 Qin Feng
Qin Feng Bin He
Bin He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 September 2019
Sec. Genitourinary Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00858
This article is part of the Research Topic Response and Resistance in Castration-Resistant Prostate Cancer View all 10 articles
Most prostate cancers are androgen-sensitive malignancies whose growths depend on the transcriptional activity of the androgen receptor (AR). In the 1940s, Charles Huggins demonstrated that the surgical removal of testes in men can result in a dramatic improvement in symptoms and can induce prostate cancer regression. Since then, androgen deprivation therapies have been the standard first-line treatment for advanced prostate cancer, including: surgical castration, medical castration, antiandrogens, and androgen biosynthesis inhibitors. These therapies relieve symptoms, reduce tumor burden, and prolong patient survival, while having relatively modest side effects. Unfortunately, hormone deprivation therapy rarely cures the cancer itself. Prostate cancer almost always recurs, resulting in deadly castration-resistant prostate cancer. The underlying escape mechanisms include androgen receptor gene/enhancer amplification, androgen receptor mutations, androgen receptor variants, coactivator overexpression, intratumoral de novo androgen synthesis, etc. Whereas, the majority of the castration-resistant prostate cancers continuously rely on the androgen axis, a subset of recurrent cancers have completely lost androgen receptor expression, undergone divergent clonal evolution or de-differentiation, and become truly androgen receptor-independent small-cell prostate cancers. There is an urgent need for the development of novel targeted and immune therapies for this subtype of prostate cancer, when more deadly small-cell prostate cancers are induced by thorough androgen deprivation and androgen receptor ablation.
The prostate is a walnut sized male reproductive gland located between the bladder and the penis. It secretes the prostatic fluid that helps to nourish and transport sperm. Androgen signaling plays a pivotal role in the development and function of a normal prostate gland. There are two native androgens in humans, testosterone (T), and 5α-dihydrotestosterone (DHT). Testosterone is produced mainly in the testis, with a small amount being produced in the adrenal glands in men. Testosterone is converted to the more potent androgen dihydrotestosterone by the enzyme 5 alpha-reductase located in the prostate, skin, scalp, etc. Both testosterone and dihydrotestosterone can bind to a single nuclear receptor protein, the androgen receptor, which is an androgen-dependent transcriptional activator and a member of nuclear receptor superfamily.
Similar to other nuclear hormone receptors, the androgen receptor protein contains three main functional domains: the NH2-terminal unstructured transcriptional activation domain, the central DNA binding domain (DBD), and the carboxyl-terminal ligand binding domain (LBD) (Figure 1A). Between DBD and LBD, there is a flexible hinge region (amino acid 624–676), which harbors a bipartite nuclear localization signal (NLS). In the classical model, the androgen receptor binds to androgen response elements (AREs) as a homodimer, and dimerization is mediated by both DBD and LBD (2, 3) (Figure 1B). Whereas, other nuclear receptors recruit LxxLL motif-containing coactivators such as the steroid receptor coactivator (SRC)/p160 family coactivators through their ligand binding domains, the androgen receptor ligand binding domain preferentially engages in the FxxLF motif-mediated NH2-terminal and carboxyl-terminal (N/C) interaction (4–7) or recruits FxxLF motif-containing coregulators (5, 8) (Figure 1B). Nevertheless, the androgen receptor can still recruit the SRC/p160 family of coactivators mainly through its unstructured NH2-terminus and LBD (9). Moreover, the androgen receptor can also recruit an AR-specific MAGE-A11 coactivator through its extended NH2-terminal FxxLF motif (10).
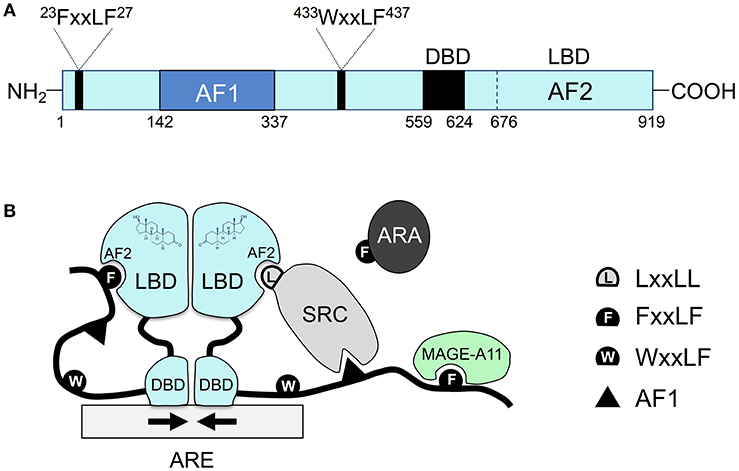
Figure 1. The unique molecular features of the androgen receptor and its coregulator recruitment. (A) The primary sequence of the androgen receptor contains several functional domains: NH2-terminal Activation Function 1 (AF1), the central DBD, the carboxyl-terminal LBD, and two AR-specific FxxLF and WxxLF motifs. (B) Schematic diagram of homodimeric androgen receptor bound to a palindromic androgen response element (ARE). Dimerization of the androgen receptor is mediated by both DBD and LBD. Shown in the diagram are FxxLF motif-mediated N/C interaction, recruitment of the SRC/p160 by AF1 and AF2, recruitment of FxxLF motif-containing ARA proteins by AF2, and recruitment of MAGE-A11 through the AR NH2-terminal extended FxxLF motif. Competition likely exists among different FxxLF, WxxLF, and LxxLF motifs for binding to the same AF2 site on AR LBD (1). SRC, steroid receptor coactivator; ARA, AR-associated protein; AF1, activation function 1; AF2, activation function 2, a hydrophobic cleft in the LBD; ARE, androgen response element; DBD, DNA binding domain; LBD, ligand binding domain.
In the absence of hormones, the androgen receptor is associated with heat shock proteins and located in the cytoplasm in an inactive conformation. Upon androgen binding, the androgen receptor quickly undergoes conformational change, nuclear translocation, recognition of androgen responsive elements in the genomic DNA, and recruitment of coactivator machineries, resulting in transcription of target genes, such as prostate-specific antigen (PSA) and transmembrane protease serine 2 (TMPRSS2).
Dihydrotestosterone is a significantly more potent androgen than testosterone both in vitro and in vivo. While this variance in potency was commonly attributed to their different binding affinities, dihydrotestosterone actually binds to the androgen receptor with similar or somewhat higher affinity compared with testosterone (11, 12). In contrast, these two androgens bind to the androgen receptor with very differing kinetics (11, 12). The rate of dissociation for dihydrotestosterone from the androgen receptor is about three to five times slower than testosterone (11, 12). Therefore, it is largely their binding kinetics, rather than affinity, which accounts for the differential androgenic activities of these two hormones. As we know, in the field of drug discovery, the notion that drug-receptor binding kinetics could be as important as affinity in determining drug efficacy is becoming more widely accepted (13). In further support of the importance of androgen binding kinetics, the unique androgen receptor inter-domain N/C interaction slows the rate of androgen dissociation without affecting androgen binding affinity and is required for optimal target gene transcription (4).
Prostate cancer occurs in the prostate gland. It is the most commonly diagnosed non-skin cancer and the second leading cause of cancer death in men in the United States. Based on his finding that the growth of prostate glands in dogs depended on testosterone, Charles Huggins demonstrated that surgical removal of testes in men, which produces more than 90 percent of testosterone, can result in a dramatic improvement in symptoms and can induce regressions of prostate cancers at any site (14). Since then, androgen deprivation therapy has been the standard first-line treatment for advanced prostate cancer (15). In addition to surgical castration, gonadotropin-releasing hormone (GnRH) analogs such as leuprolide, goserelin, and buserelin can suppress gonadotropin secretion and thus block the production of testicular androgens. As a result of its cosmetic and psychological concerns, medical castration via GnRH analogs has been the mainstay treatment for advanced prostate cancer.
Although surgical and medical castration can suppress testosterone production in the testes, the adrenal glands can still produce small amounts of androgens. To neutralize the activity of these residual androgens, antiandrogens were used to block androgen receptor signaling in prostate cancer cells (Figure 2). For example, cyproterone acetate (CPA), a synthetic steroid, was used as a prototypical antiandrogen (16). However, due to its relative ineffectiveness, CPA was replaced by more potent non-steroidal pure antiandrogens, such as Flutamide (Eulexin), bicalutamide (Casodex), and nilutamide (Nilandron). Unlike GnRH analogs, these antiandrogens do not prevent androgen production in the body. Instead, the antiandrogens bind to the androgen receptor with a relatively high affinity but lack the ability to activate transcriptional activity of the androgen receptor. Therefore, the antiandrogens function by competitively blocking testosterone and dihydrotestosterone from binding to the androgen receptor. For instance, flutamide and its active metabolite hydroxyflutamide bind to androgen receptors with a Ki of ~3,395 and ~134 nM, respectively (17). Bicalutamide is a more potent non-steroidal antiandrogen; its affinity for androgen receptors is two to four times more potent than hydroxyflutamide and nilutamide (18). Bicalutamide was thus modestly effective in prostate cancer patients who developed resistance after flutamide treatment (19). While effective on their own, antiandrogens are not usually used in monotherapy. Instead, they have proven to be used in conjunction with medical or surgical castration (20–22).
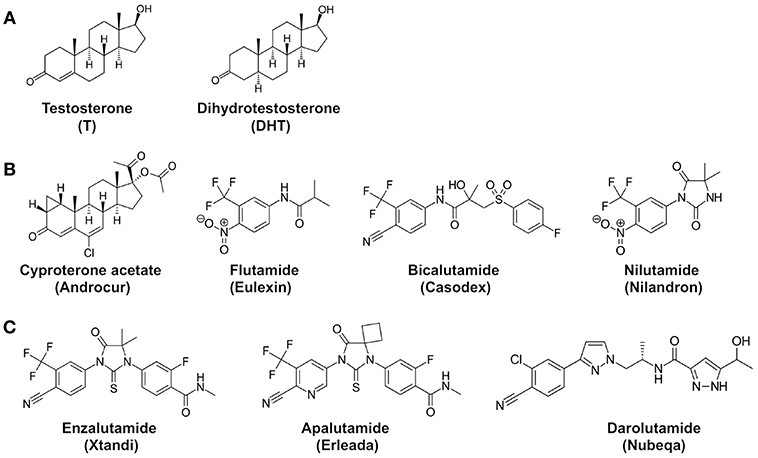
Figure 2. Chemical structures of androgens and antiandrogens. (A) Two main androgens, testosterone (T), and dihydrotestosterone (DHT). (B) First-generation antiandrogens, cyproterone acetate (Androcur), Flutamide (Eulexin), Bicalutamide (Casodex), Nilutamide (Nilandron). 2-hydroxyflutamide is the major active metabolite of flutamide in the body. (C) Representative second-generation antiandrogens, Enzalutamide (Xtandi), Apalutamide (Erleada), and Darolutamide (Nubeqa). Structures are adopted from Wikipedia.
The combination therapy of GnRH analogs and antiandrogens has promoted the survival of prostate cancer patients (21, 23). Unfortunately, most prostate cancers develop resistance to the combined androgen deprivation therapy after several years, becoming so-called “androgen-independent” prostate cancer. Surprisingly, it was found that, even after castration, the testosterone and dihydrotestosterone levels in locally recurrent prostate cancer tissues remain high enough to activate androgen receptors (24, 25). In support of this observation, the androgen receptor target gene PSA remains expressed in recurrent prostate cancer tissues, despite the castrate levels of androgens in serum (24, 25). Moreover, it has been reported that in the recurrent metastatic prostate cancers, intratumoral de novo androgen synthesis by overexpressed steroidogenic enzymes may contribute to elevated testosterone levels (26). Taken together, it becomes evident that recurrent cancers after medical or surgical castration are not truly androgen-independent (27), as they continuously depend on androgens and the androgen receptor to survive and grow. These recurrent cancers have been more appropriately classified as castration-resistant prostate cancer (28).
Subsequent studies have revealed multiple mechanisms which may contribute to the androgen receptor-dependence in castration-resistant prostate cancer. Firstly, increased androgen receptor expression can be caused by androgen receptor gene amplification (29–33) or androgen receptor enhancer amplification (34, 35). Secondly, increased expression of androgen receptor coactivators SRC1 and TIF2 stimulates androgen receptor activity in the presence of the weaker androgen androstenedione (36). The expression of the MAGE-A11 coactivator, which is recruited through androgen receptor NH2-terminal FxxLF motif, is increased in castration-resistant prostate cancer (37, 38). Thirdly, mutations in the androgen receptor ligand binding domain enable the androgen receptor to be activated by antiandrogens or other steroid hormones (39). For instance, the androgen receptor with the LNCaP mutation T877A can be activated by flutamide, estrogen, and progesterone (40, 41). The androgen receptor with L701H/T877A double mutations can be activated by glucocorticoids (42). Fourthly, constitutively active androgen receptor variants which lack ligand binding domains are another underlying mechanism of the castration resistance (43–45). Additionally, growth hormones such as epidermal growth factor (EGF) increase TIF2/GRIP1 coactivation of androgen receptor activity in recurrent cancer cells (46). Insulin-like growth factor-1 (IGF-1), keratinocyte growth factor (KGF), and EGF can all activate androgen receptor activity in the absence of androgens (47).
Interestingly, in some patients, when an antiandrogen is no longer working, simply stopping the antiandrogen treatment can stop cancer growth for a short period of time. This phenomenon is known as antiandrogen withdrawal syndrome. Decreases in PSA levels and/or clinical improvement after discontinuation of antiandrogens upon disease progression have been shown by flutamide, bicalutamide, and nilutamide withdrawal (48–50). One mechanism of antiandrogen withdrawal syndrome is acquired mutations in the androgen receptor ligand binding domain including mutation T877A and H874Y. Not surprisingly, these mutations have converted antiandrogens to androgen receptor agonists (51).
To overcome castration resistance, more potent antiandrogens, known as second-generation antiandrogens, have been designed to achieve maximal androgen blockade (52). These second-generation antiandrogens include Enzalutamide (Xtandi), Apalutamide (Erleada), and Darolutamide (Nubeqa) (Figure 2). Enzalutamide and apalutamide are structurally similar to each other, having 5- to 8-fold higher binding affinities for androgen receptors in comparison to first-generation antiandrogens. Importantly, these antiandrogens function as pure antagonists for the androgen receptor in the presence of mutations such as T877A. Darolutamide is structurally distinct and shows 8- to 10-fold higher affinity for the androgen receptor than enzalutamide and apalutamide, and can inhibit the enzalutamide-resistant mutant androgen receptor (53). Therefore, darolutamide appears to be an even more potent second-generation antiandrogen. In addition to these three FDA-approved second-generation antiandrogens, other antiandrogens are also being developed. For instance, a potent AR inhibitor JNJ-73576253 (TRC253), developed by Janssen Pharmaceuticals, is a pan-inhibitor of AR, even in the presence of certain activating mutations, and is currently in Phase 1/2A clinical trial (54).
These more potent second-generation antiandrogens have been successful in prolonging the survival of men with castration-resistant prostate cancer. For instance, in men with metastatic castration-resistant prostate cancer after chemotherapy, enzalutamide produced an overall survival benefit of 4.8 months compared to the placebo (55). For patients with metastatic prostate cancer who have not received chemotherapy, enzalutamide also significantly increased progression-free survival and overall survival (56). Moreover, enzalutamide, apalutamide, and darolutamide all had significantly prolonged metastasis-free survival in men with high-risk non-metastatic castration-resistant prostate cancer (57–59). As shown by the latest phase III trials, both enzalutamide and apalutamide could significantly increase the progression-free survival and overall survival for men with metastatic hormone-sensitive prostate cancer (60–62).
In addition to the development of second-generation antiandrogens, Abiraterone (Zytiga) was developed as an irreversible steroid inhibitor of CYP17, a key enzyme in androgen synthesis. Abiraterone acetate inhibits the production of androgens in the testes, adrenal glands, and prostate tumors. In patients with metastatic castration-resistant and chemotherapy-resistant prostate cancer, Abiraterone produced an overall survival benefit of 3.9 months in comparison to the placebo (63). More recently, the phase III LATITUDE trial has shown that the combination of Abiraterone plus prednisone with ADT conferred significant progression-free and overall survival benefits for patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (64, 65).
Unfortunately, while second-generation antigens can prolong the survival of castration-resistant prostate cancer patients, the relief is temporary. Once again, castration-resistant cancers become resistant to the newest inhibitors. The novel mutation F876L, which is evolved in the androgen receptor ligand binding domain during the treatment of enzalutamide, converts enzalutamide to an agonist (66–68). Enzalutamide-resistant prostate cancer can also bypass androgen receptor blockade by glucocorticoid receptor activation (69). Because the DNA binding domains of glucocorticoid receptor and androgen receptor are highly homologous and recognize identical DNA response elements, the glucocorticoid receptor can substitute for the androgen receptor to activate a subset of androgen receptor target genes which are required for prostate cancer survival and growth. In addition, the androgen receptor variant AR-V7 is associated with resistance to enzalutamide and Abiraterone (70, 71). Niclosamide, a novel inhibitor of AR-V7, may be able to overcome enzalutamide resistance (72). The crucial steroidogenic enzyme AKR1C3 is found to be overexpressed in enzalutamide-resistant prostate cancer cells and mediates enzalutamide resistance (73). The chemokine receptor CXCR7 is found to be overexpressed in enzalutamide-resistant prostate cancer cells and can activate MAPK to confer enzalutamide resistance (74). Up-regulation of coactivator GREB1 may also contribute to enzalutamide resistance (75).
One interesting observation is a reciprocal negative feedback regulation between AR and PI3K/Akt signaling pathways in prostate cancer. Pten loss contributed to the development of castration-resistant prostate cancer in mouse models (76, 77). It was thus postulated that combined inhibition of AR and PI3K pathways may achieve more potent inhibition of tumor growth. Indeed, in a phase Ib/II clinical trial, combination of abiraterone with Ipatasertib, an Akt inhibitor, showed more potent anticancer activity than abiraterone alone in metastatic castration-resistant prostate cancer patients (78). Moreover, the combination of Akt inhibitor AZD5363 and enzalutamide showed synergistic anti-prostate cancer effects in preclinical models (79) and has been tested in a phase I clinical trial (80). However, in another phase II clinical trial, a pan-class I PI3 kinase inhibitor BKM120 (buparlisib), with or without enzalutamide co-treatment, had only limited efficacy in men with metastatic castration-resistant prostate cancer (81).
Eukaryotic DNA wraps around histone proteins and forms an inhibitory chromatin structure. Gene activation by the androgen receptor requires assistance from other transcription factors. Among these factors, GATA2 and FoxA1 play particularly essential roles in androgen receptor signaling in prostate cancer cells. GATA2 belongs to the GATA family of transcription factors which contains six members in mammals. GATA2 factors bind to a consensus DNA sequence (A/T)GATA(A/G) and regulate gene expression. GATA factors are expressed in a tissue-specific manner and play fundamental roles in cell-fate specification (82). The role of GATA2 in androgen signaling was first implicated by the involvement of GATA2 in androgen regulation of the PSA gene (83). Binding motifs for GATA factors and Oct1 are enriched on AR binding regions in LNCaP cells, suggesting that these transcription factors cooperate with AR in mediating the androgen response (84). In addition to its co-factor function, GATA2 might directly regulate androgen receptor mRNA and protein expression in prostate cancer cells (85–87). Inhibition of GATA2 by small-molecule compounds is a potential strategy in blocking AR expression and signaling in castration-resistant prostate cancer (86).
FoxA1 is member of the forkhead family of DNA binding factors and plays a key role in androgen receptor-induced gene transcription. FoxA1 functions as a pioneer factor because it can bind to highly compacted chromatin and allows these genomic regions to be more accessible to other transcription factors. Therefore, FoxA1 functions to guide androgen receptor binding to the genomic sites in prostate cancer cells (88, 89). In normal prostate luminal epithelial cells, it plays an important role in maintaining the differentiation status. FoxA1 mutations occur frequently in primary and metastatic prostate cancers and may contribute to prostate tumorigenesis and cancer progression (90, 91). Loss of FoxA1 promotes prostate cancer progression to neuroendocrine small-cell prostate cancer (92). FoxA1 also has androgen receptor-independent function in prostate cancer (93).
Even with the latest androgen deprivation therapies, castration-resistant prostate cancers are rarely cured. They simply become resistant again. Strikingly, a substantial subset of these resistant cancers still express androgen receptors and/or their variants; their growth and survival are still dependent on androgen receptor signaling. Scientists in the field of prostate cancer research are relentless in pursuing novel strategies for more complete ablation of androgen receptor signaling.
Prompted by the clinical success of selective estrogen receptor downregulator (SERD) Faslodex (ICI 182,780 or Fulvestrant) (94), selective androgen receptor downregulators (SARDs) have been developed. For instance, a SARD compound AZD3514 (95) had undergone phase I clinical trial (96). Binding of SERD or SARD causes severe receptor conformational change, resulting in receptor degradation. Another strategy is to specifically degrade the androgen receptor protein through Proteolysis Targeting Chimeras (PROTACs). Briefly, a PROTAC molecule consists of two covalently linked ligands: one ligand binds to the target protein whereas the second ligand binds to an E3 ligase system. Several AR targeting PROTACs have been reported, including enzalutamide-derived ARCC-4 (97) and aryloxy tetramethylcyclobutane-derived ARD-69 (98, 99). ARCC-4 and ARD-69 represent a novel class of drugs which directly targets the androgen receptor protein for degradation, but their in vivo anti-prostate cancer activities remains to be established in mouse models. Similarly, small-molecule degraders of the Bromodomain and Extra-Terminal (BET) family of epigenetic regulators, which are essential for prostate cancer growth, showed in vivo anti-cancer efficacy in a castration-resistant VCaP xenograft mouse model (100).
Another strategy is to silence androgen receptor gene expression at the transcriptional level. Androgen receptor gene expression is driven by an orphan nuclear receptor RORγ in metastatic castration-resistant prostate cancer (101). RORγ antagonists XY018 and SR2211 potently suppressed the expression of the full length androgen receptor and truncated androgen receptor variants at the transcriptional level, consequently inhibiting prostate cancer growth in xenograft mouse model (101). It has been shown that enzalutamide-resistant prostate tumors are sensitive to RORγ antagonists, suggesting that such a strategy may be able to overcome resistance to second-generation antiandrogens. In comparison to older strategies, this treatment can silence the expression of both full length and truncated variant androgen receptors.
One more exciting area for cancer drug development is the use of synthetic lethality. Because a subset of cancers contains defects in their DNA repair system, they become particularly vulnerable to inhibition of DNA repair enzymes. Olaparib, an inhibitor of poly(ADP-ribose) polymerase (PARP) 1 and 2, two key enzymes involved in DNA repair, has been approved by FDA for germline BRCA-mutated metastatic breast cancer (102). In a phase II trial, olaparib produced a high response rate in castration-resistant prostate cancers with DNA-repair defects including BRCA2 loss and ATM aberrations (103). A phase II trial further shows that olaparib in combination with abiraterone increased progression-free survival in men with metastatic castration-resistant prostate cancer (104).
Although androgen deprivation therapy prolongs the survival and improves the quality of life of prostate cancer patients, it does not cure the disease. With more complete androgen deprivation therapies and androgen receptor ablation in the near future, we hypothesize that more castration-resistant prostate cancers will undergo de-differentiation, eventually lose androgen receptor expression, and become truly androgen-independent androgen receptor-negative small-cell prostate cancers (105, 106) (Figure 3). These small-cell prostate cancers have neuroendocrine markers or basal stem cell gene signatures (109), and they will no longer respond to hormone therapy or androgen receptor targeting therapy. There will be an urgent need to develop novel targeted therapies for this subtype of prostate cancer, when more small-cell prostate cancers are induced by complete androgen deprivation and androgen receptor ablation. It has been reported that these cancers contain gene amplification of AURKA and MYCN, which are promising therapeutic targets for this subtype of cancer (106).
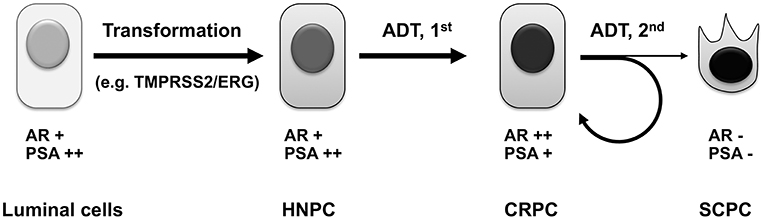
Figure 3. Evolution of prostate cancer under androgen deprivation therapy. Androgen sensitive primary prostate cancers arise from prostate luminal epithelial cells, which have undergone genetic alterations, such as mutation of PTEN tumor suppressor (107) or chromosomal rearrangement resulting in the TMPRSS2/ERG chimeric gene (108). Upon androgen deprivation including castration and the first-generation antiandrogen treatment, most HNPC will develop into CRPC, whose survival and growth still depends on androgen receptor signaling. After treatment with more potent androgen deprivation therapies such as second-generation antiandrogens, the majority of CRPC manages to develop novel mechanisms to maintain active androgen signaling axis to confer resistance, whereas a subset of CRPC will irreversibly lose androgen receptor expression, undergo divergent clonal evolution or de-differentiation, and become truly androgen-independent small-cell prostate cancer. ADT, androgen deprivation therapy; HNPC, hormone-naïve prostate cancer; CRPC, castration-resistant prostate cancer; SCPC, small-cell prostate cancer.
Recent advances in immunotherapy are revolutionizing the treatment of cancer. For example, Sipuleucel-T (Provenge) for CRPC is the first FDA-approved therapeutic cancer vaccine (110). However, while the use of Sipuleucel-T prolonged overall survival, it did not lead to PSA reduction, tumor shrinkage, or improve disease free survival. The checkpoint blockade therapies using antibodies to block CTLA-4 or PD-1 have achieved long-term clinical benefits, and even cures a subset of cancers (111). Tumor infiltrating lymphocytes (TIL) have also shown huge promise in treating cancers (112). The success of checkpoint blockade and TIL therapies are dependent on the tumor mutational burden (113, 114). With more somatic mutations, cancer cells are more likely to be recognized by T lymphocytes as “non-self” foreigners and thereby likely to be eliminated by the immune system. Prostate cancer cells are known to have low mutation rates (115, 116) and therefore the vast majority of prostate cancers are insensitive to current single checkpoint blockade immunotherapies. Only a small subset of prostate cancers with mismatch repair defects or CDK12 mutations are likely to respond to checkpoint blockade (117, 118). Nevertheless, the combination of PD-1 and CTLA4 inhibitors in a phase II CheckMate 650 trial elicited durable clinical responses in metastatic castration-resistant prostate cancers (119). It is also possible that continuous androgen deprivation therapies will cause more mutations and genomic alternations, and render prostate cancer cells more vulnerable to immunotherapy (117).
In addition, the recently emerging chimeric antigen receptor (CAR) T cell therapy is a promising strategy for treatment of castration-resistant prostate cancer. The CAR T cell immunotherapy has recently been approved by FDA for treatment of refractory pre-B cell acute lymphoblastic leukemia and diffuse large B cell lymphoma (120). Because CAR-engineered T lymphocytes recognize cancer cells through cancer cell surface antigens, their anti-cancer activity is not dependent on mutations in cancer cells. This is particularly important for prostate cancers which harbor low amount of somatic mutations. In the literature, there are several reports of PSMA-specific CAR T-cell therapies which have shown anti-prostate cancer activity in vitro and in mouse models (121, 122). Additionally, CAR-engineered natural killer (NK) cell therapy is another promising treatment for castration-resistant prostate cancer. Taken together, with these new targeted and immune therapies in sight, scientists and patients can be optimistic about eventually winning the battle against castration-resistant prostate cancer.
QF and BH conceived and wrote the manuscript.
This work was supported by National Institute of Health (NIH) National Cancer Institute R01CA211861, American Cancer Society RSG-13-061-01-TBE, and Houston Methodist Research Institute start-up funding to BH, and by NIH National Institute of Allergy and Infectious Disease R33AI122418 and R61AI133697 to QF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Adam Q. He for help with editing the manuscript.
1. He B, Bowen NT, Minges JT, Wilson EM. Androgen-induced NH2- and COOH-terminal interaction inhibits p160 coactivator recruitment by activation function 2. J Biol Chem. (2001) 276:42293–301. doi: 10.1074/jbc.M107492200
2. Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci USA. (2004) 101:4758–63. doi: 10.1073/pnas.0401123101
3. Nadal M, Prekovic S, Gallastegui N, Helsen C, Abella M, Zielinska K, et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat Commun. (2017) 8:14388. doi: 10.1038/ncomms14388
4. He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. (2000) 275:22986–94. doi: 10.1074/jbc.M002807200
5. He B, Minges JT, Lee LW, Wilson EM. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem. (2002) 277:10226–35. doi: 10.1074/jbc.M111975200
6. He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, An G, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. (2004) 16:425–38. doi: 10.1016/j.molcel.2004.09.036
7. He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J Biol Chem. (2002) 277:25631–9. doi: 10.1074/jbc.M202809200
8. Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. (2002) 23:175–200. doi: 10.1210/edrv.23.2.0460
9. He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. J Biol Chem. (1999) 274:37219–25. doi: 10.1074/jbc.274.52.37219
10. Bai S, He B, Wilson EM. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol Cell Biol. (2005) 25:1238–57. doi: 10.1128/MCB.25.4.1238-1257.2005
11. Wilson EM, French FS. Binding properties of androgen receptors. Evidence for identical receptors in rat testis, epididymis, and prostate. J Biol Chem. (1976) 251:5620–9.
12. Grino PB, Griffin JE, Wilson JD. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology. (1990) 126:1165–72. doi: 10.1210/endo-126-2-1165
13. Swinney DC. The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Discov Devel. (2009) 12:31–9.
14. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. (1941) 1:293–7.
15. Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA Cancer J Clin. (2002) 52:154–79. doi: 10.3322/canjclin.52.3.154
16. de Voogt HJ. The position of cyproterone acetate (CPA), a steroidal anti-androgen, in the treatment of prostate cancer. Prostate Suppl. (1992) 4:91–5. doi: 10.1002/pros.2990210514
17. Simard J, Luthy I, Guay J, Belanger A, Labrie F. Characteristics of interaction of the antiandrogen flutamide with the androgen receptor in various target tissues. Mol Cell Endocrinol. (1986) 44:261–70. doi: 10.1016/0303-7207(86)90132-2
18. Kolvenbag GJ, Furr BJ, Blackledge GR. Receptor affinity and potency of non-steroidal antiandrogens: translation of preclinical findings into clinical activity. Prostate Cancer Prostatic Dis. (1998) 1:307–14. doi: 10.1038/sj.pcan.4500262
19. Joyce R, Fenton MA, Rode P, Constantine M, Gaynes L, Kolvenbag G, et al. High dose bicalutamide for androgen independent prostate cancer: effect of prior hormonal therapy. J Urol. (1998) 159:149–53. doi: 10.1016/S0022-5347(01)64039-4
20. Labrie F, Dupont A, Belanger A, Cusan L, Lacourciere Y, Monfette G, et al. New hormonal therapy in prostatic carcinoma: combined treatment with an LHRH agonist and an antiandrogen. Clin Invest Med. (1982) 5:267–75.
21. Labrie F, Dupont A, Belanger A, St-Arnaud R, Giguere M, Lacourciere Y, et al. Treatment of prostate cancer with gonadotropin-releasing hormone agonists. Endocr Rev. (1986) 7:67–74. doi: 10.1210/edrv-7-1-67
22. Dijkman GA, Janknegt RA, De Reijke TM, Debruyne FM. Long-term efficacy and safety of nilutamide plus castration in advanced prostate cancer, and the significance of early prostate specific antigen normalization. International Anandron Study Group. J Urol. (1997) 158:160–3. doi: 10.1097/00005392-199707000-00051
23. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists' Collaborative Group. Lancet. (2000) 355:1491–8. doi: 10.1016/S0140-6736(00)02163-2
24. Mohler JL, Gregory CW, Ford OH III, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. (2004) 10:440–8. doi: 10.1158/1078-0432.CCR-1146-03
25. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. (2005) 11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525
26. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. (2008) 68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249
27. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. (2001) 1:34–45. doi: 10.1038/35094009
28. Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. (2004) 11:459–76. doi: 10.1677/erc.1.00525
29. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. (1995) 9:401–6. doi: 10.1038/ng0495-401
30. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. (2001) 61:3550–5.
31. Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. (1997) 57:314–9.
32. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. (2004) 10:33–9. doi: 10.1038/nm972
33. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. (2015) 161:1215–28. doi: 10.1016/j.cell.2015.06.053
34. Takeda DY, Spisak S, Seo JH, Bell C, O'Connor E, Korthauer K, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. (2018) 174:422–32.e13. doi: 10.1016/j.cell.2018.05.037
35. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. (2018) 174:758–69.e9. doi: 10.1016/j.cell.2018.06.039
36. Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. (2001) 61:4315–9.
37. Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res. (2009) 7:523–35. doi: 10.1158/1541-7786.MCR-08-0400
38. Minges JT, Su S, Grossman G, Blackwelder AJ, Pop EA, Mohler JL, et al. Melanoma antigen-A11 (MAGE-A11) enhances transcriptional activity by linking androgen receptor dimers. J Biol Chem. (2013) 288:1939–52. doi: 10.1074/jbc.M112.428409
39. Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res. (1996) 2:277–85.
40. Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. (1990) 173:534–40. doi: 10.1016/S0006-291X(05)80067-1
41. Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. (1995) 332:1393–8. doi: 10.1056/NEJM199505253322101
42. Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. (2000) 6:703–6. doi: 10.1038/76287
43. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. (2009) 69:16–22. doi: 10.1158/0008-5472.CAN-08-2764
44. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. (2009) 69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795
45. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. (2008) 68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594
46. Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. (2004) 279:7119–30. doi: 10.1074/jbc.M307649200
47. Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. (1994) 54:5474–8.
48. Kelly WK, Scher HI. Prostate specific antigen decline after antiandrogen withdrawal: the flutamide withdrawal syndrome. J Urol. (1993) 149:607–9. doi: 10.1016/S0022-5347(17)36163-3
49. Small EJ, Carroll PR. Prostate-specific antigen decline after casodex withdrawal: evidence for an antiandrogen withdrawal syndrome. Urology. (1994) 43:408–10. doi: 10.1016/0090-4295(94)90092-2
50. Huan SD, Gerridzen RG, Yau JC, Stewart DJ. Antiandrogen withdrawal syndrome with nilutamide. Urology. (1997) 49:632–4. doi: 10.1016/S0090-4295(96)00558-4
51. Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. (1997) 3:1383–8.
52. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. (2009) 324:787–90. doi: 10.1126/science.1168175
53. Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. (2015) 5:12007. doi: 10.1038/srep12007
54. Rathkopf DE, Saleh MN, Tsai FYC, Rosen LS, Adams BJ, Liu L, et al. An open-label phase 1/2a study to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of TRC253, an androgen receptor antagonist, in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. (2018) 36:TPS403. doi: 10.1200/JCO.2018.36.6_suppl.TPS403
55. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
56. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. (2014) 371:424–33. doi: 10.1056/NEJMoa1405095
57. Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. (2018) 378:1408–18. doi: 10.1056/NEJMoa1715546
58. Hussain M, Fizazi K, Saad F, Rathenborg P, Shore N, Ferreira U, et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med. (2018) 378:2465–74. doi: 10.1056/NEJMoa1800536
59. Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. (2019) 380:1235–46. doi: 10.1056/NEJMoa1815671
60. Chi KN, Agarwal N, Bjartell A, Chung BH, Pereira de Santana Gomes AJ, Given R, et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N Engl J Med. (2019) 381:13–24. doi: 10.1056/NEJMoa1903307
61. Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med. (2019) 381:121–31. doi: 10.1056/NEJMoa1903835
62. Armstrong AJ, Szmulewitz RZ, Petrylak DP, Holzbeierlein J, Villers A, Azad A, et al. ARCHES: A randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J Clin Oncol. (2019). doi: 10.1200/JCO.19.00799. [Epub ahead of print].
63. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. (2011) 364:1995–2005. doi: 10.1056/NEJMoa1014618
64. Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. (2019) 20:686–700. doi: 10.1016/S1470-2045(19)30082-8
65. Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. (2017) 377:352–60. doi: 10.1056/NEJMoa1704174
66. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. (2013) 3:1030–43. doi: 10.1158/2159-8290.CD-13-0142
67. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. (2013) 3:1020–9. doi: 10.1158/2159-8290.CD-13-0226
68. Lallous N, Volik SV, Awrey S, Leblanc E, Tse R, Murillo J, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. (2016) 17:10. doi: 10.1186/s13059-015-0864-1
69. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. (2013) 155:1309–22. doi: 10.1016/j.cell.2013.11.012
70. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. (2014) 371:1028–38. doi: 10.1056/NEJMoa1315815
71. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. (2013) 73:483–9. doi: 10.1158/0008-5472.CAN-12-3630
72. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. (2014) 20:3198–210. doi: 10.1158/1078-0432.CCR-13-3296
73. Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. (2015) 75:1413–22. doi: 10.1158/0008-5472.CAN-14-3080
74. Li S, Fong KW, Gritsina G, Zhang A, Zhao JC, Kim J, et al. Activation of MAPK signaling by CXCR7 leads to enzalutamide resistance in prostate cancer. Cancer Res. (2019) 79:2580–92. doi: 10.1158/0008-5472.CAN-18-2812
75. Lee E, Wongvipat J, Choi D, Wang P, Lee YS, Zheng D, et al. GREB1 amplifies androgen receptor output in human prostate cancer and contributes to antiandrogen resistance. Elife. (2019) 8:e41913. doi: 10.7554/eLife.41913
76. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. (2011) 19:575–86. doi: 10.1016/j.ccr.2011.04.008
77. Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. (2011) 19:792–804. doi: 10.1016/j.ccr.2011.05.006
78. de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating Akt blockade with Ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res. (2019) 25:928–36. doi: 10.1158/1078-0432.CCR-18-0981
79. Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol. (2015) 67:986–90. doi: 10.1016/j.eururo.2014.08.006
80. Kolinsky MP, Rescigno P, Bianchini D, Zafeiriou Z, Mehra N, Mateo J. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 in patients with mCRPC. J Clin Oncol. (2017) 35:135. doi: 10.1200/JCO.2017.35.6_suppl.135
81. Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer. (2017) 81:228–36. doi: 10.1016/j.ejca.2017.02.030
82. Patient RK, McGhee JD. The GATA family (vertebrates and invertebrates). Curr Opin Genet Dev. (2002) 12:416–22. doi: 10.1016/S0959-437X(02)00319-2
83. Perez-Stable CM, Pozas A, Roos BA. A role for GATA transcription factors in the androgen regulation of the prostate-specific antigen gene enhancer. Mol Cell Endocrinol. (2000) 167:43–53. doi: 10.1016/S0303-7207(00)00300-2
84. Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. (2007) 27:380–92. doi: 10.1016/j.molcel.2007.05.041
85. Bohm M, Locke WJ, Sutherland RL, Kench JG, Henshall SM. A role for GATA-2 in transition to an aggressive phenotype in prostate cancer through modulation of key androgen-regulated genes. Oncogene. (2009) 28:3847–56. doi: 10.1038/onc.2009.243
86. He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci USA. (2014) 111:18261–6. doi: 10.1073/pnas.1421415111
87. Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. (2014) 42:3607–22. doi: 10.1093/nar/gkt1382
88. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. (2013) 73:1570–80. doi: 10.1158/0008-5472.CAN-12-2350
89. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. (2011) 30:3962–76. doi: 10.1038/emboj.2011.328
90. Parolia A, Cieslik M, Chu SC, Xiao L, Ouchi T, Zhang Y, et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature. (2019). 571:413–8. doi: 10.1158/1538-7445.SABCS18-4497
91. Adams EJ, Karthaus WR, Hoover E, Liu D, Gruet A, Zhang Z, et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature. (2019) 571:408–12. doi: 10.1038/s41586-019-1318-9
92. Kim J, Jin H, Zhao JC, Yang YA, Li Y, Yang X, et al. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene. (2017) 36:4072–80. doi: 10.1038/onc.2017.50
93. Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. (2013) 73:3725–36. doi: 10.1158/0008-5472.CAN-12-3468
94. Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. (2002) 20:3396–403. doi: 10.1200/JCO.2002.10.057
95. Bradbury RH, Acton DG, Broadbent NL, Brooks AN, Carr GR, Hatter G, et al. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg Med Chem Lett. (2013) 23:1945–8. doi: 10.1016/j.bmcl.2013.02.056
96. Omlin A, Jones RJ, van der Noll R, Satoh T, Niwakawa M, Smith SA, et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer - results of two parallel first-in-human phase I studies. Invest New Drugs. (2015) 33:679–90. doi: 10.1007/s10637-015-0235-5
97. Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. (2018) 1:100. doi: 10.1038/s42003-018-0105-8
98. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. (2019) 62:941–64. doi: 10.1021/acs.jmedchem.8b01631
99. Guo C, Linton A, Kephart S, Ornelas M, Pairish M, Gonzalez J, et al. Discovery of aryloxy tetramethylcyclobutanes as novel androgen receptor antagonists. J Med Chem. (2011) 54:7693–704. doi: 10.1021/jm201059s
100. Kregel S, Malik R, Asangani IA, Wilder-Romans K, Rajendiran T, Xiao L, et al. Functional and mechanistic interrogation of BET bromodomain degraders for the treatment of metastatic castration-resistant prostate cancer. Clin Cancer Res. (2019) 78(Suppl. 13):5795. doi: 10.1158/1538-7445.AM2018-5795
101. Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, et al. ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. (2016) 22:488–96. doi: 10.1038/nm.4070
102. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
103. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
104. Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2018) 19:975–86. doi: 10.1016/S1470-2045(18)30365-6
105. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. (2016) 22:298–305. doi: 10.1038/nm.4045
106. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. (2011) 1:487–95. doi: 10.1158/2159-8290.CD-11-0130
107. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. (1997) 275:1943–7. doi: 10.1126/science.275.5308.1943
108. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. (2005) 310:644–8. doi: 10.1126/science.1117679
109. Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, et al. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci USA. (2015) 112:E6544–52. doi: 10.1073/pnas.1518007112
110. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
111. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
112. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
113. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386
114. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
115. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
116. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. (2015) 349:1483–9. doi: 10.1126/science.aab4082
117. Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. (2018) 173:1770–82.e14. doi: 10.1016/j.cell.2018.04.034
118. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
119. Anti-PD-1-CTLA4 Combo hits prostate cancer. Cancer Discov. (2019) 9:569–70. doi: 10.1158/2159-8290.CD-NB2019-039
120. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
121. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
Keywords: prostate cancer (PCa), androgen receptor (AR), androgen deprivation therapy (ADT), castration-resistant prostate cancer (CRPC), small-cell prostate cancer (SCPC), antiandrogen
Citation: Feng Q and He B (2019) Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 9:858. doi: 10.3389/fonc.2019.00858
Received: 02 June 2019; Accepted: 20 August 2019;
Published: 04 September 2019.
Edited by:
Renee de Leeuw, University of Illinois at Chicago, United StatesReviewed by:
Joaquin Mateo, Vall d'Hebron Institute of Oncology (VHIO), SpainCopyright © 2019 Feng and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Feng, cWZlbmc0QGNlbnRyYWwudWguZWR1; Bin He, YmhlQGhvdXN0b25tZXRob2Rpc3Qub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.