but metastases in draining and distant lymph nodes and became moribund due to thousands of miliary lung metastases (452). The local tumor does not, the metastasizing tumor expresses all previously listed PaCIC markers (453). While overexpression of CD44v6, Tspan8, β4, EpCAM, and cld7 supported selective metastasis-associated features, but not the full-fledged metastatic profile (242, 454–457), a kd of each of these markers was accompanied by loss or strongly reduced metastasis formation (240, 388, 458, 459). CIC being unknown at that time, our “blind” studies may convincingly demonstrate the strong impact of CIC markers in tumor progression, their interdependent activities, and importantly, the requirement for a tumor-host crosstalk, the topic of the following chapters.
Stroma Dysplasia in Pancreatic Cancer
PaCa is characterized by an exuberant desmoplastic stroma reaction (DR) that may occupy far more space than the tumor cells, which form small nodules embedded in the dense DR (460). The DR is composed of ECM proteins, PSC, FB, EC, immune cells, and neurons (461).
PSC, quiescent in the healthy pancreas, are located in the basolateral region of acinar cells (462, 463). They are characterized by GFAP1, desmin, vimentin, nestin, NGF, and NCAM1 (464). During pancreatic injury, PSC develop a myofibroblast phenotype expressing αSMA1, actively proliferate and migrate. Activation of PSC is promoted by TGFβ, HGF, FGF, EGF, and sHH1 (465) (Figures 9A,B).
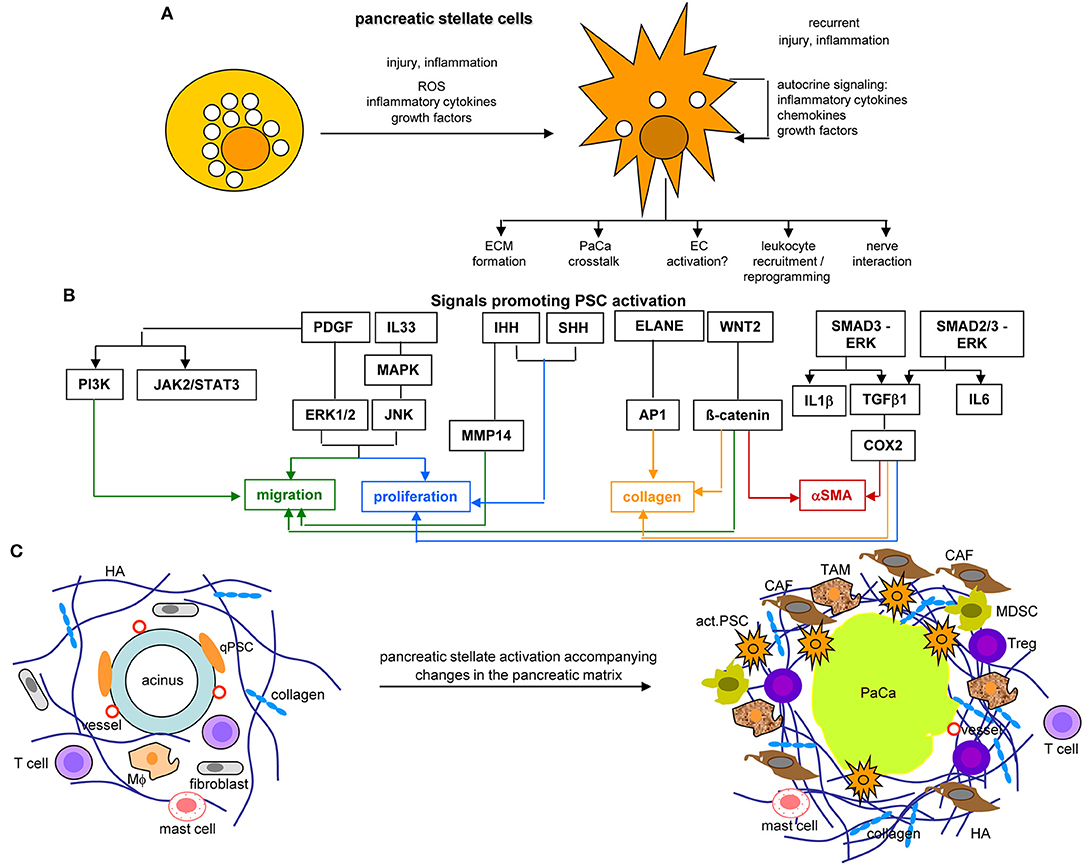
Figure 9. The core position of pancreatic stellate cells in the dysplastic stroma reaction in PaCa. (A) PSC abundantly contain lipid droplets and lay close to the acinar cells in the healthy pancreas. They become activated by injury or inflammation, with a contribution of inflammatory cytokines, growth factors and ROS. Recurrent injury promotes autokrine signaling with further provision of growth factors, inflammatory cytokines, and chemokines. They partly loose the lipid droplets and become dispersed throughout the pancreatic stroma, where they affect the ECM, PaCa cells, leukocytes, and nerves. (B) Main factors contributing to PSC activation are PDGF and IL33 that assist proliferation and migration, Wnt2-β-catenin and IHH-MMP14 also contribute to the migratory phenotype and IHH-/SHH-Cox2 to proliferation. ELANE-AP1, Wnt2-β-catenin, and Smad3-ERK-TGFß1-Cox2 support collagen secretion, the latter two also support αSMA expression. (C) PSC activation is accompanied by the generation of a very dense ECM rich in HA and collagen, the recruitment of CAF, TAM, MDSC, and Treg, but a paucity of T cells in the dense ECM. Finally, they are engaged in a most intense crosstalk with the PaCa cells. Full name of proteins are listed in Table S1. PSC become activated at an early stage of PaCa initiation. Signals promoting PSC activation contribute to PSC collagen and αSMA expression, proliferation, and migration. aPSC are supposed to account for the ECM formation, to crosstalk with the tumor cells, to recruit and reprogram of leukocytes and to interact with the intrapancreatic nerves, some of these activities are detailed in the following figures.
Activated PSC (aPSC) modulate the tumor matrix. They secrete ECM proteins including collagen I, III, and IV, FN and LN (464). Matrix deposition is supported by epithelial cell secreted SERPINE21, which activates PSC resulting in enlarged ECM protein deposits (466). PSC secrete MMP2, MMP9, MMP13, TIMP1, and TIMP2, which account for matrix modulation (467–470). aPSC also affect immune cells. They express TLR2-5, required for non-adaptive immune cell activation (471), but also TLR9, which is protumorigenic via CCL11. CCL11 recruits regulatory T-cells (Treg) and promotes myeloid-derived suppressor cell (MDSC) proliferation (472). aPSC express MHCII and present tumor antigen peptides (473). However, in the absence of costimulatory signals MHC II presentation is not sufficient for helper T-cells (Th) activation (474). Further supporting immunosuppression, aPSC express high level of CXCL10/IP101, which correlates with a Treg increase and reduced CTL (cytotoxic T lymphocyte) and NK (natural killer cell) activity (475). aPSC also express T-cell apoptosis-inducing GAL1 (476, 477). Nonetheless, the impact of PSC on the immune system is still debated, as reverting activated to resting PSC appears superior to PSC elimination (478–480) (Figure 9C).
Taken together aPSC/CAF account for the dense stroma formation and ECM modulation. The DR provides a barrier for immune cells, but also for chemotherapy by poor drug access (481). Beyond this “passive action,” aPSC/CAF contribute to the acquisition of major hallmarks of PaCa via cytokines, chemokines, growth factors, and their receptors that promote tumor cell proliferation and chemoresistance, accelerate intrapancreatic nerve invasion and distant metastatic growth and assist establishing an inflammatory milieu that forces immune destruction (482). aPSC/CAF supply essential nutrients and promote metabolic reprogramming backing tumor cell survival and proliferation (483), which is assisted by aPSC/CAF miRNA (484). These activities are briefly elaborated in the following sections. Despite overwhelming evidences for aPSC/CAF supporting PaCa growth and progression, under selected circumstances they may provide a host defense against the tumor, the hypothesis building on poorer prognosis after HH depletion and in αSMA-ko mice (485, 486).
Activated Pancreatic Stellate Cells and the Crosstalk with Tumor Cells
The extensive crosstalk between aPSC and the embedded tumor cells is pivotal for PaCa survival and progression. Provision of TGFβ, PDGF, FGF2, profibrinogenic factors, serpin2, galectins3, and 9 sustain persisting PSC activation, proliferation, migration, and collagen synthesis. The aPSC also provide growth factors and nutritients (Figure 10A). aPSC/CAF secrete SPARC1, involved in cell migration and proliferation (487), and periostin, which modulates invasion via AKT signaling and EMT (488, 489). Most abundant chemokines are CXC/CC family members CCL2/MCP11, CXCL8/IL8, CXCL11, and CXCL2/MIP21, all engaged in PaCa progression (490–492). Increased radioresistance by aPSC/CAF relies on ß1 integrin-FAK activation and DNA damage response regulation (493, 494). An impact on chemotherapy resistance hinges on accessibility (495), activation of the SDF1-CXCR4 axis with subsequent upregulation of IL6, increased HH expression, and IL1β-IRAK41 or mTOR/EIF4E1 pathway activation (496–501). Finally, aPSC/CAF support metastasis formation via the HGF/cMET/survivin pathway, which is regulated by TP531/CDKN1A1 (502) or through altered lipid metabolism, particularly oleic-, palmitoleic-, and linoleic-acid upregulation (503). Tumor progression is further supported by CAF through partial EMT induction by HH signaling (504) and through aPSC-Exo delivering tumor growth promoting miRNA and lncRNA, which liberate oncogenic/metastasis-promoting mRNA from suppressive miRNA to name only one of the lncRNA functional activities (133). Furthermore, aPSC accompanying migrating tumor cells provide in loco support in establishing premetastatic niches (505, 506).
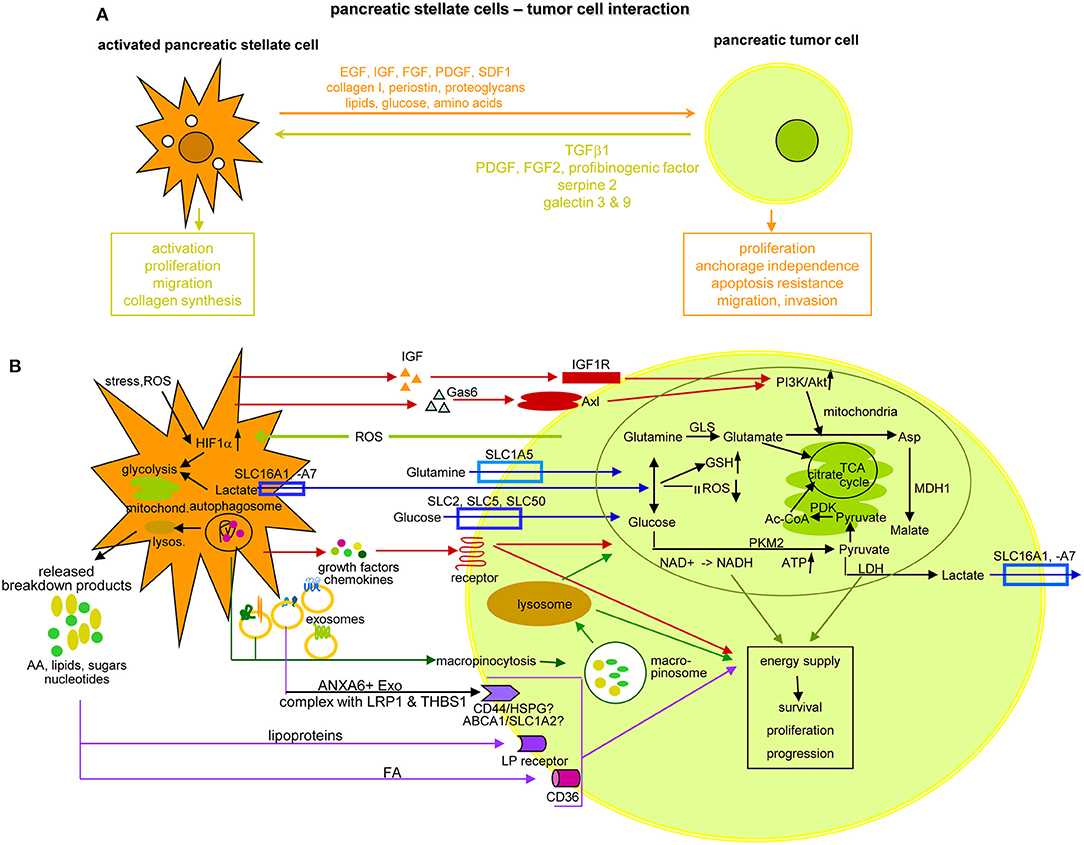
Figure 10. The crosstalk between PSC and pancreatic cancer cells. (A) Overview of the support provided by aPSC to PaCa survival, expansion and gain in aggressiveness and feedback by the tumor cell, which sustains PSC activation, expansion and matrix protein synthesis. (B) Some of the important components delivered by aPSC toward tumor cells and the initiated changes with a focus on altered metabolism. Glutamate derived from influxed glutamine can replace the TCA cycle to generate citrate, which also can derive from the pyruvate-PDK-Ac-CoA pathway. Lactate, delivered via lactate transporters supports glutamine and glucose generation, GSH upregulation and ROS reduction. Glucose also becomes enriched by glucose transporter in the tumor cell, where PKM2 via NADH and ATP promotes pyruvate generation. After lysosome degradation of aPSC autophagosomes, a plethora of AA, lipids, lipoproteins, sugars, and nucleotides is delivered that in part are taken up by specific receptors, not all being identified so far. Alternatively, autophagosomes are taken up by macropinocytosis, the macropinosome content being delivered after lysosome degradation. Lysosome degradation is also required for the delivery of the aPSC Exo content. Another option is receptor-mediated uptake of selective transmembrane complexes as ANXA61 bound LRP11 and THBS11. The predominant route of transfer from aPSC in PaCa cells is indicated by a color code: red: signaling receptor mediated uptake; blue: delivery or uptake by transporters; vesicle uptake: green; violet: receptor-mediated lipid and lipoprotein uptake; an olive circle encloses for a few of the aPSC-delivered components the pathway, whereby they contribute to the altered metabolism of PaCa cells; others may directly support PaCa survival and aggressiveness. Full name of proteins are listed in Table S1. aPSC support PaCa survival, expansion and progression, which to a considerable degree relies on their input of components initiating energy generation by altered metabolic pathways. Despite the focus on PSC-promoted metabolic adaptation of PaCa cells, the presented data cover only a minor part of the present state of knowledge and additional information can be expected by improved proteomic methodologies combined with organoid cultures.
Nutrient provision by altered metabolic pathways is another important aPSC contribution to PaCa cell progression. This proceeds through increased glycolysis, amino acid (AA) production from protein degradation, by glycosylation and fatty acid synthesis, called the metabolic switch (507). Accordingly, glycolytic enzymes such as hexokinase-2, enolase-2, LDHA, and B1 (508) and glycolytic metabolites are elevated (509). In addition, mitochondria adapt and account for energy supply. We recommend a most informative report on the different options, which tumor cells use to alter metabolic pathways (510), and give some examples on specific aPSC contributions. First, aPSC deliver cytokines that by binding to receptors initiate signaling cascades toward activation of the PI3K/Akt pathway, which is central for glycolysis, ATP level maintenance, stabilization of the mitochondrial potential, and tumor cell survival. Two examples are aPSC-derived IGF binding to the IGF1R and Gas61 binding to Axl. Gas6 is a member of the vitamin K-dependent protein family that resembles blood coagulation factors rather than typical growth factors (511). Both, IGF and Gas6 binding promote via PI3K/Akt activation Asp provision (512). Second, uptake of glucose and essential AA is facilitated by transporters either for delivery by aPSC or for uptake by PaCa cells that may also expulse unwanted byproducts, transporter families and their activities being profoundly reviewed (513). An example are glutamine transporters, which are supported by the glutamine-utilizing enzymes glutaminase GLS11, phosphoribosyl pyrophosphate synthetase PRPS21, and carbamoyl-phosphate synthetase 2 CAD converting glutamine to glutamate. Glutamate cannot exit and its accumulation replaces the TCA (tricarboxylic acid) cycle to generate citrate, which also can derive from the pyruvate-PDK-Ac-CoA pathway. Glutamate also stimulates cysteine uptake. Lactate, delivered via lactate transporters supports glutamine and glucose generation, GSH upregulation and ROS reduction. Glucose transporters in the tumor cells further assist glucose enrichment. Promoted by PKM2, NADH, and ATP support the generation of pyruvate. Excellent reviews unravel the current state of knowledge on the TCA cycle and the mitochondrial contribution in detail (508, 514–517). Autophagy accounts for a third support by CAF for nutrient supply. Autophagy is a cytoplasmic recycling process, where unfolded macromolecules, dysfunctional aggregates and organelles are sequestered in a double membrane organelle, called autophagosome, which fuses with lysosomes (518). The released breakdown products, AA, FA, nucleotides, and sugars are reused or released. One of the released AA, alanine is converted into pyruvate that is integrated into the TCA cycle (519). As far as aPSC deliver autophagosomes rather than the single components generated by lysosome degradation, autophagosomes are taken up by macropinocytosis, the nutrients becoming available after degradation in the tumor cell's lysosomes (520). Lysosome degradation is also required for access to nutrients provided by aPSC-derived Exo that modify the metabolic machinery of cancer cells increasing glycolysis and glutamine-dependent reductive carboxylation by providing AA, lipids, and TCA cycle intermediates (521). Finally, PaCa cells essentially depend on large amounts of lipids. FA uptake proceeds via different pathways. Besides gaining access by lysosome degradation of autophagosomes and Exo, the fatty acid translocase CD36 transports circulating free FA across the cell membrane (522, 523). FA sequestered in lipoproteins can be released by low density lipoprotein receptors before uptake by CD36. Alternatively and more frequently in PaCA, lipoproteins are internalized via LDL receptor-mediated endocytosis or macropinocytosis (524, 525). Notably the Exo transfer requires ANXA6+ Exo derived from CAF, where ANXA6 forms a complex with LRP11 and THBS11, the complex being only recovered in aPSC from patient with PaCa (526) (Figure 10B). Thus, though free nutrients are rare in the stroma, embedded aPSC provide a potent source.
In brief, PaCa cells express surface molecules and secrete factors that sustain PSC activation and expansion. aPSC, in turn, support PaCa proliferation, survival and progression. They promote proliferation and migration via cytokine and chemokine delivery, and apoptosis/drug resistance as well as a shift toward EMT via integrin and RTK activation. Ample provision of nutrients supports tumor cell survival and expansion mostly by sustaining altered metabolic pathways. Exo delivered by aPSC add to nutrient supply. Exo miRNA and lncRNA contribute to inactivation of tumor suppressor and liberation of metastasis-associated gene mRNA. lncRNA additionally support chromosome accessibility and transcription initiation, which adds to access of metabolism driving genes. Obviously, stress signals from PaCa cells suffice for aPCS/CAF responding with a plethora of supports.
Angiogenesis in Pancreatic Cancer
PaCa cells can support angiogenesis (527–529) and microvessel density after PaCa resection correlates with recurrence and poor survival (530). Nonetheless, PaCa are mostly hypovascular and hypoxic, due to a dominance of negative angiogenesis modulators (531, 532).
Several angiogenesis inhibitory factors, elegantly reviewed by Walia et al. (533), are enriched in PaCa. They originate from ECM degradation, poor vascularization being a secondary phenomenon to the fibrotic microenvironment (534). Angiostatin, a 38-kDa tumor cell-derived plasminogen fragment, inhibits primary and metastatic tumor growth by blocking angiogenesis (535–537). Fibstatin, another endogenous angiogenesis inhibitor, is a FN fragment containing the type III domains 12–14 (538). Fibstatin cooperates with CXCL4L1/PF4V11, inhibiting EC proliferation, migration and tubulogenesis in vitro and both angiogenesis and lymphangiogenesis in vivo (539). Endostatin, another matricellular protein regulating cell function without contributing to ECM structural integrity (533), is a collagen XVIII fragment (540, 541). MMP12 is engaged in endostatin and angiostatin generation (542), VEGF and FGF2 support secretion (543). Endostatin binds both endogenous angiogenesis inhibitors thrombospondin-1 and SPARC (544, 545) and upregulates thrombospondin-1 expression (546). Endostatin also binds VEGFR2 on EC and VEGFR3 on lymphatic vessels preventing activation and downstream signaling (533, 547, 548). By occupying integrin-ECM binding sites, initiation of the tyrosine phosphorylation cascade, src activation, and EC migration are interrupted (549, 550). Endostatin additionally prevents clustering with caveolin-1 and downstream signaling activation (551). A different mechanism underlies the antiangiogenic effect of RNASET21. Independent of its ribonuclease activity, RNASET2 arrests tube formation, accompanied by disruption of the actin network. The authors suggest RNASET2 competing or cooperating with angiogenin (552). Statins, HMGCR1 inhibitors, interfere with angiogenesis via VEGF downregulation. Moreover, statins prevent adhesion to the ECM by blocking intercellular adhesion molecules (553). There is, at least, one exception to angiogenesis/lymphangiogenesis inhibition by the PaCa stroma. Stroma embedded mast cells enhance angiogenesis by inducing pro-angiogenic VEGF, FGF2, PDGF, and angiopoietin-1 expression (554).
It may appear surprising that angiogenesis inhibition is a special features of most malignant PaCa with an intensive desmoplasia leading to hypoxia and nutrition deprivation. However, there is no evidence of cell death. PaCa being most well-equipped to cope with nutrient deficits, already outlined in the preceding section, only PaCa cell autonomous programs will be added here. Reuse of vesicle-enclosed nutrients can be liberated in the PaCa cell lysosomes (520). PaCa cell also make use of autonomous autophagy driven by a transcriptional program. Master regulators in converging autophagic and lysosomal functions are MITF1 and TFE1. A prerequisite for fulfilling these distinct functions relates to their shuttling between the surface of lysosomes, the cytoplasm, and the nucleus in response to nutrient fluctuations and various forms of cellular stress. Shuttling depends on changes in the phosphorylation of multiple conserved amino acids, phosphorylation being mainly promoted by mTOR, ERK, GSK3, and AKT, and dephosphorylation by calcineurin (555, 556). Furthermore, in contrast to most non-transformed tissue, tumor cells engage in de novo FA synthesis under hypoxic conditions (517, 557). This occurs particularly when the PI3K-Akt-mTOR pathway is constitutively active as in PaCa. mTOR signaling activates transcription factors of the sterol-regulatory element-binding protein family, which induce expression of the lipogenic genes ACACA1, FASN1, and SCD1 (558, 559).
Taken together, hypoxia-dependent and -independent mechanisms of metabolic reprogramming account for poor vascularization not hindering PaCa progression. Metabolic reprogramming is predominantly promoted by aPSC/CAF and their Exo and is supported by tumor cell autonomous programs.
Neural Invasion in Pancreatic Cancer
Innervation of the digestive tract is composed of the intrinsic, enteric nervous system, and afferent extrinsic nerves, transferring information to the central nervous system (CNS) and efferent nerves conveying commands from the CNS to the digestive organs (560). The healthy pancreas has an abundant nerve supply. Ganglia (aggregates of neural cell bodies), the intrinsic component of the pancreatic innervation, are randomly distributed throughout the parenchyma. The afferent system, thin unmyelinated fibers run with the parasympathetic vagus or the sympathetic input splanchnic nerves, the cell bodies are located in the spinal or vagal afferent ganglia. Extrinsic parasympathetic fibers derive from the vagus or the stem brain and end in the synapse of the intrapancreatic ganglia. Postganglionic parasympathetic fibers distribute with sympathetic fibers. Postganglionic sympathetic fibers mostly run with blood vessels (561, 562). Innervation is increased in PaCa (563, 564), nerve fibers forming a dense network that interacts with tumor cells and supports tumor growth and dissemination (565–567). In fact, PaCa metastasize by PNI. Also reported in other cancer, with recovery in 80–100% of patients, PNI is most frequent in PaCa and associated with poor prognosis (37, 568–571). PNI is seen in early stages of PaCa (572, 573) and is independent of lymphatic or vascular metastasis (573, 574) (Figures 11A–C).
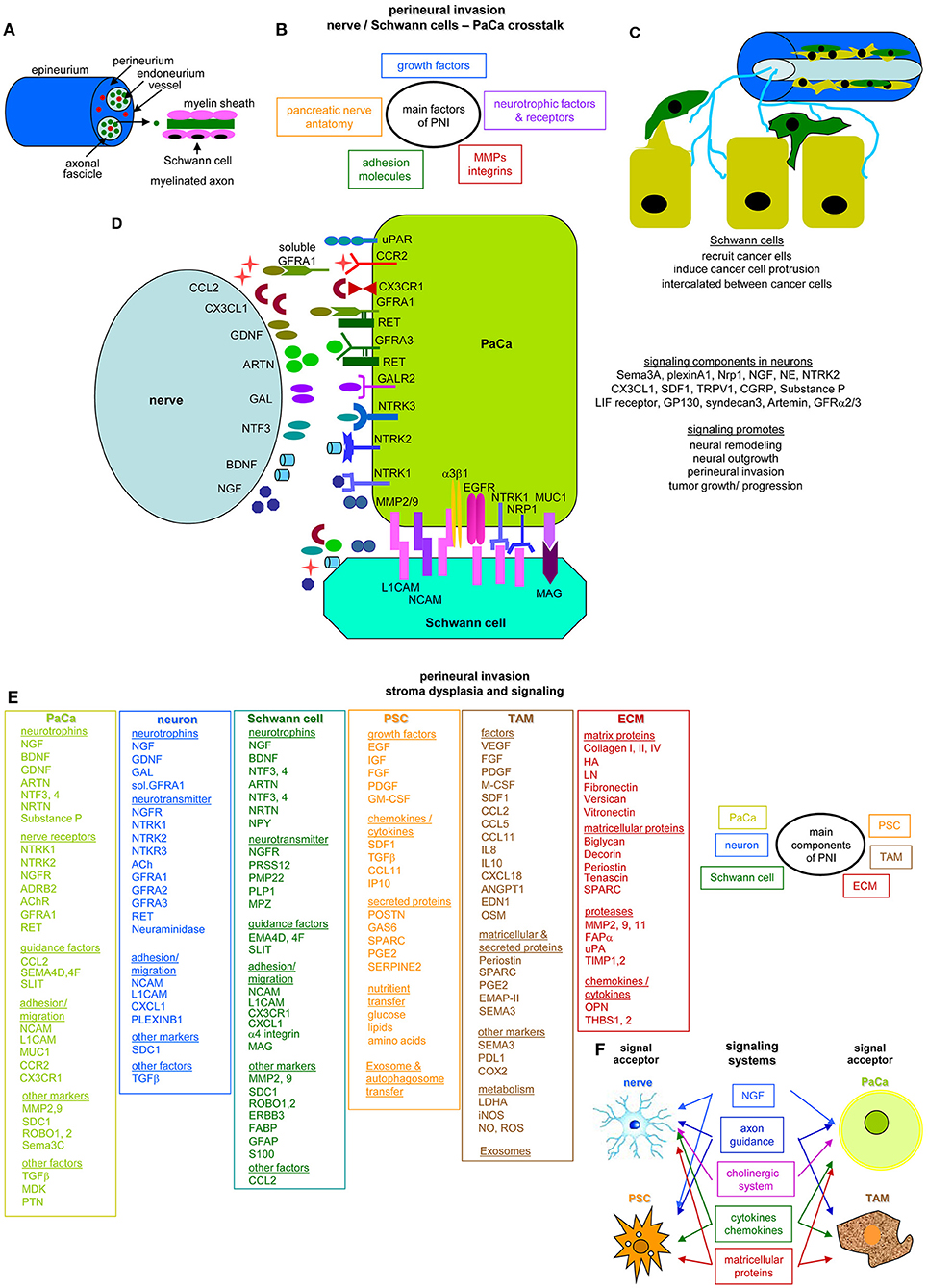
Figure 11. The nervous system and perineural invasion in pancreatic cancer. (A) Overview of nerve anatomy. The endoneurium surrounds all axons and serves to separate individual nerve fibers. The axons are covered by Schwann cells, where Schwann cells myelinate the axons. Non-myelinating axons mostly ensheath multiple small caliber axons. (B) The anatomy of the pancreatic nerves, neurotrophic factors and receptors as well as growth factors expressed by the engaged cells all contribute to perineural invasion and are supported by adhesion molecules and proteases as demonstrated in (C) for Schwann cells that intercalate between tumor cells promoting destruction of the adhesive matrix and actively recruiting tumor cells toward the nerve by signaling via adhesion molecules that promote cytoskeleton reorganization associated with acquisition of a motile phenotype. (D) Overview of abundantly delivered neurotrophic factors, cytokines, and chemokines by neurons and the corresponding receptors on PaCa tumor cells that promote tumor cell growth and invasion; dominating in the interaction between Schwann cells and tumor cells are L1CAM and NCAM. Besides homophilic binding, they bind integrins and RTK. MAG binding MUC1 on tumor cells mainly contributes to adhesion. For detailed information on signaling cascade initiation in PaCa, please see reviews mentioned in the text file. (E) Besides the direct engagement of neurons, Schwann cells and tumor cells, PSC, TAM, and the dysplastic tumor matrix contribute to PNI. Molecules predominantly contributing to PNI are listed. Selective contributions of aPSC rely predominantly on the transfer of nutrients, Exo and autophagosomes; TAM contribute by the delivery of matricellular proteins like EMAP-II and metabolism regulators such as LDHA and iNOS, the ECM supports PNI by embedded matricellular proteins and proteases. (F) All engaged cell populations are also acceptors of signaling cascade activators such as NGF, axon guidance cytokines/chemokines, and matricellular proteins. Activation of the cholinergic system is of major relevance for nerves and tumor cells. Full name of proteins are listed in Table S1. PNI is one of the dominating pathways of PaCa invasion. It is supported by neurotrophins and neurotransmitters delivered by neurons and Schwann cells, the latter in addition providing guidance factors and membrane integrated proteins that promote adhesion and migration. aPSC are essential in nutrient transfer and TAM provide cue enzymes to cope with ROS and NO. TAM and the ECM contribute by matricellular proteins and proteases that facilitate PaCa cell migration toward the nerve.
PNI is defined as the existence of tumor cells in the epineural, perineural and endoneural spaces of the neuronal sheath (566, 575) and results from mutual message transfer between nerves and tumor cells (566). Though not fully elaborated, many contributing components are known. Nerve growth factor family NGF, BDNF1, neurotrophin-3 and−4 (576) bind NTRK1/TRKA1 with high- and NGFR/p75NTR1 with low affinity (577–580), NTRK1 being highly expressed on nerves and tumor cells (581). Glial cell-derived neurotrophic factors GDNF1, NRTN, artemin and persephin are secreted by neural tissue and bind to GFRA1-A4 (582). GDNF expression strongly affects PNI in PaCa (583). This relies on RET receptor-mediated activation of downstream RAS, MAPK/ERK, JNK1, PI3K/Akt, and NFκB1 pathway activation (584–586). Anti-NGF treatment decreased expression of PNI-involved NTRK1, NGFR, TAC11, and calbindin in neural cells, reduced PNI and inhibited metastases in mice (587). The CXCR4-SDF1 axis also contributes to PNI. CXCR4 promotes tumor cell migration toward nerve cells (588, 589) and SDF1 increases NGF expression (588). Shown in an autochthonous model, PNI plays a significant role in initiation and progression of early PaCa stages, inflammation and neuronal damage in the peripheral and central nervous system already occurring in pancreatic intraepithelial neoplasia (PanIN)2, where acinar-derived cells frequently invade along sensory neurons into the spinal cord and migrate caudally to the lower thoracic and upper lumbar regions. Sensory neuron ablation prevents PNI, astrocyte activation, and neuronal damage, suggesting sensory neurons conveying inflammatory signals from the tumor to the CNS. Neuron ablation also significantly delays PanIN. These data indicate a reciprocal signaling loop between PaCa and the nervous system, including the CNS (590). Axon guidance genes semaphorins and plexins also are frequently altered in PaCa. Semaphorin3C increases PaCa proliferation, invasion, and EMT through ERK1/2 signaling pathway activation (591). Semaphorin3D secretion is regulated by AnnexinA2 phosphorylation. It acts autocrine by binding to the coreceptors plexinD1 and neuropilin-1 (592). Parakrine signaling of Semaphorin3D and plexinD1 between tumor cells and neurons mediates increased innervation, PNI and PaCa metastasis (593). Activation of the peripheral sympathetic nervous system (SNS) also assists PNI. In the healthy pancreas the SNS regulates digestive enzyme and endocrine hormone secretion (594, 595). In PaCa, β-adrenergic receptor activation of the SNS contributes to tumor progression via release of norepinephrine and epinephrine (Figure 11D). In view of the abundance of information coupled with many remaining questions we recommend readers particularly interested in PNI some recent, excellent reviews (38, 596, 597).
Beside tumor cells, nerves, Schwann cells, aPSC, TAM, and the ECM contribute to PNI. The contributing components, sorted according to molecular families and subcellular units are listed (Figure 11E). The complex contribution of dysplastic stroma elements to PNI being not fully unraveled, we only mention few examples. Tumor cells, aPSC, and TAM express GPCR β-adrenergic receptors ADRBA1,-A2, -B1, -B21 that signal via the associated trimeric G-proteins (598–600), HIF-1α (601), and ERK/MAPK (574), which in concert promote tumor growth and metastasis (39). aPSC-derived TGFβ induces NGF via the TGFBR1/ALK51 pathway and HGF-cMET activation (602, 603) that contribute to neural plasticity (604). TAM infiltration also correlates with PNI (605), where TAM-secreted IL8 assists PNI through MMP1-PAR11 signaling via ERK1/2 (606). Schwann cells highly express MAG1 (607), which is a receptor for abundant mucin-1 on PaCa (608), MAG-mucin-1 signaling promoting PNI (609). Furthermore, PaCa-derived NGF attracts Schwann cells via NGFR/p75NTR (40), which might be interpreted as the recruitment of nerve cells toward the tumor being the first step in PNI (40, 609). Finally, long distance nerve recruitment predominantly depends on Exo/MV (microvesicles) (610, 611) for several cancer (612, 613). This is best explored for glioblastoma-TEX, which are taken up by tumor cells, EC, and Mϕ, but also by healthy neural cells, and microglia (614). Furthermore, non-transformed cell-derived Exo/MV contribute to message transfer. Oligodendrocytes, glial cells in the brain accounting for axon myelination, shuttle messages between myelinating glia and neurons (615, 616) and between neurons (617). Microglia, the brain's Mϕ defense mechanism, also acts via released MV (618). Microglial MV additionally regulate neuronal excitability accompanied by neuronal ceramide and sphingosine production (618). Schwann cells, too, communicate with the peripheral nervous system via Exo (619).
In brief, the review “Splitting out the demons” is concerned about glioblastoma (620), but may well be of general relevance, particularly for PNI in PaCa. The authors demonstrate that the major signaling systems are NGF, axon guidance molecules, cytokines/chemokines, the cholinergic system, and matricellular proteins that are also delivered by several components of PaCa. Searching for signal acceptors in PaCa revealed that tumor cells, nerves, aPSC, and TAM can all be acceptors of these signaling systems creating a malicious feedback loop in PaCa (Figure 11F).
Spurred by the poor prognosis and PaCa-associated pain (620–623) and PNI being an early event in PaCa development, PNI recently received increasing attention (595). For a long time uncovered molecular pathways due to technical difficulties in culturing engaged cellular components and isolating Exo from defined subpopulations may become unraveled in the near future. Success in culturing Schwann cells particularly opens access to a hitherto inaccessible, important contributor. We consider Exo/MV as an additional promising option to interrupt PNI (618), where improved techniques for isolating and characterizing single stroma cell derived Exo will be of great help in deciphering a PNI-forcing contribution. Despite strong progress, supported by elegant autochthonous mouse models, there is still great need to unravel the complex interactions underlying PNI, which is a prerequisite for therapeutic interference (587, 624).
Pancreatic Cancer and Immunosuppression
Immune cells are abundant in the PaCa stroma (625, 626), but are immunosuppressive (627, 628), whereas effector cells are rare (629). This accounts for the innate and the adaptive immune system.
NK
NK are discussed as a therapeutic option in PaCa (630, 631). However, several constraints need clarification as NK are reduced in the juxta tumoral area compared to the stroma, possibly due to sequestration by aPSC (632) and NK apoptosis via FASL1-positive tumor cells (613). In addition, cytotoxic activity of NK cells is severely impaired (633).
Activated NK cells bind via activating receptors NKG2D1, NKp301, and NKp461 to their ligands major histocompatibility complex class I-related chain MICA/B1 and ULBP1-61 (634). NKG2D having a very short cytoplasmic tail uses the adaptor molecules DAP101 and/or DAP121 to initiate downstream signaling (635). In addition, activated NK cells secrete IFNγ, TNFα, GM-CSF1, the chemokine ligands CCL1-51, and CXCL81, which trigger activation and recruitment of other innate and adaptive immune cells, broadening and strengthening anti-tumor immune responses (636). In PaCa, instead, decreased NK activity is accompanied by low level NKp46, NKp30, granzymeB, and perforin expression (637). Lactate, a by-product of tumor metabolism also causes NKp46 downregulation (638). Another important group of NK receptors are nectin and nectin-like binding molecule DNAM11. DNAM1 downregulation on NK correlates with PaCa progression (639). Furthermore, though MICA/B is expressed in >70% of PaCa, it is also expressed on PSC (640). NK cells preferentially migrating toward PSC become sequestered in the stroma before reaching the tumor nodules (641). Moreover, ADAM10 and ADAM17 cause shedding of MICA/B and PSC inhibit NK cells via IL6 (642). Finally, NK cells tend to target (Pa)CIC due to enhanced MICA/B expression (643). In view of the CIC plasticity, it remains to be explored, whether CIC targeting by NK is of therapeutic benefit (Figure 12A).
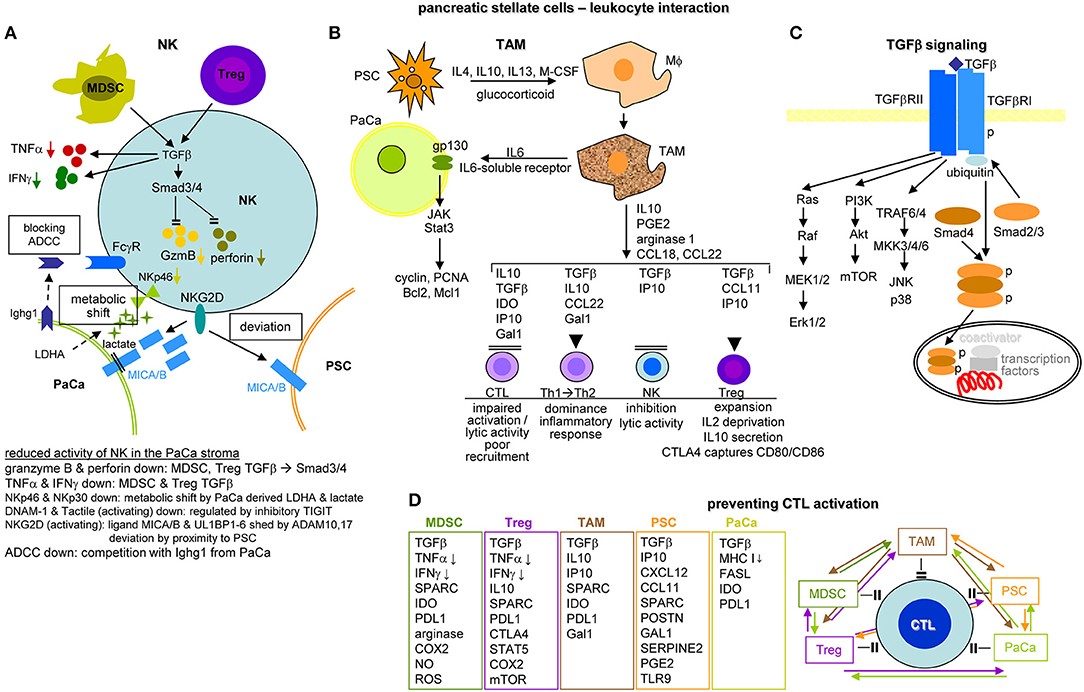
Figure 12. The impact of PSC and tumor cells on immune cells in the pancreatic cancer stroma. (A) NK cells in the stroma display reduced activity. This is mainly due to MDSC and Treg that by TGFβ delivery affect TNFα and IFNγ secretion and SMAD3/4 activation, which inhibit GzmB and perforin transcription. The activating NKG2D receptor become deviated toward PSC due to higher expression of MICAB, where MICAB in tumor cells can become shed by ADAM17, free MICAB fragments further deviating NK cells from attacking the tumor cell. The activating receptors NKp46 and NKp30 become downregulated due to a metabolic shift induced by tumor cell derived LDHA and lactate. Activating receptor can also become occupied by inhibitory receptor, like TIGIT. Finally, tumor cells deliver an IgG like molecule, Ighg1, occupying the FcγR of NK cells and thereby interfering with ADCC. (B) PSC have a strong impact on driving Mϕ into TAM by the delivery of IL4, IL10, IL13, mCSF, and glucocorticoids. TAM deliver IL6 and soluble IL6 receptor binding to gp130 on tumor cells, which activates the JAK/Stat3 pathway promoting tumor cell survival and expansion by cyclin, PCNA Bcl2, and Mcl1 expression. TAM also affect the activity of additional immune cells. Lytic NK cell activity becomes inhibited by TGFβ and IP10. A shift of Th1 to Th2 is supported by TGFβ, IL10, CCL22, and Gal1. Expansion and activity of Treg is assisted by TGFβ, IP10, and CCL11. Finally, CTL recruitment, activation and lytic activity are impaired by TAM-derived TGFβ, IL10, IP10, IDO, and Gal1. (C) A central role of TGFβ in immune deviations relies on binding to the TGFβRII, which promotes RAS, PI3K, and TRAF6/4 pathway activation and on TGFβR1 binding, where phosphorylated Smad4 forms a complex with Smad2/3, the complex migrating into the nucleus promoting together with additional coactivators and transcription factor besides other transcription of NOS, PAI-1 and PDGF. (D) CTL activation is prohibited by tumor cells, PSC and immunosuppressive MDSC, Treg and TAM. The major inhibitory factors and membrane molecules are listed. PSC particularly contribute via POSTN, GAL1, SERPINE2, PGE2, and TLR9. Low level MHCI expression on tumor cells hampers CTL activation, high FASL expression contributes to CTL lysis and IDO and PDL1 are inhibitory receptors. As shown in the overview diagram, preventing CTL activation is the result of coordinated activities between all contributing components. Full name of proteins are listed in Table S1. The dense stroma and poor angiogenesis may hamper leukocyte recruitment. However, there is no paucity of immunosuppressive leukocyte, such that changes in metabolism and activation of signaling cascades are dominating immunosuppression. Feedback circles between all contributing elements create a self-replenishing vicious circle.
Due to preferentially targeting tumor cells, NK-based immunotherapy was discussed just few years after their discovery (644), hope being fostered by their contribution to antibody-dependent cellular cytotoxicity (645). Further unraveling the impact of their surrounding, efficient use of NK cytotoxic potential may become reality in PaCa.
Mϕ
TAM are increased in the PaCa stroma (646), high numbers being associated with poor prognosis (647–649). TAM mostly exhibit the suppressive phenotype of CD163+ and CD204+ M2 (650, 651), M2 differentiation being supported by tumor- and Treg-derived IL4, IL10, and IL13 (652). TAM suppress the adaptive immune response via TGFβ, IL10, CCL17, CCL18, CCL22, and PDL11 secretion (653, 654). In addition, CCL2 and CCL201 through chemokine receptor CCR61 binding promote MMP9 upregulation and thereby invasiveness (655, 656) and can contribute to EMT (657, 658). In PaCa, TAM also secrete the serine protease FAP1, which stimulates CAF (659) and induces CDA1, contributing to drug resistance by metabolizing the active to the inactive form of Gemcitabine (660).
Briefly, the main feature of TAM is the shift to and the preponderance of immunosuppressive M2 in PaCa. Besides suppressing adaptive immune responses, TAM promote CAF and in a positive feedback loop Treg expansion. TAM also strengthen the aggressiveness of PaCa and support drug resistance. Reviews are recommended for a comprehensive overview of special TAM features in PaCa (661, 662) (Figures 12B,C).
MDSC
MDSC are a heterogeneous group of cells, characterized by myeloid origin, immature state and mostly functional activity. Two subgroups, defined as monocytic (M) and granulocytic (G) MDSC are differentiated by Ly6Chigh (M-MDSC) or Ly6Ghigh (G-MDSC), M-MDSC exerting stronger suppressive activity (663–665). MDSC are abundant in the PaCa stroma (666). MDSC are recruited toward PaCa via CAF-derived CXCL12 and tumor-derived GM-CSF (588, 667). MDSC hamper T-cell recruitment and activation, which are their major targets and promote Treg expansion (668, 669). MDSC expansion is expedited by M-CSF1, GM-CSF, SCF1, IL6, IFNγ, IL1β, VEGF, HSP72, IL13, C5a1, PGE21, and S100A8/A9 (664, 670). Inhibition of differentiation into mature myeloid cells is spurred by downstream activation of the JAK1-STAT3/STAT5 pathway with stimulation of cyclinD1, BCLXL1, survivin, c-myc, and S100A8/A9. CCL2 and SDF1 support MDSC recruitment, GM-CSF plays a major role in inflammatory milieu maintenance (667). Prominent signaling molecules engaged in MDSC activity are STAT3, COX2, HIF1α, C/EBPB1, HMOX11, and IDO1 (654, 670, 671). MDSC interfere at several levels with immune response induction (672). Downstream effector molecules arginase-1 and iNOS1 account for L-arginine depletion and ζ-chain downregulation in T-cells (673). iNOS-induced NO and ROS inhibit T-cell proliferation and promote apoptosis. HMOX1 hampers T-cell proliferation by CO production (670, 674). Membrane-bound TGFβ1 assists NK anergy (675). IL10 and TGFβ foster Treg expansion, which become recruited by CXCL10 (676). TGFβ and IL10 also account for IFNγ downregulation (670, 674). IL10 promotes TH2 deviation (677) and M2 polarization (678). Finally, MDSC Exo characterization uncovered MDSC activities being efficiently transferred by Exo (679–681).
Thus, MDSC hamper mostly T-cell, but also B-cell (682) and NK activity, at least in part by supporting Treg expansion and activation. There are several well-established options to combat MDSC induction and activities, frequently used in combination with chemotherapy whose efficacy increases by eliminating MDSC-promoted drug resistance (683, 684).
Dendritic Cells
Dendritic cells (DC) are professional antigen presenting cells, directly linking the innate and adaptive immune systems, where particularly Th activation essentially depends on processed antigen peptide presentation (685–687) and costimulatory signals provided by DC (688, 689). However, DC activity is severely impaired in cancer (690, 691). In the PaCa stroma, DC are rare and mostly located at the edge of the tumor (692). DC maturation and activation is also hindered by confrontation with immunosuppressive cytokines TGFß, IL6, IL10, and GM-CSF, which activate the STAT3 pathway (693–695). Furthermore, costimulatory molecule CD40 and CD80 expression is reduced in DC, hampering T-cell activation (696). Instead, DC produce CCL22, which recruits Treg (697, 698). Several options for coping with the DC deficit are clinically evaluated, mostly based on the transfer of antigen/peptide-loaded DC, where in PaCa mucin1 and Wilms tumor protein are promising antigen candidates. Loading DC with the patient's TEX is another option that guarantees presentation of the individual tumor's immunogenic antigens/peptides (699–701). The finding that DC-derived Exo are equipped to stimulate T-cells (702), spurred research focusing on DC transfer to overcome poor T-cell responses in PaCa (703–705).
Besides supporting Treg recruitment, DC do not actively contribute to PaCa progression. Unfortunately, their paucity in the tumor stroma, impaired antigen processing and presentation and the insufficient costimulatory molecule supply significantly hamper immune response induction. There is hope for circumventing these drawbacks by DC or DC-Exo transfer, the latter having the advantage of a technically easier implementation in the clinic.
T-Cells
The adaptive immune system, T-cells and B-cells, is the body's most specialized and efficient defense mechanism. B-cells, secreting antibodies, account for the humoral defense, T-cells for the cellular defense, where CD8+ CTL lyse their targets and CD4+ Th provide soluble factors supporting CTL, B-cells and NK. T-cells are rare in PaCa (706) and PaCa actively inhibit CD4+ T-cell proliferation and migration (707). Furthermore, PaCa tumor cells and the stroma skew Th from cell-mediated responses inducing Th1 toward Th2, which might support tolerance induction (708). The shift toward Th2 is assisted by PaCa-delivered IL10 and TGFβ (709) and by CAF-delivered lymphopoietin (710). Furthermore, lower numbers of T-cells in PaCa (706) may rely on aPSC affecting T-cell migration toward the tumor nodules (631). The Th2 cytokines IL4, IL5, IL6, MIP1α, GM-CSF, MCP11, IL17, IP10, and IL1β are dominant and are associated with poor immune responsiveness and a shorter DFS (disease free survival) (711). Moreover, PaCa inhibit CTL activity. PaCa-derived TGFβ interferes with perforin and granzyme expression (712, 713) and PDL1 on PaCa binds PD11 on CTL, spurring T-cell anergy or death (714). There are subtypes of PaCa that display higher T-cell levels, but the tumor evades the immune response due to amplification of PDL1/2 or upregulation of inhibitory cytokines and the JAK/STAT signaling pathway (715). aPSC also stimulate T-cell apoptosis, decrease IL2 and IFNγ secretion by Th1, but increase IL4 and IL5 secretion by Th2, which is linked to galectin-1 expression on PSC (716, 717).
Though mucin-16 tumor antigen-specific CTL were recovered in few long term survivors, supporting the efficacy of CTL in defending the body's integrity (718), PaCa and aPSC skew toward Th2 and promote T-cell anergy and apoptosis, low level T-cell recovery correlating with a poor prognosis (719) (Figure 12D).
Treg
Treg are CD4+CD25highFoxp3+ cells (720, 721). They contribute to immunosuppression via CD152/CTLA41 (722, 723) and TGFβ and IL10 secretion, which affects Th, CTL, Mϕ, NK, and DC (626, 724–726). In PaCa, Treg are already present at the PanIN stage, expand during tumor progression (727, 728) and are preferentially located surrounding the tumor (729). Treg promote EMT (730) and inhibit Th1 and Th17 effector functions (731). Migration toward the tumor is assisted by tumor chemokines and EC addressins and their ligands on Treg (732). PaCa secrete elevated levels of CCR5 ligands/CCL28, which increases Treg chemotaxis (733). EC in the tumor tissue express high level of mucosal VCAM-1, E-selectin and CD116/CSF2RA1, which foster Treg transmigration (734). Increased levels of Treg in the circulation (735) and the tumor stroma (731, 735) correlate with poor prognosis.
There are other unmentioned immune deviations related to PaCa. We recommend overviews focusing on cytokines and chemokines (736–739) and additional immunosuppressive molecules (740), where we only mention a few. RIP1 and 31, highly expressed in PaCa, are key mediators of necroptosis, a caspase-independent cell death. Interestingly, while an in vitro blockade of the necrosome was accompanied by increased PaCa aggressiveness, in vivo deletion was associated with increased immunogenic myeloid and T-cell infiltrates. The authors suggest that this is due to RIP1/3 signaling through CXCL1 ligation of its receptor CLEC4E/Mincle1 that is also expressed on TAM. Thus, TAM lose their immunosuppressive features in the absence of either RIP3 or CLEC4E, which is accompanied by regain of immune defense promoting signaling in T-cells (741). A clinical study showed that an IDO1 inhibitor prevented disease progression. IDO1 catalyzes the degradation of tryptophan to kynurenine (742). Tryptophan is essential for T-cells, but kynurenine supports immunosuppression. Accordingly, IDO1 suppresses effector T-cells and NK and promotes induction, activation and recruitment of Treg and MDSC, the signaling pathways differing between leukocyte subsets (743). An elegant study recently reported on Treg signaling in the tumor environment. Tumor Treg undergo apoptosis and apoptotic Treg exhibit stronger immunosuppressive features than live Treg. Treg apoptosis is due to high oxidative stress susceptibility by weak NRF21 Tf and antioxidant system-associated gene expression. Apoptotic Treg-promoted immunosuppression relies on release and conversion of a large amount of ATP to adenosine by CD39 and CD73, and ADORA2A1 pathway activation (744). Galectins are another family of secreted proteins contributing to immune evasion in PaCa (745). Galectins have high affinity for β-galactoside residues, sharing a consensus carbohydrate recognition domain (CRD) responsible for glycan binding, most of their biological functions relying on interactions with glycosylated proteins (746). aPSC account for galectin1 secretion and overexpression in the tumor microenvironment (716). Galectin1 recognizes glycoproteins on T-cells, inhibits transendothelial migration and promotes apoptosis of activated Th1 cells, tilting the immune balance toward a Th2 profile. Galectin1 also impairs NK cell recruitment, induces Treg differentiation, M2 macrophage polarization, and MDSC expansion (747, 748), suggesting galectin1 a key driver in immune evasion in PaCa (748). Galectin9 also is crucial for immune deviation in PaCa. Galectin9 is a ligand for dectin11, highly expressed in PaCa Mϕ. Dectin ligation promotes signaling via syk1, PLCγ, and the JNK pathway. The dectin1-galectin9 axis is central in directing the differentiation of TAM to a M2-like phenotype, which suffices for reprogramming CD4+ and CD8+ T-cells (749). Finally, we list some reviews helpful as starting information on PaCa-selective metabolic changes that affect immune responses in PaCa (739, 750–754).
Summarizing at least some aspects of immune modulation by the particular stroma reaction in PaCa, PSC/CAF secrete SDF1 that coats the tumor cells and prevents T-cell infiltration (640, 755). PSC also secrete galectin1 forcing T-cell apoptosis and Th2 deviation (716), but recruiting Treg (485) and supporting mononuclear cell differentiation toward MDSC (756), with suppressive myeloid cells being most abundant in PaCa, TAM accounting for 15–20% and MDSC for 5–10% (716, 757). Tumor-derived GM-CSF and MIP2 account for MDSC (716, 757), CSF1 and BAG31 for TAM (757, 758) recruitment and expansion, GM-CSF being also provided by tumor-associated mesenchymal cells (759). Both MDSC and TAM direct suppression through factors and tumor-cell-specific PDL1 expression (625, 760–762). B-cells are recruited via tumor-derived CXCL13 (763). A shift toward M2 via PI3Kγ-activated BTK1 in B-cells and TAM supports PaCa growth and progression (764).
Taken together, PaCa and the dysplastic stroma hamper leukocyte infiltration and skew toward immunosuppressive components. This accounts for the non-adaptive and the adaptive immune system. The strong impact of PaCa and the stroma is reflected by low onco-immunotherapy efficacy, which fosters research on combined therapeutic approaches. With 416 reviews total and 86 in the last 18 months, on immunotherapy in PaCa, we apologize not mentioning this aspect, which goes beyond the scope of our trial giving an overview of the particularly dense crosstalk between PaCa and the stroma. Nonetheless, the body's defense mechanism being highly efficient at maintaining health and coping with a wide range of diseases, there is some hope that after unraveling the complex and intertwined contributions of individual components and signaling pathways, immunotherapy may shortly contribute in defeating PaCa (765).
Conclusion and Outlook
PaCa has a dismal prognosis and incidence is rapidly increasing. This fostered utmost intense research aiming elaborating the underlying mechanisms, which unequivocally demonstrated the lead role of the PaCa stroma, frequently displaying rebound effects on the tumor cells and between the individual stroma elements. These features seriously aggravate pinpointing single molecular mechanisms such that despite strong progress, we are still tickling the top of a non-melting iceberg. In brief,
1. Unlike most cancer, angiogenesis is reduced in PaCa. Pressure from the dense dysplastic reaction may be partly responsible for inadequate angiogenesis. We assume an active contribution of PaCa-TEX, which interfere with EC migration, expansion and sprouting in vitro and in vivo. The underlying mechanism remains to be clarified. A comparative analysis of the proteome, coding and noncoding RNA of PaCa-TEX and TEX of a strongly vascularized tumor might be a starting point depicting active contributors to poor PaCa vascularization. Irrespective of the suggested PaCa interference with angiogenesis, the stroma provides copious nutrients and redirects the tumor cells' metabolic pathways such that hypoxia-promoted damages are completely waved.
2. PSC/CAF are central for PaCa stroma dysplasia. The dysplastic stroma strongly adds to immune defense deviation and supports PNI. Progress in suppressing the overshooting stroma reaction may be achieved by a profound analysis of signaling/metabolic pathways linked to aPSC. The discussion still being ongoing, we only mentioned few examples of aPSC/CAF-promoted metabolic reprogramming and possible contributions of aPSC/CAF miRNA and lncRNA (483, 484). Nonetheless and despite overwhelming evidences for PaCa-promoting activities of aPSC/CAF, the dysplastic stroma could serve as a protective barrier for the host against the tumor under selected circumstances. Thus, in the growing list of therapeutic reagents interfering with the metabolism and/or signaling cascades in aPSC (766), the option of reverting PSC to their quiescent state by supporting FA synthesis could be of particular interest (767).
3. PaCa shares with many tumors a paucity of immunogenic tumor-associated antigens and excessive tumor-promoted immunosuppression. These drawbacks for immunotherapy are aggravated in PaCa by the dysplastic stroma. As immunosuppressive cells are enriched in the PaCa stroma, the stroma density may not considerably contribute excluding immune cells. In fact, it is within the stroma that immune cells are killed or deviate toward immunosuppression. Tumor immunotherapy with a strong focus on the transfer of activated DC and T-cells to circumvent low tumor antigen immunogenicity, requires in depth elaboration of in loco deviation to find pathways allowing activation of transferred immune cells within PaCa. This also accounts for the transfer of DC-Exo, where physical barriers are no hindrance, and for antibody-based therapies, where BTK activation by binding to FcRγ+ TAM needs to be bypassed. However, as good progress is already achieved in MDSC elimination, there is hope that remaining hurdles may be solved.
4. PNI, though not unique, is the dominant metastatic route already at early stages of PaCa development. Elaboration of underlying mechanisms is aggravated by an active contribution of the neuronal components. Comparative analyses to brain tumors, particularly glioblastoma, may provide hints for unraveling the crosstalk between tumor cells and nerves including Schwann cells and ganglia. With strong evidence for synaptic information transfer by EV, a focus on the impact of nerve-, microglia-, and Schwann cell-derived Exo/MV on tumor cells could help unraveling the neural system contribution in diverting PaCa cells toward this particular metastatic route.
5. Many studies on PaCIC markers and the feedback on the tumor matrix, EC, the adaptive, and non-adaptive immune system point toward these markers severely affecting host matrix and cells. PaCIC markers are engaged in regulation of transcription, activation of signaling cascades, and metabolic shifts, spurring adhesion, migration, and invasion. Abundantly recovered PaCIC markers on TEX contribute to TEX biogenesis including loading, target binding, and TEX uptake (86). Intensifying studies on cooperation-based peculiarities of PaCIC-TEX markers may uncover a central switch in the PaCIC-stroma interplay, allowing for a unifying concept of PaCIC-TEX-based therapies.
6. We apologize for sparse discussion on signaling pathways in the PaCa-stroma crosstalk. First, signaling pathways are often connected and can be mutually affecting. More importantly, in vivo studies only depict the overall changes on tumor cells or stroma, even organoid cultures having some limits in depicting individual components. Nonetheless, organoid cultures provide an excellent method for unraveling the complex and mutual interactions between PaCa cells and their surrounding components (768, 769). It can be expected that continuing advancement in organoid research will markedly increase knowledge of the molecular features of the mutual crosstalk between the distinct components and pave the way for large scale therapeutic screenings that may prove reliable for clinical translation (770).
7. Though providing up-to-date references to the date of submission, for the sake of clarity and length we kindly ask scientists working on special topics gathering additional information. This request particularly applies to ncRNA, where multiple targets for most miRNA hamper coordination and the diverse range of lncRNA functions awaits comprehensive examination (86, 136, 139, 771–773). Furthermore, in view of many eminent reviews, we skipped information on therapeutic translation. Finally, we apologize for not citing numerous outstanding studies.
Author Contributions
MZ planned the organization of the review and wrote the first draft. WM and ZW helped with data collection and corrected the first draft. All authors approved the final version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81803269) (WM), the Shanghai Sailing Program (18YF1412100) (WM), and the National Natural Science Foundation of China (NSFC. 81702877) (ZW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01359/full#supplementary-material
Abbreviations
AA, amino acid; a, activated; ADCC, antibody-dependent cellular cytotoxicity; ASC, adult stem cells; BM, basal membrane; BMC, bone marrow cells; CAF, cancer-associated fibroblasts; CIC, cancer–initiating cells/cancer stem cells; CNS, central nervous system; CoCa, colorectal cancer; CRD, carbohydrate recognition domain; CTL, cytotoxic T lymphocytes; DC, dendritic cells; DFS, disease free survival; DR, desmoplastic reaction; EC, endothelial cells; ECM, extracellular matrix; EE, early endosome; EMT, epithelial mesenchymal transition; ER, endoplasmic reticulum; ERM, ezrin, radixin, moesin; eRNA, enhancer lncRNA; ESC, embryonic stem cells; ESCRT, endosomal sorting complex required for transport; EV, extracellular vesicles; Exo, exosome; FA, fatty acid; FB, fibroblast; FN, fibronectin; GPCR, G protein-coupled receptor; HCC, hepatocellular carcinoma; HNRNP, heterogeneous ribonucleoprotein; ICD, intracellular domain; ILV, intraluminal vesicle; kd, knockdown; ko, knockout; LN, laminin; lnc, long noncoding; LNC, lymph node cells; MDSC, myeloid-derived suppressor cell; MET, mesenchymal-epithelial transition; Mϕ, macrophage; MHC, major histocompatibility complex; miRNA, microRNA; MS, mass spectrometry; MV, microvesicles; MVB, multivesicular body; nc, non-coding; NEAA, non-essential amino acids; NK, natural killer cells; Non-CIC, non-metastasizing tumor cells; PaCa, pancreatic cancer; PanIN, pancreatic intraepithelial neoplasia; PNI, perineural invasion; PSC, pancreatic stellate cells; RISC, RNA induced silencing complex; RBP, RNA binding proteins; RNP, ribonucleoprotein; ROS, reactive oxygen species; RTK, receptor tyrosine kinase; SC, stem cells; SNS, sympathetic nervous system; TAM, tumor-associated macrophages; TCA, tricarboxylic acid; TEM, tetraspanin- and glycolipid-enriched membrane microdomain; TEX, tumor exosomes; Tf, transcription factor; Th, helper T cells; TJ, tight junction; Treg, regulatory T cells.
Footnote
References
1. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
2. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
3. Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. (2011) 17:320–9. doi: 10.1038/nm.2328
4. Te Boekhorst V, Friedl P. Plasticity of cancer cell invasion-mechanisms and implications for therapy. Adv Cancer Res. (2016) 132:209–64. doi: 10.1016/bs.acr.2016.07.005
5. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
6. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. (2017) 14:2611–20. doi: 10.3892/ol.2017.6497
7. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. (2016) 18:84. doi: 10.1186/s13058-016-0740-2
8. Fiorio Pla A, Gkika D. Emerging role of TRP channels in cell migration: from tumor vascularization to metastasis. Front Physiol. (2013) 4:311. doi: 10.3389/fphys.2013.00311
9. Vaahtomeri K, Karaman S, Mäkinen T, Alitalo K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev. (2017) 31:1615–34. doi: 10.1101/gad.303776.117
10. Draoui N, de Zeeuw P, Carmeliet P. Angiogenesis revisited from a metabolic perspective: role and therapeutic implications of endothelial cell metabolism. Open Biol. (2017) 7:170219. doi: 10.1098/rsob.170219
11. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
12. Hawk MA, Schafer ZT. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J Biol Chem. (2018) 293:7531–7. doi: 10.1074/jbc.TM117.000260
13. Elia I, Doglioni G, Fendt SM. Metabolic hallmarks of metastasis formation. Trends Cell Biol. (2018) 28:673–84. doi: 10.1016/j.tcb.2018.04.002
14. Lou XL, Sun J, Gong SQ, Yu XF, Gong R, Deng H. Interaction between circulating cancer cells and platelets: clinical implication. Chin J Cancer Res. (2015) 27:450–60. doi: 10.3978/j.issn.1000-9604.2015.04.10
15. Leblanc R, Peyruchaud O. Metastasis: new functional implications of platelets and megakaryocytes. Blood. (2016) 128:24–31. doi: 10.1182/blood-2016-01-636399
16. Reymond N, d'Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. (2013) 13:858–70. doi: 10.1038/nrc3628
17. Strilic B, Offermanns S. Intravascular survival and extravasation of tumor cells. Cancer Cell. (2017) 32:282–93. doi: 10.1016/j.ccell.2017.07.001
18. Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol. (2014) 28:39–50. doi: 10.1016/j.semcancer.2014.02.011
19. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
20. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
21. Lobb RJ, Lima LG, Möller A. Exosomes: key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol. (2017) 67:3–10. doi: 10.1016/j.semcdb.2017.01.004
22. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
23. Nandy SB, Lakshmanaswamy R. Cancer stem cells and metastasis. Prog Mol Biol Transl Sci. (2017) 151:137–76. doi: 10.1016/bs.pmbts.2017.07.007
24. Dhamija S, Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int J Cancer. (2016) 139:269–80. doi: 10.1002/ijc.30039
25. Brabletz T. To differentiate or not–routes towards metastasis. Nat Rev Cancer. (2012) 12:425–36. doi: 10.1038/nrc3265
26. Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen YH. Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: a comprehensive overview. J Clin Med. (2017) 7:E1. doi: 10.3390/jcm7010001
27. Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol. (2017) 11:755–69. doi: 10.1002/1878-0261.12083
28. Mareel MM, Van Roy FM, Bracke ME. How and when do tumor cells metastasize? Crit Rev Oncog. (1993) 4:559–94.
29. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. (2016) 66:7–30. doi: 10.3322/caac.21332
30. Strijker M, Chen JW, Mungroop TH, Jamieson NB, van Eijck CH, Steyerberg EW, et al. Systematic review of clinical prediction models for survival after surgery for resectable pancreatic cancer. Br J Surg. (2019) 106:342–54. doi: 10.1002/bjs.11111
31. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
32. Del Chiaro M, Segersvärd R, Lohr M, Verbeke C. Early detection and prevention of pancreatic cancer: is it really possible today? World J Gastroenterol. (2014) 20:12118–31. doi: 10.3748/wjg.v20.i34.12118
33. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. (2015) 42(Suppl. 1):S3–17. doi: 10.1053/j.seminoncol.2015.01.001
34. Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. (2016) 22:2678–700. doi: 10.3748/wjg.v22.i9.2678
35. Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. (2016) 381:194–200. doi: 10.1016/j.canlet.2015.10.035
36. Vennin C, Murphy KJ, Morton JP, Cox TR, Pajic M, Timpson P. Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology. (2018) 154:820–38. doi: 10.1053/j.gastro.2017.11.280
37. Liang D, Shi S, Xu J, Zhang B, Qin Y, Ji S, et al. New insights into perineural invasion of pancreatic cancer: more than pain. Biochim Biophys Acta. (2016) 1865:111–22. doi: 10.1016/j.bbcan.2016.01.002
38. Jurcak N, Zheng L. Signaling in the microenvironment of pancreatic cancer: transmitting along the nerve. Pharmacol Ther. (2019) 200:126–134. doi: 10.1016/j.pharmthera.2019.04.010
39. Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, Feng YJ. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am J Cancer Res. (2019) 9:1–21.
40. Deborde S, Wong RJ. How Schwann cells facilitate cancer progression in nerves. Cell Mol Life Sci. (2017) 74:4405–20. doi: 10.1007/s00018-017-2578-x
41. Parente P, Parcesepe P, Covelli C, Olivieri N, Remo A, Pancione M, et al. Crosstalk between the tumor microenvironment and immune system in pancreatic ductal adenocarcinoma: potential targets for new therapeutic approaches. Gastroenterol Res Pract. (2018) 2018:7530619. doi: 10.1155/2018/7530619
42. Foucher ED, Ghigo C, Chouaib S, Galon J, Iovanna J, Olive D. Pancreatic ductal adenocarcinoma: a strong imbalance of good and bad immunological cops in the tumor microenvironment. Front Immunol. (2018) 9:1044. doi: 10.3389/fimmu.2018.01044
43. Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. (2002) 2:569–79. doi: 10.1038/nri855
44. Boukouris S, Mathivanan S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteomics Clin Appl. (2015) 9:358–67. doi: 10.1002/prca.201400114
45. Jia S, Zocco D, Samuels ML, Chou MF, Chammas R, Skog J, et al. Emerging technologies in extracellular vesicle-based molecular diagnostics. Expert Rev Mol Diagn. (2014) 14:307–21. doi: 10.1586/14737159.2014.893828
46. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
47. Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol. (2015) 35:69–77. doi: 10.1016/j.ceb.2015.04.013
48. Javeed N, Mukhopadhyay D. Exosomes and their role in the micro-/macro-environment: a comprehensive review. J Biomed Res. (2017) 31:386–94. doi: 10.7555/JBR.30.20150162
49. Burrello J, Monticone S, Gai C, Gomez Y, Kholia S, Camussi G. stem cell-derived extracellular vesicles and immune-modulation. Front Cell Dev Biol. (2016) 4:83. doi: 10.3389/fcell.2016.00083
50. Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res. (2017) 120:1658–73. doi: 10.1161/CIRCRESAHA.117.309681
51. Rajagopal C, Harikumar KB. The origin and functions of exosomes in cancer. Front Oncol. (2018) 8:66. doi: 10.3389/fonc.2018.00066
52. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
53. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
54. Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. (2016) 36:301–12. doi: 10.1007/s10571-016-0366-z
55. Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. (2014) 28:3–13. doi: 10.1016/j.semcancer.2014.04.009
56. Choi DS, Kim DK, Kim YK, Gho YS. Proteomics of extracellular vesicles: exosomes and ectosomes. Mass Spectrom Rev. (2015) 34:474–90. doi: 10.1002/mas.21420
57. Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci USA. (2012) 109:4146–51. doi: 10.1073/pnas.1200448109
58. Egea-Jimenez AL, Zimmermann P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J Lipid Res. (2018) 59:1554–60. doi: 10.1194/jlr.R083964
59. Shen B, Fang Y, Wu N, Gould SJ. Biogenesis of the posterior pole is mediated by the exosome/microvesicle protein-sorting pathway. J Biol Chem. (2011) 286:44162–76. doi: 10.1074/jbc.M111.274803
60. Guo BB, Bellingham SA, Hill AF. The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem. (2015) 290:3455–67. doi: 10.1074/jbc.M114.605253
61. Moreno-Gonzalo O, Fernandez-Delgado I, Sanchez-Madrid F. Post-translational add-ons mark the path in exosomal protein sorting. Cell Mol Life Sci. (2018) 75:1–19. doi: 10.1007/s00018-017-2690-y
62. Vedeler A, Hollås H, Grindheim AK, Raddum AM. Multiple roles of annexin A2 in post-transcriptional regulation of gene expression. Curr Protein Pept Sci. (2012) 13:401–12. doi: 10.2174/138920312801619402
63. Janas T, Janas MM, Sapon K, Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. (2015) 589:1391–8. doi: 10.1016/j.febslet.2015.04.036
64. Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. (2013) 4:2980. doi: 10.1038/ncomms3980
65. Gezer U, Özgür E, Cetinkaya M, Isin M, Dalay N. Long non-coding RNAs with low expression levels in cells are enriched in secreted exosomes. Cell Biol Int. (2014) 38:1076–9. doi: 10.1002/cbin.10301
66. Dragomir M, Chen B, Calin GA. Exosomal lncRNAs as new players in cell-to-cell communication. Transl Cancer Res. (2018) 7(Suppl 2):S243–52. doi: 10.21037/tcr.2017.10.46
67. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. (2018) 75:193–208. doi: 10.1007/s00018-017-2595-9
68. Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res. (2010) 51:2105–20. doi: 10.1194/jlr.M003657
69. Skotland T, Hessvik NP, Sandvig K, Llorente A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J Lipid Res. (2018) 60:9–18. doi: 10.1194/jlr.R084343
70. Laulagnier K, Grand D, Dujardin A, Hamdi S, Vincent-Schneider H, Lankar D, et al. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett. (2004) 572:11–4. doi: 10.1016/j.febslet.2004.06.082
71. Carayon K, Chaoui K, Ronzier E, Lazar I, Bertrand-Michel J, Roques V, et al. Proteolipidic composition of exosomes changes during reticulocyte maturation. J Biol Chem. (2011) 286:34426–39. doi: 10.1074/jbc.M111.257444
72. Paillasse MR, de Medina P, Amouroux G, Mhamdi L, Poirot M, Silvente-Poirot S. Signaling through cholesterol esterification: a new pathway for the cholecystokinin 2 receptor involved in cell growth and invasion. J Lipid Res. (2009) 50:2203–11. doi: 10.1194/jlr.M800668-JLR200
73. Bowden KL, Bilbey NJ, Bilawchuk LM, Boadu E, Sidhu R, Ory DS, et al. Lysosomal acid lipase deficiency impairs regulation of ABCA1 gene and formation of high density lipoproteins in cholesteryl ester storage disease. J Biol Chem. (2011) 286:30624–35. doi: 10.1074/jbc.M111.274381
74. Costa Verdera H, Gitz-Francois JJ, Schiffelers RM, Vader P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J Control Release. (2017) 266:100–8. doi: 10.1016/j.jconrel.2017.09.019
75. Strauss K, Goebel C, Runz H, Möbius W, Weiss S, Feussner I, et al. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J Biol Chem. (2010) 285:26279–88. doi: 10.1074/jbc.M110.134775
76. Record M, Silvente-Poirot S, Poirot M, Wakelam MJO. Extracellular vesicles: lipids as key components of their biogenesis and functions. J Lipid Res. (2018) 59:1316–24. doi: 10.1194/jlr.E086173
77. Skotland T, Sandvig K, Llorente A. Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res. (2017) 66:30–41. doi: 10.1016/j.plipres.2017.03.001
78. Pollet H, Conrard L, Cloos AS, Tyteca D. Plasma membrane lipid domains as platforms for vesicle biogenesis and shedding? Biomolecules. (2018) 8:E94. doi: 10.3390/biom8030094
79. Choi D, Spinelli C, Montermini L, Rak J. Oncogenic regulation of extracellular vesicle proteome and heterogeneity. Proteomics. (2019) 19:e1800169. doi: 10.1002/pmic.201800169
80. Zöller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer. (2009) 9:40–55. doi: 10.1038/nrc2543
81. Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. (2014) 5:442. doi: 10.3389/fimmu.2014.00442
82. Greening DW, Xu R, Gopal SK, Rai A, Simpson RJ. Proteomic insights into extracellular vesicle biology - defining exosomes and shed microvesicles. Expert Rev Proteomics. (2017) 14:69–95. doi: 10.1080/14789450.2017.1260450
83. Rosa-Fernandes L, Rocha VB, Carregari VC, Urbani A, Palmisano G. A perspective on extracellular vesicles proteomics. Front Chem. (2017) 5:102. doi: 10.3389/fchem.2017.00102
84. Heiler S, Wang Z, Zöller M. Pancreatic cancer stem cell markers and exosomes - the incentive push. World J Gastroenterol. (2016) 22:5971–6007. doi: 10.3748/wjg.v22.i26.5971
85. Sun Z, Wang L, Dong L, Wang X. Emerging role of exosome signalling in maintaining cancer stem cell dynamic equilibrium. J Cell Mol Med. (2018). doi: 10.1111/jcmm.13676. [Epub ahead of print].
86. Wang Z, Zöller M. Exosomes, metastases, and the miracle of cancer stem cell markers. Cancer Metastasis Rev. (2019) 38:259–95. doi: 10.1007/s10555-019-09793-6
87. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. (2017) 16:10. doi: 10.1186/s12943-016-0577-4
88. Lathia JD, Liu H. Overview of cancer stem cells and stemness for community oncologists. Target Oncol. (2017) 12:387–99. doi: 10.1007/s11523-017-0508-3
89. Najafi M, Mortezaee K, Ahadi R. Cancer stem cell (a)symmetry & plasticity: tumorigenesis and therapy relevance. Life Sci. (2019) 231:116520. doi: 10.1016/j.lfs.2019.05.076
90. Rao CV, Mohammed A. New insights into pancreatic cancer stem cells. World J Stem Cells. (2015) 7:547–55. doi: 10.4252/wjsc.v7.i3.547
91. Wang Z, Zhao K, Hackert T, Zöller M. CD44/CD44v6 a reliable companion in cancer-initiating cell maintenance and tumor progression. Front Cell Dev Biol. (2018) 6:97. doi: 10.3389/fcell.2018.00097
92. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527:329–35. doi: 10.1038/nature15756
93. Paolillo M, Schinelli S. Integrins and exosomes, a dangerous liaison in cancer progression. Cancers. (2017) 9: E95. doi: 10.3390/cancers9080095
94. Ma B, Zhang L, Zou Y, He R, Wu Q, Han C, et al. Reciprocal regulation of integrin β4 and KLF4 promotes gliomagenesis through maintaining cancer stem cell traits. J Exp Clin Cancer Res. (2019) 38:23. doi: 10.1186/s13046-019-1034-1
95. Eom BW, Joo J, Park B, Jo MJ, Choi SH, Cho SJ, et al. Nomogram incorporating CD44v6 and clinicopathological factors to predict lymph node metastasis for early gastric cancer. PLoS ONE. (2016) 11:e0159424. doi: 10.1371/journal.pone.0159424
96. Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. (2017) 108:296–307. doi: 10.1111/cas.13156
97. Giovannetti E, van der Borden CL, Frampton AE, Ali A, Firuzi O, Peters GJ. Never let it go: Stopping key mechanisms underlying metastasis to fight pancreatic cancer. Semin Cancer Biol. (2017) 44:43–59. doi: 10.1016/j.semcancer.2017.04.006
98. Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. (2016) 35:816–26. doi: 10.1038/onc.2015.139
99. Li YP, Pang J, Gao S, Bai PY, Wang WD, Kong P, et al. Role of CXCR4 and SDF1 as prognostic factors for survival and the association with clinicopathology in colorectal cancer: a systematic meta-analysis. Tumour Biol. (2017) 39:1010428317706206. doi: 10.1177/1010428317706206
100. Sleightholm RL, Neilsen BK, Li J, Steele MM, Singh RK, Hollingsworth MA, et al. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol Ther. (2017) 179:158–70. doi: 10.1016/j.pharmthera.2017.05.012
101. Liu H, Jiang F, Jia X, Lan J, Guo H, Li E, et al. Cycling hypoxia affects cell invasion and proliferation through direct regulation of claudin1/claudin7 expression, and indirect regulation of P18 through claudin7. Oncotarget. (2017) 8:10298–311. doi: 10.18632/oncotarget.14397
102. Wu Z, Shi J, Song Y, Zhao J, Sun J, Chen X, et al. Claudin-7 (CLDN7) is overexpressed in gastric cancer and promotes gastric cancer cell proliferation, invasion and maintains mesenchymal state. Neoplasma. (2018) 65:349–59. doi: 10.4149/neo_2018_170320N200
103. Rassouli FB, Matin MM, Saeinasab M. Cancer stem cells in human digestive tract malignancies. Tumour Biol. (2016) 37:7–21. doi: 10.1007/s13277-015-4155-y
104. Zarour LR, Anand S, Billingsley KG, Bisson WH, Cercek A, Clarke MF, et al. Colorectal cancer liver metastasis: evolving paradigms and future directions. Cell Mol Gastroenterol Hepatol. (2017) 3:163–73. doi: 10.1016/j.jcmgh.2017.01.006
105. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. (2017) 51:1357–69. doi: 10.3892/ijo.2017.4129
106. Basu S, Haase G, Ben-Ze'ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Res. (2016) 5:F1000 Faculty Rev-699. doi: 10.12688/f1000research.7579.1
107. Rappa G, Mercapide J, Anzanello F, Pope RM, Lorico A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol Cancer. (2013) 12:62. doi: 10.1186/1476-4598-12-62
108. Li X, Zhao H, Gu J, Zheng L. Prognostic value of cancer stem cell marker CD133 expression in pancreatic ductal adenocarcinoma (PDAC): a systematic review and meta-analysis. Int J Clin Exp Pathol. (2015) 8:12084–92.
109. Mitchell SF, Parker R. Principles and properties of eukaryotic mRNPs. Mol Cell. (2014) 54:547–58. doi: 10.1016/j.molcel.2014.04.033
110. Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. (2014) 15:829–45. doi: 10.1038/nrg3813
111. Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, et al. Comprehensive identification of RNA-binding proteins by RNA interactome capture. Methods Mol Biol. (2016) 1358:131–9. doi: 10.1007/978-1-4939-3067-8_8
112. Yamashita A, Takeuchi O. Translational control of mRNAs by 3'-Untranslated region binding proteins. BMB Rep. (2017) 50:194–200. doi: 10.5483/BMBRep.2017.50.4.040
113. Castello A, Hentze MW, Preiss T. Metabolic enzymes enjoying new partnerships as RNA-binding proteins. Trends Endocrinol Metab. (2015) 26:746–57. doi: 10.1016/j.tem.2015.09.012
114. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
115. Chin A, Lécuyer E. RNA localization: making its way to the center stage. Biochim Biophys Acta Gen Subj. (2017) 1861:2956–70. doi: 10.1016/j.bbagen.2017.06.011
116. Eliscovich C, Singer RH. RNP transport in cell biology: the long and winding road. Curr Opin Cell Biol. (2017) 45:38–46. doi: 10.1016/j.ceb.2017.02.008
117. Di Liegro CM, Schiera G, Di Liegro I. Extracellular vesicle-associated RNA as a carrier of epigenetic information. Genes. (2017) 8:E240. doi: 10.3390/genes8100240
118. Sadik N, Cruz L, Gurtner A, Rodosthenous RS, Dusoswa SA, Ziegler O, et al. Extracellular RNAs: a new awareness of old perspectives. Methods Mol Biol. (2018) 1740:1–15. doi: 10.1007/978-1-4939-7652-2_1
119. Castello A, Frese CK, Fischer B, Järvelin AI, Horos R, Alleaume AM, et al. Identification of RNA-binding domains of RNA-binding proteins in cultured cells on a system-wide scale with RBDmap. Nat Protoc. (2017) 12:2447–64. doi: 10.1038/nprot.2017.106
120. Varela-Eirin M, Varela-Vazquez A, Rodríguez-Candela Mateos M, Vila-Sanjurjo A, Fonseca E, Mascareñas JL, et al. Recruitment of RNA molecules by connexin RNA-binding motifs: implication in RNA and DNA transport through microvesicles and exosomes. Biochim Biophys Acta Mol Cell Res. (2017) 1864:728–36. doi: 10.1016/j.bbamcr.2017.02.001
121. Schiera G, Di Liegro CM, Puleo V, Colletta O, Fricano A, Cancemi P, et al. Extracellular vesicles shed by melanoma cells contain a modified form of H1.0 linker histone and H1.0 mRNA-binding proteins. Int J Oncol. (2016) 49:1807–14. doi: 10.3892/ijo.2016.3692
122. Ragni E, Banfi F, Barilani M, Cherubini A, Parazzi V, Larghi P, et al. extracellular vesicle-shuttled mRNA in mesenchymal stem cell communication. Stem Cells. (2017) 35:1093–105. doi: 10.1002/stem.2557
123. Schwarzenbach H, Gahan PB. MicroRNA shuttle from cell-to-cell by exosomes and its impact in cancer. Noncoding RNA. (2019) 5:E28. doi: 10.3390/ncrna5010028
124. Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. (2009) 11:1150–6. doi: 10.1038/ncb1930
125. Kosaka N. Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. (2010) 285:17442–52. doi: 10.1074/jbc.M110.107821
126. Villarroya-Beltri C, Guerra S, Sánchez-Madrid F. ISGylation - a key to lock the cell gates for preventing the spread of threats. J Cell Sci. (2017) 130:2961–9. doi: 10.1242/jcs.205468
127. Santangelo L, Giurato G, Cicchini C, Montaldo C, Mancone C, Tarallo R, et al. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling MicroRNA sorting. Cell Rep. (2016) 17:799–808. doi: 10.1016/j.celrep.2016.09.031
128. Bhome R, Del Vecchio F, Lee GH, Bullock MD, Primrose JN, Sayan AE, et al. Exosomal microRNAs (exomiRs): small molecules with a big role in cancer. Cancer Lett. (2018) 420:228–35. doi: 10.1016/j.canlet.2018.02.002
129. Akhade VS, Pal D, Kanduri C. Long noncoding RNA: genome organization and mechanism of action. Adv Exp Med Biol. (2017) 1008:47–74. doi: 10.1007/978-981-10-5203-3_2
130. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. (2010) 3:ra8. doi: 10.1126/scisignal.2000568
131. Ng SY, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. (2012) 31:522–33. doi: 10.1038/emboj.2011.459
132. Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. (2007) 445:666–70. doi: 10.1038/nature05519
133. Fang Y, Fullwood MJ. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics. (2016) 14:42–54. doi: 10.1016/j.gpb.2015.09.006
134. Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. (2008) 32:232–46. doi: 10.1016/j.molcel.2008.08.022
135. Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. (2013) 52:101–12. doi: 10.1016/j.molcel.2013.08.027
136. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
137. Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. (2013) 24:206–214. doi: 10.1016/j.devcel.2012.12.012
138. Li XL, Wu ZQ, Fu XB, Han WD. LncRNAs: insights into their function and mechanics in underlying disorders. Mutat Res Rev Mut Res. (2014) 762:1–21. doi: 10.1016/j.mrrev.2014.04.002
139. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. (2014) 505:344–52. doi: 10.1038/nature12986
140. Xie Y, Dang W, Zhang S, Yue W, Yang L, Zhai X, et al. The role of exosomal noncoding RNAs in cancer. Mol Cancer. (2019) 18:37. doi: 10.1186/s12943-019-0984-4
141. Ahadi A, Brennan S, Kennedy PJ, Hutvagner G, Tran N. Long non-coding RNAs harboring miRNA seed regions are enriched in prostate cancer exosomes. Sci Rep. (2016) 6:24922. doi: 10.1038/srep24922
142. Kawamura Y, Yamamoto Y, Sato TA, Ochiya T. Extracellular vesicles as trans-genomic agents: emerging roles in disease and evolution. Cancer Sci. (2017) 108:824–30. doi: 10.1111/cas.13222
143. Lee TH, Chennakrishnaiah S, Meehan B, Montermini L, Garnier D, D'Asti E, et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget. (2016) 7:51991–2002. doi: 10.18632/oncotarget.10627
144. Penfornis P, Vallabhaneni KC, Whitt J, Pochampally R. Extracellular vesicles as carriers of microRNA, proteins and lipids in tumor microenvironment. Int J Cancer. (2016) 138:14–21. doi: 10.1002/ijc.29417
145. Bakhshandeh B, Kamaleddin MA, Aalishah K. A comprehensive review on exosomes and microvesicles as epigenetic factors. Curr Stem Cell Res Ther. (2017) 12:31–36. doi: 10.2174/1574888X11666160709211528
146. Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. (2014) 3. doi: 10.3402/jev.v3.24641
147. Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M, et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. (2010) 70:1668–78. doi: 10.1158/0008-5472.CAN-09-2470
148. Gesierich S, Berezovskiy I, Ryschich E, Zöller M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. (2006) 66:7083–94. doi: 10.1158/0008-5472.CAN-06-0391
149. Buck CA, Horwitz AF. Cell surface receptors for extracellular matrix molecules. Annu Rev Cell Biol. (1987) 3:179–205. doi: 10.1146/annurev.cb.03.110187.001143
150. Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, et al. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci USA. (2011) 108:4852–7. doi: 10.1073/pnas.1017667108
151. Morelli AE, Larregina AT, Shufesky WJ, Sullivan MLG, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. (2004) 104:3257–66. doi: 10.1182/blood-2004-03-0824
152. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. (2015) 17:816–26. doi: 10.1038/ncb3169
153. Han W, Duan Z. Roles of exosomes in liver metastases: novel diagnosis and treatment choices. J Cell Physiol. (2019) 234:21588–600. doi: 10.1002/jcp.28785
154. French KC, Antonyak MA, Cerione RA. Extracellular vesicle docking at the cellular port: extracellular vesicle binding and uptake. Semin Cell Dev Biol. (2017) 67:48–55. doi: 10.1016/j.semcdb.2017.01.002
155. Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. (2009) 20:1053–67. doi: 10.1681/ASN.2008070798
156. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. (2015) 523:177–82. doi: 10.1038/nature14581
157. Mu W, Rana S, Zöller M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia. (2013) 15:875–87. doi: 10.1593/neo.13786
158. Barrès C, Blanc L, Bette-Bobillo P, André S, Mamoun R, Gabius HJ, et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood. (2010) 115:696–705. doi: 10.1182/blood-2009-07-231449
159. Rana S, Malinowska K, Zöller M. Exosomal tumor MicroRNA modulates premetastatic organ cells. Neoplasia. (2013) 15:281–95. doi: 10.1593/neo.122010
160. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood. (2014) 123:208–16. doi: 10.1182/blood-2013-03-489732
161. Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. (2014) 468–70: 565–80. doi: 10.1016/j.virol.2014.09.009
162. Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. (2012) 44:1574–84. doi: 10.1016/j.biocel.2012.06.018
163. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. (2017) 546:498–503. doi: 10.1038/nature22341
164. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. (2012) 119:756–66. doi: 10.1182/blood-2011-02-338004
165. Del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. (2012) 106:1604–11. doi: 10.1182/blood-2004-03-1095
166. Tian T, Zhu YL, Hu FH, Wang YY, Huang NP, Xiao ZD. Dynamics of exosome internalization and trafficking. J Cell Physiol. (2013) 228:1487–95. doi: 10.1002/jcp.24304
167. Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci USA. (2013) 110:17380–5. doi: 10.1073/pnas.1304266110
168. Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. (2010) 11:675–87. doi: 10.1111/j.1600-0854.2010.01041.x
169. Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. (2014) 262:193–215. doi: 10.1111/imr.12212
170. Nakase I, Kobayashi NB, Takatani-Nakase T, Yoshida T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep. (2015) 5:10300. doi: 10.1038/srep10300
171. Zech D, Rana S, Büchler MW, Zöller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. (2012) 10:37. doi: 10.1186/1478-811X-10-37
172. Horibe S, Tanahashi T, Kawauchi S, Murakami Y, Rikitake Y. Mechanism of recipient cell-dependent differences in exosome uptake. BMC Cancer. (2018) 18:47. doi: 10.1186/s12885-017-3958-1
173. Nanbo A, Kawanishi E, Yoshida R, Yoshiyama H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J Virol. (2013) 87:10334–47. doi: 10.1128/JVI.01310-13
174. Zheng Y, Tu C, Zhang J, Wang J. Inhibition of multiple myeloma-derived exosomes uptake suppresses the functional response in bone marrow stromal cell. Int J Oncol. (2019) 54:1061–70. doi: 10.3892/ijo.2019.4685
175. Heusermann W, Hean J, Trojer D, Steib E, von Bueren S, Graff-Meyer A, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol. (2016) 213:173–84. doi: 10.1083/jcb.201506084
176. Holder B, Jones T, Sancho Shimizu V, Rice TF, Donaldson B, Bouqueau M, et al. Macrophage exosomes induce placental inflammatory cytokines: a novel mode of maternal-placental messaging. Traffic. (2016) 17:168–78. doi: 10.1111/tra.12352
177. Leone DA, Peschel A, Brown M, Schachner H, Ball MJ, Gyuraszova M, et al. Surface LAMP-2 is an endocytic receptor that diverts antigen internalized by human dendritic cells into highly immunogenic exosomes. J Immunol. (2017) 199:531–46. doi: 10.4049/jimmunol.1601263
178. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
179. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. (2008) 451:141–6. doi: 10.1038/nature06534
180. Daley GQ. Stem cells and the evolving notion of cellular identity. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140376. doi: 10.1098/rstb.2014.0376
181. Verga Falzacappa MV, Ronchini C, Reavie LB, Pelicci PG. Regulation of self-renewal in normal and cancer stem cells. FEBS J. (2012) 279:3559–72. doi: 10.1111/j.1742-4658.2012.08727.x
182. Liu J. The dualistic origin of human tumors. Semin Cancer Biol. (2018) 53:1–16. doi: 10.1016/j.semcancer.2018.07.004
183. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414:105–11. doi: 10.1038/35102167
184. Ratajczak MZ, Bujko K, Mack A, Kucia M, Ratajczak J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia. (2018) 32:2519–26. doi: 10.1038/s41375-018-0294-7
185. Soteriou D, Fuchs Y. A matter of life and death: stem cell survival in tissue regeneration and tumour formation. Nat Rev Cancer. (2018) 18:187–201. doi: 10.1038/nrc.2017.122
186. Bourguignon LY, Shiina M, Li JJ. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv Cancer Res. (2014) 123:255–75. doi: 10.1016/B978-0-12-800092-2.00010-1
187. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. (2015) 6:10697–711. doi: 10.18632/oncotarget.4037
188. Skvortsova I, Debbage P, Kumar V, Skvortsov S. Radiation resistance: cancer stem cells (CSCs) and their enigmatic pro-survival signaling. Semin Cancer Biol. (2015) 35:39–44. doi: 10.1016/j.semcancer.2015.09.009
189. Lipinska N, Romaniuk A, Paszel-Jaworska A, Toton E, Kopczynski P, Rubis B. Telomerase and drug resistance in cancer. Cell Mol Life Sci. (2017) 74:4121–32. doi: 10.1007/s00018-017-2573-2
190. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci. (2017) 18:E2574. doi: 10.3390/ijms18122574
191. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
192. Zhou P, Li B, Liu F, Zhang M, Wang Q, Liu Y, et al. The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol Cancer. (2017) 16:52. doi: 10.1186/s12943-017-0624-9
193. Li H, Peyrollier K, Kilic G, Brakebusch C. Rho GTPases and cancer. Biofactors. (2014) 40:226–35. doi: 10.1002/biof.1155
194. Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med. (2015) 4:1033–43. doi: 10.5966/sctm.2015-0048
195. de Lucas B, Pérez LM, Gálvez BG. Importance and regulation of adult stem cell migration. J Cell Mol Med. (2018) 22:746–54. doi: 10.1111/jcmm.13422
196. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. (2018) 18:533–48. doi: 10.1038/s41568-018-0038-z
197. Albini A, Bruno A, Gallo C, Pajardi G, Noonan DM, Dallaglio K. Cancer stem cells and the tumor microenvironment: interplay in tumor heterogeneity. Connect Tissue Res. (2015) 56:414–25. doi: 10.3109/03008207.2015.1066780
198. Correnti M, Raggi C. Stem-like plasticity and heterogeneity of circulating tumor cells: current status and prospect challenges in liver cancer. Oncotarget. (2017) 8:7094–115. doi: 10.18632/oncotarget.12569
199. Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem Sci. (2003) 28:106–12. doi: 10.1016/S0968-0004(02)00014-2
200. Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. (2005) 6:801–11. doi: 10.1038/nrm1736
201. Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology. (2005) 20:218–24. doi: 10.1152/physiol.00015.2005
202. Berditchevski F, Odintsova E. Tetraspanins as regulators of protein trafficking. Traffic. (2007) 8:89–96. doi: 10.1111/j.1600-0854.2006.00515.x
203. Yáñez-Mó M, Gutiérrez-López MD, Cabañas C. Functional interplay between tetraspanins and proteases. Cell Mol Life Sci. (2011) 68:3323–35. doi: 10.1007/s00018-011-0746-y
204. Bassani S, Cingolani LA. Tetraspanins: interactions and interplay with integrins. Int J Biochem Cell Biol. (2012) 44:703–8. doi: 10.1016/j.biocel.2012.01.020
205. Halova I, Draber P. tetraspanins and transmembrane adaptor proteins as plasma membrane organizers-mast cell case. Front Cell Dev Biol. (2016) 4:43. doi: 10.3389/fcell.2016.00043
206. Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, et al. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol. (2013) 87:3435–46. doi: 10.1128/JVI.02906-12
207. Stepanek O, Draber P, Horejsi V. Palmitoylated transmembrane adaptor proteins in leukocyte signaling. Cell Signal. (2014) 26:895–902. doi: 10.1016/j.cellsig.2014.01.007
208. Schmidt TH, Homsi Y, Lang T. Oligomerization of the tetraspanin CD81 via the flexibility of Its δ-loop. Biophys J. (2016) 110:2463–74. doi: 10.1016/j.bpj.2016.05.003
209. Termini CM, Gillette JM. Tetraspanins function as regulators of cellular signaling. Front Cell Dev Biol. (2017) 5:34. doi: 10.3389/fcell.2017.00034
210. Yue S, Zhao K, Erb U, Rana S, Zöller M. Joint features and complementarities of Tspan8 and CD151 revealed in knockdown and knockout models. Biochem Soc Trans. (2017) 45:437–47. doi: 10.1042/BST20160298
211. Greco C, Bralet MP, Ailane N, Dubart-Kupperschmitt A, Rubinstein E, Le Naour F, et al. E-cadherin/p120-catenin and tetraspanin Co-029 cooperate for cell motility control in human colon carcinoma. Cancer Res. (2010) 70:7674–83. doi: 10.1158/0008-5472.CAN-09-4482
212. Serewko-Auret MM, Mould AW, Loffler KA, Duncan R, Kay GF, Hayward NK. Alterations in gene expression in MEN1-associated insulinoma development. Pancreas. (2010) 39:1140–6. doi: 10.1097/MPA.0b013e3181dc67fc
213. Wang H, Rana S, Giese N, Büchler MW, Zöller M. Tspan8, CD44v6 and alpha6beta4 are biomarkers of migrating pancreatic cancer-initiating cells. Int J Cancer. (2013) 133:416–26. doi: 10.1002/ijc.28044
214. Madhavan B, Yue S, Galli U, Rana S, Gross W, Müller M, et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer. (2015) 136:2616–27. doi: 10.1002/ijc.29324
215. Ailane N, Greco C, Zhu Y, Sala-Valdés M, Billard M, Casal I, et al. Effect of an anti-human Co-029/tspan8 mouse monoclonal antibody on tumor growth in a nude mouse model. Front Physiol. (2014) 5:364. doi: 10.3389/fphys.2014.00364
216. Berthier-Vergnes O, El Kharbili M, de la Fouchardière A, Pointecouteau T, Verrando P, Wierinckx A, et al. Gene expression profiles of human melanoma cells with different invasive potential reveal TSPAN8 as a novel mediator of invasion. Br J Cancer. (2011) 104:155–65. doi: 10.1038/sj.bjc.6605994
217. Wei L, Li Y, Suo Z. TSPAN8 promotes gastric cancer growth and metastasis via ERK MAPK pathway. Int J Clin Exp Med. (2015) 8:8599–607.
218. Pan SJ, Wu YB, Cai S, Pan YX, Liu W, Bian LG, et al. Over-expression of tetraspanin 8 in malignant glioma regulates tumor cell progression. Biochem Biophys Res Commun. (2015) 458:476–82. doi: 10.1016/j.bbrc.2015.01.128
219. Park CS, Kim TK, Kim HG, Kim YJ, Jeoung MH, Lee WR, et al. Therapeutic targeting of tetraspanin8 in epithelial ovarian cancer invasion and metastasis. Oncogene. (2016) 35:4540–8. doi: 10.1038/onc.2015.520
220. Akiel MA, Santhekadur PK, Mendoza RG, Siddiq A, Fisher PB, Sarkar D. Tetraspanin 8 mediates AEG-1-induced invasion and metastasis in hepatocellular carcinoma cells. FEBS Lett. (2016) 590:2700–8. doi: 10.1002/1873-3468.12268
221. Wang Z, von Au A, Schnölzer M, Hackert T, Zöller M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget. (2016) 7:55409–36. doi: 10.18632/oncotarget.10580
222. Zhu Y, Ailane N, Sala-Valdés M, Haghighi-Rad F, Billard M, Nguyen V, et al. Multi-factorial modulation of colorectal carcinoma cells motility - partial coordination by the tetraspanin Co-029/tspan8. Oncotarget. (2017) 8:27454–70. doi: 10.18632/oncotarget.16247
223. Gesierich S, Paret C, Hildebrand D, Weitz J, Zgraggen K, Schmitz-Winnenthal FH, et al. Colocalization of the tetraspanins, CO-029 and CD151, with integrins in human pancreatic adenocarcinoma: impact on cell motility. Clin Cancer Res. (2005) 11:2840–52. doi: 10.1158/1078-0432.CCR-04-1935
224. Claas C, Wahl J, Orlicky DJ, Karaduman H, Schnölzer M, Kempf T, et al. The tetraspanin D6.1A and its molecular partners on rat carcinoma cells. Biochem J. (2005) 389:99–110. doi: 10.1042/BJ20041287
225. Drapkin R, Crum CP, Hecht JL. Expression of candidate tumor markers in ovarian carcinoma and benign ovary: evidence for a link between epithelial phenotype and neoplasia. Hum Pathol. (2004) 35:1014–21. doi: 10.1016/j.humpath.2004.04.014
226. Le Naour F, André M, Greco C, Billard M, Sordat B, Emile JF, et al. Profiling of the tetraspanin web of human colon cancer cells. Mol Cell Proteomics. (2006) 5:845–57. doi: 10.1074/mcp.M500330-MCP200
227. Kanatsu-Shinohara M, Takashima S, Ishii K, Shinohara T. Dynamic changes in EPCAM expression during spermatogonial stem cell differentiation in the mouse testis. PLoS ONE. (2011) 6:e23663. doi: 10.1371/journal.pone.0023663
228. Rana S, Claas C, Kretz CC, Nazarenko I, Zöller M. Activation-induced internalization differs for the tetraspanins CD9 and Tspan8: Impact on tumor cell motility. Int J Biochem Cell Biol. (2011) 43:106–19. doi: 10.1016/j.biocel.2010.10.002
229. Nievers MG, Schaapveld RQ, Sonnenberg A. Biology and function of hemidesmosomes. Matrix Biol. (1999) 18:5–17. doi: 10.1016/S0945-053X(98)00003-1
230. Margadant C, Frijns E, Wilhelmsen K, Sonnenberg A. Regulation of hemidesmosome disassembly by growth factor receptors. Curr Opin Cell Biol. (2008) 20:589–96. doi: 10.1016/j.ceb.2008.05.001
231. Pan SJ, Zhan SK, Pan YX, Liu W, Bian LG, Sun B, et al. Tetraspanin 8-rictor-integrin α3 complex is required for glioma cell migration. Int J Mol Sci. (2015) 16:5363–74. doi: 10.3390/ijms16035363
232. Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. (2014) 36:157–65. doi: 10.1016/j.semcdb.2014.08.011
233. Ladwein M, Pape UF, Schmidt DS, Schnölzer M, Fiedler S, Langbein L, et al. The cell-cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res. (2005) 309:345–57. doi: 10.1016/j.yexcr.2005.06.013
234. Heiler S, Mu W, Zöller M, Thuma F. The importance of claudin-7 palmitoylation on membrane subdomain localization and metastasis-promoting activities. Cell Commun Signal. (2015) 13:29. doi: 10.1186/s12964-015-0105-y
235. Kuhn S, Koch M, Nübel T, Ladwein M, Antolovic D, Klingbeil P, et al. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res. (2007) 5:553–67. doi: 10.1158/1541-7786.MCR-06-0384
236. Le Naour F, Zöller M. The tumor antigen EpCAM: tetraspanins and the tight junction protein claudin-7, new partners, new functions. Front Biosci. (2008) 13:5847–65. doi: 10.2741/3121
237. Wu CJ, Mannan P, Lu M, Udey MC. Epithelial cell adhesion molecule (EpCAM) regulates claudin dynamics and tight junctions. J Biol Chem. (2013) 288:12253–68. doi: 10.1074/jbc.M113.457499
238. Okada T, Nakamura T, Watanabe T, Onoda N, Ashida A, Okuyama R, et al. Coexpression of EpCAM, CD44 variant isoforms and claudin-7 in anaplastic thyroid carcinoma. PLoS ONE. (2014) 9:e94487. doi: 10.1371/journal.pone.0094487
239. Fang T, Lin J, Wang Y, Chen G, Huang J, Chen J, et al. Tetraspanin-8 promotes hepatocellular carcinoma metastasis by increasing ADAM12m expression. Oncotarget. (2016) 7:40630–43. doi: 10.18632/oncotarget.9769
240. Yue S, Mu W, Zöller M. Tspan8 and CD151 promote metastasis by distinct mechanisms. Eur J Cancer. (2013) 49:2934–48. doi: 10.1016/j.ejca.2013.03.032
241. Schmidt F, Müller M, Prox J, Arnold P, Schönherr C, Tredup C, et al. Tetraspanin 8 is an interactor of the metalloprotease meprin β within tetraspanin-enriched microdomains. Biol Chem. (2016) 397:857–69. doi: 10.1515/hsz-2016-0126
242. Claas C, Seiter S, Claas A, Savelyeva L, Schwab M, Zöller M. Association between the rat homologue of CO-029, a metastasis-associated tetraspanin molecule and consumption coagulopathy. J Cell Biol. (1998) 141:267–80. doi: 10.1083/jcb.141.1.267
243. Yao L, Damodaran G, Nikolskaya N, Gorman AM, Windebank A, Pandit A. The effect of laminin peptide gradient in enzymatically cross-linked collagen scaffolds on neurite growth. J Biomed Mater Res A. (2010) 92:484–92. doi: 10.1002/jbm.a.32359
244. Wang Z, Sun H, Provaznik J, Hackert T, Zöller M. Pancreatic cancer-initiating cell exosome message transfer into noncancer-initiating cells: the importance of CD44v6 in reprogramming. J Exp Clin Cancer Res. (2019) 38:132. doi: 10.1186/s13046-019-1129-8
245. Stewart RL, O'Connor KL. Clinical significance of the integrin α6β4 in human malignancies. Lab Invest. (2015) 95:976–86. doi: 10.1038/labinvest.2015.82
246. Ramovs V, Te Molder L, Sonnenberg A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. (2017) 57–58:213–43. doi: 10.1016/j.matbio.2016.08.007
247. Dowling J, Yu QC, Fuchs E. Beta4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol. (1996) 134:559–72. doi: 10.1083/jcb.134.2.559
248. Tsuruta D, Hashimoto T, Hamill KJ, Jones JC. Hemidesmosomes and focal contact proteins: functions and cross-talk in keratinocytes, bullous diseases and wound healing. J Dermatol Sci. (2011) 62:1–7. doi: 10.1016/j.jdermsci.2011.01.005
249. Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J Cell Biol. (1997) 139:1873–84. doi: 10.1083/jcb.139.7.1873
250. Yu PT, Babicky M, Jaquish D, French R, Marayuma K, Mose E, et al. The RON-receptor regulates pancreatic cancer cell migration through phosphorylation-dependent breakdown of the hemidesmosome. Int J Cancer. (2012) 131:1744–54. doi: 10.1002/ijc.27447
251. Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. (2006) 126:489–502. doi: 10.1016/j.cell.2006.05.047
252. Shaw LM. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol. (2001) 21:5082–93. doi: 10.1128/MCB.21.15.5082-5093.2001
253. Lee SS, Lee SJ, Lee SH, Ryu JM, Lim HS, Kim JS, et al. Netrin-1-induced stem cell bioactivity contributes to the regeneration of injured tissues via the lipid raft-dependent integrin α6β4 signaling pathway. Sci Rep. (2016) 6:37526. doi: 10.1038/srep37526
254. Laval S, Laklai H, Fanjul M, Pucelle M, Laurell H, Billon-Galés A, et al. Dual roles of hemidesmosomal proteins in the pancreatic epithelium: the phosphoinositide 3-kinase decides. Oncogene. (2014) 33:1934–44. doi: 10.1038/onc.2013.146
255. Hintermann E, Yang N, O'Sullivan D, Higgins JM, Quaranta V. Integrin alpha6beta4-erbB2 complex inhibits haptotaxis by up-regulating E-cadherin cell-cell junctions in keratinocytes. J Biol Chem. (2005) 280:8004–15. doi: 10.1074/jbc.M406301200
256. Folgiero V, Bachelder RE, Bon G, Sacchi A, Falcioni R, Mercurio AM. The α6β4 integrin can regulate ErbB-3 expression: implications for alpha6beta4 signaling and function. Cancer Res. (2007) 67:1645–52. doi: 10.1158/0008-5472.CAN-06-2980
257. O'Connor KL, Chen M, Towers LN. Integrin α6β4 cooperates with LPA signaling to stimulate Rac through AKAP-Lbc-mediated RhoA activation. Am J Physiol Cell Physiol. (2012) 302:C605–14. doi: 10.1152/ajpcell.00095.2011
258. Tan X, Xu A, Zhao T, Zhao Q, Zhang J, Fan C, et al. Simulated microgravity inhibits cell focal adhesions leading to reduced melanoma cell proliferation and metastasis via FAK/RhoA-regulated mTORC1 and AMPK pathways. Sci Rep. (2018) 8:3769. doi: 10.1038/s41598-018-20459-1
259. Tai YL, Chu PY, Lai IR, Wang MY, Tseng HY, Guan JL, et al. An EGFR/Src-dependent β4 integrin/FAK complex contributes to malignancy of breast cancer. Sci Rep. (2015) 5:16408. doi: 10.1038/srep16408
260. Fan J, Cai B, Zeng M, Hao Y, Giancotti FG, Fu BM. Integrin β4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann Biomed Eng. (2011) 39:2223–41. doi: 10.1007/s10439-011-0321-6
261. Yue S, Mu W, Erb U, Zöller M. The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget. (2015) 6:2366–84. doi: 10.18632/oncotarget.2958
262. Wang H, Jin H, Beauvais DM, Rapraeger AC. Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J Biol Chem. (2014) 289:30318–32. doi: 10.1074/jbc.M114.586438
263. Bhatia V, Mula RV, Weigel NL, Falzon M. Parathyroid hormone-related protein regulates cell survival pathways via integrin alpha6beta4-mediated activation of phosphatidylinositol 3-kinase/Akt signaling. Mol Cancer Res. (2009) 7:1119–31. doi: 10.1158/1541-7786.MCR-08-0568
264. Kawakami K, Fujita Y, Kato T, Mizutani K, Kameyama K, Tsumoto H, et al. Integrin β4 and vinculin contained in exosomes are potential markers for progression of prostate cancer associated with taxane-resistance. Int J Oncol. (2015) 47:384–90. doi: 10.3892/ijo.2015.3011
265. Jauliac S, López-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. (2002) 4:540–4. doi: 10.1038/ncb816
266. Houben AJ, Moolenaar WH. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. (2011) 30:557–65. doi: 10.1007/s10555-011-9319-7
267. Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. (2010) 176:528–35. doi: 10.2353/ajpath.2010.090526
268. Chen M, Bresnick AR, O'Connor KL. Coupling S100A4 to Rhotekin alters Rho signaling output in breast cancer cells. Oncogene. (2013) 32:3754–64. doi: 10.1038/onc.2012.383
269. Chen M, Sinha M, Luxon BA, Bresnick AR, O'Connor KL. Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J Biol Chem. (2009) 284:1484–94. doi: 10.1074/jbc.M803997200
270. Yoon SO, Shin S, Lipscomb EA. A novel mechanism for integrin-mediated ras activation in breast carcinoma cells: the alpha6beta4 integrin regulates ErbB2 translation and transactivates epidermal growth factor receptor/ErbB2 signaling. Cancer Res. (2006) 66:2732–9. doi: 10.1158/0008-5472.CAN-05-2941
271. Welser-Alves JV, Boroujerdi A, Tigges U, Wrabetz L, Feltri ML, Milner R. Endothelial β4 integrin is predominantly expressed in arterioles, where it promotes vascular remodeling in the hypoxic brain. Arterioscler Thromb Vasc Biol. (2013) 33:943–53. doi: 10.1161/ATVBAHA.112.300566
272. Feng C, Ye C, Liu X, Ma H, Li M. Beta4 integrin is involved in statin-induced endothelial cell death. Biochem Biophys Res Commun. (2004) 323:858–64. doi: 10.1016/j.bbrc.2004.08.171
273. Wang L, Dong Z, Zhang Y, Miao J. The roles of integrin β4 in vascular endothelial cells. J Cell Physiol. (2012) 227:474–8. doi: 10.1002/jcp.22769
274. Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG. Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell. (2004) 6:471–83. doi: 10.1016/j.ccr.2004.09.029
275. Mercurio AM, Lipscomb EA, Bachelder RE. Non-angiogenic functions of VEGF in breast cancer. J Mamm Gland Biol Neoplasia. (2005) 10:283–90. doi: 10.1007/s10911-006-9001-9
276. Roberts JE, Nikolopoulos SN, Oktem O, Giancotti F, Oktay K. Integrin beta-4 signaling plays a key role in mouse embryogenesis. Reprod Sci. (2009) 16:286–93. doi: 10.1177/1933719108325506
277. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. (2014) 14:342–56. doi: 10.1016/j.stem.2014.01.009
278. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. (2007) 445:106–10. doi: 10.1038/nature05372
279. Idzerda RL, Carter WG, Nottenburg C, Wayner EA, Gallatin WM, John T. Isolation and DNA sequence of a cDNA clone encoding a lymphocyte adhesion receptor for high endothelium. Proc Natl Acad Sci. USA. (1989) 86:4659–63. doi: 10.1073/pnas.86.12.4659
280. Goldstein LA, Butcher EC. Identification of mRNA that encodes an alternative form of H-CAM (CD44) in lymphoid and nonlymphoid tissues. Immunogenetics. (1990) 32:389–97. doi: 10.1007/BF00241632
281. Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc Natl Acad Sci USA. (1992) 89:12160–4. doi: 10.1073/pnas.89.24.12160
282. Peach RJ, Hollenbaugh D, Stamenkovic I, Aruffo A. Identification of hyaluronic acid binding sites in the extracellular domain of CD44. J Cell Biol. (1993) 122:257–64. doi: 10.1083/jcb.122.1.257
283. Ishii S, Ford R, Thomas P, Nachman A, Steele G Jr, Jessup JM. CD44 participates in the adhesion of human colorectal carcinoma cells to laminin and type IV collagen. Surg Oncol. (1993) 2:255–64. doi: 10.1016/0960-7404(93)90015-Q
284. Neame SJ, Isacke CM. The cytoplasmic tail of CD44 is required for basolateral localization in ephitelial MDCK cells but does not mediate association with the detergent-insoluble cytoskeleton of fibroblasts. J Cell Biol. (1993) 121:1299–310. doi: 10.1083/jcb.121.6.1299
285. Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, et al. CD44 isoforms containing exon v3 are responsible for the presentation of heparin-binding growth factor. J Cell Biol. (1995) 128:687–98. doi: 10.1083/jcb.128.4.687
286. Föger N, Marhaba R, Zöller M. Raft associated interaction of CD44 with the cytoskeleton. J Cell Sci. (2001) 114:1169–78.
287. Oliferenko S, Paiha K, Harder T, Gerke V, Schwärzler C, Schwarz H, et al. Analysis of CD44-containing lipid rafts: Recruitment of annexin II and stabilization by the actin cytoskeleton. J Cell Biol. (1999) 146:843–54. doi: 10.1083/jcb.146.4.843
288. Lokeshwar VB, Fregien N, Bourguignon LY. Ankyrin-binding domain of CD44(Gp85) is required for the expression of hyaluronic acid-mediated adhesion function. J Cell Biol. (1994) 126:1099–109. doi: 10.1083/jcb.126.4.1099
289. Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol. (1992) 116:817–25. doi: 10.1083/jcb.116.3.817
290. Toyama-Sorimachi N, Miyasaka M. A novel ligand for CD44 is sulfated proteoglycan. Int Immunol. (1994) 6:655–60. doi: 10.1093/intimm/6.4.655
291. Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. (1990) 61:1303–13. doi: 10.1016/0092-8674(90)90694-A
292. Greenfield B, Wang WC, Marquardt H, Piepkorn M, Wolff EA, Aruffo A, et al. Characterization of the heparan sulfate and chondroitin sulfate assembly sites in CD44. J Biol Chem. (1999) 274:2511–7. doi: 10.1074/jbc.274.4.2511
293. Higman VA, Briggs DC, Mahoney DJ, Blundell CD, Sattelle BM, Dyer DP, et al. A refined model for the TSG-6 link module in complex with hyaluronan: use of defined oligosaccharides to probe structure and function. J Biol Chem. (2014) 289:5619–34. doi: 10.1074/jbc.M113.542357
294. Tremmel M, Matzke A, Albrecht I, Laib AM, Olaku V, Ballmer-Hofer K, et al. A CD44v6 peptide reveals a role of CD44 in VEGFR-2 signaling and angiogenesis. Blood. (2009) 114:5236–44. doi: 10.1182/blood-2009-04-219204
295. Kim MS, Park MJ, Moon EJ, Kim SJ, Lee CH, Yoo H, et al. Hyaluronic acid induces osteopontin via the phosphatidylinositol 3-kinase/Akt pathway to enhance the motility of human glioma cells. Cancer Res. (2005) 65:686–91.
296. Yuan J, Gu K, He J, Sharma S. Preferential up-regulation of osteopontin in primary central nervous system lymphoma does not correlate with putative receptor CD44v6 or CD44H expression. Hum Pathol. (2013) 44:606–11. doi: 10.1016/j.humpath.2012.07.007
297. Orian-Rousseau V, Morrison H, Matzke A, Kastilan T, Pace G, Herrlich P, et al. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Mol Biol Cell. (2007) 18:76–83. doi: 10.1091/mbc.e06-08-0674
298. Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthases: potential therapeutic targets in cancer. Curr Drug Targets Cardiovasc Haematol Disord. (2005) 5:3–14. doi: 10.2174/1568006053005056
299. Misra S, Toole BP, Ghatak S. Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J Biol Chem. (2006) 281:34936–41. doi: 10.1074/jbc.C600138200
300. Orian-Rousseau V. CD44 Acts as a signaling platform controlling tumor progression and metastasis. Front Immunol. (2015) 6:154. doi: 10.3389/fimmu.2015.00154
301. Xu H, Tian Y, Yuan X, Wu H, Liu Q, Pestell RG, et al. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. (2015) 8:3783–92. doi: 10.2147/OTT.S95470
302. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. (2010) 11:276–87. doi: 10.1038/nrm2866
303. Stamenkovic I, Yu Q. Merlin, a “Magic” linker between extracellular cues and intracellular signaling pathways that regulate cell. motility, proliferation, and survival. Curr Protein Pept Sci. (2010) 11:471–84. doi: 10.2174/138920310791824011
304. Mori T, Kitano K, Terawaki S, Maesaki R, Fukami Y, Hakoshima T. Structural basis for CD44 recognition by ERM proteins. J Biol Chem. (2008) 283:29602–12. doi: 10.1074/jbc.M803606200
305. Horta S, Agostinho AL, Mateus R, Pereira L, Pereira C, Capinha L, et al. Looking out for cancer stem cells' properties: the value-driving role of CD44 for personalized medicines. Curr Cancer Drug Targets. (2015) 14:832–49. doi: 10.2174/1568009614666141111154713
306. Ingley E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. (2008) 1784:56–65. doi: 10.1016/j.bbapap.2007.08.012
307. Ekyalongo RC, Nakayama H, Kina K, Kaga N, Iwabuchi K. Organization and functions of glycolipid-enriched microdomains in phagocytes. Biochim Biophys Acta. (2015) 1851:90–7. doi: 10.1016/j.bbalip.2014.06.009
308. Korcsmaros T, Schneider MV, Superti-Furga G. Next generation of network medicine: interdisciplinary signaling approaches. Integr Biol. (2017) 9:97–108. doi: 10.1039/c6ib00215c
309. Cooper JA, Qian H. A mechanism for SRC kinase-dependent signaling by noncatalytic receptors. Biochemistry. (2008) 47:5681–8. doi: 10.1021/bi8003044
310. Nastase MV, Janicova A, Wygrecka M, Schaefer L. Signaling at the crossroads: matrix-derived proteoglycan and reactive oxygen species signaling. Antioxid Redox Signal. (2017) 27:855–73. doi: 10.1089/ars.2017.7165
311. Heldin P, Basu K, Kozlova I, Porsch H. HAS2 and CD44 in breast tumorigenesis. Adv Cancer Res. (2014) 123:211–29. doi: 10.1016/B978-0-12-800092-2.00008-3
312. Binder MJ, McCoombe S, Williams ED, McCulloch DR, Ward AC. The extracellular matrix in cancer progression: role of hyalectan proteoglycans and ADAMTS enzymes. Cancer Lett. (2017) 385:55–64. doi: 10.1016/j.canlet.2016.11.001
313. Vitale D, Kumar Katakam S, Greve B, Jang B, Oh ES, Alaniz L, et al. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. (2019) 286:2870–82. doi: 10.1111/febs.14967
314. Grass GD, Dai L, Qin Z, Parsons C, Toole BP. CD147: regulator of hyaluronan signaling in invasiveness and chemoresistance. Adv Cancer Res. (2014) 123:351–73. doi: 10.1016/B978-0-12-800092-2.00013-7
315. Bates RC, Elith CA, Thorne RF, Burns GF. Engagement of variant CD44 confers resistance to anti-integrin antibody-mediated apoptosis in a colon carcinoma cell line. Cell Adhes Commun. (1998) 6:21–38. doi: 10.3109/15419069809069758
316. Teoh SL, Das S. The emerging role of the hippo pathway in lung cancers: clinical implications. Curr Drug Targets. (2017) 18:1880–92. doi: 10.2174/1389450117666160907153338
317. Fan F, Xiuwen Z, Yongyi L, Weiping C, Lu G, Yueqin L, et al. The CD44 variant induces K562 cell acquired with resistance to adriamycin via NF-κB/Snail/Bcl-2 pathway. Med Hypotheses. (2018) 121:142–8. doi: 10.1016/j.mehy.2018.09.002
318. Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol. (2009) 185:949–57. doi: 10.1083/jcb.200812060
319. Zhang Y, Han X, Wu H, Zhou Y. Bioinformatics analysis of transcription profiling of solid pseudopapillary neoplasm of the pancreas. Mol Med Rep. (2017) 16:1635–42. doi: 10.3892/mmr.2017.6800
320. Bourguignon LY, Xia W, Wong G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J Biol Chem. (2009) 284:2657–71. doi: 10.1074/jbc.M806708200
321. Ibrahim SA, Gadalla R, El-Ghonaimy EA, Samir O, Mohamed HT, Hassan H, et al. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol Cancer. (2017) 16:57. doi: 10.1186/s12943-017-0621-z
322. Sun H, Rana S, Wang Z, Schnölzer M, Provaznik J, Hackert T, et al. The pancreatic cancer stem cell marker CD44v6 affects Tspan8 transcription: consequences on exosome composition and delivery. J Oncol. (2019) 2019:3516973. doi: 10.1155/2019/3516973
323. Karousou E, Misra S, Ghatak S, Dobra K, Götte M, Vigetti D, et al. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. (2017) 59:3–22. doi: 10.1016/j.matbio.2016.10.001
324. Schmitt M, Metzger M, Gradl D, Davidson G, Orian-Rousseau V. CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ. (2015) 22:677–89. doi: 10.1038/cdd.2014.156
325. Yong X, Tang B, Xiao YF, Xie R, Qin Y, Luo G, et al. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. (2016) 374:292–303. doi: 10.1016/j.canlet.2016.02.032
326. Kashyap T, Pramanik KK, Nath N, Mishra P, Singh AK, Nagini S, et al. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. (2018) 86:234–43. doi: 10.1016/j.oraloncology.2018.09.028
327. Senbanjo LT, Chellaiah MA. cd44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. (2017) 5:18. doi: 10.3389/fcell.2017.00018
328. Zhang NH, Li J, Li Y, Zhang XT, Liao WT, Zhang JY, et al. Co-expression of CXCR4 and CD133 proteins is associated with poor prognosis in stage II-III colon cancer patients. Exp Ther Med. (2012) 3:973–82. doi: 10.3892/etm.2012.527
329. Popple A, Durrant LG, Spendlove I, Rolland P, Scott IV, Deen S, et al. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br J Cancer. (2012) 106:1306–13. doi: 10.1038/bjc.2012.49
330. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. (2010) 330:1066–71. doi: 10.1126/science.1194396
331. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. (2007) 1768:952–63. doi: 10.1016/j.bbamem.2006.11.002
332. Cheng M, Huang K, Zhou J, Yan D, Tang YL, Zhao TC, et al. A critical role of Src family kinase in SDF-1/CXCR4-mediated bone-marrow progenitor cell recruitment to the ischemic heart. J Mol Cell Cardiol. (2015) 81:49–53. doi: 10.1016/j.yjmcc.2015.01.024
333. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. (2010) 16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329
334. Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. (2002) 277:49212–9. doi: 10.1074/jbc.M207294200
335. Cojoc M, Peitzsch C, Trautmann F, Polishchuk L, Telegeev GD, Dubrovska A. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther. (2013) 6:1347–61. doi: 10.2147/OTT.S36109
336. Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. (1998) 38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289
337. Liebick M, Henze S, Vogt V, Oppermann M. Functional consequences of chemically-induced β-arrestin binding to chemokine receptors CXCR4 and CCR5 in the absence of ligand stimulation. Cell Signal. (2017) 38:201–11. doi: 10.1016/j.cellsig.2017.07.010
338. Penela P, Murga C, Ribas C, Lafarga V, Mayor F Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. (2010) 160:821–32. doi: 10.1111/j.1476-5381.2010.00727.x
339. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. (2007) 69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749
340. Mandawat A, Fiskus W, Buckley KM, Robbins K, Rao R, Balusu R, et al. Pan-histone deacetylase inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic antimyeloid activity in combination with CXCR4 antagonists. Blood. (2010) 116:5306–15. doi: 10.1182/blood-2010-05-284414
341. Wysoczynski M, Reca R, Ratajczak J, Kucia M, Shirvaikar N, Honczarenko M, et al. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood. (2005) 105:40–8. doi: 10.1182/blood-2004-04-1430
342. Chinni SR, Yamamoto H, Dong Z, Sabbota A, Bonfil RD, Cher ML. CXCL12/CXCR4 transactivates HER2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol Cancer Res. (2008) 6:446–57. doi: 10.1158/1541-7786.MCR-07-0117
343. Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. (2014) 6:219ra7. doi: 10.1126/scitranslmed.3007563
344. Singh AP, Arora S, Bhardwaj A, Srivastava SK, Kadakia MP, Wang B, et al. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor κB: implications for bidirectional tumor-stromal interactions. J Biol Chem. (2012) 287:39115–24. doi: 10.1074/jbc.M112.409581
345. Wang Z, Ma Q, Liu Q, Yu H, Zhao L, Shen S, et al. Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br J Cancer. (2008) 99:1695–703. doi: 10.1038/sj.bjc.6604745
346. Pan F, Ma S, Cao W, Liu H, Chen F, Chen X, et al. SDF-1α upregulation of MMP-2 is mediated by p38 MAPK signaling in pancreatic cancer cell lines. Mol Biol Rep. (2013) 40:4139–46. doi: 10.1007/s11033-012-2225-4
347. Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. (2012) 322:169–76. doi: 10.1016/j.canlet.2012.02.035
348. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. (2007) 1:313–23. doi: 10.1016/j.stem.2007.06.002
349. Weekes CD, Song D, Arcaroli J, Wilson LA, Rubio-Viqueira B, Cusatis G, et al. Stromal cell-derived factor 1α mediates resistance to mTOR-directed therapy in pancreatic cancer. Neoplasia. (2012) 14:690–701. doi: 10.1593/neo.111810
350. Pozzobon T, Goldoni G, Viola A, Molon B. CXCR4 signaling in health and disease. Immunol Lett. (2016) 177:6–15. doi: 10.1016/j.imlet.2016.06.006
351. Teixidó J, Martínez-Moreno M, Díaz-Martínez M, Sevilla-Movilla S. The good and bad faces of the CXCR4 chemokine receptor. Int J Biochem Cell Biol. (2018) 95:121–31. doi: 10.1016/j.biocel.2017.12.018
352. Tu TC, Nagano M, Yamashita T, Hamada H, Ohneda K, Kimura K, et al. A chemokine receptor, CXCR4, which is regulated by hypoxia-inducible factor 2α, is crucial for functional endothelial progenitor cells migration to ischemic tissue and wound repair. Stem Cells Dev. (2016) 25:266–76. doi: 10.1089/scd.2015.0290
353. Billadeau DD, Chatterjee S, Bramati P, Sreekumar R, Shah V, Hedin K, et al. Characterization of the CXCR4 signaling in pancreatic cancer cells. Int J Gastrointest Cancer. (2006) 37:110–9. doi: 10.1007/s12029-007-0011-7
354. Kim SJ, Lee Y, Kim NY, Hwang Y, Hwang B, Min JK, et al. Pancreatic adenocarcinoma upregulated factor, a novel endothelial activator, promotes angiogenesis and vascular permeability. Oncogene. (2013) 32:3638–47. doi: 10.1038/onc.2012.366
355. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. (2013) 73:3927–37. doi: 10.1158/0008-5472.CAN-12-4479
356. Chen HC, Chou AS, Liu YC, Hsieh CH, Kang CC, Pang ST, et al. Induction of metastatic cancer stem cells from the NK/LAK-resistant floating, but not adherent, subset of the UP-LN1 carcinoma cell line by IFN-γ. Lab Invest. (2011) 91:1502–13. doi: 10.1038/labinvest.2011.91
357. Shi J, Wei Y, Xia J, Wang S, Wu J, Chen F, et al. CXCL12-CXCR4 contributes to the implication of bone marrow in cancer metastasis. Future Oncol. (2014) 10:749–59. doi: 10.2217/fon.13.193
358. Unlu S, Tang S, Wang E, Martinez I, Tang D, Bianchi ME, et al. Damage associated molecular pattern molecule-induced microRNAs (DAMPmiRs) in human peripheral blood mononuclear cells. PLoS ONE. (2012) 7:e38899. doi: 10.1371/annotation/18ec64ae-e086-446c-b63c-a71fa2bba046
359. Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, et al. Intracellular Hmgb1 inhibits inflammatory nuleosome release and limits acute pancreatitis in mice. Gastroenterology. (2014) 146:1097–107. doi: 10.1053/j.gastro.2013.12.015
360. Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. (2013) 4:e966. doi: 10.1038/cddis.2013.493
361. Shakir M, Tang D, Zeh HJ, Tang SW, Anderson CJ, Bahary N, et al. The chemokine receptors CXCR4/CXCR7 and their primary heterodimeric ligands CXCL12 and CXCL12/high mobility group box 1 in pancreatic cancer growth and development: finding flow. Pancreas. (2015) 44:528–34. doi: 10.1097/MPA.0000000000000298
362. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. (2007) 104:10158–63. doi: 10.1073/pnas.0703478104
363. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. (2007) 445:111–5. doi: 10.1038/nature05384
364. D'Agostino G, Cecchinato V, Uguccioni M. Chemokine heterocomplexes and cancer: a novel chapter to be written in tumor immunity. Front Immunol. (2018) 9:2185. doi: 10.3389/fimmu.2018.02185
365. Xu D, Li R, Wu J, Jiang L, Zhong HA. Drug design targeting the CXCR4/CXCR7/CXCL12 pathway. Curr Top Med Chem. (2016) 16:1441–51. doi: 10.2174/1568026615666150915120218
366. Xue LJ, Mao XB, Ren LL, Chu XY. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. (2017) 6:1424–36. doi: 10.1002/cam4.1085
367. Günzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol. (2012) 2:1819–52. doi: 10.1002/cphy.c110045
368. Nelson GM, Padera TP, Garkavtsev I, Shioda T, Jain RK. Differential gene expression of primary cultured lymphatic and blood vascular endothelial cells. Neoplasia. (2007) 9:1038–45. doi: 10.1593/neo.07643
369. Yano T, Kanoh H, Tamura A, Tsukita S. Apical cytoskeletons and junctional complexes as a combined system in epithelial cell sheets. Ann N Y Acad Sci. (2017) 1405:32–43. doi: 10.1111/nyas.13432
370. Rosenthal R, Günzel D, Theune D, Czichos C, Schulzke JD, Fromm M. Water channels and barriers formed by claudins. Ann N Y Acad Sci. (2017) 1397:100–9. doi: 10.1111/nyas.13383
371. Tsukita S, Tanaka H, Tamura A. The claudins: from tight junctions to biological systems. Trends Biochem Sci. (2019) 44:141–52. doi: 10.1016/j.tibs.2018.09.008
372. Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, et al. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. (2012) 142:305–15. doi: 10.1053/j.gastro.2011.10.025
373. Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. (2015) 64:1529–38. doi: 10.1136/gutjnl-2014-308419
374. Günzel D. Claudins: vital partners in transcellular and paracellular transport coupling. Pflugers Arch. (2017) 469:35–44. doi: 10.1007/s00424-016-1909-3
375. Baumholtz AI, Gupta IR, Ryan AK. Claudins in morphogenesis: forming an epithelial tube. Tissue Barriers. (2017) 5:e1361899. doi: 10.1080/21688370.2017.1361899
376. Tanaka H, Tamura A, Suzuki K, Tsukita S. Site-specific distribution of claudin-based paracellular channels with roles in biological fluid flow and metabolism. Ann N Y Acad Sci. (2017) 1405:44–52. doi: 10.1111/nyas.13438
377. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. (2018) 50:103. doi: 10.1038/s12276-018-0126-x
378. Hagen SJ. Non-canonical functions of claudin proteins: beyond the regulation of cell-cell adhesions. Tissue Barriers. (2017) 5:e1327839. doi: 10.1080/21688370.2017.1327839
379. Stamatovic SM, Johnson AM, Sladojevic N, Keep RF, Andjelkovic AV. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. (2017) 1397:54–65. doi: 10.1111/nyas.13346
380. Van Itallie CM, Anderson JM. Claudin interactions in and out of the tight junction. Tissue Barriers. (2013) 1:e25247. doi: 10.4161/tisb.25247
381. Sjö A, Magnusson KE, Peterson KH. Protein kinase C activation has distinct effects on the localization, phosphorylation and detergent solubility of the claudin protein family in tight and leaky epithelial cells. J Membr Biol. (2010) 236:181–9. doi: 10.1007/s00232-010-9289-7
382. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. (2013) 145:407–15. doi: 10.1053/j.gastro.2013.04.011
383. Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. (2012) 1258:9–18. doi: 10.1111/j.1749-6632.2012.06613.x
384. Manabe A, Furukawa C, Endo S, Marunaka K, Nishiyama T, Fujii N, et al. Chlorpheniramine increases paracellular permeability to marker fluorescein lucifer yellow mediated by internalization of occludin in murine colonic epithelial cells. Biol Pharm Bull. (2017) 40:1299–305. doi: 10.1248/bpb.b17-00244
385. Li J, Li YX, Chen MH, Li J, Du J, Shen B, et al. Changes in the phosphorylation of claudins during the course of experimental colitis. Int J Clin Exp Pathol. (2015) 8:12225–33.
386. Shigetomi K, Ono Y, Inai T, Ikenouchi J. Adherens junctions influence tight junction formation via changes in membrane lipid composition. J Cell Biol. (2018) 217:2373–81. doi: 10.1083/jcb.201711042
387. Thuma F, Heiler S, Schnölzer M, Zöller M. Palmitoylated claudin7 captured in glycolipid-enriched membrane microdomains promotes metastasis via associated transmembrane and cytosolic molecules. Oncotarget. (2016) 7:30659–77. doi: 10.18632/oncotarget.8928
388. Philip R, Heiler S, Mu W, Büchler MW, Zöller M, Thuma F. Claudin-7 promotes the epithelial-mesenchymal transition in human colorectal cancer. Oncotarget. (2015) 6:2046–63. doi: 10.18632/oncotarget.2858
389. Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H, Simpson RJ. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics. (2013) 12:587–98. doi: 10.1074/mcp.M112.021303
390. Kowal J, Tkach M, Théry C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. (2014) 29:116–25. doi: 10.1016/j.ceb.2014.05.004
391. Kyuno D, Bauer N, Schnölzer M, Provaznik J, Ryschich E, Hackert T, et al. Distinct origin of claudin7 in early tumor endosomes affects exosome assembly. Int J Biol Sci. (2019) 15:2224–39. doi: 10.7150/ijbs.35347
392. Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. (2012) 38:68–75. doi: 10.1016/j.ctrv.2011.04.002
393. Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. (2009) 69:5627–9. doi: 10.1158/0008-5472.CAN-09-0654
394. Litvinov SV, Velders MP, Bakker HA, Fleuren GJ, Warnaar SO. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J Cell Biol. (1994) 125:437–46. doi: 10.1083/jcb.125.2.437
395. Boesch M, Spizzo G, Seeber A. Concise review: aggressive colorectal cancer: role of epithelial cell adhesion molecule in cancer stem cells and epithelial-to-mesenchymal transition. Stem Cells Transl Med. (2018) 7:495–501. doi: 10.1002/sctm.17-0289
396. Maetzel D, Denzel S, Mack B, Eggert C, Bärr G, Gires O. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. (2009) 11:162–71. doi: 10.1038/ncb1824
397. Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC. Epithelial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial-mesenchymal transition genes expression in colon cancer. J Biol Chem. (2012) 287:39449–59. doi: 10.1074/jbc.M112.386235
398. Gires O, Stoecklein NH. Dynamic EpCAM expression on circulating and disseminating tumor cells: causes and consequences. Cell Mol Life Sci. (2014) 71:4393–402. doi: 10.1007/s00018-014-1693-1
399. Hsu YT, Gu F, Huang YW, Liu J, Ruan J, Huang RL, et al. Promoter hypomethylation of EpCAM-regulated bone morphogenetic protein gene family in recurrent endometrial cancer. Clin Cancer Res. (2013) 19:6272–85. doi: 10.1158/1078-0432.CCR-13-1734
400. Sankpal NV, Fleming TP, Sharma PK, Wiedner HJ, Gillanders WE. A double-negative feedback loop between EpCAM and ERK contributes to the regulation of epithelial-mesenchymal transition in cancer. Oncogene. (2017) 36:3706–17. doi: 10.1038/onc.2016.504
401. Herreros-Pomares A, Aguilar-Gallardo C, Calabuig-Fariñas S, Sirera R, Jantus-Lewintre E, Camps C. EpCAM duality becomes this molecule in a new Dr. Jekyll and Mr. Hyde tale. Crit Rev Oncol Hematol. (2018) 126:52–63. doi: 10.1016/j.critrevonc.2018.03.006
402. Denzel S, Mack B, Eggert C, Massoner P, Stöcklein N, Kemming D, et al. MMP7 is a target of the tumour-associated antigen EpCAM. Int J Exp Pathol. (2012) 93:341–53. doi: 10.1111/j.1365-2613.2012.00826.x
403. Imrich S, Hachmeister M, Gires O. EpCAM and its potential role in tumor-initiating cells. Cell Adh Migr. (2012) 6:30–8. doi: 10.4161/cam.18953
404. Maghzal N, Vogt E, Reintsch W, Fraser JS, Fagotto F. The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling. J Cell Biol. (2010) 191:645–59. doi: 10.1083/jcb.201004074
405. Huang L, Yang Y, Yang F, Liu S, Zhu Z, Lei Z, et al. Functions of EpCAM in physiological processes and diseases (Review). Int J Mol Med. (2018) 42:1771–85. doi: 10.3892/ijmm.2018.3764
406. Yahyazadeh Mashhadi SM, Kazemimanesh M, Arashkia A, Azadmanesh K, Meshkat Z, Golichenari B, et al. Shedding light on the EpCAM: an overview. J Cell Physiol. (2019) 234:12569–80. doi: 10.1002/jcp.28132
407. Gao J, Liu X, Yang F, Liu T, Yan Q, Yang X. By inhibiting Ras/Raf/ERK and MMP-9, knockdown of EpCAM inhibits breast cancer cell growth and metastasis. Oncotarget. (2015) 6:27187–98. doi: 10.18632/oncotarget.4551
408. Wang H, Stoecklein NH, Lin PP, Gires O. Circulating and disseminated tumor cells: diagnostic tools and therapeutic targets in motion. Oncotarget. (2017) 8:1884–912. doi: 10.18632/oncotarget.12242
409. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. (2011) 71:5317–26. doi: 10.1158/0008-5472.CAN-11-1059
410. Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer. (2009) 9:143. doi: 10.1038/nrc2499-c1
411. Driemel C, Kremling H, Schumacher S, Will D, Wolters J, Lindenlauf N, et al. Context-dependent adaption of EpCAM expression in early systemic esophageal cancer. Oncogene. (2014) 33:4904–15. doi: 10.1038/onc.2013.441
412. González B, Denzel S, Mack B, Conrad M, Gires O. EpCAM is involved in maintenance of the murine embryonic stem cell phenotype. Stem Cells. (2009) 27:1782–91. doi: 10.1002/stem.97
413. Lu TY, Lu RM, Liao MY, Yu J, Chung CH, Kao CF, et al. Epithelial cell adhesion molecule regulation is associated with the maintenance of the undifferentiated phenotype of human embryonic stem cells. J Biol Chem. (2010) 285:8719–32. doi: 10.1074/jbc.M109.077081
414. van der Gun BTF, Wasserkort R, Monami A, Jeltsch A, Raskó T, Slaska-Kiss K, et al. Persistent downregulation of the pancarcinoma-associated epithelial cell adhesion molecule via active intranuclear methylation. Int J Cancer. (2008) 123:484–9. doi: 10.1002/ijc.23476
415. Yu G, Zhang X, Wang H, Rui D, Yin A, Qiu G, et al. CpG island methylation status in the EpCAM promoter region and gene expression. Oncol Rep. (2008) 20:1061–7.
416. van der Gun BT, de Groote ML, Kazemier HG, Arendzen AJ, Terpstra P, Ruiters MH, et al. Transcription factors and molecular epigenetic marks underlying EpCAM overexpression in ovarian cancer. Br J Cancer. (2011) 105:312–9. doi: 10.1038/bjc.2011.231
417. Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, et al. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers. (2013) 5:334–56. doi: 10.3390/cancers5020334
418. Ji J, Tang J, Deng L, Xie Y, Jiang R, Li G, et al. LINC00152 promotes proliferation in hepatocellular carcinoma by targeting EpCAM via the mTOR signaling pathway. Oncotarget. (2015) 6:42813–24. doi: 10.18632/oncotarget.5970
419. Qian NS, Liu WH, Lv WP, Xiang X, Su M, Raut V, et al. Upregulated microRNA-92b regulates the differentiation and proliferation of EpCAM-positive fetal liver cells by targeting C/EBPß. PLoS ONE. (2013) 8:e68004. doi: 10.1371/journal.pone.0068004
420. Ostenfeld MS, Jensen SG, Jeppesen DK, Christensen LL, Thorsen SB, Stenvang J, et al. miRNA profiling of circulating EpCAM(+) extracellular vesicles: promising biomarkers of colorectal cancer. J Extracell Vesicles. (2016) 5:31488. doi: 10.3402/jev.v5.31488
421. Koo BK, Clevers H. Stem cells marked by the R-spondin receptor LGR5. Gastroenterology. (2014) 147:289–302. doi: 10.1053/j.gastro.2014.05.007
422. Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol. (2013) 5:a015081. doi: 10.1101/cshperspect.a015081
423. Kemper K, Prasetyanti PR, De Lau W, Rodermond H, Clevers H, Medema JP. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells. (2012) 30:2378–86. doi: 10.1002/stem.1233
424. de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. (2011) 476:293–7. doi: 10.1038/nature10337
425. de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. (2014) 28:305–16. doi: 10.1101/gad.235473.113
426. de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. (2017) 543:676–80. doi: 10.1038/nature21713
427. Leung C, Tan SH, Barker N. Recent advances in Lgr5+ stem cell research. Trends Cell Biol. (2018) 28:380–91. doi: 10.1016/j.tcb.2018.01.010
428. Corbeil D, Röper K, Hellwig A, Tavian M, Miraglia S, Watt SM, et al. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. (2000) 275:5512–20. doi: 10.1074/jbc.275.8.5512
429. Ren F, Sheng WQ, Du X. CD133: a cancer stem cells marker, is used in colorectal cancers. World J Gastroenterol. (2013) 19:2603–11. doi: 10.3748/wjg.v19.i17.2603
430. Chen S, Song X, Chen Z, Li X, Li M, Liu H, et al. CD133 expression and the prognosis of colorectal cancer: a systematic review and meta-analysis. PLoS ONE. (2013) 8:e56380. doi: 10.1371/journal.pone.0056380
431. Röper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. (2000) 2:582–92. doi: 10.1038/35023524
432. Giebel B, Corbeil D, Beckmann J, Höhn J, Freund D, Giesen K, et al. Segregation of lipid raft markers including CD133 in polarized human hematopoietic stem and progenitor cells. Blood. (2004) 104:2332–8. doi: 10.1182/blood-2004-02-0511
433. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. (2000) 1:31–9. doi: 10.1038/35036052
434. Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep. (2012) 2:951–63. doi: 10.1016/j.celrep.2012.09.016
435. Shimozato O, Waraya M, Nakashima K, Souda H, Takiguchi N, Yamamoto H, et al. Receptor-type protein tyrosine phosphatase κ directly dephosphorylates CD133 and regulates downstream AKT activation. Oncogene. (2015) 34:1949–60. doi: 10.1038/onc.2014.141
436. Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene. (2011) 30:97–105. doi: 10.1038/onc.2010.383
437. Zhang M, Liu Y, Feng H, Bian X, Zhao W, Yang Z, et al. CD133 affects the invasive ability of HCT116 cells by regulating TIMP-2. Am J Pathol. (2013) 182:565–76. doi: 10.1016/j.ajpath.2012.10.015
438. Li C, Wang C, Xing Y, Zhen J, Ai Z. CD133 promotes gallbladder carcinoma cell migration through activating Akt phosphorylation. Oncotarget. (2016) 7:17751–9. doi: 10.18632/oncotarget.7474
439. Liu C, Li Y, Xing Y, Cao B, Yang F, Yang T, et al. The interaction between cancer stem cell marker CD133 and Src protein promotes Focal Adhesion Kinase (FAK) phosphorylation and cell migration. J Biol Chem. (2016) 291:15540–50. doi: 10.1074/jbc.M115.712976
440. Song S, Pei G, Du Y, Wu J, Ni X, Wang S, et al. Interaction between CD133 and PI3K-p85 promotes chemoresistance in gastric cancer cells. Am J Transl Res. (2018) 10:304–14.
441. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. (2013) 23:171–85. doi: 10.1016/j.ccr.2012.12.021
442. Fonseca AV, Bauer N, Corbeil D. The stem cell marker CD133 meets the endosomal compartment–new insights into the cell division of hematopoietic stem cells. Blood Cells Mol Dis. (2008) 41:194–5. doi: 10.1016/j.bcmd.2008.04.004
443. Shmelkov SV, St Clair R, Lyden D, Rafii S. AC133/CD133/Prominin-1. Int J Biochem Cell Biol. (2005) 37:715–9. doi: 10.1016/j.biocel.2004.08.010
444. Corbeil D, Joester A, Fargeas CA, Jászai J, Garwood J, Hellwig A, et al. Expression of distinct splice variants of the stem cell marker prominin-1 (CD133) in glial cells. Glia. (2009) 57:860–74. doi: 10.1002/glia.20812
445. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. (2008) 319:1244–7. doi: 10.1126/science.1153124
446. Badawy SMM, Okada T, Kajimoto T, Hirase M, Matovelo SA, Nakamura S, et al. Extracellular α-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J Biol Chem. (2018) 293:8208–16. doi: 10.1074/jbc.RA118.001986
447. Izumi H, Li Y, Shibaki M, Mori D, Yasunami M, Sato S, et al. Recycling endosomal CD133 functions as an inhibitor of autophagy at the pericentrosomal region. Sci Rep. (2019) 9:2236. doi: 10.1038/s41598-019-39229-8
448. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. (2015) 527:525–30. doi: 10.1038/nature16064
449. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. (2009) 69:2400–7. doi: 10.1158/0008-5472.CAN-08-4312
450. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. (2017) 19:518–29. doi: 10.1038/ncb3513
451. Zöller M, Matzku S, Goerttler K. High incidence of spontaneous transplantable tumours in BDX rats. Br J Cancer. (1978) 37:61–6. doi: 10.1038/bjc.1978.9
452. Matzku S, Komitowski D, Mildenberger M, Zöller M. Characterization of BSp73, a spontaneous rat tumor and its in vivo selected variants showing different metastasizing capacities. Invasion Metast. (1983) 3:109–23.
453. Matzku S, Wenzel A, Liu S, Zöller M. Antigenic differences between metastatic and nonmetastatic BSp73 rat tumor variants characterized by monoclonal antibodies. Cancer Res. (1989) 49:1294–9.
454. Günthert U, Hofmann M, Rudy W, Reber S, Zöller M, Haussmann I, et al. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. (1991) 65:13–24. doi: 10.1016/0092-8674(91)90403-L
455. Herlevsen M, Schmidt DS, Miyazaki K, Zöller M. The association of the tetraspanin D6.1A with the alpha6beta4 integrin supports cell motility and liver metastasis formation. J Cell Sci. (2003) 116:4373–90. doi: 10.1242/jcs.00760
456. Würfel J, Rösel M, Seiter S, Claas C, Herlevsen M, Weth R, et al. Metastasis-association of the rat ortholog of the human epithelial glycoprotein antigen EGP314. Oncogene. (1999) 18:2323–34. doi: 10.1038/sj.onc.1202542
457. Nübel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, et al. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res. (2009) 7:285–99. doi: 10.1158/1541-7786.MCR-08-0200
458. Klingbeil P, Marhaba R, Jung T, Kirmse R, Ludwig T, Zöller M. CD44 variant isoforms promote metastasis formation by a tumor cell-matrix cross-talk that supports adhesion and apoptosis resistance. Mol Cancer Res. (2009) 7:168–79. doi: 10.1158/1541-7786.MCR-08-0207
459. Thuma F, Zöller M. EpCAM-associated claudin-7 supports lymphatic spread and drug resistance in rat pancreatic cancer. Int J Cancer. (2013) 133:855–66. doi: 10.1002/ijc.28085
460. Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum: Transworld Research Network (2012). p. 1–6.
461. Xu M, Zhou BP, Tao M, Liu J, Li W. The role of stromal components in pancreatic cancer progression. Anticancer Agents Med Chem. (2016) 16:1117–24. doi: 10.2174/1871520616666160404115532
462. Pang TCY, Wilson JS, Apte MV. Pancreatic stellate cells: what's new? Curr Opin Gastroenterol. (2017) 33:366–73. doi: 10.1097/MOG.0000000000000378
463. Allam A, Thomsen AR, Gothwal M, Saha D, Maurer J, Brunner TB. Pancreatic stellate cells in pancreatic cancer: in focus. Pancreatology. (2017) 17:514–22. doi: 10.1016/j.pan.2017.05.390
464. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. (1998) 115:421–32. doi: 10.1016/S0016-5085(98)70209-4
465. Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. (2009) 28:3513–25. doi: 10.1038/onc.2009.220
466. Neesse A, Wagner M, Ellenrieder V, Bachem M, Gress TM, Buchholz M. Pancreatic stellate cells potentiate proinvasive effects of SERPINE2 expression in pancreatic cancer xenograft tumors. Pancreatology. (2007) 7:380–5. doi: 10.1159/000107400
467. Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. (2007) 6:1186–97. doi: 10.1158/1535-7163.MCT-06-0686
468. Shields MA, Dangi-Garimella S, Krantz SB, Bentrem DJ, Munshi HG. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J Biol Chem. (2011) 286:10495–504. doi: 10.1074/jbc.M110.195628
469. Tjomsland V, Pomianowska E, Aasrum M, Sandnes D, Verbeke CS, Gladhaug IP. Profile of MMP and TIMP expression in human pancreatic stellate cells: regulation by IL-1α and TGFβ and implications for migration of pancreatic cancer cells. Neoplasia. (2016) 18:447–56. doi: 10.1016/j.neo.2016.06.003
470. Li Y, Song T, Chen Z, Wang Y, Zhang J, Wang X. Pancreatic stellate cells activation and matrix metallopeptidase 2 expression correlate with lymph node metastasis in pancreatic carcinoma. Am J Med Sci. (2019) 357:16–22. doi: 10.1016/j.amjms.2018.10.001
471. Masamune A, Kikuta K, Watanabe T, Satoh K, Satoh A, Shimosegawa T. Pancreatic stellate cells express Toll-like receptors. J Gastroenterol. (2008) 43:352–62. doi: 10.1007/s00535-008-2162-0
472. Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. (2015) 212:2077–94. doi: 10.1084/jem.20142162
473. Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. (2007) 26:117–29. doi: 10.1016/j.immuni.2006.11.011
474. Shimizu K, Hashimoto K, Tahara J, Imaeda H, Andoh A, Shiratori K. Pancreatic stellate cells do not exhibit features of antigen-presenting cells. Pancreas. (2012) 41:422–7. doi: 10.1097/MPA.0b013e31822e673b
475. Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 attracts regulatory T cells: implication for pancreatic cancer. Oncoimmunology. (2015) 4:e1027473. doi: 10.1080/2162402X.2015.1027473
476. Tang D, Gao J, Wang S, Yuan Z, Ye N, Chong Y, et al. Apoptosis and anergy of T cell induced by pancreatic stellate cells-derived galectin-1 in pancreatic cancer. Tumour Biol. (2015) 36:5617–26. doi: 10.1007/s13277-015-3233-5
477. Jiang ZJ, Shen QH, Chen HY, Yang Z, Shuai MQ, Zheng S. Galectin-1 restores immune tolerance to liver transplantation through activation of hepatic stellate cells. Cell Physiol Biochem. (2018) 48:863–79. doi: 10.1159/000491955
478. Saison-Ridinger M, DelGiorno KE, Zhang T, Kraus A, French R, Jaquish D, et al. Reprogramming pancreatic stellate cells via p53 activation: a putative target for pancreatic cancer therapy. PLoS ONE. (2017) 12:e0189051. doi: 10.1371/journal.pone.0189051
479. Pothula SP, Xu Z, Goldstein D, Merrett N, Pirola RC, Wilson JS, et al. Targeting the HGF/c-MET pathway: stromal remodelling in pancreatic cancer. Oncotarget. (2017) 8:76722–39. doi: 10.18632/oncotarget.20822
480. Schnittert J, Heinrich MA, Kuninty PR, Storm G, Prakash J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett. (2018) 420:247–58. doi: 10.1016/j.canlet.2018.01.072
481. Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV, et al. Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol. (2017) 23:382–405. doi: 10.3748/wjg.v23.i3.382
482. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics. (2018) 8:5072–87. doi: 10.7150/thno.26546
483. Fu Y, Liu S, Zeng S, Shen H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer. (2018) 17:62. doi: 10.1186/s12943-018-0815-z
484. Schoepp M, Ströse AJ, Haier J. Dysregulation of miRNA expression in cancer associated fibroblasts (CAFs) and its consequences on the tumor microenvironment. Cancers. (2017) 9:E54. doi: 10.3390/cancers9060054
485. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
486. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
487. Seux M, Peuget S, Montero MP, Siret C, Rigot V, Clerc P, et al. TP53INP1 decreases pancreatic cancer cell migration by regulating SPARC expression. Oncogene. (2011) 30:3049–61. doi: 10.1038/onc.2011.25
488. Kanno A, Satoh K, Masamune A, Hirota M, Kimura K, Umino J, et al. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int J Cancer. (2008) 122:2707–18. doi: 10.1002/ijc.23332
489. Saini F, Argent RH, Grabowska AM. Sonic hedgehog ligand: a role in formation of a mesenchymal niche in human pancreatic ductal adenocarcinoma. Cells. (2019) 8:E424. doi: 10.3390/cells8050424
490. Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. (2004) 64:6950–6. doi: 10.1158/0008-5472.CAN-04-0677
491. Qian D, Tian L, Lu Z, Miao Y. Cytokine profiling and orthotopic xenografing of pancreatic stellate cells. Methods Mol Biol. (2019) 1882:157–60. doi: 10.1007/978-1-4939-8879-2_14
492. Tang D, Wu Q, Yuan Z, Xu J, Zhang H, Jin Z, et al. Identification of key pathways and genes changes in pancreatic cancer cells (BXPC-3) after cross-talked with primary pancreatic stellate cells using bioinformatics analysis. Neoplasma. (2019) 66:681–93. doi: 10.4149/neo_2018_181020N786
493. Al-Assar O, Demiciorglu F, Lunardi S, Gaspar-Carvalho MM, McKenna WG, Muschel RM, et al. Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother Oncol. (2014) 111:243–51. doi: 10.1016/j.radonc.2014.03.014
494. Zhang H, Yue J, Jiang Z, Zhou R, Xie R, Xu Y, et al. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. (2017) 8:e2790. doi: 10.1038/cddis.2017.180
495. Amrutkar M, Aasrum M, Verbeke CS, Gladhaug IP. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer. (2019) 19:596. doi: 10.1186/s12885-019-5803-1
496. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F, et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med. (2015) 7:735–53. doi: 10.15252/emmm.201404346
497. Liu Y, Li F, Gao F, Xing L, Qin P, Liang X, et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour Biol. (2016) 37:15283–91. doi: 10.1007/s13277-016-5321-6
498. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, et al. Tumor-stroma IL1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res. (2018) 78:1700–12. doi: 10.1158/0008-5472.CAN-17-1366
499. Neumann CCM, von Hörschelmann E, Reutzel-Selke A, Seidel E, Sauer IM, Pratschke J, et al. Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat Dis Int. (2018) 17:461–72. doi: 10.1016/j.hbpd.2018.09.004
500. Hernandez-Unzueta I, Benedicto A, Romayor I, Herrero A, Sanz E, Arteta B, et al. Ocoxin oral solution exerts an antitumoral effect in pancreatic cancer and reduces the stromal-mediated chemoresistance. Pancreas. (2019) 48:555–67. doi: 10.1097/MPA.0000000000001277
501. Hwang HJ, Oh MS, Lee DW, Kuh HJ. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J Exp Clin Cancer Res. (2019) 38:258. doi: 10.1186/s13046-019-1225-9
502. Yang XP, Liu SL, Xu JF, Cao SG, Li Y, Zhou YB. Pancreatic stellate cells increase pancreatic cancer cells invasion through the hepatocyte growth factor /c-Met/survivin regulated by P53/P21. Exp Cell Res. (2017) 357:79–87. doi: 10.1016/j.yexcr.2017.04.027
503. Okumura T, Ohuchida K, Sada M, Abe T, Endo S, Koikawa K, et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget. (2017) 8:18280–95. doi: 10.18632/oncotarget.15430
504. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J, et al. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int J Oncol. (2017) 50:121–8. doi: 10.3892/ijo.2016.3779
505. Suetsugu A, Snyder CS, Moriwaki H, Saji S, Bouvet M, Hoffman RM. Imaging the interaction of pancreatic cancer and stellate cells in the tumor microenvironment during metastasis. Anticancer Res. (2015) 35:2545–51.
506. Pang TCY, Xu Z, Pothula S, Becker T, Goldstein D, Pirola RC, et al. Circulating pancreatic stellate (stromal) cells in pancreatic cancer-a fertile area for novel research. Carcinogenesis. (2017) 38:588–91. doi: 10.1093/carcin/bgx030
507. Vaziri-Gohar A, Zarei M, Brody JR, Winter JM. Metabolic dependencies in pancreatic cancer. Front Oncol. (2018) 8:617. doi: 10.3389/fonc.2018.00617
508. Cameron ME, Yakovenko A, Trevino JG. Glucose and lactate transport in pancreatic cancer: glycolytic metabolism revisited. J Oncol. (2018) 2018:6214838. doi: 10.1155/2018/6214838
509. Chan AK, Bruce JI, Siriwardena AK. Glucose metabolic phenotype of pancreatic cancer. World J Gastroenterol. (2016) 22:3471–85. doi: 10.3748/wjg.v22.i12.3471
510. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
511. Sasaki T, Knyazev PG, Clout NJ, Cheburkin Y, Göhring W, Ullrich A, et al. Structural basis for Gas6-Axl signalling. EMBO J. (2006) 25:80–7. doi: 10.1038/sj.emboj.7600912
512. Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S, et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. (2018) 17:20. doi: 10.1186/s12943-018-0769-1
513. Kandasamy P, Gyimesi G, Kanai Y, Hediger MA. Amino acid transporters revisited: new views in health and disease. Trends Biochem Sci. (2018) 43:752–89. doi: 10.1016/j.tibs.2018.05.003
514. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. (2016) 61:667–76. doi: 10.1016/j.molcel.2016.02.011
515. Corbet C, Feron O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta Rev Cancer. (2017) 1868:7–15. doi: 10.1016/j.bbcan.2017.01.002
516. Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. (2017) 74:1999–2017. doi: 10.1007/s00018-016-2451-3
517. Michalopoulou E, Bulusu V, Kamphorst JJ. Metabolic scavenging by cancer cells: when the going gets tough, the tough keep eating. Br J Cancer. (2016) 115:635–40. doi: 10.1038/bjc.2016.256
518. Chun Y, Kim J. autophagy: an essential degradation program for cellular homeostasis and life. Cells. (2018) 7:E278. doi: 10.3390/cells7120278
519. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. (2016) 536:479–83. doi: 10.1038/nature19084
520. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. (2015) 524:361–5. doi: 10.1038/nature14587
521. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. (2016) 5:e10250. doi: 10.7554/eLife.10250
522. Glatz JF, Luiken JJ. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie. (2017) 136:21–6. doi: 10.1016/j.biochi.2016.12.007
523. Wang R, Tao B, Fan Q, Wang S, Chen L, Zhang J, et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine. (2019) 45:108–23. doi: 10.1016/j.ebiom.2019.06.037
524. Mattijssen F, Georgiadi A, Andasarie T, Szalowska E, Zota A, Krones-Herzig A, et al. Hypoxia-inducible lipid droplet-associated (HILPDA) is a novel peroxisome proliferator-activated receptor (PPAR) target involved in hepatic triglyceride secretion. J Biol Chem. (2014) 289:19279–93. doi: 10.1074/jbc.M114.570044
525. Vasseur S, Guillaumond F. LDL receptor: an open route to feed pancreatic tumor cells. Mol Cell Oncol. (2015) 3:e1033586. doi: 10.1080/23723556.2015.1033586
526. Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. (2016) 126:4140–56. doi: 10.1172/JCI87734
527. Yamazaki M, Nakamura K, Mizukami Y, Ii M, Sasajima J, Sugiyama Y, et al. Sonic hedgehog derived from human pancreatic cancer cells augments angiogenic function of endothelial progenitor cells. Cancer Sci. (2008) 99:1131–8. doi: 10.1111/j.1349-7006.2008.00795.x
528. Patel MB, Pothula SP, Xu Z, Lee AK, Goldstein D, Pirola RC, et al. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis. (2014) 35:1891–900. doi: 10.1093/carcin/bgu122
529. Zhou R, Curry JM, Roy LD, Grover P, Haider J, Moore LJ, et al. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: role in induction of VEGF signaling and angiogenesis. Oncogene. (2016) 35:5608–18. doi: 10.1038/onc.2015.516
530. Ntellas P, Dadouli K, Perivoliotis K, Sogka E, Pentheroudakis G, Ioannou M, et al. Microvessel density and impact of angiogenesis on survival of resected pancreatic cancer patients: a systematic review and meta-analysis. Pancreas. (2019) 48:233–41. doi: 10.1097/MPA.0000000000001237
531. Erkan M, Kurtoglu M, Kleeff J. The role of hypoxia in pancreatic cancer: a potential therapeutic target? Expert Rev Gastroenterol Hepatol. (2016) 10:301–16. doi: 10.1586/17474124.2016.1117386
532. Longo V, Brunetti O, Gnoni A, Cascinu S, Gasparini G, Lorusso V, et al. Angiogenesis in pancreatic ductal adenocarcinoma: a controversial issue. Oncotarget. (2016) 7:58649–58. doi: 10.18632/oncotarget.10765
533. Walia A, Yang JF, Huang YH, Rosenblatt MI, Chang JH, Azar DT. Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim Biophys Acta. (2015) 1850:2422–38. doi: 10.1016/j.bbagen.2015.09.007
534. Ruge T, Carlsson AC, Larsson A, Ärnlöv J. Endostatin: a promising biomarker in the cardiovascular continuum? Biomark Med. (2017) 11:905–16. doi: 10.2217/bmm-2017-0025
535. O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. (1994) 79:315–28. doi: 10.1016/0092-8674(94)90200-3
537. Yadav L, Puri N, Rastogi V, Satpute P, Sharma V. Tumour angiogenesis and angiogenic inhibitors: a review. J Clin Diagn Res. (2015) 9:XE01–5. doi: 10.7860/JCDR/2015/12016.6135
538. Bossard C, Van den Berghe L, Laurell H, Castano C, Cerutti M, Prats AC, et al. Antiangiogenic properties of fibstatin, an extracellular FGF-2-binding polypeptide. Cancer Res. (2004) 64:7507–12. doi: 10.1158/0008-5472.CAN-04-0287
539. Prats AC, Van den Berghe L, Rayssac A, Ainaoui N, Morfoisse F, Pujol F, et al. CXCL4L1-fibstatin cooperation inhibits tumor angiogenesis, lymphangiogenesis and metastasis. Microvasc Res. (2013) 89:25–33. doi: 10.1016/j.mvr.2013.05.005
540. Sasaki T, Hohenester E, Timpl R. Structure and function of collagen-derived endostatin inhibitors of angiogenesis. IUBMB Life. (2002) 53:77–84. doi: 10.1080/15216540211466
541. Heljasvaara R, Aikio M, Ruotsalainen H, Pihlajaniemi T. Collagen XVIII in tissue homeostasis and dysregulation - Lessons learned from model organisms and human patients. Matrix Biol. (2017) 57–58:55–75. doi: 10.1016/j.matbio.2016.10.002
542. Tsuji T, Kelly NJ, Takahashi S, Leme AS, Houghton AM, Shapiro SD. Macrophage elastase suppresses white adipose tissue expansion with cigarette smoking. Am J Respir Cell Mol Biol. (2014) 51:822–9. doi: 10.1165/rcmb.2014-0083OC
543. Yan C, Wang C, Dong M, Liu S, Qi C, Zhao Y. RNA interference-mediated silencing of VEGF and bFGF suppresses endostatin secretion in pancreatic carcinoma cells. Oncol Lett. (2013) 5:1031–5. doi: 10.3892/ol.2013.1102
544. Ramirez MU, Stirling ER, Emenaker NJ, Roberts DD, Soto-Pantoja DR. Thrombospondin-1 interactions regulate eicosanoid metabolism and signaling in cancer-related inflammation. Cancer Metastasis Rev. (2018) 37:469–76. doi: 10.1007/s10555-018-9737-x
545. Rossi MK, Gnanamony M, Gondi CS. The 'SPARC' of life: analysis of the role of osteonectin/SPARC in pancreatic cancer (Review). Int J Oncol. (2016) 48:1765–71. doi: 10.3892/ijo.2016.3417
546. Faye C, Chautard E, Olsen BR, Ricard-Blum S. The first draft of the endostatin interaction network. J Biol Chem. (2009) 284:22041–7. doi: 10.1074/jbc.M109.002964
547. Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, et al. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. (2002) 277:27872–9. doi: 10.1074/jbc.M202771200
548. Han KY, Chang JH, Dugas-Ford J, Alexander JS, Azar DT. Involvement of lysosomal degradation in VEGF-C-induced down-regulation of VEGFR-3. FEBS Lett. (2014) 588:4357–63. doi: 10.1016/j.febslet.2014.09.034
549. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA. (2003) 100:4766–71. doi: 10.1073/pnas.0730882100
550. Morais C, Ebrahem Q, Anand-Apte B, Parat MO. Altered angiogenesis in caveolin-1 gene-deficient mice is restored by ablation of endothelial nitric oxide synthase. Am J Pathol. (2012) 180:1702–14. doi: 10.1016/j.ajpath.2011.12.018
551. Hu J, Cheng T, Zhang L, Sun B, Deng L, Xu H. Anti-tumor peptide AP25 decreases cyclin D1 expression and inhibits MGC-803 proliferation via phospho-extracellular signal-regulated kinase-, Src-, c-Jun N-terminal kinase- and phosphoinositide 3-kinase-associated pathways. Mol Med Rep. (2015) 12:4396–402. doi: 10.3892/mmr.2015.3912
552. Nesiel-Nuttman L, Schwartz B, Shoseyov O. Human recombinant truncated RNASET2, devoid of RNase activity) A potential cancer therapeutic agent. Oncotarget. (2014) 5:11464–78. doi: 10.18632/oncotarget.2562
553. Vallianou NG, Kostantinou A, Kougias M, Kazazis C. Statins and cancer. Anticancer Agents Med Chem. (2014) 14:706–12. doi: 10.2174/1871520613666131129105035
554. Longo V, Tamma R, Brunetti O, Pisconti S, Argentiero A, Silvestris N, et al. Mast cells and angiogenesis in pancreatic ductal adenocarcinoma. Clin Exp Med. (2018) 18:319–23. doi: 10.1007/s10238-018-0493-6
555. Yang M, Liu E, Tang L, Lei Y, Sun X, Hu J, et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal. (2018) 16:31. doi: 10.1186/s12964-018-0242-1
556. Puertollano R, Ferguson SM, Brugarolas J, Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. (2018) 37:e98804. doi: 10.15252/embj.201798804
557. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. (2019) 120:1090–8. doi: 10.1038/s41416-019-0451-4
558. Yin F, Sharen G, Yuan F, Peng Y, Chen R, Zhou X, et al. TIP30 regulates lipid metabolism in hepatocellular carcinoma by regulating SREBP1 through the Akt/mTOR signaling pathway. Oncogenesis. (2017) 6:e347. doi: 10.1038/oncsis.2017.49
559. Siqingaowa Sekar S, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun. (2017) 488:136–40. doi: 10.1016/j.bbrc.2017.05.023
560. Salvioli B, Bovara M, Barbara G, DePont F, Stanghellini V, Tomini M, et al. Neurology and neuropathology of the pancreatic innervation. JOP. (2002) 3:26–33.
561. Niebergall-Roth E, Singer MV. Central and peripheral neural control of pancreatic exocrine secretion. J Physiol Pharmacol. (2001) 52:523–38.
562. Grundy D. Neuroanatomy of visceral nociception: vagal and splanchnic afferent. Gut. (2002) 51 (Suppl. 1):i2–5. doi: 10.1136/gut.51.suppl_1.i2
563. Stopczynski RE, Normolle DP, Hartman DJ, Ying H, DeBerry JJ, Bielefeldt K, et al. Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res. (2014) 74:1718–27. doi: 10.1158/0008-5472.CAN-13-2050
564. Demir IE, Friess H, Ceyhan GO. Neural plasticity in pancreatitis and pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2015) 12:649–59. doi: 10.1038/nrgastro.2015.166
565. Lindsay TH, Halvorson KG, Peters CM, Ghilardi JR, Kuskowski MA, Wong GY, et al. A quantitative analysis of the sensory and sympathetic innervation of the mouse pancreas. Neuroscience. (2006) 137:1417–26. doi: 10.1016/j.neuroscience.2005.10.055
566. Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, et al. Pancreatic neuropathy and neuropathic pain—A comprehensive pathomorphological study of 546 cases. Gastroenterology. (2009) 136:177–86. doi: 10.1053/j.gastro.2008.09.029
567. Iwasaki T, Hiraoka N, Ino Y, Nakajima K, Kishi Y, Nara S, et al. Reduction of intrapancreatic neural density in cancer tissue predicts poorer outcome in pancreatic ductal carcinoma. Cancer Sci. (2019) 110:1491–502. doi: 10.1111/cas.13975
568. Chatterjee D, Katz MH, Rashid A, Wang H, Iuga AC, Varadhachary GR, et al. Perineural and intraneural invasion in posttherapy pancreaticoduodenectomy specimens predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Am J Surg Pathol. (2012) 36:409–17. doi: 10.1097/PAS.0b013e31824104c5
569. Lee SR, Kim HO, Son BH, Yoo CH, Shin JH. Prognostic factors associated with long-term survival and recurrence in pancreatic adenocarcinoma. Hepatogastroenterology. (2013) 60:358–62.
570. Chang ST, Jeffrey RB, Patel BN, DiMaio MA, Rosenberg J, Willmann JK, et al. preoperative multidetector ct diagnosis of extrapancreatic perineural or duodenal invasion is associated with reduced postoperative survival after pancreaticoduodenectomy for pancreatic adenocarcinoma: preliminary experience and implications for patient care. Radiology. (2016) 281:816–25. doi: 10.1148/radiol.2016152790
571. Schorn S, Demir IE, Haller B, Scheufele F, Reyes CM, Tieftrunk E, et al. The influence of neural invasion on survival and tumor recurrence in pancreatic ductal adenocarcinoma - A systematic review and meta-analysis. Surg Oncol. (2017) 26:105–15. doi: 10.1016/j.suronc.2017.01.007
572. Pour PM, Bell RH, Batra SK. Neural invasion in the staging of pancreatic cancer. Pancreas. (2003) 26:322–5. doi: 10.1097/00006676-200305000-00002
573. Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer. (2009) 115:3379–91. doi: 10.1002/cncr.24396
574. Chang A, Kim-Fuchs C, Le CP, Hollande F, Sloan EK. Neural regulation of pancreatic cancer: a novel target for intervention. Cancers. (2015) 7:1292–312. doi: 10.3390/cancers7030838
575. Amit M, Na'Ara S, Gil Z. Mechanisms of cancer dissemination along nerves. Nat Rev Cancer. (2016) 16:399–408. doi: 10.1038/nrc.2016.38
576. Liu H, Li X, Xu Q, Lv S, Li J, Ma Q. Role of glial cell line-derived neurotrophic factor in perineural invasion of pancreatic cancer. Biochim Biophys Acta. (2012) 1826:112–20. doi: 10.1016/j.bbcan.2012.03.010
577. Farina AR, Cappabianca L, Ruggeri P, Gneo L, Pellegrini C, Fargnoli MC, et al. The oncogenic neurotrophin receptor tropomyosin-related kinase variant, TrkAIII. J Exp Clin Cancer Res. (2018) 37:119. doi: 10.1186/s13046-018-0786-3
578. Griffin N, Faulkner S, Jobling P, Hondermarck H. Targeting neurotrophin signaling in cancer: the renaissance. Pharmacol Res. (2018) 135:12–17. doi: 10.1016/j.phrs.2018.07.019
579. Wislet S, Vandervelden G, Rogister B. From neural crest development to cancer and vice versa: how p75NTR and (Pro)neurotrophins could act on cell migration and invasion? Front Mol Neurosci. (2018) 11:244. doi: 10.3389/fnmol.2018.00244
580. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. (2013) 341:1236361. doi: 10.1126/science.1236361
581. Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, et al. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J Natl Cancer Inst. (2010) 102:107–18. doi: 10.1093/jnci/djp456
582. Fielder GC, Yang TW, Razdan M, Li Y, Lu J, Perry JK, et al. The GDNF family: a role in cancer? Neoplasia. (2018) 20:99–117. doi: 10.1016/j.neo.2017.10.010
583. Ceyhan GO, Schäfer KH, Kerscher AG, Rauch U, Demir IE, Kadihasanoglu M, et al. Nerve growth factor and artemin are paracrine mediators of pancreatic neuropathy in pancreatic adenocarcinoma. Ann Surg. (2010) 251:923–31. doi: 10.1097/SLA.0b013e3181d974d4
584. Okada Y, Eibl G, Duffy JP, Reber HA, Hines OJ. Glial cell-derived neurotrophic factor upregulates the expression and activation of matrix metalloproteinase-9 in human pancreatic cancer. Surgery. (2003) 134:293–9. doi: 10.1067/msy.2003.239
585. Veit C, Genze F, Menke A, Hoeffert S, Gress TM, Gierschik P, et al. Activation of phosphatidylinositol 3-kinase and extracellular signal-regulated kinase is required for glial cell line-derived neurotrophic factor-induced migration and invasion of pancreatic carcinoma cells. Cancer Res. (2004) 64:5291–300. doi: 10.1158/0008-5472.CAN-04-1112
586. Takahashi H, Funahashi H, Sawai H, Sakamoto M, Matsuo Y, Yamamoto M, et al. Glial cell line-derived neurotrophic factor enhances nuclear factor-kappaB activity and invasive potential in human pancreatic cancer cells. Pancreas. (2004) 29:22–7. doi: 10.1097/00006676-200407000-00051
587. Saloman JL, Singhi AD, Hartman DJ, Normolle DP, Albers KM, Davis BM. systemic depletion of nerve growth factor inhibits disease progression in a genetically engineered model of pancreatic ductal adenocarcinoma. Pancreas. (2018) 47:856–63. doi: 10.1097/MPA.0000000000001090
588. Xu Q, Wang Z, Chen X, Duan W, Lei J, Zong L, et al. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget. (2015) 6:4717–32. doi: 10.18632/oncotarget.3069
589. Wang J, Wang H, Cai J, Du S, Xin B, Wei W, et al. Artemin regulates CXCR4 expression to induce migration and invasion in pancreatic cancer cells through activation of NF-κB signaling. Exp Cell Res. (2018) 365:12–23. doi: 10.1016/j.yexcr.2018.02.008
590. Saloman JL, Albers KM, Li D, Hartman DJ, Crawford HC, Muha EA, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci USA. (2016) 113:3078–83. doi: 10.1073/pnas.1512603113
591. Xu X, Zhao Z, Guo S, Li J, Liu S, You Y, et al. Increased semaphorin 3c expression promotes tumor growth and metastasis in pancreatic ductal adenocarcinoma by activating the ERK1/2 signaling pathway. Cancer Lett. (2017) 397:12–22. doi: 10.1016/j.canlet.2017.03.014
592. Foley K, Rucki AA, Xiao Q, Zhou D, Leubner A, Mo G, et al. Semaphorin 3D autocrine signaling mediates the metastatic role of annexin A2 in pancreatic cancer. Sci Signal. (2015) 8:ra77. doi: 10.1126/scisignal.aaa5823
593. Jurcak NR, Rucki AA, Muth S, Thompson E, Sharma R, Ding D, et al. Axon guidance molecules promote perineural invasion and metastasis of orthotopic pancreatic tumors in mice. Gastroenterology. (2019) 157:838–50.e6. doi: 10.1053/j.gastro.2019.05.065
594. Sabbatini ME, Villagra A, Davio CA, Vatta MS, Fernandez BE, Bianciotti LG. Atrial natriuretic factor stimulates exocrine pancreatic secretion in the rat through NPR-C receptors. Am J Physiol Gastrointest Liver Physiol. (2003) 285:G929–37. doi: 10.1152/ajpgi.00010.2003
595. Rossi J, Santamäki P, Airaksinen MS, Herzig KH. Parasympathetic innervation and function of endocrine pancreas requires the glial cell line-derived factor family receptor alpha2 (GFRalpha2). Diabetes. (2005) 54:1324–30. doi: 10.2337/diabetes.54.5.1324
596. Haas SL, Fitzner B, Jaster R, Wiercinska E, Gaitantzi H, Jesnowski R, et al. Transforming growth factor-beta induces nerve growth factor expression in pancreatic stellate cells by activation of the ALK-5 pathway. Growth Factors. (2009) 27:289–99. doi: 10.1080/08977190903132273
597. Gasparini G, Pellegatta M, Crippa S, Lena MS, Belfiori G, Doglioni C, et al. Nerves and pancreatic cancer: new insights into a dangerous relationship. Cancers. (2019) 11:E893. doi: 10.3390/cancers11070893
598. Al-Wadei HA, Ullah MF, Al-Wadei M. GABA (γ-aminobutyric acid), a non-protein amino acid counters the β-adrenergic cascade-activated oncogenic signaling in pancreatic cancer: a review of experimental evidence. Mol Nutr Food Res. (2011) 55:1745–58. doi: 10.1002/mnfr.201100229
599. Lin X, Luo K, Lv Z, Huang J. Beta-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology. (2012) 59:584–8.
600. Shan T, Ma J, Ma Q, Guo K, Guo J, Li X, et al. β2-AR-HIF-1α: a novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr Mol Med. (2013) 13:1023–34. doi: 10.2174/15665240113139990055
601. Lucki A, Klein E, Karry R, Ben-Shachar D. Dexamethasone in the presence of desipramine enhances MAPK/ERK1/2 signaling possibly via its interference with β-arrestin. J Neural Transm. (2014) 121:289–98. doi: 10.1007/s00702-013-1099-5
602. Nan L, Qin T, Xiao Y, Qian W, Li J, Wang Z, et al. Pancreatic stellate cells facilitate perineural invasion of pancreatic cancer via HGF/c-Met pathway. Cell Transplant. (2019) 28:1289–98. doi: 10.1177/0963689719851772
603. Demir IE, Ceyhan GO, Rauch U, Altintas B, Klotz M, Müller MW, et al. The microenvironment in chronic pancreatitis and pancreatic cancer induces neuronal plasticity. Neurogastroenterol Motil. (2010) 22:480–90. doi: 10.1111/j.1365-2982.2009.01428.x
604. Zeng L, Guo Y, Liang J, Chen S, Peng P, Zhang Q, et al. Perineural invasion and TAMs in pancreatic ductal adenocarcinomas: review of the original pathology reports using immunohistochemical enhancement and relationships with clinicopathological features. J Cancer. (2014) 5:754–60. doi: 10.7150/jca.10238
605. Cavel O, Shomron O, Shabtay A, Vital J, Trejo-Leider L, Weizman N, et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. (2012) 72:5733–43. doi: 10.1158/0008-5472.CAN-12-0764
606. Swanson BJ, McDermott KM, Singh PK, Eggers JP, Crocker PR, Hollingsworth MA. MUC1 is a counter-receptor for myelin-associated glycoprotein (Siglec-4a) and their interaction contributes to adhesion in pancreatic cancer perineural invasion. Cancer Res. (2007) 67:10222–9. doi: 10.1158/0008-5472.CAN-06-2483
607. Stolt CC, Wegner M. Schwann cells and their transcriptional network: evolution of key regulators of peripheral myelination. Brain Res. (2016) 1641:101–10. doi: 10.1016/j.brainres.2015.09.025
608. Demir IE, Boldis A, Pfitzinger PL, Teller S, Brunner E, Klose N, et al. Investigation of Schwann cells at neoplastic cell sites before the onset of cancer invasion. J Natl Cancer Inst. (2014) 106:dju184. doi: 10.1093/jnci/dju184
609. Madeo M, Colbert PL, Vermeer DW, Lucido CT, Cain JT, Vichaya EG, et al. Cancer exosomes induce tumor innervation. Nat Commun. (2018) 9:4284. doi: 10.1038/s41467-018-06640-0
610. Vermeer PD. Exosomal induction of tumor innervation. Cancer Res. (2019) 79:3529–35. doi: 10.1158/0008-5472.CAN-18-3995
611. Lucido CT, Wynja E, Madeo M, Williamson CS, Schwartz LE, Imblum BA, et al. Innervation of cervical carcinoma is mediated by cancer-derived exosomes. Gynecol Oncol. (2019) 154:228–35. doi: 10.1016/j.ygyno.2019.04.651
612. Zhang Z, Yan C, Li B, Li L. Potential biological functions of microvesicles derived from adenoid cystic carcinoma. Oncol Lett. (2018) 15:7900–8.
613. Ohta T, Elnemr A, Kitagawa H, Kayahara M, Takamura H, Fujimura T, et al. Fas ligand expression in human pancreatic cancer. Oncol Rep. (2004) 12:749–54. doi: 10.3892/or.12.4.749
614. Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. (2016) 17:160–72. doi: 10.1038/nrn.2015.29
615. Fruhbeis C, Fröhlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte–neuron communication. PLoS Biol. (2013) 11:e1001604. doi: 10.1371/journal.pbio.1001604
616. Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles. (2014) 3:24722. doi: 10.3402/jev.v3.24722
617. Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1β release from microglia. J Immunol. (2005) 174:7268–77. doi: 10.4049/jimmunol.174.11.7268
618. Antonucci F, Turola E, Riganti L, Caleo M, Gabrielli M, Perrotta C, et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. (2012) 31:1231–40. doi: 10.1038/emboj.2011.489
619. Lopez-Verrilli MA, Court FA. Transfer of vesicles from schwann cells to axons: a novel mechanism of communication in the peripheral nervous system. Front Physiol. (2012) 3:205. doi: 10.3389/fphys.2012.00205
620. André-Grégoire G, Gavard J. Spitting out the demons: extracellular vesicles in glioblastoma. Cell Adh Migr. (2017) 11:164–72. doi: 10.1080/19336918.2016.1247145
621. Seufferlein T, Hammel P, Delpero JR, Macarulla T, Pfeiffer P, Prager GW, et al. Optimizing the management of locally advanced pancreatic cancer with a focus on induction chemotherapy: expert opinion based on a review of current evidence. Cancer Treat Rev. (2019) 77:1–10. doi: 10.1016/j.ctrv.2019.05.007
622. Springfeld C, Jäger D, Büchler MW, Strobel O, Hackert T, Palmer DH, et al. Chemotherapy for pancreatic cancer. Presse Med. (2019) 48(3 Pt 2):e159–74. doi: 10.1016/j.lpm.2019.02.025
623. Boldrini L, Cusumano D, Cellini F, Azario L, Mattiucci GC, Valentini V. Online adaptive magnetic resonance guided radiotherapy for pancreatic cancer: state of the art, pearls and pitfalls. Radiat Oncol. (2019) 14:71. doi: 10.1186/s13014-019-1275-3
624. Alcántara-Hernández R, Hernández-Méndez A. Adrenergic signaling molecular complexes. Gac Med Mex. (2018) 154:223–35. doi: 10.24875/GMM.M18000135
625. Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine. (2016) 95:e5541. doi: 10.1097/MD.0000000000005541
626. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. (2013) 108:914–23. doi: 10.1038/bjc.2013.32
627. Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA. (2005) 102:419–24. doi: 10.1073/pnas.0408197102
628. Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. (2005) 65:11743–51. doi: 10.1158/0008-5472.CAN-05-0045
629. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. (2015) 38:1–11. doi: 10.1097/CJI.0000000000000062
630. Van Audenaerde JRM, Roeyen G, Darcy PK, Kershaw MH, Peeters M, Smits ELJ. Natural killer cells and their therapeutic role in pancreatic cancer: a systematic review. Pharmacol Ther. (2018) 189:31–44. doi: 10.1016/j.pharmthera.2018.04.003
631. Davis M, Conlon K, Bohac GC, Barcenas J, Leslie W, Watkins L, et al. Effect of pemetrexed on innate immune killer cells and adaptive immune T cells in subjects with adenocarcinoma of the pancreas. J Immunother. (2012) 35:629–40. doi: 10.1097/CJI.0b013e31826c8a4f
632. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. (2013) 145:1121–32. doi: 10.1053/j.gastro.2013.07.025
633. Peng YP, Zhu Y, Zhang JJ, Xu ZK, Qian ZY, Dai CC, et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J Transl Med. (2013) 11:262. doi: 10.1186/1479-5876-11-262
634. Wensveen FM, Jelenčić V, Polić B. NKG2D: a master regulator of immune cell responsiveness. Front Immunol. (2018) 9:441. doi: 10.3389/fimmu.2018.00441
635. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
636. Peng YP, Zhang JJ, Liang WB, Tu M, Lu ZP, Wei JS, et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer. (2014) 14:738. doi: 10.1186/1471-2407-14-738
637. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
638. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. (2016) 22:5183–8. doi: 10.1158/1078-0432.CCR-16-0933
639. Peng YP, Xi CH, Zhu Y, Yin LD, Wei JS, Zhang JJ, et al. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget. (2016) 7:66586–94. doi: 10.18632/oncotarget.11953
640. Van Audenaerde JRM, De Waele J, Marcq E, Van Loenhout J, Lion E, Van den Bergh JMJ, et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget. (2017) 8:56968–79. doi: 10.18632/oncotarget.18185
641. Lu Y, Hu J, Sun W, Duan X, Chen X. Hypoxia-mediated immune evasion of pancreatic carcinoma cells. Mol Med Rep. (2015) 11:3666–72. doi: 10.3892/mmr.2015.3144
642. Huang Q, Huang M, Meng F, Sun R. Activated pancreatic stellate cells inhibit NK cell function in the human pancreatic cancer microenvironment. Cell Mol Immunol. (2019) 16:87–9. doi: 10.1038/s41423-018-0014-2
643. Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. J Immunol. (2015) 195:4010–9. doi: 10.4049/jimmunol.1500447
644. Herberman RB, Holden HT, Ting CC, Lavrin DL, Kirchner H. Cell-mediated immunity to leukemia virus- and tumor-associated antigens in mice. Cancer Res. (1976) 36:615–21.
645. Aya T, Matsuo T, Takada K, Osato T, Mizuno F. Antibody-dependent autologous lymphocyte cytotoxicity against cells freshly transformed by Epstein-Barr virus. IARC Sci Publ. (1978) 24:711–9.
646. Karnevi E, Andersson R, Rosendahl AH. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol Cell Biol. (2014) 92:543–52. doi: 10.1038/icb.2014.22
647. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
648. Knudsen ES, Vail P, Balaji U, Ngo H, Botros IW, Makarov V, et al. Stratification of pancreatic ductal adenocarcinoma: combinatorial genetic, stromal, and immunologic markers. Clin Cancer Res. (2017) 23:4429–40. doi: 10.1158/1078-0432.CCR-17-0162
649. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R+macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep. (2018) 23:1448–60. doi: 10.1016/j.celrep.2018.03.131
650. Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE. (2013) 8:e80908. doi: 10.1371/journal.pone.0080908
651. Attri KS, Mehla K, Singh PK. Evaluation of macrophage polarization in pancreatic cancer microenvironment under hypoxia. Methods Mol Biol. (2018) 1742:265–76. doi: 10.1007/978-1-4939-7665-2_23
652. Sun W, Wei FQ, Li WJ, Wei JW, Zhong H, Wen YH, et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br J Cancer. (2017) 117:1631–43. doi: 10.1038/bjc.2017.329
653. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
654. Jones KI, Tiersma J, Yuzhalin AE, Gordon-Weeks AN, Buzzelli J, Im JH, et al. Radiation combined with macrophage depletion promotes adaptive immunity and potentiates checkpoint blockade. EMBO Mol Med. (2018) 10:e9342. doi: 10.15252/emmm.201809342
655. Kimsey TF, Campbell AS, Albo D, Wilson M, Wang TN. Co-localization of macrophage inflammatory protein-3alpha (Mip-3alpha) and its receptor, CCR6, promotes pancreatic cancer cell invasion. Cancer J. (2004) 10:374–80. doi: 10.1097/00130404-200411000-00007
656. Lee S, Lee E, Ko E, Ham M, Lee HM, Kim ES, et al. Tumor-associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1-α and MMP-9. Cancer Lett. (2018) 437:25–34. doi: 10.1016/j.canlet.2018.08.025
657. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. (2013) 93:844–54. doi: 10.1038/labinvest.2013.69
658. Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I, et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer. (2014) 135:843–61. doi: 10.1002/ijc.28736
659. Arnold JN, Magiera L, Kraman M, Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol Res. (2014) 2:121–6. doi: 10.1158/2326-6066.CIR-13-0150
660. Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. (2014) 33:3812–9. doi: 10.1038/onc.2013.357
661. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage polarity in cancer: a review. J Cell Biochem. (2019) 120:2756–65. doi: 10.1002/jcb.27646
662. Yu M, Guan R, Hong W, Zhou Y, Lin Y, Jin H, et al. Prognostic value of tumor-associated macrophages in pancreatic cancer: a meta-analysis. Cancer Manag Res. (2019) 11:4041–58. doi: 10.2147/CMAR.S196951
663. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
664. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. (2015) 5:e1004983. doi: 10.1080/2162402X.2015.1004983
665. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
666. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
667. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
668. Boros P, Ochando J, Zeher M. Myeloid derived suppressor cells and autoimmunity. Hum Immunol. (2016) 77:631–6. doi: 10.1016/j.humimm.2016.05.024
669. Chen W, Jiang J, Xia W, Huang J. Tumor-related exosomes contribute to tumor-promoting microenvironment: an immunological perspective. J Immunol Res. (2017) 2017:1073947. doi: 10.1155/2017/1073947
670. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
671. Trikha P, Carson WE 3rd. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta. (2014) 1846:55–65. doi: 10.1016/j.bbcan.2014.04.003
672. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology. (2015) 4:e954829. doi: 10.4161/21624011.2014.954829
673. Shi H, Dong G, Yan F, Zhang H, Li C, Ma Q, et al. Arctigenin ameliorates inflammation by regulating accumulation and functional activity of MDSCs in endotoxin shock. Inflammation. (2018) 41:2090–100. doi: 10.1007/s10753-018-0852-1
674. Chen J, Ye Y, Liu P, Yu W, Wei F, Li H, et al. Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol. (2017) 78:113–9. doi: 10.1016/j.humimm.2016.12.001
675. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
676. Hatziioannou A, Alissafi T, Verginis P. Myeloid-derived suppressor cells and T regulatory cells in tumors: unraveling the dark side of the force. J Leukoc Biol. (2017) 102:407–21. doi: 10.1189/jlb.5VMR1116-493R
677. Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology. (2016) 148:377–86. doi: 10.1111/imm.12616
678. Roussel M, Ferrell PB Jr, Greenplate AR, Lhomme F, Le Gallou S, Diggins KE, et al. Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J Leukoc Biol. (2017) 102:437–47. doi: 10.1189/jlb.5MA1116-457R
679. Chauhan S, Danielson S, Clements V, Edwards N, Ostrand-Rosenberg S, Fenselau C. Surface glycoproteins of exosomes shed by myeloid-derived suppressor cells contribute to function. J Proteome Res. (2017) 16:238–46. doi: 10.1021/acs.jproteome.6b00811
680. Geis-Asteggiante L, Belew AT, Clements VK, Edwards NJ, Ostrand-Rosenberg S, El-Sayed NM, et al. Differential content of proteins, mRNAs, and miRNAs suggests that MDSC and their exosomes may mediate distinct immune suppressive functions. J Proteome Res. (2018) 17:486–98. doi: 10.1021/acs.jproteome.7b00646
681. Zöller M. Janus-faced myeloid-derived suppressor cell exosomes for the good and the bad in cancer and autoimmune disease. Front Immunol. (2018) 9:137. doi: 10.3389/fimmu.2018.00137
682. Roussel M, Irish JM, Menard C, Lhomme F, Tarte K, Fest T. Regulatory myeloid cells: an underexplored continent in B-cell lymphomas. Cancer Immunol Immunother. (2017) 66:1103–11. doi: 10.1007/s00262-017-2036-5
683. Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. (2017) 24:100–5. doi: 10.1038/cgt.2016.65
684. Wang Z, Liu Y, Zhang Y, Shang Y, Gao Q. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget. (2016) 7:4760–9. doi: 10.18632/oncotarget.6734
685. Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. (2010) 22:109–17. doi: 10.1016/j.coi.2010.01.022
686. Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. (2013) 1:145–9. doi: 10.1158/2326-6066.CIR-13-0102
687. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. (2011) 11:823–36. doi: 10.1038/nri3084
688. Verboogen DR, Dingjan I, Revelo NH, Visser LJ, ter Beest M, van den Bogaart G. The dendritic cell side of the immunological synapse. Biomol Concepts. (2016) 7:17–28. doi: 10.1515/bmc-2015-0028
689. Ugur M, Mueller SN. T cell and dendritic cell interactions in lymphoid organs: more than just being in the right place at the right time. Immunol Rev. (2019) 289:115–28. doi: 10.1111/imr.12753
690. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
691. Bandola-Simon J, Roche PA. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol Immunol. (2019) 113:31–7. doi: 10.1016/j.molimm.2018.03.025
692. Dallal RM, Christakos P, Lee K, Egawa S, Son YI, Lotze MT. Paucity of dendritic cells in pancreatic cancer. Surgery. (2002) 131:135–8. doi: 10.1067/msy.2002.119937
693. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. (2004) 10:48–54. doi: 10.1038/nm976
694. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. (2004) 4:941–52. doi: 10.1038/nri1498
695. Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res. (2007) 67:5479–88. doi: 10.1158/0008-5472.CAN-06-3963
696. Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol. (2006) 177:3448–60. doi: 10.4049/jimmunol.177.5.3448
697. Wiedemann GM, Knott MM, Vetter VK, Rapp M, Haubner S, Fesseler J, et al. Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology. (2016) 5:e1175794. doi: 10.1080/2162402X.2016.1175794
698. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+ CD25+ regulatory T cell proliferation. J Exp Med. (2005) 202:919–29. doi: 10.1084/jem.20050463
699. Murgaski A, Bardet PMR, Arnouk SM, Clappaert EJ, Laoui D. Unleashing tumour-dendritic cells to fight cancer by tackling their three A's: abundance, activation and antigen-delivery. Cancers. (2019) 11:E670. doi: 10.3390/cancers11050670
700. Deicher A, Andersson R, Tingstedt B, Lindell G, Bauden M, Ansari D. Targeting dendritic cells in pancreatic ductal adenocarcinoma. Cancer Cell Int. (2018) 18:85. doi: 10.1186/s12935-018-0585-0
701. Xiao L, Erb U, Zhao K, Hackert T, Zöller M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology. (2017) 6:e1319044. doi: 10.1080/2162402X.2017.1319044
702. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. (1998) 4:594–600. doi: 10.1038/nm0598-594
703. Shenoda BB, Ajit SK. Modulation of immune responses by exosomes derived from antigen-presenting cells. Clin Med Insights Pathol. (2016) 9(Suppl. 1):1–8. doi: 10.4137/CPath.S39925
704. Tkach M, Kowal J, Zucchetti AE, Enserink L, Jouve M, Lankar D, et al. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. (2017) 36:3012–28. doi: 10.15252/embj.201696003
705. Lindenbergh MFS, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol. (2018) 36:435–59. doi: 10.1146/annurev-immunol-041015-055700
706. Helm O, Mennrich R, Petrick D, Goebel L, Freitag-Wolf S, Röder C, et al. Comparative characterization of stroma cells and ductal epithelium in chronic pancreatitis and pancreatic ductal adenocarcinoma. PLoS ONE. (2014) 9:e94357. doi: 10.1371/journal.pone.0094357
707. Fogar P, Basso D, Fadi E, Greco E, Pantano G, Padoan A, et al. Pancreatic cancer alters human CD4+ T lymphocyte function: a piece in the immune evasion puzzle. Pancreas. (2011) 40:1131–7. doi: 10.1097/MPA.0b013e31822077b8
708. Suzuki D, Furukawa K, Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, et al. Effects of perioperative immunonutrition on cell-mediated immunity, T helper type 1 (Th1)/Th2 differentiation, and Th17 response after pancreaticoduodenectomy. Surgery. (2010) 148:573–81. doi: 10.1016/j.surg.2010.01.017
709. Pu N, Zhao G, Gao S, Cui Y, Xu Y, Lv Y, et al. Neutralizing TGF-β promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Cent Eur J Immunol. (2018) 43:123–31. doi: 10.5114/ceji.2018.77381
710. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. (2011) 208:469–78. doi: 10.1084/jem.20101876
711. Piro G, Simionato F, Carbone C, Frizziero M, Malleo G, Zanini S, et al. A circulating TH2 cytokines profile predicts survival in patients with resectable pancreaticadenocarcinoma. Oncoimmunology. (2017) 6:e1322242. doi: 10.1080/2162402X.2017.1322242
712. Trapani JA. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell. (2005) 8:349–50. doi: 10.1016/j.ccr.2005.10.018
713. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
714. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
715. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. (2018) 18:139–47. doi: 10.1038/nrc.2017.117
716. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. (2012) 130:2337–48. doi: 10.1002/ijc.26290
717. Seicean A, Popa D, Mocan T, Cristea V, Neagoe I. Th1 and Th2 profiles in patients with pancreatic cancer compared with chronic pancreatitis. Pancreas. (2009) 38:594–5. doi: 10.1097/MPA.0b013e31819313d0
718. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identifica-tion of unique neoantigen qualities in long-term survivorsof pancreatic cancer. Nature. (2017) 551:512–6. doi: 10.1038/nature24462
719. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. (2017) 8:15095. doi: 10.1038/ncomms15095
720. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
721. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
722. Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol. (2017) 49:14–19. doi: 10.1016/j.coi.2017.07.014
723. Mitsuiki N, Schwab C, Grimbacher B. What did we learn from CTLA-4 insufficiency on the human immune system? Immunol Rev. (2019) 287:33–49. doi: 10.1111/imr.12721
724. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
725. Azuma T, Takahashi T, Kunisato A, Azuma T, Takahashi T. Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res. (2003) 63:4516–20.
726. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
727. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369
728. Kobayashi N, Kubota K, Kato S, Watanabe S, Shimamura T, Kirikoshi H, et al. FOXP3+ regulatory T cells and tumoral indoleamine 2,3-dioxygenase expression predicts the carcinogenesis of intraductal papillary mucinous neoplasms of the pancreas. Pancreatology. (2010) 10:631–40. doi: 10.1159/000308966
729. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE. (2014) 9:e91551. doi: 10.1371/journal.pone.0091551
730. Wartenberg M, Zlobec I, Perren A, Koelzer VH, Lugli A, Karamitopoulou E. Accumulation of FOXP3+T-cells in the tumor microenvironment is associated with an epithelial-mesenchymal-transition-type tumor budding phenotype and is an independent prognostic factor in surgically resected pancreatic ductal adenocarcinoma. Oncotarget. (2015) 6:4190–201. doi: 10.18632/oncotarget.2775
731. Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother. (2013) 62:1249–60. doi: 10.1007/s00262-013-1429-3
732. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. (2009) 182:1746–55. doi: 10.4049/jimmunol.182.3.1746
733. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
734. Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst. (2007) 99:1188–99. doi: 10.1093/jnci/djm064
735. Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Hirooka S, Yamaki S, et al. Circulating CD4+CD25+ regulatory T cells in patients with pancreatic cancer. Pancreas. (2012) 41:409–15. doi: 10.1097/MPA.0b013e3182373a66
736. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
737. Roshani R, McCarthy F, Hagemann T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. (2014) 345:157–63. doi: 10.1016/j.canlet.2013.07.014
738. Wörmann SM, Diakopoulos KN, Lesina M, Algül H. The immune network in pancreatic cancer development and progression. Oncogene. (2014) 33:2956–67. doi: 10.1038/onc.2013.257
739. Padoan A, Plebani M, Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int J Mol Sci. (2019) 20:E676. doi: 10.3390/ijms20030676
740. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
741. Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. (2016) 532:245–9. doi: 10.1038/nature17403
742. Beatty GL, O'Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patientswith advanced solid malignancies. Clin Cancer Res. (2017) 23:3269–76. doi: 10.1158/1078-0432.CCR-16-2272
743. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the Tryptophan-Kynurenine-Aryl hydrocarbon axis. Clin Cancer Res. (2019) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
744. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
745. Martinez-Bosch N, Vinaixa J, Navarro P. Immune evasion in pancreatic cancer: from mechanisms to therapy. Cancers. (2018) 10:E6. doi: 10.3390/cancers10010006
746. Cerliani JP, Blidner AG, Toscano MA, Croci DO, Rabinovich GA. Translating the “Sugar Code” into immune and vascular signaling programs. Trends Biochem Sci. (2017) 42:255–73. doi: 10.1016/j.tibs.2016.11.003
747. Martínez-Bosch N, Fernández-Barrena MG, Moreno M, Ortiz-Zapater E, Munné-Collado J, Iglesias M, et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and hedgehog signaling activation. Cancer Res. (2014) 74:3512–24. doi: 10.1158/0008-5472.CAN-13-3013
748. Méndez-Huergo SP, Blidner AG, Rabinovich GA. Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr Opin Immunol. (2017) 45:8–15. doi: 10.1016/j.coi.2016.12.003
749. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. (2017) 23:556–67. doi: 10.1038/nm.4314
750. Zeligs KP, Neuman MK, Annunziata CM. Molecular pathways: the balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-κB signaling for cancer treatment. Clin Cancer Res. (2016) 22:4302–8. doi: 10.1158/1078-0432.CCR-15-1374
751. Flint TR, Fearon DT, Janowitz T. Connecting the metabolic and immune responses to cancer. Trends Mol Med. (2017) 23:451–64. doi: 10.1016/j.molmed.2017.03.001
752. Mohamed E, Cao Y, Rodriguez PC. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: a promising opportunity for cancer immunotherapy. Cancer Immunol Immunother. (2017) 66:1069–78. doi: 10.1007/s00262-017-2019-6
753. Wang K, Baldwin GS, Nikfarjam M, He H. p21-activated kinase signalling in pancreatic cancer: new insights into tumour biology and immune modulation. World J Gastroenterol. (2018) 24:3709–23. doi: 10.3748/wjg.v24.i33.3709
754. Daniel SK, Sullivan KM, Labadie KP, Pillarisetty VG. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin Transl Med. (2019) 8:10. doi: 10.1186/s40169-019-0226-9
755. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
756. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. (2013) 73:3007–18. doi: 10.1158/0008-5472.CAN-12-4601
757. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. (2013) 73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731
758. Rosati A, Basile A, D'Auria R, d'Avenia M, De Marco M, Falco A, et al. BAG3 promotes pancreatic ductal adenocarcinoma growth by activating stromal macrophages. Nat Commun. (2015) 6:8695. doi: 10.1038/ncomms9695
759. Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. (2014) 25:605–20. doi: 10.1016/j.ccr.2014.03.021
760. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. (2017) 66:124–36. doi: 10.1136/gutjnl-2016-312078
761. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2Axis. Clin Cancer Res. (2013) 19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525
762. Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, et al. Chemo-therapy-derived inflammatory responses accelerate the formation of immunosuppressive myeloid cells in the tissue microenvironment of human pancreatic cancer. Cancer Res. (2015) 75:2629–40. doi: 10.1158/0008-5472.CAN-14-2921
763. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov. (2016) 6:247–55. doi: 10.1158/2159-8290.CD-15-0843
764. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Brutontyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
765. Tan E, El-Rayes B. Pancreatic cancer and immunotherapy: resistance mechanisms and proposed solutions. J Gastrointest Cancer. (2019) 50:1–8. doi: 10.1007/s12029-018-0179-z
766. Heinemann V, Reni M, Ychou M, Richel DJ, Macarulla T, Ducreux M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat Rev. (2014) 40:118–28. doi: 10.1016/j.ctrv.2013.04.004
767. Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers. (2017) 10:E3. doi: 10.3390/cancers10010003
768. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. (2018) 18:335. doi: 10.1186/s12885-018-4238-4
769. Tiriac H, Plenker D, Baker LA, Tuveson DA. Organoid models for translational pancreatic cancer research. Curr Opin Genet Dev. (2019) 54:7–11. doi: 10.1016/j.gde.2019.02.003
770. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science. (2019) 364:952–5. doi: 10.1126/science.aaw6985
771. Ali S, Suresh R, Banerjee S, Bao B, Xu Z, Wilson J, et al. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am J Cancer Res. (2015) 5:1251–64
772. Li Y, Yang X, Kang X, Liu S. The regulatory roles of long noncoding RNAs in the biological behavior of pancreatic cancer. Saudi J Gastroenterol. (2019) 25:145–51. doi: 10.4103/sjg.SJG_465_18
Keywords: pancreatic cancer, metastasis, exosomes, cancer-initiating cell markers, stellate cells, metabolism, perineural invasion, immunosuppression
Citation: Mu W, Wang Z and Zöller M (2019) Ping-Pong—Tumor and Host in Pancreatic Cancer Progression. Front. Oncol. 9:1359. doi: 10.3389/fonc.2019.01359
Received: 09 August 2019; Accepted: 18 November 2019;
Published: 16 December 2019.
Edited by:
Mohit Kumar Jolly, Indian Institute of Science (IISc), IndiaReviewed by:
Sabarish Ramachandran, Texas Tech University Health Sciences Center, United StatesAbhinav Achreja, University of Michigan, United States
Dongya Jia, Rice University, United States
Copyright © 2019 Mu, Wang and Zöller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Mu, d2VpbXVAc2hzbXUuZWR1LmNu