 Rintu Thomas
Rintu Thomas Zhang Weihua*
Zhang Weihua*- Department of Biology and Biochemistry, College of Natural Science and Mathematics, University of Houston, Houston, TX, United States
The epidermal growth factor receptor (EGFR) is one of most potent oncogenes that are commonly altered in cancers. As a receptor tyrosine kinase, EGFR's kinase activity has been serving as the primary target for developing cancer therapeutics, namely the EGFR inhibitors including small molecules targeting its ATP binding pocket and monoclonal antibodies targeting its ligand binding domains. EGFR inhibitors have produced impressive therapeutic benefits to responsive types of cancers. However, acquired and innate resistances have precluded current anti-EGFR agents from offering sustainable benefits to initially responsive cancers and benefits to EGFR-positive cancers that are innately resistant. Recent years have witnessed a realization that EGFR possesses kinase-independent (KID) pro-survival functions in cancer cells. This new knowledge has offered a different angle of understanding of EGFR in cancer and opened a new avenue of targeting EGFR for cancer therapy. There are already many excellent reviews on the role of EGFR with a focus on its kinase-dependent functions and mechanisms of resistance to EGFR targeted therapies. The present opinion aims to initiate a fresh discussion about the function of EGFR in cancer cells by laying out some unanswered questions pertaining to EGFR in cancer cells, by rethinking the unmet therapeutic challenges from a view of EGFR's KID function, and by proposing novel approaches to target the KID functions of EGFR for cancer treatment.
Highlights
- EGFR possesses oncogenic pro-survival functions independent of its tyrosine kinase activity.
- Targeting EGFR's kinase independent functions may overcome cancer resistance to current EGFR inhibitors.
Introduction
Structure-function based studies have firmly established the foundation of our knowledge about the canonical function of epidermal growth factor receptor (EGFR), a receptor tyrosine kinase that can dimerize, autocross-phosphorylate, and initiate a cascade of down-stream signals (1). Assuming that elevation of the default tyrosine kinase function of EGFR, owning to over-expression or kinase activating mutations, is all that cancer cells depend on in driving malignancy, the canonical tyrosine kinase function of EGFR has served as a beacon directing the design of EGFR targeted therapies for cancer. However, current potent EGFR inhibitors, small molecules of tyrosine kinase inhibitors (TKI) competing with ATP for kinase activation and monoclonal antibody inhibitors (mAb) preventing EGFR from being activated by its ligands, have exhibited limited efficacies and have been challenged by innate and acquired resistance in the clinic (2, 3). The majority of EGFR positive cancers do not respond to TKIs nor to mAbs, e.g., non-small lung cancers expressing wild-type EGFR, representing the innate resistance to TKIs. A fraction of EGFR positive cancers expressing EGFR with kinase activating mutation, non-small cell lung cancer (NSCLC) in particular, transiently respond to TKIs, however, these cancers develop acquired resistance to TKIs within about 1 year of therapy without exception, which exemplifies the acquired resistance (4–7). The exact mechanism underlying sensitivity to anti-EGFR mAbs remains undefined. Only a small fraction of EGFR positive cancers represented by advanced colorectal cancers expressing wild type KRAS respond to anti-EGFR mAbs although acquired resistance also commonly occurs (8, 9). The mechanism responsible for the innate resistance is largely unexplored.
The realization that EGFR possesses pro-survival functions independent of its kinase activity over the past decade has opened a new window for a better understanding the role of EGFR in cancer and offered a novel approach of targeting this powerful oncogene for cancer therapy.
Alterations of EGFR in Cancer
EGFR is one of the most frequently altered oncogenes in solid cancers (1, 10). There are two types of pathological alterations of EGFR in cancers, one is kinase-activating mutation in EGFR and the other is over-expression of the EGFR protein. The kinase-activating mutations, which lead to increased tyrosine kinase activity of EGFR, can be primary or secondary to anti-EGFR therapies (11–13). Over-expression of EGFR protein can be associated with/without EGFR gene amplifications (14–21). Primary kinase-activating mutations in EGFR occur often in NSCLC and glioblastoma, but rarely in other types of cancers. In NSCLCs, EGFR is mutated in about 30–40% of East Asian patients and about 5–15% in non-East Asian patients (22, 23). In about 30% of glioblastomas, the 2–7 exons of EGFR are deleted which gives rise to an extracellular domain truncated EGFR named EGFRvIII whose tyrosine kinase is constantly active due to its ligand independent dimerization (24). As to secondary mutations contributing to the acquired resistance to anti-EGFR therapies, the T790M mutation accounts for 50% of resistance in NSCLC patients treated with first and second-generation TKIs (25–27). The C797S mutation is seen in T790M selective TKI treated NSCLC patients, however, its incidence remains unknown (28, 29). Mutations in the extracellular domain of EGFR were found in a few resistant colorectal cancer (CRC) patients after treatment with monoclonal antibody Cetuximab (30).
Unlike the EGFR kinase-activating mutations that occur mainly in NSCLC patients, wild-type EGFR protein is commonly over-expressed in many types of solid cancers and is often associated with negative prognosis (31–47), i.e., over-expression of wild-type EGFR is a more common phenomenon than EGFR mutations in solid cancers and promotes disease progression. Alterations of EGFR in 13 types of solid cancers and their responses to anti-EGFR agents are summarized in Table 1. It is worth noting that the majority of EGFR positive cancers do not respond to current anti-EGFR agents. Anti-EGFR therapies are mainly used for treating three types of cancers, which are NSCLC bearing kinase-activating mutations in EGFR for TKIs (4, 88, 89), about 10% of advanced metastatic colorectal cancers (CRCs) for anti-EGFR mAbs (90, 91), and locoregional advanced head and neck cancers (HNCs) for combination of mAbs with radiotherapy (92, 93).
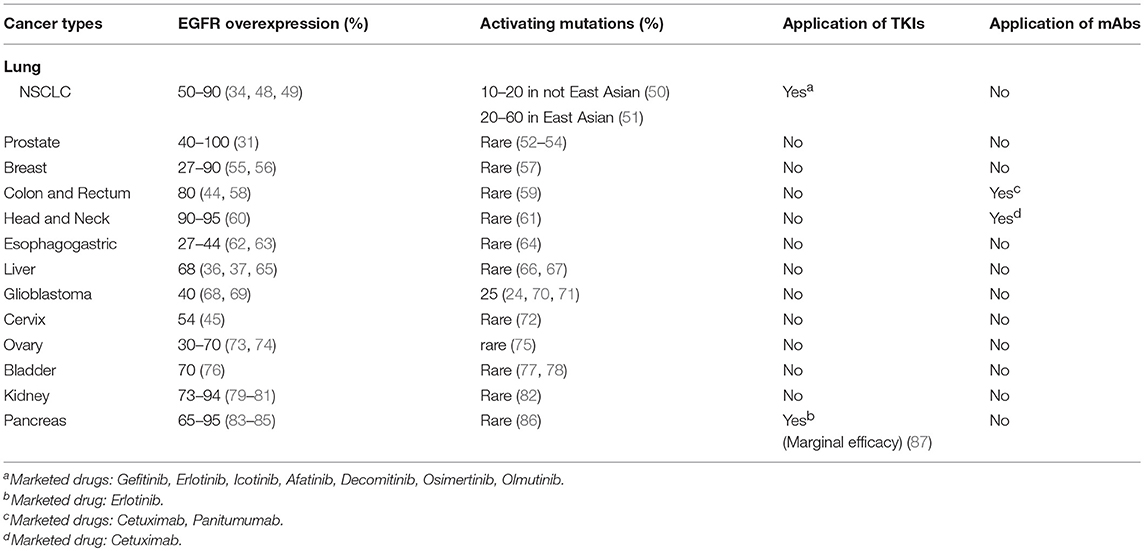
Table 1. Alterations of EGFR in cancers and application of EGFR inhibitors.
The Mechanistic Basis for Current Anti-EGFR Cancer Therapies
Our understanding of EGFR started from the purification of the epidermal growth factor (EGF) (94), the default ligand of EGFR, the discovery of the intrinsic tyrosine kinase activity of EGFR (95), and the cloning of the EGFR gene (96). The canonical function of EGFR is initiated by ligand binding, which results in EGFR dimerization, cross-phosphorylation of its counterpart in the dimer at a few tyrosyl residues located at the carboxyl intracellular domain of EGFR, and subsequently these phosphorylated microdomains serve as docking sites for signal transductors to trigger downstream signaling cascades (1). This canonical mechanism of EGFR function has served as the authentic guidance for designing EGFR targeted therapeutics.
With regard to TKIs, there has been so called four generations of them. The first generation TKIs are represented by Gefitinib and Erlotinib, each of which reversibly competes with ATP to bind to EGFR (97). The second generation of TKI is represented by Afatinib that covalently binds to the ATP binding pocket to irreversibly inhibit EGFR's kinase activity regardless of EGFR mutations (98–100). The third generation of TKIs are represented by Osimertinib and Olmutinib that preferentially and covalently inhibit the T970M mutant of EGFR that is responsible for about 50% of acquired resistance to the earlier generation of TKIs (101–103). The fourth generation of TKIs, which preferentially inhibit the T790M/C797S EGFR mutant that leads to some resistance to the 3rd generation TKIs, are under early phases of preclinical development. The first 4th generation of TKI is represented by an allosteric inhibitor EAI045 that is effective in inhibiting the kinase activity of T790M/C797S only in combination with anti-EGFR mAb Cetuximab but not as a single agent (104), and its underlying mechanism is unknown. Updated molecular principals of design, action, and clinical impact of these TKIs have been comprehensively reviewed (105). Regardless of the mutational selectivity of the TKIs, their effectiveness is determined by their capability of inhibiting the tyrosine kinase activity of a given form of EGFR, i.e., the tyrosine kinase activity of EGFR is the primary target.
Currently, there are two FDA approved anti-EGFR mAbs for cancer therapies in the USA, Cetuximab and Panitumumab for metastatic colorectal cancer (9, 106, 107), and Cetuximab for locoregional head and neck cancer (92). The exact mechanisms underlying the therapeutic effects of the anti-EGFR mAbs remain to be defined, although the rationale for the design of these mAb is primarily rooted at blocking EGFR from being activated by its ligands (108–111). Proposed mechanisms mediating the therapeutic effects of anti-EGFR mAbs include inhibition of EGFR's kinase dependent downstream signals (108), down-regulation of membranous EGFR by induction of EGFR internalization and subsequent degradation in late endosomes (112), and induction of antibody-dependent cell-mediated cytotoxicity (113). However, neither the phosphorylation status nor the expression levels of EGFR in cancer tissues is predictive for efficacy of anti-EGFR mAbs (107, 114, 115). The therapeutic effect of anti-EGFR Abs cannot be solely attributed to inhibition of EGFR's tyrosine kinase activity.
Persistent Challenges to Current Anti-EGFR Cancer Therapies
EGFR TKIs have clearly been clinically efficacious in responsive types of cancers (expressing kinase-activating mutations in EGFR), however, the benefits are often limited to improving the progression free survival (PSF) and quality of life rather than the overall survival (OS) (116–121). EGFR mAbs alone or in combination with chemotherapies have achieved an increase in unsustainable OS to <10% of metastatic colorectal cancer (122), and EGFR mAb in combination with radiotherapy has been shown to increase the 5 year OS rate by about 10% to regionally advanced head and neck cancers (123). Overall, meaningful clinical benefits offered by the current anti-EGFR agents are limited. Two major unmet challenges have stymied the efficacy of EGFR targeted cancer therapies.
The first challenge is the acquired resistance toward the anti-EGFR drugs, which has also been the research focus of EGFR targeted therapy. Expectedly, molecular adaptations at two levels, adaptive mutations in EGFR gene and adaptive gain-of-function of alternative survival and growth pathways play important roles in the development of acquired resistance to anti-EGFR drugs. As to TKI acquired resistance, reported resistant mechanisms include secondary amplification of and mutations in EGFR such as the T790M and C797S, gain-of-activities of alternative oncogenic pathways such as RAF/MEK/MAPK/ERK, PI3K/Akt, and MET regulated signal pathways, which has been extensively reviewed (4, 5, 88, 124, 125). Regarding anti-EGFR mAbs, mechanisms of acquired resistance are largely unclear, which is understandable given that the exact mechanism of action of these drugs has not been fully understood. Nevertheless, secondary mutations in the extracellular domain of EGFR, mutations in KRAS, NRAS and C-Met, loss of PTEN and activating mutations in PIK3CA, and gain-of-activity in the IGFR pathway have been associated with acquired resistances of some cases of colorectal cancer (8, 9, 126, 127).
The second challenge is the innate resistance to anti-EGFR drugs, which is much more prevalent than the acquired resistance. Although EGFR TKIs are potent in inhibiting the kinase activity of wild-type EGFR, cancers expressing wild-type EGFR, such as lung cancer (128–131), head and neck cancers (132), prostate cancer (133), and ovarian cancer (134), do not respond to TKIs regardless of the expression level of EGFR. In addition, NSCLCs with certain kinase activating exon 20 insertions are often insensitive to TKIs (135–138). There is about more than 80% of advanced colorectal cancers that do not respond to anti-EGFR mAbs (127). Many other types of EGFR positive cancers, such as prostate cancer (139, 140), and ovarian cancer (33) are innately resistant to anti-EGFR mAbs. One speculation has been that EGFR is simply unimportant for those cancers that are innately resistant to EGFR kinase inhibitor. This assumption has been negated by the observations of severe cell death upon down-regulating EGFR proteins in cancer cells of cancers innately resistant to EGFR kinase inhibitors, e.g., prostate cancer cells (141, 142), breast cancer, ovarian cancer cells, wild-type EGFR expressing lung cancer cells, wild-type EGFR expressing colon cancer cells (142–144), renal cancer (79), and glioma (145). In other words, EGFR is indispensable for the survival of cancer cells that are innately resistant to EGFR kinase inhibitors.
Unanswered Questions Pertaining to EGFR's Kinase Dependent (KD) Role in Cancer Cells, and Their Impacts on Current Anti-EGFR Cancer Therapies
It has been more than a half century since the finding of EGF (94), the default physiological ligand of EGFR, from which the whole field of growth factors stemmed (95, 96). There is a large body of literature on EGFR biology which has firmly established the molecular mechanisms underlying its tyrosine kinase function and the canonical signal cascades governed by its kinase. However, when it comes to the utilization of these well-established theories to target EGFR for cancer therapy, the reality has raised some questions challenging the comprehensiveness of our knowledge on EGFR in cancer.
Question 1: Given the fact that increased protein expression level of EGFR correlates with cancer progression and over-expression of wild-type EGFR is tumorigenic, why do wild-type EGFR expressing cancers not respond to TKIs?
Our understanding of EGFR began from studying the function of wild-type EGFR using cancer cells (146, 147) and non-cancerous cells (148–151). Regardless of the cell types being used, TKIs have exhibited potent in vitro and in vivo effects on inhibiting the tyrosine kinase activity of wild-type EGFR. Over-expression of wild-type EGFR is tumorigenic in several types of cells (152–155), validating that wild-type EGFR is oncogenic. On one hand, protein levels of EGFR, but not its phosphorylation status, is strongly associated with disease progression and poor prognosis of many types of cancers that rarely express mutated EGFR (31, 32, 34, 38, 55, 76, 80, 156–160). Examples of cancers that exhibit increased EGFR expression along with disease progress and do not respond to TKIs include prostate cancer (133), ovarian cancer (157), pancreatic cancer (161), colorectal cancer (162), head and neck cancer (40), cervical cancer (163), and lung cancers expressing wild-type EGFR (128, 152, 164). On the other hand, EGFR mutations but not protein expression levels are associated with responsiveness to EGFR TKIs (165). There is no doubt that wild-type EGFR protein promotes cancer progression, but why do cancers expressing wild-type EGFR not respond to EGFR TKIs?
Explanation to this puzzling phenomenon has been that the wild-type EGFR expressing/overexpressing cancers are not addicted to EGFR function for growth/survival, however, this assertion is challenged by observations that TKIs are potent in inhibiting the growth of wild-type EGFR expressing cells (166–171) and by studies showing wild-type EGFR expressing cells cannot survive after EGFR knockdown by siRNA (142, 145, 172–174). These observations suggest that the EGFR wild-type cancer cells may be not addicted to EGFR's kinase activity but rely on the existence EGFR for survival, i.e., the survival of cancer cells is sustained by EGFR without involving its kinase activity.
Question 2: Why does the phosphorylation status of EGFR not correlate with cancer progression nor with responsiveness to anti-EGFR drugs?
There is no doubt that activation of the tyrosine kinase activity of EGFR, regardless of its mutational status, promotes cell proliferation and tumor growth of experimental models, which has served as the scientific basis supporting the targeting of the kinase activity of EGFR for cancer therapies (175). However, on one hand the level of total EGFR protein expression is closely associated with poor prognosis of many types of cancers including those cancers resistant to anti-EGFR agents (31, 32, 34, 38, 39, 80, 156–159) and gene copy number of EGFR is currently one of the most reliable predictors for sensitivity to anti-EGFR therapeutics; on the other hand, the level of phosphorylated EGFR is not a reliable predictor of NSCLC's sensitivity to TKIs and mutational status of EGFR is (89, 176, 177)—why?
The lack of association between pEGFR levels and disease status has been hypothetically attributed to a sum of technical inconsistences among studies, such as technical variations in performing immunohistochemistry, qualities of anti-pEGFR antibodies, procedures of cancer tissue preservation in the clinic, and patient sample size employed for analysis. These possibilities portray a virtually impossible mission to having these issues resolved. However, it does not stop the proposition of an untested concept that if the kinase activity of EGFR is indeed not critically involved in progression of cancers expressing wild-type EGFR but the total level of EGFR protein is, shouldn't we start considering a possibility that EGFR may own powerful oncogenic functions independent of its tyrosine kinase activity? This possibility is supported by a recent study that loss-of-function mutations of all the phosphorylatable tyrosyl residues of the C-terminal domain of a kinase-activating EGFR mutant retains its oncogenic function (178), i.e., the kinase dependent down-stream signaling of EGFR is not required for its oncogenic function.
Question 3: Why do the TKI responsive cancers not overlap with the anti-EGFR mAbs responsive cancers?
Both TKIs and anti-EGFR mAbs are potent in inhibiting the tyrosine kinase activity of EGFR in cancer cells, however, oddly the responsive cancer types of these two kinds of anti-EGFR reagents do not overlap at all. TKIs are approved for NSCLC especially for cancers expressing mutated EGFR (179, 180), whereas anti-EGFR mAbs are approved for KRAS wild-type colorectal cancer and local regional head and neck cancers (40, 106). Currently, there is no positive biomarker available for selection of cancer types that are favorable to anti-EGFR mAbs, although KRAS mutations are a negative predictor for anti-EGFR mAbs in treating colorectal cancer (181). An obvious question raised by this discrepancy between suitable cancer types of TKIs vs. mAbs is: Is inhibition of the kinase activity of EGFR primarily accountable for the efficacy of anti-EGFR mAbs?
Question 4: Is the tyrosine kinase activity of EGFR the shared primary driver of EGFR's pro-growth and pro-survival functions?
It has been a conventional statement that EGFR as a receptor tyrosine kinase plays important roles in promoting cell growth and survival without differentiating its weight on growth vs. survival; what is even more equivocal is that the EGFR regulated cell growth and survival has never been mechanistically differentiated. Cell growth and cell survival are totally different biological events, the former refers to increase in numbers or in size of individual entity whereas the latter refers to the ability to cope with stresses in order to stay alive. Growth depends on survival, however, survival is independent of growth. Cancer is a disease driven by abnormal cell growth and cell survival, thus treatment strategies ought to be differentially directed toward growth and survival.
Accumulated data over the past two decades strongly suggest that the tyrosine kinase activity of EGFR is predominantly involved in promoting cell proliferation (175) compared to cell survival. Consistently, EGFR TKIs and mAbs have constantly exhibited anti-proliferative effects under physiologically relevant conditions (182–185), which are often accompanied by surrogate makers of cell survival but not direct evidence of cell death of in vitro cultured cells (47, 166, 171, 186–196). Given the current understanding that the apoptosis process is reversible even at stages of the activation of caspases (197) and that therapeutic stresses can cause secretion of DNA fragment containing exosomes by cancer cells (198, 199), which can interfere the interpretation of the increase of sub-G0 cells (used to represent apoptotic cells using flow cytometry) caused by TKI treatments. Furthermore, TKIs or mAbs do not cause DNA fragmentation in many types of EGFR-positive cancer cells while their growth inhibition effects are obvious (166, 167, 169, 200–202). Regarding the impact of TKIs on cell survival, recent studies have revealed that TKIs are potent in inducing cytoprotective autophagy that in turn promotes cell survival (203–207).
It is critical to differentiate EGFR's pro-growth from its pro-survival functions, because if the kinase activity of EGFR is not pivotal for sustaining cancer cell survival, it becomes explainable that the current anti-EGFR reagents aiming to block the kinase activity of EGFR are unable to significantly induce death of cancer cells but are good at transiently inhibiting growth of cancer cells before cells develop alternative pro-growth signal pathways resulting in resistance. Re-growth associated gain of kinase activity mutations in EGFR (such as T709M and C797S) strongly suggests that the kinase activity of EGFR is important for cell growth. The dependence on EGFR for survival and the impact of its kinase activity on cell proliferation raises another important question: Are the pro-growth and pro-survival functions of EGFR divergent at its tyrosine kinase activity?
Current Standing of Our Knowledge of EGFR Biology and EGFR Targeted Cancer Therapy
Our current understanding of EGFR's canonical function and status of its tyrosine kinase targeted cancer therapy can be summarized as follows:
Validated canonical functions and mechanisms of EGFR action:
1. The tyrosine kinase activity of EGFR and mechanisms connecting with its down-stream signal cascades
2. The growth promoting role of the tyrosine kinase activity of EGFR
3. The oncogenic capacity of EGFR
4. The positive association of EGFR expression with progression of certain cancers
5. The dependence of kinase-activating mutations in EGFR for therapeutic effect of TKIs
6. The dependence of kinase-activating mutations in EGFR for a portion of acquired TKI resistance
7. The fact of unavoidable resistance to current anti-EGFR therapeutics
8. The clinical benefit of increased progression free survival but not overall survival for patients suitable for treatment of TKIs.
Overarching challenges:
1. Why do wild-type EGFR expressing/overexpressing cancers not response to EGFR TKIs?
2. What is the exact mechanism underlying anti-EGFR mAbs' therapeutic effect?
3. What is the exact mechanism underlying EGFR's pro-survival function in cancer cells?
Realization of the Existence of Kinase Independent Pro-survival Function of EGFR in Cancer Cells
As discussed above, when it comes to the question pertaining to EGFR's pro-survival function, the kinase activity of EGFR does not offer a full accountability. The past 10 years have witnessed a growing body of evidence indicating that EGFR possesses pro-survival functions that are independent of its tyrosine kinase activity in cancer cells.
In cancer cells, by comparison of the effects of an EGFR TKI and EGFR siRNA, it was found that EGFR maintained survival of prostate cancer cells independent of its kinase activity, i.e., TKI inhibited cell proliferation without effecting on cell survival whereas loss-of-EGFR expression induced by siRNA knockdown led to severe autophagic cell death that could be rescued by a kinase-dead EGFR (142). Furthermore, this study found that the sodium/glucose co-transporter 1 (SGLT1) played a critical role in mediating the KID pro-survival function of EGFR by maintaining active glucose uptake of cancer cells (142).
The existence of KID pro-survival function of EGFR has also been revealed in different types of cancer cells involving several cellular functional domains that include the plasma membrane, the autophagic machinery, and the mitochondrion. Within the plasma membrane, kinase independently, EGFR interacts with SGLT1 to maintain active glucose uptake (142), interacts with the system x antiporter to maintain cystine import (145), interacts with fatty acid synthase to maintain de novo fatty acids synthesis (208), and interacts with the mTORC2 complex to suppress Akt (143). Within the autophagy domain, inhibition of the kinase activity of EGFR promotes pro-survival autophagy (205, 207, 209) and endosomal kinase inactive EGFR interacts with LAPTM4B to promote pro-survival autophagy under nutrient starvation stress (210). As for mitochondrion, kinase independently, EGFR inhibits mitophagy via repressing intracellular activation of Akt (143), and EGFR interacts with PUMA to inhibit apoptosis (211). An update on the kinase-independent functions of EGFR in cancer cells is summarized in Figure 1.
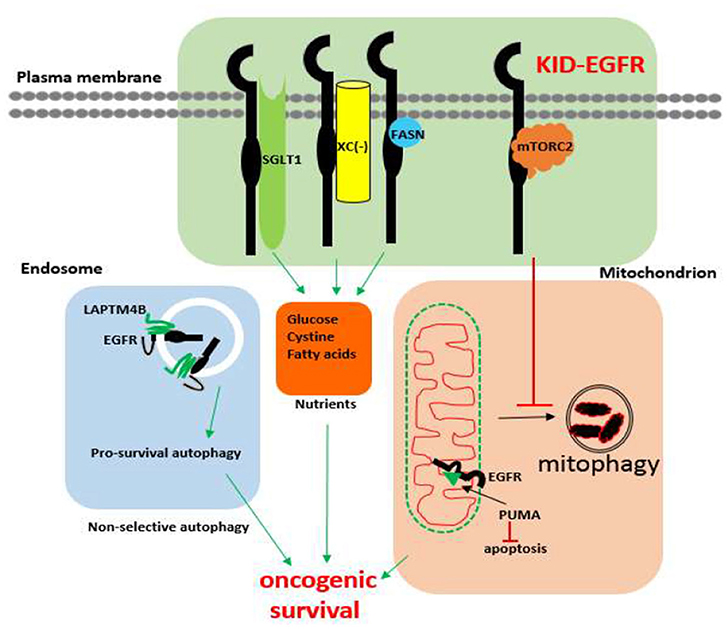
Figure 1. Known kinase-independent functions of EGFR in cancer cells. Currently known kinase-independent (KID) functions of EGFR locate at three functional domains of cancer cell. One is in the plasma membrane where EGFR interacts with SGLT1, Xc−, fatty acid synthase (FASN), and the mTORC2 complex to support transportation of glucose, cystine, de novo fatty acid synthase, and repressing mitophagy, respectively. The second function domain is the endosomal autophagy machinery where kinase inactive EGFR promotes pro-survival autophagy. The third domain is the mitochondrial domain where kinase inactive EGFR interacts with PUMA to inhibit apoptosis. KID-EGFR is oncogenic and pro-survival.
In non-cancerous cells, one of two kinase-impaired EGFR mutants was found to be able to oppose IL3-removal induced apoptosis of an EGFR negative noncancerous hematopoietic 32D by undefined mechanisms (212), and knockout of EGFR in mice is lethal (213) but mice with a loss-of-kinase mutation in EGFR are viable with only mild defects in the eyes and skin (214). The discrepant phenotypes of EGFR knockout mice and mice bearing loss-of-kinase mutant EGFR argues that EGFR also exhibits KID functions in non-cancerous cells, however, this is beyond the scope of this review. The existence of KID pro-survival function of EGFR is undeniable.
Implication of KID Functions of EGFR in Advancing Our Understanding the Role of EGFR in Cancer
While much more research effort is needed to fully unveil the KID pro-survival function of EGFR in cancer cells, the discovery and realization of EGFR's KID pro-survival function bears a profound implication on overcoming the aforementioned long-lasting challenges of EGFR targeted cancer therapies.
First of all, it offers an alternative interpretation to the clinical failures of EGFR kinase inhibitors. Regarding cancers expressing/overexpressing wild-type EGFR, such as head and neck, prostate, and ovarian cancer, which are innately resistant to EGFR TKIs, a new interpretation is that these types of cancers are more dependent on EGFR's KID function for survival rather than on its kinase activity for growth. This possibility is supported by the fact that, without TKI treatment, the phosphorylation status of EGFR does not correlate with disease progression nor with prognosis of many cancers but the total EGFR protein level does (31, 32, 34, 38, 39, 80, 156–159) and further that the innate TKI resistant cancer cells cannot survive without EGFR (141–143, 215), and that disconnecting the EGFR's kinase activity from its downstream kinase cascades does not affect EGFR's oncogenic function (178). Regarding the acquired TKI resistance, an alternative interpretation is that TKI treatment shifts EGFR's kinase dependent function toward its KID pro-survival function that offers cancer cells addicted to EGFR's kinase activity for growth an adaptive window to develop alternative proliferative mechanisms circumventing the EGFR kinase dominated pathway under the constant exposure to TKIs. This possibility is supported by the observation showing that in cancer cells EGFR exists in two types of status, a kinase activatable one and a kinase unactivatable one (216). The kinase activatable EGFR refers to the EGFRs that behave according to the canonical mechanisms, whereas the kinase unactivatable EGFR refers to the EGFRs physically interacting with other proteins at its C-terminal kinase domain, such as the EGFRs interacting with SGLT1 (216), thus cannot be autophosphorylated. Supportively, it has been recently reported that autophosphorylation of the C-terminal domain of EGFR is not required for EGFR's oncogenic activity (178). The shift toward KID function of EGFR by TKI in TKI sensitive cancer cells is also supported by the observations that TKIs shift EGFR from non-lipid raft regions to lipid rafts (217) where many cell survival dependent proteins, such as mTORC2, Na+/K+ ATPase, fatty acid synthase reside (218). Additionally, TKIs, especially the first generation of TKIs (Gefitinib and Erlotinib), are capable of causing dimerization of EGFR without significantly altering the level of EGFR protein (219–221) in a manner that is dependent on EGFR palmitoylation and independent of EGFR's kinase activity (222), which implicates that the TKI induced kinase inactivated EGFR dimer may gain new functions by recruiting novel interacting proteins. Further supports for the hypothesis that the kinase activity of EGFR is more critically involved in the proliferation than in the survival of cancer cells are offered by two studies: one is a study using rat models showing that inhibition of EGFR's kinase activity by TKI was able to inhibit growth but not the incidence of chemical or hormonal induced liver cancer (223), and consistently another study shows that the phosphorylation of the C-terminal tail (a hub domain that connects the kinase function of EGFR with its down-stream kinase dependent signaling cascades) is not required for the oncogenic function of EGFR mutant derived from lung cancer (178). Thus, we propose a new model of EGFR function: EGFR exists in two types of functional nodes, a kinase dependent functional node (the canonical functional node) that predominantly oversees cell proliferation and a kinase independent functional node that predominantly oversees cell survival, which is depicted by Figure 2.

Figure 2. A model of two functional statuses of EGFR in cancer cells. The kinase activatable EGFRs are mainly involved in promoting cell growth, and the kinase unactivatable EGFRs, which are blocked from autocross-phosphorylation by interacting proteins, are mainly in charge of promoting oncogenic cell survival.
The KID pro-survival function of EGFR also explains the worse prognosis of patients treated with a combination of EGFR TKI and chemotherapeutics than those treated with chemotherapeutics alone (164, 224–227). One explanation to it is that, in these scenario, the KID oncogenic function of EGFR might be enhanced by TKIs and thus survivability of cancer cells, which hampers the cytotoxic effect of the chemotherapeutics.
Secondly, the KID function of EGFR offers an alternative interpretation to experimental observations that cannot be fully explained by the kinase function of EGFR. For example, the contrasting phenotypes between EGFR knockout mice (213) and loss-of-kinase EGFR mutant mice (214), in the former model where the EGFR gene was systemically knocked out, the homozygous EGFR−/− mice die within a week after birth due to failures of multiple organs especially the lung and the heart, whereas the Waved-2 mice who lose more than 99% of EGFR's tyrosine kinase activity, survive and develop well with only a minor defect in the hair follicles that give rise curved hairs. The discrepancies between these two animal models of EGFR mutant argue that the tyrosine kinase activity is not the sole physiological function of EGFR and the KID function of EGFR is critical for the survival, although a proof-of-concept definitive experiment of rescuing the EGFR knockout mice with a kinase dead form EGFR needs to be performed.
Thirdly, the KID function of EGFR offers a partial explanation for the unique therapeutic effect of the anti-EGFR mAbs. Neither the phosphorylation status nor that for the total EGFR expression are predictive of responses to anti-EGFR mAbs (107, 115, 177), suggesting that repression of the kinase activity of EGFR by these mAbs might not be the primary mechanism underlying the therapeutic effect of anti-EGFR mAbs. Unlike the TKIs that can induce EGFR dimerization without activation (219, 220, 228) and are only effective in cancers bearing kinase-activating mutations in EGFR, anti-EGFR mAbs are capable of reducing EGFR proteins by shifting the ligand induced EGFR endocytosis toward the non-recyclable stage, the late-endosomal stage where EGFR is to be degraded rather than being recycled back to the plasma membrane as most of the early-endosome localized EGFR are programed to do (9). Many studies have proposed that the mAb binding induced EGFR endocytosis and subsequent degradation is a key mechanism as compared to antigen dependent cellular cytotoxicity (ADCC) by which anti-EGFR mAbs execute their therapeutic effect. This is supported by the observation that both Cetuximab and Panitumumab are capable of reducing EGFR protein levels, however, unlike Cetuximab (229–231), Panitumumab, as an IgG2 is less capable of inducing ADCC (231). The EGFR endocytosis induced by anti-EGFR mAbs is not free of EGFR recycling, although the balance between degradation and recycling is tilted toward degradation as compared to the EGFR ligand binding induced endocytosis (232). A better understanding of mechanistic differences between ligand induced EGFR endocytosis and that induced by anti-EGFR mAb may lead to discovery of novel actionable targets to enhance the effect of mAb induced reduction of EGFR protein and the therapeutic efficacy of anti-EGFR mAbs.
While the specific mechanisms underlying the KID pro-survival function of EGFR remains to be fully revealed, existing evidence is sufficient in concluding that the pro-survival function of EGFR is regulated by mechanisms that are largely independent of EGFR's kinase function. It is proposed that targeting the KID pro-survival function of EGFR by reducing its protein levels or interrupting the mechanisms mediating its KID pro-survival function may lead to novel and more effective approaches of targeting EGFR for cancer therapy. In this regard, a proof-of-concept synthetic peptide that can cause degradation of EGFR has been shown to be effective in treating orthotopic ovarian cancers in mice by inducing mitophagic cell death of cancer cells (143).
Perspective on EGFR Targeted Cancer Therapies
EGFR is the most commonly expressed/overexpressed membranous oncogenic protein in cancer. The majority of EGFR overexpressing cancer patients are yet to benefit from current anti-EGFR therapeutics. Targeting the kinase activity of EGFR is preordained to acquired and innate resistance. Given its frequent expression in cancers, its powerful oncogenic function, and easy accessibility for targeting, EGFR remains an ideal therapeutic target for cancers. A growing body of evidence has revealed that hijacking kinases for non-kinase usages by cells is a common phenomenon (233, 234). For cancers, besides EGFR, it has been found that, kinase independently, AKT promotes cancer cell survival (235), AURKA (Aurora kinase A) enhances stemness of breast cancer cells (236), cyclin-dependent kinase 6 promotes tumorigenesis of lymphoma (237), cyclin-dependent kinase 19 promotes cell proliferation of osteosarcoma cells (238), cyclin E promotes proliferation of liver cancer cells (239), EphA2 (ephrin type-A receptor 2) promotes invasion and metastasis of prostate cancer (240), ERKs promote cell cycle entry of retinoblastoma cells (241), PAK4 (P21-activated kinase 4) promotes adhesion and migration of breast cancer cells (242), and RIPK1 (receptor-interacting protein kinase 1) promotes liver carcinogenesis (243). The time is now to step out the box of the tyrosine kinase function of EGFR and explore new ways of targeting EGFR.
With the KID functions of EGFR on board, a hypothesis pertaining to EGFR's divergent roles in regulating growth vs. survival of cancer cells in relevant to TKI resistance is proposed as the following (Figure 3): EGFR exists in two types of status, one is kinase activatable and the other is kinase unactivatable (functions as a scaffold protein), the former is mainly in charge of cell growth, the latter is mainly in charge of survival. In cancer cells expressing kinase-activating mutations, the role of EGFR is shifted toward its kinase-dependent functions; while in cancer cells over-expressing wild type EGFR, the role of EGFR is shifted toward its kinase-independent functions; at situation of TKI treatment, the role of EGFR is also tilted toward its kinase-independent functions that allows cancer cells to survive and develop alternative growth-promoting mechanisms to counteract with TKI's inhibitory effect.
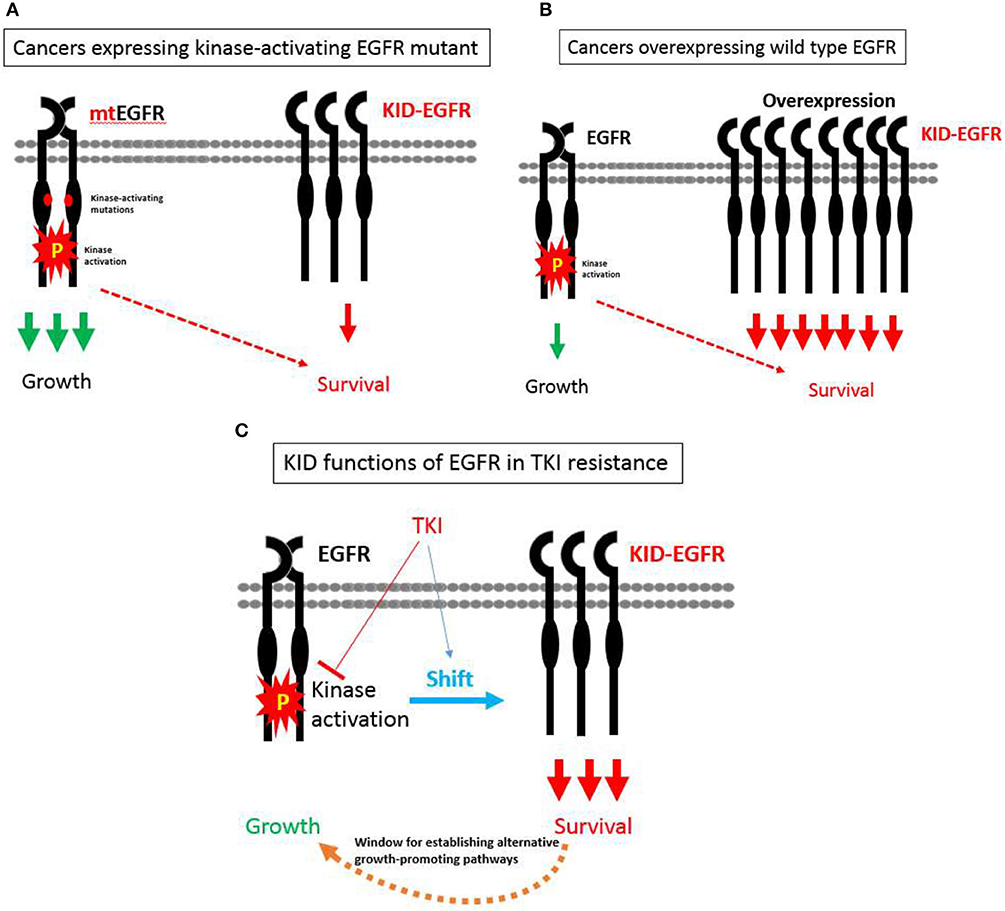
Figure 3. A hypothesis pertaining to EGFR's divergent roles in regulating growth vs. survival of cancer cells in relevant to TKI resistance. (A) In cancer cells expressing kinase-activating mutations, the role of EGFR is shifted toward its kinase-dependent functions, which sensitizes these cancers cells to TKI. (B) In cancer cells over-expressing wild type EGFR, the role of EGFR is shifted toward its kinase-independent functions, which promotes the progression of cancers rather desensitizes these cancers to TKI. (C) At situation of TKI treatment, the role of EGFR is also tilted toward its kinase-independent functions that allows cancer cells to survive and develop alternative growth-promoting mechanisms to counteract with TKI's inhibitory effect.
The evidence of KID function of EGFR is somewhat scattering however undeniable, and more researches on the KID functions of EGFR are warranted. Targeting EGFR's KID functions by either decreasing EGFR protein levels or interfering with the mechanisms underlying EGFR's KID functions forecast significant promise. Currently proposed approaches may include disrupting the protein-protein interacting complex of KID EGFR, down-regulating EGFR protein using synthetic molecules (143, 222), siRNA or protein targeting chimeras (PROTAC) technologies (244), and manipulating signal pathways controlled by KID EGFR such as simultaneously activating mTORC2 and inhibiting mTORC1 (143).
Author Contributions
ZW formulated the concept. ZW and RT co-wrote the manuscript.
Conflict of Interest Statement
ZW is a co-founder and a shareholder of Metabocentric Biotechnologies Inc., a spinoff startup company of the University of Houston, which focuses on developing metabolic therapeutics for cancer.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. We apologize to the authors whose works could not be cited due to space constraints. The author thank all the previous and current laboratorial members, Lakshmi Reddy Bollu, Rajasekhara Reddy Katreddy, Jinrong Wang, Jiangong Ren, Fei Su, Alicia Blessing, Shivangi Srivastava, RT, Shuping Xu, Jinyu Chen, Lei Xu, Xuefeng Wu, and Guang Gao, for their hard work and trust in the science of EGFR kinase independent function. The author is grateful to friends, Dr. Ganxiong Huang, Dr. Xiongbin Lu, Dr. Pratip Bhattacharya, Dr. Steven Bark, Dr. Tasneem Bawa, Dr. Chin-Yo Lin, Dr. Mehmet Sen, Dr. William Widger, Dr. Michael Rea, Dr. James Briggs, Dr. Margaret Warner, and the chairman of the department Dr. Amy Sater, for sharing their knowledge and cordial supports.
References
1. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. (2012) 12:553–63. doi: 10.1038/nrc3309
2. Gusterson BA, Hunter KD. Should we be surprised at the paucity of response to EGFR inhibitors? Lancet Oncol. (2009) 10:522–7. doi: 10.1016/S1470-2045(09)70034-8
3. Dancey JE, Freidlin B. Targeting epidermal growth factor receptor–are we missing the mark? Lancet. (2003) 362:62–4. doi: 10.1016/S0140-6736(03)13810-X
4. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. (2014) 11:473–81. doi: 10.1038/nrclinonc.2014.104
5. Cortot AB, Jänne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev. (2014) 23:356–66. doi: 10.1183/09059180.00004614
6. Landi L, Cappuzzo F. Pharmacotherapy targeting the EGFR oncogene in NSCLC. Expert Opin Pharmacother. (2014) 15:2293–305. doi: 10.1517/14656566.2014.957179
7. Minuti G, D'Incecco A, Landi L, Cappuzzo F. Protein kinase inhibitors to treat non-small-cell lung cancer. Expert Opin Pharmacother. (2014) 15:1203–13. doi: 10.1517/14656566.2014.909412
8. Bertotti A, Sassi F. Molecular pathways: sensitivity and resistance to Anti-EGFR antibodies. Clin Cancer Res. (2015) 21:3377–83. doi: 10.1158/1078-0432.CCR-14-0848
9. Capdevila J, Elez E, Macarulla T, Ramos FJ, Ruiz-Echarri M, Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat Rev. (2009) 35:354–63. doi: 10.1016/j.ctrv.2009.02.001
10. Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. (2010) 10:59–64. doi: 10.1038/nrc2771
11. Inal C, Yilmaz E, Piperdi B, Perez-Soler R, Cheng H. Emerging treatment for advanced lung cancer with EGFR mutation. Expert Opin Emerg Drugs. (2015) 20:597–612. doi: 10.1517/14728214.2015.1058778
12. Siegelin MD, Borczuk AC. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab Invest. (2014) 94:129–37. doi: 10.1038/labinvest.2013.147
13. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. (2009) 28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198
14. De Luca A, Normanno N. Predictive biomarkers to tyrosine kinase inhibitors for the epidermal growth factor receptor in non-small-cell lung cancer. Curr Drug Targets. (2010) 11:851–64. doi: 10.2174/138945010791320773
15. Li AR, Chitale D, Riely GJ, Pao W, Miller VA, Zakowski MF, et al. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagn. (2008) 10:242–8. doi: 10.2353/jmoldx.2008.070178
16. Jung MJ, Woo CG, Lee S, Chin S, Kim HK, Kwak JJ, et al. Gene copy number variation and protein overexpression of EGFR and HER2 in distal extrahepatic cholangiocarcinoma. Pathology. (2017) 49:582–8. doi: 10.1016/j.pathol.2017.06.001
17. Birkman EM, Ålgars A, Lintunen M, Ristamäki R, Sundström J, Carpén O. EGFR gene amplification is relatively common and associates with outcome in intestinal adenocarcinoma of the stomach, gastro-oesophageal junction and distal oesophagus. BMC Cancer. (2016) 16:406. doi: 10.1186/s12885-016-2456-1
18. Yang X, Wang W, Wang C, Wang L, Yang M, Qi M, et al. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol Rep. (2014) 32:700–8. doi: 10.3892/or.2014.3261
19. Rossi E, Villanacci V, Danesino C, Donato F, Nascimbeni R, Bassotti G. Epidermal growth factor receptor overexpression/amplification in adenocarcinomas arising in the gastrointestinal tract. Rev Esp Enferm Dig. (2011) 103:632–9. doi: 10.4321/S1130-01082011001200005
20. Huang SF, Cheng SD, Chien HT, Liao CT, Chen IH, Wang HM, et al. Relationship between epidermal growth factor receptor gene copy number and protein expression in oral cavity squamous cell carcinoma. Oral Oncol. (2012) 48:67–72. doi: 10.1016/j.oraloncology.2011.06.511
21. Itakura Y, Sasano H, Shiga C, Furukawa Y, Shiga K, Mori S, et al. Epidermal growth factor receptor overexpression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer. (1994) 74:795–804.
22. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. (2008) 359:1367–80. doi: 10.1056/NEJMra0802714
23. Zhang YL, Yuan JQ, Wang KF, Fu XH, Han XR, Threapleton D, et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. (2016) 7:78985–93. doi: 10.18632/oncotarget.12587
24. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. (2007) 25:2288–94. doi: 10.1200/JCO.2006.08.0705
25. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. (2013) 19:2240–7. doi: 10.1158/1078-0432.CCR-12-2246
26. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
27. Campo M, Gerber D, Gainor JF, Heist RS, Temel JS, Shaw AT, et al. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J Thorac Oncol. (2016) 11:2022–6. doi: 10.1016/j.jtho.2016.06.032
28. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. (2015) 21:3924–33. doi: 10.1158/1078-0432.CCR-15-0560
29. Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, et al. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. (2015) 1:982–4. doi: 10.1001/jamaoncol.2015.1066
30. Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. (2015) 21:2157–66. doi: 10.1158/1078-0432.CCR-14-2821
31. Di Lorenzo G, Tortora G, D'Armiento FP, De Rosa G, Staibano S, Autorino R, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. (2002) 8:3438–44.
32. Pryczynicz A, Guzinska-Ustymowicz K, Kemona A, Czyzewska J. Expression of EGF and EGFR strongly correlates with metastasis of pancreatic ductal carcinoma. Anticancer Res. (2008) 28:1399–404.
33. Gui T, Shen K. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol. (2012) 36:490–6. doi: 10.1016/j.canep.2012.06.005
34. Hirsch FR, Scagliotti GV, Langer CJ, Varella-Garcia M, Franklin WA. Epidermal growth factor family of receptors in preneoplasia and lung cancer: perspectives for targeted therapies. Lung Cancer. (2003) 41(Suppl 1):S29–42. doi: 10.1016/S0169-5002(03)00137-5
35. Liu TC, Jin X, Wang Y, Wang K. Role of epidermal growth factor receptor in lung cancer and targeted therapies. Am J Cancer Res. (2017) 7:187–202.
36. Kira S, Nakanishi T, Suemori S, Kitamoto M, Watanabe Y, Kajiyama G. Expression of transforming growth factor alpha and epidermal growth factor receptor in human hepatocellular carcinoma. Liver. (1997) 17:177–82. doi: 10.1111/j.1600-0676.1997.tb00803.x
37. Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K, et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer. (2001) 84:1377–83. doi: 10.1054/bjoc.2000.1580
38. Dordević G, Matušan Ilijaš K, HadŽisejdić I, Maričić A, Grahovac B, Jonjić N. EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J Biomed Sci. (2012) 19:40. doi: 10.1186/1423-0127-19-40
39. Wei Q, Sheng L, Shui Y, Hu Q, Nordgren H, Carlsson J. EGFR, HER2, and HER3 expression in laryngeal primary tumors and corresponding metastases. Ann Surg Oncol. (2008) 15:1193–201. doi: 10.1245/s10434-007-9771-3
40. Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. (2016) 9:1927–43. doi: 10.2147/OTT.S93720
41. Thorne AH, Zanca C, Furnari F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. (2016) 18:914–8. doi: 10.1093/neuonc/nov319
42. Okines A, Cunningham D, Chau I. Targeting the human EGFR family in esophagogastric cancer. Nat Rev Clin Oncol. (2011) 8:492–503. doi: 10.1038/nrclinonc.2011.45
43. Dragovich T, Campen C. Anti-EGFR-targeted therapy for esophageal and gastric cancers: an evolving concept. J Oncol. (2009) 2009:804108. doi: 10.1155/2009/804108
44. Spindler KL, Lindebjerg J, Nielsen JN, Olsen DA, Bisgård C, Brandslund I, et al. Epidermal growth factor receptor analyses in colorectal cancer: a comparison of methods. Int J Oncol. (2006) 29:1159–65. doi: 10.3892/ijo.29.5.1159
45. Kersemaekers AM, Fleuren GJ, Kenter GG, Van den Broek LJ, Uljee SM, Hermans J, et al. Oncogene alterations in carcinomas of the uterine cervix: overexpression of the epidermal growth factor receptor is associated with poor prognosis. Clin Cancer Res. (1999) 5:577–86.
46. Matsuda N, Lim B, Wang X, Ueno NT. Early clinical development of epidermal growth factor receptor targeted therapy in breast cancer. Expert Opin Investig Drugs. (2017) 26:463–79. doi: 10.1080/13543784.2017.1299707
47. Costa V, Fregnani ER, Fonseca FP, Abreu Alves F, Pinto CAL, Kaminagakura E. EGFR is not amplified in ameloblastoma. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 125:454–8. doi: 10.1016/j.oooo.2018.02.014
48. Nishio M, Taguchi F, Ohyanagi F, Horikike A, Ishikawa Y, Satoh Y, et al. Gefitinib efficacy associated with multiple expression of HER family in non-small cell lung cancer. Anticancer Res. (2006) 26:3761–5.
49. Franklin WA, Carbone DP. Molecular staging and pharmacogenomics. clinical implications: from lab to patients and back. Lung Cancer. (2003) 41(Suppl 1):S147–54. doi: 10.1016/S0169-5002(03)00158-2
50. Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol. (2005) 23:2556–68. doi: 10.1200/JCO.2005.07.799
51. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol. (2014) 9:154–62. doi: 10.1097/JTO.0000000000000033
52. Fu M, Zhang W, Shan L, Song J, Shang D, Ying J, et al. Mutation status of somatic EGFR and KRAS genes in Chinese patients with prostate cancer (PCa). Virchows Arch. (2014) 464:575–81. doi: 10.1007/s00428-014-1566-x
53. Peraldo-Neia C, Migliardi G, Mello-Grand M, Montemurro F, Segir R, Pignochino Y, et al. Epidermal growth factor receptor (EGFR) mutation analysis, gene expression profiling and EGFR protein expression in primary prostate cancer. BMC Cancer. (2011) 11:31. doi: 10.1186/1471-2407-11-31
54. de Muga S, Hernández S, Agell L, Salido M, Juanpere N, Lorenzo M, et al. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod Pathol. (2010) 23:703–12. doi: 10.1038/modpathol.2010.45
55. Gonzalez-Conchas GA, Rodriguez-Romo L, Hernandez-Barajas D, Gonzalez-Guerrero JF, Rodriguez-Fernandez IA, Verdines-Perez A, et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: a systematic review and a meta-analysis. Cancer Treat Rev. (2018) 62:1–8. doi: 10.1016/j.ctrv.2017.10.008
56. Suo Z, Nesland JM. Type 1 protein tyrosine kinases in breast carcinoma: a review. Ultrastruct Pathol. (2002) 26:125–35. doi: 10.1080/01913120290076784
57. Kim A, Jang MH, Lee SJ, Bae YK. Mutations of the epidermal growth factor receptor gene in triple-negative breast cancer. J Breast Cancer. (2017) 20:150–9. doi: 10.4048/jbc.2017.20.2.150
58. Italiano A, Saint-Paul MC, Caroli-Bosc FX, François E, Bourgeon A, Benchimol D, et al. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors correlates with EGFR expression in related metastatic sites: biological and clinical implications. Ann Oncol. (2005) 16:1503–7. doi: 10.1093/annonc/mdi282
59. Zhang X, Nagahara H, Mimori K, Inoue H, Sawada T, Ohira M, et al. Mutations of epidermal growth factor receptor in colon cancer indicate susceptibility or resistance to gefitinib. Oncol Rep. (2008) 19:1541–4. doi: 10.3892/or.19.6.1541
60. Rogers SJ, Harrington KJ, Rhys-Evans P, O-Charoenrat P, Eccles SA. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. (2005) 24:47–69. doi: 10.1007/s10555-005-5047-1
61. Perisanidis C. Prevalence of EGFR tyrosine kinase domain mutations in head and neck squamous cell carcinoma: cohort study and systematic review. In Vivo. (2017) 31:23–34. doi: 10.21873/invivo.11020
62. Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK, Kim WH. EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology. (2008) 52:738–46. doi: 10.1111/j.1365-2559.2008.03021.x
63. Galizia G, Lieto E, Orditura M, Castellano P, Mura AL, Imperatore V, et al. Epidermal growth factor receptor (EGFR) expression is associated with a worse prognosis in gastric cancer patients undergoing curative surgery. World J Surg. (2007) 31:1458–68. doi: 10.1007/s00268-007-9016-4
64. Pan X, Ji X, Zhang R, Zhou Z, Zhong Y, Peng W, et al. Landscape of somatic mutations in gastric cancer assessed using next-generation sequencing analysis. Oncol Lett. (2018) 16:4863–70. doi: 10.3892/ol.2018.9314
65. Daveau M, Scotte M, François A, Coulouarn C, Ros G, Tallet Y, et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. (2003) 36:130–41. doi: 10.1002/mc.10103
66. Lee SC, Lim SG, Soo R, Hsieh WS, Guo JY, Putti T, et al. Lack of somatic mutations in EGFR tyrosine kinase domain in hepatocellular and nasopharyngeal carcinoma. Pharmacogenet Genomics. (2006) 16:73–4. doi: 10.1097/01.fpc.0000184959.82903.02
67. Ikeda S, Tsigelny IF, Skjevik ÅA, Kono Y, Mendler M, Kuo A, et al. Next-generation sequencing of circulating tumor DNA reveals frequent alterations in advanced hepatocellular carcinoma. Oncologist. (2018) 23:586–93. doi: 10.1634/theoncologist.2017-0479
68. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. (2013) 19:764–72. doi: 10.1158/1078-0432.CCR-12-3002
69. Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci USA. (1987) 84:6899–903. doi: 10.1073/pnas.84.19.6899
70. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA. (1990) 87:8602–6. doi: 10.1073/pnas.87.21.8602
71. Del Vecchio CA, Giacomini CP, Vogel H, Jensen KC, Florio T, Merlo A, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. (2013) 32:2670–81. doi: 10.1038/onc.2012.280
72. Wei H, Wang XW, Chen KM, Ling SR, Yi CJ. Analysis of gene mutation associated with tyrosine kinase inhibitor sensitivity of epidermal growth factor receptor in cervical cancer patients. Eur Rev Med Pharmacol Sci. (2018) 22:6280–7. doi: 10.26355/eurrev_201810_16036
73. Maihle NJ, Baron AT, Barrette BA, Boardman CH, Christensen TA, Cora EM, et al. EGF/ErbB receptor family in ovarian cancer. Cancer Treat Res. (2002) 107:247–58. doi: 10.1007/978-1-4757-3587-1_11
74. Stadlmann S, Gueth U, Reiser U, Diener PA, Zeimet AG, Wight E, et al. Epithelial growth factor receptor status in primary and recurrent ovarian cancer. Mod Pathol. (2006) 19:607–10. doi: 10.1038/modpathol.3800575
75. Showeil R, Romano C, Valganon M, Lambros M, Trivedi P, Van Noorden S, et al. The status of epidermal growth factor receptor in borderline ovarian tumours. Oncotarget. (2016) 7:10568–77. doi: 10.18632/oncotarget.7257
76. Carlsson J, Wester K, De La Torre M, Malmström PU, Gårdmark T. EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides. Radiol Oncol. (2015) 49:50–8. doi: 10.2478/raon-2014-0015
77. El Hamdani W, Hadami K, Bensaid M, El Ahanidi H, Ameur A, Filali Maltouf A, et al. Identification of G2607A mutation in EGFR gene with a significative rate in Moroccan patients with bladder cancer. Cell Mol Biol. (2017) 63:75–81. doi: 10.14715/cmb/2017.63.5.14
78. Chaux A, Cohen JS, Schultz L, Albadine R, Jadallah S, Murphy KM, et al. High epidermal growth factor receptor immunohistochemical expression in urothelial carcinoma of the bladder is not associated with EGFR mutations in exons 19 and 21: a study using formalin-fixed, paraffin-embedded archival tissues. Hum Pathol. (2012) 43:1590–5. doi: 10.1016/j.humpath.2011.11.016
79. Cossu-Rocca P, Muroni MR, Sanges F, Sotgiu G, Asunis A, Tanca L, et al. EGFR kinase-dependent and kinase-independent roles in clear cell renal cell carcinoma. Am J Cancer Res. (2016) 6:71–83.
80. Uhlman DL, Nguyen P, Manivel JC, Zhang G, Hagen K, Fraley E, et al. Epidermal growth factor receptor and transforming growth factor alpha expression in papillary and nonpapillary renal cell carcinoma: correlation with metastatic behavior and prognosis. Clin Cancer Res. (1995) 1:913–20.
81. Moch H, Sauter G, Buchholz N, Gasser TC, Bubendorf L, Waldman FM, et al. Epidermal growth factor receptor expression is associated with rapid tumor cell proliferation in renal cell carcinoma. Hum Pathol. (1997) 28:1255–9. doi: 10.1016/S0046-8177(97)90198-2
82. Fiorentino M, Gruppioni E, Massari F, Giunchi F, Altimari A, Ciccarese C, et al. Wide spetcrum mutational analysis of metastatic renal cell cancer: a retrospective next generation sequencing approach. Oncotarget. (2017) 8:7328–35. doi: 10.18632/oncotarget.12551
83. Boeck S, Jung A, Laubender RP, Neumann J, Egg R, Goritschan C, et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br J Cancer. (2013) 108:469–76. doi: 10.1038/bjc.2012.495
84. Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig Surg. (2006) 23:74–9. doi: 10.1159/000093497
85. Dancer J, Takei H, Ro JY, Lowery-Nordberg M. Coexpression of EGFR and HER-2 in pancreatic ductal adenocarcinoma: a comparative study using immunohistochemistry correlated with gene amplification by fluorescencent in situ hybridization. Oncol Rep. (2007) 18:151–5. doi: 10.3892/or.18.1.151
86. Tzeng CW, Frolov A, Frolova N, Jhala NC, Howard JH, Buchsbaum DJ, et al. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery. (2007) 141:464–9. doi: 10.1016/j.surg.2006.09.009
87. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of canada clinical trials group. J Clin Oncol. (2007) 25:1960–6. doi: 10.1200/JCO.2006.07.9525
88. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
89. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. (2007) 7:169-81. doi: 10.1038/nrc2088
90. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. (2004) 351:337–45. doi: 10.1056/NEJMoa033025
91. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. (2007) 25:1658–64. doi: 10.1200/JCO.2006.08.1620
92. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. (2010) 11:21–8. doi: 10.1016/S1470-2045(09)70311-0
93. Cohen RB. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat Rev. (2014) 40:567–77. doi: 10.1016/j.ctrv.2013.10.002
94. Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem. (1962) 237:1555–62.
95. Cohen S, Fava RA, Sawyer ST. Purification and characterization of epidermal growth factor receptor/protein kinase from normal mouse liver. Proc Natl Acad Sci USA. (1982) 79:6237–41. doi: 10.1073/pnas.79.20.6237
96. Downward J, Yarden Y, Mayes E, Scrace G, Totty N, Stockwell P, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. (1984) 307:521–7. doi: 10.1038/307521a0
97. Seymour L. Epidermal growth factor receptor inhibitors: an update on their development as cancer therapeutics. Curr Opin Investig Drugs. (2003) 4:658–66.
98. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. (2015) 6:6377. doi: 10.1038/ncomms7377
99. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. (2008) 27:4702–11. doi: 10.1038/onc.2008.109
100. Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. (2007) 67:11924–32. doi: 10.1158/0008-5472.CAN-07-1885
101. Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. (2009) 462:1070–4. doi: 10.1038/nature08622
102. Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. (2013) 3:1404–15. doi: 10.1158/2159-8290.CD-13-0314
103. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. (2014) 4:1046–61. doi: 10.1158/2159-8290.CD-14-0337
104. Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. (2017) 385:51–4. doi: 10.1016/j.canlet.2016.11.008
105. Roskoski RJr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res. (2019) 139:395–411. doi: 10.1016/j.phrs.2018.11.014
106. Troiani T, Napolitano S, Della Corte CM, Martini G, Martinelli E, Morgillo F, et al. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open. (2016) 1:e000088. doi: 10.1136/esmoopen-2016-000088
107. Cheng L, Ren W, Xie L, Li M, Liu J, Hu J, et al. Anti-EGFR MoAb treatment in colorectal cancer: limitations, controversies, and contradictories. Cancer Chemother Pharmacol. (2014) 74:1–13. doi: 10.1007/s00280-014-2489-6
108. Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. (1984) 44:1002–7.
109. Vincenzi B, Santini D, Tonini G. Biological interaction between anti-epidermal growth factor receptor agent cetuximab and magnesium. Expert Opin Pharmacother. (2008) 9:1267–9. doi: 10.1517/14656566.9.8.1267
110. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. (2005) 7:301–11. doi: 10.1016/j.ccr.2005.03.003
111. Arteaga CL. ErbB-targeted therapeutic approaches in human cancer. Exp Cell Res. (2003) 284:122–30. doi: 10.1016/S0014-4827(02)00104-0
112. Okada Y, Kimura T, Nakagawa T, Okamoto K, Fukuya A, Goji T, et al. EGFR Downregulation after Anti-EGFR therapy predicts the antitumor effect in colorectal cancer. Mol Cancer Res. (2017) 15:1445–54. doi: 10.1158/1541-7786.MCR-16-0383
113. Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. (2010) 10:80–95. doi: 10.2174/156800910790980241
114. Carcereny E, Maurel J. Monoclonal antibodies against epidermal growth factor receptor in advanced colorectal carcinoma: clinical efficacy and markers of sensitivity. Rev Recent Clin Trials. (2006) 1:113–8. doi: 10.2174/157488706776876463
115. Hecht JR, Mitchell E, Neubauer MA, Burris HA, Swanson P, Lopez T, et al. Lack of correlation between epidermal growth factor receptor status and response to Panitumumab monotherapy in metastatic colorectal cancer. Clin Cancer Res. (2010) 16:2205–13. doi: 10.1158/1078-0432.CCR-09-2017
116. Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai CM, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst. (2013) 105:595–605. doi: 10.1093/jnci/djt072
117. Lee CK, Davies L, Wu YL, Mitsudomi T, Inoue A, Rosell R, et al. Gefitinib or erlotinib vs chemotherapy for EGFR mutation-positive lung cancer: individual patient data meta-analysis of overall survival. J Natl Cancer Inst. (2017) 109:djw279. doi: 10.1093/jnci/djw279
118. Dempke WC. Gefitinib in non-small-cell lung cancer-an old lesson new re-visited. Transl Lung Cancer Res. (2013) 2:435–8. doi: 10.3978/j.issn.2218-6751.2013.10.01
119. Rupp T, Zuckerman D. Quality of life, overall survival, and costs of cancer drugs approved based on surrogate endpoints. JAMA Intern Med. (2017) 177:276–7. doi: 10.1001/jamainternmed.2016.7761
120. Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. (2013) 31:3327–34. doi: 10.1200/JCO.2012.44.2806
121. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. (2014) 15:213–22. doi: 10.1016/S1470-2045(13)70604-1
122. Yazdi MH, Faramarzi MA, Nikfar S, Abdollahi M. A comprehensive review of clinical trials on EGFR inhibitors such as cetuximab and panitumumab as monotherapy and in combination for treatment of metastatic colorectal cancer. Avicenna J Med Biotechnol. (2015) 7:134–44.
123. Moreira J, Tobias A, O'Brien MP, Agulnik M. Targeted therapy in head and neck cancer: an update on current clinical developments in epidermal growth factor receptor-targeted therapy and immunotherapies. Drugs. (2017) 77:843–57. doi: 10.1007/s40265-017-0734-0
124. Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Treat Rev. (2018) 65:1–10. doi: 10.1016/j.ctrv.2018.02.006
125. Husain H, Scur M, Murtuza A, Bui N, Woodward B, Kurzrock R. Strategies to overcome bypass mechanisms mediating clinical resistance to EGFR tyrosine kinase inhibition in lung cancer. Mol Cancer Ther. (2017) 16:265–72. doi: 10.1158/1535-7163.MCT-16-0105
126. Yamaoka T, Ohba M, Ohmori T. Molecular-Targeted Therapies for Epidermal Growth Factor Receptor and Its Resistance Mechanisms. Int J Mol Sci. (2017) 18:E2420. doi: 10.3390/ijms18112420
127. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. (2010) 28:1254–61. doi: 10.1200/JCO.2009.24.6116
128. Tomasini P, Brosseau S, Mazières J, Merlio JP, Beau-Faller M, Mosser J, et al. EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR wild-type pre-treated advanced nonsmall cell lung cancer in daily practice. Eur Respir J. (2017) 50:1700514. doi: 10.1183/13993003.00514-2017
129. Itchins M, Clarke S, Pavlakis N. Do EGFR tyrosine kinase inhibitors (TKIs) still have a role in EGFR wild-type pre-treated advanced non-small cell lung cancer (NSCLC)?-the shifting paradigm of therapeutics. Transl Lung Cancer Res. (2018) 7(Suppl 1):S39–45. doi: 10.21037/tlcr.2018.01.06
130. Lisberg A, Garon EB. Epidermal growth factor tyrosine kinase inhibitor therapy inferior to second-line chemotherapy in EGFR wild-type non-small cell lung cancer patients: results of French nationwide observational study. Transl Lung Cancer Res. (2017) 6:S39–S40. doi: 10.21037/tlcr.2017.10.16
131. Gelsomino F, Agustoni F, Niger M, Valota M, Haspinger ER. Epidermal growth factor receptor tyrosine kinase inhibitor treatment in patients with EGFR wild-type non-small-cell lung cancer: the never-ending story. J Clin Oncol. (2013) 31:3291–3. doi: 10.1200/JCO.2013.50.2617
132. Wen Y, Grandis JR. Emerging drugs for head and neck cancer. Expert Opin Emerg Drugs. (2015) 20:313–29. doi: 10.1517/14728214.2015.1031653
133. Ojemuyiwa MA, Madan RA, Dahut WL. Tyrosine kinase inhibitors in the treatment of prostate cancer: taking the next step in clinical development. Expert Opin Emerg Drugs. (2014) 19:459–70. doi: 10.1517/14728214.2014.969239
134. Wilken JA, Badri T, Cross S, Raji R, Santin AD, Schwartz P, et al. EGFR/HER-targeted therapeutics in ovarian cancer. Future Med Chem. (2012) 4:447–69. doi: 10.4155/fmc.12.11
135. Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. (2013) 12:220–9. doi: 10.1158/1535-7163.MCT-12-0620
136. Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. (2013) 8:179–84. doi: 10.1097/JTO.0b013e3182779d18
137. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. (2012) 13:e23–31. doi: 10.1016/S1470-2045(11)70129-2
138. Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. (2013) 5:216ra177. doi: 10.1126/scitranslmed.3007205
139. Guérin O, Fischel JL, Ferrero JM, Bozec A, Milano G. EGFR Targeting in hormone-refractory prostate cancer: current appraisal and prospects for treatment. Pharmaceuticals (Basel). (2010) 3:2238–47. doi: 10.3390/ph3072238
140. Jakobovits A. Monoclonal antibody therapy for prostate cancer. Handb Exp Pharmacol. (2008) 181:237–56. doi: 10.1007/978-3-540-73259-4_11
141. Day KC, Lorenzatti Hiles G, Kozminsky M, Dawsey SJ, Paul A, Broses LJ, et al. HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. (2017) 77:74–85. doi: 10.1158/0008-5472.CAN-16-1656
142. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. (2008) 13:385–93. doi: 10.1016/j.ccr.2008.03.015
143. Katreddy RR, Bollu LR, Su F, Xian N, Srivastava S, Thomas R, et al. Targeted reduction of the EGFR protein, but not inhibition of its kinase activity, induces mitophagy and death of cancer cells through activation of mTORC2 and Akt. Oncogenesis. (2018) 7:5. doi: 10.1038/s41389-017-0021-7
144. Satpathy M, Mezencev R, Wang L, McDonald JF. Targeted in vivo delivery of EGFR siRNA inhibits ovarian cancer growth and enhances drug sensitivity. Sci Rep. (2016) 6:36518. doi: 10.1038/srep36518
145. Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, et al. The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(-). Cancer Res. (2016) 76:2954–63. doi: 10.1158/0008-5472.CAN-15-2121
146. Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. (1984) 309:418–25. doi: 10.1038/309418a0
147. Gullick WJ, Marsden JJ, Whittle N, Ward B, Bobrow L, Waterfield MD. Expression of epidermal growth factor receptors on human cervical, ovarian, and vulval carcinomas. Cancer Res. (1986) 46:285–92.
148. Yano S, Kondo K, Yamaguchi M, Richmond G, Hutchison M, Wakeling A, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. (2003) 23:3639–50.
149. Hollenberg MD, Cuatrecasas P. Insulin and epidermal growth factor. Human fibroblast receptors related to deoxyribonucleic acid synthesis and amino acid uptake. J Biol Chem. (1975) 250:3845–53.
150. Carpenter G, Lembach KJ, Morrison MM, Cohen S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J Biol Chem. (1975) 250:4297–304.
151. Cohen S, Carpenter G, Lembach KJ. Interaction of epidermal growth factor (EGF) with cultured fibroblasts. Adv Metab Disord. (1975) 8:265–84. doi: 10.1016/B978-0-12-027308-9.50024-X
152. Xu N, Fang W, Mu L, Tang Y, Gao L, Ren S, et al. Overexpression of wildtype EGFR is tumorigenic and denotes a therapeutic target in non-small cell lung cancer. Oncotarget. (2016) 7:3884–96. doi: 10.18632/oncotarget.6461
153. Velu TJ, Beguinot L, Vass WC, Willingham MC, Merlino GT, Pastan I, et al. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science. (1987) 238:1408–10. doi: 10.1126/science.3500513
154. Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. (2005) 2:e313. doi: 10.1371/journal.pmed.0020313
155. Merlino GT, Xu YH, Ishii S, Clark AJ, Semba K, Toyoshima K, et al. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science. (1984) 224:417–9. doi: 10.1126/science.6200934
156. Shah RB, Ghosh D, Elder JT. Epidermal growth factor receptor (ErbB1) expression in prostate cancer progression: correlation with androgen independence. Prostate. (2006) 66:1437–44. doi: 10.1002/pros.20460
157. Reyes HD, Thiel KW, Carlson MJ, Meng X, Yang S, Stephan JM, et al. Comprehensive profiling of EGFR/HER receptors for personalized treatment of gynecologic cancers. Mol Diagn Ther. (2014) 18:137–51. doi: 10.1007/s40291-013-0070-3
158. Tang X, Varella-Garcia M, Xavier AC, Massarelli E, Ozburn N, Moran C, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev Res (Phila). (2008) 1:192–200. doi: 10.1158/1940-6207.CAPR-08-0032
159. Komposch K, Sibilia M. EGFR Signaling in Liver Diseases. Int J Mol Sci. (2015) 17:E30. doi: 10.3390/ijms17010030
160. Templeton AJ, Diez-Gonzalez L, Ace O, Vera-Badillo F, Seruga B, Jordán J, et al. Prognostic relevance of receptor tyrosine kinase expression in breast cancer: a meta-analysis. Cancer Treat Rev. (2014) 40:1048–55. doi: 10.1016/j.ctrv.2014.08.003
161. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. (2012) 483:570–5. doi: 10.1038/nature11005
162. Chan DLH, Segelov E, Wong RS, Smith A, Herbertson RA, Li BT, et al. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst Rev. (2017) 6:CD007047. doi: 10.1002/14651858.CD007047.pub2
163. Vici P, Mariani L, Pizzuti L, Sergi D, Di Lauro L, Vizza E, et al. Emerging biological treatments for uterine cervical carcinoma. J Cancer. (2014) 5:86–97. doi: 10.7150/jca.7963
164. Jassem J, Dziadziuszko R. EGFR inhibitors for wild-type EGFR NSCLC: to use or not to use? Lancet Oncol. (2013) 14:916–7. doi: 10.1016/S1470-2045(13)70352-8
165. Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res. (2006) 12:7117–25. doi: 10.1158/1078-0432.CCR-06-0760
166. Emlet DR, Brown KA, Kociban DL, Pollice AA, Smith CA, Ong BB, et al. Response to trastuzumab, erlotinib, and bevacizumab, alone and in combination, is correlated with the level of human epidermal growth factor receptor-2 expression in human breast cancer cell lines. Mol Cancer Ther. (2007) 6:2664–74. doi: 10.1158/1535-7163.MCT-07-0079
167. Magné N, Fischel JL, Tiffon C, Formento P, Dubreuil A, Renée N, et al. Molecular mechanisms underlying the interaction between ZD1839 ('Iressa') and cisplatin/5-fluorouracil. Br J Cancer. (2003) 89:585–92. doi: 10.1038/sj.bjc.6601131
168. Sieghart W, Pinter M, Dauser B, Rohr-Udilova N, Piguet AC, Prager G, et al. Erlotinib and sorafenib in an orthotopic rat model of hepatocellular carcinoma. J Hepatol. (2012) 57:592–9. doi: 10.1016/j.jhep.2012.04.034
169. Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Jänne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. (2004) 64:7241–4. doi: 10.1158/0008-5472.CAN-04-1905
170. Vicentini C, Festuccia C, Gravina GL, Angelucci A, Marronaro A, Bologna M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J Cancer Res Clin Oncol. (2003) 129:165–74. doi: 10.1007/s00432-003-0420-3
171. Ioannou N, Dalgleish AG, Seddon AM, Mackintosh D, Guertler U, Solca F, et al. Anti-tumour activity of afatinib, an irreversible ErbB family blocker, in human pancreatic tumour cells. Br J Cancer. (2011) 105:1554–62. doi: 10.1038/bjc.2011.396
172. Nagy P, Arndt-Jovin DJ, Jovin TM. Small interfering RNAs suppress the expression of endogenous and GFP-fused epidermal growth factor receptor (erbB1) and induce apoptosis in erbB1-overexpressing cells. Exp Cell Res. (2003) 285:39–49. doi: 10.1016/S0014-4827(02)00050-2
173. Kang CS, Pu PY, Li YH, Zhang ZY, Qiu MZ, Huang Q, et al. An in vitro study on the suppressive effect of glioma cell growth induced by plasmid-based small interference RNA (siRNA) targeting human epidermal growth factor receptor. J Neurooncol. (2005) 74:267–73. doi: 10.1007/s11060-004-8322-z
174. Chen G, Kronenberger P, Teugels E, Umelo IA, De Grève J. Effect of siRNAs targeting the EGFR T790M mutation in a non-small cell lung cancer cell line resistant to EGFR tyrosine kinase inhibitors and combination with various agents. Biochem Biophys Res Commun. (2013) 431:623–9. doi: 10.1016/j.bbrc.2012.12.070
175. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. (2017) 9:E52. doi: 10.3390/cancers9050052
176. Balko JM, Potti A, Saunders C, Stromberg A, Haura EB, Black EP. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC Genomics. (2006) 7:289. doi: 10.1186/1471-2164-7-289
177. Haas-Kogan DA, Prados MD, Lamborn KR, Tihan T, Berger MS, Stokoe D. Biomarkers to predict response to epidermal growth factor receptor inhibitors. Cell Cycle. (2005) 4:1369–72. doi: 10.4161/cc.4.10.2105
178. Cho J, Kim S, Du J, Meyerson M. Autophosphorylation of the carboxyl-terminal domain is not required for oncogenic transformation by lung-cancer derived EGFR mutants. Int J Cancer. (2018) 143:679-85. doi: 10.1002/ijc.31332
179. Russo A, Franchina T, Ricciardi GR, Picone A, Ferraro G, Zanghì M, et al. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): old successes and future perspectives. Oncotarget. (2015) 6:26814–25. doi: 10.18632/oncotarget.4254
180. Mahipal A, Kothari N, Gupta S. Epidermal growth factor receptor inhibitors: coming of age. Cancer Control. (2014) 21:74–9. doi: 10.1177/107327481402100111
181. Lee MS, Kopetz S. Current and future approaches to target the epidermal growth factor receptor and its downstream signaling in metastatic colorectal cancer. Clin Colorectal Cancer. (2015) 14:203–18. doi: 10.1016/j.clcc.2015.05.006
182. Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. (2003) 9:2316–26.
183. Morelli MP, Cascone T, Troiani T, De Vita F, Orditura M, Laus G, et al. Sequence-dependent antiproliferative effects of cytotoxic drugs and epidermal growth factor receptor inhibitors. Ann Oncol. (2005) 16(Suppl 4):iv61–68. doi: 10.1093/annonc/mdi910
184. Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. (2000) 6:2053–63.
185. Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. (1999) 59:1935–40.
186. Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. (2009) 119:3000–10. doi: 10.1172/JCI38746
187. Cha MY, Lee KO, Kim M, Song JY, Lee KH, Park J, et al. Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int J Cancer. (2012) 130:2445–54. doi: 10.1002/ijc.26276
188. Ochi N, Isozaki H, Takeyama M, Singer JW, Yamane H, Honda Y, et al. Synergistic effect of pacritinib with erlotinib on JAK2-mediated resistance in epidermal gowth factor receptor mutation-positive non-small cell lung cancer. Exp Cell Res. (2016) 344:194–200. doi: 10.1016/j.yexcr.2016.05.008
189. Huether A, Höpfner M, Baradari V, Schuppan D, Scherübl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol. (2005) 70:1568–78. doi: 10.1016/j.bcp.2005.09.007
190. Huether A, Höpfner M, Sutter AP, Schuppan D, Scherübl H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J Hepatol. (2005) 43:661–9. doi: 10.1016/j.jhep.2005.02.040
191. Chang GC, Yu CT, Tsai CH, Tsai JR, Chen JC, Wu CC, et al. An epidermal growth factor inhibitor, Gefitinib, induces apoptosis through a p53-dependent upregulation of pro-apoptotic molecules and downregulation of anti-apoptotic molecules in human lung adenocarcinoma A549 cells. Eur J Pharmacol. (2008) 600:37–44. doi: 10.1016/j.ejphar.2008.10.024
192. Diaz Miqueli A, Blanco R, Garcia B, Badia T, Batista AE, Alonso R, et al. Biological activity in vitro of anti-epidermal growth factor receptor monoclonal antibodies with different affinities. Hybridoma (Larchmt). (2007) 26:423–31. doi: 10.1089/hyb.2007.0516
193. Poindessous V, Ouaret D, El Ouadrani K, Battistella A, Mégalophonos VF, Kamsu-Kom N, et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin Cancer Res. (2011) 17:6522–30. doi: 10.1158/1078-0432.CCR-11-1607
194. Balin-Gauthier D, Delord JP, Rochaix P, Mallard V, Thomas F, Hennebelle I, et al. In vivo and in vitro antitumor activity of oxaliplatin in combination with cetuximab in human colorectal tumor cell lines expressing different level of EGFR. Cancer Chemother Pharmacol. (2006) 57:709–18. doi: 10.1007/s00280-005-0123-3
195. Skvortsov S, Sarg B, Loeffler-Ragg J, Skvortsova I, Lindner H, Werner Ott H, et al. Different proteome pattern of epidermal growth factor receptor-positive colorectal cancer cell lines that are responsive and nonresponsive to C225 antibody treatment. Mol Cancer Ther. (2004) 3:1551–8.
196. Giannopoulou E, Antonacopoulou A, Matsouka P, Kalofonos HP. Autophagy: novel action of panitumumab in colon cancer. Anticancer Res. (2009) 29:5077–82.
197. Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. (2012) 23:2240–52. doi: 10.1091/mbc.e11-11-0926
198. Li XQ, Liu JT, Fan LL, Liu Y, Cheng L, Wang F, et al. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget. (2016) 7:24585–95. doi: 10.18632/oncotarget.8358
199. Wojtuszkiewicz A, Schuurhuis GJ, Kessler FL, Piersma SR, Knol JC, Pham TV, et al. Exosomes secreted by apoptosis-resistant acute myeloid leukemia (AML) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol Cell Proteomics. (2016) 15:1281–98. doi: 10.1074/mcp.M115.052944
200. Kuboki Y, Matsusaka S, Minowa S, Shibata H, Suenaga M, Shinozaki E, et al. Circulating tumor cell (CTC) count and epithelial growth factor receptor expression on CTCs as biomarkers for cetuximab efficacy in advanced colorectal cancer. Anticancer Res. (2013) 33:3905–10.
201. Markaverich BM, Vijjeswarapu M, Shoulars K, Rodriguez M. Luteolin and gefitinib regulation of EGF signaling pathway and cell cycle pathway genes in PC-3 human prostate cancer cells. J Steroid Biochem Mol Biol. (2010) 122:219–31. doi: 10.1016/j.jsbmb.2010.06.006
202. Sutter AP, Höpfner M, Huether A, Maaser K, Scherübl H. Targeting the epidermal growth factor receptor by erlotinib (Tarceva) for the treatment of esophageal cancer. Int J Cancer. (2006) 118:1814–22. doi: 10.1002/ijc.21512
203. Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE. (2011) 6:e18691. doi: 10.1371/journal.pone.0018691
204. Jutten B, Rouschop KM. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. (2014) 13:42–51. doi: 10.4161/cc.27518
205. Henson E, Chen Y, Gibson S. EGFR family members' regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers. (2017) 9:E27. doi: 10.3390/cancers9040027
206. Cai J, Sun M, Ge X, Sun Y. EGFR tyrosine kinase inhibitors differentially affect autophagy in head and neck squamous cell carcinoma. Biochem Biophys Res Commun. (2017) 486:1027–33. doi: 10.1016/j.bbrc.2017.03.157
207. Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE. (2013) 8:e76503. doi: 10.1371/journal.pone.0076503
208. Bollu LR, Ren J, Blessing AM, Katreddy RR, Gao G, Xu L, et al. Involvement of de novo synthesized palmitate and mitochondrial EGFR in EGF induced mitochondrial fusion of cancer cells. Cell Cycle. (2014) 13:2415–30. doi: 10.4161/cc.29338
209. Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. (2013) 154:1269–84. doi: 10.1016/j.cell.2013.08.015
210. Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell. (2015) 160:145–60. doi: 10.1016/j.cell.2014.12.006
211. Zhu H, Cao X, Ali-Osman F, Keir S, Lo HW. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Lett. (2010) 294:101–10. doi: 10.1016/j.canlet.2010.01.028
212. Ewald JA, Wilkinson JC, Guyer CA, Staros JV. Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Exp Cell Res. (2003) 282:121–31. doi: 10.1016/S0014-4827(02)00014-9
213. Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. (1995) 269:230–4. doi: 10.1126/science.7618084
214. Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkins NA, et al. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. (1994) 8:399–413. doi: 10.1101/gad.8.4.399
215. Mueller KL, Yang ZQ, Haddad R, Ethier SP, Boerner JL. EGFR/Met association regulates EGFR TKI resistance in breast cancer. J Mol Signal. (2010) 5:8. doi: 10.1186/1750-2187-5-8
216. Ren J, Bollu LR, Su F, Gao G, Xu L, Huang WC, et al. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate. (2013) 73:1453–61. doi: 10.1002/pros.22692
217. Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol. (2011) 226:2316–28. doi: 10.1002/jcp.22570
218. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
219. Bjorkelund H, Gedda L, Barta P, Malmgvist M, Andersson K, Buday L. Gefitinib induces epidermal growth factor receptor dimers which alters the interaction characteristics with (1)(2)(5)I-EGF. PLoS ONE. (2011) 6:e24739. doi: 10.1371/journal.pone.0024739
220. Bublil EM, Pines G, Patel G, Fruhwirth G, Ng T, Yarden Y. Kinase-mediated quasi-dimers of EGFR. FASEB J. (2010) 24:4744–55. doi: 10.1096/fj.10-166199
221. Lu C, Mi LZ, Schürpf T, Walz T, Springer TA. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J Biol Chem. (2012) 287:38244–53. doi: 10.1074/jbc.M112.414391
222. Thomas R, Srivastava S, Katreddy RR, Sobieski J, Weihua Z. Kinase-Inactivated EGFR Is Required for the Survival of Wild-Type EGFR-Expressing Cancer Cells Treated with Tyrosine Kinase Inhibitors. Int J Mol Sci. (2019) 20:E2125. doi: 10.3390/ijms20102515
223. Yang H, Wang R, Peng S, Chen L, Li Q, Wang W. The epidermal growth factor receptor (EGFR) inhibitor gefitinib reduces but does not prevent tumorigenesis in chemical and hormonal induced hepatocarcinogenesis rat models. Int J Mol Sci. (2016) 17:E1618. doi: 10.3390/ijms17101618
224. Tang J, Guo F, Du Y, Liu X, Qin Q, Liu X, et al. Continuous exposure of non-small cell lung cancer cells with wild-type EGFR to an inhibitor of EGFR tyrosine kinase induces chemoresistance by activating STAT3. Int J Oncol. (2015) 46:2083–95. doi: 10.3892/ijo.2015.2898
225. Gandara DR, Gumerlock PH. Epidermal growth factor receptor tyrosine kinase inhibitors plus chemotherapy: case closed or is the jury still out? J Clin Oncol. (2005) 23:5856–8. doi: 10.1200/JCO.2005.05.030
226. Harari PM, Huang SM. Combining EGFR inhibitors with radiation or chemotherapy: will preclinical studies predict clinical results? Int J Radiat Oncol Biol Phys. (2004) 58:976–83. doi: 10.1016/j.ijrobp.2003.09.097
227. Garassino MC, Martelli O, Broggini M, Farina G, Veronese S, Rulli E, et al. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol. (2013) 14:981–8. doi: 10.1016/S1470-2045(13)70310-3
228. Chiba M, Togashi Y, Bannno E, Kobayashi Y, Nakamura Y, Hayashi H, et al. Efficacy of irreversible EGFR-TKIs for the uncommon secondary resistant EGFR mutations L747S, D761Y, and T854A. BMC Cancer. (2017) 17:281. doi: 10.1186/s12885-017-3263-z
229. Holubec L, Polivka J, Safanda M, Karas M, Liska V. The role of cetuximab in the induction of anticancer immune response in colorectal cancer treatment. Anticancer Res. (2016) 36:4421–6. doi: 10.21873/anticanres.10985
230. Lattanzio L, Denaro N, Vivenza D, Varamo C, Strola G, Fortunato M, et al. Elevated basal antibody-dependent cell-mediated cytotoxicity (ADCC) and high epidermal growth factor receptor (EGFR) expression predict favourable outcome in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy. Cancer Immunol Immunother. (2017) 66:573–9. doi: 10.1007/s00262-017-1960-8
231. Monteverde M, Milano G, Strola G, Maffi M, Lattanzio L, Vivenza D, et al. The relevance of ADCC for EGFR targeting: a review of the literature and a clinically-applicable method of assessment in patients. Crit Rev Oncol Hematol. (2015) 95:179–90. doi: 10.1016/j.critrevonc.2015.02.014
232. Jaramillo ML, Leon Z, Grothe S, Paul-Roc B, Abulrob A, O'Connor McCourt M. Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF receptor endocytosis and sorting. Exp Cell Res. (2006) 312:2778–90. doi: 10.1016/j.yexcr.2006.05.008
233. Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal. (2011) 9:23. doi: 10.1186/1478-811X-9-23
234. Munoz L. Non-kinase targets of protein kinase inhibitors. Nat Rev Drug Discov. (2017) 16:424–40. doi: 10.1038/nrd.2016.266
235. Vivanco I, Chen ZC, Tanos B, Oldrini B, Hsieh WY, Yannuzzi N, et al. A kinase-independent function of AKT promotes cancer cell survival. Elife. (2014) 3:e03751. doi: 10.7554/eLife.03751
236. Zheng F, Yue C, Li G, He B, Cheng W, Wang X, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. (2016) 7:10180. doi: 10.1038/ncomms10180
237. Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. (2016) 30:359–60. doi: 10.1016/j.ccell.2016.07.003
238. Audetat KA, Galbraith MD, Odell AT, Lee T, Pandey A, Espinosa JM, et al. A Kinase-independent role for cyclin-dependent kinase 19 in p53 response. Mol Cell Biol. (2017) 37:e00626-16. doi: 10.1128/MCB.00626-16
239. Geng Y, Michowski W, Chick JM, Wang YE, Jecrois ME, Sweeney KE, et al. Kinase-independent function of E-type cyclins in liver cancer. Proc Natl Acad Sci USA. (2018) 115:1015–20. doi: 10.1073/pnas.1711477115
240. Taddei ML, Parri M, Angelucci A, Onnis B, Bianchini F, Giannoni E, et al. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am J Pathol. (2009) 174:1492–503. doi: 10.2353/ajpath.2009.080473
241. Rodríguez J, Calvo F, González JM, Casar B, Andrés V, Crespo P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin a complexes. J Cell Biol. (2010) 191:967–79. doi: 10.1083/jcb.201004067
242. Dart AE, Box GM, Court W, Gale ME, Brown JP, Pinder SE, et al. PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J Cell Biol. (2015) 211:863–79. doi: 10.1083/jcb.201501072
243. Van TM, Polykratis A, Straub BK, Kondylis V, Papadopoulou N, Pasparakis M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J Clin Invest. (2017) 127:2662–77. doi: 10.1172/JCI92508
Keywords: EGFR, kinase independent function, mitophagy, cell survival, cancer
Citation: Thomas R and Weihua Z (2019) Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Front. Oncol. 9:800. doi: 10.3389/fonc.2019.00800
Received: 29 April 2019; Accepted: 06 August 2019;
Published: 23 August 2019.
Edited by:
Wafik S. El-Deiry, Brown University, United StatesReviewed by:
Mohane S. Coumar, Pondicherry University, IndiaKevin Xueying Sun, Harbin Medical University, China
Copyright © 2019 Thomas and Weihua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhang Weihua, d3poYW5nMTMmI3gwMDA0MDt1aC5lZHU=