 Jiali Cheng
Jiali Cheng Lei Zhao1†
Lei Zhao1† Yuanyuan Zhang
Yuanyuan Zhang Yun Qin
Yun Qin Yuqi Guan
Yuqi Guan Chaohong Liu
Chaohong Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 21 November 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01237
This article is part of the Research Topic Molecular and Immunological Advances in Hematological Malignancies View all 11 articles
Taking advantage of the immune system to exert an antitumor effect is currently a novel approach in cancer therapy. Adoptive transfer of T cells engineered to express chimeric antigen receptors (CARs) targeting a desired antigen has shown extraordinary antitumor activity, especially in refractory and relapsed B-cell malignancies. The most representative in this respect, as well as the most successful example, is CD19 CAR T-cell therapy in B-cell acute lymphoblastic leukemia (B-ALL). However, with the widespread use of CAR T-cell therapy, problems of resistance and relapse are starting to be considered. This review provides a comprehensive picture of the mechanisms of resistance to CAR T-cell therapy from three aspects, namely, CAR T-cell factors, tumor factors, and tumor microenvironment factors, offering insights for improving CAR T-cell therapy.
With changes in lifestyles and environments, the incidence of tumors, especially malignant tumors, has increased, threatening human health and society. In the centuries of battle against tumors, treatment strategies have evolved from surgery, radiotherapy, and chemotherapy to immunotherapy, which is more efficient and precise. Currently, immunotherapy includes antibodies, vaccines, immune checkpoint inhibitors, and adoptive cell transfer (ACT), such as T-cell receptor (TCR)-expressing T-cell infusion and chimeric antigen receptor (CAR)-expressing T-cell infusion (1–4). A common characteristic of active immunotherapy is that it utilizes the patient's own immune system to attack tumor cells. Among the available immunotherapies, the most encouraged is CAR T-cell therapy, which involves the genetic engineering of a patient's own T cells to kill tumor cells; it was first proposed and administered by the Israeli immunologist Zelig Eshlar in 1993 but has undergone a long and tortuous journey (5, 6). CAR T cells, a type of genetically engineered peripheral T cell, have a special antigen receptor whose extracellular single-chain variable fragment (scFv) can directly recognize a specific antigen independent of the major histocompatibility complex (MHC), an intracellular CD3ζ domain that conveys the T-cell activation signal, and a CD28 or 4-1BB domain that provides a costimulatory signal to facilitate the proliferation of CAR T cells and enable them to persistently attack tumor cells (7). First-generation CAR T cells contain an antigen-recognition domain and a CD3ζ domain; thus, although these cells can be activated, they are unable to proliferate (8). Second-generation CAR T cells introduce a costimulatory signal that enables T cells to proliferate after activation, making them a living drug in vivo (9). Thus far, second-generation CAR T cells have been widely used in hematological malignancies, including B-cell acute lymphoblastic leukemia (B-ALL), B-cell Non-Hodgkin lymphoma (B-NHL), B-cell chronic lymphoblastic leukemia (B-CLL), and multiple myeloma (MM) (10–14), and have shown significant efficacy, as summarized in Table 1. The U.S. Food and Drug Administration (FDA) has approved anti-CD19 CAR T-cell therapy for patients with relapsed/refractory B-ALL and diffuse large B-cell lymphoma (DLBCL). Despite the impressive remission rates, some patients still relapse or are resistant to CAR T-cell therapy (15). Thus, when understanding the extraordinary efficacy, it is important for us to focus on unresponsive and relapsed cases to improve CAR T-cell therapy and facilitate the treatment of tumors. This article briefly reviews the efficacy and toxicity of CAR T-cell therapy, comprehensively analyzes the possible mechanisms of resistance to this therapy, and proposes possible solutions.
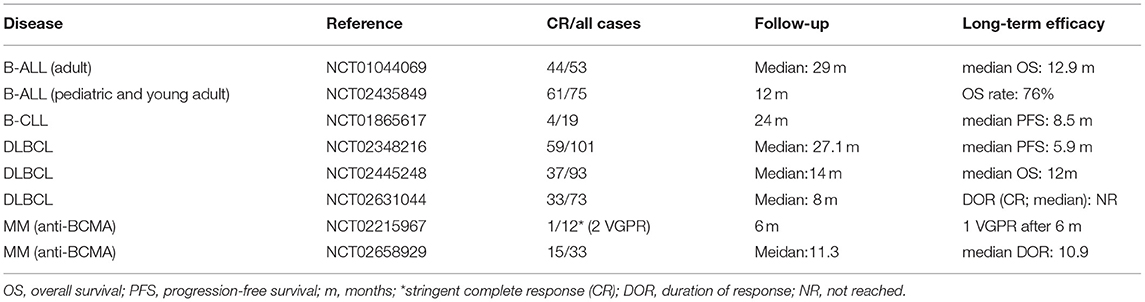
Table 1. Efficacy of CAR T-cell therapy in B-cell malignancies.
As summarized in Table 1, CAR T-cell therapy has shown impressive efficacy in B-cell malignancies, with a complete response (CR) rate of 81–90% for B-ALL and of approximately 50% for B-NHL (15–19). The cellular kinetics of CAR T cells and tumor burden are two important factors affecting the efficacy of CAR T-cell therapy in B-ALL (15, 20). AUC0-28d (area under the curve from the beginning to 28 days post-infusion), representing the expansion of CAR T cells, is a valuable parameter for predicting response: the larger the AUC0-28d, the better the response (21). The persistence of genetically engineered T cells is also an important factor affecting the prognosis of hematological malignancies. Integrated CD19 CAR transgene sequences can remain detectable over several years after infusion in peripheral blood, indicating that CAR T cells are able to survive in the body over a long period of time. Therefore, persistent remission after therapy is promising (21, 22). In addition, tumor burden is another important factor responsible for the prognosis of B-ALL patients. Heavy tumor burden, defined as 5% or more bone marrow blasts or extramedullary disease at the time of CAR T-cell infusion, is associated with a relatively higher chance of minimal residual disease (MRD)-positive remission (23). Research has indicated that the probability of relapse of a patient with CAR T-cell-induced MRD-positive CR is 100% (9 cases) and the probability of relapse of a patient with MRD-negative CR is 50% (16/32) (15). It is speculated that the ratio of AUC0-28d to tumor burden is a good indicator of the status of MRD and therefore the long-term prognosis of patients. Apart from the cellular kinetics of CAR T cells and tumor burden, the density of the targeted antigen, the tumor microenvironment, the status of donor T cells and the characteristic of disease, which can impact the recognition or cytotoxicity of CAR T cells, all have an impact on response to CAR T-cell therapy in B-cell malignancies (24).
The toxicity of CAR T-cell therapy mainly comprises the on-target effect, cytokine release syndrome (CRS), and neurologic toxicity. For anti-CD19 CAR T-cell therapy, B cell aplasia is a predictable on-target side-effect that impairs the humoral immunity and makes patients more susceptible to viral infections. Patients receiving CAR T-cell therapy may develop symptoms that differ from fever to hypotension, hypoxemia, and even multiple organ failure, usually with elevated cytokines like IL-6 and ferritin in serum, which is what is known as CRS. Park and collaborators reported that 45 out of 53 patients developed CRS during CAR T-cell therapy, 14 patients with grade 3 or higher CRS (15). Most cases of CRS can be managed through supportive care, tocilizumab, or corticosteroids. It is reported that the severity of CRS may be related to the tumor burden and the expansion of CAR T cells rather than the infusion dose of CAR T cells (23, 25). How CRS develops is still under research. Recently, a study by Li et al. suggested that TNF-α released by activated lymphocyte is key to inducing IL-6 and IL-1β secretion by monocytes and macrophages in the treatment of anti-HER2/CD3 bispecific antibody, which possibly exists in CAR T-cell therapy (26). Moreover, CAR T-cell therapy may result in neurotoxicity, manifested as cognitive defects, seizures, cerebral edema, etc. The mechanism of CAR T-cell therapy-related neurotoxicity also remains unclear. A study suggested that it may be due to endothelial damage and increased blood-brain barrier permeability (27).
In summary, AUC0-28d and tumor burden together with CAR T-cell persistence in peripheral blood affect the prognosis of patients. Most of the toxicities of CAR T-cell therapy are controllable, but the mechanisms of CRS and neurotoxicity remains to be further elucidated.
Despite the impressive efficacy of CAR T-cell therapy in refractory/relapsed B-cell malignancies, the problem of relapse has gradually come to light with prolonged follow-up periods. According to data from different clinical trials, the relapse rate varies from 21 to 45% in B-ALL and increases with the follow-up time (23, 28, 29). We will discuss the mechanisms of resistance to CAR T-cell therapy, as shown in Figure 1, based on three aspects.
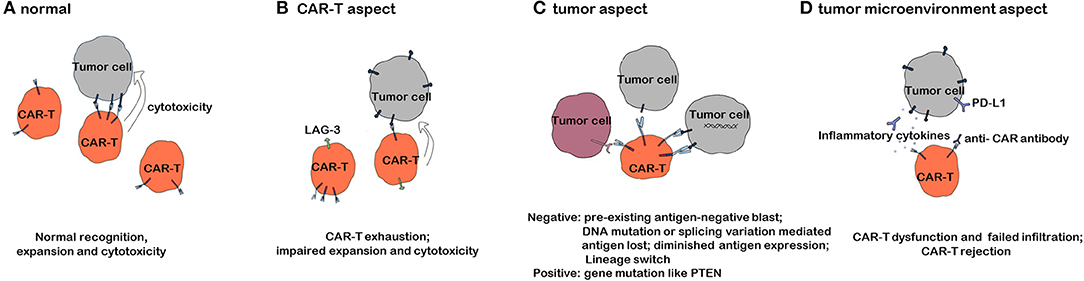
Figure 1. Mechanisms of resistance to CAR T-cell therapy. (A) CAR T cells exerting an antitumor effect under normal circumstances. (B) Mechanisms of resistance to CAR T-cell therapy with respect to CAR T cells. (C) Mechanisms of resistance to CAR T-cell therapy with respect to tumor cells. (D) Mechanisms of resistance to CAR T-cell therapy with respect to the tumor microenvironment.
The response to tumor immunotherapy largely depends on the status of the immune function, suggesting that any defect in the immune system potentially attenuates the prognosis of patients. As shown in Table 1, the efficacy of CAR T-cell therapy in chronic lymphoblastic leukemia (CLL) is much worse than that in B-ALL, and this effect is proposed to be related to innate T-cell defects in CLL patients (13). In a phase I clinical trial of B-ALL (NCT01044069), 2 of 67 patients failed to produce CAR T cells, and in another clinical trial of DLBCL (NCT02445248), 12 of 165 patients experienced the same problem, suggesting that T cell defects have an impact on CAR T-cell production (15, 30). Expansion, persistence, and tumor cytotoxicity are the three main characteristics of CAR T cells that influence treatment efficacy. T cells from a cancer patient can often be deficient in their intrinsic cytotoxicity (31). CAR T cells derived from these cancer patients will thus have diminished cytotoxicity, resulting in a relatively poor prognosis. Transcriptome analysis has revealed that enhanced expression levels of key regulators of late memory/effector T-cell differentiation and aerobic glycolysis are associated with poor response to CAR T-cell therapy (32). Inhibition of glycolysis with 2-deoxy-D-glucose can facilitate the differentiation of central memory CAR T cells. Furthermore, studies indicate that the activation of interleukin (IL)-6/signal transducer and activator of transcription (STAT)-3 signaling pathways promotes central memory T-cell differentiation, which may play an important role in regulating the proliferation of CAR T cells (33). These findings suggest that the IL-6/STAT3 signaling pathway and glycometabolism can modulate the proliferation-dependent expansion of CAR T cells by regulating CAR T-cell differentiation. Furthermore, the constitution of the T-cell pool is constantly changing with age, shifting from undifferentiated naïve T cells to differentiated effector/memory T cells, which lack CD28 expression and have decreased proliferation ability when stimulated by antigens (34, 35). Whether the condition of the T-cell pool can influence the proliferation of CAR T cells has not been reported. In addition to impaired cytotoxicity and expansion, CAR T-cell exhaustion can lead to the failure of CAR T-cell therapy. T-cell exhaustion refers to a state of dysfunction characterized by a decrease in effectors and increased expression of inhibitory receptors, usually induced by chronic stimulation, such as in cancer (36, 37). B-cell recovery in peripheral blood is a marker of CD19 CAR T-cell dysfunction in the body. In pediatric B-ALL, B-cell recovery in peripheral blood within 3 months indicates a high risk of relapse, which may be due to CAR T-cell exhaustion (38). In DLBCL, a high percentage of LAG3+ T cells, a biomarker of T-cell exhaustion, is correlated with limited responses to CD19 CAR T-cell therapy (30). The mechanisms of CAR T-cell exhaustion are poorly understood. One study suggested that CARs on CAR T cells can spontaneously cluster in an antigen-independent manner, generating tonic CAR-CD3ζ signaling that can induce CAR T-cell exhaustion (39). Additionally, the endogenous TCR signal of CAR T cells in the presence of a specific antigen has been shown to induce T-cell exhaustion (40).
Tumor factors are generally divided into two categories: relapse with positive target antigen expression and relapse with negative target antigen expression. Here, we discuss the two categories separately.
Targeted antigen-negative relapse is one of the main reasons for resistance to CAR T-cell therapy and accounts for approximately 9–25% of cases of relapse in B-ALL according to several different clinical trials (15, 16, 20, 41). As recently reported at the 2018 ASH Meeting, CD19-negative and CD19-positive relapses occurred in 7/21 and 14/21 relapses, respectively, among DLBCL patients (42). The mechanisms of target antigen-negative relapse mainly involve four aspects: the preexistence of target antigen-negative tumor cells, diminished expression of target antigens, mutation-, splicing variation-, or lineage switching-mediated target antigen loss, and failure of presentation of target antigens. The efficacy of targeted therapy is thought to be tightly associated with the density of target antigens on the cell membrane. CD19, uniquely and broadly expressed in B-linage cells, is a favorable target antigen for CAR T-cell therapy (43). A research group from the Children's Hospital of Philadelphia (CHOP) investigated flow cytometric data from 628 cases of relapsed or refractory B-ALL and showed that before treatment, approximately 17% of cases had CD19-negative tumor cells (defined as the presence of more than 1% of negative cells), which may lead to relapse after anti-CD19 CAR T-cell therapy. Additionally, compared to healthy people, 7% of patients displayed diminished CD19 expression, and 24% of patients had low-normal CD19 expression (43). In addition to preexisting CD19− or CD19dim tumor cells, splicing variations and mutations partially accounted for CD19-negative relapse in CTL019 therapy (44, 45). Researchers in CHOP identified an SRSF3-involved alternative splicing of exon 2 of CD19 messenger RNA (mRNA) in CD19-negative relapsed B-ALL, which resulted in the loss of the targeted epitope in the membrane and consequent escape from the attack of anti-CD19 CAR T cells (44). Exon 5 and exon 6 deletion-mediated deficiency of the transmembrane domain of CD19 also induces CD19-negative relapse in response to anti-CD19 CAR T-cell administration. Mutations in exons 2–5, such as frameshift in exon 2, 3, or 4, insertion in exon 3, and nonsynonymous mutations in exon 4, lead to the loss of CD19 expression in the membrane and relapse post-CD19 CAR T-cell therapy (45). Lineage switching, referred to as conversions of leukemic cell lineage (46), can lead to CD19-negative relapse after CAR T-cell therapy. Research has shown that B-ALL cases with initial clearance of blasts post-infusion of CTL019 displayed CD19-negative relapse with a myeloid phenotype (47, 48). Experiments based on murine ALL models showed that in E2a:PBX1 B-ALL, the relapsed cells lost CD19 expression but expressed myeloid antigens post-CD19 CAR T-cell treatment, and the frequency was much lower in Eμ-RET B-ALL, suggesting that lineage switching-mediated CAR T-cell therapy resistance is related to genetic background (47). The exact mechanisms of lineage switching remain elusive, but there are two mainstream theories: drug-induced reprogramming of the original tumor stem cell and expansion of a different phenotypic clone of tumor cells during the process of targeted immunotherapy (49). Furthermore, deficient maturation and translocation of CD19 post-translation is a possible mechanism of CD19-negative resistance. A study identified three CD81 deficiency-related CD19-negative relapsed B-ALL cases post-blinatumomab therapy. Further investigation indicated a deficiency in CD81 expression resulting in failed formation of the CD19/CD21/CD81 coreceptor complex and hindered maturation and translocation of CD19 from the Golgi body to the cell membrane (50). Slightly different from CD19, the density of membrane CD22 differs considerably in normal situations, indicating that diminished CD22 site density is an important problem leading to antigen escape-mediated CAR T-cell therapy resistance. Additionally, approximately 22% of B-ALL patients are negative for CD22 according to a report from CHOP (43, 51).
In clinical practice, target antigen-positive relapses can result from CAR T-cell defects. However, herein we will only discuss the tumor-related factors leading to target antigen-positive resistance. CAR T cells exert antitumor effects, which are dependent not only on the recognition of specific antigens but also on the induced apoptosis of tumor cells. Signals that induce apoptosis of tumor cells include tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), Fas ligand (FasL), and cytokines such as interferon (IFN)-γ (52–54). The mechanism of tumor antigen-positive resistance to CAR T-cell therapy underlies changes in tumor cell survival or apoptosis. Experiments have shown that even when the function of CD19 CAR T cells as measured using the secretion of type I cytokines is normal, a TRAIL inhibitor can suppress the cytotoxic effect of CAR T cells when the CAR T cells are cocultured with sensitive cells, indicating that a lack of TRAIL signaling in tumor cells can lead to tumor antigen-positive resistance to CAR T-cell therapy (55). To date, research on the relationship between tumor mutations and CAR T-cell therapy resistance remains limited. However, data from programmed cell death-1 (PD-1) therapy can provide some insights. In PD-1 immunotherapy of melanoma, PTEN deficiency leads to reduced tumor infiltration and decreased cytotoxicity of T cells, resulting in a poor response. Concurrent administration of PD-1/cytotoxic T-lymphocyte (CTL)-associated protein 4 (CTLA-4) inhibitor and PI3K-beta inhibitor can ameliorate the poor prognosis, indicating that PI3K-AKT over-activation is an important step in the process of antigen-positive resistance or relapse (56, 57). In addition, loss-of-function mutations in Janus kinase 1 (JAK1) or Janus kinase 2 (JAK2) can lead to resistance to PD-1 therapy in melanoma by blocking the IFN-γ signal (58). Whether the mechanisms of resistance to CAR T-cell therapy are similar to those to PD-1 therapy remains unknown.
In summary, target antigen-negative relapse mainly results from the preexistence or generation of targeted antigen-negative or targeted antigen-diminished tumor cells. As far as tumor-related factors are concerned, target antigen-positive resistance mainly results from tumor mutations such as those in PTEN and JAK1/JAK2, and this mechanism requires further investigation.
Studies on the role of the tumor microenvironment in CAR T-cell therapy are rare, which is likely due to the fact that CAR T-cell therapy is mainly used in hematological cancers such as B-ALL. The tumor microenvironment is mainly comprised of various cell types, including tumor-infiltrating immune cells, fibroblasts, and endothelial cells, and the extra-cellular cytokines, matrix, chemokine, etc., which modulate the development of tumor and the response to immunotherapy (59). The immunosuppressive tumor microenvironment in solid tumors is one of the most important factors impairing the efficacy of immunotherapy. The microenvironment interferes with the function and infiltration of immune effector cells. The phenomenon that tumor cells upregulate programmed cell death-ligand 1 (PD-L1) expression on the membrane to induce apoptosis in immune effector cells has been extensively studied in recent years (60–62). Recently, the Wei Gao group demonstrated that melanoma cells not only express PD-L1 but also release PD-L1 into the tumor microenvironment and blood circulation, leading to a poor response to cancer immunotherapy (63). As well as T-cell checkpoint blockage, hypoxia and glucose depletion result in lactic acid accumulation, consequently leading to low pH values in the tumor microenvironment, which suppresses the function of effector T cells, characterized by reduced IL-2 and IFN-γ secretion and lytic activity (64, 65). Additionally, specific components of the inflammatory tumor environment, for instance, Prostaglandin E2 (PGE2) produced by tumor cells in a mouse model, can affect the antitumor activity of T cells depending on IL-6, chemokine (C-X-C motif) ligand 1 (CXCL1), and granulocyte-colony stimulating factor (G-CSF) (66). Apart from cytotoxic T lymphocyte (CTL) dysfunction, cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), and M2 subtypes of tumor-associated macrophages (TAMs) in the tumor microenvironment are reported to restrict infiltration of CTLs (67). On the other hand, the microenvironment trains tumor cells to disguise themselves to avoid recognition by immune cells. A clinical trial of Melan-A (MART-1)-targeted adoptive T-cell transfer therapy for metastatic melanoma revealed that TNF-α secreted by infiltrated CTLs induced dedifferentiation of melanoma cells to lose MART-1 and gp100 expression and acquire NGFR expression, leading to resistance to cancer immunotherapy (68).
An important concern in CAR T-cell therapy is the production of antibodies and CTLs against murine CAR scFv, which may result in CAR T-cell rejection. Most B-ALL patients who relapse after CD19 CAR T-cell therapy had no response to reinfusion of CD19 CAR T cells, even when CD19 expression is positive (31). In 2006, Kershaw reported that during anti-folic acid receptor CAR T-cell therapy for ovarian cancer, three of six cases displayed the presence of factors inhibiting CAR T-cell function in serum. After protein G administration, the inhibition was relieved (69). In 2013, Maus reported that during murine anti-mesothelin CAR T-cell therapy, after multiple reinfusion of CAR T cells, one patient developed an acute allergic reaction and died of cardiac arrest within a few minutes, which could be ascribed to the persistence of anti-CAR IgE in the body (70). According to the results reported by Turtle and collaborators, cytotoxic CD8+ T-cell responses to CAR T cells occurred in five CAR T-cell therapy-resistant patients, and the murine scFv FMC63 was identified to contain the immunogenic epitope (31). Together, these data suggest that anti-murine scFv CAR antibodies and CTLs are generated after CAR T-cell infusion, which can lead to CAR T-cell rejection. However, Mueller and collaborators reported that although 84.8% of patients generated anti-murine CAR antibodies after tisagenlecleucel treatment, it did not affect the efficacy of CAR T-cell therapy (22). Thus, the presence of an immune response to murine scFv and the consequence of this reaction remain unclear.
In summary, the tumor microenvironment is considered to be the bottleneck of CAR T-cell therapy in solid tumors, mainly due to its effect on defective CTL infiltration and dysfunction. Other important factors include anti-murine scFv antibodies and effector T cells, which may induce CAR T-cell rejection.
We can improve CAR T-cell engineering to overcome the deficient cytotoxicity, expansion, and persistence of CAR T cells. To address an intrinsic deficiency in T cells, universal CAR T cells or haploidentical CAR T cells can be alternatively used (71). Generating universal CAR-T cells from allogeneic healthy donors required additional genetic modification to effectively abolish GVHD and/or CAR-T cell rejection. Universal CAR-T cell product offers a way to overcome the problem of quantitatively insufficient CAR-T cells from infants or highly treated patients who are profoundly lymphopenic owing to multiple previous chemotherapies. Qasim et al. demonstrated that two infants with relapsed refractory acute lymphocytic leukemia achieved molecular remission when treated with universal CAR-T cells (72). Besides, universal CAR T cells will make CAR T-cell therapy an off-the-shelf treatment, reduce the cost and time required to manipulate the patient's own T cells, and exclude the possible quality problems in T cells (73). To improve the proliferation of CAR T cells, modification of the costimulatory signal of CARs may also be an option. Currently, the most commonly used costimulatory signals of CARs involve 4-1BB and CD28. The 4-1BB signal induces moderate expansion and prolonged persistence of CAR T cells, but the CD28 signal induces robust expansion and relatively short persistence of CAR T cells (74), indicating that the costimulatory signal of CAR T cells can control the proliferation and persistence of these cells. Recently, third-generation CAR T-cells have been produced by incorporation of both the CD28 and 4-1BB co-stimulatory signals, expecting to obtain good anti-tumor potency and prolonged persistence at the same time. Preclinical data showed that third-generation CAR T-cells had balanced anti-tumor efficacy, improved persistence and decreased exhaustion compared with second-generation CAR T-cells (75). Clinical data confirmed the superior expansion and persistence of third-generation CAR T-cells (76). In a phase I/IIa clinical trial of third-generation CAR T-cells, 4 of 11 r/r B-cell lymphoma patients had initial CR, and another 3 patients achieved remission within 3 months. Two of 4 B-ALL patients had initial CR (77). More clinical data are required to inspect the efficacy of third-generation CAR T-cell therapy. Nevertheless, a better costimulatory signal remains to be discovered. Alternatively, we can infuse a specific composition of CAR T cells to improve proliferation and persistence. Studies have shown that an increased frequency of CD27+CD45RO−CD8+ CAR T cells, with a memory cell-like phenotype, can contribute to complete remission and prolonged event-free survival (78). Interestingly, biallelic inactivation of the gene Tet methylcytosine dioxygenase 2 (TET2) improves the persistence of CAR T cells, indicating the importance for identifying genes that determine the persistence of CAR T cells for the long-term prognosis in response to CAR T-cell therapy (79).
To circumvent antigen escape-mediated relapse, we can use CAR T cells targeting another antigen. A phase I trial reported that CD22 CAR T cells induced CR in 73% (11/15) of patients who had received CD19 CAR T-cell therapy and experienced a CD19-negative relapse or resistance, indicating that targeting another antigen may work in target antigen-negative relapse or resistance. However, 7 of 11 patients relapsed again with CD22− or CD22dim lymphoblasts, indicating that targeting another antigen is an effective but not a radical solution (51). Target escapes still happen against new agents. Another way to reduce target escape is to target multiple antigens at the same time or to sequentially infuse CAR T cells targeting different antigens in the beginning (80). To date, no data have been published to test the efficacy of these strategies. It is unknown whether the efficacy of sequential infusion of CD19 and CD22 CAR T cells is superior to that of infusion of CD19 CAR T cells alone at the beginning of therapy followed by infusion of CD22 CAR T cells after relapse. It should be noted that designing new target antigens is not always easy. Concerning the existence of CD19dim or CD22dim leukemic cells, researchers have tried to elevate the affinity between CAR and antigens to reduce the density of antigens required for CAR T-cell activation (81, 82). Nevertheless, the efficacy and safety of high-affinity CAR T cells remain to be evaluated. Immunogenic cell death is another potential strategy to overcome target antigen-negative relapse. Under certain conditions, cell death will activate the adaptive immune response (immunogenic cell death). The initiation of adaptive immune response mainly relies on two factors: adjuvants that can release a danger signal to trigger the immune response [it should be noted that some damage associated molecular patterns (DAMPs) suppress immune response instead of activating it (83)] and antigens that do not induce central and peripheral tolerance (84). Cancer cell death resulting in exposure of neoantigens to the immune system can, however, avoid its activation by limiting the danger signal (85). On the other hand, cell death induced by conditions such as chemotherapy or radiotherapy has been shown to release DAMPs, like CALR and HSP70, that promote the activation of the immune system (86, 87). Therefore, a combination of CAR T-cell therapy with radiotherapy, immune checkpoint inhibition, or vaccine may exert a synergistic anti-tumor effect because of the ICD-induced activation of the immune response. It remains unknown whether novel strategies, such as induction of tumor antigen expression, inhibition of targeted antigen loss, and targeting of a tumor marker at the DNA level, can help us win the battle against antigen-negative relapse in the future.
With regard to antigen-positive resistance, the main issue is that the cytotoxic signals emitted by CAR T cells fail to overcome the survival signals of tumor cells due to enhanced survival, proliferation, or cytotoxic signal shielding by tumor cells. Therefore, we can use target-specific drugs, such as PI3K-beta inhibitor, in combination with CAR T cells to regulate these signaling pathways and to counterbalance the abnormal proliferation and apoptosis signals in tumor cells. In vitro experiments have shown that the administration of the bcl-2 family apoptosis inhibitor ABT-737 can increase apoptosis in tumor cells induced by CAR T cells (88). Histone deacetylase inhibitors such as SAHA and LBH589 can also promote the sensitivity of resistant NHL cell lines toward CD19 CAR T cells by regulating apoptotic gene expression (55). Moreover, we can take advantage of the targeting ability of CAR T cells to accurately deliver drugs, thereby improving treatment efficacy and reducing side effects. In addition, hematopoietic stem cell transplantation (HSCT) is an alternative method, although there is still controversy as to whether HSCT after complete remission induced by CAR T-cell therapy benefits patients. Summers et al. reported that consolidative HSCT after CAR T-cell therapy in those ALL patients who have never received HSCT tends to improve the PFS, with a p-value of 0.059 (89). However, Park et al. reported that HSCT after CR induced by CAR T-cell therapy did not improve the PFS and OS, with a p-value of 0.64 for all CR patients and of 0.89 for MRD-negative CR patients (15). More clinical data are required to define whether HSCT is a beneficial consolidative treatment after CAR T-cell therapy.
The most attractive solution to overcome resistance due to the tumor microenvironment is to genetically engineer CAR T cells to secrete specific cytokines, such as IL-2 and IL-12. A phase I trial in 2005 reported that IL-12-secreting CAR T cells displayed stronger cytotoxicity and longer persistence during treatment in six cases of MUC16ecto+ ovarian cancer (NCT01457131). IL-12 is a proinflammatory factor that can activate the innate and adaptive immune systems to exert an antitumor effect and reduce the activity of regulatory T (Treg) cells and myeloid-derived immunosuppressive cells to counteract the immunosuppressive microenvironment (90). Based on the immune checkpoint theory, a more direct approach is to inactivate the immunosuppressive signal inside CAR T cells through gene-editing technology, to engineer CAR T cells to secrete PD-1 inhibitors, or to combine PD-1 blocking antibodies with CAR T cells (NCT02926833). It has been reported that knocking down PDCD1, the gene encoding PD-1, can increase the antitumor activity of CAR T cells (91). CAR T cells can also be engineered to secrete some enzymes or chemokines, such as heparanase, to promote the infiltration of immune effector cells into tumor, especially in solid tumors. For antibodies against murine CAR scFv, the application of humanized CAR T cells is the best solution.
The emergence of CAR T-cell therapy has altered the landscape of cancer immunotherapy, showing an impressive outcome in B-cell malignancies. Two CD19 CAR T-cell therapies have been approved for the treatment of B-ALL and DLBCL. However, resistance, both primary and acquired, to CAR T-cell therapy can still emerge. One of the most important goals of the field is to determine the signals triggered by CAR stimulation, which is fundamental for advancing CAR T-cell therapy. Immune escape of target antigen-negative tumor cells also occurs in CAR T-cell therapy, which could be managed by targeting another antigen. Nevertheless, resistance to the new target antigen can also occur in theory. This situation is similar to a race, i.e., if immune effector cells can find all tumor cells before they are masked, the tumor loses; otherwise, the treatment is unsuccessful. Additionally, the tumor microenvironment, a complicated and dynamic environment, can hamper the efficacy of CAR T-cell therapy, especially in solid tumors. Advances in gene-editing technology and cell culture technology may facilitate the efficacy of CAR T-cell therapy. Nonetheless, tumor cells are evolving, and, thus, mechanisms to radically avoid immune escape remain to be explored. There is still a long way for humans to go to defeat cancer. Finally, apart from the accessibility of technology, the heavy economic burden of CAR T-cell therapy has limited the use of the therapy for cancer. In the USA, the price of Tiga-Cel (“Kymriah”) is $475,000 and that Axi-Cel (“Yescarta”) is $350,000, which most countries in the world cannot afford. In all, CAR T-cell therapy represents one of the most effective and advanced treatments in B-cell malignancies, although it still faces some challenges, namely that defects in CAR T-cell, targeted antigen escape, tumor mutation, and the tumor microenvironment can result in resistance or relapse to CAR T-cell therapy. However, as the development of science and technology continues, CAR-based cellular immunotherapy will become more powerful.
JC wrote the initial draft. LZ revised the review. YZ, YQ, YG, TZ, and CL discussed the review and approved the final manuscript. JZ conducted the whole process. All authors checked and approved the final version.
This work was supported by the National Science Foundation of China (Grant No. 81670150).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully thank Jingwen Li for editing the figure.
ACT, adoptive cell transfer; AUC, area under the curve; B-NHL, Non-Hodgkin lymphoma; B-CLL, B-cell chronic lymphoblastic leukemia; B-ALL, B-cell acute lymphoblastic leukemia; CAR, chimeric antigen receptor; CD3, cluster of differentiation 3; CR, complete remission; CRS, cytokine release syndrome; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CTL, cytotoxic T lymphocyte; CAF, cancer-associated fibroblast; CXCL1, chemokine (C-X-C motif) ligand 1; Dim, diminished; DNA, deoxyribonucleic acid; DLBCL, diffused large B cell lymphoma; FDA, U.S. Food and Drug Administration; G-SCF, granulocyte-colony stimulating factor; HSCT, hematopoietic stem cell transplantation; IL-6, Interleukin 6; ICD, immunogenic cell death; IFN, interferon; JAK, janus kinase; LAG-3, Lymphocyte-activation gene 3; MHC, major histocompatibility complex; MM, multiple myeloma; mRNA, messenger RNA; MLL, Histone-lysine N-methyltransferase; MRD, minimal residual disease; MART-1, melan-A; MDSC, myeloid-derived suppressor cell; p53, cellular tumor antigen p53; PR, partial remission PI3K-AKT, phosphatidylinositide 3-kinases-Protein kinase B; PD-1, programmed cell death-1; PD-L1, programmed cell death-ligand 1; PGE2, prostaglandin E2; scFv, single-chain variable fragment; STAT3, transducer and activator of transcription 3; TCR, T cell receptor; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TNF, tumor necrosis factor; Treg, regulatory T cell; TET2, Tet methylcytosine dioxygenase 2; TAM2, tumor-associated macrophage.
1. Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. (2016) 15:235–47. doi: 10.1038/nrd.2015.35
2. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
3. Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J Exp Med. (1955) 102:157–77. doi: 10.1084/jem.102.2.157
4. Hedrick SM. Chimeric T cell receptor-immunoglobulin molecules: function and applications. Int Rev Immunol. (1993) 10:279–90. doi: 10.3109/08830189309061702
5. Rosenbaum L. Tragedy, perseverance, and chance - the story of CAR-T therapy. N Engl J Med. (2017) 377:1313–5. doi: 10.1056/NEJMp1711886
6. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
7. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
8. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
9. Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. (2002) 20:70–5. doi: 10.1038/nbt0102-70
10. Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified t cells after failure of ibrutinib. J Clin Oncol. (2017) 35:3010–20. doi: 10.1200/JCO.2017.72.8519
11. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
12. Tang XY, Sun Y, Zhang A, Hu GL, Cao W, Wang DH, et al. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ Open. (2016) 6:e013904. doi: 10.1136/bmjopen-2016-013904
13. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
14. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
15. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
16. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
17. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
18. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in Relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
19. Abramson JS, Siddiqi T, Palomba ML, Gordon LI, Lunning MA, Arnason JE, et al. High durable CR rates and preliminary safety profile for JCAR017 in R/R aggressive b-NHL (TRANSCEND NHL 001 Study): a defined composition CD19-directed CAR T-cell product with potential for outpatient administration. J Clin Oncol. (2018) 36:120. doi: 10.1200/JCO.2018.36.5_suppl.120
20. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
21. Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. (2017) 130:2317–25. doi: 10.1182/blood-2017-06-786129
22. Mueller KT, Waldron E, Grupp SA, Levine JE. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2018) 24:6175–84. doi: 10.1158/1078-0432.CCR-18-0758
23. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
24. Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science. (2016) 352:658–60. doi: 10.1126/science.aaf2834
25. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
26. Li J, Piskol R, Ybarra R, Chen YJ, Li J, Slaga D, et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. (2019) 11:eaax8861. doi: 10.1126/scitranslmed.aax8861
27. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
28. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
29. Park JH, Geyer MB. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. (2016) 127:3312–20. doi: 10.1182/blood-2016-02-629063
30. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
31. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
32. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
33. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. (2011) 35:806–18. doi: 10.1016/j.immuni.2011.09.016
34. Chou JP, Effros RB. T cell replicative senescence in human aging. Curr Pharm Des. (2013) 19:1680–98. doi: 10.2174/138161213805219711
35. van Deursen JM. The role of senescent cells in ageing. Nature. (2014) 509:439–46. doi: 10.1038/nature13193
36. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
38. Maude SL, Barrett DM, Rheingold SR, Aplenc R, Teachey DT, Callahan C, et al. Efficacy of humanized CD19-targeted chimeric antigen receptor (CAR)-modified T cells in children with relapsed ALL. J Clin Oncol. (2016) 34:3007. doi: 10.1200/JCO.2016.34.15_suppl.3007
39. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
40. Yang Y, Kohler ME, Chien CD. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. (2017) 9:eaag1209. doi: 10.1126/scitranslmed.aag1209
41. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent to treat leukemia remission by CD19CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
42. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/s1470-2045(18)30864-7
43. Rosenthal J, Naqvi AS, Luo M, Wertheim G, Paessler M, Thomas-Tikhonenko A, et al. Heterogeneity of surface CD19 and CD22 expression in B lymphoblastic leukemia. Am J Hematol. (2018) 93:E352–5. doi: 10.1002/ajh.25235
44. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
45. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. (2018) 24:1504–6. doi: 10.1038/s41591-018-0146-z
46. Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D. Lineage switch in acute leukemia. Blood. (1984) 64:701–6. doi: 10.1182/blood.V64.3.701.bloodjournal643701
47. Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. (2016) 7:12320. doi: 10.1038/ncomms12320
48. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. (2016) 127:2406–10. doi: 10.1182/blood-2015-08-665547
49. Dorantes-Acosta E, Pelayo R. Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res. (2012) 2012:406796. doi: 10.1155/2012/406796
50. Braig F, Brandt A, Goebeler M, Tony HP, Kurze AK, Nollau P, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. (2017) 129:100–4. doi: 10.1182/blood-2016-05-718395
51. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
52. Corazza N, Kassahn D, Jakob S, Badmann A, Brunner T. TRAIL-induced apoptosis: between tumor therapy and immunopathology. Ann N Y Acad Sci. (2009) 1171:50–8. doi: 10.1111/j.1749-6632.2009.04905.x
53. Seino K, Kayagaki N, Okumura K, Yagita H. Antitumor effect of locally produced CD95 ligand. Nat Med. (1997) 3:165–70. doi: 10.1038/nm0297-165
54. Platanias LC. Interferons and their antitumor properties. J Interferon Cytokine Res. (2013) 33:143–4. doi: 10.1089/jir.2013.0019
55. Torres-Collado AX, Jazirehi AR. Overcoming resistance of human non-Hodgkin's lymphoma to CD19-CAR CTL therapy by celecoxib and histone deacetylase inhibitors. Cancers. (2018) 10:E200. doi: 10.3390/cancers10060200
56. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
57. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. (2008) 454:776–9. doi: 10.1038/nature07091
58. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
59. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. (2017) 171:934–49.e16. doi: 10.1016/j.cell.2017.09.028
60. Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. (2018) 131:68–83. doi: 10.1182/blood-2017-07-740993
61. Fang X, Xiu B, Yang Z, Qiu W, Zhang L, Zhang S, et al. The expression and clinical relevance of PD-1, PD-L1, and TP63 in patients with diffuse large B-cell lymphoma. Medicine. (2017) 96:e6398. doi: 10.1097/MD.0000000000006398
62. Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. (2017) 14:203–20. doi: 10.1038/nrclinonc.2016.168
63. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
64. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
65. Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Bashir AH, Elhassan GO, et al. Erratum: glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. (2015) 2:317. doi: 10.18632/oncoscience.158
66. Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. (2010) 16:399–403. doi: 10.1097/PPO.0b013e3181eacbd8
67. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
68. Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B, et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. (2018) 8:935–43. doi: 10.1158/2159-8290.CD-17-1178
69. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
70. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. (2013) 1:26–31. doi: 10.1158/2326-6066.CIR-13-0006
71. Li T, Zhang Y, Peng D, Mao X, Zhou X, Zhou J. A good response of refractory mantel cell lymphoma to haploidentical CAR T cell therapy after failure of autologous CAR T cell therapy. J Immunother Cancer. (2019) 7:51. doi: 10.1186/s40425-019-0529-9
72. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aaj2013
73. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. (2018) 173:1426–38.e11. doi: 10.1016/j.cell.2018.03.038
74. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. (2018) 11:eaat6753. doi: 10.1126/scisignal.aat6753
75. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
76. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin's lymphomas. Mol Ther. (2018) 26:2727–37. doi: 10.1016/j.ymthe.2018.09.009
77. Enblad G, Karlsson H, Gammelgard G, Wenthe J, Lovgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. (2018) 24:6185–94. doi: 10.1158/1078-0432.CCR-18-0426
78. Kueberuwa G, Gornall H, Alcantar-Orozco EM, Bouvier D, Kapacee ZA, Hawkins RE, et al. CCR7(+) selected gene-modified T cells maintain a central memory phenotype and display enhanced persistence in peripheral blood in vivo. J Immunother Cancer. (2017) 5:14. doi: 10.1186/s40425-017-0216-7
79. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. (2018) 558:307–312. doi: 10.1038/s41586-018-0178-z
80. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. (2016) 126:3814–26. doi: 10.1172/JCI87366
81. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. (2012) 42:3174–9. doi: 10.1002/eji.201242606
82. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. (2015) 75:3505–18. doi: 10.1158/0008-5472.CAN-15-0139
83. Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. (2013) 14:668–75. doi: 10.1038/ni.2635
84. Matzinger P. Tolerance, danger, and the extended family. Ann Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
85. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Ann Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
86. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
87. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. (2011) 71:4821–33. doi: 10.1158/0008-5472.CAN-11-0950
88. Karlsson H, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, Essand M, et al. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. (2013) 20:386–93. doi: 10.1038/cgt.2013.35
89. Summers C, Annesley C, Bleakley M, Dahlberg A, Jensen MC, Gardner R. Long term follow-up after SCRI-CAR19v1 reveals late recurrences as well as a survival advantage to consolidation with HCT after CAR T cell induced remission. Blood. (2018) 132:967–967. doi: 10.1182/blood-2018-99-115599
90. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
Keywords: CAR T-cell therapy, resistance mechanism, T-cell defect, tumor factor, tumor microenvironment
Citation: Cheng J, Zhao L, Zhang Y, Qin Y, Guan Y, Zhang T, Liu C and Zhou J (2019) Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 9:1237. doi: 10.3389/fonc.2019.01237
Received: 02 April 2019; Accepted: 28 October 2019;
Published: 21 November 2019.
Edited by:
Ritu Gupta, All India Institute of Medical Sciences, IndiaReviewed by:
Yago Nieto, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2019 Cheng, Zhao, Zhang, Qin, Guan, Zhang, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianfeng Zhou, amZ6aG91QHRqaC50am11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.