
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 14 August 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00772
This article is part of the Research Topic Spectrum and clinical implications of chromatin structure and epigenetic changes in leukemia and lymphoma View all 8 articles
 Stéphanie Mourad1,2†
Stéphanie Mourad1,2† Mélanie Bilodeau1†Mathieu Roussy1,3,4Louise Laramée1
Mélanie Bilodeau1†Mathieu Roussy1,3,4Louise Laramée1 Luc Boulianne5Alexandre Rouette6
Luc Boulianne5Alexandre Rouette6 Loubna Jouan6Patrick Gendron7Michel Duval1,3Pierre Teira1,3Josée Hébert3,8,9,10Henrique Bittencourt1,3
Loubna Jouan6Patrick Gendron7Michel Duval1,3Pierre Teira1,3Josée Hébert3,8,9,10Henrique Bittencourt1,3 Yves Pastore1,3*
Yves Pastore1,3* Josette-Renée Landry1,3,11*
Josette-Renée Landry1,3,11* Sonia Cellot1,3,9*
Sonia Cellot1,3,9*Shwachman-Diamond syndrome (SDS) is a rare and systemic disease mostly caused by mutations in the SBDS gene and characterized by pancreatic insufficiency, skeletal abnormalities, and a bone marrow dysfunction. In addition, SDS patients are predisposed to develop myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), typically during adulthood and associated with TP53 mutations. Although most SDS diagnoses are established in childhood, the nature and frequency of serial bone marrow cell investigations during the patients' lifetime remain a debatable topic. The precise molecular mechanisms leading to AML progression in SDS patients have not been fully elucidated because the patient cohorts are small and most disease monitoring is conducted using standard histological and cytogenetic approaches. Here we report a rare case of a patient with SDS who was diagnosed with AML at 5 years of age and survived. Intermittent neutropenia preceded the AML diagnostic but serial bone marrow monitoring according to the standard of care revealed no cytogenetic anomalies nor signs of clonal hematopoiesis. Using next generation sequencing approaches to find cytogenetically cryptic pathogenic mutations, we identified the cancer hotspot mutation c.394C>T/p.Arg132Cys in IDH1 with high variant allelic frequency in bone marrow cells, suggesting clonal expansion of a major leukemic clone karyotypically normal, in the SDS-associated AML. The mutation was somatic and likely occurred at the leukemic transformation stage, as it was not detected in a matched normal tissue nor in bone marrow smear prior to AML diagnosis. Gain-of-function mutations in IDH1, such as c.394C>T/p.Arg132Cys, create a neo-activity of isocitrate dehydrogenase 1 converting α-ketoglutarate into the oncometabolite D-2-hydroxyglutarate, inhibiting α-ketoglutarate-dependent enzymes, such as histone and DNA demethylases. Overall, our results suggest that along with previously described abnormalities such as TP53 mutations or monosomy7, 7q-, which are all absent in this patient, additional mechanisms including IDH1 mutations drive SDS-related AML and are likely associated with variable outcomes. Sensitive techniques complementary to standard cytogenetics, such as unbiased or targeted panel-based next generation sequencing approaches, warrant testing for monitoring of myelodysplasia, clonal hematopoiesis, and leukemia in the context SDS. Such analyses would also assist treatment decisions and allow to gain insight into the disease biology.
Shwachman-Diamond syndrome (SDS) is a rare autosomal recessive bone marrow failure syndrome characterized by exocrine pancreatic insufficiency, bone abnormalities, progressive cytopenia, and predispositions to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). The risk of worsening cytopenia(s), clonal myeloid evolution or AML is estimated at 20% by age 18 (1). The Severe Chronic Neutropenia International Registry reported a 1% per year progression rate to MDS/AML in patients with SDS, with a cumulative risk of MDS/AML reaching 36% by 30 years of age (2).
As the median age at diagnosis of SDS-associated AML is reported to be between 18 and 20 years and as the risk of AML increases with age (3), there are very few pediatric cases reported in the literature (<10 to our knowledge in the last 25 years, Table 1). As such, the molecular events driving malignant transformation in SDS are poorly described, and recent evidence points to a leukemia promoting bone marrow niche in SDS (6). The scant reported pediatric cases of SDS-related AML in the literature are mostly characterized by patient age, cytogenetic profile, and outcome. To our knowledge, all pediatric reported cases of SDS-associated AML occurred between 2.5 and 13 years of age (2, 4, 5) and had cytogenetic abnormalities discernable on karyotype, with frequent chromosome seven involvement (2, 4, 5). Furthermore, these patients had exceedingly poor outcome with only one reported case of survival in a 13-year-old child with SDS following hematopoietic stem cell transplant for AML [(5); Table 1].
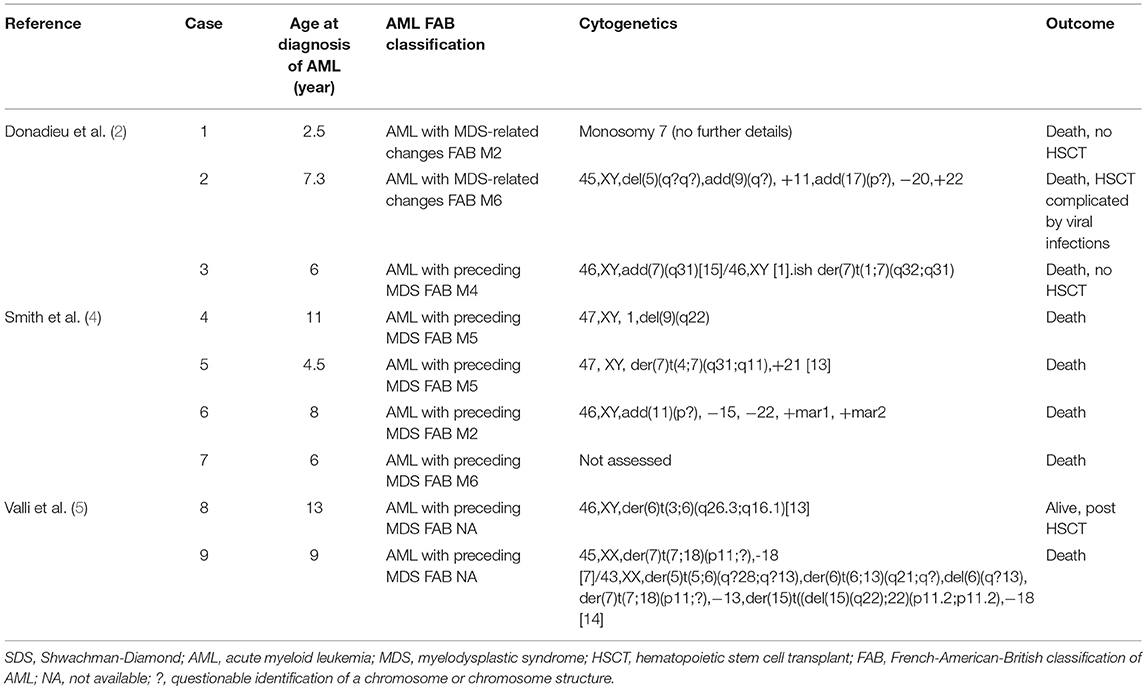
Table 1. Reported cases of pediatric SDS-related AML in the literature between 1994 and 2019.
In addition to the paucity of pediatric cases of SDS-related AML, limited molecular profiling data are available. A recent report suggests that acquisition of TP53 mutations are initiating events contributing to the development of MDS/AML in SDS and may confer a poor prognosis (7). However, no other alteration is known to have any prognostic value.
Here we report a unique case of SDS-associated AML in a young pediatric patient, in absence of cytogenetic abnormalities. Using next-generation sequencing approaches to identify cytogenetically cryptic pathogenic mutations, we reveal an acquired somatic mutation in the Isocitrate Dehydrogenase 1 gene (IDH1), which is a rare cooperating event in pediatric AML and has never been reported in conjunction with SDS. Gain-of-function mutations in Isocitrate Dehydrogenase 1, an enzyme involved in cell metabolism, are known to modify the epigenetic landscape and to promote cancer progression (8). We propose that besides TP53 mutations, additional cytogenetically silent mechanisms drive SDS-related myelodysplastic syndrome or AML, such as IDH1 mutations, and next generation sequencing approaches may benefit the hematological evaluation and risk classification of patients.
A male infant was investigated at 7 months of life with failure to thrive secondary to exocrine pancreatic insufficiency, hypotonia, musculoskeletal congenital anomalies (syndactyly of toes, abnormally short ribs) and intermittent neutropenia with an absolute neutrophil count fluctuating between 0.8 ×109/L and 4.2 ×109/L, suggesting a diagnosis of SDS. The patient had an unremarkable medical family history and was referred to genetic counseling at the age of 1 year. The genetic basis of SDS was confirmed by sequencing of blood/buccal swab-derived DNA revealing two compound heterozygous mutations of the SBDS gene (intron 2 c.258+2T>C and exon 1 c.120delG (p.Ser41fs) (Figures 1A,B). He was followed with serial bone marrow aspirations, biopsies, and cytogenetic analysis, until the age of 5.5 years when he developed worsening cytopenia and his bone marrow evaluation revealed infiltration by 47.4% myeloblasts with underlying multilineage dysplasia (Figure 2A). These changes were compatible with a diagnosis of AML (M0, per FAB classification), with blasts expressing CD45dim, CD34+, CD33+, CD117+, DR+, MPO+, CD15+, CD13partial on flow cytometry and with a normal karyotype (46,XY) on cytogenetic analysis (Figures 2B,C).
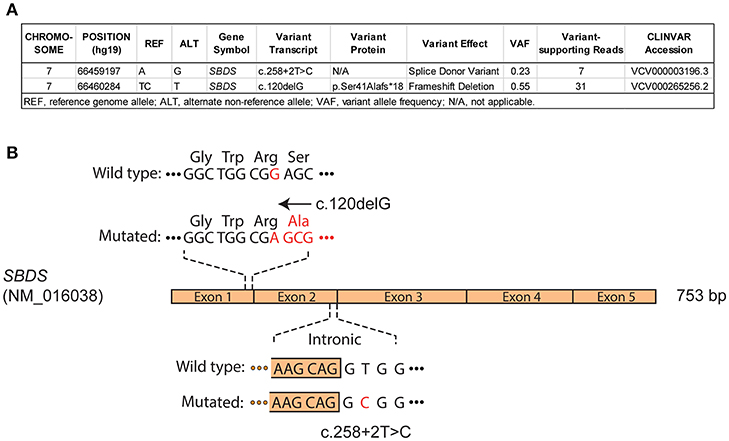
Figure 1. Molecular diagnosis of Shwachman-Diamond syndrome in a pediatric patient. Targeted sequencing of pre-leukemia blood DNA and (A) whole exome sequencing analysis of buccal swab DNA at AML diagnosis reveal compound heterozygous mutations impacting SBDS gene, consistent with an autosomal recessive Shwachman-Diamond syndrome. (B) Schematic representation of SBDS coding sequence (NM_016038), with confirmed c.120delG (exonic), and c.258+2T>C (intronic/splicing) mutations.
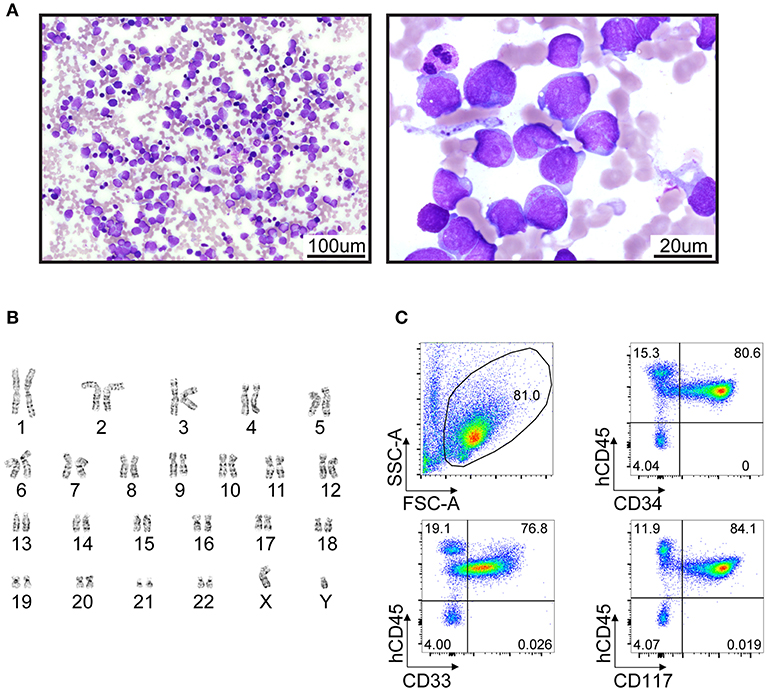
Figure 2. Clinical presentation of AML in a 5-year-old patient with SDS syndrome. (A) Wright-Giemsa-stained cytologic preparation showing leukemic blasts collected from a bone marrow aspiration. Imaging was performed with an upright microscope (Zeiss AxioScope A.1) equipped with 20X/0.5 plan-neofluar and 100x/1.4 oil objectives and an Axiocam 105 color camera. Left and right panels, 200X and 1000X magnification, respectively. (B) Standard GTG-banded karyotype revealing no cytogenetic abnormality. (C) Flow cytometry analyses of bone marrow cells suggest a myeloid leukemia (CD45+ CD33+ CD34+CD117+) with high infiltration (>80%).
He was treated with a first cycle of induction chemotherapy (cytarabine, daunorubicin, etoposide), but minimal residual disease (MRD) remained positive at 0.4%, as assessed by flow cytometry at the end of the cycle. He pursued treatment with a second cycle consisting of high-dose cytarabine and etoposide, after which he achieved MRD negativity and proceeded to hematopoietic stem cell transplant. A first attempt with intra-osseous cord blood transplant following busulfan, fludarabine, and anti-thymocyte globulin (ATG) was unsuccessful with persistent aplasia. A second attempt with an alternate cord blood unit also failed to engraft after conditioning with total body irradiation, cyclophosphamide and fludarabine. Finally, the patient underwent a matched unrelated donor bone marrow transplant following conditioning with cyclophosphamide, fludarabine, and ATG. Hematopoietic engraftment was successful and the patient remains in first complete remission 5.5 years post-transplant (11-year-old).
Given his young age at diagnosis of SDS-associated AML and the lack of clonal cytogenetic anomalies, we searched for discrete oncogenic alterations such as indels and point mutations. First, array-based comparative genomic hybridization (aCGH) analysis did not reveal chromosomal copy-number abnormalities (data not shown) in leukemic cells, consistent with a normal karyotype. Then, whole exome and transcriptome sequencing were performed using bone marrow cells at diagnosis. A point mutation in IDH1 (c.394C>T/p.Arg132Cys), reported to be pathogenic and recurrent in adult AML and other cancer types (8), was identified both by exome and transcriptome sequencing, with both wild-type and mutant alleles expressed at 45 and 55%, respectively (Figure 3A, Figure S1 and data not shown). The mutation was confirmed by Sanger sequencing (Figure 3B). Whole exome sequencing of buccal swab DNA confirmed that the IDH1 mutation was somatic in origin as it was not found in a matched normal tissue (Figure S1). Somatic mutation of IDH1 defined a major leukemic clone (variant allele fraction of 0.38 for a specimen with ~80% leukemic infiltration) while a minor clone with a mutation in BRCA2 (variant allele fraction of 0.08) was also detected (Figure 3A). No fusion transcripts were identified from RNA sequencing reads using two different fusion detection tools (data not shown). The TP53 gene was devoid of somatic alterations according to whole exome sequencing analysis (420X and 70X average coverage for leukemic bone marrow sample and buccal swab DNA, respectively, not shown). To estimate the timing of IDH mutation appearance, we performed NGS on DNA material obtained from scraping fixed bone-marrow smears, archived 6 months prior to AML diagnosis. IDH1 c.394C>T (p.Arg132Cys) mutation was not detected in any of the 44 sequencing reads obtained spanning that position, indicating that the mutation was not present or below our detection threshold 6 months prior to AML diagnosis (Figure S1). These results suggest a cooperative role of IDH1 somatic mutation in SDS-related AML progression, and warrant further mechanistic and multicenter clinical studies. Overall, these molecular analyses suggest that next-generation sequencing may reveal clonal evolution in cytogenetically normal SDS-associated myelodysplasia/leukemia as well as cooperating mutations.
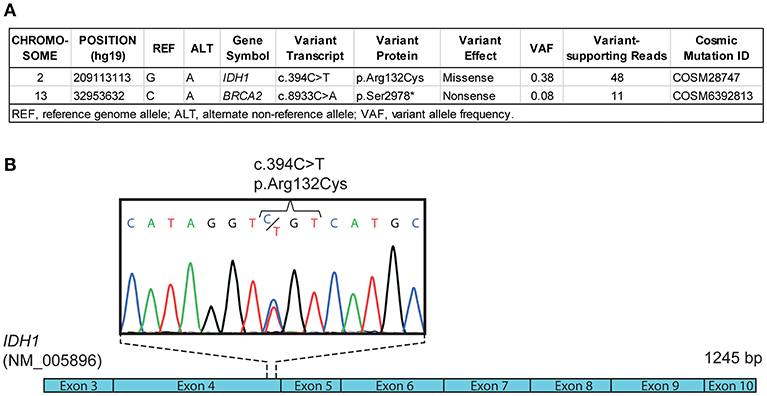
Figure 3. Molecular profiling of SDS-associated leukemia. (A) Differential whole exome sequencing analyses of leukemic bone marrow cell DNA compared with normal control (e.g., buccal swab DNA) reveal a somatic mutation (c.394C>T) in IDH1 coding sequence, corresponding to the recurrent p.Arg132Cys mutation reported in adult AML. (B) Schematic representation of IDH1 coding sequence (NM_005896) and Sanger sequencing chromatogram confirming the c.394C>T (p.Arg132Cys) mutation in leukemic bone marrow cell DNA.
SDS is a ribosomopathy characterized by genomic instability contributing to the occurrence of chromosomal abnormalities and therefore to the risk of malignant transformation to MDS/AML. Characterization of high-risk clonal chromosomal abnormalities remains a challenge, as some patients only develop transient genetic alterations devoid of malignant potential. The most commonly encountered chromosomal anomalies in SDS comprise isochromosome 7 (i(7)(q10)), resulting in a gain of long arm of chromosome 7 (7q), and deletion 20q (del(20)(q)) (5), not typically associated with initiating events in leukemogenesis. Deletion 20q has not yet been reported to have malignant transformation potential, unless associated with other cytogenetic changes (9). Overall, the risk of MDS/AML in patients with SDS increases with age and varies considerably according to the acquired cytogenetic abnormalities, with complex karyotypes seemingly conferring an increased risk of malignant transformation (2).
SDS being a rare disorder, thorough understanding of SDS-related AML has remained limited due to the small number of patients, especially in pediatrics. Given the heterogeneity of MDS/AML progression in patients with SDS, we postulated that acquired cooperating somatic mutations must contribute to malignant transformation. This should be especially true for pediatric patients in whom the risk of SDS-related MDS/AML is considerably lower than in adults, prompting our investigations for this case. The unique presentation of an SDS-associated AML in a young pediatric patient lacking chromosomal rearrangements and any pathogenic TP53 mutation confirmed this hypothesis and revealed a cooperating genetic mutation in the Isocitrate Dehydrogenase 1 gene (IDH1), associated with promoting leukemia.
IDH1 encodes a key cytoplasmic and peroxisomal enzyme of the Krebs cycle responsible for the catalyzation of isocitrate to α-ketoglutarate while reducing NADP to NADPH and liberating CO2 (8). The mutant IDH1 enzyme acquires a novel molecular function by which it converts α-ketoglutarate to the oncometabolite D-2-hydroxyglutarate (8). D-2-hydroxyglutarate inhibits the α-ketoglutarate-dependent enzymes that regulate epigenetic modeling, collagen synthesis and cell signaling (10). IDH1 mutations have been found to be rare in pediatric AML (~1–2%) (11–13), but to be present at a higher frequency in pediatric AML with a normal karyotype (~5%) (11). They have also been reported at the leukemic transformation of myeloproliferative diseases (14, 15) and, combined with IDH2 mutations, to be present in ~5% of MDS patients (16). Somatic mosaic mutations of IDH1 have been detected in hematologic malignancies (BCP-ALL and AML) of children with Maffucci syndrome, a non-hereditary disorder characterized by multiple enchondromas and hemangiomas (17, 18). In adult, detection of somatic mutations in IDH1 in peripheral blood DNA is highly predictive of a future leukemia development (19).
To the best of our knowledge, we present the first case of oncogenic IDH1 mutation identified in SDS-associated AML. Pediatric AML associated with IDH1 mutations present a favorable overall survival (13) compared with adult AML with TP53 alterations (20), but large multicentric cohort of patients will be necessary to better stratify the genetic subsets and outcomes of SDS-associated leukemia. This study reports one of the only cases of pediatric SDS-related AMLs who achieved complete remission and survived, raising the interesting hypothesis that it is ultimately the genetic makeup of the leukemia that impacts prognosis in the context of SDS, and not the underlying cancer predisposition syndrome in itself.
The discovery of a cooperating IDH1 mutation in SDS-associated AML illustrates the necessity to refine surveillance for malignant transformation in this condition. Standard cytogenetic analyses may fail to detect single nucleotide genetic alterations driving clonal hematopoiesis and leukemic progression. Additional sensitive techniques, such as unbiased or targeted panel-based next generation sequencing approaches, would benefit the monitoring of myelodysplasia, clonal hematopoiesis, and leukemia in the context SDS. The genetic landscape of SDS-related AML should be evaluated to establish the disease-risk stratification, to assist treatment decisions and ultimately to gain insight into the biology of a rare malignancy.
This manuscript contains previously unpublished data. The name of the repository and accession number(s) are not available at the time of publication but will be available upon request.
The Institutional Review Board of CHU Sainte-Justine approved the study and informed consent was obtained from the parents.
SM, J-RL, MB, AR, and SC wrote the manuscript. SM, J-RL, MB, MR, LL, and AR generated the figures and table. SM, J-RL, MB, MR, LL, LB, AR, LJ, and PG contributed to experiments and analyses. YP, MD, PT, HB, and SC were in charge of the patient. JH and SC supervised the biobanking resource. All authors revised and approved the manuscript.
This work was supported through a CCSRI impact grant (#705047) (SC) and a CCS-Cole Foundation Grant (#705474) (SC, JH). SC was recipient of a Charles Bruneau Foundation grant. The Quebec Leukemia Cell Bank was supported by the Cancer Research Network of the Fonds de Recherche du Québec - Santé. J-RL was the recipient of the Fonds de recherche du Québec (FRQ) Innovator in Residence fellowship and a Rare Disease Foundation Microgrant. MR was a recipient of a Cole Foundation Ph.D. Fellowship.
J-RL is a founder and has ownership stake in Streamline Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are sincerely grateful for the support received from the patients and their families, as well as from the members of the pediatric hematology-oncology team of CHU Sainte-Justine. We thank Virginie Saillour, Charles Privé, and René Allard of the Integrated Center for Pediatric Clinical Genomics at CHU Sainte-Justine for their contribution with whole exome sequencing and Jennifer Huber at IRIC (Institute for Research in Immunology and Cancer) for her appreciated assistance with RNA sequencing. We are grateful to colleagues of the hematology-oncology division of CHU Sainte-Justine and members of the BCLQ (Banque de cellules leucémiques du Québec) for their contribution to biobanking initiative. We thank Dr. Emmanuelle Lemyre and France Léveillé for the clinical cytogenetics and the team of genetic counseling at CHU Sainte-Justine.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00772/full#supplementary-material
1. Hashmi SK, Allen C, Klaassen R, Fernandez CV, Yanofsky R, Shereck E, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin Genet. (2011) 79:448–58. doi: 10.1111/j.1399-0004.2010.01468.x
2. Donadieu J, Fenneteau O, Beaupain B, Beaufils S, Bellanger F, Mahlaoui N, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. (2012) 97:1312–9. doi: 10.3324/haematol.2011.057489
3. Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. (2013) 27:117–28. doi: 10.1016/j.hoc.2012.10.003
4. Smith OP, Hann IM, Chessells JM, Reeves BR, Milla P. Haematological abnormalities in Shwachman-Diamond syndrome. Br J Haematol. (1996) 94:279–84. doi: 10.1046/j.1365-2141.1996.d01-1788.x
5. Valli R, De Paoli E, Nacci L, Frattini A, Pasquali F, Maserati E. Novel recurrent chromosome anomalies in Shwachman-Diamond syndrome. Pediatr Blood Cancer. (2017) 64:e26454. doi: 10.1002/pbc.26454
6. Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. (2016) 19:613–27. doi: 10.1016/j.stem.2016.08.021
7. Xia J, Miller CA, Baty J, Ramesh A, Jotte MRM, Fulton RS, et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood. (2018) 131:408–16. doi: 10.1182/blood-2017-08-801985
8. Raineri S, Mellor J. IDH1: linking metabolism and epigenetics. Front Genet. (2018) 9:493. doi: 10.3389/fgene.2018.00493
9. Maserati E, Pressato B, Valli R, Minelli A, Sainati L, Patitucci F, et al. The route to development of myelodysplastic syndrome/acute myeloid leukaemia in Shwachman-Diamond syndrome: the role of ageing, karyotype instability, and acquired chromosome anomalies. Br J Haematol. (2009) 145:190–7. doi: 10.1111/j.1365-2141.2009.07611.x
10. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. (2013) 3:730–41. doi: 10.1158/2159-8290.CD-13-0083
11. Andersson AK, Miller DW, Lynch JA, Lemoff AS, Cai Z, Pounds SB, et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia. (2011) 25:1570–7. doi: 10.1038/leu.2011.133
12. Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:103–12. doi: 10.1038/nm.4439
13. Damm F, Thol F, Hollink I, Zimmermann M, Reinhardt K, van den Heuvel-Eibrink MM, et al. Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia. (2011) 25:1704–10. doi: 10.1038/leu.2011.142
14. Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. (2010) 24:1302–9. doi: 10.1038/leu.2010.113
15. Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med. (2010) 362:369–70. doi: 10.1056/NEJMc0910063
16. Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. (2017) 17:5–19. doi: 10.1038/nrc.2016.112
17. Akiyama M, Yamaoka M, Mikami-Terao Y, Ohyama W, Yokoi K, Arakawa Y, et al. Somatic mosaic mutations of IDH1 and NPM1 associated with cup-like acute myeloid leukemia in a patient with Maffucci syndrome. Int J Hematol. (2015) 102:723–8. doi: 10.1007/s12185-015-1892-z
18. Hirabayashi S, Seki M, Hasegawa D, Kato M, Hyakuna N, Shuo T, et al. Constitutional abnormalities of IDH1 combined with secondary mutations predispose a patient with Maffucci syndrome to acute lymphoblastic leukemia. Pediatr Blood Cancer. (2017) 64:e26647. doi: 10.1002/pbc.26647
19. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. (2018) 24:1015–23. doi: 10.1038/s41591-018-0081-z
Keywords: Shwachman-Diamond syndrome, predisposition syndrome, pediatric patient, leukemia, molecular profiling, cooperating mutation
Citation: Mourad S, Bilodeau M, Roussy M, Laramée L, Boulianne L, Rouette A, Jouan L, Gendron P, Duval M, Teira P, Hébert J, Bittencourt H, Pastore Y, Landry J-R and Cellot S (2019) IDH1 as a Cooperating Mutation in AML Arising in the Context of Shwachman-Diamond Syndrome. Front. Oncol. 9:772. doi: 10.3389/fonc.2019.00772
Received: 02 June 2019; Accepted: 30 July 2019;
Published: 14 August 2019.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Michael Alexander Morgan, Hannover Medical School, GermanyCopyright © 2019 Mourad, Bilodeau, Roussy, Laramée, Boulianne, Rouette, Jouan, Gendron, Duval, Teira, Hébert, Bittencourt, Pastore, Landry and Cellot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Pastore, eXZlcy5wYXN0b3JlQHVtb250cmVhbC5jYQ==; Josette-Renée Landry, am9zZXR0ZUBzdHJlYW1saW5lZ2Vub21pY3MuY29t; Sonia Cellot, c29uaWEuY2VsbG90QHVtb250cmVhbC5jYQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.