 Emily N. Arner1
Emily N. Arner1 Rolf A. Brekken
Rolf A. Brekken
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 October 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01049
This article is part of the Research Topic Oncogenic RAS-dependent Reprogramming of Cellular Plasticity View all 10 articles
Cellular plasticity, a feature associated with epithelial-to-mesenchymal transition (EMT), contributes to tumor cell survival, migration, invasion, and therapy resistance. Phenotypic plasticity of the epithelium is a critical feature in multiple phases of human cancer in an oncogene- and tissue-specific context. Many factors can drive epithelial plasticity, including activating mutations in KRAS, which are found in an estimated 30% of all cancers. In this review, we will introduce cellular plasticity and its effect on cancer progression and therapy resistance and then summarize the drivers of EMT with an emphasis on KRAS effector signaling. Lastly, we will discuss the contribution of cellular plasticity to metastasis and its potential clinical implications. Understanding oncogenic KRAS cellular reprogramming has the potential to reveal novel strategies to control metastasis in KRAS-driven cancers.
KRAS is mutated in an estimated 30% of all cancers. In fact, the small GTPase KRAS has an activating point mutation in over 90% of pancreatic cancer patients (1), ~35% of lung cancer patients, and ~40% of colorectal cancer patients (2). As such, oncogenic KRAS is established as a driver of cancer initiation, progression, metastasis, therapy resistance, and immune suppression in multiple cancers (3). KRAS is an alluring therapeutic target, yet strategies targeting KRAS have been largely unsuccessful. However, understanding downstream effectors of KRAS signaling might provide alternative strategies to indirectly target KRAS and the cellular reprogramming driven by oncogenic KRAS signaling.
Recent evidence suggests that individual KRAS mutations activate distinct signaling pathways (2, 4). For example, gene expression analysis of primary human NSCLCs expressing G12C or G12V activating mutations in KRAS showed distinct gene expression profiles compared to cell lines expressing other KRAS activating point mutations (5). Similarly, Hammond et al. (6) engineered SW48 colorectal cancer cells, which are KRAS wild-type, to express KRAS point mutations: G12V, G12D, or G13D. Subsequent phosphoprotein expression analysis revealed the activation of differential signaling pathways in distinct KRAS mutational contexts. In support of these results, a large-scale screening effort using RNAi, small-molecules, and genetic analysis of cell lines and TCGA analysis revealed that KRAS binds to different effector proteins depending on the cellular context, which was determined by cell lineage, secondary mutations, and metabolic state (7). To further study context-dependent KRAS signaling in cancer, Brubaker et al. (4) developed a statistical approach to humanize multiplexed quantitative proteomic data from mouse models of colon and pancreatic cancer. Through the integration of proteomics and mutation data from human PDAC cohorts they identified synthetic lethal partners with oncogenic KRAS and mutant KRAS tissue-specific and cross-tissue signaling. Each of these studies indicate that the signaling outcome and thus cellular phenotype driven by KRAS mutation is deeply dependent on cellular context.
Epithelial plasticity or an epithelial-to-mesenchymal transition (EMT) is a key cellular program that can be activated by KRAS. EMT contributes to tumor progression by enhancing tumor cell survival and therapy resistance and by facilitating success in the metastatic cascade. In this review, we will introduce cellular plasticity and its effect on cancer progression and therapy resistance and then summarize drivers of EMT with an emphasis on KRAS signaling. Lastly, we will discuss the contribution of cellular plasticity to metastasis and its potential clinical implications.
Cellular plasticity serves as a mechanism of tissue adaptation and regeneration in normal tissues and can also predispose tissue to cancer transformation (8). In the pancreas, pancreatic epithelial and acinar cells display robust plasticity, enabling adaptation to metabolic and environmental stress. In pancreatic cancer, tumor cells alter their phenotype as a result of exposure to diverse metabolic conditions, signaling molecules, stromal elements, and therapeutic agents. This plastic state in tumor cells can facilitate tumor progression, including metastasis, chemoresistance, and immune evasion (8).
Acinar-to-ductal metaplasia (ADM) (9), describes a process where normal pancreatic acinar cells assume a duct-like state in the setting of chronic injury, such as pancreatitis. When pancreatitis resolves in normal/non-malignant pancreatic tissue, ADM lesions revert to acinar morphology. However, if KRAS-transformed acinar cells are subjected to the stress of pancreatitis, precancerous pancreatic intraepithelial neoplasia often forms (10–14). This suggests that pancreatic ductal adenocarcinomas (PDACs) may arise from acinar cells that have undergone transdifferentiation to a duct-like state. Normal pancreatic cells are sensitive to the transforming effects of mutant KRAS and the loss of phosphatase and tensin homolog (15), indicating that the likelihood of tumor formation and eventual histologic tumor type depends on the specific drivers that are present as well as the cellular compartments in which they are expressed (16–20).
EMT is another example of cellular plasticity program that is used by cells and tissues to adapt to cues or cellular stress. EMT classically defined is a developmental program that is instrumental in early embryo patterning during gastrulation (21, 22) and is characterized by epithelial cells losing cell-to-cell adhesion, epithelial tight junctions, and desmosomes. These changes are thought to occur through coordinated genetic reprogramming induced by EMT-transcription factors (EMT-TFs) that are activated in response to extracellular cues (21). These cues include growth factors such as transforming growth factor-β (TGFβ), epidermal growth factor (EGF), hepatocyte growth factor (HGF), and insulin-like growth factor 1 (IGF1) (21, 23–26). This essential developmental program can be hijacked during tumorigenesis to promote increased cell migration and survival.
EMT in tumor cells can also be induced by cellular stress such as inflammation or nutrient/oxygen deprivation (27), and transforming oncogenes including oncogenic KRAS (28, 29). The genetic reprogramming associated with EMT in normal tissue or cancer leads to a shift from an epithelial to a mesenchymal phenotype. Epithelial cells often have polygonal shapes in monolayer culture, are polarized along their apical-basal axis and are tightly joined to one another laterally through adherens junctions. In contrast, mesenchymal cells exhibit spindle-like morphology and are loosely attached to the surrounding stroma through focal adhesions, which contributes to increased motility and invasive behavior (30) (Figure 1).
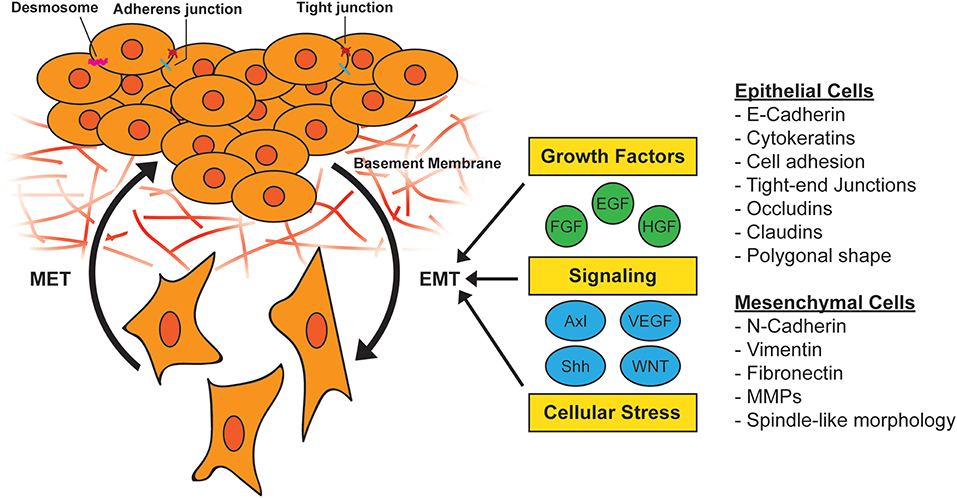
Figure 1. Activation of epithelial-to-mesenchymal transition (EMT). During EMT, epithelial cells lose their cell-to-cell adhesion and adopt a more spindle-like morphology due to the expression of mesenchymal markers. This morphology change results in the ability to escape the basement membrane and invade and survive stressful situations, including therapy. EMT can be induced by a variety of growth factors, signaling pathways, and cellular stress such as hypoxia and nutrient deprivation. MET, mesenchymal-to-epithelial transition; MMP, matrix metallopeptidase.
In epithelial tumors, the manifestation of an EMT program is associated with tumor grade. High-grade cancer is aggressive and characterized by a loss of normal tissue structure and architecture. High-grade tumors are often described as poorly differentiated and mesenchymal, displaying tumor cells that have undergone EMT. In contrast, low-grade tumors are characterized as well-differentiated cancers that retain an epithelial phenotype. Across human cancer, tumors that are high grade and poorly differentiated carry a worse prognosis with a high likelihood of metastasizing to distant organs (8).
EMT is a common feature associated with tumor progression and is thought to be critical to cancer cell dissemination in some tumors (31–33). The metastasis of epithelial tumors, such as PDAC, requires the cancer cells to escape epithelial nests, invade surrounding stroma, intravasate into blood or lymphatic vessels, survive circulation, and extravasate at the secondary site, where successful cells form micrometastases and eventually macrometastases (34). The escape of tumor cells from tumor cell nests encapsulated by a basement membrane can be facilitated by tumor cell epithelial plasticity, which results in epithelial tumor cells losing contact with the basement membrane and nearby cells while adopting mesenchymal-like features that enable cell migration and invasion. This is a common feature in mouse models of PDAC (35–37). While epithelial plasticity alters morphology and cell-cell contact it also enhances tumor cell survival under stressful environmental conditions, such as chemotherapy and radiation (32, 38–40). EMT and metastasis are generally considered to be late events in tumorigenesis; however, EMT and the metastatic cascade has been shown to occur even in “preinvasive” stages of PDAC (35). Thus, the concept that EMT is driven by the oncogenotype of a tumor is worthy of consideration.
In KRAS-driven tumors, such as PDAC, tumorigenesis and epithelial plasticity programs are often intertwined. For example, in genetically engineered mouse models (GEMMs) of PDAC harboring mutant KRAS, EMT was found to be an early event after tumor formation (35). Furthermore, co-expression of mutant KRAS and a polycomb-group repressor complex protein, Bmi1, in normal human pancreatic duct-derived cells (HPNE) induces partial EMT via upregulation of the EMT-TF Snail (28, 41–43). In addition, multiple receptor tyrosine kinases (RTKs) implicated in the induction of EMT activate RAS and the resulting signaling cascade induces the expression of EMT-TFs in a RAS-dependent manner (43–46). Other pathways have also been shown to interact with mutant KRAS to drive EMT. For example, the EMT-TF, Snail has been shown to induce TGFβ signaling in a mutant KRAS dependent manner to drive EMT (47). Other studies revealed that signal transducer and activator of transcription 3 (STAT3) can mediate a synergistic interaction between TGFβ and RAS resulting to enhance Snail driven EMT (48). Other small GTPases, RAC, and RHO, are also activated by RAS via PI3K to drive EMT by regulating adherens junctions and focal adhesions (49). Thus, while mutant KRAS driven tumors are often dependent on RAS activity for development and maintenance (28, 41, 42) the prominent oncogenic mutation also is a critical component of epithelial plasticity.
Epithelial plasticity is a key chemoresistance and immune surveillance evasion strategy exploited by tumor cells (50, 51). Plastic tumor cells exhibit increased rates of resistance to therapy including radio-, chemo-, targeted, and immunotherapy (39, 40, 52–54). Stress, such as inflammation, nutrient/oxygen deprivation, and therapy can induce epithelial plasticity in cancer cells (27). A common consequence of EMT is reduced drug uptake by tumor cells. For example, the expression of equilibrative nucleoside transporter 1 (ENT1), which can transport nucleoside analog chemotherapy into cells, is often reduced in tumor cells that have undergone EMT. However, tumors engineered to lack EMT transcription factors (EMT-TFs), such as Snail and Twist, showed elevated ENT1 expression and increased sensitivity to gemcitabine, a nucleoside analog (55). Consistent with these results, Ludwig et al. (54) found that inhibition of AXL reduced epithelial plasticity in models of PDAC, increased ENT1 expression and enhanced sensitivity to gemcitabine when compared to gemcitabine alone or control treated animals. To combat chemoresistance in cancer patients, intermittent dosing or “drug holidays” have been suggested, although recent studies have revealed that resistance driven by oncogenic KRAS is not reversible (56). In human cancer cell lines, therapy resistance driven by mutant KRAS was found to irreversibly drive ZEB1-dependent EMT and chemoresistance through the hyperactivation of ERK1/2 (56), arguing against the use of intermittent dosing in tumors driven by oncogenic KRAS. Fischer et al. (57) showed in a spontaneous breast-to-lung metastasis model that EMT contributes to chemotherapy resistance, as mesenchymal-like tumor cells survived cyclophosphamide treatment, demonstrating reduced proliferation, apoptotic tolerance, and increased expression of chemoresistance-related genes. These observations highlight the potential increase in therapeutic efficacy that might result from combining standard therapy with strategies to combat epithelial plasticity.
The hypoxic state of pancreatic tumors increases tumor cell migration and chemoresistance (58). In fact, EMT can be driven by hypoxia often via the induction of TGFβ (59). Additionally, in human pancreatic cancer cell lines, hypoxia has been shown to drive EMT in an NFκB dependent manner through the stability of hypoxia-inducible factor 1 alpha (HIF-1α) and subsequent activation of RelA (p. 65) (60–63), a subunit of the NFκB family of transcription factors (64, 65). NFκB is considered a crucial component of drug resistance in mutant KRAS driven tumors such as pancreatic cancer and colorectal cancer, which typically expresses high levels of the protein (66). The activation of NFκB has been shown to upregulate anti-apoptosis proteins such as Bcl-XL and Bcl-2, promoting chemoresistance (67, 68). As such, NFκB inhibition might be an approach to combat chemoresistance in tumors with KRAS-driven EMT.
Resistance to targeted therapy has also been associated with a mesenchymal state. In non-small cell lung cancer (NSCLC), the expression of an EMT gene signature, which included AXL expression, was associated with resistance to treatment with epidermal growth factor receptor (EGFR) and phosphatidylinositol 3-kinase (PI3K) inhibitors (69–73). Similarly, in vitro studies suggested that epithelial NSCLC cell lines are more sensitive to EGFR inhibitors than mesenchymal cell lines (74), and that when AXL is inhibited, sensitivity to EGFR inhibitors is increased (75, 76). In breast cancer patients, the EMT program also serves as a major driver of drug resistance, disease occurrence, and systemic dissemination (52, 77, 78).
In addition to targeted and chemotherapy, EMT has been associated with resistance to immunotherapy (79). In murine melanoma cells, Snail, a canonical EMT-TF, was found to be necessary and sufficient for resistance to cytotoxic T-cell–mediated killing via the induction of regulatory T cells. The effect was driven by immunosuppressive CD11c+ dendritic cells, which were generated in response to Snail-expressing melanoma cells (40). Similarly, immune therapy-resistant melanomas display a mesenchymal gene signature, including the downregulation of E-cadherin and upregulation of factors involved in extracellular matrix (ECM) remodeling, angiogenesis, and wound healing (80). Additionally, the immune system is a key component of chemotherapy responses, as many chemotherapeutic agents directly affect the immune landscape of tumors (81). Therefore, identification of key signaling pathways involved in epithelial plasticity could reveal overlap with tumor immune evasion and new therapeutic targets, inhibition of which increases the efficacy of chemo- and immunotherapy.
Metabolic alterations are associated with mutant KRAS-induced EMT. Cancer cells often increase glycolytic flux to meet the high energy demand to support rapid cell growth and division (82). In contrast to normal cells that typically generate energy via the breakdown of pyruvate, cancer cells generate energy by the non-oxidative breakdown of glucose with tumor cells displaying glycolytic rates up to 200 times higher than normal cells in the body (83). This preferential activation of glycolysis for energy supply is referred to as the “Warburg Effect” (83). In pre-clinical models as well as human patient samples, oncogenic Kras signaling can transcriptionally upregulate the glucose transporter GLUT1, as well as multiple enzymes in the glycolytic pathway [e.g., Hexokinase1 (HK1), Hexokinase2 (HK2), Phosphofructokinase1 (PFK-1), and Lactate dehydrogenase A (LDHA)] (82, 84, 85). Hypoxia, a common environmental condition in solid tumors, triggers O-linked β-N-acetylglucosamine (O-GlcNAcylation) at S529 of PFK-1, inducing glycolysis and giving a selective growth advantage to the cancer cells (86, 87). Cancer induced HIF-1α and MUC1 have also been shown to upregulate the expression of key glucose transporters and glycolytic enzymes, including GLUT1 and aldolase A, which leads to increased glucose uptake and glycolysis (82, 84, 88). In addition to glycolysis, recent evidence suggests oncogenic KRAS drives glucose into the hexosamine biosynthetic pathway (HBP), which is required for multiple glycosylation events (89, 90). Taparra et al. (91), recently showed in models of lung tumorigenesis, that KRAS and the EMT program coordinated elevated expression of key enzymes within the HBP pathway. Additionally, they showed that elevated O-GlcNAcylation of intracellular proteins such as the EMT-TF Snail results in suppressed oncogenic-induced senescence and accelerated lung tumorigenesis (91). Understanding the evident metabolic changes driven by oncogenic KRAS and reinforced by epithelial plasticity may reveal novel therapeutic targets for KRAS-driven tumorigenesis.
A variety of stimuli can induce EMT, including soluble factors, ECM components, environmental conditions, and oncogenic transcriptional programs (92). These stimuli, which include signaling factors such as TGFβ, Wnt, Notch, and Sonic hedgehog (Shh), as well as growth factors such as EGF and platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), serve as ligands for the signaling pathways they activate (Figure 1). EMT programs can also be activated in response to several paracrine signals in parallel (21). These networks activate signal cascades and intermediates that include mitogen-activated protein kinases (MAPKs), PI3K, AKT, Smads, RhoB, c-Fos, and RAS (93), which then regulate EMT-TFs. RTKs are common initiation sites for signaling that induces EMT-TF activity.
AXL is an archetypal RTK associated with EMT (94–96) and with worse outcomes in multiple tumor types (71, 94, 97, 98). Consistent with poor outcomes, AXL expression also is associated with metastasis and resistance to therapy (54, 96). AXL is a member of the TAM (Tyro3, AXL, MerTK) family of RTKs (99). Its ligand, growth arrest-specific gene 6 (GAS6) induces AXL signaling by stimulating the auto-phosphorylation of several tyrosine residues of AXL, which function as docking sites for multiple substrates including PI3K, phospholipase C, and c-SRC (100, 101). Additionally, AXL can be activated by forming heterodimers with non-TAM family proteins, such as EGFR, PDGFR, or another TAM family member (71). Elevated AXL expression is found in multiple cancer types, including lung, breast, ovarian, gastric, colon, pancreatic, and prostate (71–73, 94, 95, 97, 102, 103). AXL expression is induced by drivers of EMT, for example TGFβ, and is generally associated with markers of EMT including N-cadherin and vimentin (104, 105).
Our lab and others have shown that AXL expression in RAS-driven cancers, such as PDAC, maintains epithelial plasticity (96). GAS6-AXL signal transduction is required to maintain epithelial-mesenchymal plasticity traits of PDAC (96). When AXL was inhibited in GEMMs of pancreatic cancer, Ludwig et al. (54) observed an increase of epithelial differentiated tumor cells. In addition to chemotherapy resistance, AXL has been strongly implicated in resistance to targeted therapy such as EGFR and PI3K/AKT inhibitors (72, 73).
RAS genes (HRAS, KRAS, and NRAS) are the most frequently mutated gene family in cancer (106). Of these, KRAS is the most mutated (86% of all RAS-mutant cancers), followed by NRAS (12%), and HRAS (4%) (107). KRAS mutations are frequent in PDAC, lung, and colorectal cancers, and also occur in other cancers such as multiple myeloma (2, 108).
KRAS, a small GTPase, functions as a molecular switch, cycling between an active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound states (109). In non-transformed cells, RAS is typically GDP-bound and inactive, but upon activation of RTKs, there is a rapid activation of RAS-GTP, leading to the activation of intracellular signaling networks that promote growth, proliferation, and migration (110) (Figure 2). Because KRAS-activating mutations cluster around the nucleotide-binding pocket (2), these mutations cause RAS to be persistently GTP-bound and constitutively active, resulting in the hyperactivation of signaling networks to drive cancer growth and progression (111).
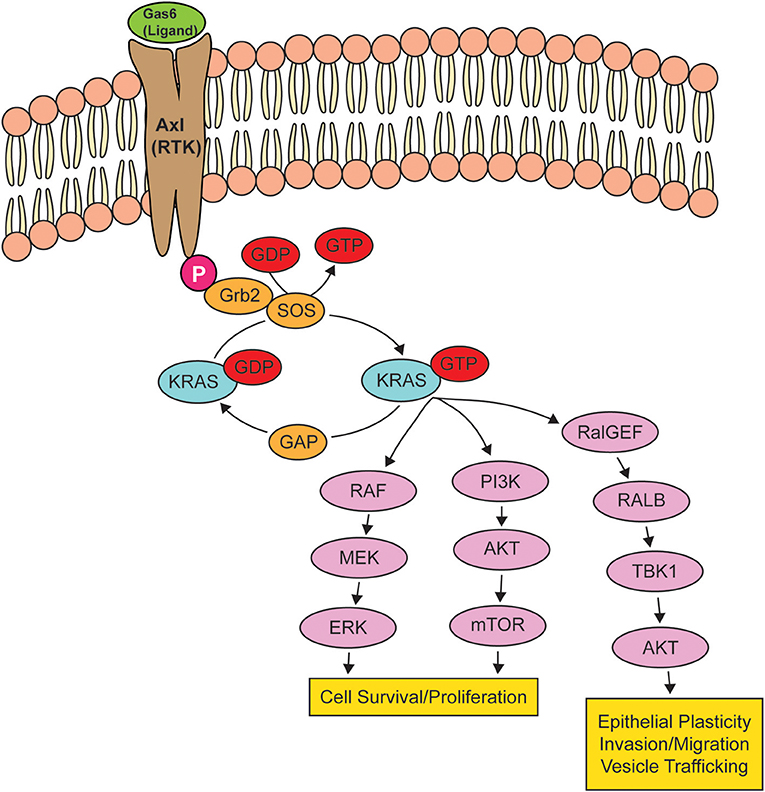
Figure 2. Oncogenic KRAS effector pathways. When a receptor tyrosine kinase (RTK) is activated by its ligand, KRAS binds to GTP, rendering it active until the GTP hydrolyzes to GDP, turning KRAS off. When KRAS is mutated, KRAS remains bound to GTP, leading to the overstimulation of KRAS signaling pathways, resulting in cell survival and proliferation, epithelial plasticity, and migration. The activation of RTK AXL by GAS6 is shown as a potential signaling pathway that can drive an epithelial-to-mesenchymal transition via the activation of KRAS.
Multiple RTKs, including AXL and EGFR, can activate KRAS (112). Signaling networks downstream of RAS such as ERK/MAPK and PI3K/AKT can mediate mutant Ras-induced EMT, such that the inhibition of MEK1 or AKT (113) can reverse RAS-stimulated epithelial plasticity. Genovese et al. (114) completed a gene set enrichment analysis of highly metastatic and poorly metastatic clonal cells lines isolated from a GEMM of PDAC, i.e., KPfC mice (KRASLSLG12D/+; Trp53Lox/Lox; Pdx1Cre/+). Their analysis revealed that “metastasis-low” clones exhibited a downregulating of KRAS signature genes, whereas “metastasis-high” clones exhibited a higher expression of KRAS signature genes (114). After validation through in vivo lineage tracing, their study demonstrated that in PDAC, cells reside in a spectrum of epithelial-mesenchymal states where mesenchymal cells activate KRAS signaling at a higher level.
Other genome-sequencing studies revealed genetic heterogeneity beyond a few frequently mutated drivers in human PDAC (115–121). The heterogeneity in genomic changes makes it challenging to link definitive genomic alterations to biological, morphological, or clinical phenotypes (116, 121). Despite these challenges, Mueller et al. (37), found that the gene dosage of KRAS G12D in human and mouse PDAC correlated with a markedly increased metastatic potential and a mesenchymal phenotype. These results link the aggressive mesenchymal PDAC subtype with the highest dosage of mutant KRAS and Ras-related transcriptional programs. Additionally, oncogenic Ras is closely associated with resistance to drug therapy and pathways that drive PDAC initiation, progression, and metastasis.
Although the majority of RAS effector-targeted therapies inhibit the RAF and PI3K signaling networks, the RALGEF pathway encompassing RALA and RALB GTPases are more consistently activated than RAF or PI3K in human PDAC (122, 123). Additionally, it has been demonstrated in human cell lines that RALGTPase activation is essential for RAS-induced transformation in a spectrum of human epithelial cells and that RALGTPase activation alone is sufficient to induce a tumorigenic phenotype in some settings (124, 125). Given that RAS signaling is a driver of epithelial plasticity and that the RALGEF pathway is a critical effector of RAS, investigating RALGEF signaling has the potential to reveal novel targets involved in epithelial plasticity, metastasis, and therapy resistance in RAS-mutant tumors.
The serine/threonine protein kinase TANK-binding kinase 1 (TBK1) is an atypical Ikβ kinase, that together with its homolog, IKKε, contributes to innate immunity by activating interferon regulatory factor 3/7 (IRF3/7) thereby inducing type 1 interferon gene expression in response to pathogen exposure (126, 127). Additionally, TBK1 kinase activity supports cell growth, self-renewal, pathogen clearance, and organelle function (128–131). TBK1 is a constituent of the RAL pathway and is crucial to the induction and progression of RAS-driven cancers (105, 130, 132, 133). Additionally, TBK1 has been linked to the survival of mutant KRAS-expressing cells (128) and can directly activate AKT (130). The importance of RALB and TBK1 to RAS-induced lung cancer was confirmed in a RNA inhibitor screen of synthetic lethal partners of oncogenic KRAS, where RALB and TBK1 were identified as top targets (132). Further, Cooper et al. (134) screened 100 NSCLC lines for sensitivity to TBK1 inhibitors Bx795 and compound II to tease out biological features of TBK1-dependent cell lines. Sensitivity profiles correlated strongly with profiles of multiple inhibitors of the AKT/mTOR pathway, particularly in mutant KRAS NSCLC lines, suggesting a mechanistic interaction between TBK1 and the mTOR pathway (134). Further analysis of TBK1 inhibitor (TBK1i)-sensitive cell lines revealed mutations in RAS family members and increased mesenchymal gene expression compared to TBK1i-resistant cell lines, which had a more differentiated gene expression profile.
In support of the contribution of TBK1 to RAS-induced EMT, we reported that TBK1 expression is associated with a poor prognosis in pancreatic cancer patients (135). Furthermore, we found that the loss of TBK1 function resulted in reduced invasion, migration, and tumor growth, and reduced metastatic events in preclinical models of mutant KRAS PDAC, indicating that TBK1 actively contributes to pancreatic cancer progression (105). In fact, one of the most significant and top dysregulated gene networks distinguishing TBK1 WT and TBK1-mutant tumors was the cancer/cellular movement networks, including many genes involved in EMT. In comparison with TBK1 WT tumors, tumors from TBK1 mutant mice showed a trend toward higher expression of epithelial markers and lower expression of mesenchymal markers; this trend was confirmed at the protein level (105). Mechanistic studies established that TBK1 promotes EMT downstream of AXL in PDAC, in a RAS-RALB dependent manner (105). Although the precise mechanism of how TBK1 promotes EMT is unclear, evidence suggests that TBK1 can directly activate AKT (130), which can drive EMT via the induction of EMT-TFs (e.g., Snail and Slug) (38, 136, 137). Further studies are needed to delineate the whether the interaction between TBK1 and AKT is critical to the mesenchymal phenotype of tumor cells in PDAC. The identification of additional TBK1 substrates that might promote EMT programs is also needed.
In contrast, knockdown of TBK1 in estrogen receptor α-positive (ERα) breast cancer cells resulted in enhanced tumorigenesis and lung metastasis in part by increasing EMT (138). Further studies are required to investigate if this pathway is dependent on oncogenic RAS. Another group observed that TBK1 is active in mutant NRAS melanoma and promoted migration and invasion of these cells (139), suggesting that RAS-driven epithelial plasticity may be active in the presence of other RAS isoform-driven cancers. Regardless, these studies suggest that therapies targeting TBK1 could be used to reduce EMT in Ras-mutant tumors.
In agreement with the concept that TBK1 loss affects antitumor immunity, studies by the Cantley (140) and Barbie (133) groups have reported that immune evasion and metastatic behavior are associated with the cGAS/STING/TBK1 innate immune pathway in cancer cells (133, 140, 141). Canadas et al. (133) revealed that mesenchymal tumor subpopulations with high AXL expression and low histone-lysine N-methyltransferase levels trigger the expression of a specific set of interferon-stimulated antisense endogenous retroviruses (ERVs). These ERVs were present in human cancer cells that produced tumors with hyperactive innate immune signaling, myeloid cell infiltration, and utilized immune checkpoint pathways. Therapeutically, this may have important implications for immune oncology drug combinations. In the second study, Bakhoum et al. (140) found that chromosomal instability (CIN) of cancer cells, promoted cellular invasion and metastasis through the presence of double-stranded DNA in the cytosol. Clustering of tumor cells via EMT genes accurately classified most cells according to their CIN status and revealed that CIN-high cells expressed mesenchymal markers. This CIN-high population also exhibited increased migratory and invasive behavior in vitro, underwent actin cytoskeletal reorganization, and stained positive for mesenchymal markers such as vimentin and β-catenin. Additionally, cells derived from metastases more frequently exhibited cytoplasmic micronuclei than CIN-low or primary tumor-derived cells. These studies showed that cytosolic DNA activates the cGAS/STING pathway to mediate EMT, invasion, and metastasis (140). Under normal conditions, the cGAS-STING pathway functions as an innate cellular defense mechanism against viral infections. Once STING activates TBK1, TFs such as IRF3 and NF-κB are phosphorylated and translocate to the nucleus (142), where they mediate the transcription of inflammatory genes (143–146). In human breast and lung cancer-derived cell lines, chronic cGAS-STING activity resulting from chromosome instability has been shown to drive migration, invasion, and metastasis (140). Additionally, CIN can result in elevated mutant KRAS gene dosage in pancreatic cancer, which can drive higher expression of EMT genes and increase metastasis (37).
Similar to epithelial plasticity, CIN has been implicated in treatment resistance by generating heterogeneity within the tumor that enhances natural selection, thereby promoting tumor cell survival, immune evasion, drug resistance, and metastasis (37, 147–152). Given the widespread nature of CIN in human cancer, therapies targeting CIN and cGAS/STING have therapeutic potential to reduce therapy resistance and reduce metastasis.
EMT is thought to be regulated largely through changes in the expression of genes necessary for the epithelial state, such as adherens junctions and tight junction components, which are transcriptionally repressed through the activation of EMT TFs including Snail, Twist, and Zeb (153). As previously mentioned, EMT can be induced by many signaling factors, such as TGFβ, EGF, FGF, HGF, NOTCH, and Wnt ligands. These factors initiate signaling cascades, leading to the expression of one or more EMT-TFs, which inhibit E-cadherin transcription by binding to E-boxes within the E-cadherin promoter region (154, 155).
EMT-TFs are often associated with poor patient outcomes. In resected PDAC, nearly 80% of tumors expressed moderate to strong levels of SNAI1, while only 50% showed SNAI2 expression, and very few expressed TWIST (156). Additionally, ZEB1 expression in pathologic specimens correlated with advanced tumor grade and worse outcomes (157, 158). Functions for individual EMT-TFs in different cancers have been described: for ZEB1 and ZEB2 in melanoma (159, 160), Snail and Slug in breast cancer (161), and for Sox4 (162), and Prrx (163) in PDAC. These functions can be tissue-specific, as demonstrated by the different functions of Snail in the metastasis of breast cancer (164) and PDAC (55). Such functional diversity of EMT-TFs suggests that distinct EMT programs operate in different tissues during tumor progression. With this in mind, therapeutic strategies targeting EMT-TFs should consider tissue context and target multiple factors simultaneously (112).
ZEB1 is a zinc finger/homeodomain protein that is associated with EMT and tumor progression. ZEB1 functions as a transcriptional activator by binding to CtBP co-repressors, histone acetyl-transferase TIP60, chromatin remodeling ATPase BRG1, and SIRT1, a histone deacetylase (21). Larsen et al. (165) found that ZEB1-induced EMT was crucial for the development of NSCLC but required premalignant oncogenic mutations such those for KRAS. Moreover, they found that ZEB1-driven EMT was a crucial early event in the progression of human bronchial epithelial cells to malignancy (165). These results supported previous in vitro (166) and in vivo (167–170) studies that established ZEB1 as a driver of EMT in lung cancer tumorigenesis. In PDAC, Krebs et al. (112) demonstrated that ZEB1 is a key driver of PDAC progression from early tumorigenesis to late-stage metastasis, highlighting the important contribution of EMT activation in these processes (112).
Beyond the levels of mRNAs, EMT-TFs can alter chromatin to achieve the stable, long-term silencing of epithelial genes required for complete EMT (171). Snail, an EMT-TF, can recruit a series of chromatin-modifying enzymes to the E-cadherin promotor to erase a mark of active transport and replace it with a trimethylated H3K9 mark that promotes the recruitment of DNA methyltransferases, causing CpG methylation of the promoter and formation of a constitutive heterochromatin resistant to transcription activation (172). Additionally, TFs of the Zeb family form a double-negative feedback loop with the miR-200 family of microRNAs (miRNA), causing this regulatory loop to operate as a switch between epithelial and mesenchymal states in a variety of tumor types (173–175). Similarly, Snail represses the expression of miR-34, a miRNA that binds to the 3′ UTR of Snail mRNA to mark it for degradation (176).
Targeting RAS proteins was first attempted when the proteins were shown to be modified and rendered functional by farnesylation (177–179). This initiated the launch of identifying compounds that block farnesyl transferase activity. Farnesyl transferase inhibitors were developed with impressive potency and selectivity, but they failed to show efficacy in the clinic (180). Another approach that has been considered is the development of a GTP antagonist. However, due to the picomolar affinity of GTP and RAS and the millimolar concentration of GTP in the cell, GTP antagonists had long been deemed impossible (111) until recently. In 2013, the dream of directly targeting RAS was re-imagined when Shokat and colleagues identified compounds that bind covalently and specifically to KRAS G12C (181). Lead compounds were further developed by Wellspring Biosciences, who showed that the compounds ARS853 and ARS1620 inhibit KRAS G12C effectively and specifically in cells and animals (182, 183). The first KRAS G12C inhibitor to enter clinical trials is Amgen 510 (Table 1). Multiple groups are working to create improved G12C-targeted compounds with better RAS-GTP destabilizing activity (184, 185). These studies have reinvigorated the field and initiated research efforts, such as the NCI-supported RAS initiative.
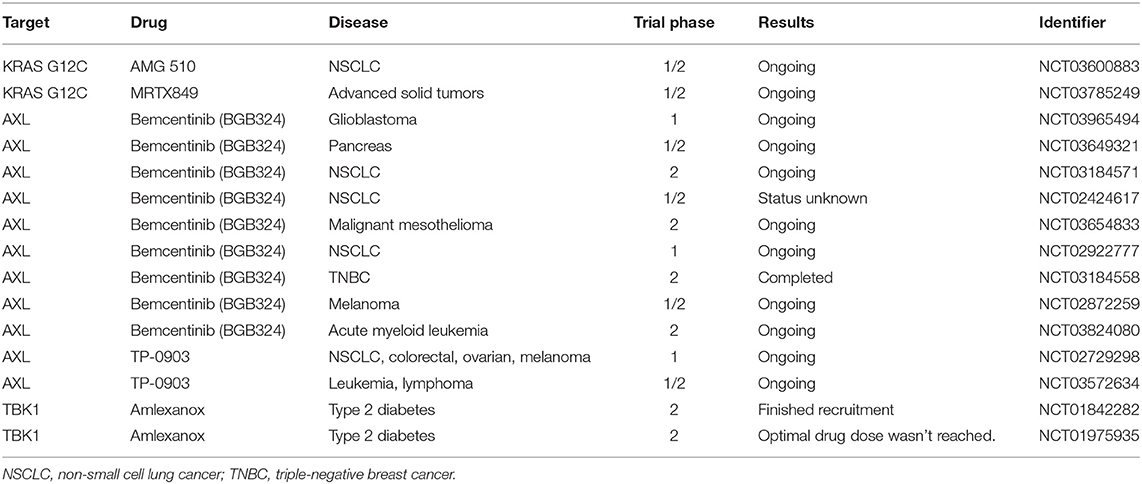
Table 1. Clinical trials targeting KRAS, AXL, and TBK1.
Although this recent breakthrough suggests that targeting KRAS G12C may be effective, it is possible that this targetable allele may be an outlier (186). KRAS G12C is rarely mutated in KRAS-addicted cancers and it is likely that KRAS G12D and G12V, the most common mutant KRAS alleles, will be more challenging to specifically inhibit (187). As a result, the development of therapeutic strategies that either inhibit RAS effector signaling elements, such as TBK1, or inhibit elements that can activate RAS, such as AXL, remain an attractive therapeutic alternative.
Due to its implication in metastasis, EMT, and drug therapy resistance, large efforts are focused on pharmacologically inhibiting AXL. In fact, multiple strategies are being tested clinically, including blocking GAS6 or AXL with monoclonal antibodies and small molecules (99, 188). One of the most advanced selective AXL inhibitors to date is bemcentinib (BGB324), developed by BerGenBio ASA. BGB324 has been investigated by our group in preclinical models of late-stage PDAC and shown promising therapeutic effects in enhancing gemcitabine efficacy and reducing metastasis (54). Other groups have also investigated BGB324, where it has been found to have antitumor, antimetastatic, and therapy-sensitizing effects in preclinical models of pancreatic cancer, breast cancer, glioblastoma, prostate cancer, chronic myeloid leukemia, ovarian cancer, and uterine serous cancer (189–195). Recently phase II clinical trials have begun to enroll patients using bemcentinib in multiple cancer types as a single agent or in combination with targeted or chemo- and immunotherapies (Table 1). Another selective AXL inhibitor is TP-0903, developed by Tolero Pharmaceuticals. In preclinical models, TP-0903 has been shown to have antitumor and therapy-sensitizing effects on multiple cancers, including neuroblastoma, leukemia, and lung cancer (196–199). TP-0903 is currently being evaluated clinically in multiple indications (Table 1).
For TBK1 to be a relevant target in the clinic, it will be necessary to evaluate the therapeutic efficacy of TBK1 inhibition in preclinical cancer models. Currently there are at least six distinct small molecules that inhibit TBK1, including BX795, compound II, CYT387, MRT67307, GSK2292978A, and Amlexanox, although none are highly selective. Currently, Amlexanox is the only TBK1i known to enter clinical testing, which is in a phase 2 study for the treatment of type 2 diabetes, non-alcoholic fatty liver disease, or obesity (Table 1). Further investigations and better inhibitors will be needed before TBK1 can be directly targeted in RAS-driven cancer in preclinical and clinical settings. Moving forward, it will be vital to understand the distinct function of TBK1 in each relevant cell type within tumors. As mesenchymal tumor cells express high levels of active TBK1 (105) and are associated with aggressive disease, metastasis, and poor patient outcomes (30), targeting TBK1 in RAS-driven cancers is a promising alternative strategy to reduce the tumor-promoting effects of KRAS-driven EMT.
EMT is a key cellular program that is activated by KRAS and thus contributes to tumor progression by enhancing tumor cell survival, tumor cell dissemination, and therapy resistance and has a strong association with worse clinical prognosis in many KRAS-driven cancers. Because KRAS is not currently an amenable target for many of these KRAS-driven cancers, targeting KRAS effector signaling is an attractive alternative. With this in mind, pharmacologically targeting the pathways that contribute to KRAS-driven EMT is worth considering as a strategy to improve response to standard therapy and reduce clinical progression, therapy resistance, and metastasis.
Despite significant evidence that EMT directly contributes to tumor progression, several studies have suggested EMT is not required for the metastatic spread of PDAC and breast cancer (55, 57, 200, 201). For example, most metastatic lesions are known to exhibit epithelial features, an observation that seems to be at odds with EMT as a prerequisite for metastasis (30, 202, 203). As such, the importance of EMT in cancer biology has long been questioned (204).
Epithelial plasticity not only includes the process of EMT, but also the reverse, mesenchymal-to-epithelial transition or MET. Recent evidence suggests that MET is required for successful metastatic colonization, although it remains unknown whether the tissue-specific adaptations are acquired thorough epigenetic or genetic means. Distant metastases in carcinoma patients often present with epithelial features having a similar histology as the tissue of origin (205, 206). These observations support that epithelial plasticity lies at the heart of tumor development and progression, and that such plasticity is necessary for tumor cell survival and colonization. It has become increasingly evident that EMT encompasses a range of hybrid plastic states, a phenotype coined as “partial EMT” (36, 207, 208). Because partial EMT is not well-defined, it is unclear whether this hybrid status signifies a transitional phase during EMT or represents its own state. Similarly, using a mouse model of PDAC, the Stanger group has shown that individual tumors can activate different plasticity programs, such as “classical EMT” which involves transcriptional repression and an alternative program in which the epithelial state is lost post-transcriptionally (36). These plasticity programs were associated with either single-cell invasion or collective invasion, respectively (36). It is unclear what underlies this phenotypic heterogeneity, considering the tumors investigated in this study had the same oncogenic drivers (TP53 and KRAS). Perhaps the only difference between the states is the tumor microenvironment, as Aiello et al. found that when partial EMT cells are exposed to TGFβ, they execute a classic EMT program (36, 209). This constant plastic state may partially explain the intratumoral heterogeneity that is often seen in carcinomas such as PDAC (210–212).
The chronic activation of an EMT program within a tumor may depend on paracrine signals within the tumor microenvironment, dictating whether the tumor cells undergo EMT or MET. Because these cells exist in a plastic state, it is possible that these tumor cells readily revert their phenotype based on a microenvironment-specific context and factors (36, 205, 213, 214). One challenge impeding current in vivo studies is the difficulty of distinguishing carcinoma cells that have undergone EMT from fibroblasts or other mesenchymal cells that are normally found in the tumor stroma. To combat this, many labs have begun to use single-cell sequencing technology in KRAS-driven cancers such as PDAC to investigate EMT in vivo (215). Additionally, current in vivo lineage-tracing technology has not settled the debate between the importance of collective migration and/or EMT for metastatic dissemination. Additionally, the mechanisms of invasion and metastatic potential and their correlation with clinical outcome has yet to be defined. Regardless, epithelial plasticity remains as an indispensable feature in multiple phases of human cancer in an oncogene- and tissue-specific manner.
EA and WD wrote the manuscript. RB reviewed and revised the manuscript.
The work was supported by NIH grants R01 CA192381 and U54 CA210181 Project 2 to RB and support from the Effie Marie Cain Scholarship in Angiogenesis Research.
RB receives research support from BerGenBio ASA and Tolero, companies developing Axl inhibitors.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to the colleagues whose work could not be included due to space restrictions. We thank the members of the Brekken laboratory for critical discussion and thank Dave Primm for editorial assistance.
1. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. (2003) 4:437–50. doi: 10.1016/S1535-6108(03)00309-X
2. Haigis KM. KRAS Alleles: the devil is in the detail. Trends Cancer. (2017) 3:686–97. doi: 10.1016/j.trecan.2017.08.006
3. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. (2008) 9:517–31. doi: 10.1038/nrm2438
4. Brubaker DK, Paulo JA, Sheth S, Poulin EJ, Popow O, Joughin BA, et al. Proteogenomic network analysis of context-specific KRAS signaling in mouse-to-human cross-species translation. Cell Systems. (2019) 9:258–70.e6. doi: 10.1016/j.cels.2019.07.006
5. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. (2012) 104:228–39. doi: 10.1093/jnci/djr523
6. Hammond DE, Mageean CJ, Rusilowicz EV, Wickenden JA, Clague MJ, Prior IA. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J Proteome Res. (2015) 14:1535–46. doi: 10.1021/pr501191a
7. Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, et al. Differential effector engagement by oncogenic KRAS. Cell Rep. (2018) 22:1889–902. doi: 10.1016/j.celrep.2018.01.051
8. Yuan M, Cottrell CA, Ozorowski G, van Gils MJ, Kumar S, Wu NC, et al. Conformational plasticity in the HIV-1 fusion peptide facilitates recognition by broadly neutralizing antibodies. Cell Host Microbe. (2019) 25:873–83.e5. doi: 10.1016/j.chom.2019.04.011
9. Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest. (2011) 121:4572–8. doi: 10.1172/JCI57131
10. Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. (2003) 63:2016–9.
11. Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. (2007) 133:1999–2009. doi: 10.1053/j.gastro.2007.09.009
12. De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci USA. (2008) 105:18907–12. doi: 10.1073/pnas.0810111105
13. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 22:737–50. doi: 10.1016/j.ccr.2012.10.025
14. Yamaguchi J, Yokoyama Y, Kokuryo T, Ebata T, Nagino M. Cells of origin of pancreatic neoplasms. Surg Today. (2018) 48:9–17. doi: 10.1007/s00595-017-1501-2
15. Kopp JL, Dubois CL, Schaeffer DF, Samani A, Taghizadeh F, Cowan RW, et al. Loss of Pten and activation of Kras synergistically induce formation of intraductal papillary mucinous neoplasia from pancreatic ductal cells in mice. Gastroenterology. (2018) 154:1509–23.e5. doi: 10.1053/j.gastro.2017.12.007
16. Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. (2012) 122:2911–5. doi: 10.1172/JCI63212
17. Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest. (2012) 122:3914–8. doi: 10.1172/JCI63065
18. Guest RV, Boulter L, Kendall TJ, Minnis-Lyons SE, Walker R, Wigmore SJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. (2014) 74:1005–10. doi: 10.1158/0008-5472.CAN-13-1911
19. Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, Matsubara D, et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci Rep. (2016) 6:23899. doi: 10.1038/srep23899
20. Hill MA, Alexander WB, Guo B, Kato Y, Patra K, O'Dell MR, et al. Kras and Tp53 Mutations cause cholangiocyte- and hepatocyte-derived cholangiocarcinoma. Cancer Res. (2018) 78:4445–51. doi: 10.1158/0008-5472.CAN-17-1123
21. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
22. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. (2018) 18:128–34. doi: 10.1038/nrc.2017.118
23. Kamei T, Matozaki T, Sakisaka T, Kodama A, Yokoyama S, Peng YF, et al. Coendocytosis of cadherin and c-Met coupled to disruption of cell-cell adhesion in MDCK cells–regulation by Rho, Rac and Rab small G proteins. Oncogene. (1999) 18:6776–84. doi: 10.1038/sj.onc.1203114
24. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. (2004) 23:1739–48. doi: 10.1038/sj.emboj.7600136
25. Bryant DM, Wylie FG, Stow JL. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol Biol Cell. (2005) 16:14–23. doi: 10.1091/mbc.e04-09-0845
26. Perrais M, Chen X, Perez-Moreno M, Gumbiner BM. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell. (2007) 18:2013–25. doi: 10.1091/mbc.e06-04-0348
27. Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, et al. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. (2011) 8:27–33. doi: 10.1038/nrgastro.2010.188
28. Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. (2009) 15:489–500. doi: 10.1016/j.ccr.2009.03.022
29. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. (2014) 158:171–84. doi: 10.1016/j.cell.2014.06.004
30. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. (2015) 25:675–86. doi: 10.1016/j.tcb.2015.07.012
31. Gaianigo N, Melisi D, Carbone C. EMT and treatment resistance in pancreatic cancer. Cancers. (2017) 9:E122. doi: 10.3390/cancers9090122
32. Puls TJ, Tan X, Whittington CF, Voytik-Harbin SL. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE. (2017) 12:e0188870. doi: 10.1371/journal.pone.0188870
33. Wang S, Huang S, Sun YL. Epithelial-mesenchymal transition in pancreatic cancer: a review. Biomed Res Int. (2017) 2017:2646148. doi: 10.1155/2017/2646148
34. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. (2013) 18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
35. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
36. Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell. (2018) 45:681–95.e4. doi: 10.1016/j.devcel.2018.05.027
37. Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. (2018) 554:62–8. doi: 10.1038/nature25459
38. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. (2005) 24:7443–54. doi: 10.1038/sj.onc.1209091
39. Cates JM, Byrd RH, Fohn LE, Tatsas AD, Washington MK, Black CC. Epithelial-mesenchymal transition markers in pancreatic ductal adenocarcinoma. Pancreas. (2009) 38:e1–6. doi: 10.1097/MPA.0b013e3181878b7f
40. Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during snail-induced EMT of cancer cells. Cancer Cell. (2009) 15:195–206. doi: 10.1016/j.ccr.2009.01.023
41. Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. (2002) 2:243–7. doi: 10.1016/S1535-6108(02)00122-8
42. Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. (2005) 8:381–92. doi: 10.1016/j.ccr.2005.10.014
43. Chen SJ, Chen YT, Zeng LJ, Zhang QB, Lian GD, Li JJ, et al. Bmi1 combines with oncogenic KRAS to induce malignant transformation of human pancreatic duct cells in vitro. Tumour Biol. (2016) 37:11299–309. doi: 10.1007/s13277-016-4840-5
44. Garg M. Epithelial-mesenchymal transition - activating transcription factors - multifunctional regulators in cancer. World J Stem Cells. (2013) 5:188–95. doi: 10.4252/wjsc.v5.i4.188
45. Garg M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev Mol Med. (2017) 19:e3. doi: 10.1017/erm.2017.6
46. Tripathi K, Garg M. Mechanistic regulation of epithelial-to-mesenchymal transition through RAS signaling pathway and therapeutic implications in human cancer. J Cell Commun Signal. (2018) 12:513–27. doi: 10.1007/s12079-017-0441-3
47. Shields MA, Ebine K, Sahai V, Kumar K, Siddiqui K, Hwang RF, et al. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol Cancer Res. (2013) 11:1078–87. doi: 10.1158/1541-7786.MCR-12-0637
48. Saitoh M, Endo K, Furuya S, Minami M, Fukasawa A, Imamura T, et al. STAT3 integrates cooperative Ras and TGF-beta signals that induce Snail expression. Oncogene. (2016) 35:1049–57. doi: 10.1038/onc.2015.161
49. Edme N, Downward J, Thiery JP, Boyer B. Ras induces NBT-II epithelial cell scattering through the coordinate activities of Rac and MAPK pathways. J Cell Sci. (2002) 115(Pt. 12):2591–601.
50. Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. (2017) 77:3982–9. doi: 10.1158/0008-5472.CAN-16-3292
51. Voon DC, Huang RY, Jackson RA, Thiery JP. The EMT spectrum and therapeutic opportunities. Mol Oncol. (2017) 11:878–91. doi: 10.1002/1878-0261.12082
52. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
53. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. (2010) 29:4741–51. doi: 10.1038/onc.2010.215
54. Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, et al. Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. (2018) 78:246–55. doi: 10.1158/0008-5472.CAN-17-1973
55. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. (2015) 527:525–30. doi: 10.1038/nature16064
56. Sale MJ, Balmanno K, Saxena J, Ozono E, Wojdyla K, McIntyre RE, et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat Commun. (2019) 10:2030. doi: 10.1038/s41467-019-09438-w
57. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. (2015) 527:472–6. doi: 10.1038/nature15748
58. Yuen A, Diaz B. The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia. (2014) 2:91–106. doi: 10.2147/HP.S52636
59. Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. (2014) 74:1032–44. doi: 10.1158/0008-5472.CAN-13-2800
60. Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM, Zhou HX, et al. Nuclear factor-kappaB-dependent epithelial to mesenchymal transition induced by HIF-1alpha activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE. (2011) 6:e23752. doi: 10.1371/journal.pone.0023752
61. Zhu GH, Huang C, Feng ZZ, Lv XH, Qiu ZJ. Hypoxia-induced snail expression through transcriptional regulation by HIF-1alpha in pancreatic cancer cells. Dig Dis Sci. (2013) 58:3503–15. doi: 10.1007/s10620-013-2841-4
62. Cheng ZX, Wang DW, Liu T, Liu WX, Xia WB, Xu J, et al. Effects of the HIF-1alpha and NF-kappaB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol Rep. (2014) 31:1891–8. doi: 10.3892/or.2014.3022
63. Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. (2014) 74:2455–64. doi: 10.1158/0008-5472.CAN-13-3009
64. Ryseck RP, Weih F, Carrasco D, Bravo R. RelB, a member of the Rel/NF-kappa B family of transcription factors. Braz J Med Biol Res. (1996) 29:895–903.
65. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. (2013) 12:86. doi: 10.1186/1476-4598-12-86
66. Karin M. Nuclear factor-κB in cancer development and progression. Nature. (2006) 441:431–6. doi: 10.1038/nature04870
67. Greten FR, Weber CK, Greten TF, Schneider G, Wagner M, Adler G, et al. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology. (2002) 123:2052–63. doi: 10.1053/gast.2002.37075
68. Li Y, Wang Y, Li L, Kong R, Pan S, Ji L, et al. Hyperoside induces apoptosis and inhibits growth in pancreatic cancer via Bcl-2 family and NF-kappaB signaling pathway both in vitro and in vivo. Tumour Biol. (2016) 37:7345–55. doi: 10.1007/s13277-015-4552-2
69. Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. (2009) 15:68–74. doi: 10.1038/nm.1908
70. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
71. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. (2012) 44:852–60. doi: 10.1038/ng.2330
72. Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. (2013) 19:279–90. doi: 10.1158/1078-0432.CCR-12-1558
73. Wu F, Li J, Jang C, Wang J, Xiong J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol. (2014) 7:6653–61.
74. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. (2011) 17:500–3. doi: 10.1038/nm.2344
75. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin Cancer Res. (2015) 21:2601–12. doi: 10.1158/1078-0432.CCR-14-2648
76. Choi YJ, Kim SY, So KS, Baek IJ, Kim WS, Choi SH, et al. AUY922 effectively overcomes MET- and AXL-mediated resistance to EGFR-TKI in lung cancer cells. PLoS ONE. (2015) 10:e0119832. doi: 10.1371/journal.pone.0119832
77. Oliveras-Ferraros C, Corominas-Faja B, Cufi S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM, et al. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle. (2012) 11:4020–32. doi: 10.4161/cc.22225
78. Cheng Q, Chang JT, Gwin WR, Zhu J, Ambs S, Geradts J, et al. A signature of epithelial-mesenchymal plasticity and stromal activation in primary tumor modulates late recurrence in breast cancer independent of disease subtype. Breast Cancer Res. (2014) 16:407. doi: 10.1186/s13058-014-0407-9
79. Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, et al. New insights into the role of EMT in tumor immune escape. Mol Oncol. (2017) 11:824–46. doi: 10.1002/1878-0261.12093
80. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
81. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
82. Wang F, Liu H, Hu L, Liu Y, Duan Y, Cui R, et al. The Warburg effect in human pancreatic cancer cells triggers cachexia in athymic mice carrying the cancer cells. BMC Cancer. (2018) 18:360. doi: 10.1186/s12885-018-4271-3
83. Alfarouk KO. Tumor metabolism, cancer cell transporters, and microenvironmental resistance. J Enzyme Inhib Med Chem. (2016) 31:859–66. doi: 10.3109/14756366.2016.1140753
84. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. (2017) 31:5–19. doi: 10.1016/j.ccell.2016.12.006
85. Biancur DE, Kimmelman AC. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim Biophys Acta Rev Cancer. (2018) 1870:67–75. doi: 10.1016/j.bbcan.2018.04.011
86. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA III, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. (2012) 337:975–80. doi: 10.1126/science.1222278
87. Gomez LS, Zancan P, Marcondes MC, Ramos-Santos L, Meyer-Fernandes JR, Sola-Penna M, et al. Resveratrol decreases breast cancer cell viability and glucose metabolism by inhibiting 6-phosphofructo-1-kinase. Biochimie. (2013) 95:1336–43. doi: 10.1016/j.biochi.2013.02.013
88. Vaziri-Gohar A, Zarei M, Brody JR, Winter JM. Metabolic dependencies in pancreatic cancer. Front Oncol. (2018) 8:617. doi: 10.3389/fonc.2018.00617
89. Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. (2012) 13:312–21. doi: 10.1038/nrm3334
90. Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. (2015) 208:869–80. doi: 10.1083/jcb.201501101
91. Taparra K, Wang H, Malek R, Lafargue A, Barbhuiya MA, Wang X, et al. O-GlcNAcylation is required for mutant KRAS-induced lung tumorigenesis. J Clin Invest. (2018) 128:4924–37. doi: 10.1172/JCI94844
92. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
93. Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. (2007) 101:816–29. doi: 10.1002/jcb.21215
94. Koorstra JB, Karikari CA, Feldmann G, Bisht S, Rojas PL, Offerhaus GJ, et al. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol Ther. (2009) 8:618–26. doi: 10.4161/cbt.8.7.7923
95. Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci USA. (2010) 107:1124–9. doi: 10.1073/pnas.0909333107
96. Kirane A, Ludwig KF, Sorrelle N, Haaland G, Sandal T, Ranaweera R, et al. Warfarin blocks gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. (2015) 75:3699–705. doi: 10.1158/0008-5472.CAN-14-2887-T
97. Song X, Wang H, Logsdon CD, Rashid A, Fleming JB, Abbruzzese JL, et al. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer. (2011) 117:734–43. doi: 10.1002/cncr.25483
98. Leconet W, Larbouret C, Chardes T, Thomas G, Neiveyans M, Busson M, et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene. (2014) 33:5405–14. doi: 10.1038/onc.2013.487
99. Du W, Brekken RA. Does Axl have potential as a therapeutic target in pancreatic cancer? Expert Opin Ther Targets. (2018) 22:955–66. doi: 10.1080/14728222.2018.1527315
100. Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. (1997) 14:2619–31. doi: 10.1038/sj.onc.1201123
101. Weinger JG, Gohari P, Yan Y, Backer JM, Varnum B, Shafit-Zagardo B. In brain, Axl recruits Grb2 and the p85 regulatory subunit of PI3 kinase; in vitro mutagenesis defines the requisite binding sites for downstream Akt activation. J Neurochem. (2008) 106:134–46. doi: 10.1111/j.1471-4159.2008.05343.x
102. Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. (2014) 33:1316–24. doi: 10.1038/onc.2013.57
103. Del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, Chakravarty P, et al. Mesenchymal cancer cell-stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep. (2015) 13:2456–69. doi: 10.1016/j.celrep.2015.11.025
104. Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis. (2014) 35:769–75. doi: 10.1093/carcin/bgt372
105. Cruz VH, Arner EN, Du W, Bremauntz AE, Brekken RA. Axl-mediated activation of TBK1 drives epithelial plasticity in pancreatic cancer. JCI Insight. (2019) 5:126117. doi: 10.1172/jci.insight.126117
106. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
107. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. (2014) 13:828–51. doi: 10.1038/nrd4389
108. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
109. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. (2010) 10:842–57. doi: 10.1038/nrc2960
110. Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. (2010) 1:2–27. doi: 10.4161/sgtp.1.1.12178
111. Waters AM, Der CJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med. (2018). 8:a031435. doi: 10.1101/cshperspect.a031435
112. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. (2017) 19:518–29. doi: 10.1038/ncb3513
113. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. (2002) 156:299–313. doi: 10.1083/jcb.200109037
114. Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. (2017) 542:362–6. doi: 10.1038/nature21064
115. Jones J, Bentas W, Blaheta RA, Makarevic J, Hudak L, Wedel S, et al. Modulation of adhesion and growth of colon and pancreatic cancer cells by the histone deacetylase inhibitor valproic acid. Int J Mol Med. (2008) 22:293–9. doi: 10.3892/ijmm_00000022
116. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. (2010) 467:1109–13. doi: 10.1038/nature09460
117. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. (2012) 491:399–405. doi: 10.1038/nature11547
118. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. (2015) 518:495–501. doi: 10.1038/nature14169
119. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. (2015) 6:6744. doi: 10.1038/ncomms7744
120. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
121. Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. (2017) 49:358–66. doi: 10.1038/ng.3764
122. Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. (2006) 16:2385–94. doi: 10.1016/j.cub.2006.10.023
123. Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral effector signaling network: the road less traveled for anti-ras drug discovery. Genes Cancer. (2011) 2:275–87. doi: 10.1177/1947601911407329
124. Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. (2002) 16:2045–57. doi: 10.1101/gad.993902
125. Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. (2004) 6:171–83. doi: 10.1016/j.ccr.2004.07.009
126. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. (2003) 300:1148–51. doi: 10.1126/science.1081315
127. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. (2004) 199:1651–8. doi: 10.1084/jem.20040528
128. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. (2006) 127:157–70. doi: 10.1016/j.cell.2006.08.034
129. Radtke AL, Delbridge LM, Balachandran S, Barber GN, O'Riordan MX. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. (2007) 3:e29. doi: 10.1371/journal.ppat.0030029
130. Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. (2011) 41:458–70. doi: 10.1016/j.molcel.2011.01.019
131. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. (2011) 333:228–33. doi: 10.1126/science.1205405
132. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. (2009) 462:108–12. doi: 10.1038/nature08460
133. Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. (2018) 24:1143–50. doi: 10.1038/s41591-018-0116-5
134. Cooper JM, Ou YH, McMillan EA, Vaden RM, Zaman A, Bodemann BO, et al. TBK1 provides context-selective support of the activated AKT/mTOR pathway in lung cancer. Cancer Res. (2017) 77:5077–94. doi: 10.1158/0008-5472.CAN-17-0829
135. Cruz VH, Brekken RA. Assessment of TANK-binding kinase 1 as a therapeutic target in cancer. J Cell Commun Signal. (2018) 12:83–90. doi: 10.1007/s12079-017-0438-y
136. Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. (2003) 63:2172–8.
137. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. (2015) 9:317–24. doi: 10.1080/19336918.2015.1016686
138. Yang KM, Jung Y, Lee JM, Kim W, Cho JK, Jeong J, et al. Loss of TBK1 induces epithelial-mesenchymal transition in the breast cancer cells by ERalpha downregulation. Cancer Res. (2013) 73:6679–89. doi: 10.1158/0008-5472.CAN-13-0891
139. Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res. (2014) 12:1509–19. doi: 10.1158/1541-7786.MCR-14-0204
140. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
141. Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell. (2018) 174:1347–60. doi: 10.1016/j.cell.2018.08.027
142. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
143. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
144. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
145. Galluzzi L, Vanpouille-Box C, Bakhoum SF, Demaria S. SnapShot: CGAS-STING signaling. Cell. (2018) 173:276–.e1. doi: 10.1016/j.cell.2018.03.015
146. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. (2018) 9:1249. doi: 10.1038/s41467-018-03555-8
147. Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. (2010) 468:321–5. doi: 10.1038/nature09529
148. Chen G, Bradford WD, Seidel CW, Li R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature. (2012) 482:246–50. doi: 10.1038/nature10795
149. Potapova TA, Zhu J, Li R. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. (2013) 32:377–89. doi: 10.1007/s10555-013-9436-6
150. Laughney AM, Elizalde S, Genovese G, Bakhoum SF. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. (2015) 12:809–20. doi: 10.1016/j.celrep.2015.06.065
151. Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. (2016) 538:378–82. doi: 10.1038/nature19823
152. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. (2017) 355:eaaf8399. doi: 10.1126/science.aaf8399
153. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
154. Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci. (2013) 116:317–36. doi: 10.1016/B978-0-12-394311-8.00014-5
155. Wong TS, Gao W, Chan JY. Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors. Biomed Res Int. (2014) 2014:921564. doi: 10.1155/2014/921564
156. Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. (2007) 13:4769–76. doi: 10.1158/1078-0432.CCR-06-2926
157. Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. (2007) 6:532–41. doi: 10.1158/1535-7163.MCT-06-0462
158. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. (2009) 69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819
159. Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. (2013) 24:466–80. doi: 10.1016/j.ccr.2013.08.018
160. Denecker G, Vandamme N, Akay O, Koludrovic D, Taminau J, Lemeire K, et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. (2014) 21:1250–61. doi: 10.1038/cdd.2014.44
161. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. (2015) 525:256–60. doi: 10.1038/nature14897
162. Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. (2013) 23:768–83. doi: 10.1016/j.ccr.2013.04.020
163. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. (2012) 22:709–24. doi: 10.1016/j.ccr.2012.10.012
164. Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. (2014) 74:6330–40. doi: 10.1158/0008-5472.CAN-14-0923
165. Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J Clin Invest. (2016) 126:3219–35. doi: 10.1172/JCI76725
166. Takeyama Y, Sato M, Horio M, Hase T, Yoshida K, Yokoyama T, et al. Knockdown of ZEB1, a master epithelial-to-mesenchymal transition (EMT) gene, suppresses anchorage-independent cell growth of lung cancer cells. Cancer Lett. (2010) 296:216–24. doi: 10.1016/j.canlet.2010.04.008
167. Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. (2009) 23:2140–51. doi: 10.1101/gad.1820209
168. Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP, et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest. (2012) 122:3170–83. doi: 10.1172/JCI63608
169. Liu Y, Zhang N, Wang Y, Xu M, Liu N, Pang X, et al. Zinc finger E-box binding homeobox 1 promotes invasion and bone metastasis of small cell lung cancer in vitro and in vivo. Cancer Sci. (2012) 103:1420–8. doi: 10.1111/j.1349-7006.2012.02347.x
170. Yang Y, Ahn YH, Chen Y, Tan X, Guo L, Gibbons DL, et al. ZEB1 sensitizes lung adenocarcinoma to metastasis suppression by PI3K antagonism. J Clin Invest. (2014) 124:2696–708. doi: 10.1172/JCI72171
171. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. (2013) 19:1438–49. doi: 10.1038/nm.3336
172. Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des. (2014) 20:1698–705. doi: 10.2174/13816128113199990512
173. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. (2008) 9:582–9. doi: 10.1038/embor.2008.74
174. Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. (2010) 11:670–7. doi: 10.1038/embor.2010.117
175. Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. (2013) 132:745–54. doi: 10.1002/ijc.27708
176. Brabletz T. MiR-34 and SNAIL: another double-negative feedback loop controlling cellular plasticity/EMT governed by p53. Cell Cycle. (2012) 11:215–6. doi: 10.4161/cc.11.2.18900
177. Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. (1984) 310:583–6. doi: 10.1038/310583a0
178. Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci USA. (1989) 86:8323–7. doi: 10.1073/pnas.86.21.8323
179. Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. (1989) 57:1167–77. doi: 10.1016/0092-8674(89)90054-8
180. Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. (2015) 21:1819–27. doi: 10.1158/1078-0432.CCR-14-3214
181. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. (2013) 503:548–51. doi: 10.1038/nature12796
182. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. (2016) 351:604–8. doi: 10.1126/science.aad6204
183. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. (2016) 6:316–29. doi: 10.1158/2159-8290.CD-15-1105
184. Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. (2014) 53:199–204. doi: 10.1002/anie.201307387
185. Nnadi CI, Jenkins ML, Gentile DR, Bateman LA, Zaidman D, Balius TE, et al. Novel K-Ras G12C switch-II covalent binders destabilize Ras and accelerate nucleotide exchange. J Chem Inf Model. (2018) 58:464–71. doi: 10.1021/acs.jcim.7b00399
186. McCormick F. Progress in targeting RAS with small molecule drugs. Biochem J. (2019) 476:365–74. doi: 10.1042/BCJ20170441
187. McGregor LM, Jenkins ML, Kerwin C, Burke JE, Shokat KM. Expanding the scope of electrophiles capable of targeting K-Ras oncogenes. Biochemistry. (2017) 56:3178–83. doi: 10.1021/acs.biochem.7b00271
188. Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight. (2018) 3:124184. doi: 10.1172/jci.insight.124184
189. Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. (2010) 70:1544–54. doi: 10.1158/0008-5472.CAN-09-2997
190. Vouri M, An Q, Birt M, Pilkington GJ, Hafizi S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget. (2015) 6:16183–97. doi: 10.18632/oncotarget.3952
191. Antony J, Tan TZ, Kelly Z, Low J, Choolani M, Recchi C, et al. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci Signal. (2016) 9:ra97. doi: 10.1126/scisignal.aaf8175
192. Ben-Batalla I, Erdmann R, Jorgensen H, Mitchell R, Ernst T, von Amsberg G, et al. Axl blockade by BGB324 inhibits BCR-ABL tyrosine kinase inhibitor-sensitive and -resistant chronic myeloid leukemia. Clin Cancer Res. (2017) 23:2289–300. doi: 10.1158/1078-0432.CCR-16-1930
193. Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, et al. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget. (2017) 8:41064–77. doi: 10.18632/oncotarget.17026
194. Palisoul ML, Quinn JM, Schepers E, Hagemann IS, Guo L, Reger K, et al. Inhibition of the receptor tyrosine kinase AXL restores paclitaxel chemosensitivity in uterine serous cancer. Mol Cancer Ther. (2017) 16:2881–91. doi: 10.1158/1535-7163.MCT-17-0587
195. Sadahiro H, Kang KD, Gibson JT, Minata M, Yu H, Shi J, et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. (2018) 78:3002–13. doi: 10.1158/0008-5472.CAN-17-2433
196. Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. (2015) 29:2382–9. doi: 10.1038/leu.2015.147
197. Myers SH, Brunton VG, Unciti-Broceta A. AXL inhibitors in cancer: a medicinal chemistry perspective. J Med Chem. (2016) 59:3593–608. doi: 10.1021/acs.jmedchem.5b01273
198. Aveic S, Corallo D, Porcu E, Pantile M, Boso D, Zanon C, et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur J Pharmacol. (2018) 818:435–48. doi: 10.1016/j.ejphar.2017.11.016
199. Sinha S, Boysen JC, Chaffee KG, Kabat BF, Slager SL, Parikh SA, et al. Chronic lymphocytic leukemia cells from ibrutinib treated patients are sensitive to Axl receptor tyrosine kinase inhibitor therapy. Oncotarget. (2018) 9:37173–84. doi: 10.18632/oncotarget.26444
200. Zhao Z, Zhu X, Cui K, Mancuso J, Federley R, Fischer K, et al. In vivo visualization and characterization of epithelial-mesenchymal transition in breast tumors. Cancer Res. (2016) 76:2094–104. doi: 10.1158/0008-5472.CAN-15-2662
201. Chen Y, LeBleu VS, Carstens JL, Sugimoto H, Zheng X, Malasi S, et al. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol Med. (2018) 10:e9085. doi: 10.15252/emmm.201809085
202. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA. (2001) 98:10356–61. doi: 10.1073/pnas.171610498
203. Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. (2001) 23:912–23. doi: 10.1002/bies.1132
204. Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. (2005) 65, 5996–6000. discussion: 6000–5991. doi: 10.1158/0008-5472.CAN-05-0699
205. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. (2002) 2:442–54. doi: 10.1038/nrc822
206. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. (2011) 481:85–9. doi: 10.1038/nature10694
207. Grigore AD, Jolly MK, Jia D, Farach-Carson MC, Levine H. Tumor budding: the name is EMT. Partial EMT J Clin Med. (2016) 5:E51. doi: 10.3390/jcm5050051
208. Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol. (2017) 11:755–69. doi: 10.1002/1878-0261.12083
209. Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. (2009) 11:1287–96. doi: 10.1038/ncb1973
210. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. (2011) 108:7950–5. doi: 10.1073/pnas.1102454108
211. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. (2011) 146:633–44. doi: 10.1016/j.cell.2011.07.026
212. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. (2013) 154:61–74. doi: 10.1016/j.cell.2013.06.005
213. Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. (2002) 70:537–46. doi: 10.1046/j.1432-0436.2002.700907.x
214. Jechlinger M, Grunert S, Beug H. Mechanisms in epithelial plasticity and metastasis: insights from 3D cultures and expression profiling. J Mammary Gland Biol Neoplasia. (2002) 7:415–32. doi: 10.1023/A:1024090116451
Keywords: EMT, KRAS, metastasis, TBK1, AXL, drug resistance
Citation: Arner EN, Du W and Brekken RA (2019) Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front. Oncol. 9:1049. doi: 10.3389/fonc.2019.01049
Received: 26 July 2019; Accepted: 26 September 2019;
Published: 11 October 2019.
Edited by:
Georgia Konstantinidou, University of Bern, SwitzerlandReviewed by:
Mitsuo Sato, Nagoya University, JapanCopyright © 2019 Arner, Du and Brekken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rolf A. Brekken, cm9sZi5icmVra2VuQHV0c291dGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.