 Massimiliano Bonifacio
Massimiliano Bonifacio Fabio Stagno
Fabio Stagno Luigi Scaffidi
Luigi Scaffidi Mauro Krampera
Mauro Krampera Francesco Di Raimondo
Francesco Di Raimondo- 1Department of Medicine, Section of Hematology, University of Verona, Verona, Italy
- 2Division of Hematology With BMT, AOU Policlinico “Vittorio Emanuele”, University of Catania, Catania, Italy
Management of chronic myeloid leukemia (CML) in advanced phases remains a challenge also in the era of tyrosine kinase inhibitors (TKIs) treatment. Cytogenetic clonal evolution and development of resistant mutations represent crucial events that limit the benefit of subsequent therapies in these patients. CML is diagnosed in accelerated (AP) or blast phase (BP) in <5% of patients, and the availability of effective treatments for chronic phase (CP) has dramatically reduced progressions on therapy. Due to smaller number of patients, few randomized studies are available in this setting and evidences are limited. Nevertheless, three main scenarios may be drawn: (a) patients diagnosed in AP are at higher risk of failure as compared to CP patients, but if they achieve optimal responses with frontline TKI treatment their outcome may be similarly favorable; (b) patients diagnosed in BP may be treated with TKI alone or with TKI together with conventional chemotherapy regimens, and subsequent transplant decisions should rely on kinetics of response and individual transplant risk; (c) patients in CP progressing under TKI treatment represent the most challenging population and they should be treated with alternative TKI according to the mutational profile, optional chemotherapy in BP patients, and transplant should be considered in suitable cases after return to second CP. Due to lack of validated and reliable markers to predict blast crisis and the still unsatisfactory results of treatments in this setting, prevention of progression by careful selection of frontline treatment in CP and early treatment intensification in non-optimal responders remains the main goal. Personalized evaluation of response kinetics could help in identifying patients at risk for progression.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder characterized by the neoplastic transformation of the hematopoietic stem cell and the pathognomonic presence of the Philadelphia (Ph) chromosome arising from a reciprocal translocation between chromosomes 9 and 22. This balanced chromosomal alteration causes the fusion of the Abelson oncogene (ABL) from chromosome 9q34 with the breakpoint cluster region (BCR) on chromosome 22q11.2, t(9;22)(q34;q11.2) and induces the formation of a distinct chimeric BCR-ABL1 fusion gene, which in turn translates into a Bcr-Abl oncoprotein. This oncoprotein most frequently has a molecular weight of 210 kD (p. 210) and displays increased tyrosine kinase activity which causes growth factor independence and leukemic cell growth in hematopoietic cell lines, contributes also to the clonal evolution of the disease and leads to its evolution toward acute leukemia (1). CML usually presents in chronic phase (CP), characterized by the clonal expansion of mature myeloid cells. Indeed, all untreated patients will eventually progress to a lethal blast phase (BP) that is sometimes preceded by an accelerated phase (AP). The development of Tyrosine Kinase Inhibitors (TKIs) in the last 20 years has represented an outstanding revolution in the management and outcome of CML, and a paradigm for targeted therapy of cancer (2). Although life expectancy for patients diagnosed with CP-CML nowadays is similar to that of general healthy population (3), the onset of disease in advanced phase, or progression from CP to AP or BP following TKI failure still represent a complex challenge. In fact, advanced phases are typically resistant to treatment and have a worse prognosis, with death occurring from infection and bleeding complications similar to acute leukemia (4, 5).
In this review, we will focus on the biological characteristics of CML in advanced phase, the main results of the available treatments, the options for improving outcome, and, finally, we will briefly discuss on the optimal management of CP-CML in order to prevent disease evolution.
Definition, Epidemiology and Biological Determinants of Advanced Phase
Definition of advanced phase is controversial (6). There are four main classifications, provided by the International Blood and Marrow Transplant Registry [IBMTR; (7)], the MD Anderson Cancer Center [MDACC; (8)], the World Health Organization [WHO; (9)], and the European LeukemiaNet [ELN; (10)]. Details about the different criteria are summarized in Table 1. The IBMTR criteria have been mostly used in studies of bone marrow transplantation (BMT) and are the most comprehensive, but include some parameters which are somewhat subjective (leukocytosis, thrombocytosis or splenomegaly unresponsive to treatment, or thrombocytopenia and anemia unrelated to therapy); the significance of these findings as determinants of advanced phase seems to shrink in the TKI era. One of the most striking differences among the classification systems is the threshold of blast percentage used for defining AP and BP (15–29% according to ELN and 10–19% according to WHO for AP; ≥30% according to ELN and ≥20% according to WHO for BP). These different cutoffs should be kept in mind when results of new strategies have to be evaluated. In a retrospective cohort of 809 patients treated with imatinib, those who had a blast count between 20 and 29% had better complete cytogenetic response (CCyR) and 3-year overall survival (OS) compared to patients with blasts ≥30%, thus validating the threshold proposed by ELN (11). Recently, the WHO has added “provisional” criteria for AP based on the response to TKI (absence of complete hematologic response to the first TKI, or absence of response to two sequential TKIs, or development of two or more BCR-ABL1 mutations while on TKI treatment), which require further validation (12).
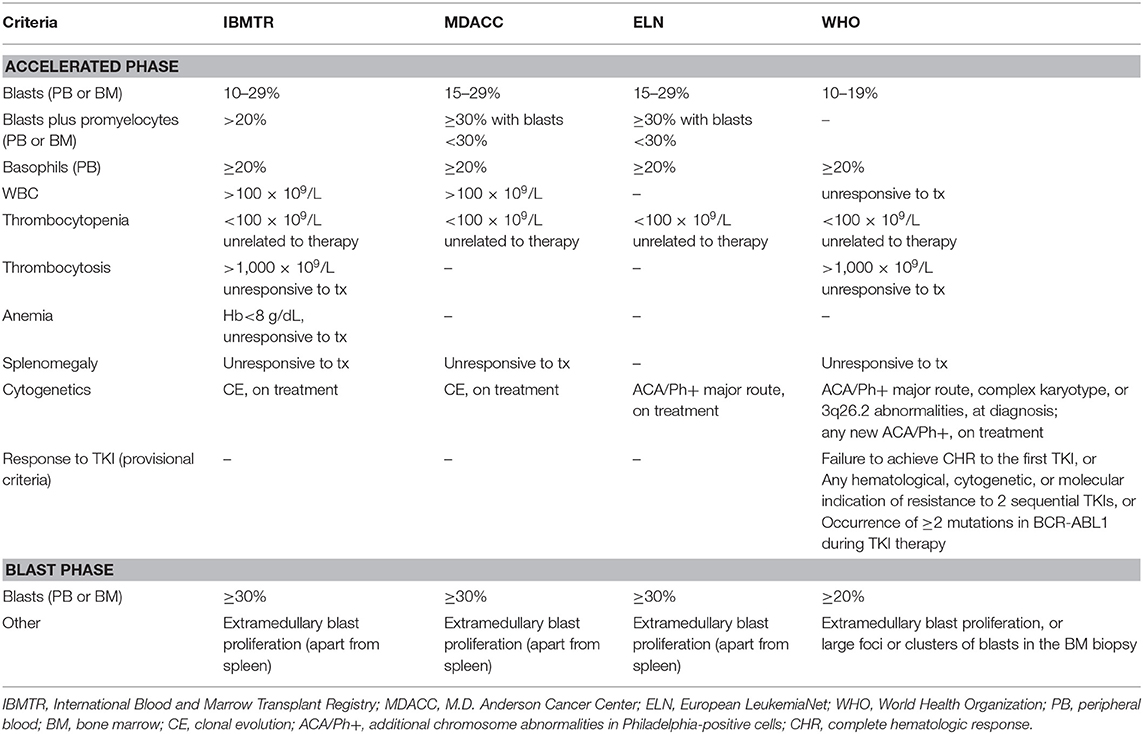
Table 1. Definitions of accelerated and blast phase of chronic myeloid leukemia.
The incidence of advanced phase at diagnosis is not really defined. A proportion of patients with AP or BP features since first referral was reported as high as 6–11% and 8–16%, respectively, in large monocentric series (13, 14). National multicenter registries showed lower incidences, around or inferior to 5% each: specifically, advanced phase at diagnosis was reported in 3% of patients in France (2% AP and 1% BP) (15), 7% in Czech Republic and Slovakia (5% AP and 2% BP) (16), 5% in Turkey (4% AP and 1% BP) (17), 6% in Sweden (4% AP and 2% BP) (18), and 1% in Italy (19). Among the EUTOS population-based registry (2,904 patients), the incidence of AP and BP at diagnosis were 3.5% and 2.2%, respectively (20). These differences may be partly related to the different application of diagnostic procedures. For example, a bone marrow (BM) core biopsy has been found either essential or helpful in correctly defining the disease phase and in evaluating the presence of BM fibrosis, a feature related with advanced phase (21), however indication for performing routine BM biopsy at CML diagnosis is not provided by current guidelines (22, 23). A recent study, while assessing minimal residual disease in children with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+-ALL), found that, in some patients, from 12 to 83% of nonmalignant B cells, T cells, and myeloid cells were positive for BCR-ABL1, suggesting that the translocation probably have occurred in a multipotent hematopoietic cell, thus concluding that these patients were actually affected by CML in blast crisis rather than de-novo Ph+-ALL (24). Although a similar study has not been performed in adult patients, the higher incidence of Ph+-ALL in the adult setting may suggests that presentation of CML in blast crisis could be more common than usually reported (25).
The incidence of progression from CP to blast crisis has dramatically decreased after the introduction of TKI therapy (26). In the pre-imatinib era progression rates were around 1.5–3.7% per year and decreased to 0.3–2.2% per year in the imatinib-based CML study IV (27). The same picture was seen in the imatinib arm of the pivotal IRIS trial, were the estimated 10-year cumulative incidence of blast crisis was 7.9% and appeared to be higher in the first 4 years after diagnosis, then decreasing around zero as soon as patients reached a molecular response (28). The introduction of 2nd generation TKI as frontline treatment of CP-CML further reduced the incidence of progression, although the difference vs. imatinib was statistically significant for the nilotinib arms only of the ENESTnd trial (0.7% for nilotinib 300 mg twice daily vs. 1.3% for nilotinib 400 mg twice daily vs. 4.8% for imatinib 400 mg daily at 5 years, p < 0.05 for both comparisons) (29) while there was a trend toward less progression rates in the dasatinib arm of the DASISION trial (3.0% for dasatinib 100 mg daily vs. 5.7% for imatinib 400 mg daily at 5 years) (30) and the bosutinib arm of the BFORE trial (1.6% for bosutinib 400 mg daily vs. 2.5% for imatinib 400 mg daily at 12 months) (31). In a non-academic healthcare setting investigated within the Swedish CML registry, the cumulative incidence of progression at 2 years from diagnosis was 4.3%. Of note, all patients undergoing progression had been treated with imatinib frontline, high-risk EUTOS score was associated to the risk of progression, and insufficient cytogenetic and/or molecular monitoring was found in 33% of them (32).
A detailed discussion about the mechanisms of evolution to advanced phase is beyond the scope of this article and there are many beautiful reviews on this topic (33–35). Here, we will focus on cytogenetic clonal evolution (CE) and on development of BCR-ABL1 mutations, two determinants of progression that may have a relevant impact on treatment choices and outcomes.
Cytogenetic CE is considered an AP-defining characteristic according to various classification systems (Table 1). A favorable outcome of patients displaying cytogenetic CE as the single feature of AP (i.e., not associated with high blast count, or other AP abnormalities) was demonstrated in patients treated with interferon (36), allogeneic BMT (37), imatinib (38) and 2nd generation TKI after imatinib failure (39). However, compared to patients with standard karyotype, those with cytogenetic CE have inferior responses to imatinib (40, 41) and the presence of additional chromosomal abnormalities (ACA) other than Ph chromosome at diagnosis are recognized as a warning feature by ELN (22). In the German CML Study IV the occurrence of trisomy 8 (+8), isochromosome i(17q), trisomy 19 (+19), or an extra copy of Ph (+Ph) had a striking unfavorable clinical impact on response to imatinib and prognosis, and these abnormalities were identified as “major route” ACA (42). In contrast, other cytogenetic aberrations, like the loss of Y-chromosome or other sporadic abnormalities, were called “minor route” ACA and they were considered as mere indicators of genomic instability instead of determinants of progression (43). In a comprehensive study from the MDACC, the unfavorable prognostic role of the isolated +8 was not confirmed and two previously considered minor-route ACA (3q26.2 rearrangement and monosomy 7/7q deletion) were associated to poor treatment response and dismal survival (44). Moreover, different ACA were associated to a lineage-specific progression to BP, being +8, 3q26.2 rearrangement, i(17q) and +19 significantly more common in myeloid BP, and−7/7q- more common in lymphoid BP (45). On this basis, a cytogenetic-based model for predicting the risk of progression to BP was recently proposed: patients without ACA represented the standard risk group, patients with +8, +Ph, or other single ACA the intermediate-1 risk group, patients with other complex ACA the intermediate-two risk group, and patients with isolated 3q26.2 rearrangement, −7/7q–, i(17q), or with these abnormalities in the context of a complex karyotype represented the high-risk group. This model predicted different probabilities of CE while on TKI treatment, with high-risk patients considered as candidates for transplant in first CP due to the high rate of rapid progression even when treated with 2nd generation TKI (46).
BCR-ABL1 kinase domain mutations have been detected in 26–37% of imatinib-naïve patients diagnosed in advanced phase (47), prompting the recommendation of performing mutation analysis in any case of AP or BP at diagnosis, but not in the large majority of CML patients presenting in CP (48). In patients failing imatinib, frequency and number of mutations correlate to the risk of progression to advanced phase, thus resistant mutations act at least in part as a determinant of disease evolution (49, 50). Moreover, time to progression to advanced phase and survival were significantly shorter in imatinib-resistant patients harboring BCR-ABL1 mutations compared to patients without detectable mutations (51). Mutational analysis is therefore recommended both in case of failure or suboptimal response to frontline treatment (48). Direct sequencing of the BCR-ABL1 gene is still the reference method for mutation detection, but its sensitivity is low. Using more sensitive techniques such as mass spectrometry (52) or next-generation sequencing (53), a higher number of low-level mutations were found in patients with inadequate response to their treatment, and predicted for lower rates of response to subsequent lines of treatment, especially if patients received TKIs to whom they were insensitive. Testing longitudinal samples of patients resistant to imatinib or 2nd generation TKIs, NGS technique revealed pathogenic BCR-ABL1 mutations in about half of cases 3 months before the same mutations could be detected by conventional sequencing (54), including cases harboring the highly resistant T315I mutation (55).
The Role of TKI in Advanced Phase
Evidences about the optimal treatment of CML patients in advanced phase are much less solid than in CP patients, but the evolving concepts in the use of TKI have been also redirected in the management of AP and BP patients. Current indications of BCR-ABL1 inhibitors for CML in advanced phase are summarized in Table 2.
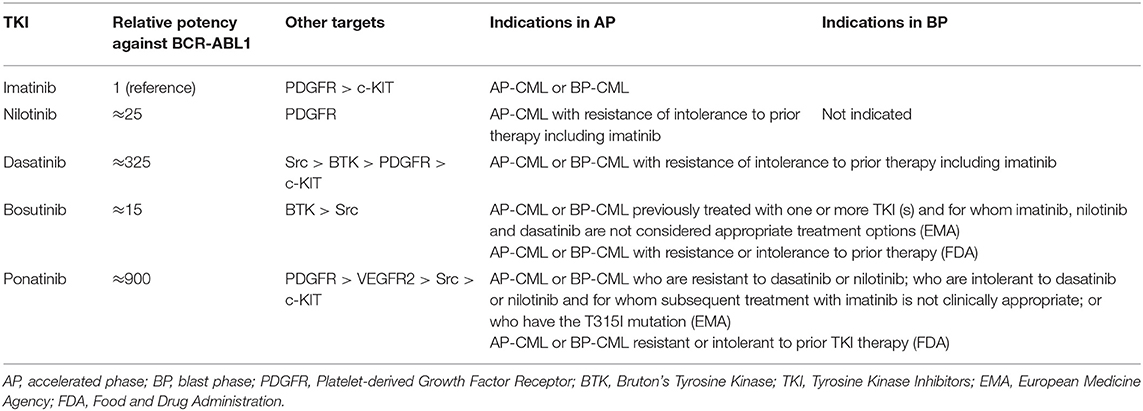
Table 2. Indications of currently available TKI in advanced phase of chronic myeloid leukemia.
Imatinib
Imatinib was the first targeted agent used in patients with advanced disease. Main results of imatinib clinical trials in AP and BP patients are reported in Tables 3, 4.
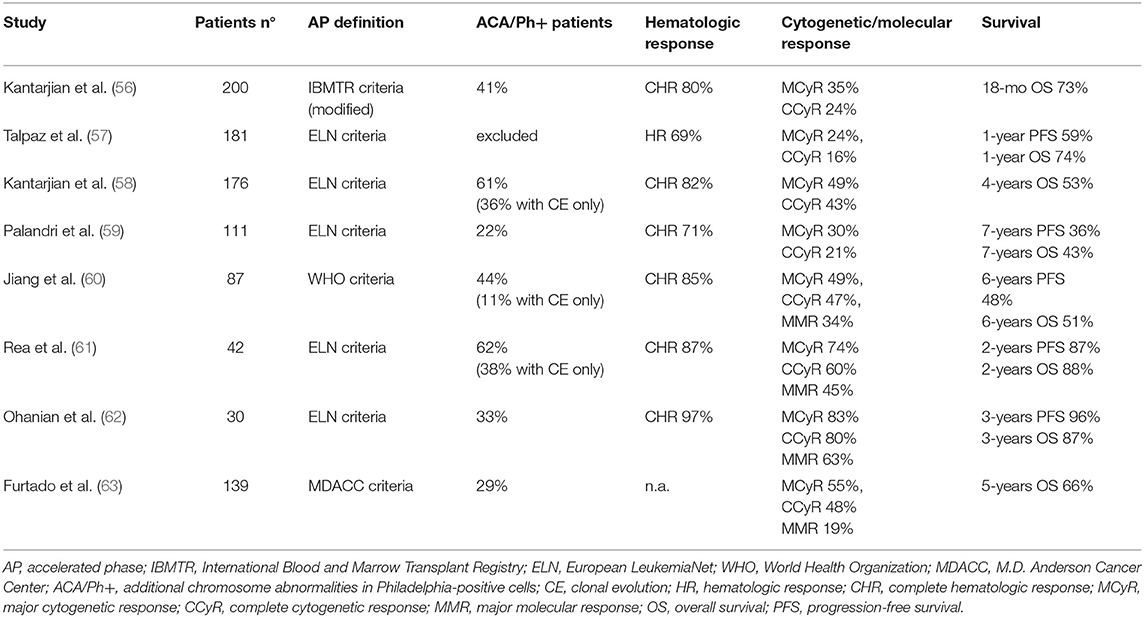
Table 3. Imatinib in accelerated phase of chronic myeloid leukemia.
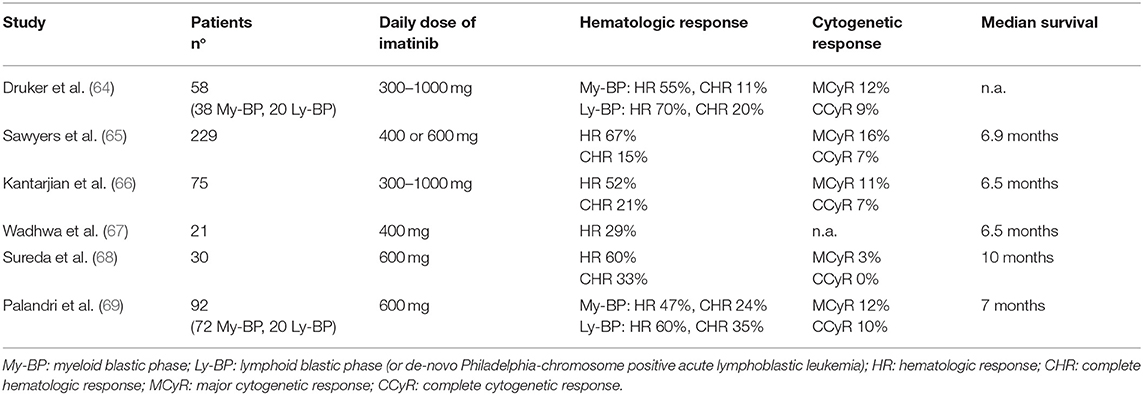
Table 4. Imatinib in blast phase of chronic myeloid leukemia.
Early studies in AP-CML patients not previously exposed to other TKIs showed that imatinib determined 60–85% rates of complete hematologic response (CHR, 16–45% of complete cytogenetic response (CCyR) and 19–34% of major molecular response (MMR) (56–60, 63). OS ranged from 74% at 12 months to around 40–50% at 5–7 years, an outcome clearly less favorable as compared to CP patients.
Notably, these patients were mainly in late AP, as a result of progression from CP treated for years with chemotherapy or interferon. Studies in newly diagnosed AP-CML patients treated with imatinib frontline showed higher rates of CCyR (60–80%) and MMR (45–63%), and better survival (61, 62). Other reasons for the different results observed in these trials might reside in the criteria of patient selection (including or not patients with 10–15% blasts in blood or marrow) and the proportion of patients defined as AP-CML due to hematologic criteria, cytogenetic criteria, or both. Patients with CE at diagnosis but lacking hematologic signs of progression show a more favorable response to imatinib (40), while patients presenting with CE associated to hematologic criteria of AP have a worse outcome (61).
Efficacy rates of imatinib in BP-CML were around 50–70% for hematologic response (defined as return to CP, i.e., blasts <30%), 15–35% for CHR (defined as blasts <5%, normalization of blood counts and absence of extramedullary disease), and <10% for CCyR (64–69). However, these responses were largely transient, although the achievement of some degrees of cytogenetic response correlated to a better outcome. In a large international study and in a multicentric Italian study by the GIMEMA CML Working Party the median survival was 6 months for patients without major cytogenetic response (MCyR) and ranged from 12 to 20 months for patients achieving this response (65, 69). Overall, the benefit of imatinib single-agent was inferior in BP than in AP patients, since none of the BP-CML trials demonstrated a median OS longer than 1 year.
Nilotinib
Nilotinib is a 2nd generation TKI with greater potency and BCR-ABL1 selectivity, and is effective against the majority of BCR-ABL1 mutants which confer resistance to imatinib. Main results of nilotinib clinical trials in AP and BP patients are reported in Tables 5, 6.
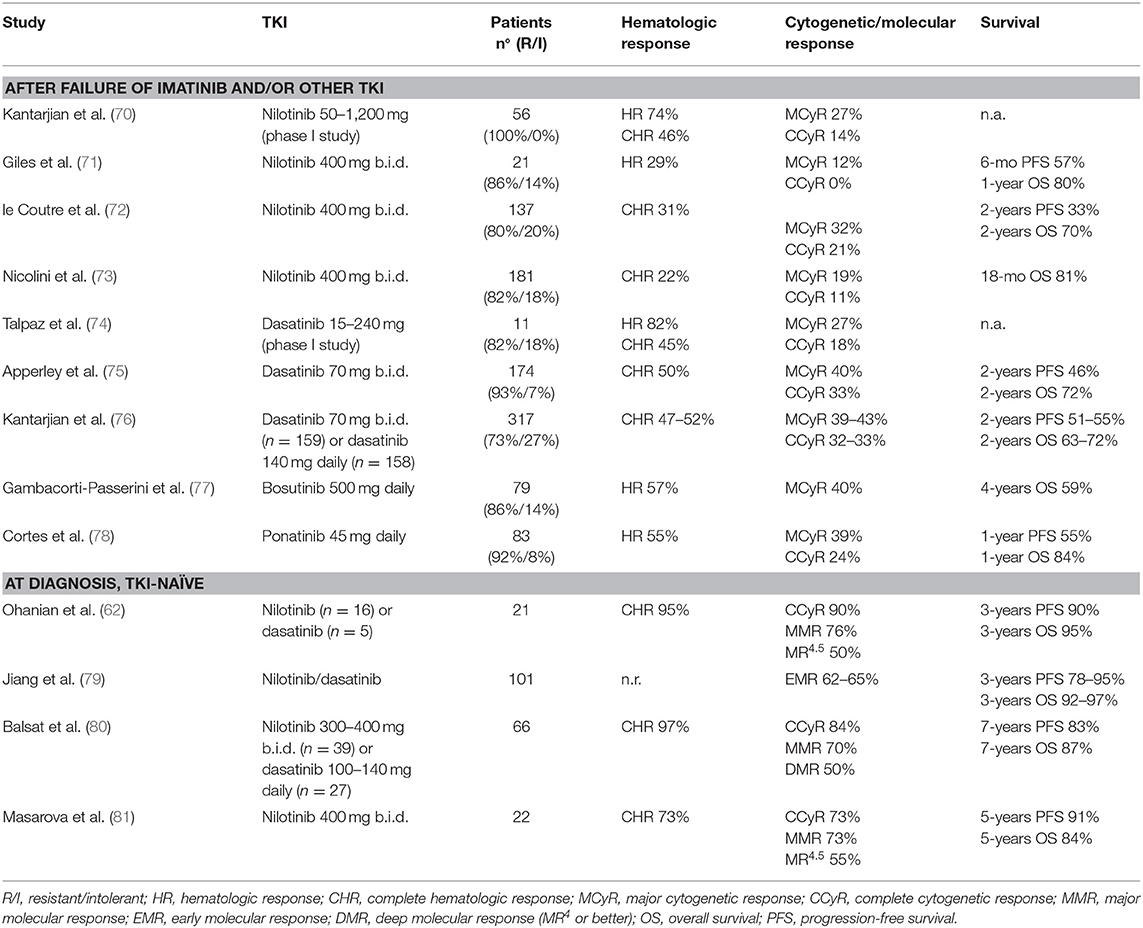
Table 5. 2nd/3rd generation TKI in accelerated phase of chronic myeloid leukemia.
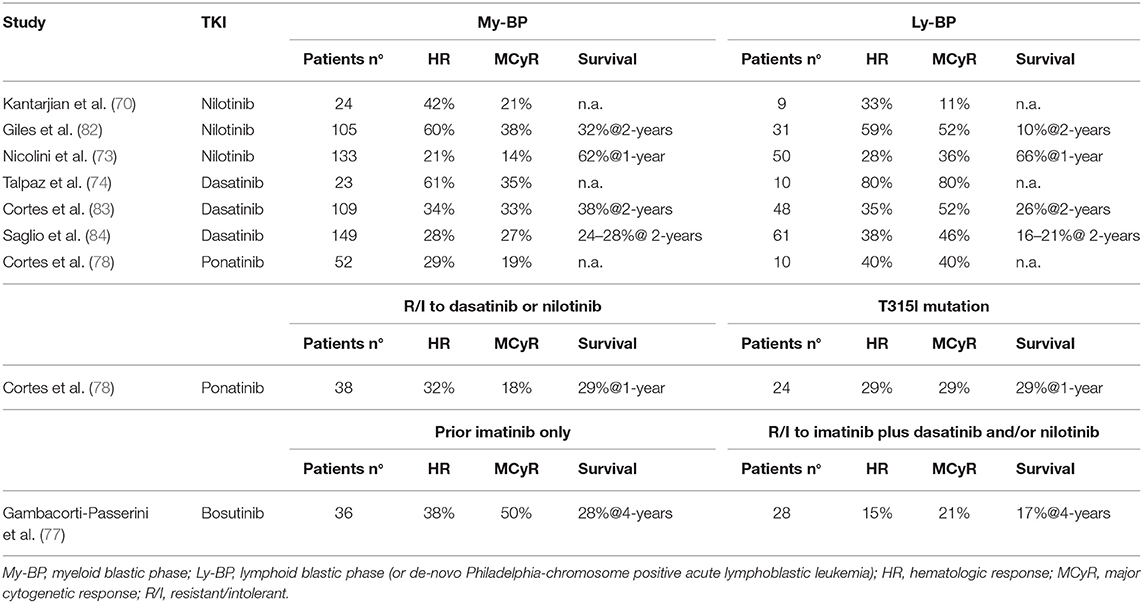
Table 6. 2nd/3rd generation TKI in blastic phase of chronic myeloid leukemia, after imatinib failure.
Two main trials investigated the role of nilotinib in AP-CML patients failing imatinib: a phase 2 registration study (72) and a phase 3b expanded-access study recruiting also patients resistant/intolerant to dasatinib or after stem cell transplant (73). In both studies the median duration of CML was around 5–6 years and median duration of previous imatinib treatment was more than 2 years. As expected for a heavily pretreated population, only a proportion of patients obtained clinical meaningful benefits: sustained CHR and CCyR were achieved by 22–31 and 11–21% of patients, respectively. Response rates were slightly better for patients who switched from imatinib due to intolerance than for resistance (72).
In two contemporary trials (a registration phase 2 study and an expanded access phase 3b study) nilotinib 400 mg twice daily was administered to BP-CML resistant (82%) or intolerant (18%) to imatinib (73, 82). CHR rates were 7–24 and 14–41% in myeloid and lymphoid BP-CML, respectively. Cytogenetic responses were obtained rapidly, but were transient. Few patients received an allogeneic stem cell transplant (SCT) after nilotinib. OS was similar in the two studies (10 months for myeloid BP-CML and 8 months for lymphoid BP-CML). Nilotinib was not further developed in BP-CML and did not receive regulatory approval for this setting of patients.
Dasatinib
Dasatinib is a 2nd generation multitargeted inhibitor of BCR-ABL1 and SRC-family kinases, which differs from imatinib in its ability to bind to both the active and inactive conformations of the ABL kinase. Dasatinib is 325-fold more potent than imatinib against wild-type BCR-ABL1 and is effective against the majority of BCR-ABL1 mutants. Main results of dasatinib clinical trials in AP and BP patients are reported in Tables 5, 6.
Dasatinib was studied in a series of clinical trials in patients with CML in all phases of disease after resistance or intolerance to imatinib (the SCR/ABL Tyrosine Kinase Inhibition Activity Research Trial of Dasatinib [START] program). Results of the phase 2 study in AP-CML (START-A) were initially reported on the first 107 patients (85) and then on the full population of 174 patients (75). These patients had a long history of CML prior to dasatinib therapy (median 6.5 years) and were extensively pretreated with high doses of imatinib (59%) and multiple other therapies for CML, including transplant (18%). More than 50% of patients had BCR-ABL1 mutations. Hematologic and cytogenetic responses were obtained both in mutated and unmutated patients, except for the T315I mutation. CCyR was achieved by one third of the overall population. A randomized phase 3 trial (CA180-035) compared two schedules of dasatinib administration (70 mg twice daily vs. 140 mg once daily) in a large population of patients with CML in advanced phase after imatinib failure, including 317 AP-CML patients (76). Efficacy rates were similar across the two groups and consistent with the previous phase 2 study (75). Single daily 140 mg dose of dasatinib was associated with a better safety profile. A recent long-term update showed that the estimated 5-years OS rates were 45 and 57% for patients randomized to 140 mg once daily vs. 70 mg twice daily, respectively. Although numerically higher, the different OS rate does not suggest a higher efficacy of the twice daily regimen, but it is likely to be related to the subsequent therapies, since only 40 patients (13% of the original cohort) continued to receive their assigned treatment beyond 5 years (86).
Patients with myeloid or lymphoid BP-CML were enrolled in two parallel phase 2 studies (START-B/START-L) and received dasatinib at the dose of 70 mg twice daily with dose escalation to 100 mg twice daily in case of inadequate response (83, 87). A total of 157 patients were enrolled, all after imatinib failure (52% received imatinib at 800 mg daily dose). Of note, 42% of myeloid and 65% of lymphoid BP-CML had BCR-ABL1 mutations. McyR rate was inferior in myeloid than in lymphoid BP-CML (34 vs. 52%, respectively) but the median duration of response was higher (16.8 vs. 4.1 months) and translated in a longer median OS (11.8 vs. 5.3 months). As previously mentioned, a large randomized study compared two different schedules of dasatinib in advanced CML patients, including 209 BP-CML patients (84). Efficacy was similar in the two treatment arms and, consistently with the previous phase 2 study, patients with lymphoid BP-CML resulted to have higher cytogenetic responses than myeloid BP-CML (MCyR 46 vs. 27%, CCyR 37 vs. 18%, respectively) but these responses were transient. Median OS was 7.7–7.9 months for myeloid BP-CML and 9-11.4 months for lymphoid BP-CML. As in AP-CML patients, also in BP-CML population the once daily regimen was better tolerated, resulting in fewer dose adjustments and fewer treatment interruptions.
Overall, these studies demonstrated that dasatinib could determine hematologic and cytogenetic responses in a relevant proportion of patients with a long history of CML and extensive pre-treatments. No studies formally addressed the role of dasatinib as frontline treatment in BP-CML patients, although it is likely that these patients could benefit of more potent TKI treatment earlier rather than after failure of other therapies.
Nilotinib and Dasatinib in AP-CML Patients, Frontline
In a monocentric series of 51 patients with AP features at the time of diagnosis, frontline treatment with nilotinib (n = 16) or dasatinib (n = 5) was superior to imatinib (n = 30) although the difference was not statistically significant. CCyR and MMR rates were 80 and 63% in the imatinib group, and 90 and 76% in the 2nd generation TKI group, respectively. The estimated 3-year OS was 87% with imatinib and 95% with 2nd generation TKIs (62). A retrospective comparison of 101 AP-CML and 656 CP-CML treated with frontline imatinib (n = 660), nilotinib (n = 85), or dasatinib (n = 11) showed that an early molecular response (i.e., BCR-ABL1 <10% at 3 months) was attained by 62–65% of AP-CML, similar to Sokal high-risk (58%) and Sokal intermediate-risk (66%) patients, but inferior to Sokal low-risk (83%) CP-CML patients. With a median follow-up of 39 months PFS and OS were 78 and 92% for AP-CML with blasts ≥15%, while the corresponding survival rates in AP-CML with basophils ≥20% were 95 and 97% (79). Preliminary results of two studies employing 2nd generation TKI in newly diagnosed AP-CML patients were recently presented. In a French multicenter cohort of 66 patients, treatment with nilotinib (n = 39) or dasatinib (n = 27) achieved CHR, CCyR, and MMR rates of 97, 84, and 70%, respectively. Long-term survival was excellent, with 7-year PFS and OS rates of 83.4 and 87.1%, respectively. Survival was similar in the group of 33 patients classified as AP for cytogenetic clonal evolution only and in the group of 33 patients with hematologic AP features, but the first group had a significantly higher probability of attaining a deep molecular response (DMR, MR4 or better, 66 vs. 33%, respectively) (80). Similar results were reported in 22 patients treated at the M.D. Anderson Cancer Center with nilotinib 400 mg twice daily. Rates of cytogenetic and molecular response were high, but 18% of patients lost their best achieved response while on study due to acquired BCR-ABL1 mutations. One electively discontinued nilotinib after a sustained DMR lasting for 107 months. After a median follow-up of 5.7 years, the estimated 5-year OS was 84% (81). Overall, these studies showed that nilotinib and dasatinib had similar efficacy in TKI-naïve AP-CML patients, probably superior to imatinib. Of note, in all these studies more than 50% of patients reached a DMR, demonstrating that an early treatment with potent TKI can counterbalance the negative prognostic impact of the advanced disease.
Bosutinib
Bosutinib is a dual Src/Abl 2nd generation TKI. Its activity and tolerability was studied in a phase 1/2 trial enrolling CML patients in all phases of disease with resistance/intolerance to imatinib only or resistance/intolerance to imatinib plus dasatinib and/or nilotinib (77). Patients in AP-CML were 79. CHR and CCyR rates were 33 and 31%. Cytogenetic responses were higher in patients treated with bosutinib after imatinib only than in patients receiving bosutinib in ≥3rd line of treatment (MCyR 48 vs. 27%, CCyR 35 vs. 23%, respectively). Responses were durable in around 50% of patients. Considering patients receiving 2nd generation TKI in second-line only, 4 years OS rate with bosutinib (66%) compared favorably to the 60–70% 2-years OS rate of nilotinib (72) and dasatinib (76). Patients in BP-CML were 64, including 23 myeloid BP-CML, 10 lymphoid BP-CML and 31 BP-CML of unspecified lineage. CHR and CCyR rates were 28 and 22% and responses were higher in patients treated with bosutinib after imatinib only than in patients receiving bosutinib in ≥3rd line of treatment (MCyR 50 vs. 21%, CCyR 37 vs. 17%, respectively). Responses were achieved across various baseline BCR-ABL1 mutations both in AP and BP cohorts, except for patients with T315I (n = 13) for whom only one response was achieved. Median OS was 10.9 months with two patients (3%) still receiving bosutinib at 4 years.
Ponatinib
Ponatinib is a potent 3rd generation BCR-ABL1 inhibitor, rationally designed to overcome resistance to other TKIs due to BCR-ABL1 mutations, including the T315I mutation. A large international phase 2 study (Ponatinib Ph-positive Acute lymphoblastic leukemia and CML Evaluation, PACE) was conducted to determine efficacy of ponatinib in heavily pretreated CML patients, resistant to many lines of treatment or harboring the T315I mutation (78). The study enrolled 449 patients, including 83 AP-CML and 62 BP-CML. In the AP-CML cohort 55% of patients had a major HR which was sustained after at least 1 year in half of them; MCyR was attained by 39% of patients and sustained after at least 1 year in 73% of them. Patients who received fewer previous treatments tended to have higher response rates. No single mutation conferring resistance to ponatinib was observed. Interestingly, estimated PFS and OS at 5 years were 22 and 49%, respectively (88). Seventeen AP-CML patients, stopped ponatinib due to resistance or intolerance: after ponatinib failure the outcome was poor, since one of four patients treated with SCT achieved MMR and none of the non-transplanted patients responded to subsequent therapy (89). In the BP-CML cohort 31% of patients had a major HR and 23% a MCyR by 6 months, but few patients maintained their best response after at least 1 year, and median OS was only 7 months, without substantial differences between patients resistant to dasatinib/nilotinib or with T315I mutation. Estimated OS at 3 years was 9% (88).
TKI and Chemotherapy for the Treatment of Advanced Phase
Chemotherapy has been used for many years in BP-CML since it appeared logical to employ induction protocols designed for ALL or acute myeloid leukemia (AML) also in lymphoid and myeloid BP-CML, respectively (90). Rates of response of BP-CML to chemotherapy were around 30% but remission duration was short and intensive regimens determined high rates of myelosuppression and induction deaths (91–94). Less intensive treatments were associated to fewer toxicities but the outcome was similarly dismal (95). Median survival was 6–8 months and increased to 2 years or more in a subset of patients who responded to intensive chemotherapy and could subsequently proceed to allogeneic SCT (96). Hypomethylating agents (alone or in combination with low-dose chemotherapy) have been tried in patients with myeloid BP-CML and encouraging results have been observed especially in elderly patients (97, 98). In a retrospective comparison of different treatment modalities in 162 myeloid BP-CML patients, response rates were similar among patients treated with intensive chemotherapy or with decitabine, but the latter group had less toxicity and response duration was overall longer, with a highly significant benefit on survival when only older patients were considered (99).
After the advent of TKI, all these treatment modalities were tested in combination with BCR-ABL1 inhibitors. Main results of these studies are reported in Table 7. Overall, the majority of these trials was very small and did not produce convincing evidence that any chemotherapy combination was superior to TKI alone (100–103). Of note, in many cases patients continued to receive the same TKI they had been previously exposed while in the CP, and this could have limited the benefit of treatment combination. Outcome was better when patients were treated with chemotherapy and 2nd generation TKI (104, 105), or when only de-novo BP-CML were considered, a group of patients with a reported median OS of 3 years or longer (103, 106). Results of different treatment strategies were analyzed in a large monocentric series of 477 patients with primary BP-CML (15%) or progressed after CP-CML or AP-CML (85%). Initial therapy for blast crisis was represented by TKI alone, a combination of TKI and chemotherapy, and non-TKI treatment in 35, 46, and 19% of patients, respectively. TKI (alone or in combination) was mainly imatinib (189 patients), followed by dasatinib (110 patients), nilotinib, bosutinib, and ponatinib (<30 patients each). Rates of response were slightly superior in patients treated with TKI and chemotherapy, but benefits seemed to be better when comparing 2nd generation TKIs vs. imatinib than comparing the same TKI with or without chemotherapy. In the whole cohort, factors significantly associated to a better survival in multivariate analysis were lymphoid immunophenotype, de-novo BP-CML, age <58 years, and the realization of transplant after induction treatment (106). Smaller studies explored the combination of hypomethylating agents or other agents (omacetaxine) and TKI. Results of imatinib combination were disappointing (107, 108), partly because the majority of these patients had already failed imatinib (111). Combination of azacytidine and dasatinib, nilotinib or ponatinib determined better hematologic, cytogenetic and molecular responses, and OS of 2 years or more were observed (109, 110).
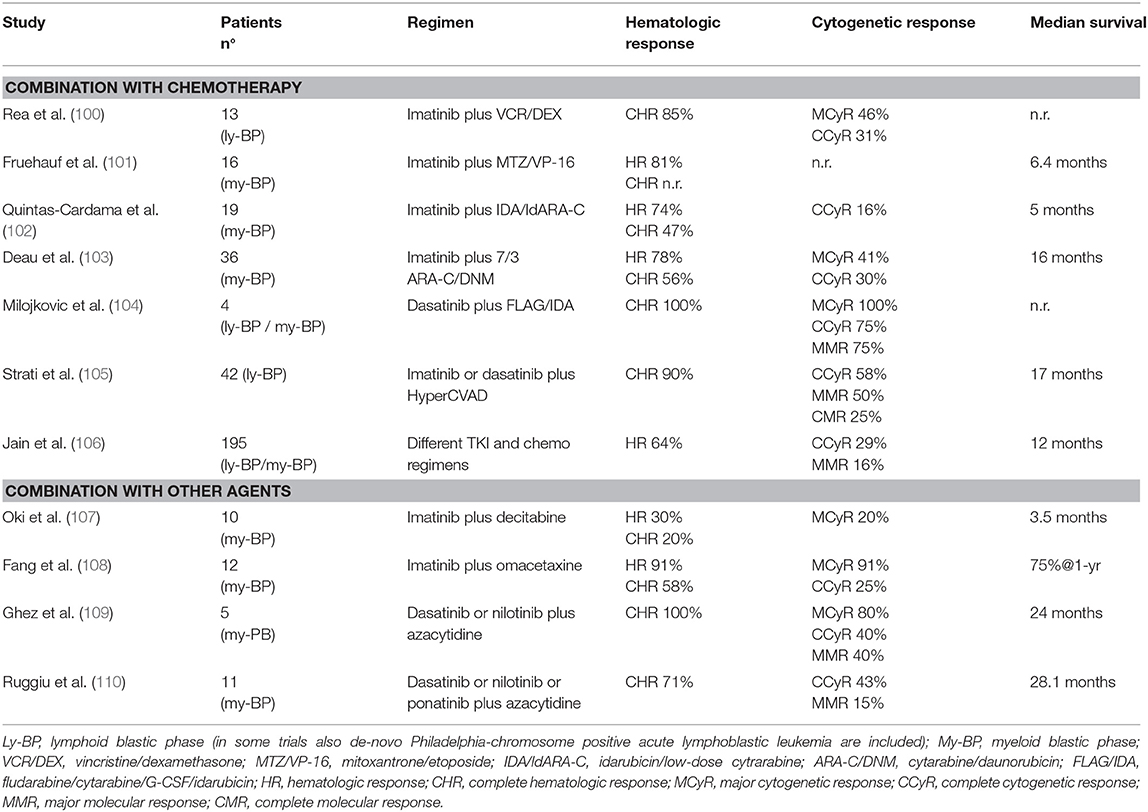
Table 7. TKI and chemotherapy or other agents in blastic phase of chronic myeloid leukemia.
Allogeneic SCT for the Treatment of Advanced Phase
The number of CML patients undergoing allogeneic SCT has dramatically reduced over years as a consequence of the TKI efficacy (112, 113). Current indications for allogeneic SCT in CML are listed in Table 8 (22, 23, 114). Pre-transplant treatment with TKI has no negative impact on transplant outcome (115). In AP-CML patients a randomized study showed that allogeneic SCT determined a superior outcome compared to imatinib in patients with at least one of the following characteristics: disease duration >12 months, anemia, and peripheral blood blasts >5% (60). In BP-CML a retrospective comparison showed a significantly higher 4-year OS for patients who underwent allogeneic SCT after TKI therapy as compared to those treated with TKI alone (47 vs. 10%; p < 0.001) (116). A consistent analysis from the CIBMTR reported a disease-free survival of 26–27% for AP-CML and 8–11% for BP-CML (117). OS rates ranged from 40 to 60% after 3–5 years from transplant (118, 119). Of note, advanced phase represents in se a risk factor for transplant outcome and was identified as one of the 5 predictors of survival after allogeneic SCT in CML by the EBMT, the other being donor type, patient age, donor/recipient sex, and time from diagnosis to transplant (120). A retrospective, indirect comparison among patients with T315I mutation who received ponatinib in the PACE study and those who were transplanted in the EBMT registry showed that in advanced phase allogeneic SCT represents an important and curative option, especially for BP-CML and might be considered early in patients developing clinical progression, after a trial of ponatinib therapy (121). In the setting of allogeneic SCT both myeloablative and non-myeloablative strategies have been used and which is the best regimen option still remains to be determined (122). After allogeneic SCT conventional molecular monitoring of BCR-ABL1 transcripts should be recommended whilst the role of a post-transplant TKI-therapy warrant further investigation.

Table 8. Indications for allogeneic stem cell transplant in chronic myeloid leukemia.
Emerging Treatment for CML in Advanced Phase
A growing genomic instability is the hallmark of advanced disease in CML and novel drugs, both targeting the BCR-ABL1-dependent and -independent mechanisms of resistance to TKI, are now considered in pre-clinical or clinical investigation and tested for combination efficacy (123–125).
Asciminib (ABL001) is a selective allosteric inhibitor of BCR-ABL1 (126). Differently from other TKIs, asciminib binds to the myristoyl pocket of ABL1 kinase, induces the formation of an inactive kinase conformation and blocks leukemic cells proliferation. It is currently tested in clinical trials for relapsed/refractory Philadelphia chromosome-positive leukemia patients (NCT02081378). The rational of asciminib design represents a major advance in target therapy since it allows a possible combination strategy (dual-drug targeting) with other BCR-ABL1 inhibitors (127).
Histone Deacetilase Inhibitors are small molecules that block HDAC enzymes involved in epigenetic modifications (128) and different HDAC isoforms have been found overexpressed in cancer cells. Since HDAC up-regulation has been associated with a reduction in both overall and disease-free survival, a possible role for HDAC inhibitors as antitumor drugs has been suggested (129). Pracinostat, vorinostat and panobinostat have been diversely evaluated in CML (130–134).
The BCL2-inhibitor venetoclax (ABT-199) has shown a BCL2-selective antagonism, presenting however a modest activity against CML progenitors when used as single agent but seeming to enhance imatinib cytotoxicity when used in combination (135, 136).
Following the evidence that JAK2 interacts with the ABL C-terminal, leading to its constitutive activation (137), JAK2 inhibitors have been combined with imatinib, nilotinib, and dasatinib with the aim of eliminating resistant CML cells and restoring TKI-sensitivity in resistant CML cell lines (138). On this basis, ruxolitinib is being evaluated in clinical trials alone or in combination with different TKIs in patients with advanced or resistant disease (NCT01702064, NCT02253277, NCT01751425, NCT01914484, NCT02973711).
Other molecules such as Aurora kinase inhibitors (tozasertib, danusertib, alisertib) have been used in advanced phases of CML and showed some degrees of clinical efficacy but also remarkable levels of toxicities (139–144).
Open Questions and Perspectives
A schematic view of the modern management of CML in advanced phase is presented in Figure 1.
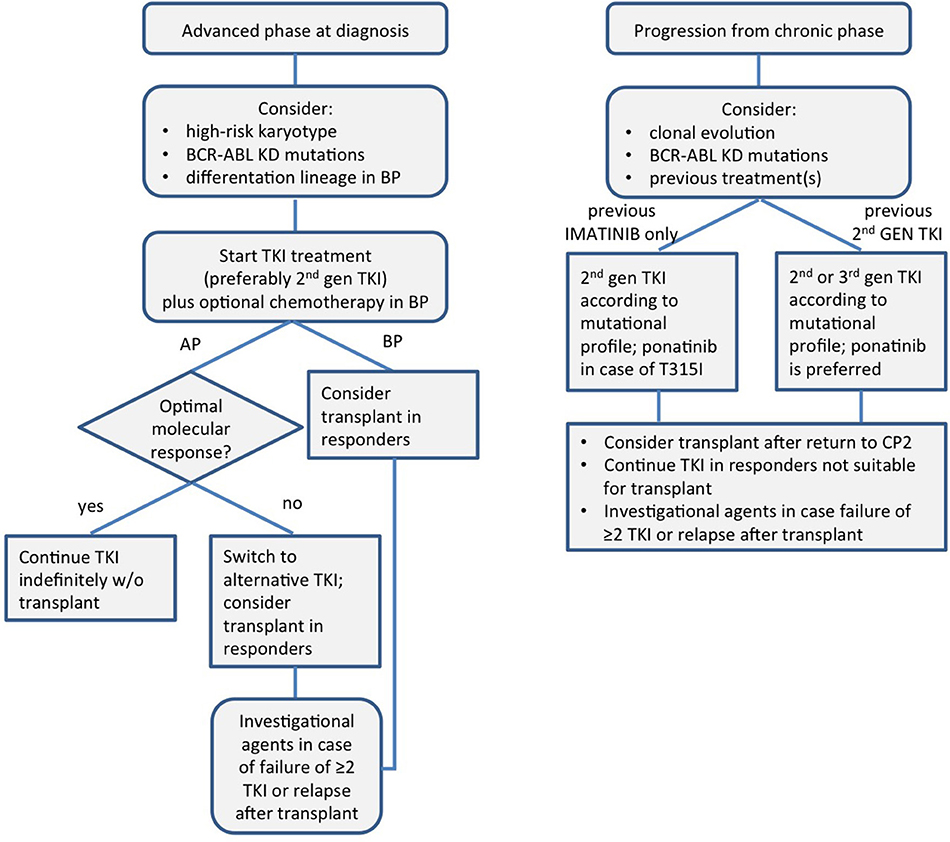
Figure 1. A schematic view of the modern management of chronic myeloid leukemia in advanced phase. KD, kinase domain; TKI, tyrosine kinase inhibitor; AP, accelerated phase; BP, blast phase; CP, chronic phase.
How to Treat Patients in Advanced Phase in 2019
Earlier ELN treatment guidelines did not provide recommendations on treatment of advanced phase (10) or suggested allogeneic SCT for all patients in advanced phase preceded by a TKI treatment (145). More recent guidelines recognized the value of frontline TKI treatment without the need for subsequent transplant, especially for AP patients. In newly diagnosed AP patients treatment with TKI alone is recommended by ELN (22) and NCCN (23), and transplant is considered an option only for patients not achieving an optimal response. In the European Union, only imatinib 600 mg daily has a market authorization for frontline use in newly diagnosed patients in advanced phase, but its efficacy is limited by the development of BCR-ABL1 kinase domain mutations that occur more frequently in this setting than in CP. High efficacy of frontline treatment with nilotinib or dasatinib, including remarkable DMR rates, has been demonstrated in both retrospective (79) and prospective studies (80, 81). Interestingly, the magnitude of benefit of 2nd generation TKIs over imatinib in the randomized prospective trials on CP patients was more evident in high-risk patients, underlying thus the limits of imatinib in controlling a more aggressive disease (29–31).
For patients with de-novo BP, transplant is recommended in all patients after return to CP with TKI alone or in combination with chemotherapy (22, 114). No specific chemotherapy regimen can be suggested since studies are few, heterogeneous, and not controlled. In some retrospective series of patients treated with different modalities, the benefits of chemotherapy in addition to TKI were uncertain or minimal (32, 106). Moreover, in the setting of Ph+-ALL, the possibility of a rapid achievement of deeper rates of molecular response and long-term remission after TKI with minimal or no induction chemotherapy has been demonstrated (146–148). The use of frontline TKI followed by targeted immunotherapy (blinatumomab) is also actively investigated in prospective trials (NCT02744768, NCT03263572) and may represent an attractive option also for de-novo lymphoid BP-CML.
Disease progression to advanced phase while on TKI treatment has a worse prognosis than de-novo AP- or BP-CML. Treatment with alternative TKI, according to the BCR-ABL1 mutational status, participation in clinical trials, and evaluation for allogeneic SCT is recommended by current guidelines for all progressing patients (22, 23, 114).
In our opinion, the distinction between AP and high-risk CP features has limited value when 2nd generation TKIs are used in the frontline setting and early attainment of molecular response should be viewed as the only determinant for prognosis, irrespectively from disease characteristics at presentation. A chemo- and transplant-free approach for de-novo BP patients is also intriguing but there are very limited and indirect data to support such a strategy, and prospective trials are needed to address this issue. Patients progressing after multi-TKI failure represent a challenging setting in which further treatment with TKI alone has a limited value (149), and novel treatment options are eagerly awaited.
Of note, frontline treatment of advanced phase patients with TKI instead of chemotherapy and allogeneic SCT will further increase the economic burden of CML treatment, which still represents a relevant issue for sustainability of health systems (150).
Which Are the Optimal Endpoints in the Treatment of AP/BP?
The optimal depth of response to frontline TKI at different time points is already well-defined for patients in CP (22, 23), but evidence for defining optimal response in advanced phases is still lacking. In earlier trials, it was observed that rates of MCyR often exceeded those of CHR in BP-CML patients (Tables 4, 5) and it was suggested that achievement of MCyR without CHR was associated to inferior survival (151). More recently, a direct correlation between the depth of response and survival was seen in a cohort of 386 patients with BP-CML: 5-year OS rates were 72, 34, 12, and 11% for patients with complete molecular response (CMR), MMR but not CMR, CCyR but not MMR, and HR only, respectively (152). Interestingly, no differences in survival were seen between patients who underwent or not to allogeneic SCT once they had obtained a CMR, reinforcing the concept that rapid kinetics of response to TKI therapy could allow long-term disease control even without transplant.
Should Patients With Myeloid and Lymphoid BP-CML Receive Different Treatments?
Response to TKI is similar in myeloid and lymphoid BP-CML, although in the large retrospective experience of MDACC a lymphoid phenotype was associated with a better survival (106). Targeted immunotherapy, i.e., with the anti-CD33 antibody gemtuzumab ozogamicin in myeloid BP or the anti-CD19 bispecific antibody blinatumomab for lymphoid BP, may represent an attracting distinctive treatment strategy but reports are still anecdotal (153, 154). Another concern is about the utility of distinguishing lymphoid BP-CML from Ph+-ALL, since the differential diagnosis is often challenging. Patients with no preceding history of CP-CML may be considered as de-novo lymphoid BP-CML instead of Ph+-ALL if they have morphologic features of CML, such as left-shifted myeloid predominance, eosinophilia, and/or basophilia. The surface marker CD26 (dipeptidylpeptidase-IV) has been proposed as a specific marker of CML Leukemic Stem Cell (LSC) (155) and the assessment of CD26+ LSC in peripheral blood by flow cytometry has been suggested as a rapid tool for CML diagnosis (156) or as a marker for minimal residual disease during TKI treatment and treatment-free remission (157). Notably, in Ph+-ALL CD26 expression was found on LSC in patients with major BCR-ABL1 transcript encoding p210 BCR-ABL1 protein but not in patients with minor BCR-ABL1 transcript encoding p190 BCR-ABL1 protein (158). This finding may have therapeutic implications, since CD19-negative myeloid lineage relapses after blinatumomab have been observed in p210 Ph+-ALL patients as a result of the selection of preexisting CD19-negative malignant progenitor of myeloid origin (159). Although suggestive, these biological distinctions are not yet useful in guiding treatment selection and their clinical significance still remains to be elucidated.
Is It Possible to Prevent Blast Crisis by the Optimization of Treatment in Chronic Phase?
Despite the groundbreaking results obtained with TKI in CP-CML, ~5–10% of CML patients eventually progress to advanced phase while on treatment. The mechanisms underlining TKI failure, disease progression and cytogenetic evolution remain largely unknown. Mutations in the ABL1 kinase domain, amplification of the BCR-ABL1 oncogene and high expression levels of the BCR-ABL1 mRNA represent BCR-ABL1 dependent mechanisms responsible for TKI failure (160). Then, uncontrolled BCR-ABL1 signaling leads to genetic instability and a more disorganized state until the anaplastic threshold is reached and other oncogenes ultimately lead to progression in a BCR-ABL1-independent way (161). Both quantitative and qualitative mechanisms can underlie the more aggressive behavior of CML clones expressing high BCR-ABL1 levels. According to the quantitative hypothesis, higher BCR-ABL1 transcripts translate into higher expression of the BCR-ABL1 oncoprotein and increased tyrosine kinase activity that would ultimately strengthen canonical BCR-ABL1-dependent signaling, resulting in a less responsive leukemic population. Higher BCR-ABL1 transcripts at diagnosis measured using GUS as a reference gene identified patients with inferior probability of response to frontline standard dose imatinib (162). On the other hand, the qualitative hypothesis assumes that higher BCR-ABL1 activity leads to “leakage” of BCR-ABL1 signaling to downstream targets that are usually not involved in BCR-ABL1-dependent transformation.
Many baseline factors at CML diagnosis have been correlated to a different probability of progression. Patients identified as high-risk by current prognostic models have a higher likelihood of disease transformation to AP-CML or BP-CML. In particular, the Eutos Long-Term Survival (ELTS) score identifies three risk groups with significantly different probabilities of death due to progression in advanced phase (163). Using the standard Sokal model, it was seen that within the same risk group younger patients had a higher risk of sudden progression and death (164). Biological variables associated to a high risk of progression included CIP2A levels (165), the expression levels of the polycomb group BMI1 gene (166), the activation of beta-catenin (167), specific gene signatures (168), and mutations in cancer-associated genes such as ASXL1, IKZF1, RUNX1, SETD1B, GATA2, MLL, and UBE2A (169). None of these variables has been extensively validated or is easily available in clinical practice.
The most relevant predictor of progression is the kinetics of response to treatment (170). It has been extensively demonstrated that not achieving a reduction <10% BCR-ABL1 after 3 months is linked to a higher risk of progression to advanced phase and reduced survival both with frontline imatinib and 2nd generation TKIs (29, 171–173). Measurement of the BCR-ABL1 transcript halving time during the first months of treatment may increase sensitivity and specificity of response measurement (174, 175). Finally, those patients who are regularly monitored according to guidelines have a lower risk of progression than patients monitored less frequently (176). However, sudden onset of BP despite adequate monitoring and apparently adequate response to TKI may occasionally occur (177, 178).
In our opinion, ELTS scoring system should be preferably used for frontline treatment decisions in all newly diagnosed CP-CML patients, and non low-risk patients should be considered for 2nd generation TKIs, or carefully monitored for early molecular response when imatinib is chosen as frontline treatment.
Conclusion
Management of CML in advanced phase remains challenging. However, prognosis for patients diagnosed in AP improved clearly over years and presently the majority of patients with AP features at diagnosis can be managed as high-risk CP patients. Patients in blast crisis have inferior outcomes due to emergent resistance to TKI. Rational combination of TKI and chemotherapy or, preferably, novel agents including immunotherapy could improve remission rates and duration.
Frontline imatinib results challenged the concept of transplantation in CP-CML patients; nowadays the use of more potent TKI might modify the same concept also in patients presenting with advanced disease. However, optimal management of patients in CP represents the best way to avoid disease evolution and to allow a quite normal life duration for all patients.
Due to the limited evidences and the still numerous unmet needs, it would be desirable that a dedicated expert panel would provide updated recommendations for the management of CML in advanced phase.
Author Contributions
All authors reviewed the literature, wrote the paper, and agree to be accountable for the content of the work.
Conflict of Interest
MB declares research funding from Novartis and consultancy fees (advisory board honoraria) from Amgen, Incyte, Novartis, and Pfizer, outside the present work; FS and FD declare consultancy fees (advisory board honoraria) from Bristol Myers Squibb, Incyte, Novartis, and Pfizer, outside the present work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Goldman JM, Melo JV. Chronic myeloid leukemia - advances in biology and new approaches to treatment. N Engl J Med. (2003) 349:1451–64. doi: 10.1056/NEJMra020777
2. Stagno F, Stella S, Spitaleri A, Pennisi MS, Di Raimondo F, Vigneri P. Imatinib mesylate in chronic myeloid leukemia: frontline treatment and long-term outcomes. Exp Rev Anticanc Ther. (2016) 16:273–8. doi: 10.1586/14737140.2016.1151356
3. Bower H, Björkholm M, Dickman PW, Höglund M, Lambert PC, et al. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. (2016) 34:2851–7. doi: 10.1200/JCO.2015.66.2866
4. Cortes J, Kantarjian H. Advanced-phase chronic myeloid leukemia. Semin Hematol. (2003) 40:79–86. doi: 10.1053/shem.2003.50005
5. Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. (2015) 94:s107–21. doi: 10.1007/s00277-015-2325-z
6. Hehlmann R. How I treat CML blast crisis. Blood. (2012) 120:737–47. doi: 10.1182/blood-2012-03-380147
7. Speck B, Bortin MM, Champlin R, Goldman JM, Herzig RH, McGlave PB, et al. Allogeneic bone-marrow transplantation for chronic myelogenous leukaemia. Lancet. (1984) 1:665–8. doi: 10.1016/S0140-6736(84)92179-2
8. Kantarjian HM, Dixon D, Keating MJ, Talpaz M, Walters RS, McCredie KB, et al. Characteristics of accelerated disease in chronic myelogenous leukemia. Cancer. (1988) 61:1441–6. doi: 10.1002/1097-0142(19880401)61:7<1441::AID-CNCR2820610727>3.0.CO;2-C
9. Vardiman JW, Pierre R, Thiele J, Imbert M, Brunning RD, Flandrin G. Chronic myelogenous leukemia. In: Jaffe ES, Harris NL, Stein H, Wardiman JW, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopotietic and Lymphoid Tissues. (2001).Lyon: International Agency for Research on Cancer Press (2001).
10. Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. (2006) 108:1809–20. doi: 10.1182/blood-2006-02-005686
11. Cortes JE, Talpaz M, O'Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. (2006) 106:1306–15. doi: 10.1002/cncr.21756
12. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
13. Wang AH, Wang YY, Yao Y, Xu ZZ, Zhou L, Wang L, et al. Summary of 615 patients of chronic myeloid leukemia in Shanghai from 2001 to 2006. J Exp Clin Cancer Res. (2010) 29:20. doi: 10.1186/1756-9966-29-20
14. Kantarjan H, O'Brien S, Jabbour E, Garcia-Manero G, Quintas-Cardama A, Shan J, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. (2012) 119:1981–7. doi: 10.1182/blood-2011-08-358135
15. Tardieu S, Brun-Strang C, Berthaud P, Michallet M, Guilhot F, Rousselot P, et al. Management of chronic myeloid leukemia in France: a multicentered cross-sectional study on 538 patients. Pharmacoepidemiol Drug Saf. (2005) 14:545–53. doi: 10.1002/pds.1046
16. Faber E, MuŽik J, Koza V, Demečková E, Voglová J, Demitrovičová L, et al. Treatment of consecutive patients with chronic myeloid leukaemia in the cooperating centres from the Czech Republic and the whole of Slovakia after 2000 – a report from the population-based CAMELIA registry. Eur J Haematol. (2011) 87:157–68. doi: 10.1111/j.1600-0609.2011.01637.x
17. Sahin F, Saydam G, Cömert M, Uz B, Yavuz AS, Turan E, et al. Turkish chronic myeloid leukemia study: retrospective sectional analysis of CML patients. Turk J Haematol. (2013) 30:351–8. doi: 10.4274/Tjh.2013.0151
18. Höglund M, Sandin F, Hellström K, Björeman M, Björkholm M, Brune M, et al. Tyrosine kinase inhibitor usage, treatment outcome, and prognostic scores in CML: report from the population-based Swedish CML registry. Blood. (2013) 122:1284–92. doi: 10.1182/blood-2013-04-495598
19. Specchia G, Pregno P, Nicolosi M, Castagnetti F, Martino B, Breccia M, et al. Chronic myeloid leukemia Italian multicenter observational study (CML-IT-MOS): clinical characteristics of chronic myeloid leukemia (CML) patients treated in real-life between 2012 and 2016 in 66 Italian hematology centers of the Gimema study group. Blood. (2018) 132:45. doi: 10.1182/blood-2018-99-116648
20. Hoffmann VS, Baccarani M, Hasford J, Lindoerfer D, Burgstaller S, Sertic D, et al. The EUTOS population-based registry: incidence and clinical characteristics of 2904 CML patients in 20 European countries. Leukemia. (2015) 29:1336–43. doi: 10.1038/leu.2015.73
21. Hidalgo-Lopez JE, Kanagal-Shamanna R, Quesada AE, Gong Z, Wang W, Hu S, et al. Bone marrow core biopsy in 508 consecutive patients with chronic myeloid leukemia: assessment of potential value. Cancer. (2018) 124:3849–55. doi: 10.1002/cncr.31663
22. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. (2013) 122:872–84. doi: 10.1182/blood-2013-05-501569
23. Radich JP, Deininger M, Abboud CN, Altman JK, Berman E, Bhatia R, et al. Chronic myeloid leukemia, version 1.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2018) 16:1108–35. doi: 10.6004/jnccn.2018.0071
24. Hovorkova L, Zaliova M, Venn NC, Bleckmann K, Trkova M, Potuckova E, et al. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood. (2017) 129:2771–81. doi: 10.1182/blood-2016-11-749978
25. Hunger SP. CML in blast crisis: more common than we think? Blood. (2017) 129:2713–4. doi: 10.1182/blood-2017-04-776369
26. Saussele S, Silver RT. Management of chronic myeloid leukemia in blast crisis. Ann Hematol. (2015) 94:S159–65. doi: 10.1007/s00277-015-2324-0
27. Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ, et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia. (2017) 31:2398–406. doi: 10.1038/leu.2017.253
28. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. (2017) 376:917–27. doi: 10.1056/NEJMoa1609324
29. Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. (2016) 30:1044–54. doi: 10.1038/leu.2016.5
30. Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boquè C, et al. Final 5-year study results of DASISION: the Dasatinib vs Imatinib study in treatment-naïve chronic myeloid leukemia patients trial. J Clin Oncol. (2016) 34:2333–40. doi: 10.1200/JCO.2015.64.8899
31. Cortes JE, Gambacorti-Passerini C, Deininger MW, Mauro MJ, Chuah C, Kim DW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol. (2018) 36:231–7. doi: 10.1200/JCO.2017.74.7162
32. Söderlund S, Dahlén T, Sandin F, Olsson-Strömberg U, Creignou M, Dreimane A, et al. Advanced phase chronic myeloid leukemia (CML) in the tyrosine kinase inhibitor era – a report from the Swedish CML register. Eur J Haematol. (2017) 98:57–66. doi: 10.1111/ejh.12785
33. Radich JP. The biology of CML blast crisis. Hematology Am Soc Hematol Educ Program. (2007) 212:384–91. doi: 10.1182/asheducation-2007.1.384
34. Crews LA, Jamieson CH. Chronic myeloid leukemia stem cell biology. Curr Hematol Malig Rep. (2012) 7:125–32. doi: 10.1007/s11899-012-0121-6
35. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. (2014) 103:4010–22. doi: 10.1182/blood-2003-12-4111
36. Cortes J, Talpaz M, O'Brien S, Rios MB, Majlis A, Keating M, et al. Suppression of cytogenetic clonal evolution with interferon alfa therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia. J Clin Oncol. (1998) 16:3279–85. doi: 10.1200/JCO.1998.16.10.3279
37. Przepiorka D, Thomas ED. Prognostic significance of cytogenetic abnormalities in patients with chronic myelogenous leukemia. Bone Marrow Transplant. (1988) 3:113–9.
38. Cortes JE, Talpaz M, Giles F, O'Brien S, Rios MB, Shan J, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. (2003) 101:3794–800. doi: 10.1182/blood-2002-09-2790
39. Verma D, Kantarjian H, Shan J, O'Brien S, Estrov Z, Garcia-Manero G, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer. (2010) 116:2673–81. doi: 10.1002/cncr.25015
40. O'Dwyer ME, Mauro MJ, Kurilik G, Mori M, Balleisen S, Olson S, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood. (2002) 100:1628–33. doi: 10.1182/blood-2002-03-0777
41. Luatti S, Castagnetti F, Marzocchi G, Baldazzi C, Gugliotta G, Iacobucci I, et al. Additional chromosomal abnormalities in Philadelphia-positive clone: adverse prognostic influence on frontline imatinib therapy: a GIMEMA Working Party on CML analysis. Blood. (2012) 120:761–7. doi: 10.1182/blood-2011-10-384651
42. Fabarius A, Leitner A, Hochhaus A, Muller MC, Hanfstein B, Haferlach C, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML study IV. Blood. (2011) 118:6760–8. doi: 10.1182/blood-2011-08-373902
43. Fabarius A, Kalmanti L, Dietz CT, Lauseker M, Rinaldetti S, Haferlach C, et al. Impact of unbalanced minor route versus major route karyotypes at diagnosis on prognosis of CML. Ann Hematol. (2015) 94:2015–24. doi: 10.1007/s00277-015-2494-9
44. Wang W, Cortes JE, Tang G, Khoury JD, Wang S, Bueso-Ramos CE, et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood. (2016) 127:2742–50. doi: 10.1182/blood-2016-01-690230
45. Chen Z, Shao C, Wang W, Zuo Z, Mou X, Hu SJ, et al. Cytogenetic landscape and impact in blast phase of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. (2017) 31:585–92. doi: 10.1038/leu.2016.231
46. Gong Z, Medeiros LJ, Cortes JE, Chen Z, Zheng L, Li Y, et al. Cytogenetic-based risk prediction of blastic transformation of chronic myeloid leukemia in the era of TKI therapy. Blood Adv. (2017) 1:2541–52. doi: 10.1182/bloodadvances.2017011858
47. Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. (2005) 106:2128–37. doi: 10.1182/blood-2005-03-1036
48. Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. (2011) 118:1208–15. doi: 10.1182/blood-2010-12-326405
49. Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. (2002) 2:117–25. doi: 10.1016/S1535-6108(02)00096-X
50. Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. (2002) 100:1014–8. doi: 10.1182/blood.V100.3.1014
51. Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. (2005) 23:4100–9. doi: 10.1200/JCO.2005.05.531
52. Parker WT, Lawrence RM, Ho M, Irwin DL, Scott HS, Hughes TP, et al. Sensitive detection of BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. J Clin Oncol. (2011) 29:4250–9. doi: 10.1200/JCO.2011.35.0934
53. Soverini S, De Benedittis C, Polakova KM, Linhartova J, Castagnetti F, Gugliotta G, et al. Next-generation sequencing for sensitive detection of BCR-ABL1 mutations relevant to tyrosine kinase inhibitor choice in imatinib-resistant patients. Oncotarget. (2016) 7:21982–90. doi: 10.18632/oncotarget.8010
54. Soverini S, De Benedittis C, Castagnetti F, Gugliotta G, Mancini M, Bavaro L, et al. In chronic myeloid leukemia patients on second-line tyrosine kinase inhibitor therapy, deep sequencing of BCR-ABL1 at the time of warning may allow sensitive detection of emerging drug-resistant mutants. BMC Cancer. (2016) 16:572. doi: 10.1186/s12885-016-2635-0
55. Baer C, Kern W, Koch S, Nadarajah N, Schindela S, Meggendorfer M, et al. Ultra-deep sequencing leads to earlier and more sensitive detection of the tyrosine kinase inhibitor resistance mutation T315I in chronic myeloid leukemia. Haematologica. (2016) 101:830–8. doi: 10.3324/haematol.2016.145888
56. Kantarjian HM, O'Brien S, Cortes JE, Smith TL, Rios MB, Shan J, et al. Treatment of Philadelphia chromosome-positive, accelerated-phase chronic myelogenous leukemia with imatinib mesylate. Clin Cancer Res. (2002) 8:2167–76.
57. Talpaz M, Silver RT, Druker BJ, Goldamn JM, Gambacorti-Passerini C, Guilhot F, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. (2002) 99:1928–37. doi: 10.1182/blood.V99.6.1928
58. Kantarjian H, Talpaz M, O'Brien S, Giles F, Faderl S, Vertovsek S, et al. Survival benefit with imatinib mesylate therapy in patients with accelerated-phase chronic myelogenous leukemia – comparison with historic experience. Cancer. (2005) 103:2099–108. doi: 10.1002/cncr.21032
59. Palandri F, Castagnetti F, Alimena G, Testoni N, Breccia M, Luatti S, et al. The long-term durability of cytogenetic responses in patients with accelerated phase chronic myeloid leukemia treated with imatinib 600 mg: the GIMEMA CML Working Party experience after a 7-year follow-up. Haematologica. (2009) 94:205–12. doi: 10.3324/haematol.13529
60. Jiang Q, Xu LP, Liu DH, Liu KY, Chen SS, Jiang B, et al. Imatinib mesylate versus allogeneic hematopoietic stem cell transplantation for patients with chronic myelogenous leukemia in the accelerated phase. Blood. (2011) 117:3032–40. doi: 10.1182/blood-2010-09-308510
61. Rea D, Etienne G, Nicolini F, Cony-Morkhoul P, Johnson-Ansah H, Legros L, et al. First-line imatinib mesylate in patients with newly diagnosed accelerated phase-chronic myeloid leukemia. Leukemia. (2012) 26:2254–9. doi: 10.1038/leu.2012.92
62. Ohanian M, Kantarjian HM, Quintas-Cardama A, Jabbour E, Abruzzo L, Vertovsek S, et al. Tyrosine kinase inhibitors as initial therapy for patients with chronic myeloid leukemia in accelerated phase. Clin Lymphoma Myeloma Leuk. (2014) 14:155–62.e1. doi: 10.1016/j.clml.2013.08.008
63. Furtado VF, Santos GR, de Carvalho DS, Staziaki PV, Pasquini R, Funke VA. Accelerated phase chronic myeloid leukemia: evaluation of clinical criteria as predictors of survival, major cytogenetic response and progression to blast phase. Rev Bras Hematol Hemoter. (2015) 37:341–7. doi: 10.1016/j.bjhh.2015.07.004
64. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. (2001) 344:1038–42. doi: 10.1056/NEJM200104053441402
65. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. (2002) 99:3530–9. doi: 10.1182/blood.V99.10.3530
66. Kantarjian HM, Cortes J, O'Brien S, Giles FJ, Albitar M, Rios MB, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome positive chronic myelogenous leukemia in blast phase. Blood. (2002) 99:3547–53. doi: 10.1182/blood.V99.10.3547
67. Wadhwa J, Szydlo RM, Apperley JF, Chase A, Bua M, Marin D, et al. Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood. (2002) 99:2304–9. doi: 10.1182/blood.V99.7.2304
68. Sureda A, Carrasco M, de Miguel M, Martinez JA, Conde E, Sanz MA, et al. Imatinib mesylate as treatment for blastic transformation of Philadelphia chromosome positive chronic myelogenous leukemia. Haematologica. (2003) 88:1213–20.
69. Palandri F, Castagnetti F, Testoni N, Luatti S, Marzocchi G, Bassi S, et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: outcome of the patients alive after a 6-year follow-up. Haematologica. (2008) 93:1792–6. doi: 10.3324/haematol.13068
70. Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, et al. Nilotinib in imatinib- resistant CML and philadelphia chromosome-positive ALL. N Engl J Med. (2006) 354:2542–51. doi: 10.1056/NEJMoa055104
71. Giles FJ, Abruzzese E, Rosti G, Kim DW, Bhatia R, Bosly A, et al. Nilotinib is active in chronic and accelerated phase chronic myeloid leukemia following failure of imatinib and dasatinib therapy. Leukemia. (2010) 24:1299–301. doi: 10.1038/leu.2010.110
72. le Coutre PD, Giles FJ, Hochhaus A, Apperley JF, Ossenkoppele GJ, Blakesley R, et al. Nilotinib in patients with Ph+ chronic myeloid leukemia in accelerated phase following imatinib resistance or intolerance: 24-month follow-up results. Leukemia. (2012) 26:1189–94. doi: 10.1038/leu.2011.323
73. Nicolini FE, Masszi T, Shen Z, Gallagher NJ, Jootar S, Powell BL, et al. Expanding Nilotinib Access in Clinical Trials (ENACT), an open-label multicenter study of oral nilotinib in adult patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase or blast crisis. Leuk Lymphoma. (2012) 53:907–14. doi: 10.3109/10428194.2011.627480
74. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoli J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. (2006) 354:2531–41. doi: 10.1056/NEJMoa055229
75. Apperley JF, Cortes JE, Kim DW, Roy L, Roboz GJ, Rosti G, et al. Dasatinib in the treatment of chronic myeloid leukemia in accelerated phase after imatinib failure: the START A trial. J Clin Oncol. (2009) 27:3472–9. doi: 10.1200/JCO.2007.14.3339
76. Kantarjian H, Cortes J, Kim DW, Dorlhiac-Llacer P, Pasquini R, DiPersio J, et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood. (2009) 113:6322–9. doi: 10.1182/blood-2008-11-186817
77. Gambacorti-Passerini C, Kantarjian HM, Kim DW, Khoury HJ, Turkina AG, Brummendorf TH, et al. Long-term efficacy and safety of bosutinib in patients with advanced leukemia following resistance/intolerance to imatinib and other tyrosine kinase inhibitors. Am J Hematol. (2015) 90:755–68. doi: 10.1002/ajh.24034
78. Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. (2013) 369:1783–96. doi: 10.1056/NEJMoa1306494
79. Jiang Q, Yu L, Qin Y, Lai Y, Huang X. Comparison of the outcome between newly diagnosed patients in the accelerated phase and chronic phase with chronic myeloid leukemia treated with frontline tyrosine kinase inhibitors. Blood. (2017) 130:2876.
80. Balsat M, Alcazer V, Etienne G, Fossard G, Huguet F, Berger MG, et al. First-line second generation tyrosine kinase inhibitors in newly diagnosed accelerated phase chronic myeloid leukemia. Blood. (2018) 132:48. doi: 10.1182/blood-2018-99-113058
81. Masarova L, Cortes JE, Patel KP, O'Brien SM, Nogueras Gonzalez GM, Konopleva MY, et al. Phase 2 study of nilotinib 400 mg twice daily in newly diagnosed patients with accelerated phase of chronic myeloid leukemia, results after 5.7 years of follow-up. Blood. (2018) 132:3011. doi: 10.1182/blood-2018-99-120155
82. Giles FJ, Kantarjian HM, le Coutre PD, Baccarani M, Mahon FX, Blakesley RE, et al. Nilotinib is effective in imatinib-resistant or-intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. (2012) 26:959–62. doi: 10.1038/leu.2011.355
83. Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. (2007) 109:3207–13. doi: 10.1182/blood-2006-09-046888
84. Saglio G, Hochhaus A, Goh YT, Masszi T, Pasquini R, Maloisel F, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. (2010) 116:3852–61. doi: 10.1002/cncr.25123
85. Guilhot F, Apperley J, Kim DW, Bullorsky EO, Baccarani M, Roboz GJ, et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood. (2007) 109:4143–50. doi: 10.1182/blood-2006-09-046839
86. Ottmann O, Saglio G, Apperley JF, Arthur C, Bullorsky E, Charbonnier A, et al. Long-term efficacy and safety of dasatinib in patients with chronic myeloid leukemia in accelerated phase who are resistant to or intolerant of imatinib. Blood Cancer J. (2018) 8:88. doi: 10.1038/s41408-018-0122-3
87. Cortes J, Kim DW, Raffoux E, Martinelli G, Ritchie E, Roy L, et al. Efficacy and safety of dasatinib in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blast phase. Leukemia. (2008) 22:2176–83. doi: 10.1038/leu.2008.221
88. Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. (2018) 132:393–404. doi: 10.1182/blood-2016-09-739086
89. Boddu P, Shah AR, Borthakur G, Verstovsek G, Garcia-Manero G, Daver N, et al. Life after ponatinib failure: outcomes of chronic and accelerated phase CML patients who discontinued ponatinib in the salvage setting. Leuk Lymphoma. (2018) 59:1312–22. doi: 10.1080/10428194.2017.1379076
90. Marks SM, Baltimore D, McCaffrey R. Terminal transferase as a predictor of initial responsiveness to vincristine and prednisone in blastic chronic myelogenous leukemia. N Engl J Med. (1978) 298:812–4. doi: 10.1056/NEJM197804132981503
91. Canellos GP, DeVita VT, Whang-Peng J, Carbone PP. Hematologic and cytogenetic remission of blastic transformation in chronic granulocytic leukemia. Blood. (1971) 38:671–9. doi: 10.1182/blood.V38.6.671.671
92. Iacoboni SJ, Plunkett W, Kantarjian HM, Estey E, Keating MJ, McCredie KB, et al. High-dose cytosine arabinoside: treatment and cellular pharmacology of chronic myelogenous leukemia blast crisis. J Clin Oncol. (1986) 4:1079–88. doi: 10.1200/JCO.1986.4.7.1079
93. Lambertenghi-Deliliers G, Annaloro C, Cortellaro M, Pozzoli E, Oriani A, Polli EE. Idarubicin in blastic crisis of chronic myelogenous leukemia. Haematologica. (1991) 76:406–8.
94. Kantarjian HM, Talpaz M, Kontoyiannis D, Gutterman J, Keating MJ, Estey EH, et al. Treatment of chronic myelogenous leukemia in accelerated and blastic phase with daunorubicin, high-dose cytarabine and granulocyte macrophage colony stimulating factor. J Clin Oncol. (1992) 10:398–405. doi: 10.1200/JCO.1992.10.3.398
95. Di Raimondo F, Milone G, Guglielmo P, Cacciola E, Giustolisi R. Treatment of CML blast crisis with low dose ARA-C. Br J Haematol. (1985) 60:773–4. doi: 10.1111/j.1365-2141.1985.tb07486.x
96. Axdorph U, Stenke L, Grimfors G, Carneskog J, Hansen J, Linder O, et al. Intensive chemotherapy in patients with chronic myelogenous leukaemia (CML) in accelerated or blastic phase – a report from the Swedish CML group. Br J Haematol. (2002) 118:1048–54. doi: 10.1046/j.1365-2141.2002.03765.x
97. Schiffer CA, DeBellis R, Kasdorf H, Wiernik PH. Treatment of the blast crisis of chronic myelogenous leukemia with 5-azacitidine and VP-16-213. Cancer Treat Rep. (1982) 66:267–71.
98. Kantarjian HM, O'Brien S, Cortes J, Giles FJ, Faderl S, Issa JP, et al. Results of decitabine (5-aza-2'deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.98:522–8. doi: 10.1002/cncr.11543
99. Sacchi S, Kantarjian HM, O'Brien S, Cortes J, Rios MB, Giles FJ, et al. Chronic myelogenous leukemia in nonlymphoid blastic phase: analysis of the results of first salvage therapy with three different treatment approaches for 162 patients. Cancer. (1999) 86:2632–41. doi: 10.1002/(SICI)1097-0142(19991215)86:12<2632::AID-CNCR7>3.0.CO;2-A
100. Rea D, Legros L, Raffoux E, Thomas X, Turlure P, Maury S, et al. High-dose imatinib mesylate combined with vincristine and dexamethasone (DIV regimen) as induction therapy in patients with resistant Philadelphia-positive acute lymphoblastic leukemia and lymphoid blast crisis of chronic myeloid leukemia. Leukemia. (2006) 20:400–3. doi: 10.1038/sj.leu.2404115
101. Fruehauf S, Topaly J, Buss EC, Fischer T, Ottmann OG, Emmerich B, et al. Imatinib combined with mitoxantrone/etoposide and cytarabine is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis. Cancer. (2007) 109:1543–9. doi: 10.1002/cncr.22535
102. Quintas-Cardama A, Kantarjian H, Garcia-Manero G, O'Brien S, Faderl S, Ravandi F, et al. A pilot study of imatinib, low-dose cytarabine and idarubicin for patients with chronic myeloid leukemia in myeloid blast phase. Leuk Lymphoma. (2007) 48:283–9. doi: 10.1080/10428190601075973
103. Deau B, Nicolini FE, Guilhot J, Huguet F, Guerci A, Legros L, et al. The addition of daunorubicin to imatinib mesylate in combination with cytarabine improves the response rate and the survival of patients with myeloid blast crisis chronic myelogenous leukemia (AFR01 study). Leuk Res. (2011) 35:777–82. doi: 10.1016/j.leukres.2010.11.004
104. Milojkovic D, Ibrahim A, Reid A, Foroni L, Apperley J, Marin D. Efficacy of combining dasatinib and FLAG-IDA for patients with chronic myeloid leukemia in blastic transformation. Haematologica. (2012) 97:473–4. doi: 10.3324/haematol.2011.057513
105. Strati P, Kantarjian H, Thomas D, O'Brien S, Konoplev S, Jorgensen JL, et al. HCVAD plus imatinib or dasatinib in lymphoid blastic phase chronic myeloid leukemia. Cancer. (2014) 120:373–80. doi: 10.1002/cncr.28433
106. Jain P, Kantarjian HM, Gorab A, Sasaki K, Jabbour EJ, Nogueras Gonzalez G, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: cohort study of 477 patients. Cancer. (2017) 123:4391–402. doi: 10.1002/cncr.30864
107. Oki Y, Kantarjian HM, Gharibyan V, Jones D, O'Brien S, Verstovsek S, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. (2007) 109:899–906. doi: 10.1002/cncr.22470
108. Fang B, Li N, Song Y, Han Q, Zhao RC. Standard-dose imatinib plus low-dose homoharringtonine and granulocyte colony-stimulating factor is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis who have failed prior single-agent therapy with imatinib. Ann Hematol. (2010) 89:1099–105. doi: 10.1007/s00277-010-0991-4
109. Ghez D, Micol JB, Pasquier F, Auger N, Saada V, Spentchian M, et al. Clinical efficacy of second generation tyrosine kinase inhibitor and 5-azacytidine combination in chronic myelogenous leukaemia in myeloid blast crisis. Eur J Cancer. (2013) 49:3666–70. doi: 10.1016/j.ejca.2013.07.147
110. Ruggiu M, Oberkampf F, Ghez D, Cony-Makhoul P, Beckeriche F, Cano I, et al. Azacytidine in combination with tyrosine kinase inhibitors induced durable responses in patients with advanced phase chronic myelogenous leukemia. Leuk Lymphoma. (2018) 59:1659–65. doi: 10.1080/10428194.2017.1397666
111. Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. (2005) 23:3948–56. doi: 10.1200/JCO.2005.11.981
112. Barrett AJ, Ito S. The role of stem cell transplantation for chronic myelogenous leukemia in the 21st century. Blood. (2015) 125:3230–5. doi: 10.1182/blood-2014-10-567784
113. Craddock FC. We do still transplant CML, don't we? Hematology Am Soc Hematol Educ Program. (2018) 2018:177–84. doi: 10.1182/asheducation-2018.1.177
114. Hochhaus A, Saussele S, Rosti G, Mahon FX, Jannsen JJWM, Hjorth-Hansen H, et al. Chronic myeloid leukemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2017) 28(suppl. 4):iv41–51. doi: 10.1093/annonc/mdx219
115. Lee SJ, Kukreja M, Wang T, Giralt SA, Szer J, Arora M, et al. Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood. (2008) 112:3500–7. doi: 10.1182/blood-2008-02-141689
116. Jiang H, Xu LP, Liu DH, Liu KY, Chen SS, Jiang B, et al. Allogeneic hematopoietic SCT in combination with tyrosine kinase inhibitor treatment compared with TKI treatment alone in CML blast crisis. Bone Marrow Transpl. (2014) 49:1146–54. doi: 10.1038/bmt.2014.146
117. Khoury HJ, Kukreja M, Goldman JM, Wang T, Halter J, Arora M, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transpl. (2012) 47:810–6. doi: 10.1038/bmt.2011.194
118. Saussele S, Lauseker M, Gratwohl A, Beelen DW, Bunjes D, Schwerdtfeger R, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study, I. V. Blood. (2010) 115:1880–5. doi: 10.1182/blood-2009-08-237115
119. Oyekunle A, Zander AR, Binder M, Ayuk F, Zabelina T, Christopeit M, et al. Outcome of allogeneic SCT in patients with chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Ann Hematol. (2013) 92:487–96. doi: 10.1007/s00277-012-1650-8
120. Passweg JR, Walker I, Sobocinski KA, Klein JP, Horowitz MM, Giralt SA, et al. Validation and extension of the EBMT risk score for patients with chronic myeloid leukemia (CML) receiving allogeneic haematopoietic stem cell transplants. Br J Haematol. (2004) 125:613–20. doi: 10.1111/j.1365-2141.2004.04955.x
121. Nicolini FE, Basak GW, Kim DW, Olavarria E, Pinilla-Ibarz J, Apperley JF, et al. Overall survival with ponatinib versus allogeneic stem cell transplantation in Philadelphia chromosome-positive leukemias with the T315I mutation. Cancer. (2017) 123:2875–80. doi: 10.1002/cncr.30558
122. Chhabra S, Ahn KW, Hu ZH, Jain S, Assal A, Cerny J, et al. Myeloablative vs reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chronic myeloid leukemia. Blood Adv. (2018) 2:2922–36. doi: 10.1182/bloodadvances.2018024844
123. Buffa P, Romano C, Pandini A, Massimino M, Tirro E, Di Raimondo F, et al. BCR-ABL residues interacting with ponatinib are critical to preserve the tumorigenic potential of the oncoprotein. FASEB J. (2014) 28:1221–6. doi: 10.1096/fj.13-236992
124. Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, et al. Association between imatinib resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. (2008) 100:926–39. doi: 10.1093/jnci/djn188
125. Wagle M, Eiring AM, Wongchenko M, Lu S, Guan Y, Wang Y, et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. (2016) 30:1493–501. doi: 10.1038/leu.2016.51
126. Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature. (2017) 543:733–7. doi: 10.1038/nature21702
127. Hughes TP, Goh YT, Ottmann OG, Minami H, Rea D, Lang F, et al. Expanded phase 1 study of ABL001, a potent, allosteric inhibitor of BCR-ABL, reveals significant and durable responses in patients with CML-chronic phase with failure of prior TKI therapy. Blood. (2016) 128:625. http://www.bloodjournal.org/content/128/22/625.
128. Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer. (2016) 114:605–11. doi: 10.1038/bjc.2016.36
129. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. (2014) 124:30–9. doi: 10.1172/JCI69738
130. Zaritskey GA, Konopka L, Shamsazar J, Bourquelot PM, Jalaluddin M, Li M, et al. A phase II study of oral Panobinostat (LBH589) for chronic phase chronic myeloid leukemia (CML) with resistance to ≥2 BCR-ABL tyrosine kinase inhibitors. Blood. (2008) 112:4254. http://www.bloodjournal.org/content/112/11/4254
131. Okabe S, Tauchi T, Tanaka Y, Kimura S, Maekawa T, Ohyashiki K. Activity of histone deacetylase inhibitors and an aurora kinase inhibitor in BCR-ABL expressing leukemia cells: combination of HDAC and aurora inhibitors in BCR-ABL-expressing cells. Cancer Cell Int. (2013) 13:32. doi: 10.1186/1475-2867-13-32
132. Okabe S, Tauchi T, Kimura S, Maekawa T, Kitahara T, Tanaka Y, et al. Combining the ABL1 kinase inhibitor ponatinib and the histone deacetylase inhibitor vorinostat: a potential treatment for BCR-ABL-positive leukemia. PLoS ONE. (2014) 9:e89080. doi: 10.1371/journal.pone.0089080
133. Matsuda Y, Yamauchi T, Hosono N, Uzui K, Negoro E, Morinaga K, et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant CML cells. Cancer Sci. (2016) 107:1029–38. doi: 10.1111/cas.12965
134. Rauzan M, Chuah CT, Ko TK, Ong ST. The HDAC inhibitor SB939 overcomes resistance to BCR-ABL kinase inhibitors conferred by the BIM deletion polymorphism in chronic myeloid leukemia. PLoS ONE. (2017) 12:e0174107. doi: 10.1371/journal.pone.0174107
135. Goff DJ, Court Recart A, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, et al. A pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. J Stem Cell. (2013) 12:316–28. doi: 10.1016/j.stem.2012.12.011
136. Ko TK, Huang JWJ, Ng KP, Tiong Ong S. The BCL2 inhibitor ABT-199 significantly enhances imatinib induced cell death in chronic myeloid leukemia progenitors. Oncotarget. (2014) 5:9033–8. doi: 10.18632/oncotarget.1925
137. Danial NN, Rothman P. JAK-STAT signaling activated by Abl oncogenes. Oncogene. (2000) 19:2523–31. doi: 10.1038/sj.onc.1203484
138. Chen M, Gallipoli P, DeGeer D, Sloma I, Forrest DL, Chan M, et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst. (2013) 105:405–23. doi: 10.1093/jnci/djt006
139. Gontarewicz A, Balabanov S, Keller G, Colombo R, Graziano A, Pesenti E, et al. Simultaneous targeting of aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood. (2008) 111:4355–64. doi: 10.1182/blood-2007-09-113175
140. Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, et al. The novel aurora a kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. (2011) 15:2057–70. doi: 10.1111/j.1582-4934.2010.01218.x
141. Hielke J, Meulenbeld RHM, Verweij J, de Wit R, de Jonge MJA. Danusertib, an aurora kinase inhibitor. Expert Opin Investig Drugs. (2012) 21:383–93. doi: 10.1517/13543784.2012.652303
142. Giles FJ, Swords RT, Nagler A, Hochhaus A, Ottmann OG, Rizzieri DA, et al. MK-0457, an aurora kinase and BCR-ABL inhibitor, is active in patients with BCR-ABL T315I leukemia. Leukemia. (2013) 27:113–7. doi: 10.1038/leu.2012.186
143. Seymour JF, Kim DW, Rubin E, Haregewoin A, Clark J, Watson P, et al. A phase 2 study of MK-0457 in patients with BCR-ABL T315I mutant chronic myelogenous leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia. Blood Cancer J. (2014) 4:e238. doi: 10.1038/bcj.2014.60
144. Borthakur G, Dombret H, Schafhausen P, Brummendorf TH, Boissel N, Jabbour E, et al. A phase I study of danusertib (PHA-739358) in adult patients with accelerated or blastic phase chronic myeloid leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia resistant or intolerant to imatinib and/or other second generation c-ABL therapy. Haematologica. (2015) 100:898–904. doi: 10.3324/haematol.2014.115279
145. Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. (2009) 27:6041–51. doi: 10.1200/JCO.2009.25.0779
146. Foà R, Vitale A, Vignetti M, Meloni G, Guarini A, De Propris MS, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. (2011) 118:6521–8. doi: 10.1182/blood-2011-05-351403
147. Chalandon Y, Thomas X, Hayette S, Cayuela JM, Abbal C, Huguet F, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. (2015) 125:3711–9. doi: 10.1182/blood-2015-02-627935
148. Martinelli G, Piciocchi A, Papayannidis C, Paolini S, Robustelli V, Soverini S, et al. First report of the Gimema LAL1811 phase II prospective study of the combination of steroids with ponatinib as frontline therapy of elderly or unfit patients with Philadelphia-chromosome positive acute lymphoblastic leukemia. Blood. (2017) 130(suppl. 1):99.
149. Haznedaroglu IC. Drug therapy in the progressed CML patient with multi-TKI failure. Meditter J Hematol Infect Dis. (2015) 7:e2015014. doi: 10.4084/mjhid.2015.014
150. Abboud C, Berman E, Cohen A, Cortes J, DeAngelo D, Deininger M, et al. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. Blood. (2013) 121:4439–42. doi: 10.1182/blood-2013-03-490003
151. Fava C, Kantarjian HM, Jabbour E, O'Brien S, Jain N, Rios MB, et al. Failure to achieve a complete hematologic response at the time of a major cytogenetic response with second-generation tyrosine kinase inhibitors is associated with a poor prognosis among patients with chronic myeloid leukemia in accelerated or blast phase. Blood. (2009) 113:5058–63. doi: 10.1182/blood-2008-10-184960
152. Chen Z, Medeiros LJ, Kantarjian HM, Zheng L, Gong Z, Patel KP, et al. Differential depth of treatment response required for optimal outcome in patients with blast phase versus chronic phase of chronic myeloid leukemia. Blood Cancer J. (2017).7:e521. doi: 10.1038/bcj.2017.4
153. Jabbour E, Garcia-Manero G, Cortes J, Ravandi F, Plunkett W, Gandhi V, et al. Twice-daily fludarabine and cytarabine combination with or without gemtuzumab ozogamicin is effective in patients with relapsed/refractory acute myeloid leukemia, high-risk myelodysplastic syndrome, and blast-phase chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. (2012) 12:244–51. doi: 10.1016/j.clml.2012.03.003
154. Assi R, Kantarjian H, Short NJ, Daver N, Takahashi K, Garcia-Manero G, et al. Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome-positive leukemia. Cin Lymphoma Myeloma Leuk. (2017) 17:897–901. doi: 10.1016/j.clml.2017.08.101
155. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. (2014) 123:3951–62. doi: 10.1182/blood-2013-10-536078
156. Raspadori D, Pacelli P, Sicuranza A, Abruzzese E, Iurlo A, Cattaneo D, et al. Flow cytometry assessment of CD26+ leukemic stem cells in peripheral blood: a simple and rapid new diagnostic tool for chronic myeloid leukemia. Cytometry B Clin Cytom. (2019) 98:294–9. doi: 10.1002/cyto.b.21764
157. Bocchia M, Sicuranza A, Abruzzese E, Iurlo A, Sirianni S, Gozzini A, et al. Residual peripheral blood CD26+ leukemic stem cells in chronic myeloid leukemia patients during TKI therapy and during treatment-free remission. Front Oncol. (2018) 8:194. doi: 10.3389/fonc.2018.00194
158. Blatt K, Menzl I, Eisenwort G, Cerny-Reiterer S, Herrmann H, Herndlhofer S, et al. Phenotyping and target expression profiling of CD34+/CD38− and CD34+/CD38+ stem- and progenitor cells in acute lymphoblastic leukemia. Neoplasia. (2018) 20:632–42. doi: 10.1016/j.neo.2018.04.004
159. Nagel I, Bartels M, Duell J, Oberg HH, Ussat S, Bruckmueller H, et al. Hematopoietic stem cell involvement in BCR-ABL1-positive ALL as a potential mechanism of resistance to blinatumomab therapy. Blood. (2017) 130:2027–31. doi: 10.1182/blood-2017-05-782888
160. Patel AB, O'Hare T, Deininger MW. Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am. (2017) 31:589–612. doi: 10.1016/j.hoc.2017.04.007
161. Savona M, Talpaz M. Getting to the stem of chronic myeloid leukemia. Nat Rev Cancer. (2008) 8:341–50. doi: 10.1038/nrc2368
162. Vigneri P, Stagno F, Stella S, Cupri A, Forte S, Massimino M, et al. High BCR–ABL/GUSIS levels at diagnosis of chronic phase CML are associated with unfavorable responses to standard-dose imatinib. Clin Cancer Res. (2017) 23:7189–98. doi: 10.1158/1078-0432.CCR-17-0962
163. Pfirrmann M, Baccarani M, Saussele S, Guilhot J, Cervantes F, Ossenkoppele G, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. (2016) 30:48–56. doi: 10.1038/leu.2015.261
164. Castagnetti F, Gugliotta G, Baccarani M, Breccia M, Specchia G, Levato L, et al. Differences among young adults, adults and elderly chronic myeloid leukemia patients. Ann Oncol. (2015) 26:185–92. doi: 10.1093/annonc/mdu490
165. Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. (2011) 117:6660–8. doi: 10.1182/blood-2010-08-304477
166. Mothy M, Yong AS, Szydlo RM, Apperley JF, Melo JV. The polycomb group BMI1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood. (2007) 110:380–3. doi: 10.1182/blood-2006-12-065599
167. Jamieson CH, Ailles LE, Dylla SJ, Muljtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. (2004) 351:657–67. doi: 10.1056/NEJMoa040258
168. Radich JP, Dai H, Mao M, Oehler V, Schelter J, Druker B, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA. (2006) 103:2794–9. doi: 10.1073/pnas.0510423103
169. Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. (2018) 132:948–61. doi: 10.1182/blood-2018-02-832253
170. Hanfstein B, Muller MC, Hochhaus A. Response-related predictors of survival in CML. Ann Hematol. (2015) 94(suppl. 2):S227–39. doi: 10.1007/s00277-015-2327-x
171. Marin D, Ibrahin AR, Lucas C, Gerrard G, Wang L, Szydlo RM, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. (2012) 30:232–8. doi: 10.1200/JCO.2011.38.6565
172. Hanfstein B, Muller MC, Hehlmann R, Erben P, Lauseker M, Fabarius A, et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (CML). Leukemia. (2012) 26:2096–102. doi: 10.1038/leu.2012.85
173. Jabbour E, Kantarjian HM, Saglio G, Steegmann JL, Shah NP, Boquè C, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood. (2014) 123:494–500. doi: 10.1182/blood-2013-06-511592
174. Branford S, Yeung DT, Parker WT, Roberts ND, Purins L, Braley JA, et al. Prognosis for patients with CML and >10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood. (2014) 124:511–8. doi: 10.1182/blood-2014-03-566323
175. Hanfstein B, Shlyakhto V, Lauseker M, Hehlmann R, Saussele S, Dietz C, et al. Velocity of early BCR-ABL transcript elimination as an optimized predictor of outcome in chronic myeloid leukemia (CML) patients in chronic phase on treatment with imatinib. Leukemia. (2014) 28:1988–92. doi: 10.1038/leu.2014.153
176. Goldberg SL, Chen L, Guerin A, Macalalad AR, Liu N, Kaminsky M, et al. Association between molecular monitoring and long-term outcomes in chronic myelogenous leukemia patients treated with first line imatinib. Curr Med Res Opin. (2013) 29:1075–82. doi: 10.1185/03007995.2013.812034
177. Kantarjian H, O'Brien S, Cortes J, Giles F, Thomas D, Kornblau S, et al. Sudden onset of the blastic phase of chronic myelogenous leukemia: patterns and implications. Cancer. (2003) 98:81–5. doi: 10.1002/cncr.11477
Keywords: accelerated phase, blast phase, clonal evolution, molecular monitoring, allogeneic stem cell transplant
Citation: Bonifacio M, Stagno F, Scaffidi L, Krampera M and Di Raimondo F (2019) Management of Chronic Myeloid Leukemia in Advanced Phase. Front. Oncol. 9:1132. doi: 10.3389/fonc.2019.01132
Received: 08 April 2019; Accepted: 10 October 2019;
Published: 25 October 2019.
Edited by:
Massimo Breccia, Sapienza University of Rome, ItalyReviewed by:
Elisabetta Abruzzese, University of Rome Tor Vergata, ItalyIbrahim C. Haznedaroglu, Hacettepe University Medical School, Turkey
Copyright © 2019 Bonifacio, Stagno, Scaffidi, Krampera and Di Raimondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimiliano Bonifacio, bWFzc2ltaWxpYW5vLmJvbmlmYWNpb0B1bml2ci5pdA==