 Amelia McCartney1
Amelia McCartney1 Ilenia Migliaccio2Martina Bonechi2Chiara Biagioni3Dario Romagnoli3Francesca De Luca2Francesca Galardi2Emanuela Risi1Irene De Santo1
Ilenia Migliaccio2Martina Bonechi2Chiara Biagioni3Dario Romagnoli3Francesca De Luca2Francesca Galardi2Emanuela Risi1Irene De Santo1 Matteo Benelli3Luca Malorni1,2Angelo Di Leo1*
Matteo Benelli3Luca Malorni1,2Angelo Di Leo1*- 1“Sandro Pitigliani” Department of Medical Oncology, Hospital of Prato, Prato, Italy
- 2“Sandro Pitigliani” Translational Research Unit, Hospital of Prato, Prato, Italy
- 3Bioinformatics Unit, Hospital of Prato, Prato, Italy
The recent arrival of CDK4/6 inhibitor agents, with an approximate doubling of progression-free survival (PFS) associated with their use in hormone receptor-positive, HER2-negative advanced breast cancer (BC), has radically changed the approach to managing this disease. However, resistance to CDK4/6 inhibitors is considered a near-inevitability in most patients. Mechanisms of resistance to these agents are multifactorial, and research in this field is still evolving. Biomarkers with the ability to identify early resistance, or to predict the likelihood of successful treatment using CDK4/6 inhibitors are yet to be identified, and represent an area of unmet clinical need. Here we present selected mechanisms of resistance to CDK4/6 inhibitors, largely focussing on roles of Rb, cyclin E1, and the PIK3CA pathway, with discussion of associated biomarkers which have been investigated and applied in recent pre-clinical and clinical studies. These biological drivers may furthermore influence clinical treatment strategies adopted beyond CDK4/6 resistance.
In normal cell signaling, mitogenic signaling via pathways including ER and PI3K/Akt/mTOR activates the cyclin D1-cyclin-dependent kinases 4 and 6 (CDK4/6) complex. Once activated, CDK4/6 phosphorylates the retinoblastoma protein (Rb), an event which causes Rb to lose the ability to bind to the E2F family of transcription factors. E2F is therefore released, activating gene transcription and thus initiating progression of the cell cycle from G1 to S phase (resulting in DNA synthesis) (1). Dysregulation of the CDK4/6 pathway, occasioning unchecked cell-cycle progression and proliferation, has been implicated in breast cancer (BC) via various mechanisms, including amplification of cyclin D1 (2), gain of CDK4, low expression of p18/high expression of RB1 (3) and inactivation of p16, the tumor suppressor protein and negative moderator of the cyclin D-CDK4/6 complex (4). The p16 protein, encoded by the INK4a gene, is a common target of inactivating mutations and deletions, operating upstream from RB (5).
The past decade has seen a radical shift in the management of advanced or metastatic hormone receptor-positive (HR+), human epidermal growth factor receptor-2 (HER2) negative BC. Endocrine therapy (ET) as monotherapy, previously thought to be the gold-standard in first and often subsequent lines of management, has been augmented and displaced in the treatment hierarchy by the emergence of selective, small-molecule inhibitors of CDK4/6. Three such agents are currently in widespread clinical use, with all three, when given in combination with ET, resulting in an approximate doubling of progression-free survival (PFS) in patients with advanced HR+/HER2–negative BC, compared to ET plus placebo. These consistent positive PFS results have been demonstrated in large phase III trials in the upfront setting [PALOMA-2 (6) for palbociclib plus letrozole; MONALEESA-2 (7) for ribociclib plus letrozole and MONARCH-3 (8) for abemaciclib plus letrozole or anastrozole], as well as in populations previously treated with ET for advanced disease [PALOMA-3 for palbociclib plus fulvestrant (9); MONARCH-2 for abemaciclib and fulvestrant (10); and MONALEESA-3 for ribociclib plus fulvestrant (11)]. However, de novo or acquired resistance to CDK4/6 inhibitors is an almost ubiquitous inevitability, which has stimulated substantial interest in examining potential mechanisms of resistance, ways to overcome it, and methods of identifying it. Recently published reviews offer detailed insight into the myriad of likely mechanisms of resistance (12–14); conversely, we focus primarily on selected mechanisms and associated biomarkers that are of particular clinical interest, of whose potential has been validated in recent studies.
Mechanisms of Resistance—Avenues for Possible Molecular Biomarkers
Loss of Retinoblastoma Susceptibility Gene Product (Rb) Function
Loss of Rb, the main target of CDK4/6, has been implicated by multiple preclinical studies in being a driver of resistance to CDK4/6 inhibitors (15–17). Without the inhibitory influence of Rb, transcription factors of the E2F family continue unchecked, thus facilitating unregulated cellular progression to S-phase entry. Conversely, higher levels of RB1, the gene encoding for Rb, and cyclin D1 (and lower levels of p16) are observed in human BC cell lines sensitive to palbociclib (18). Clinical evidence of acquired RB1 mutations leading to CDK4/6 inhibitor resistance was recently reported in a case-series of three patients with metastatic BC who had genotyping performed in both tissue and blood samples before and after commencing a CDK4/6 inhibitor (19). Each somatic mutation was detected via ctDNA analyses performed at the point of disease progression, and was not present prior to initiation of CDK4/6 inhibition. Polyclonal RB1 mutations were identified in patients assigned to the palbociclib arm of PALOMA-3, albeit at comparatively low frequency (4.7%) (20). RBsig, a gene expression signature of Rb loss-of-function, has been validated in identifying between palbociclib-sensitive and resistant BC cell lines (21), and has been associated with sensitivity to abemaciclib monotherapy in tumors derived from the neoMONARCH study (NCT02441948) (22). Similarly, a gene set containing E2F targets, the E2F regulon, was significantly associated with lack of PFS improvement from palbociclib combination in the PALOMA-3 trial (23). Of note, no interaction was found between treatment and RB1 expression in the same study, indicating that a wider analysis of RB pathway might be needed to identify resistant patients. Recent data emerging from genomic analysis of ER-positive BCs treated with CDK4/6 inhibitors revealed—not unsurprisingly—that loss of RB1 was associated with treatment resistance. However, also implicated in resistance to CDK4/6 inhibition were loss-of-function mutations of FAT1, leading to cellular proliferation mediated via activation of the Hippo signaling pathway and elevations in CDK6, thus revealing an additional and intriguing potential mechanism of resistance (24).
Cyclin E1 (CCNE1)
CCNE1, the gene that encodes cyclin E1, is upregulated in models with resistance to CDK4/6 inhibitors (25, 26). Data emerging from PALOMA-3 suggests that CCNE1 expression is associated with benefit from palbociclib (23), in line with previous pre-clinical data which suggested CCNE1 amplification is associated with acquired resistance to palbociclib (27), as well as exploratory data derived in the neoadjuvant NeoPalAna trial, associating high levels of CCNE1 with palbociclib resistance (28). Tumor tissue procured from recurrent disease on trial was assessed via mRNA profiling, assessing a range of cell cycle-related genes. Although all biomarker groups derived benefit from palbociclib, those with low tumor CCNE1 expression had a greater response (median PFS for those receiving palbociclib plus fulvestrant, 14.1 months; vs. 4.8 months in those receiving fulvestrant plus placebo) than those with high CCNE1 expression (7.6 months vs. 4.0; palbociclib plus fulvestrant vs. fulvestrant plus placebo, respectively). The predictive power of CCNE1 mRNA was stronger in metastatic biopsies (interaction p < 0.001) than archived primary biopsy samples (interaction p = 0.09). Investigators provided further validation in an independent cohort (N = 61) drawn from the Preoperative Palbociclib (POP) Clinical Trial (NCT02008734), wherein high CCNE1 mRNA expression correlated with a significantly lower anti-proliferative response to palbociclib. Contrastingly, no such association with CCNE1 expression and PFS was found in the biomarker analyses of MONALEESA-2, with near-identical hazard ratios observed across expression groups (HR 0.54 95% CI 0.38–0.78 for CCNE1 high expression; HR 0.53 95% CI 0.34–0.83 for low expression) (29). An earlier analysis of tumor specimens from PALOMA-2 also failed to demonstrate an association between CCNE1 expression and benefit from palbociclib (30).
Combining Rb and CCNE1—Possible Biomarker of CDK4/6 Sensitivity
A recent analysis of cell cycle-related markers found in a large panel of HR+ BC cell lines was recently reported, describing findings identified by gene-expression profiles and western blot (31). Both modalities identified that concurrent overexpression of CCNE1 and down-regulation of Rb occurred at the time of palbociclib resistance. Subsequent in silico analyses, correlating the ratio between CCNE1 and RB1 expression levels (CCNE1/RB1) with palbociclib IC50 in a large dataset of cell lines, showed the ratio outperformed both CCNE1 and/or RB1 when they were utilized as sole markers. Furthermore, retrospective analyses showed CCNE1/RB1 to be an adverse prognostic factor, with the ability to differentiate between palbociclib sensitive and resistant patients enrolled in the neoadjuvant NeoPalAna trial (28).
Initially Promising Targets but Negative Data: Cyclin D and p16
CCND1, the gene coding for cyclin D1 is amplified in approximately 15% of all BCs, and overexpression of cyclin D1 is observed in around 50% (32). Given the crucial role cyclin D1 plays in cell cycle mediation and its interplay with CDK4/6, it has been hypothesized that expression levels or dysregulation of cyclin D1 may relate to response to CDK4/6 inhibition. Similarly, intuitively, loss of p16INK4A and the consequent deficit of its usual inhibitory action on cyclin D1 would appear to be a reasonable premise of CDK4/6 inhibitor resistance. However, this is largely not been borne out in the clinical setting. In a phase II study of single-agent palbociclib, low p16 did not correlate with clinical outcome in Rb-positive, heavily pre-treated advanced BC (33). In the same study, amplification of cyclin D1 was also not associated with clinical benefit or PFS. PALOMA-1 failed to show any significant difference in PFS in patients whose tumors harbored evidence of a loss of p16 or CCND1 amplification, compared to unselected patients (34). Expression levels of cyclin D1 was not associated with benefit from palbociclib in PALOMA-3 (23).
PIK3CA—Activation of Growth Factor Signaling Pathways
Mutations in the phosphatidylinositol 3-kinase (PI3K) catalytic subunit (PIK3CA) are found in approximately 40% estrogen receptor-positive BC (3). The PI3K/mTOR pathway has been shown to be upregulated in response to chronic exposure to CDK4/6 inhibitors, which in turn upregulates cyclin D. In the absence of CDK4 and CDK6, activated cyclin D can activate CDK2, which subsequently drives cell cycle progression (27). Circulating tumor DNA sequencing was performed on 195 patients enrolled in the PALOMA-3 study, comparing baseline and end-of-treatment analyses (20), demonstrating the emergence of driver mutations in PIK3CA and ESR1. Patients with a history of greater drug exposure appeared more likely to develop driver gene mutations, perhaps underlining the role that drug pressure plays in clonal expansion. Contrastingly, PIK3CA mutations were detected in the circulating DNA of 129 patients enrolled in PALOMA-3, with no significant association observed with response to treatment (9). Similarly, biomarker analysis of MONALEESA-3 demonstrated consistent benefit from ribociclib plus fulvestrant, irrespective of PIK3CA alteration status, as detected in baseline circulating tumor DNA (35). Functioning downstream of PI3K is 3-phosphoinositide-dependent protein kinase 1 (PDK1), a vital requisite for the full activation of AKT (36). The PI3K-PDK1 signaling pathway has been implicated in mediating resistance to CDK4/6 inhibitors, with ribociclib-resistant BC cell lines demonstrating an increase in PDK1 levels following drug exposure, resulting in activation of the AKT pathway (37).
Prediction of Sensitivity or Early Response to CDK4/6 Inhibition—A Possible Role for TK1
Thymidine kinase-1 (TK1) is an enzyme in the pyrimidine salvage pathway that plays a critical role in the synthesis of DNA and in cell proliferation (38). High TK1 levels and activity in primary BC tissue correlate with poor prognosis (39, 40). Malignant cells can secrete pathological levels of TK1 detectable in blood, whereas in disease-free controls, levels are low or undetectable (41), with similar patterns reported in membrane expression of TK1 (42). TK1 as a marker of cell proliferation has been known and studied for some decades, but until recently, widespread, reliable quantification of absolute levels and activity have been limited, with most historical tests being radioimmunoassay-based. DiviTum (Biovica International, Sweden) is a refined ELISA-based assay capable of estimating TK1 activity (TKa) in cell lines, plasma and serum. Previous studies have suggested baseline and repeated assessments of TKa during the course of treatment may provide prognostic information (43–45).
TK1 as a Biomarker—Founding Data Within Endocrine Therapy Studies
Previous studies have validated the use of DiviTum, both as a prognostic marker, and as one of response to ET. A pilot study of 31 women with advanced HR+/HER2 negative BC commencing on a new line of palliative endocrine therapy showed that those with low baseline levels of plasma TKa had a median PFS of 25.9 months, vs. 5.9 months in those with high baseline levels (p = 0.012) (46). Furthermore, patients whose TKa levels dropped after 1 month of ET demonstrated a significantly higher median PFS than those in whom TKa levels increased on treatment (14.5 months vs. 3.8, respectively; p = 0.0026).
These findings were upheld by a second retrospective study of a larger, more heavily pre-treated population derived from the cohort of EFECT, a landmark study which originally compared head-to-head palliative exemestane vs. fulvestrant (47). Again, baseline TKa levels proved prognostic: patients with low baseline readings had a median TTP (mTPP) of 5.03 vs. 2.57 in those with high baseline readings (p < 0.001). Patients whose TKa increased from baseline after 3 months of treatment had a significantly shorter mTTP (3.39 months, 95% CI: 2.14–4.11) than those whose TKa did not increase (5.39 months, 95% CI: 4.01–6.68) (P = 0.0045). After adjusting for major prognostic factors, TKa remained an independent marker (48).
TK1 and CDK4/6 Inhibitors
Evidence of the prognostic role TK1 may play in HR+ BC patients has provided a proof-of-concept to justify moving investigation forward into the field of CDK4/6 inhibitors. Recent data suggests potential utility for TKa as a marker of CDK4/6 inhibition in patients receiving neoadjuvant ET plus palbociclib (49). DiviTum was employed in an analysis of serum samples derived from NeoPalAna, a neoadjuvant trial of 4 weeks of anastrozole monotherapy followed by four cycles of additional palbociclib, followed by a subsequent palbociclib washout in all but eight patients (28, 49). TKa was shown to markedly reduce with the introduction of palbociclib, rising at washout (but remaining suppressed in patients who did not receive washout). There was high concordance between changes in TKa and tumor Ki67 in the same direction from baseline to C1D15 and from C1D15 to point of curative surgery. This led to the conjecture that TKa may be seen as a dynamic marker that signifies the presence or absence of palbociclib activity. However, there is some pre-clinical evidence suggesting that TKa may precede a significant reduction in cellular proliferation. In a panel of HR+ BC models—both with sensitivity to palbociclib, and with acquired resistance to the drug—exposure to escalating levels of palbociclib and its relation to cellular proliferation and TKa was examined (50). In palbociclib-sensitive models, TKa significantly reduced after 3 days of drug exposure compared to control (p < 0.05). Concurrently, cellular proliferation (as assessed by methylene blue assay) was observed to drop significantly after a minimum of 6 days, suggesting TKa may be an early marker of proliferative inhibition in response to palbociclib. This phenomenon was not observed in models with acquired resistance to palbociclib. The prognostic ability of TKa has been clinically validated in planned translational studies of plasma derived from the TREnd study (NCT02549430), a phase II trial which tested the activity and safety of single-agent palbociclib against palbociclib combined with the ET the patient had received (and progressed on) most recently before enrollment (51). Not unlike to previous findings in ET-based TK studies, TREnd patients with a low baseline TKa had a significantly longer PFS compared to those whose levels were high at study commencement. Similarly, on treatment, those patients whose TKa levels rose had a shorter time to disease progression compared to those patients whose levels remained stable or dropped in response to treatment (52). The prognostic role of serum TK1 assessed at baseline and on treatment is being further explored in two ongoing clinical trials of luminal BC patients treated with CDK4/6 inhibitors and ET: BIOITALEE (NCT03439046), a Phase 3b biomarker study of ribociclib plus letrozole in the first-line setting and PYTHIA (NCT02536742), a phase 2 biomarker discovery trial of palbociclib and fulvestrant in patients with endocrine resistant disease.
Implications on Therapeutic Approaches Following Progression on CDK4/6 Inhibitors
Primary Resistance to CDK4/6 Inhibitors
Approximately 10% of patients will have primary resistance to CDK4/6 inhibitors. Biomarkers may have future potential to identify such patients at baseline or soon after commencing treatment, thus facilitating an early switch to a more efficacious treatment. For instance, patients with evidence of functional Rb loss at baseline are not likely to benefit from CDK4/6 inhibition. Similarly, baseline evidence of increased cyclin E1 expression, or the CCNE1/RB ratio may also play a role in identifying these patients. Peripheral evidence of ongoing neoplastic proliferation, as manifested by a rise in TK1 activity within a month of commencing therapy, may also provide a marker of early resistance.
Secondary Resistance to CDK4/6 Inhibitors
An unanswered question regards the continuation of CDK4/6 inhibitors beyond progression on these agents. The premise that continuing a CDK4/6 inhibitor beyond progression may prove an effective strategy is being tested by several ongoing Phase 1 and 2 trials (MAINTAIN NCT02632045, PACE NCT03147287, NCT01857193, NCT 02871791, and TRINITI-1 NCT 02732119). Mutations in RB1, resulting in activation of other cell cycle factors, such as E2F and the Cyclin E-CDK2 axis, has been demonstrated in cases of acquired resistance (19, 53). This in turn results in independence from the CDK4/6 pathway for cell cycle progression from G1 to S phase. In such cases, in the setting of disease progression on a CDK4/6 inhibitor, concurrent biomarker evidence of a functional loss of Rb may support a switch to a new agent, rather than continuing CDK4/6 agents beyond progression.
A Potential Role for PIK3CA Inhibitors?
PI3K-dependent activation of non-canonical cyclin D1-CDK2 and resultant recovery of Rb phosphorylation and S phase entry has been implicated in early resistance to CDK4/6 inhibition, with combined PI3K and CDK4/6 inhibition demonstrating the ability to overcome resistance to CDK4/6 inhibitors in BC cell lines (27). Hence, the role that PIK3CA inhibitors may play in overcoming resistance is of relevant interest. The SOLAR-1 trial randomly assigned 572 patients with pre-treated HR+/HER2–negative advanced BC to receive the oral PIK3CA inhibitor alpelisib plus fulvestrant or fulvestrant plus placebo (54). The primary endpoint was PFS in patients with PIK3CA mutations detectable in tumor tissue (n = 341). After a median follow up of 20 months, the median PFS was almost double in mutation-positive patients receiving alpelisib compared to those receiving placebo (11.0 months vs. 5.7 months, respectively; HR 0.65 95% CI 0.50–1.25 p = 0.00065). Further data, reporting the efficacy of alpelisib according to mutational status evaluated by ctDNA, suggested an even greater clinical benefit than tissue analysis (55). In patients with a PIK3CA mutation detected via liquid biopsy (n = 186), there was a 45% risk reduction in PFS (HR 0.55 95% CI 0.39–0.79). In the small number of patients who had previously received CDK4/6 inhibition (n = 20), there was a 52% risk reduction in PFS in favor of alpelisib over placebo (HR 0.48 95% CI 0.17–1.36). Alpelisib is selective for the alpha isoform of PI3K, which has so far set it apart from pan-PI3K inhibitors, which have reported notably poor safety profiles (56, 57). Nevertheless, data from SOLAR-1 still reflect considerable toxicity. All-grade hyperglycaemia occurred in 64% of patients receiving alpelisib (37% occurring at grade 3/4), 58% reported diarrhea, 45% nausea and 36% developed rash (10% at grade 3/4). Five percent of patients discontinued from the alpelisib arm due to adverse events (54).
Whilst the number of patients with prior exposure to CDK4/6 inhibitors subsequently enrolled in SOLAR-1 was small, it is not unreasonable to consider—in patients harboring an actionable mutation—PIK3CA inhibition following disease progression on a CDK4/6 agent. This is particularly relevant, given the likelihood of emergence of driver mutations of PIK3CA secondary to previous ET and CDK4/6-targeted therapies (20). Whilst some pre-clinical data suggest that triplet therapy, combining ET plus CDK4/6 inhibitors with PIK3CA agents may be better in preventing acquired CDK4/6 resistance than doublet regimens (27), this approach may potentially come at the cost of increased toxicity (58). Clinical trials investigating the feasibility and utility of combining CDK4/6 and PI3K inhibition are ongoing (Table 1). Another alternative may be to expose patients to these agents sequentially rather than simultaneously, reserving PIK3CA inhibition for those harboring a druggable mutation following exposure to CDK4/6 inhibition. This tactic is being tested in a currently-recruiting phase II study, which will assess the efficacy and safety of combining alpelisib and ET in patients with PIK3CA mutations whose disease has progressed on or after receiving a CDK4/6 inhibitor plus ET (NCT03056755). A summary of clinical trials of PIK3CA agents, currently recruiting patients with endocrine receptor-positive advanced BC, is presented in Table 2.
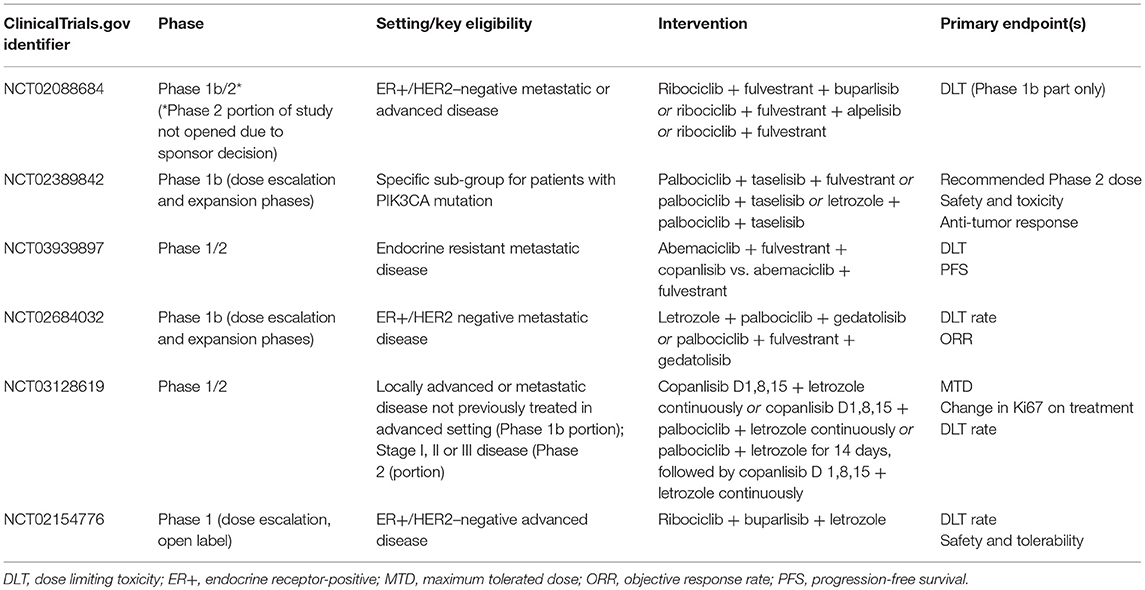
Table 1. Ongoing trials evaluating the combination of CDK4/6 inhibitors with PIK3CA agents in breast cancer [ClinicalTrials.gov June 2019].
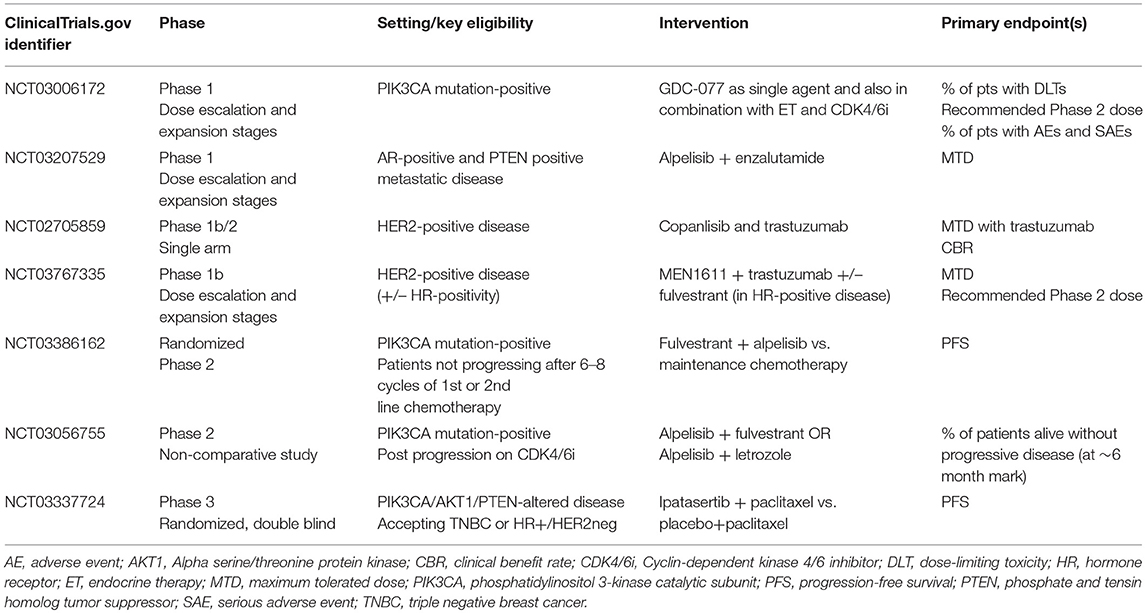
Table 2. Currently enrolling trials recruiting patients with hormone receptor-positive metastatic breast cancer, evaluating the role of PIK3CA agents [ClinicalTrials.gov June 2019].
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was partially supported by the Breast Cancer Research Foundation [grant number BCRF 18-037].
Conflict of Interest Statement
AD has received honoraria from, and served as an advisory board member for, Pfizer, Lilly, AstraZeneca, and Novartis. AD has received research funding from Pfizer, AstraZeneca, and Novartis. LM has received consultation fees from Novartis and research support from Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the Sandro Pitigliani Foundation, Prato, Italy, for their ongoing support.
References
1. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. (2008) 8:671–82. doi: 10.1038/nrc2c399
2. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Cancer Rev. (2011) 11:558–72. doi: 10.1038/nrc3c090
3. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature1e1412
4. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. (2006) 24:1770–83. doi: 10.1200/JCO.2005.03.7689
5. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4K and CDK6K: from discovery to therapy. Cancer Discov. (2016) 6:353–67. doi: 10.1158/2159-8290.CD-15-0894
6. Finn RS, Martin M, Rugo HS, Jones S, Seock-Ah I, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. (2016) 375:1925–36. doi: 10.1056/NEJMoa1a607303
7. Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. (2016) 375:1738–48. doi: 10.1056/NEJMoa1609709
8. Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH3H: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. (2017) 35:3638–46. doi: 10.1200/JCO.2017.75.6155
9. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Seock-Ah I, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2R-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA 3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
10. Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2R- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
11. Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Seock-Ah I, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. (2018) 36:2465–72. doi: 10.1200/JCO.2018.78.9909
12. Guarducci C, Bonechi M, Boccalini G, Benelli M, Risi E, Di Leo A, et al. Mechanisms of resistance to CDK4K/6 inhibitors in breast cancer and potential biomarkers of response. Breast Care. (2017) 12:304–8. doi: 10.1159/000484167
13. Pandey K, An H-J, Kim SK, Lee SA, Kim S, Lim SM, et al. Molecular mechanisms of resistance to CDK4K/6 inhibitors in breast cancer: a review. Int J Cancer. (2018) 145:1179–88. doi: 10.1002/ijc.32020
14. Portman N, Alexandrou S, Carson E, Wang S, Lim E, Caldon CE. Overcoming CDK4K/6 inhibitor resistance in ER-positive breast cancer. Endocr Relat Cancer. (2019) 26:R1R5–30. doi: 10.1530/ERC-18-0317
15. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD0D332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. (2004) 3:1427–38.
16. Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CK4K/6 inhibition to breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. (2012) 11:2756–61. doi: 10.4161/cc.21195
17. Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine and therapeutic interventions. Breast Cancer Res. (2014) 16:207. doi: 10.1186/bcr3r652
18. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991,2991 a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. (2009) 11:R77. doi: 10.1186/bcr2r419
19. Condorelli R, Spring L, O'Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1B mutations and acquired resistance to CDK4K/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. (2018) 29:640–5. doi: 10.1093/annonc/mdx7x84
20. O'Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. (2018) 8:1390–403. doi: 10.1158/2159-8290.CD-18-0264
21. Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget. (2016) 7:68012–22. doi: 10.18632/oncotarget.12010
22. Hurvitz S, Martin M, Press M, Wijayawardana S, Brahmachary M, Ebert PJ, et al. Abstract PD2D-10: treatment with abemaciclib modulates the immune response in gene expression analysis of the neoMONARCH neoadjuvant study of abemaciclib in postmenopausal women with HR+, HER2R negative breast cancer. Cancer Res. (2019) 79(Suppl. 4):Abstract nr PD2-10. doi: 10.1158/1538-7445.SABCS18-PD2-10
23. Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1E expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol. (2019) 37:1169–78. doi: 10.1200/JCO.18.00925
24. Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4K/6 inhibitors via the hippo pathway. Cancer Cell. (2018) 34:893–905. doi: 10.1016/j.ccell.2018.11.006
25. Taylor-Harding B, Aspuria PJ, Agadjanian H, Cheon DJ, Mizuno T, Greenberg D, et al. Cyclin E1E and RTK/RAS signalling drive CDK inhibitor resistance via activation of E2EF22F and ETS. Oncotarget. (2015) 6:696–714. doi: 10.18632/oncotarget.2673
26. Knudsen ES, Witkiewicz AK. The strange case of CDK4K/6 inhibitors: mechanisms, resistance and combination strategies. Trends Cancer. (2017) 3:39–55. doi: 10.1016/j.trecan.2016.11.006
27. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early adaption and acquired resistance to CDK4K/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. (2016) 76:2301–13. doi: 10.1158/0008-5472.CAN-15-0728
28. Ma CX, Gao F, Luo J, Northfelt DW, Goetz M, Forero A, et al. NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor-positive breast cancer. Clin Cancer Res. (2017) 23:4055–65. doi: 10.1158/1078-0432.CCR-16-3206
29. Hortobagyi GN, Paluch-Shimon S, Petrakova K, Villaneuva C, Chan A, Nusch A, et al. First-line ribociclib (RIB) + letrozole (LET) in hormone receptor-positive (HR+), HER2R-negative (HER2R-) advanced breast cancer (ABC): MONALEESA-2 biomarker analyses. J Clin Oncol. (2018) 36(Suppl. 15):1022. doi: 10.1200/JCO.2018.36.15_suppl.1022
30. Finn RS, Yiu Y, Martin M, et al. Abstract P2P-09-10: comprehensive gene expression biomarker analysis of CDK4K/6 and endocrine pathways from the PALOMA-2 study. Cancer Res. (2018) 78 (Suppl. 4):P2-09-10-P2-09-10. doi: 10.1158/1538-7445.SABCS1S7-P2P-09-10
31. Guarducci C, Bonechi M, Benelli M, Biagioni C, Boccalini G, Romagnoli D, et al. Cyclin E1E and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer. (2018) 4:38. doi: 10.1038/s4s1523-018-0092-4
32. Gillett C, Fantl V, Smith R, Risher C, Bartek J, Dickson C, et al. Amplification and overexpression of cyclin D1D in breast cancer detected by immunohistochemical staining. Cancer Res. (1994) 54:1812–17.
33. DeMichele A, Clare AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK4K/6 inhibitor palbociclib (PD0D332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. (2015) 21:995–1001. doi: 10.1158/1078-0432.CCR-14-2258
34. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2R-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. (2015) 16:25–35. doi: 10.1016/S1S470-2045(14)71159-3
35. Neven P, Petrakova K, Val Bianchi G. Abstract PD2D-05: biomarker analysis by baseline circulating tumor DNA alternations in the MONALEESA-3 study. Cancer Res. (2019) 79 (Suppl. 4):PD2-05-PD2-05. doi: 10.1158/1538-7445.SABCS1S8-PD2D-05
36. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr Biol. (1997) 7:261–9. doi: 10.1016/S0S960-9822(06)00122-9
37. Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee K-M, Hutchinson KE, et al. Kinome-wide RNA interference screen reveals a role for PDK1K in acquired resistance to CDK4K/6 inhibition in ER-positive breast cancer. Cancer Res. (2017) 77:2488–99. doi: 10.1158/0008-5472.CAN-16-2653
38. Topolcan O, Holubec L. The role of thymidine kinase in cancer diseases. Expert Opin Med Diagn. (2008) 2:129–41. doi: 10.1517/17530059.2.2.129
39. Romain S, Spyratos F, Descortes F, Daver A, Rostaing-Puissant B, Bougnoux P, et al. Prognostic role of DNA-synthesising enzyme activity (thymidine kinase and thymidylate synthase) in 908 T1T-T2T N0N-N1N, M0M breast cancers: a retrospective multicenter study. Int J Cancer. (2000) 87:860–8.
40. Broët P, Romain S, Daver A, Ricolleau G, Quillien V, Rallet A, et al. Thymidine kinase as a proliferative marker: clinical relevance in 1692 primary breast cancer patients. J Clin Oncol. (2001) 19:2778–87. doi: 10.1200/JCO.2001.19.11.2778
41. Robertson JF, O'Neill KL, Thomas MW, McKenna PG, Blamey RW. Thymidine kinase in breast cancer. Br J Cancer. (1990) 62:663–7. doi: 10.1038/bjc.1990.352
42. Weagel EG, Burrup W, Kovtun R, Velazquez EJ, Felsted AM, Townsend MH, et al. Membrane expression of thymidine kinase 1 and potential clinical relevance in lung, breast and colorectal malignancies. Cancer Cell Int. (2018) 10:135. doi: 10.1186/s1s2935-018-0633-9
43. Nisman B, Allweis T, Kadouri L, Mali B, Hamburger T, Baras M, et al. Comparison of diagnostic and prognostic performance of two assays measuring thymidine kinase 1 activity in serum of breast cancer patients. Clin Chem Lab Med. (2013) 51:439–47. doi: 10.1515/cclm-2012-0162
44. Bolayirli M, Papila C, Korkmaz GG, Papila B, Aydogan K, Karatas A, et al. Serum thymidine kinase 1 activity in solid tumour (breast and colorectal cancer) patients treated with adjuvant chemotherapy. J Clin Lab Anal. (2013) 27:220–6. doi: 10.1002/jcla.21587
45. Bjohle J, Bergqvist J, Gronowitz JS, Johansson H, Carlsson L, Einbeigi Z, et al. Serum thymidine kinase activity compared with CA 15-3 in locally advanced and metastataic breast cancer within a randomised trial. Breast Cancer Res Treat. (2013) 139:751–8. doi: 10.1007/s1s0549-013-2579-x
46. Bonechi M, Galardi F, Biagioni C, De Luca F, Bergqvist M, Neumüller M, et al. Plasma thymidine kinase-1 predicts outcome in patients with hormone receptor positive and HER2R negative metastatic breast cancer treated with endocrine therapy. Oncotarget. (2018) 27:16389–99. doi: 10.18632/oncotarget.24700
47. Chia S, Gradishar W, Bauriac L, Bines J, Amant F, Federico M, et al. Double-blind, randomized placebo-controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: Results from EFECT. J Clin Oncol. (2008) 26:1664–70. doi: 10.1200/JCO.2007.13.5822
48. McCartney A, Biagioni C, Schiavon G, Bergqvist M, Mattsson K, Miagliaccio I, et al. Prognostic role of serum thymidine kinase 1 activity in patients with hormone receptor-positive metastatic breast cancer: analysis of the randomised phase III Evaluation of Faslodex versus Exemestane Clinical Trial (EFECT). Eur J Cancer. (2019) 114:55–66. doi: 10.1016/j.ejca.2019.04.002
49. Bagegni N, Thomas S, Liu N, Luo J, Hoog J, Dorthfelt DW, et al. Serum thymidine kinase 1 activity as a pharmacodynamic marker of cyclin-dependent kinase 4/6 inhibition in patients with early-stage breast cancer receiving neoadjuvant palbociclib. Breast Can Res. (2017) 19:123. doi: 10.1186/s1s3058-017-0913-7
50. Bonechi M, Migliaccio I, Benelli M, Romagnoli D, Bergqvist M, Mattsson K, et al. Effects of palbociclib on thymidine kinase-1 (TK1K) in hormone receptor positive (HR+) breast cancer cell lines. Cancer Res. (2019) 79 (Suppl. 4):P6-09-02-P6-09-02. doi: 10.1158/1538-7445.SABCS1S8-P6P-09-02
51. Malorni L, Curigliano G, Minisini AM, Cinieri S, Tondini CA, D'Hollander K, et al. Palbociclib as single agent or in combination with the endocrine therapy received before disease progression for estrogen receptor-positive, HER2R-negative metastatic breast cancer: TREnd trial. Ann Oncol. (2018) 29:1748–54. doi: 10.1093/annonc/mdy2y14
52. Malorni L, Biagioni C, De Luca F, Bonechi M, Curigliano G, Minisini AM, et al. Abstract 4416/10 plasma thymidine kinase activity in patients with luminal metastatic breast cancer treated with palbociclib within the phase II TREnd trial. In: AACR Annual Meeting (Atlanta, GA).
53. Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, et al. p1p6-CDK4K-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0D332991 in glioblastoma xenograft cells. Neuro Oncol. (2012) 14:870–81. doi: 10.1093/neuonc/nos1s14
54. André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase III SOLAR-1 trial. Ann Oncol. (2018) 29:abstract LBA3_PR. doi: 10.1093/annonc/mdy4y24.010
55. Juric D, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib + fulvestrant for advanced breast cancer: subgroup analyses from the phase III SOLAR-1 trial. Cancer Res. (2019) doi: 10.1158/1538-7445.SABCS1S8-GS3S-08. [Epub ahead of print].
56. Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2R-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2018) 19:P87–100. doi: 10.1016/S1S470-2045(17)30688-5
57. Baselga J, Dent SF, Cortés J, Young-Hyuck I, Diéras V, Harbeck N, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULVE in patients (pts) with estrogen receptor (ER)-positive, PIK3KC33CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clin Oncol. (2018) 36:18. doi: 10.1200/JCO.2018.36.18_suppl.LBA1006
Keywords: CDK4/6 inhibitors, biomarker, thymidine kinase-1, PIK3CA, resistance, palbociclib, ribociclib, abemaciclib
Citation: McCartney A, Migliaccio I, Bonechi M, Biagioni C, Romagnoli D, De Luca F, Galardi F, Risi E, De Santo I, Benelli M, Malorni L and Di Leo A (2019) Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 9:666. doi: 10.3389/fonc.2019.00666
Received: 10 May 2019; Accepted: 08 July 2019;
Published: 23 July 2019.
Edited by:
Mothaffar Rimawi, Baylor College of Medicine, United StatesReviewed by:
Tomás Pascual Martinez, Hospital Clínic de Barcelona, SpainNuria Chic, Hospital Clínic de Barcelona, Spain
Copyright © 2019 McCartney, Migliaccio, Bonechi, Biagioni, Romagnoli, De Luca, Galardi, Risi, De Santo, Benelli, Malorni and Di Leo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Di Leo, YW5nZWxvLmRpbGVvQHVzbGNlbnRyby50b3NjYW5hLml0