 Mirco Galiè
Mirco Galiè
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 12 November 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01199
This article is part of the Research Topic Oncogenic RAS-dependent Reprogramming of Cellular Plasticity View all 10 articles
Oncogenic activation of RAS isoforms leads tumor initiation and progression in many types of cancers and is gaining increasing interest as target for novel therapeutic strategies. In sharp contrast with other types of cancer, the importance of RAS in breast tumorigenesis has long been undermined by the low frequency of its oncogenic mutation in human breast lesions. Nevertheless, a wealth of studies over the last years have revealed how the engagement of RAS function might be mandatory downstream varied oncogenic alterations for the progression, metastatic dissemination, and therapy resistance in breast cancers. We review herein the major studies over the last three decades which have explored the controversial role of RAS proteins and their mutation status in breast tumorigenesis and have contributed to reveal their role as supporting actors, instead of as primary cause, in breast cancer.
RAS G-proteins mediate the signal transduction through the transmembrane receptors. In humans, there are four highly homologous ≈21 KDa RAS isoforms: HRAS, encoded by the Harvey rat sarcoma viral oncogene homolog (HRAS), NRAS, encoded by neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS), and KRAS4A and KRAS4B, alternative splice variants of the Kristen rat sarcoma viral oncogene homolog (KRAS). RAS proteins function as binary switches that cycle between an active (“on”) GDP-bound to an inactive (“off”) GTP-bound state depending on the activation status of the upstream receptors. The switch between “on” and “off” states is modulated by the complementary action of enzymes that promote either the GDP to GTP exchange (guanine exchange factors, GEFs) or the conversion back to GDP-bound form (GTPase-activating proteins, GAPs). The multiplicity of GTPases and GAPs allows the function of RAS to be finely regulated depending on the variety of extracellular and intracellular signal inputs. RAS proteins activate a hierarchical cascade of intersecting pathways which modulate biological functions such as cell proliferation, apoptosis, motility, metabolism, immune evasion. Dysregulation of RAS function is largely associated with tumorigenesis. This may rely either on genomic mutations which alter the RAS-intrinsic structure or on alteration of RAS regulating factors, which enhances RAS expression and activity.
This review is an effort to recapitulate more than 30 years of studies on RAS oncogenes and breast cancer, with the aim to reconcile two apparently conflicting evidences arisen by these studies: (1) experimental studies on cancer cells and murine models have demonstrated that RAS oncogenes and their mutations have a strong potential in breast cancer initiation and progression as it does in other type of cancers; (2) clinical studies have demonstrated that, actually, the incidence of tumorigenic RAS mutations in human breast cancers is marginal, in sharp contrast with other types of cancer.
RAS genes were the first mutated genes identified in human cancer (1–3). The discovery in the late 1970s that their gain-of-function mutations were able to trigger tumorigenesis inaugurated the modern molecular oncology and posed the basis of the molecularly targeted anticancer drug discovery (4). To date, hundreds of genes have been identified which harbor oncogenic mutations, but the RAS genes still remain amongst the most frequently mutated oncogene families in cancer. The oncogenic activation of RAS genes is usually caused by a single point mutation (5–7) which impair the RAS responsiveness to the GAP-mediated modulation and locks RAS and their downstream pathways in a persistently active state. Traditional studies (first carried out for the HRAS oncogenic allele) have shown that a single oncogenic RAS gene could transform cells in vitro and could provide them with the ability to induce tumors in mice (3). Twenty five percent of human cancers display missense gain-of-function mutations in at least one of the RAS genes and in 98% of the cases mutations are found at one of the mutational hotspots G12, G13, and Q61 (COSMIC v75). Not all RAS isoforms are mutated equally, with KRAS displaying the highest frequency. Also, mutations of specific RAS isoforms exhibit marked preferences for different tumor types and different impact on clinical outcome (Figure 1).
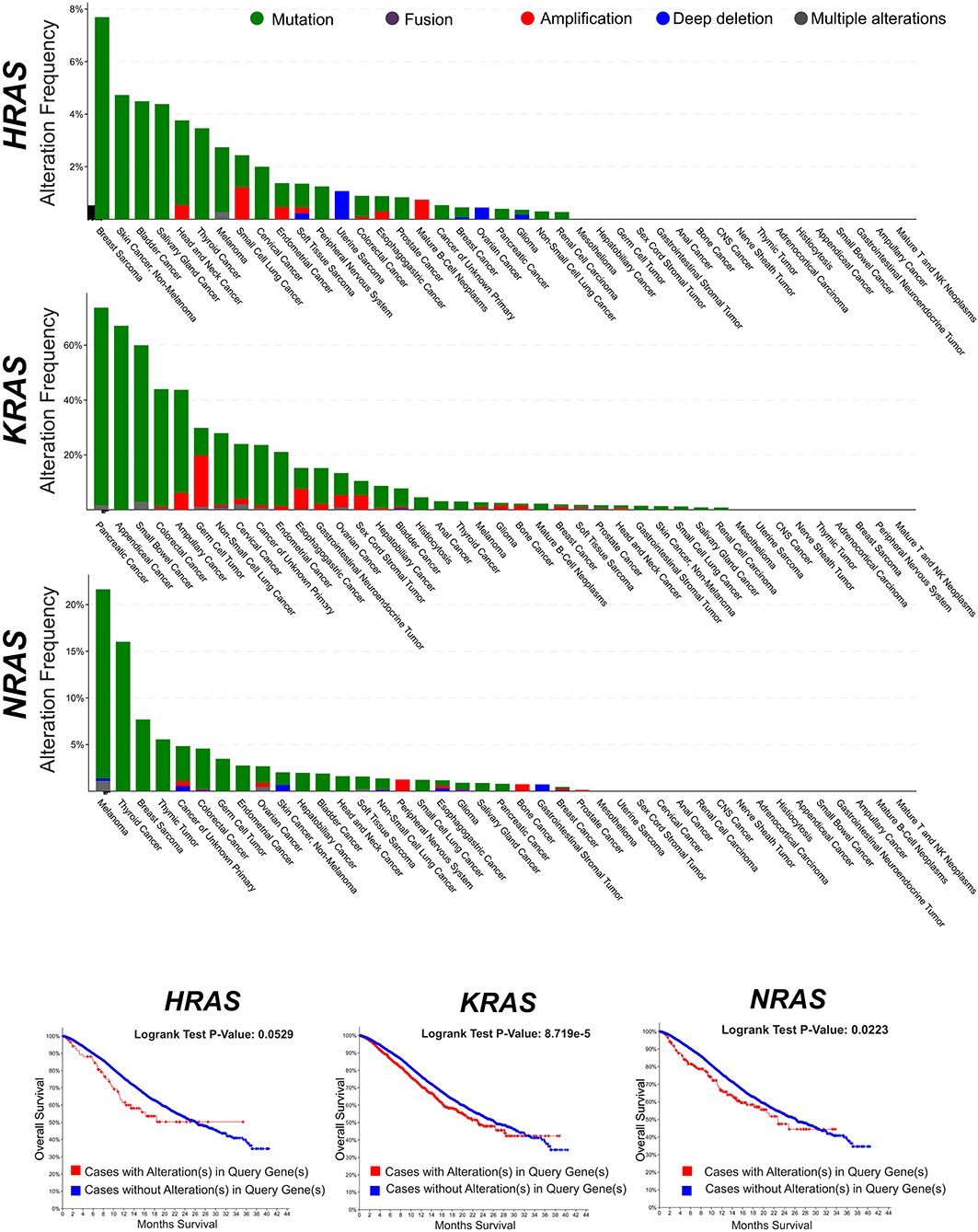
Figure 1. Frequency of genomic alterations (mutation, fusion, amplification, deep deletion, multiple alterations) of the RAS genes (HRAS, KRAS, NRAS) across different tumor types of the MSK-IMPACT Clinical Sequencing Cohort (8). Data have been accessed through cBioportal for Cancer Genomics website (https://www.cbioportal.org).
Mammary cell lines have served as tumor models for many seminal studies which demonstrated the tumorigenic potential of RAS oncogenes. These studies have shown that oncogenic RAS mutations constitutively enhance mammary cell interaction with basement membrane, alter the tridimensional growth in collagen gel, induce anchorage-independent phenotype, invasiveness, tumorigenic potential, secretion of TGF-β and IGF-1, activation of EGFR, mitogen-activated protein kinase (MAPK), and estrogen-insensitivity (9–18). Single copies of mutant KRAS cooperate with mutant PIK3CA to induce tumor transformation in immortalized human epithelial cells (19). Conditional expression of Ki-RasG12V in the mammary cells induces estrogen receptor alpha (ERα)-positive adenocarcinoma in mice (20), while HRAS Q61 drives breast adenomyoepitheliomas (21).
Several pathways and downstream effectors have been identified which mediate the tumorigenic phenotype induced by oncogenic RAS mutations in mammary cells. Activated NRAS oncogene and its homolog NRAS proto-oncogene act through the same pathway for in vivo tumorigenesis (22). Oncogenic RAS mutations support cancer progression and metastatic dissemination through the modulation of the ΔNp63, a amino-terminal truncated isoform of p63, a member of the p53 family of transcription factors (23, 24). Oncogenic RAS mutations promotes TFG-β-induced epithelial-mesenchymal transition through the activation of leukotriene B4 receptor-2-linked cascade (25). Mutated RAS associates with the induction of cyclooxygenase-2 (COX-2) expression in human breast cancer cell lines (26). Activated HRAS induces the invasive phenotype in breast epithelial cell lines through the recruitment of p38 (27, 28). Invasion of breast carcinoma cells also relies on activated Ras-mediated stimulation of E2F and a consequent E2F-mediated modulation of integrin α6β4 (29). Oncogenic RAS mutation regulates the activity of CXCL10 and its receptor splice variant CXCR-B (30). Id1 and activated RAS cooperate to subvert the cellular senescence response and to induce metastatic dissemination in mammary carcinoma (31). Focal adhesion kinase signaling is required for activated RAS and PI3K-dependent breast tumorigenesis in mice and humans (32). Dominant negative Ras activates the Raf-Mek-Erk signal transduction pathway and induces lactogenic hormone-induced differentiation (33). Activated RAS signals centrosome amplification through cyclin D1/Cdk4 and Nek2 (34). Autophagy is critically implicated in malignant transformation by oncogenic KRAS mutations and is promoted by the reactive-oxygen species-mediated JNK activation through up-regulation of ATG5 and ATG7 (35). RAS oncogenesis is accelerated by p21WAF1/CIP1 depletion in mammary cancer (36), while p21CIP attenuates RAS- and c-MYC-dependent epithelial-to-mesenchymal transition and cancer stem cell-like transcriptional profile in vivo (37). Gadd45a induces apoptosis and senescence in Ras-driven mammary cancers through activation of c-jun NH2-terminal kinase and p38 stress signaling (38). HMGA1a regulates genes involved in the RAS/ERK mitogenic signaling pathway, including KIT ligand and caveolin 1 and 2 (39). Oncogenic RAS mutations induce metabolic rearrangement in breast cancer as part of their tumorigenic program. Activated c-ha-Ras induces loss of fatty-acid delta desaturating ability in human mammary epithelial cells (40). Moderate restriction of energy intake hampers v-Ha-ras-induced mammary tumorigenesis (41). PI3K and KRAS cooperate to stimulate de novo lipid synthesis through mTORC1 and SREBP (42).
After the first identification of the tumorigenic potential of oncogenic RAS mutations in vitro, a great effort has been made in search for RAS mutations in human cancers, and their role in driving tumorigenesis (43). The most remarkable finding was the discovery of the stricking incidence of oncogenic KRAS mutations in colon (44, 45), lung (46), and pancreatic carcinomas (47) (Figure 1). According to what found in other tumor types, KRAS confirms to be the most frequently mutated RAS isoform in breast cancer (Figure 2A) and its mutation, unlike mutations of HRAS and NRAS, is strongly associated with the poor clinical outcome (Figure 2B). Nevertheless, the frequency of RAS mutations in human breast cancer proven to be much lower than expected (49) (Figures 1, 2). This stands against a critical role of RAS oncogenic activation as the primary driver of the breast cancer initiation and progression in humans and has discouraged for many years the effort to investigate RAS proteins as potential targets for breast cancer therapies (Figure 2A). Also, human RAS oncogene, unlike their retroviral counterpart, cannot transform primary cells without the cooperation by a second oncogene such as MYC or adenovirus E1A.
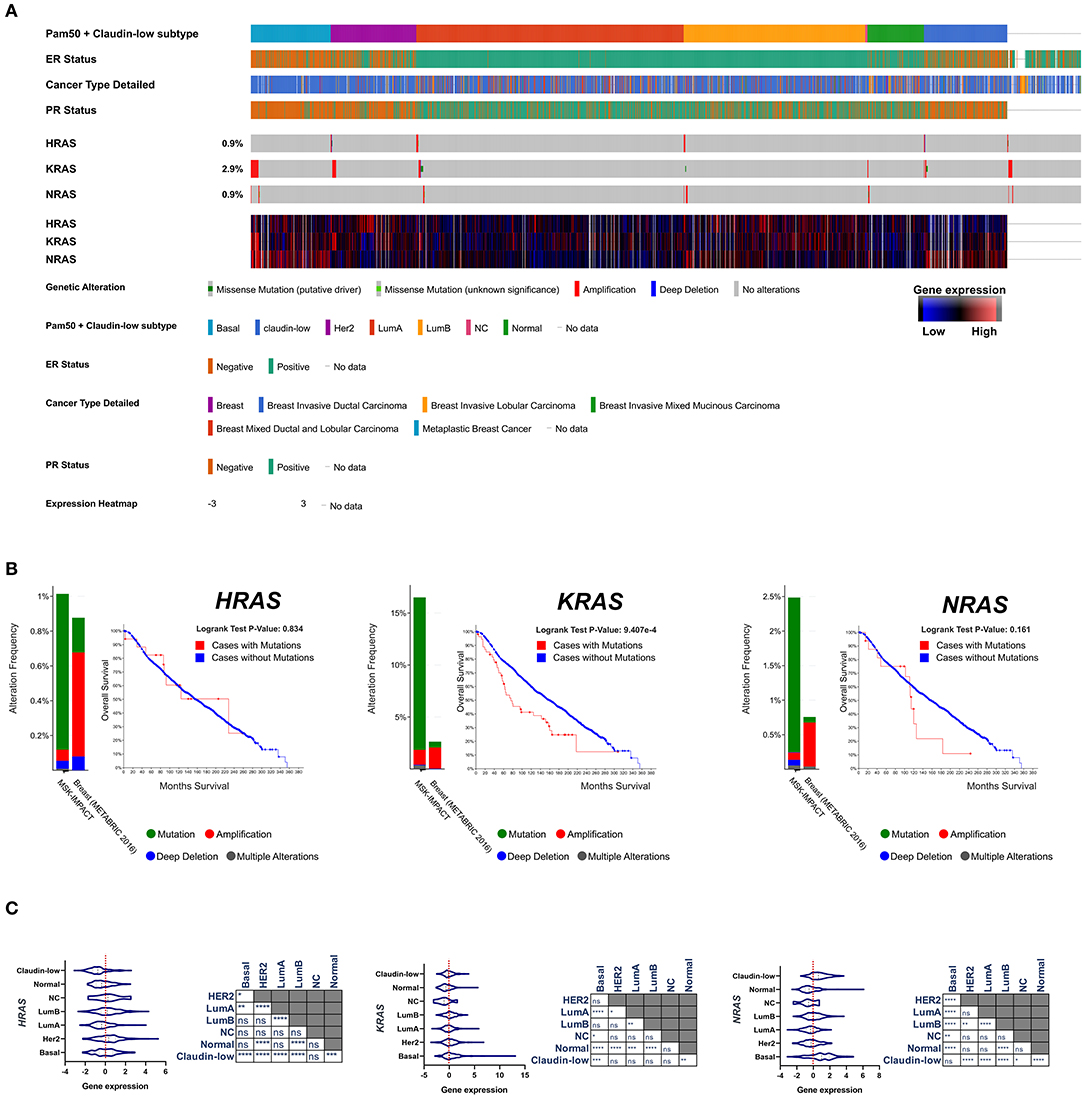
Figure 2. (A) Frequency of genomic alterations and heatmap of gene expression of the RAS genes (HRAS, KRAS, NRAS) across 2,509 breast tumor samples of the METABRIC cohort (48), assigned to the major intrinsic subtypes (on the basis of the PAM50 profile). (B) The fraction of breast tumors (cases) with genomic alterations (METABRIC cohort, 2,509 breast tumors) of the RAS genes are reported in comparison with the fraction of tumor with genomic alteration in a pan-tumor cohort (MSK-IMPACT, 10945). Kaplan-Mayer curve are reported for each RAS isoform comparing the overall survival of the breast tumors with or without genomic alterations (METABRIC cohort, 2,509 breast tumors). (C) Gene expression level of RAS isoforms across the different subtypes of breast cancer are compared and statistically evaluated by ANOVA t-test (Turkey post-hoc) *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Data have been accessed through cBioportal for Cancer Genomics website (https://www.cbioportal.org).
However, the oncogenic function of RAS proteins does not rely completely on gene mutations. RAS protein overexpression, hyperactivation of upstream RAS activators, such as receptor tyrosine kinases, perturbation in the activity of RAS regulators, such as GEFs and GAPS, all may contribute to promote and sustain tumorigenicity (50, 51).
There are a wealth of evidence that stratified over the last 3 decades which have established a role of RAS as supporting actor in breast cancer downstream the dysregulated action of oncogenic pathways and effectors. RAS proteins serve as hub of the major intracellular signaling pathways which govern cell growth, motility, angiogenesis, immune escape. Hence, it is quite clear that the engagement of RAS function is mandatory for many oncogenic factors to be able to propagate their signals and execute their aberrant programs, while its inhibition may dampen upstream tumorigenic signals. Studies in the early 1990s reported that in 71% of human breast cancers the expression of RAS proteins was higher than in normal breast tissues and correlated with that of p185/HER-2. Interestingly, NRAS and HRAS result to be overexpressed in basal-like and HER2 tumors, the most aggressive subtypes of breast cancer (52, 53) (Figure 2A). HER2, as well as its cognate epidermal growth factor receptor (EGFR), is coupled to the Ras signaling by interaction with the adaptor protein Grb2, and Sos, a Ras GDP-GTP exchange factor. The overexpression of these receptors in breast cancer cells amplifies the RAS signaling pathway (54). Consistently, the tyrosine kinase inhibitors have been shown to hamper breast cancer cell proliferation at least in part by the inhibition of signal transduction processes potentially mediated through RAS (55). RAS overexpression associates with p53 loss, HER2 amplification/overexpression and aneuploidy in infiltrating ductal carcinomas (56). RAS is required also for the mammary tumorigenesis induced by the oncogene MYC, although in an inducible mouse model of c-MYC-driven mammary tumorigenesis the spontaneous occurrence of secondary RAS mutations was necessary to prevent the full regression of tumors upon c-MYC deinduction (57). Pin1, a prolyl isomerase which regulates the conformation of a subset of phosphorylated Ser/Thr-Pro motifs, is overexpressed in human tumors and interacts with Ras signaling in increasing c-Jun transcriptional activity toward cyclin D1 (58). Breast cancer displays the downregulation of the RAS/MAPK inhibitor proteins sprouty 1 and 2 (59). RAS functions downstream Rab-coupling protein RCP (also known as RAB11FIP1), a breast cancer-related oncogene (60). Bone Morphogenetic Protein 1 (BMP1) cooperates with HRAS to induce metastatic breast cancer (61). RAS signaling amplification has been reported to play a crucial role in metastatic progression and poor clinical outcome of luminal breast cancer patients (62). MicroRNA-382-5p accelerates breast cancer progression by modulating the RERG/RAS/ERK signaling axis (63). Pharmacological inhibition of SHP2 phosphatase has been recently shown to reduce the proliferation rate of receptor-tyrosine-kinase-driven human cancer cells in vitro and in vivo through the inhibition of the RAS-MAPK signaling (64). BCL-XL directly modulates RAS signaling to favor cancer cell stem-like phenotype (65).
RAS hyperfunction with tumorigenic effects can be induced by the altered activity of RAS-specific regulators. R-RasGTPase activating protein mediates the interaction between estrogen and insulin signaling pathways in breast cancer cells (66) and affects the motile phenotype of breast epithelial cells through the modulation of Rho/Rho-kinase (67). On the other hand, RAS-GTPase inhibition promotes apoptosis in tumor cells (68). The RasGAP gene, RASAL2, functions as a tumor and metastasis suppressor in human luminal breast cancer (69) but promote triple-negative breast cancer progression through RAC1 activation (70). The Rho GTPase Rnd1 dampens mammary tumor progression and EMT by restraining RAS-MAPK signaling (71, 72). R-Ras2, a transforming GTPase that shares downstream effector with Ras proteins, promotes tumor progression in a PI3K-dependent and signaling autonomous manner although its prometastatic role requires other priming oncogenic signals and downstream effectors (73). Transposon insertion in one of two RASGAP genes, neurofibromin1 (Nf1) and RAS p21 protein activator (Rasa1), might function as the causal role of the mammary tumor development in a tumor mouse model generated by the activation of a mutagenic T2Onc2 transposon via expression of a transposase driven by the keratin K5 promoter in a p53+/− background (74).
Triple negative breast cancer (TNBC) is a heterogeneous group of tumors defined on the basis of their negativity for Estrogen receptor, Progesterone Receptor and HER2. They account for ≈15% of breast tumors and are statistically associated to poor prognosis. TNBC phenotype and clinical outcome partially overlap those of the basal-like breast cancer subtype previously identified on the basis of the gene expression profiling (52, 53), although the identification between these two categories of breast tumors is controversial (75). RAS activity and its regulators have been reported to play a role in the progression of TNBC/basal-like tumors. A 3′-untranslated region of KRAS variant has been identified which regulates the development of TNBC (76). KRAS(G12D) provides human mammary basal cells and luminal progenitors with the ability to produce serially transplantable, polyclonal, invasive ductal carcinomas into immunodeficient mice, which display a dramatic clonal diversification (77). miR-143/145 loss-of-function amplifies the tumorigenic potential of PTEN-deficient basal-like breast tumor cells at least partially through the induction of RAS signaling. In humans, miR-145 deficiency correlates with enhanced RAS-pathway activity in basal-like breast cancer, and patient with combined PTEN/miR-145 loss or PTEN-loss/high RAS-pathway activity exhibit poor clinical outcome (78). Also, wild-type NRAS, upregulated in basal-like breast cancer (Figure 2A), promotes tumorigenesis through IL-8 secretion via JAK2 activation (79). RAS-MAPK pathway activation promotes immune-evasion in triple negative breast cancer (80). High level of ERK1/2 phosphorylation, a readout of Ras signaling activation, has been found in metastatic sites relative to primary breast tumors and is more common in TNBC/basal-like cancers (81). Transcriptional signature of RAS/MAPK pathway activation is highly prevalent in TNBC/basal-like cancers compared to other subtypes of breast cancer (82, 83).
Other than being a potent mediator of tumor transformation and progression, RAS might also confer resistance to therapies in breast cancer (84). RAS induces resistance to Cis-platinum by increasing GST-pi expression (85) and ERCC1 (86). Oncogenic RAS mutations cause resistance to the growth inhibitor insulin-like growth factor binding protein-3 (IGFBP-3) (87). Also, RAS induces resistance to lapatinib which might be overcome by MEK inhibition (88). RAS/Raf-1/MAPK pathway affects response to tamoxifen but not chemotherapy in breast cancer patients (89). Raf-1 functions as an effector of RAS in the radiation-response (90).
The role of RAS in breast tumorigenesis and resistance to therapies provides the rationale to assess RAS as target in breast cancer treatment. Three decades of studies contributed to rise the notion that RAS oncogenes are “undruggable,” due to its conformational architecture, which lacks of pockets to facilitate the binding of small inhibitors, and its picomolar affinity for the nucleotide substrate. However, recent technologies and approaches have renewed the challenge to thwart cancer by targeting RAS directly or through its downstream signaling pathways. Direct approaches currently under investigation are addressed to enhance GTP hydrolytic activity of RAS, to inhibit its nucleotide exchange function or to prevent its interaction with downstream effectors (91, 92). These approaches are providing encouraging results at preclinical stages, but none of them have entered clinical practice thus far.
A reliable alternative approach consists in blocking the RAS downstream pathways (93). As for breast cancer, it holds great promise the therapeutic use of inhibitors of the Ras/MAPK pathway. FDA-approved Inhibitors of MEK, a central node in the Ras/MAPK pathway, specifically inhibit proliferation of TNBC/Basal-like cancer cell lines (83) and may complement chemotherapeutic treatments in xenograft models (82). MEK inhibition has been shown to prevent epithelial-mesenchymal transition and metastatic potential of tumor cells by targeting cancer stem cell compartment (94). Although the phase I studies have shown a scarce efficacy of MEK in humans, the combination with neoadjuvant or post-operative treatments might represents a promising alternative (95, 96).
Decades of studies have contributed to unveil the primary role of RAS oncogenes in leading tumor initiation in many types of cancers. For reasons that are still unknown, breast cancer is not amongst them. Although oncogenic RAS is able to transform mammary cancer cell lines in vitro, the marginal incidence of RAS mutations in clinics does not support a primary role of RAS proteins in breast tumor etiology. Nevertheless, a wealth of studies over many years have demonstrated the importance of RAS function in the progression, metastatic dissemination and therapy resistance in breast cancers, regardless the molecular trigger they are initiated by, thus contributing to draw for RAS proteins a crucial role as supporting actors in breast tumorigenesis.
MG: conceptualization, writing, and financial support.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Parada LF, Tabin CJ, Shih C, Weinberg RA Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. (1982) 297:474–8. doi: 10.1038/297474a0
2. Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. (1982) 298:343–7. doi: 10.1038/298343a0
3. Shih C, Shilo BZ, Goldfarb MP, Dannenberg A, Weinberg RA. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc Natl Acad Sci USA. (1979) 76:5714–8. doi: 10.1073/pnas.76.11.5714
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature. (1982) 300:149–52. doi: 10.1038/300149a0
6. Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, et al. Mechanism of activation of a human oncogene. Nature. (1982) 300:143–9. doi: 10.1038/300143a0
7. Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature. (1982) 300:762–5. doi: 10.1038/300762a0
8. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703–13. doi: 10.1038/nm.4333
9. Albini A, Graf J, Kitten GT, Kleinman HK, Martin GR, Veillette A, et al. 17 beta-estradiol regulates and v-Ha-ras transfection constitutively enhances MCF7 breast cancer cell interactions with basement membrane. Proc Natl Acad Sci USA. (1986) 83:8182–6. doi: 10.1073/pnas.83.21.8182
10. Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog. (1991) 4:25–35. doi: 10.1002/mc.2940040106
11. Choi C, Helfman DM. The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene. (2014) 33:3668–76. doi: 10.1038/onc.2013.341
12. Dickson RB, Kasid A, Huff KK, Bates SE, Knabbe C, Bronzert D, et al. Activation of growth factor secretion in tumorigenic states of breast cancer induced by 17 beta-estradiol or v-Ha-ras oncogene. Proc Natl Acad Sci USA. (1987) 84:837–41. doi: 10.1073/pnas.84.3.837
13. Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. (2001) 15:50–65. doi: 10.1101/gad.828901
14. Kasid A, Knabbe C, Lippman ME. Effect of v-rasH oncogene transfection on estrogen-independent tumorigenicity of estrogen-dependent human breast cancer cells. Cancer Res. (1987) 47:5733–8.
15. Keely PJ. Ras and Rho protein induction of motility and invasion in T47D breast adenocarcinoma cells. Methods Enzymol. (2001) 333:256–66. doi: 10.1016/S0076-6879(01)33061-6
16. Liu E, Dollbaum C, Scott G, Rochlitz C, Benz C, Smith HS. Molecular lesions involved in the progression of a human breast cancer. Oncogene. (1988) 3:323–7.
17. Martínez-Lacaci I, Kannan S, De Santis M, Bianco C, Kim N, Wallace-Jones B, et al. RAS transformation causes sustained activation of epidermal growth factor receptor and elevation of mitogen-activated protein kinase in human mammary epithelial cells. Int J Cancer. (2000) 88:44–52. doi: 10.1002/1097-0215(20001001)88:1<44::AID-IJC7>3.0.CO;2-8
18. Worland PJ, Bronzert D, Dickson RB, Lippman ME, Hampton L, Thorgeirsson SS, et al. Secreted and cellular polypeptide patterns of MCF-7 human breast cancer cells following either estrogen stimulation or v-H-ras transfection. Cancer Res. (1989) 49:51–7.
19. Wang GM, Wong HY, Konishi H, Blair BG, Abukhdeir AM, Gustin JP, et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res. (2013) 73:3248–61. doi: 10.1158/0008-5472.CAN-12-1578
20. Andò S, Malivindi R, Catalano S, Rizza P, Barone I, Panza S, et al. Conditional expression of Ki-Ras. Oncogene. (2017) 36:6420–31. doi: 10.1038/onc.2017.252
21. Geyer FC, Li A, Papanastasiou AD, Smith A, Selenica P, Burke KA, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun. 9:1816. doi: 10.1038/s41467-018-04128-5
22. Mangues R, Symmans WF, Lu S, Schwartz S, Pellicer A. Activated N-ras oncogene and N-ras proto-oncogene act through the same pathway for in vivo tumorigenesis. Oncogene. (1996) 13:1053–63.
23. Hu L, Liang S, Chen H, Lv T, Wu J, Chen D, et al. ΔNp63α is a common inhibitory target in oncogenic PI3K/Ras/Her2-induced cell motility and tumor metastasis. Proc Natl Acad Sci USA. (2017) 114:E3964–73. doi: 10.1073/pnas.1617816114
24. Vasilaki E, Morikawa M, Koinuma D, Mizutani A, Hirano Y, Ehata S, et al. Ras and TGF-β signaling enhance cancer progression by promoting the ΔNp63 transcriptional program. Sci Signal. (2016) 9:ra84. doi: 10.1126/scisignal.aag3232
25. Kim H, Choi JA, Kim JH. Ras promotes transforming growth factor-β (TGF-β)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem. (2014) 289:22151–60. doi: 10.1074/jbc.M114.556126
26. Gilhooly EM, Rose DP. The association between a mutated ras gene and cyclooxygenase-2 expression in human breast cancer cell lines. Int J Oncol. (1999) 15:267–70. doi: 10.3892/ijo.15.2.267
27. Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. (2003) 63:5454–61.
28. Shin I, Kim S, Song H, Kim HR, Moon A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: its critical role in invasion and migration of breast epithelial cells. J Biol Chem. (2005) 280:14675–83. doi: 10.1074/jbc.M411625200
29. Yoon SO, Shin S, Mercurio AM. Ras stimulation of E2F activity and a consequent E2F regulation of integrin alpha6beta4 promote the invasion of breast carcinoma cells. Cancer Res. (2006) 66:6288–95. doi: 10.1158/0008-5472.CAN-06-0826
30. Datta D, Flaxenburg JA, Laxmanan S, Geehan C, Grimm M, Waaga-Gasser AM, et al. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: relevance for the development of human breast cancer. Cancer Res. (2006) 66:9509–18. doi: 10.1158/0008-5472.CAN-05-4345
31. Swarbrick A, Roy E, Allen T, Bishop JM. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc Natl Acad Sci USA. (2008) 105:5402–7. doi: 10.1073/pnas.0801505105
32. Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. (2009) 119:252–66. doi: 10.1172/JCI37160
33. Cerrito MG, Galbaugh T, Wang W, Chopp T, Salomon D, Cutler ML. Dominant negative Ras enhances lactogenic hormone-induced differentiation by blocking activation of the Raf-Mek-Erk signal transduction pathway. J Cell Physiol. (2004) 201:244–58. doi: 10.1002/jcp.20077
34. Zeng X, Shaikh FY, Harrison MK, Adon AM, Trimboli AJ, Carroll KA, et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. (2010) 29:5103–12. doi: 10.1038/onc.2010.253
35. Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. (2011) 286:12924–32. doi: 10.1074/jbc.M110.138958
36. Adnane J, Jackson RJ, Nicosia SV, Cantor AB, Pledger WJ, Sebti SM. Loss of p21WAF1/CIP1 accelerates Ras oncogenesis in a transgenic/knockout mammary cancer model. Oncogene. (2000) 19:5338–47. doi: 10.1038/sj.onc.1203956
37. Liu M, Casimiro MC, Wang C, Shirley LA, Jiao X, Katiyar S, et al. p21CIP1 attenuates Ras- and c-Myc-dependent breast tumor epithelial mesenchymal transition and cancer stem cell-like gene expression in vivo. Proc Natl Acad Sci USA. (2009) 106:19035–9. doi: 10.1073/pnas.0910009106
38. Tront JS, Hoffman B, Liebermann DA. Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res. (2006) 66:8448–54. doi: 10.1158/0008-5472.CAN-06-2013
39. Treff NR, Pouchnik D, Dement GA, Britt RL, Reeves R. High-mobility group A1a protein regulates Ras/ERK signaling in MCF-7 human breast cancer cells. Oncogene. (2004) 23:777–85. doi: 10.1038/sj.onc.1207167
40. Grammatikos S, Harvey M, Subbaiah P, Victor T, Miller W. Loss of fatty-acid delta(6) desaturating ability in human mammary epithelial-cells that express an activated C-ha-ras oncogene. Int J Oncol. (1995) 6:1039–46. doi: 10.3892/ijo.6.5.1039
41. Fernandes G, Chandrasekar B, Troyer DA, Venkatraman JT, Good RA. Dietary lipids and calorie restriction affect mammary tumor incidence and gene expression in mouse mammary tumor virus/v-Ha-ras transgenic mice. Proc Natl Acad Sci USA. (1995) 92:6494–8. doi: 10.1073/pnas.92.14.6494
42. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. (2016) 35:1250–60. doi: 10.1038/onc.2015.179
44. Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. (1987) 327:293–7. doi: 10.1038/327293a0
45. Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. (1987) 327:298–303. doi: 10.1038/327298a0
46. Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med. (1987) 317:929–35. doi: 10.1056/NEJM198710083171504
47. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. (1988) 53:549–54. doi: 10.1016/0092-8674(88)90571-5
48. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 7:11479. doi: 10.1038/ncomms11479
49. Rochlitz CF, Scott GK, Dodson JM, Liu E, Dollbaum C, Smith HS, et al. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. (1989) 49:357–60.
50. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. (1983) 304:596–602. doi: 10.1038/304596a0
51. Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. (1983) 304:648–51. doi: 10.1038/304648a0
52. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. (2000) 406:747–52. doi: 10.1038/35021093
53. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
54. Janes PW, Daly RJ, deFazio A, Sutherland RL. Activation of the Ras signalling pathway in human breast cancer cells overexpressing erbB-2. Oncogene. (1994) 9:3601–8.
55. Clark JW, Santos-Moore A, Stevenson LE, Frackelton AR. Effects of tyrosine kinase inhibitors on the proliferation of human breast cancer cell lines and proteins important in the ras signaling pathway. Int J Cancer. (1996) 65:186–91. doi: 10.1002/(SICI)1097-0215(19960117)65:2<186::AID-IJC10>3.3.CO;2-F
56. Smith CA, Pollice AA, Gu LP, Brown KA, Singh SG, Janocko LE, et al. Correlations among p53, Her-2/neu, and ras overexpression and aneuploidy by multiparameter flow cytometry in human breast cancer: evidence for a common phenotypic evolutionary pattern in infiltrating ductal carcinomas. Clin Cancer Res. (2000) 6:112–26.
57. D'Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. (2001) 7:235–9. doi: 10.1038/84691
58. Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. (2001) 20:3459–72. doi: 10.1093/emboj/20.13.3459
59. Lo TL, Yusoff P, Fong CW, Guo K, McCaw BJ, Phillips WA, et al. The ras/mitogen-activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res. (2004) 64:6127–36. doi: 10.1158/0008-5472.CAN-04-1207
60. Zhang J, Liu X, Datta A, Govindarajan K, Tam WL, Han J, et al. RCP is a human breast cancer-promoting gene with Ras-activating function. J Clin Invest. (2009) 119:2171–83. doi: 10.1172/JCI37622
61. Hoenerhoff MJ, Chu I, Barkan D, Liu ZY, Datta S, Dimri GP, et al. BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene. (2009) 28:3022–32. doi: 10.1038/onc.2009.165
62. Wright KL, Adams JR, Liu JC, Loch AJ, Wong RG, Jo CE, et al. Ras signaling is a key determinant for metastatic dissemination and poor survival of luminal breast cancer patients. Cancer Res. (2015) 75:4960–72. doi: 10.1158/0008-5472.CAN-14-2992
63. Ho JY, Hsu RJ, Liu JM, Chen SC, Liao GS, Gao HW, et al. MicroRNA-382-5p aggravates breast cancer progression by regulating the RERG/Ras/ERK signaling axis. Oncotarget. (2017) 8:22443–59. doi: 10.18632/oncotarget.12338
64. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. (2016) 535:148–52. doi: 10.1038/nature18621
65. Carné Trécesson S, Souazé F, Basseville A, Bernard AC, Pécot J, Lopez J, et al. BCL-X. Nat Commun. (2017) 8:1123. doi: 10.1038/s41467-017-01079-1
66. Yu Y, Hao Y, Feig LA. The R-Ras GTPase mediates cross talk between estrogen and insulin signaling in breast cancer cells. Mol Cell Biol. (2006) 26:6372–80. doi: 10.1128/MCB.00509-05
67. Jeong HW, Nam JO, Kim IS. The COOH-terminal end of R-Ras alters the motility and morphology of breast epithelial cells through Rho/Rho-kinase. Cancer Res. (2005) 65:507–15.
68. Leblanc V, Delumeau I, Tocqué B. Ras-GTPase activating protein inhibition specifically induces apoptosis of tumour cells. Oncogene. (1999) 18:4884–9. doi: 10.1038/sj.onc.1202855
69. McLaughlin SK, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell. (2013) 24:365–78. doi: 10.1016/j.ccr.2013.08.004
70. Feng M, Bao Y, Li Z, Li J, Gong M, Lam S, et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J Clin Invest. (2014) 124:5291–304. doi: 10.1172/JCI76711
71. Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, et al. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. (2015) 17:81–94. doi: 10.1038/ncb3082
72. Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, et al. Author correction: the Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. (2019) 21:534. doi: 10.1038/s41556-019-0288-3
73. Larive RM, Moriggi G, Menacho-Márquez M, Cañamero M, de Álava E, Alarcón B, et al. Contribution of the R-Ras2 GTP-binding protein to primary breast tumorigenesis and late-stage metastatic disease. Nat Commun. 5:3881. doi: 10.1038/ncomms4881
74. Suárez-Cabrera C, Quintana RM, Bravo A, Casanova ML, Page A, Alameda JP, et al. A Transposon-based Analysis Reveals. Cancer Res. (2017) 77:1357–68. doi: 10.1158/0008-5472.CAN-16-1586
75. Lerma E, Barnadas A, Prat J. Triple negative breast carcinomas: similarities and differences with basal like carcinomas. Appl Immunohistochem Mol Morphol. (2009) 17:483–94. doi: 10.1097/PAI.0b013e3181a725eb
76. Paranjape T, Heneghan H, Lindner R, Keane FK, Hoffman A, Hollestelle A, et al. A 3'-untranslated region KRAS variant and triple-negative breast cancer: a case-control and genetic analysis. Lancet Oncol. (2011) 12:377–86. doi: 10.1016/S1470-2045(11)70044-4
77. Nguyen LV, Pellacani D, Lefort S, Kannan N, Osako T, Makarem M, et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature. (2015) 528:267–71. doi: 10.1038/nature15742
78. Wang S, Liu JC, Ju Y, Pellecchia G, Voisin V, Wang DY, et al. microRNA-143/145 loss induces Ras signaling to promote aggressive Pten-deficient basal-like breast cancer. JCI Insight. (2017) 2:93313. doi: 10.1172/jci.insight.93313
79. Zheng ZY, Tian L, Bu W, Fan C, Gao X, Wang H, et al. Wild-type N-Ras, overexpressed in basal-like breast cancer, promotes tumor formation by inducing IL-8 secretion via JAK2 activation. Cell Rep. (2015) 12:511–24. doi: 10.1016/j.celrep.2015.06.044
80. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. (2016) 22:1499–509. doi: 10.1158/1078-0432.CCR-15-1125
81. Adeyinka A, Nui Y, Cherlet T, Snell L, Watson PH, Murphy LC. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin Cancer Res. (2002) 8:1747–53.
82. Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. (2012) 18:1052–9. doi: 10.1038/nm.2795
83. Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. (2009) 15:4649–64. doi: 10.1158/1078-0432.CCR-09-0317
84. Jin W, Wu L, Liang K, Liu B, Lu Y, Fan Z. Roles of the PI-3K and MEK pathways in Ras-mediated chemoresistance in breast cancer cells. Br J Cancer. (2003) 89:185–91. doi: 10.1038/sj.bjc.6601048
85. Di Simone D, Galimberti S, Basolo F, Ciardiello F, Petrini M, Scheper RJ. c-Ha-ras transfection and expression of MDR-related genes in MCF-10A human breast cell line. Anticancer Res. (1997) 17:3587–3592.
86. Youn CK, Kim MH, Cho HJ, Kim HB, Chang IY, Chung MH, et al. Oncogenic H-Ras up-regulates expression of ERCC1 to protect cells from platinum-based anticancer agents. Cancer Res. (2004) 64:4849–57. doi: 10.1158/0008-5472.CAN-04-0348
87. Martin JL, Baxter RC. Oncogenic ras causes resistance to the growth inhibitor insulin-like growth factor binding protein-3 (IGFBP-3) in breast cancer cells. J Biol Chem. (1999) 274:16407–11. doi: 10.1074/jbc.274.23.16407
88. Zoppoli G, Moran E, Soncini D, Cea M, Garuti A, Rocco I, et al. Ras-induced resistance to lapatinib is overcome by MEK inhibition. Curr Cancer Drug Targets. (2010) 10:168–75. doi: 10.2174/156800910791054211
89. McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, et al. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. (2009) 15:1487–95. doi: 10.1158/1078-0432.CCR-07-4967
90. Suy S, Anderson WB, Dent P, Chang E, Kasid U. Association of Grb2 with Sos and Ras with Raf-1 upon gamma irradiation of breast cancer cells. Oncogene. (1997) 15:53–61. doi: 10.1038/sj.onc.1201165
91. O'Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res. (2019) 139:503–11. doi: 10.1016/j.phrs.2018.10.021
92. Spencer-Smith R, O'Bryan JP. Direct inhibition of RAS: quest for the holy grail? Semin Cancer Biol. (2019) 54:138–48. doi: 10.1016/j.semcancer.2017.12.005
93. Papke B, Der CJ. Drugging RAS: know the enemy. Science. (2017) 355:1158–63. doi: 10.1126/science.aam7622
94. Balko JM, Schwarz LJ, Bhola NE, Kurupi R, Owens P, Miller TW, et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. (2013) 73:6346–58. doi: 10.1158/0008-5472.CAN-13-1385
95. Jing J, Greshock J, Holbrook JD, Gilmartin A, Zhang X, McNeil E, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. (2012) 11:720–9. doi: 10.1158/1535-7163.MCT-11-0505
Keywords: RAS, breast cancer, oncogene, mutations, signal transduction
Citation: Galiè M (2019) RAS as Supporting Actor in Breast Cancer. Front. Oncol. 9:1199. doi: 10.3389/fonc.2019.01199
Received: 17 August 2019; Accepted: 22 October 2019;
Published: 12 November 2019.
Edited by:
Alessandro Rimessi, University of Ferrara, ItalyReviewed by:
Kenneth Beaman, Rosalind Franklin University of Medicine and Science, United StatesCopyright © 2019 Galiè. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirco Galiè, bWlyY28uZ2FsaWVAdW5pdnIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.