
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 17 May 2019
Sec. Cancer Molecular Targets and Therapeutics
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00417
 Danielle Golub1,2*
Danielle Golub1,2* Nishanth Iyengar3
Nishanth Iyengar3 Siddhant Dogra3Taylor Wong1Devin Bready1Karen Tang2,4Aram S. Modrek5Dimitris G. Placantonakis1,6,7,8,9*
Siddhant Dogra3Taylor Wong1Devin Bready1Karen Tang2,4Aram S. Modrek5Dimitris G. Placantonakis1,6,7,8,9*The identification of heterozygous neomorphic isocitrate dehydrogenase (IDH) mutations across multiple cancer types including both solid and hematologic malignancies has revolutionized our understanding of oncogenesis in these malignancies and the potential for targeted therapeutics using small molecule inhibitors. The neomorphic mutation in IDH generates an oncometabolite product, 2-hydroxyglutarate (2HG), which has been linked to the disruption of metabolic and epigenetic mechanisms responsible for cellular differentiation and is likely an early and critical contributor to oncogenesis. In the past 2 years, two mutant IDH (mutIDH) inhibitors, Enasidenib (AG-221), and Ivosidenib (AG-120), have been FDA-approved for IDH-mutant relapsed or refractory acute myeloid leukemia (AML) based on phase 1 safety and efficacy data and continue to be studied in trials in hematologic malignancies, as well as in glioma, cholangiocarcinoma, and chondrosarcoma. In this review, we will summarize the molecular pathways and oncogenic consequences associated with mutIDH with a particular emphasis on glioma and AML, and systematically review the development and preclinical testing of mutIDH inhibitors. Existing clinical data in both hematologic and solid tumors will likewise be reviewed followed by a discussion on the potential limitations of mutIDH inhibitor monotherapy and potential routes for treatment optimization using combination therapy.
The discovery of mutations in isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) in over 80% of low-grade gliomas (LGGs) and secondary glioblastomas has revolutionized pharmaceutical approaches to targeted therapies and the overall glioma classification schema (1, 2). Driver mutations in IDH1 and IDH2 have been likewise identified in acute myeloid leukemia (AML), chondrosarcoma, myelodysplastic syndromes, and cholangiocarcinoma (3–6). Limitations in current treatment options, particularly in LGG and AML, due to both inefficacy and systemic toxicity, make mutant IDH (mutIDH), and its associated molecular pathways attractive therapeutic targets (7–9). Major strides in developing and testing candidates for mutIDH inhibition have been made in the past few years with the FDA approvals of Ivosidenib (Tibsovo®) and Enasidenib (Idhifa®), selective mutIDH1 and mutIDH2 inhibitors, respectively (10, 11). While these agents have had some preliminary success in AML, utility in the treatment of IDH-mutant glioma or other IDH-mutated cancers has not been established (12, 13).
IDH1 and IDH2 are homodimeric isoenzymes involved in a major pathway for cellular NADPH generation through the oxidative decarboxylation of isocitrate to α-ketoglutarate. IDH1 is found in the cytosol and in peroxisomes, while IDH2 is a mitochondrial enzyme. Mutations in IDH3 isoforms, which form heterotetrameric complexes in mitochondria, are rarely seen in cancer, but there is some evidence that upregulation of wild-type IDH3 may contribute to various tumorigenic metabolic pathways (14, 15). The IDH1/2 mutations are heterozygous and neomorphic in that they establish a pathway for the NADPH-dependent conversion of the wild-type IDH product, α-ketoglutarate, to 2-hydroxyglutarate (2HG) (16). Simultaneously, significant decreases in NADPH production are also seen (17). Early structural and pharmacokinetic studies show that mutant IDH develops an increased affinity for both the cofactor NADPH and substrate α-ketoglutarate (16, 18). In the most common IDH1/2 mutants, the wild-type IDH function of oxidative decarboxylation of isocitrate to α-ketoglutarate is lost due to mutation of critical amino acid residues in the catalytic domain, IDH1 R132 and IDH2 R172, which are normally responsible for binding the β-carboxyl group of isocitrate and initiating catalysis (1, 16, 18). Interestingly, there is some evidence that, unlike the IDH1 mutant, the IDH2 mutant may not depend on heterodimerization with an IDH wild-type partner for 2HG production (19). Nevertheless, while the mutant IDH enzyme can exist either as a homodimer or as a heterodimer with the wild-type IDH within cancer cells, all reported oncogenic IDH mutations to date are genetically heterozygous, suggesting that the critical role of mutant IDH is related to its gain-of-function for conversion of the wild-type IDH product, α-ketoglutarate, to 2HG (20).
Accumulation of 2HG, increasingly well-characterized as an oncometabolite, disrupts multiple regulatory cellular pathways involving α-ketoglutarate-dependent dioxygenases including those involved in epigenetic remodeling and DNA repair (Figure 1) (21–23). Structural similarities between α-ketoglutarate and 2HG allow the latter to competitively occupy the same pockets as α-ketoglutarate in α-ketoglutarate-dependent dioxygenases (of which over 60 have been described in humans), without promoting enzymatic activation (22, 24–26). Changes in the epigenetic landscape brought on by 2HG-mediated disruption of the ten-eleven translocation (TET) family of 5-methylcytosine (5 mC) hydroxylases (DNA demethylases) and the JmJC domain-containing histone lysine demethylases (KDMs) are hypothesized to promote oncogenesis through DNA and histone hypermethylation and resultant transcriptional dysregulation (22, 27). The resulting global increase in DNA methylation in the mutIDH context is aptly named the CpG Island Methylator Phenotype (CIMP) (28, 29). Manipulating and reversing the oncogenic IDH-mutant methylome is the primary molecular endpoint for therapeutic IDH inhibition and 2HG reduction in both glioma and AML. It remains to be seen, however, if 2HG reduction alone will be sufficient to reverse oncogenic changes to the methylome, as epigenetic memory persists through daughter cells via methyltransferases, a topic we explore further in our discussion (30, 31).
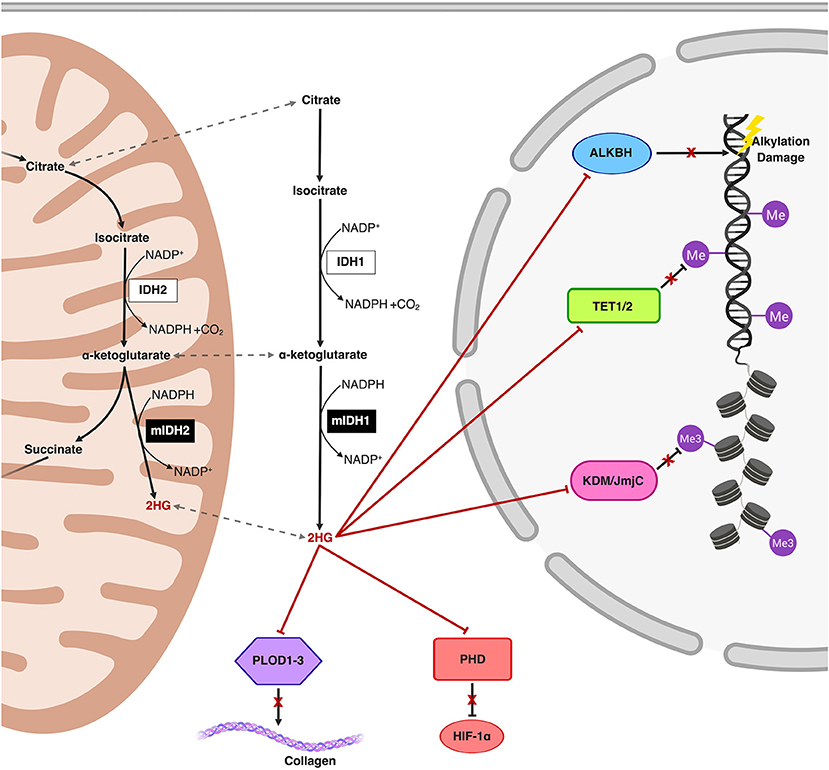
Figure 1. Schematic representation of the mutIDH1 and mutIDH2 pathways and molecular mechanisms related to oncogenesis. The neomorphic enzyme, mutIDH1/2, converts the wild-type IDH product, α-ketoglutarate, to the oncometabolite, 2-hydroxyglutarate (2HG) both in the cytosol and in the mitochondria. 2HG competitively inhibits α-ketoglutarate-dependent dioxygenases both in the cytosol and in the nucleus. 2HG-mediated inhibition of the activity of Ten-Eleven Translocation (TET) enzymes and histone lysine demethylases (KDM) result in global epigenetic modifications on DNA and histones, respectively, resulting in a hypermethylator phenotype. Inhibition of prolyl hydroxylases and lysyl hydroxylases (such as PLOD1-3) interferes with both collagen maturation and with the degradation pathway of Hypoxia-Inducible Factor 1α (HIF-1α), thereby post-translationally stabilizing HIF-1α. Additionally, ALKBH, responsible for repair of oxidative DNA damage, is also inhibited by 2HG, an effect which potentially introduces risk for increased mutational burden.
Here, we provide an overview of the current literature on IDH mutations in cancer with a particular emphasis on glioma and AML and the potential for mutIDH as a therapeutic target in these contexts. We describe the current evidence for the various generations of mutIDH inhibitors through the drug-discovery, preclinical, and clinical stages and systematically review related past and ongoing clinical trials. We furthermore describe the possible adverse effects of IDH inhibitors, such as “differentiation syndrome,” and conclude with a discussion on the potential for enhancing the efficacy of IDH inhibitors in combination with epigenetic modification-based therapies.
Ten years ago, our understanding of the molecular landscape in glioma was transformed by the first genome-wide analysis of somatic mutations in glioblastoma (GBM) and the identification of recurrent mutations in IDH1 nearly exclusively in secondary GBM (2). Mutations in IDH1 and IDH2 are seen in over 80% of lower-grade gliomas (WHO grades II and III) and secondary GBMs that are thought to later develop from lower-grade lesions (2, 32, 33). The vast majority of somatic IDH mutations (>95%) are seen in IDH1, and the most commonly observed IDH1 mutation occurs at the R132 residue (1, 34). IDH2 mutations, which are mutually exclusive with those in IDH1 and found at a functionally analogous R172 residue, only represent a minority of somatic IDH mutations in glioma (35, 36).
IDH-mutant gliomas are generally further categorized into two major subtypes: those with chromosome 1p/19q co-deletion, historically termed oligodendrogliomas; and those without 1p/19q co-deletion, also known as astrocytomas (37). These two groups are biologically and clinically distinct. Up to 94% of IDH-mutant non-1p/19q co-deleted gliomas harbor loss-of-function TP53 mutations and 86% have inactivating ATRX mutations (37). Only few IDH mutant astrocytomas carry IDH wild-type driver mutations or copy number alterations, and those who do (for example CDKN2A or CDKN2B loss) are usually classified as IDH mutant GBM (1). These robust genomic differences are highly suggestive of a unique mechanism of oncogenesis in the IDH-mutant subgroup and furthermore imply that the IDH mutation is likely an early player in a cell-of-origin, which in its native state is capable of giving rise to both astrocyte and oligodendrocyte lineages. Clinically, IDH-mutant lesions present in a younger age group (median age in the fourth vs. the sixth decade of life), when compared to IDH-wild type gliomas (33). Furthermore, IDH mutations are well-known to be an independent favorable prognostic factor at all stages of glioma progression; for example, the median survival in IDH-mutant GBM is 31 months, over twice as long as the median 15-months survival in the wild-type counterpart (1).
Consistent with other IDH-mutant cancers, IDH-mutant glioma is characterized by high levels of 2HG and the resulting “CIMP” hypermethylator phenotype described previously. In glioma specifically, these genome-wide DNA methylation changes have been shown to establish “insulator dysfunction” or disruption of topologically-associated domains (TADs) and thereby directly influence key transcriptional regulatory pathways related to gliomagenesis (38, 39). As previously mentioned, analyses of clonality among glioma tumor samples suggests that the IDH mutation is a tumor-initiating event in a common progenitor cell, hypothesized by many to be derived from the subventricular zone stem cell niche (7, 40–42). Despite our enhanced understanding of the molecular pathogenesis of IDH-mutant glioma, however, effective treatments have yet to be developed and clinicians remain reliant on maximal safe surgical resection and various chemotherapeutic agents and radiation treatments to prolong survival (7). Furthermore, a unique characteristic of LGG is its diffuse and highly infiltrative phenotype, making surgical resection rarely curative in the long term. To compound the complexity of these tumors, and historically popular chemotherapeutic agents have been shown to induce hypermutant recurrent tumors (7). Recent efforts in developing small molecule inhibitors that target IDH mutation provide a new opportunity for progress in glioma treatment.
Around the same time as the identification of recurrent IDH mutations in glioma, Mardis et al. published the results of a landmark study in which they sought to pinpoint recurrent mutations in AML that may be associated with the pathogenesis of the malignancy (43). In this study, the investigators identified for the first time the presence of IDH1 mutations in AML (43). 8.5% of analyzed samples had an IDH1 mutation at the R132 residue (mutated to either cysteine, histidine, or serine), which is also the site of the overwhelming majority of somatic IDH mutations in glioma (1, 34). Shortly after the discovery of IDH1 mutations in AML, another landmark study reported the first case of IDH2-mutated AML, in which the R172 residue was mutated to lysine (18). Further investigation of AML DNA samples revealed the existence of several additional cases of AML where the IDH2R172 residue was mutated (18). Interestingly, this study also found that a majority of the analyzed samples had IDH2 mutations (compared to IDH1 mutations) (18). This is in stark contrast to glioma, where the majority of IDH mutations are in IDH1.
Nearly one in five cases of AML is IDH-mutant, with IDH2-mutant AML being more prevalent than IDH1-mutant AML (11–13, 44–50). The IDH2R140 mutation (in particular, the R140Q variant) is the most common, with the IDH1R132 and IDH2R172 mutations also appearing frequently in the literature (3, 12, 45, 46, 50–52). Other mutations include, but are not limited to, IDH1V71 and IDH1 SNP rs11554137, a GGC to GGT transversion at the glycine residue at codon position 105 with unknown significance (47, 48, 53). Clinical and pathologic characteristics associated with IDH-mutant AML include normal karyotype (intermediate-risk cytogenetics), increased patient age, elevated platelet count, increased bone marrow blast percentage at initial presentation, increased peripheral blast percentage, decreased absolute neutrophil count (especially in IDH1-mutant AML), and concurrent mutations such as NPM1 and FLT3-ITD (44–47, 54). IDH1 and IDH2 mutations in AML are mutually exclusive, as in glioma. Likewise, in AML, IDH mutations are almost entirely mutually exclusive with TET2 mutations, suggesting that, mechanistically, these genes aref both involved in DNA hypermethylation as a driver of leukemogenesis (3, 45–47, 54).
It has been suggested that testing AML patients for IDH mutation status is simple and should be performed universally; however, the relationship between IDH mutation status and prognosis is considerably less clear and more controversial in AML than it is in other cancers such as glioma (46, 55). Most studies of IDH-mutant AML have suggested that mutIDH either foreshadows an adverse prognosis (given an association with increased blast percentage and older age at diagnosis) or is of little prognostic value (45, 48, 52, 55). Reported 2–3-years overall survival in IDH-mutant AML ranges between 51 and 89% in the literature; discrepancies are thought to be related to differences in cohort age, but some authors also argue that different specific IDH mutations may carry varied prognostic implications (3, 44, 45, 47, 54, 56, 57). Interestingly, IDH mutation status may also be useful for the detection of residual disease and prognostication following treatment; several studies investigating the value of serum 2HG during remission in AML have found that elevated serum 2HG levels actually predict shortened overall survival (55, 58, 59).
While induction/consolidation chemotherapy has revolutionized AML treatment strategy in the past 20 years, this standard-of-care universal treatment has evolved minimally since its introduction and is often contraindicated in elderly or otherwise frail patients (44). Given our enhanced understanding of the molecular and genetic subtypes of AML and the potential for targeted treatment, manipulation of these markers with small molecules may provide significant benefit. Drugs targeted to the mutIDH isotypes are one such example; for almost a decade, mutIDH inhibitors have been a focus of laboratory and clinical research in AML with great recent success leading to two FDA approvals specifically for AML indications.
Multiple mutIDH inhibitors, including one pan-inhibitor and several specific to one mutIDH isoform, have been developed over the last several years. A handful of these are in use in clinical trials, but only two have been approved by the FDA; Enasidenib and Ivosidenib (10, 11). A detailed review of the structural and pharmacokinetic properties and relevant preclinical data for both FDA approved inhibitors will follow a brief discussion of other mutIDH inhibitors with demonstrated and repeated preclinical efficacy (Table 1).
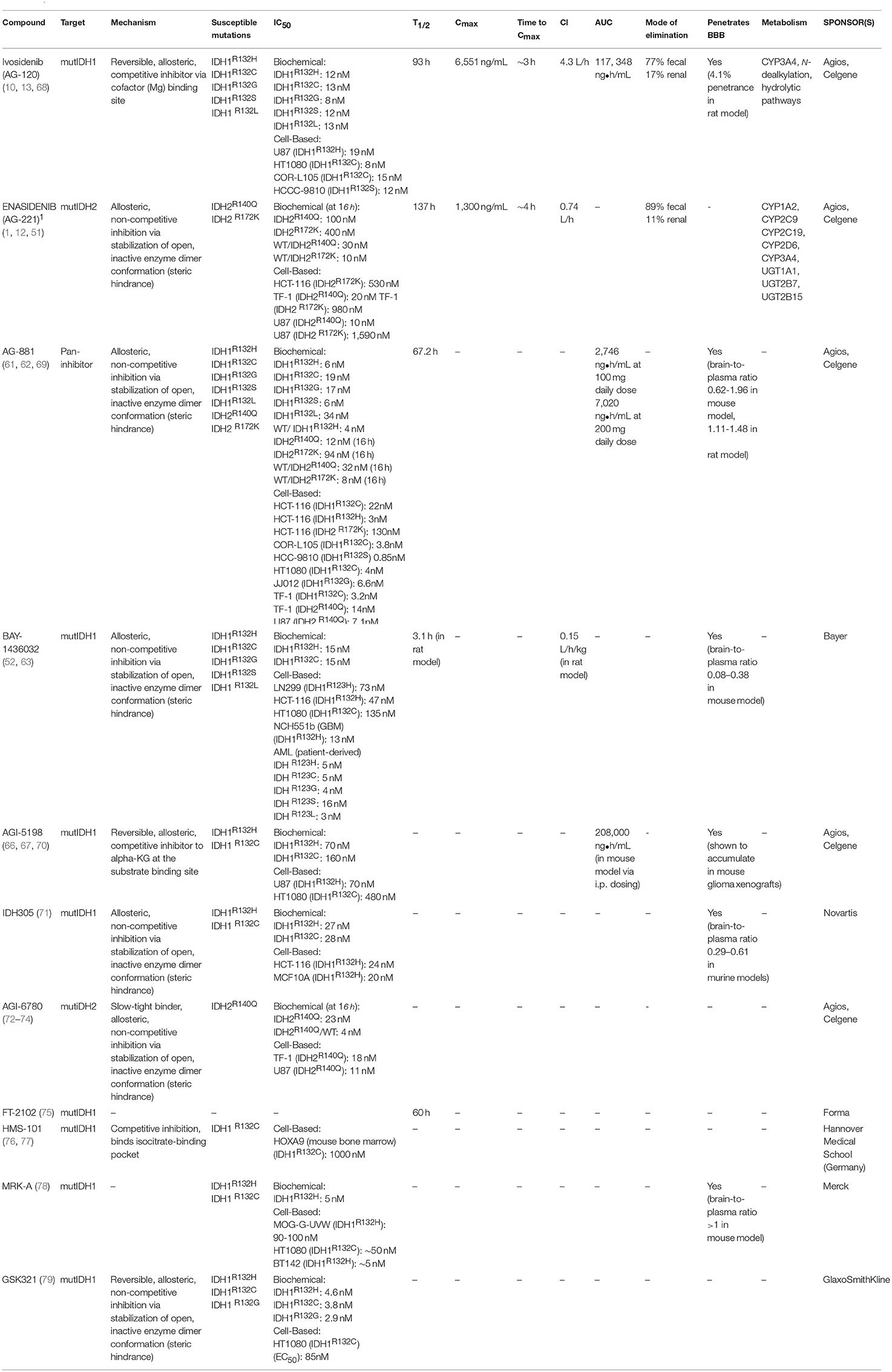
Table 1. MutIDH inhibitors.
AG-881 (Vorasidenib) is an orally available pan-inhibitor of both mutIDH1 and mutIDH2 and was the first pan-inhibitor developed under the Celgene and Agios Pharmaceuticals collaboration (Figure 2) (60–62). AG-881 contains a triazine moiety responsible for its allosteric inhibitory activity, and crystallography studies show that AG-881 binds mutIDH1 and mutIDH2 using the same allosteric pocket at the dimer interface, causing steric hindrance that locks the enzymes in an open, inactive conformation (61). Notably, the association of AG-881 with mutIDH1, in particular with IDH1R132H, is more efficient than its interaction with mutIDH2 as it achieves maximal potency in vitro after significantly shorter incubation periods (61). IC50 for inhibition of 2-HG formation following 1 h of preincubation ranged from 6 to 34 nM in both patient-derived and genetically- engineered cell lines expressing IDH1R132C, IDH1R132G, IDH1R132H, IDH1R132L, or IDH1R132S. For U87 and TF-1 cells transfected with IDH2R140Q or IDH2R172K by lentiviral vector, the IC50 values following 1 h of preincubation were 118 nM and 32 nM, respectively (62). In the same study, it was demonstrated that ex vivo treatment of primary human AML blasts with AG-881 induced myeloid differentiation (62). AG-881 has also been shown to effectively penetrate the blood-brain barrier in rodents, implicating its potential to treat both IDH-mutant AML and glioma patients (62). Based on this preclinical evidence, two multicenter clinical trials investigating the safety and efficacy of AG-881, one in solid tumors and the other in hematologic malignancies, are currently ongoing (60, 61).
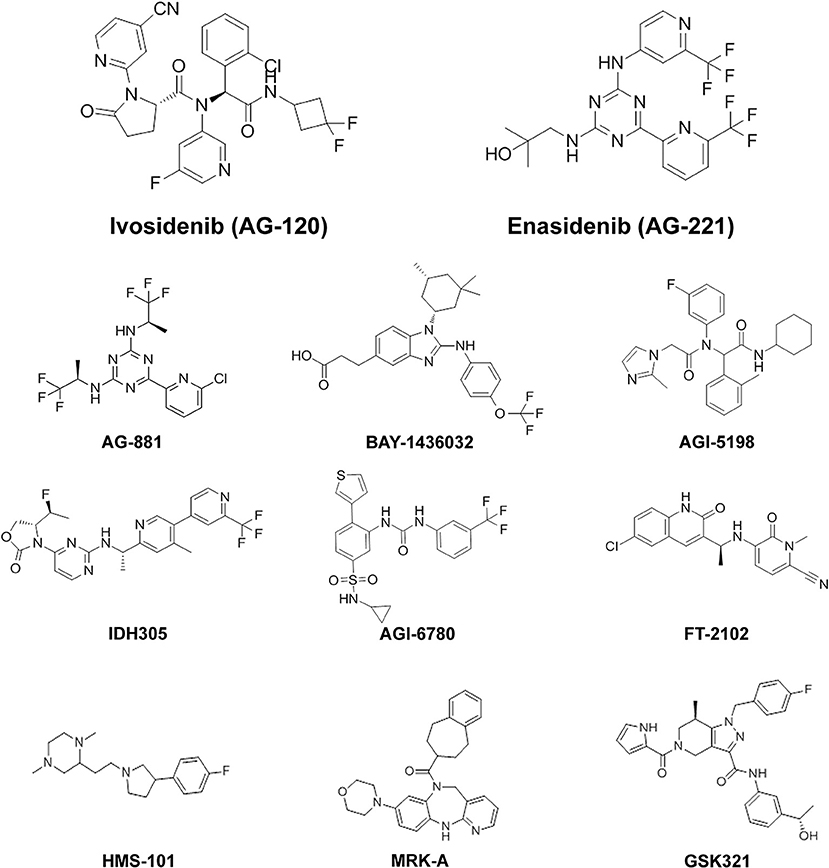
Figure 2. Chemical structures of mutIDH inhibitor compounds reviewed. MutIDH1 inhibitors: Ivosidenib (AG-120), BAY-1436032, AGI-5198, IDH305, FT-2102, HMS-101, MRK-A, GSK321. MutIDH2 inhibitors: Enasidenib (AG-221), AGI-6780. Pan-inhibitors: AG-881.
One of the first mutIDH1-specific inhibitors to show preclinical efficacy in both AML and glioma models is BAY-1436032, developed by Bayer. An initial screen of over 3 million compounds based on mutIDH enzymatic activity generated a small group of compounds—with IC50 ranging from 0.6 to 17.1 μM—for further evaluation. Optimization of a lead compound based on differential inhibition of mutIDH1 and wild-type IDH1 enzymes resulted in BAY-1436032, an allosteric inhibitor that binds at the IDH dimer interface (Figure 2) (63). Interestingly, BAY-1436032 demonstrates potent inhibition of all known IDH1R132 mutants with nearly equal efficacy in both human-derived AML cells (IC50 3–16 nM) and genetically engineered cell lines representative of solid tumors (IC50 13–135 nM) (52, 63). Additionally, reduced proliferation and induction of differentiation was seen in vitro in both IDH-mutant AML and glioma cell lines. In AML cell lines, BAY-1436032 demonstrated some efficacy in reducing histone methylation as well, but multiple studies have failed to show changes in histone or DNA methylation status in glioma models (52, 63). In vivo, however, BAY-1436032 effectively penetrates the blood-brain barrier and has shown prolonged survival in mice with IDHR132H astrocytoma xenographs (52, 64). Two dose escalation and expansion phase I trials, for AML and solid tumors (including glioma) respectively, are currently ongoing but initial results have yet to be reported (ClinicalTrials.gov NCT03127735, NCT02746081).
A collaboration between Celgene Corporation and Agios Pharmaceuticals for research in cancer metabolism-based therapeutics starting in early 2010 has generated a host of mutIDH inhibitors through a high-throughput screening campaign (65). One of the earliest, and most well-studied mutIDH1-specific inhibitors is AG-5198, a phenyl-glycine-based compound (Figure 2) (66). In early characterization studies, AGI-5198 showed up to 90% 2HG reduction in IDH1R132H U87 xenografts (IC50 0.07 μM in vitro) (66, 67). Rohle et al. re-demonstrated the efficacy of AG-5198 in inhibiting 2HG production in patient-derived glioma xenografts and additionally showed that AGI-5198 promotes expression of markers for differentiation, decreases cellular proliferation and decreases histone methylation in the same cell line. However, based on methylation array data, global DNA methylation contributing to the glioma CIMP phenotype was notably unchanged after AGI-5198 treatment (67). A key study by Johannessen et al. using AGI-5198 in an inducible mutIDH1 knock-in human astrocyte model by Johannessen et al. puts these mixed findings into larger context: mutIDH1 inhibition appears to have a small effective time frame, since the role in of mutIDH in gliomagenesis likely changes from “driver” to “passenger.” (80). Early and persistent exposure to AGI-5198 prior to inducing mutIDH1 resulted in reduced 2HG, blocked histone modifications such as methylation, and decreased cellular proliferation; however, just 4 days after the oncogenic insult, the drug was rendered incapable of reversing or blocking the genetic and phenotypic changes rendered by mutIDH (80). Work by Tateishi et al. complements these findings: In multiple patient-derived IDH1R132H glioma tumorsphere lines, treatment with the S-enantiomer of AGI-5198 counterintuitively resulted in modest but consistent increases in cellular proliferation, despite successful depletion of 2HG. Furthermore, it was observed that mice with recurrent mutIDH glioblastoma xenografts had equitable survival and developed similar size tumors between those treated and not treated with the AGI-5198 S-enantiomer (81). The presentation of mixed results in glioma models is likely due to the fact that certain tumorigenic processes are uncoupled from the IDH1 mutation, and further studies into combination treatment (discussed in a later section) may be warranted. Popular hypotheses to explain this changing role of mutIDH include the generation of an epigenetic memory that is indelible by its enzymatic inhibition alone and the accumulation of additional mutations during tumor evolution. The poor pharmacodynamic profile of AGI-5198 due to its rapid metabolism and clearance has precluded its use in clinical trials (68).
Another mutIDH1-specific inhibitor with demonstrated preclinical efficacy is IDH305, a pyrimidin-5-yl-oxazolidine-2-one compound recently developed by Novartis (Figure 2) (71). IDH305 was developed from efforts to optimize Novartis's first published mutIDH inhibitor, IDH889, as this “parent” drug's high intrinsic clearance, high plasma protein binding, and poor solubility posed significant challenges to further clinical development (82). X-ray crystallography reveals that IDH305 binds to an allosteric binding pocket to stabilize the mutIDH1 enzyme in a catalytically inactive conformation (71). In preclinical characterization testing, IDH305 demonstrated efficacious 2HG reduction in an IDH1R132H colorectal cancer cell line (IC50 24 nM), low liver microsomal clearance values, and substantial brain penetrance in murine models (71). IDH305 has moved into clinical testing in humans with IDH-mutant glioma, AML/MDS, and other solid tumors, and phase 1 safety data in all tumor types is promising (ClinicalTrials.gov NCT02381886) (83).
The first mutIDH2-specific inhibitor to come out of development was AGI-6780, developed as part of the Celgene/Agios collaboration (72). AGI-6780 is a urea sulfonamide inhibitor of the IDH2R140Q mutant enzyme specifically and exhibits non-competitive inhibition with respect to substrate, and uncompetitive inhibition with respect to the NADPH cofactor, operating at an allosteric site at the enzyme's dimer interface (Figure 2) (72, 73). In early pharmacokinetic studies, AGI-6780 demonstrated time-dependent potency for 2HG reduction in both mutant homodimer and mutant/wild-type heterodimer enzyme contexts as well as in more organic cellular contexts (IC50 11nM in IDH2R140Q-overexpressing U87 cells) (72). The study of AGI-6780's biological effects is limited to AML-related contexts. The capability for erythropoietin-induced differentiation of TF-1 erythroleukemia cells with IDH2R140Q overexpression is restored after AGI-6780 treatment in vitro, and the same effect was seen in patient-derived primary IDH2-mutant AML blood and bone marrow samples cultured ex vivo (72). Additional studies have demonstrated that AGI-6780 is not only capable of promoting the expression of differentiation markers such as hemoglobin gamma (HBG) and Kruppel-like factor 1 (KLF1), but that it does so by reversing 2HG-induced DNA and histone hypermethylation in AML cellular models—thereby suggesting that AGI-6780 can manipulate key mechanisms of oncogenesis (72, 74). Lack of in vivo evidence and the subsequent development of the broader mutIDH2 inhibitor Enasidenib, discussed below, stunted AGI-6780's further clinical development.
The remaining group of latest-generation compounds described in the literature include FT-2102, HMS-101, MRK-A, and GSK321—all mutIDH1-specific inhibitors (Figure 2). Despite little preclinical information and an as of yet undisclosed mechanism, FT-2102 is in clinical trials as monotherapy and as combination therapy with azacitidine for AML and MDS, with favorable safety and efficacy data in Phase 1/2 (ClinicalTrials.gov NCT02719574) (75). HMS-101 was an early candidate identified by in silico computational screening and validated in vitro in murine bone marrow cells transduced with IDH1R132C to deplete 2HG production with an IC50 of 1μM and in vivo by prolonging survival in a leukemia mouse model (76, 77). MRK-A, developed by Merck, demonstrated effective 2HG reduction in both in vitro and in vivo glioma models, but yielded mixed results in terms of survival response across various patient-derived glioma xenografts and showed a limited effect on cellular proliferation (78). Lastly, GSK321, developed by GlaxoSmithKline, has been shown to decrease intracellular 2HG in primary IDH1-mutant AML cells ex vivo and consequently inhibit cellular proliferation, promote differentiation, and induce global hypomethylation (79). Poor pharmacokinetic properties, especially bioavailability, of GSK321 has limited its clinical use, but modified versions of this compound, such as GSK864, are already undergoing preclinical testing in the developmental pipeline (79, 84).
The first mutIDH inhibitor given FDA approval was Enasidenib (Idhifa®) or AG-221, developed by Celgene Corporation under a license from Agios Pharmaceuticals. Enasidenib is a first-in-class, oral, selective inhibitor of the mutIDH2 enzyme. A precursor form of Enasidenib was originally selected from a high-throughput screen for inhibitors of the most prevalent form of mutIDH2 in AML, IDH2R140Q (47, 51). With triazine substructure-based inhibitory activity, the precursor compound binds mutIDH2 at an allosteric site within the heterodimer interface of the enzyme, much like its predecessor, AGI-6780 (72). X-ray crystallography revealed that this binding at the heterodimer interface forces mutIDH2 to adopt a non-catalytic open conformation, a mechanism of inhibition consistent with conformational functional changes also described in IDH1R132H inhibition (51, 85). Modification of peripheral chemical substituents with additional polar moieties optimized the potency, solubility, clearance and bioavailability profiles of the molecule, yielding Enasidenib (Figure 2) (51).
In initial characterization testing, Enasidenib demonstrated time-dependent potency in reducing 2HG levels in biochemical assays with an IC50 of 0.03 μM for the most prevalent oncogenic enzyme, the IDH2R140Q/WT heterodimer, and also for the IDH2R172K/WT enzyme (IC50 = 0.01 μM) (51, 86). Robust 2HG depletion was seen in both in transgenic IDH2R140Q TF-1 erythroleukemia cells and in patient-derived primary AML blast cells with either IDH2R140Q or IDH2R172K mutations (51). Furthermore, treatment of patient-derived blasts with Enasidenib also inhibited cellular proliferation and reversed the histone hypermethylation profile associated with the IDH2 mutation (51). In vivo, within 10 to 20 days, Enasidenib treatment in mouse models with patient-derived IDH2R140Q AML produced differentiated cells expressing markers such as CD11b, CD14, CD15, and CD24 with a decrease in expression of the immature CD117 marker. Enasidenib treatment also significantly prolonged survival in these models (51). These robust preclinical successes supported further development of the molecule, eventually culminating in demonstration of clinical efficacy in relapsed or refractory AML and FDA approval (11, 12).
Ivosidenib (Tibsovo®), the mutIDH1-specific counterpart to Enasidenib, was developed (also under the Celegene/Agios cancer metabolome research license agreement) out of efforts to optimize AGI-5198 for human therapeutic applications (10). Poor pharmacokinetics of AGI-5198, in particular its prohibitively high metabolic clearance rate, precluded its use in clinical studies despite some success in preclinical testing (66–68). Broad structure-activity relationship profiling directed the substitution of fluorinated cycloalkyl groups in place of cyclohexyl moieties, preventing extensive oxidation of the molecule in the human liver microsome (68). The further addition of a pyrimidine ring restored enzymatic potency after initial modifications, resulting in a metabolically-optimized prototype, AG-14100 (68). However, assessment in early screening however revealed that AG-14100 was a relatively potent cytochrome P450 (CYP) 3A4-inducer, with approximately 70% of the activity of rifampicin. Further functional group substitutions to increase polarity resulted in a molecule with low CYP 3A4 activation, and adequate clearance and potency profiles: AG-120 (Ivosidenib) (Figure 2) (68).
Ivosidenib is a highly specific, allosteric, reversible inhibitor of mutIDH1. It competes for binding with the enzyme's essential cofactor, the magnesium ion, thereby preventing the formation of a catalytically active site (87). It possesses equitable potency across various IDH1R132 mutants in a series of cell lines in a selective manner, showing no inhibition of wild-type or mutIDH2 isoforms (68). Testing in animal models with an intact blood-brain barrier suggests a modest level of brain penetration (4.1%), which is hypothesized to be increased in glioma patients with some intrinsic blood-brain barrier disruption (68). Robust, dose- and time-dependent 2HG depletion has been observed across a host of cell types including human chondrosarcoma cells and mouse-model xenografts, primary human AML myeloblasts, and mutIDH1R132H glioma xenografts (IC50 range 5–13 nM for various IDH1 mutants in vitro) (68, 88, 89). Furthermore, preclinical evidence that Ivosidenib modulates oncogenic properties of cancer cells (in addition to 2HG reduction) has been repeatedly demonstrated by induction of cellular differentiation in AML myeloblasts and through inhibition of cellular migration and invasion in a chondrosarcoma cell line (88–90). These early preclinical successes, followed by phase I clinical trials demonstrating a favorable safety profile, earned Ivosidenib orphan drug status for glioma in January 2018, and its first global approval by the FDA in July 2018 for adults with refractory or relapsed AML (10).
A summary of the completed, ongoing, recruiting, and planned clinical trials of mutIDH inhibitors in AML and other hematological malignancies can be found in Table 2 and will be reviewed below.
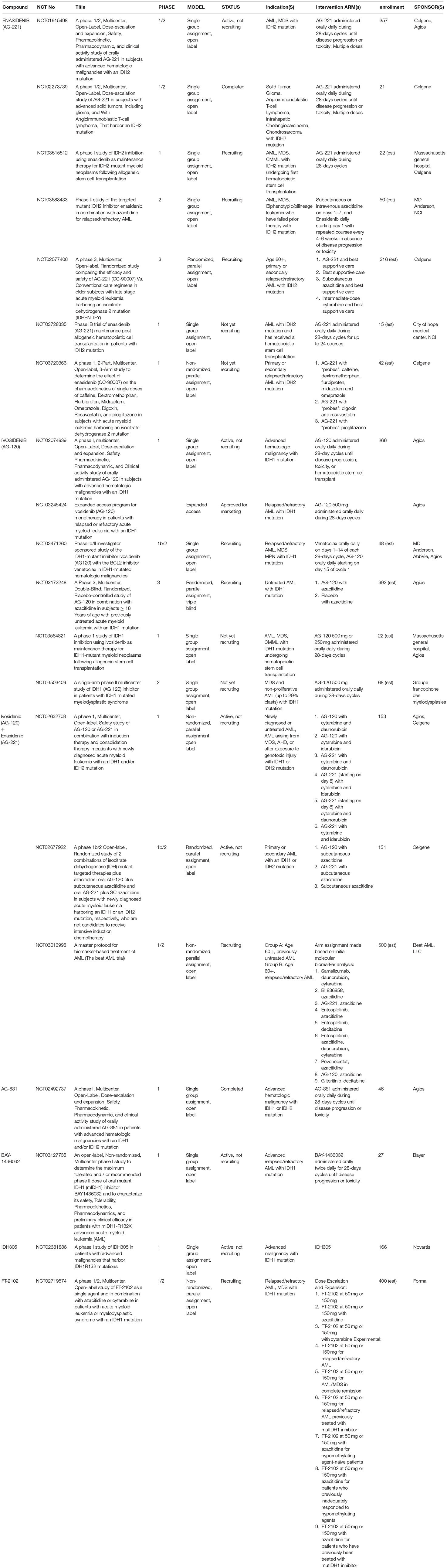
Table 2. Clinical trials in AML/hematologic malignancies.
The safety and efficacy of Enasidenib demonstrated in a phase 1/2 trial in IDH2-mutant advanced myeloid malignancies (including AML and MDS) paved the way for FDA approval of the first small molecule mutIDH inhibitor in cancer and propelled global clinical research efforts for IDH-based targeted cancer treatments (ClinicalTrials.gov NCT01915498) (12). In this landmark first-in-human dose-escalation and expansion study, the pharmacokinetic and pharmacodynamic profiles of Enasidenib were first established along with evidence for clinical efficacy in patients with relapsed/refractory AML. In the initial dose-escalation part of the study, 176 patients were administered doses of Enasidenib ranging from 50 to 650 mg daily in 28-days cycles and a maximum tolerated dose (MTD) was not reached. A 100 mg daily dose was chosen for the 109 patients in the dose expansion arm based on a maximized plasma depletion of 2HG of up to 99% in IDH2R140Q patients and up to 94% in IDH2R172K patients. In patients with relapsed or refractory AML, 38.5% of patients receiving the expansion dose demonstrated a clinical response with a median 5.6 months response duration (8.8 months in those who initially achieved a complete response) (12). Unlike in standard cytoreductive chemotherapy, treatment response occurred without a period of bone marrow hypoplasia, and bone marrow aspirates revealed that responders were characterized by robust myeloid differentiation and trilineage hematopoietic recovery. The most common treatment-related adverse events were indirect hyperbilirubinemia (38%) and nausea (23%), and IDH-inhibitor-associated differentiation syndrome (IDH-DS) was one of the most frequent high-grade adverse events (6%) (12). A similarly designed trial of Enasidenib in IDH2-mutant solid tumors, including glioma, was completed in 2016, but data from the trial has not yet been published (ClinicalTrials.gov NCT02273739).
Since the success of the Enasidenib phase 1/2 trial in myeloid malignancies and FDA approval for the indication of relapsed/refractory IDH2-mutant AML was obtained in 2017, clinical trials investigating the efficacy of mutIDH2 inhibitors, mostly Enasidenib, in various AML subpopulations have become abundant. One, which is actively recruiting, is analyzing the safety of Enasidenib when given in combination with standard AML induction and consolidation chemotherapy in newly diagnosed AML (ClinicalTrials.gov NCT02632708). Another recruiting phase 1 trial is investigating Enasidenib as a maintenance therapy for IDH2-mutant AML or CML following allogeneic hematopoietic stem cell transplantation (ClinicalTrials.gov NCT03515512). Two trials, a phase 1b/2 and a phase 2, are evaluating Enasidenib in combination with a DNA methyltransferase (DNMT) inhibitor, azacitidine, in newly-diagnosed and relapsed/refractory AML, respectively (ClinicalTrials.gov NCT02677922, NCT03683433). Another phase 1b/2 trial is studying biomarker-based treatment of AML more broadly by using genomic screening to identify specific subtypes of AML (including IDH2-mutant) and assign each subtype to a distinct treatment regimen—IDH2-mutant AML will be assigned to Enasidenib treatment (ClinicalTrials.gov NCT03013998). There is one phase 3 randomized, open-label study comparing Enasidenib to conventional care specifically in patients age 60 or older with relapsed/refractory IDH2-mutant advanced AML, also known as IDHENTIFY (ClinicalTrials.gov NCT02577406). The IDHENTIFY protocol was presented at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting (91). A few clinical trials involving Enasidenib are still in the planning stages and are not yet actively recruiting (ClinicalTrials.gov NCT03728335, NCT03720366), and one phase 1 trial investigating pan-inhibitor AG-881 (ClinicalTrials.gov NCT02492737) in advanced hematologic malignancies with either IDH1 or IDH2 mutations concluded in 2018, but no trial results have been released.
Shortly after the approval of Enasidenib, its mutIDH1 counterpart, Ivosidenib, similarly demonstrated safety and clinical efficacy in a phase 1/2 dose-escalation/dose-expansion trial of relapsed/refractory IDH1-mutant AML (ClinicalTrials.gov NCT02074839) (13). In this study, the primary efficacy group included 125 patients with relapsed/refractory IDH1-mutant AML who received 500 mg of Ivosidenib daily for at least 6 months. The 500 mg daily dose was chosen during the dose-escalation phase as it was noted that no additional inhibition of 2HG in plasma was observed at higher doses, and at this dose, a decrease in plasma 2HG to the level of healthy persons was achieved in most patients. In the primary efficacy group, complete remission or complete remission with partial hematologic recovery was seen in 30.4% of patients and the overall response rate was 41.6%. The median duration of overall response was 6.5 months and overall survival was 8.8 months. Interestingly, of the 30.4% of patients with complete remission, 21% had no detectible residual IDH1 mutations on digital polymerase chain reaction assay. These patients had significantly longer durations of remission (11.1 vs. 6.5 months) and overall survival (14.5 vs. 10.2 months) than those who did not clear the mutation (13). However, no preexisting gene mutation or other genetic status significantly predicted treatment success or failure, although increased mutations in receptor tyrosine kinase pathway genes were more strongly associated with non-responders. As in the Enasidenib trials, induction of myeloblast differentiation, as seen on bone marrow aspirates, seemed to drive the clinical efficacy of Ivosidenib. Most frequently observed high-grade treatment-related adverse events included QT prolongation (7.8%) and IDH-DS (3.9%) (13). Shortly after the publication of these results, the FDA approved its second mutIDH inhibitor, Ivosidenib, for IDH1-mutant relapsed or refractory AML (10). Additionally, Ivosidenib now also has an expanded access program for this patient population (ClinicalTrials.gov NCT03245424).
There are several trials studying Ivosidenib in AML in various treatment contexts. The previously mentioned currently recruiting phase 1b/2 study investigating biomarker-based treatment of AML also includes an Ivosidenib arm for those patients with IDH1 mutations (ClinicalTrials.gov NCT03013998). Another trial in the recruitment phase is examining simultaneous treatment with Ivosidenib and the chronic lymphocytic leukemia drug venetoclax in patients with relapsed or refractory AML (NCT03471260). In another currently-recruiting phase 3 study azacitidine is combined with either Ivosidenib or placebo as therapy for previously untreated AML with IDH1 mutations (NCT03173248). This study is planning on enrolling 392 patients, but data as of September 2017 indicated that out of 11 patients who received both Ivosidenib and azacitidine, 8 of them achieved a response, including 4 who had complete remission (92). As mentioned above, another active phase 1 study is examining treatment with either Ivosidenib or Enasidenib, depending on IDH1 vs. IDH2 mutational status, alongside standard induction or consolidation therapy for AML (ClinicalTrials.gov NCT02632708). In preliminary results looking at patients treated with Ivosidenib, 12 of 14 patients with primary AML and 4 of 9 patients with secondary AML achieved either complete remission, complete remission with incomplete platelet recovery, or complete remission with incomplete hematologic recovery (93). Preliminary results of another phase 1b/2 study looking at Ivosidenib with subcutaneous azacitidine in newly diagnosed AML were presented at the 2018 ASCO Annual Meeting; of 23 patients taking both Ivosidenib and azacitidine, 18 (78%) received a response, of whom 10 had complete remission and the median time to response was 1.8 months (ClinicalTrials.gov NCT02677922) (94). There are also a handful of additional trials of Ivosidenib for hematological malignancies that are still in the planning stages (ClinicalTrials.gov NCT03564821, NCT03503409).
Clinical trials experimenting with other mutIDH1 inhibitors in hematologic malignancy have also taken off. An open-label phase 1 trial of BAY-1436032 in patients with IDH1R132-mutant AML started in 2017 with no results published to date (ClinicalTrials.gov NCT03127735). A phase 1/2 study is evaluating the use of FT-2102 alone or with azacitidine in patients with AML or MDS carrying IDH1 mutations (ClinicalTrials.gov NCT02719574). Results presented at the 2018 ASCO Annual Meeting from data on 57 patients (31 treated with FT-2102, 27 treated with both FT-2102 and azacitidine) demonstrated a complete response rate of 38% with monotherapy and 27% with combination therapy (75). Lastly, there is an ongoing phase 1 study looking at IDH305 in patients with diverse IDH1 mutation-harboring malignancies, including glioma, AML/MDS, and other solid tumors (ClinicalTrials.gov NCT02381886). In the AML/MDS cohort (n = 24), complete remission was achieved in 2 patients (9.5%), complete remission with incomplete recovery in 1 patient (4.8%), and partial remission in 4 patients (19%) (83). Notably, early adverse event data from this study has halted clinical development of IDH305 because of dose-limiting toxicities such as hyperbilirubinemia and transaminitis (64).
Several clinical trials studying mutIDH inhibitors in glioma and other solid tumors are ongoing or in planning stages and involve both pan and specific inhibitors (Table 3). Early results from ongoing phase 1 safety and dose-escalation trials continue to be updated annually during national conferences (69, 95–100).
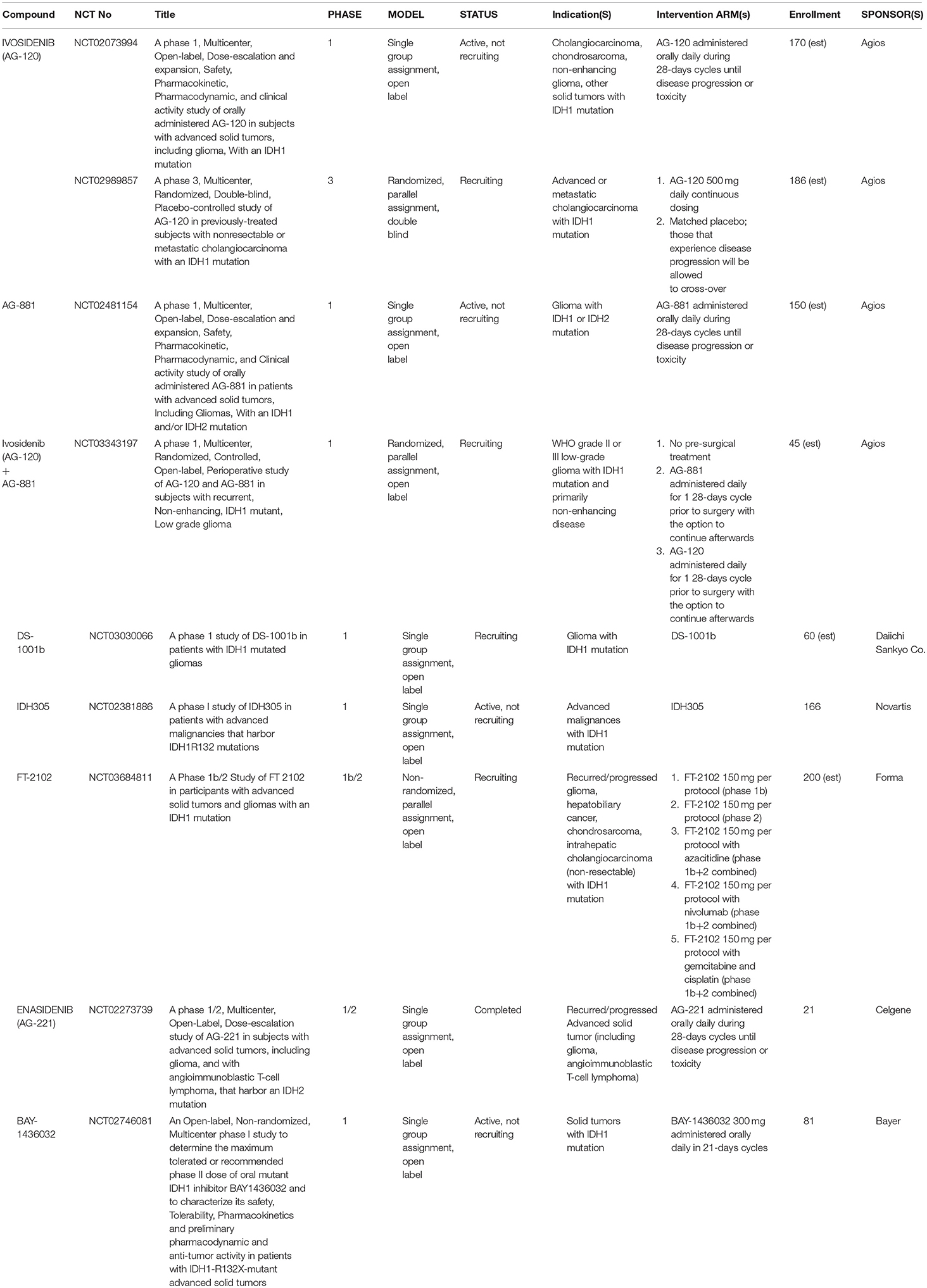
Table 3. Clinical trials in glioma and other solid tumors.
In 2015, the first phase 1 data on the safety of Ivosidenib in patients with advanced glioma and other IDH-mutant solid tumors was presented at the annual EORTC-NCI-AAR Molecular Targets and Cancer Therapeutics Symposium and indicated that Ivosidenib treatment was well-tolerated with a positive pharmacokinetic profile (ClinicalTrials.gov NCT02073994) (95). This multicenter, open-label study was designed as a dose-escalation study with a following dose-expansion cohort. Ivosidenib was administered daily over 28-days cycles at doses ranging from 100 mg up to 1200 mg. Doses beyond 500 mg did not result in increased plasma 2HG reduction; therefore the 500 mg dose was selected for the dose-expansion arms, which included both enhancing and non-enhancing IDH-mutant gliomas (97). Most recent efficacy data reported at the 2016 Society for Neuro-Oncology Annual Meeting indicated a 35% clinical benefit rate (stable disease or better) based on imaging at 6-months follow-up (96). By 2017, 168 patients were enrolled in the dose-escalation arm and 108 patients in the 500 mg daily dose-expansion arm (97). Throughout the study period, Ivosidenib has continued to demonstrate good oral bioavailability, a lengthy half-life (mean 40–102 h after a single dose), and a persistent, robust response in 2HG depletion in both plasma and tumor tissue (97). No treatment-related serious adverse events have been reported; other adverse events included headache, nausea, fatigue, and gastrointestinal symptoms (96).
AG-881 (Vorasidenib), a pan-inhibitor, is the only other mutIDH inhibitor currently with supporting clinical data in glioma. In a similarly designed, phase 1, multicenter, open-label, dose-escalation and expansion trial, the safety and pharmacokinetic profiles of AG-881 is being investigated in both mutIDH1 and mutIDH2 gliomas and other solid tumors (ClinicalTrials.gov NCT02481154) (69, 98). According to the most recent data presented at the 2018 Society for Neuro-Oncology Annual Meeting, 52 glioma patients have received treatment with AG-881 on a 28-days cycle either as part of the dose-escalation arm (dose range 25–300 mg daily) or as part of the dose-expansion arm (10 or 50 mg daily) (69). While preliminary efficacy data has yet to be published, the most frequently observed adverse events included elevation of transaminases (ALT 44.2 and AST 38.5%) and headache (34.6%). Furthermore, at doses >100 mg, five subjects experienced dose-limiting toxicity presenting as liver injury (69). Forthcoming work with AG-881 includes an additional ongoing phase 1 randomized open-label trial evaluating the ability of pre-treatment with either AG-881 or AG-120 to suppress intra-tumoral 2HG levels in surgical pathology specimens as a measure of pharmacological efficacy (ClinicalTrials.gov NCT03343197). This trial will use the 10 or 50 mg daily dosing (69, 98, 101). The protocol for the latter study was likewise recently presented at the 2018 Society for Neuro-Oncology Annual Meeting (101).
Other ongoing clinical trials of mutIDH inhibitor compounds in glioma (currently without early results) are also in phase 1 and are mostly in the early recruitment phase. These studies are evaluating safety and pharmacology of mutIDH-specific inhibitors such as DS-1001b, IDH305, and FT-2012, Enasidenib, and BAY-1436032 (ClinicalTrials.gov NCT03030066, NCT02381886, NCT03684811, NCT02273739, NCT02746081). There is a single phase 3, multicenter, randomized, double-blind study in solid tumors comparing Ivosidenib (500 mg daily) to placebo in advanced or metastatic mutIDH1 cholangiocarcinoma, also known as the “ClarIDHy” trial (ClinicalTrials.gov NCT02989857) (100). The “ClarIDHy” protocol was presented at the 2017 ASCO Annual Meeting based on previous phase 1 trial findings of 6% partial improvement and 56% stable tumor response, and a progression-free survival rate of 40% at 6 months in a similar patient population (ClinicalTrials.gov NCT02073994) (100).
It should be noted that mutIDH inhibitors are not without significant side effects to which patients have differential susceptibilities depending on whether they are being treated for a solid or a hematologic malignancy. Reported adverse events in trials for solid tumors are fewer and far more manageable: In the phase 1 studies for AG-881, the most common adverse events included fatigue (30.8–38.7%), nausea (26.9–35.5%), and dose-dependent transaminase elevation (without elevated bilirubin) (34.4–44.2%) (69, 98). All dose-related toxicities were reversible with dose modification to the eventual expansion arm dose of 50 mg or with a short period of discontinuation (98). Likewise, in the Ivosidenib studies in both glioma and cholangiocarcinoma, the most frequently occurring adverse events included fatigue (41%), nausea (21.8–36%), and diarrhea (16.4–30%) (95, 102). Only one patient in the dose-escalation arm of the cholangiocarcinoma cohort required a dose reduction to the expansion arm dose (1,200 mg reduced to 500 mg) for suspected drug-related muscle cramps (102).
Patients with hematologic malignancy, on the other hand, experience a greater incidence of more critical treatment-related adverse events that are more critical in nature. As previously discussed, 2017 and 2018 saw the respective publications of the results of two landmark clinical trials in AML: first, a phase 1/2 study of the selective IDH2 mutant inhibitor Enasidenib, and second, a phase 1 dose-escalation and dose-expansion study of the selective IDH1 mutant inhibitor Ivosidenib (12, 13). In the Enasidenib trial, approximately four of every five patients had a treatment-related adverse event, the most common overall being indirect hyperbilirubinemia (38%) and nausea (23%). Indirect hyperbilirubinemia was presumed to be the result of a benign, Gilbert syndrome—like mechanism, as no evidence was found in these patients of Enasidenib-induced hepatotoxicity. Additionally, as previously discussed, the IDH-inhibitor—associated differentiation syndrome (IDH-DS) was also one of the most frequent high-grade treatment-related adverse events (6%) (12). Similarly, in the Ivosidenib trial, approximately one in five patients experienced at least one severe adverse event, the most common being QT interval prolongation (7.8%) and IDH-DS (3.9%). Notably, no patients were required to permanently discontinue Ivosidenib as a result of these adverse events and there were no Ivosidenib-related fatalities, but two patients in the phase 1 Enasidenib trial died due to complications of IDH-DS (12, 13). IDH-DS has been reported as a severe adverse effect of interest in AML trials across nearly all of the mutIDH inhibitors, and warrants further discussion (12, 13, 64, 75).
Differentiation syndrome (DS), previously called “retinoic acid syndrome,” was originally described over a quarter-century ago as an adverse event in patients with acute promyelocytic leukemia (APL) undergoing treatment with all-trans retinoic acid (ATRA) (103). This life-threatening syndrome is typically seen within 3 weeks of ATRA treatment initiation and consists primarily of fever and respiratory distress, with other notable findings including weight gain, lower extremity edema, pleural or pericardial effusions, and episodic hypotension. In the originally described cohort, three patients died from complications of “retinoic acid syndrome,” while high-dose dexamethasone administered early in the course of the syndrome was found to be efficacious in treating those patients who survived (103). A similar differentiation syndrome has also been described following arsenic trioxide treatment in APL (104).
IDH-DS can first develop up to several months following the initiation of IDH mutant inhibitor treatment for hematologic malignancy and shares distinctive symptomology with the retinoic acid syndrome: culture-negative fever, rapid weight gain or edema, respiratory symptoms with or without infiltrates, pleural or pericardial effusions, hypotension, and acute renal failure (105, 106). Diagnosis is often challenging because symptomology is also similar to that of leukemic progression (105, 107–109). Shortly after the 2017 phase 1 Enasidenib trial in AML, an independent differentiation syndrome review committee (DSRC) retrospectively analyzed possible cases of IDH-DS, identified distinguishing characteristics of patients who developed IDH-DS, such as fewer previous anticancer therapies and higher baseline peripheral blasts and lactate dehydrogenase levels, and outlined an easy-to-follow protocol for IDH-DS diagnosis and management (105). Similar to treatment for DS in APL, treatment for IDH-DS is dexamethasone 10 mg twice daily until IDH-DS symptoms have significantly improved. If associated with severe pulmonary or renal symptoms, mutIDH inhibitor therapy should be halted until IDH-DS symptom resolution (105). Table 4 summarizes current knowledge and consensus concerning diagnosis and management of DS more generally (103, 105, 106, 110–115).
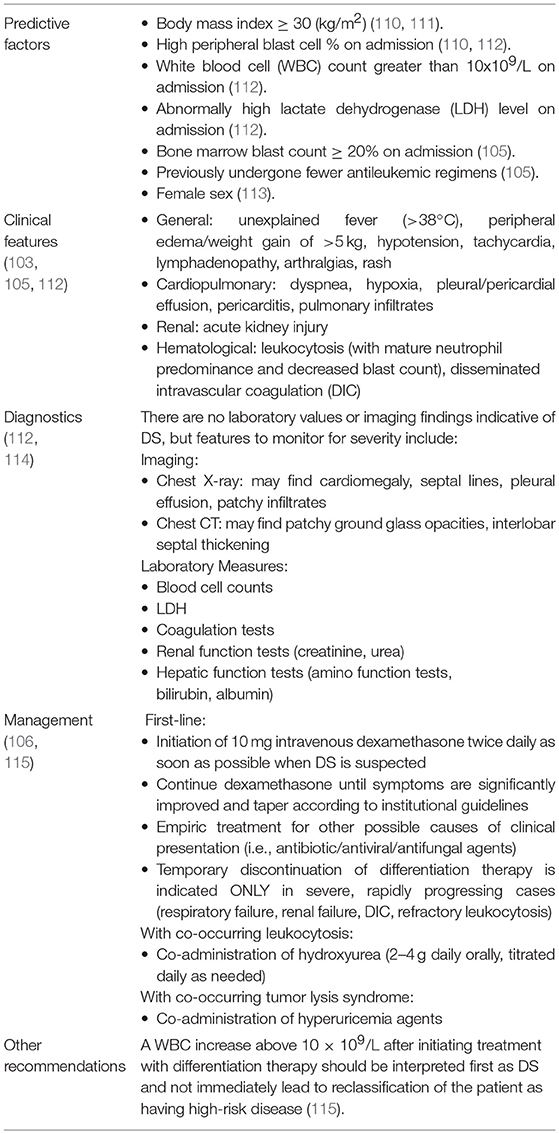
Table 4. Differentiation syndrome: clinical features and management.
While exact mechanisms and pathophysiology remain poorly understood, differentiation syndromes are generally thought to be inflammatory phenomena resulting from cytokine release during widespread drug-induced differentiation of immature blasts into mature cell types (51). While not yet proven in IDH-DS, it has been suggested that given its inflammatory pathogenesis, prophylactic administration of corticosteroids can reduce the incidence of retinoic acid-induced DS in APL (116). Leading mechanistic hypotheses suggest that the continuous generation of cytokines and adhesion molecules during differentiation promotes extravascular extravasation of fluid into compartments such as the lung, pleura, and pericardium in a “two-step model.” In the first step, the “initiation phase,” migration of differentiating leukemic cells is promoted by increased expression of cell surface β2 integrins and adhesion molecules such as ICAM-1 and ICAM-2—both on the surface of differentiating leukemic cells and on the pulmonary vascular endothelium. In the second step, the “aggravation phase,” there is increased production of pro-migratory chemokines by the lung and concomitant upregulation of corresponding surface receptors on the differentiating leukemic cells (117). Other studies have suggested that in addition to pulmonary infiltration of differentiating leukemic cells, there is migration of normal leukocytes into the lung as a consequence of systemically increased levels of tumor necrosis factor-alpha, interleukin (IL)-1β, IL-6, and IL-8 (118). Interestingly, a more recent study has implicated a highly-conserved nuclear protein called high-mobility group box 1 (HMGB1) in APL DS pathogenesis via the MEK/ERK signaling pathway and has suggested that HMGB1 may be a possible future target in the treatment of DS (119). Continued research is required to further elucidate the pathogenesis of DS, especially in association with the novel mutIDH inhibitors, since the overwhelming majority of existing DS research remains limited to the context of APL.
MutIDH inhibitors have demonstrated some efficacy in IDH-mutant AML and early results have validated their safety in glioma patients. However, given the extent of hypermethylation induced early in oncogenesis by the IDH mutation, mutIDH inhibitor monotherapy may be insufficient to thoroughly undo these preexisting and persistent global epigenetic alterations keeping the tumor cells in a dedifferentiated state. Preclinical evidence, particularly in glioma, supports the hypothesis that mutIDH assumes a “passenger,” rather than a “driver,” role soon after the development of the IDH mutation likely due to initiating methylation changes being preserved by epigenetic memory and consequent acquisition of additional driver mutations (80, 81).
The addition of hypomethylating agents, such as DNA methyltransferase (DNMT) inhibitors, to a mutIDH inhibitor regimen may generate a synergistic response by dually targeting the CIMP phenotype. In AML, it is known that patients with IDH mutations are up to 14 times more likely to response to DNMT inhibitors (120). In preclinical studies, in vitro human IDH1R132H TF-1 erythroleukemia cells demonstrated increased cell differentiation by a factor of 25 after Ivosidenib treatment alone, which improved to a 46-fold increase with concurrent azacitidine treatment (90). Several clinical trials involving mutIDH inhibitors and DNMT inhibitors in AML are ongoing; early data was presented at the 2018 ASCO Annual Meeting from a phase 1b/2 study of combination treatment in adults with newly diagnosed IDH-mutant AML testing both Ivosidenib and Enasidenib in combination with azacitidine (ClinicalTrials.gov NCT02677922) (94). In the Ivosidenib + azacitidine group, 78% achieved a response with a 44% complete response rate, and in the Enasidenib + azacitidine group, 67% achieved a response with a 50% complete response rate. Incidence of serious adverse events were similar to those reported with azacitidine monotherapy (94). A phase 3 multicenter randomized controlled trial entitled “AGILE” will evaluate the efficacy of the Ivosidenib + azacitidine regimen compared to a placebo + azacitidine control in a similar patient population (92).
In IDH-mutant glioma, multiple DNMT inhibitors have demonstrated efficacy in preclinical studies to induce cellular differentiation, reduce global methylation and inhibit growth (121, 122). A recent study by Yamashita et al. in human IDH-mutant glioma cell lines found not only that azacitidine reduced cellular proliferation and induced increased expression of astroglial differentiation markers like GFAP, but also that azacitidine worked synergistically with temozolomide to inhibit tumor growth (123). Based on this evidence, a single phase 2 non-comparative non-randomized single center study of azacitidine in adults with recurrent IDH-mutant glioma is in the planning stages (ClinicalTrials.gov NCT03666559). However, evidence supporting combination therapy involving DNMT inhibitors and mutIDH inhibitors in glioma remains limited to early and mixed evidence from preclinical data alone (121, 123). Nevertheless, given both the synergistic response to dual therapy seen in AML and the limited efficacy of mutIDH inhibitor monotherapy in glioma trials, further investigation of combination DNMT inhibitor and mutIDH inhibitor therapy in glioma is warranted.
Epigenetic memory in IDH-mutant cancers is likely not limited to DNA methylation alone. RNA methylation patterns have also been shown to be critical for glioblastoma stem cell self-renewal and tumorigenesis specifically, and as previously discussed, the early molecular insults of IDH mutations and 2HG include competitive inhibition of histone demethylases (22, 23, 124, 125). While there is a paucity of research on RNA methylation status as a therapeutic target, histone-modifying agents such as histone deacetylase inhibitors and histone methyltransferase inhibitors are already in phase 1/2 clinical testing for AML, glioma, cholangiocarcinoma, and myelodysplastic syndromes (126–129). Early preclinical data in intrahepatic cholangiocarcinoma in particular suggests that bromodomain and extraterminal domain (BET) (histone regulatory complexes) inhibitors such as JQ1 may have an enhanced antiproliferative effect in IDH-mutant cancer cell lines (130). Furthermore, downstream oncogenic effects of histone dysregulation in IDH-mutant cancers, such as downregulation of the ATM gene causing an increase in spontaneous DNA damage, continue to become more well understood and suggest that a parallel effort to understand and target epigenetic memory carried in histones, in addition to DNA methylation, should be pursued (131).
Lastly, acquired resistance to isotype-specific treatment as a consequence of mutIDH isoform switching has recently been described as a potentially critical limitation of mutIDH inhibitor monotherapy. A small case series described two cases of IDH1R132C-mutant AML that, after achieving remission on Ivosidenib, both recurred with new IDH2R140Q mutations (99). A third case described a similar switch from an IDH1 to IDH2 mutation in cholangiocarcinoma and a fourth case described a switch from IDH2R140Q-mutant AML to IDHR132C after Enasidenib treatment that was then responsive to second-line treatment with pan-inhibitor, AG-881 (99). Additionally, the same research group defined another potential avenue for resistance through mutational changes in conformation of the mutIDH enzyme trans-dimer interface—the precise target of these small molecule therapeutics (132). Together, these findings suggest that despite the possibility of conversion of mutIDH from “driver” to “passenger” in supporting some chromatin modifications and cellular growth, other biological pressures for 2HG production persist over time in IDH-mutant cancers (80, 99). Future clinical studies investigating the comparative long-term efficacy of pan- and specific-mutIDH inhibition, as well as combination treatments with relevant epigenetic modifiers as previously described, may inform and improve current treatment strategies.
The identification of neomorphic mutations in IDH in several cancer types including glioma and AML has generated robust research and drug discovery efforts to both elucidate critical pathways in oncogenesis and create effective, targeted molecular therapies. A host of existing mutIDH inhibitors are continually being redesigned for pharmacokinetic and pharmacodynamic optimization to permit entry into clinical trials. Two such compounds, Enasidenib (AG-221), a mutIDH2-specific inhibitor, and Ivosidenib (AG-120), a mutIDH1-specific inhibitor, have demonstrated safety and efficacy in phase 1 clinical trials for relapsed or refractory AML and recently earned FDA approval for this indication (12, 13). They are also being tested widely in other IDH-mutant cancers, such as glioma and cholangiocarcinoma (95–97). Significant early preclinical and clinical success in AML, as compared to solid tumors, may be a consequence of differences in the biological role of and oncogenic dependencies on mutIDH across these cancer subtypes. In glioma, it has been hypothesized that the mutIDH enzyme rapidly converts from an oncogenic “driver” to “passenger,” and distinctive mutIDH-associated global DNA hypermethylation patterns (CIMP) may not be readily reversible solely with mutIDH inhibition and depletion of its product, 2HG (80). In contrast, several studies in AML cellular models have demonstrated an ability for mutIDH inhibition to readily reverse DNA hypermethylation (51, 72, 74, 90). Future efforts to improve mutIDH inhibitor efficacy should focus on biologically-based combination treatment strategies, in particular with demethylating agents such as DNMT inhibitors or histone-modifying agents, which may improve response rate and duration across IDH-mutant cancers.
In the context of this narrative review article, no consent, or IRB approval was required by the authors' institution given that no human subjects were involved in this research.
DG, NI, SD, TW, KT, and AM manuscript drafting. DG, NI, SD, TW, and DB figure and table design. DG, NI, SD, TW, DB, KT, AM, and DP manuscript editing and revision. DG, NI, SD, and TW literature review. DG, NI, and SD clinical trials review. DG, DB, KT, AM, and DP research team management and oversight.
This research is supported in part by the following grant: NYU CTSA/NCATS: UL1TR001445.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Yan H, Parsons DW, Jin G, McLendon R, Rasheed A, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. (2009) 360:765–73. doi: 10.1056/NEJMoa0808710
2. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. (2008) 321:1807–12. doi: 10.1126/science.1164382
3. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
4. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. (2011) 224:334–43. doi: 10.1002/path.2913
5. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): an update. Mutat Res Rev Mutat Res. (2016) 769:47–62. doi: 10.1016/j.mrrev.2016.04.009
6. Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. (2017) 18:2780–94. doi: 10.1016/j.celrep.2017.02.033
7. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. (2014) 343:189–93. doi: 10.1126/science.1239947
8. Dombret H, Itzykson R. How and when to decide between epigenetic therapy and chemotherapy in patients with AML. Hematology Am Soc Hematol Educ Program. (2017) 2017:45–53. doi: 10.1182/asheducation-2017.1.45
9. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
10. Dhillon S. Ivosidenib: first global approval. Drugs. (2018) 78:1509–16. doi: 10.1007/s40265-018-0978-3
11. Kim ES. Enasidenib: first global approval. Drugs. (2017) 77:1705–11. doi: 10.1007/s40265-017-0813-2
12. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. (2017) 130:722–31. doi: 10.1182/blood-2017-04-779405
13. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
14. May JL, Kouri FM, Hurley LA, Liu J, Tommasini-Ghelfi S, Ji Y, et al. IDH3alpha regulates one-carbon metabolism in glioblastoma. Sci Adv. (2019) 5:eaat0456. doi: 10.1126/sciadv.aat0456
15. Krell D, Assoku M, Galloway M, Mulholland P, Tomlinson I, Bardella C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH mutations in glioblastoma. PLoS ONE. (2011) 6:e19868. doi: 10.1371/journal.pone.0019868
16. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. (2009) 462:739–44. doi: 10.1038/nature08617
17. Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. (2010) 119:487–94. doi: 10.1007/s00401-010-0645-6
18. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
19. Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. (2013) 288:3804–15. doi: 10.1074/jbc.M112.435495
20. Brooks E, Wu X, Hanel A, Nguyen S, Wang J, Zhang JH, et al. Identification and characterization of small-molecule inhibitors of the R132H/R132H mutant isocitrate dehydrogenase 1 homodimer and R132H/wild-type heterodimer. J Biomol Screen. (2014) 19:1193–200. doi: 10.1177/1087057114541148
21. Madala HR, Punganuru SR, Arutla V, Misra S, Thomas TJ, Srivenugopal KS. Beyond brooding on oncometabolic havoc in IDH-mutant gliomas and AML: current and future therapeutic strategies. Cancers. (2018) 10:E49. doi: 10.3390/cancers10020049
22. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
23. Clark O, Yen K, Mellinghoff IK. Molecular pathways: isocitrate dehydrogenase mutations in cancer. Clin Cancer Res. (2016) 22:1837–42. doi: 10.1158/1078-0432.CCR-13-1333
24. Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J Inorg Biochem. (2006) 100:644–69. doi: 10.1016/j.jinorgbio.2006.01.024
25. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. (2011) 12:463–9. doi: 10.1038/embor.2011.43
26. Joberty G, Boesche M, Brown JA, Eberhard D, Garton NS, Humphreys PG, et al. Interrogating the druggability of the 2-oxoglutarate-dependent dioxygenase target class by chemical proteomics. ACS Chem Biol. (2016) 11:2002–10. doi: 10.1021/acschembio.6b00080
27. Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. (2018) 50:62–72. doi: 10.1038/s41588-017-0001-z
28. Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. (2018) 20:608–20. doi: 10.1093/neuonc/nox183
29. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. (2012) 483:479–83. doi: 10.1038/nature10866
30. Morita R, Hirohashi Y, Suzuki H, Takahashi A, Tamura Y, Kanaseki T, et al. DNA methyltransferase 1 is essential for initiation of the colon cancers. Exp Mol Pathol. (2013) 94:322–9. doi: 10.1016/j.yexmp.2012.10.004
31. Wongtrakoongate P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J Stem Cells. (2015) 7:137–48. doi: 10.4252/wjsc.v7.i1.137
32. Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. (2012) 26:756–84. doi: 10.1101/gad.187922.112
33. Yan H, Bigner DD, Velculescu V, Parsons DW. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. (2009) 69:9157–9. doi: 10.1158/0008-5472.CAN-09-2650
34. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. (2009) 30:7–11. doi: 10.1002/humu.20937
35. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. (2009) 118:469–74. doi: 10.1007/s00401-009-0561-9
36. Koh J, Cho H, Kim H, Kim SI, Yun S, Park CK, et al. IDH2 mutation in gliomas including novel mutation. Neuropathology. (2015) 35:236–44. doi: 10.1111/neup.12187
37. Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. (2015) 372:2481–98. doi: 10.1056/NEJMoa1402121
38. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. (2016) 529:110–4. doi: 10.1038/nature16490
39. Modrek AS, Golub D, Khan T, Bready D, Prado J, Bowman C, et al. Low-grade astrocytoma mutations in IDH1, P53, and ATRX cooperate to block differentiation of human neural stem cells via repression of SOX2. Cell Rep. (2017) 21:1267–80. doi: 10.1016/j.celrep.2017.10.009
40. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. (2009) 174:1149–53. doi: 10.2353/ajpath.2009.080958
41. Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1(R132H) in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell. (2016) 30:578–94. doi: 10.1016/j.ccell.2016.08.017
42. Pirozzi CJ, Carpenter AB, Waitkus MS, Wang CY, Zhu H, Hansen LJ, et al. Mutant IDH1 disrupts the mouse subventricular zone and alters brain tumor progression. Mol Cancer Res. (2017) 15:507–20. doi: 10.1158/1541-7786.MCR-16-0485
43. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. (2009) 361:1058–66. doi: 10.1056/NEJMoa0903840
44. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
45. DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. (2015) 90:732–6. doi: 10.1002/ajh.24072
46. Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. (2014) 28:1774–83. doi: 10.1038/leu.2014.124
47. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. (2010) 28:2348–55. doi: 10.1200/JCO.2009.27.3730
48. Wagner K, Damm F, Göhring G, Görlich K, Heuser M, Schäfer I, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol. (2010) 28:2356–64. doi: 10.1200/JCO.2009.27.6899
49. Thol F, Damm F, Wagner K, Göhring G, Schlegelberger B, Hoelzer D, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. (2010) 116:614–6. doi: 10.1182/blood-2010-03-272146
50. Thomas D, Majeti R. Optimizing next-generation AML therapy: activity of mutant IDH2 inhibitor AG-221 in preclinical models. Cancer Discov. (2017) 7:459–61. doi: 10.1158/2159-8290.CD-17-0270
51. Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. (2017) 7:478–93. doi: 10.1158/2159-8290.CD-16-1034
52. Chaturvedi A, Herbst L, Pusch S, Klett L, Goparaju R, Stichel D, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. (2017) 31:2020–8. doi: 10.1038/leu.2017.46
53. Ho PA, Kopecky KJ, Alonzo TA, Gerbing RB, Miller KL, Kuhn J, et al. Prognostic implications of the IDH1 synonymous SNP rs11554137 in pediatric and adult AML: a report from the Children's Oncology Group and SWOG. Blood. (2011) 118:4561–6. doi: 10.1182/blood-2011-04-348888
54. Mondesir J, Willekens C, Touat M, de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. (2016) 7:171–80. doi: 10.2147/JBM.S70716
55. Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. (2017) 31:272–81. doi: 10.1038/leu.2016.275
56. Green CL, Evans CM, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood. (2010) 116:2779–82. doi: 10.1182/blood-2010-02-270926
57. Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. (2011) 118:409–12. doi: 10.1182/blood-2010-12-322479
58. DiNardo CD, Propert KJ, Loren AW, Paietta E, Sun Z, Levine RL, et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood. (2013) 121:4917–24. doi: 10.1182/blood-2013-03-493197
59. Wang JH, Chen WL, Li JM, Wu SF, Chen TL, Zhu YM, et al. Prognostic significance of 2-hydroxyglutarate levels in acute myeloid leukemia in China. Proc Natl Acad Sci USA. (2013) 110:17017–22. doi: 10.1073/pnas.1315558110
60. Chen J, Yang J, Cao P. The evolving landscape in the development of isocitrate dehydrogenase mutant inhibitors. Mini Rev Med Chem. (2016) 16:1344–58. doi: 10.2174/1389557516666160609085520
61. Ma R, Yun CH. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem Biophys Res Commun. (2018) 503:2912–7. doi: 10.1016/j.bbrc.2018.08.068
62. Yen K, Konteatis Z, Sui Z, Artin E, Dang L, Straley K, et al. Abstract B126: AG-881, a brain penetrant, potent, pan-mutant IDH (mIDH) inhibitor for use in mIDH solid and hematologic malignancies. Mol Cancer Ther. (2018) 17(suppl. 1):B126. doi: 10.1158/1535-7163.TARG-17-B126
63. Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. (2017) 133:629–44. doi: 10.1007/s00401-017-1677-y
64. DiNardo CD, Stein EM. SOHO state of the art update and next questions: IDH therapeutic targeting in AML. Clinical Lymphoma Myeloma Leuk. (2018) 18:769–72. doi: 10.1016/j.clml.2018.10.007
65. Corporation C. Celgene Corporation and Agios Pharmaceuticals Announce Global Strategic Collaboration to Advance Unique Science of Cancer Metabolism [Media Release]. (2010).
66. Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett. (2012) 3:850–5. doi: 10.1021/ml300225h
67. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. (2013) 340:626–30. doi: 10.1126/science.1236062
68. Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. (2018) 9:300–5. doi: 10.1021/acsmedchemlett.7b00421
69. Mellinghoff I, Penas-Prado M, Peters K, Cloughesy T, Burris H, Maher E, et al. ACTR-31. Phase 1 study of AG-881, an inhibitor of mutant IDH1 and IDH2: results from the recurrent/progressive glioma population. Neuro Oncol. (2018) 20(suppl. 6):vi18. doi: 10.1093/neuonc/noy148.064
70. Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS ONE. (2015) 10:e0133813. doi: 10.1371/journal.pone.0133813
71. Cho YS, Levell JR, Liu G, Caferro T, Sutton J, Shafer CM, et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. (2017) 8:1116–21. doi: 10.1021/acsmedchemlett.7b00342
72. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. (2013) 340:622–6. doi: 10.1126/science.1234769
73. Chen J, Yang J, Sun X, Wang Z, Cheng X, Lu W, et al. Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/R140Q. Sci Rep. (2017) 7:16458. doi: 10.1038/s41598-017-16427-w
74. Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. (2015) 125:296–303. doi: 10.1182/blood-2013-10-533604
75. Watts JM, Baer MR, Lee S, Yang J, Dinner SN, Prebet T, et al. A phase 1 dose escalation study of the IDH1m inhibitor, FT-2102, in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). J Clin Oncol. (2018) 36(suppl. 15):7009. doi: 10.1200/JCO.2018.36.15_suppl.7009
76. Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. (2013) 122:2877–87. doi: 10.1182/blood-2013-03-491571
77. Chaturvedi A, Araujo Cruz MM, Goparaju R, Jyotsana N, Baehre H, Goerlich K, et al. A novel inhibitor of mutant IDH1 induces differentiation in vivo and prolongs survival in a mouse model of leukemia. Blood. (2014) 124:3598. Available online at: http://www.bloodjournal.org/content/124/21/3598/tab-article-info
78. Kopinja J, Sevilla RS, Levitan D, Dai D, Vanko A, Spooner E, et al. A brain penetrant mutant IDH1 inhibitor provides in vivo survival benefit. Sci Rep. (2017) 7:13853. doi: 10.1038/s41598-017-14065-w
79. Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. (2015) 11:878–86. doi: 10.1038/nchembio.1930
80. Johannessen TA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R, et al. Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol Cancer Res. (2016) 14:976–83. doi: 10.1158/1541-7786.MCR-16-0141
81. Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. (2015) 28:773–84. doi: 10.1016/j.ccell.2015.11.006
82. Levell JR, Caferro T, Chenail G, Dix I, Dooley J, Firestone B, et al. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1. ACS Med Chem Lett. (2017) 8:151–6. doi: 10.1021/acsmedchemlett.6b00334
83. DiNardo CD, Schimmer AD, Yee KWL, et al. A Phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 Mutations. Blood. (2016) 128:1073. Available online at: http://www.bloodjournal.org/content/128/22/1073
84. Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. (2017) 19:1858–73. doi: 10.1016/j.celrep.2017.05.014
85. Yang B, Zhong C, Peng Y, Lai Z, Ding J. Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. (2010) 20:1188–200. doi: 10.1038/cr.2010.145
86. Stein EM. Enasidenib, a targeted inhibitor of mutant IDH2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. (2018) 14:23–40. doi: 10.2217/fon-2017-0392
87. Deng G, Shen J, Yin M, McManus J, Mathieu M, Gee P, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. (2015) 290:762–74. doi: 10.1074/jbc.M114.608497
88. Heredia V. 1524P - AG-120, a novel IDH1 targeted molecule, inhibits invasion and migration of chondrosarcoma cells in vitro. Ann Oncol. (2017) 28:v521–38. doi: 10.1093/annonc/mdx387.049
89. Nicolay B, Narayanaswamy R, Aguado E, Nagaraja R, Murtie J, Liu G, et al. EXTH-59. The IDH1 mutant inhibitor AG-120 shows strong inhibition of 2-HG production in an orthotopic IDH1 mutant glioma model in vivo. Neuro Oncol. (2017) 19(suppl. 6):vi86. doi: 10.1093/neuonc/nox168.351
90. Yen K, Chopra VS, Tobin E, Avanzino B, Mavrommatis K, Dimartino J, et al. Abstract 4956: functional characterization of the ivosidenib (AG-120) and azacitidine combination in a mutant IDH1 AML cell model. Cancer Res. (2018) 78(suppl. 13):4956. doi: 10.1158/1538-7445.AM2018-4956
91. Tallman MS, Knight RD, Glasmacher AG, Dohner H. Phase III randomized, open-label study comparing the efficacy and safety of AG-221 vs conventional care regimens (CCR) in older patients with advanced acute myeloid leukemia (AML) with isocitrate dehydrogenase (IDH)-2 mutations in relapse or refractory to multiple prior treatments: the idhentify trial. J Clin Oncol. (2016) 34(suppl. 15):TPS7074. doi: 10.1200/JCO.2016.34.15_suppl.TPS7074
92. Stein E, Dinardo CD, Jang JH, Miyazaki Y, Martinez RO, Auer J, et al. AGILE: A phase 3, multicenter, randomized, placebo-controlled study of ivosidenib in combination with azacitidine in adult patients with previously untreated acute myeloid leukemia with an IDH1 mutation. J Clin Oncol. (2018) 36(suppl. 15):TPS7074. doi: 10.1200/JCO.2018.36.15_suppl.TPS7074
93. Stein EM, DiNardo CD, Mims AS, Savona MR, Pratz K, Stein AS, et al. Ivosidenib or enasidenib combined with standard induction chemotherapy is well tolerated and active in patients with newly diagnosed AML with an IDH1 or IDH2 mutation: initial results from a Phase 1 trial. Blood. (2017) 130(suppl. 1):726. Available online at: http://www.bloodjournal.org/content/130/Suppl_1/726/tab-article-info
94. Dinardo CD, Stein AS, Stein EM, Fathi AT, Montesinos P, Odenike O, et al. Mutant IDH (mIDH) inhibitors, ivosidenib or enasidenib, with azacitidine (AZA) in patients with acute myeloid leukemia (AML). J Clin Oncol. (2018) 36(suppl. 15):7042. doi: 10.1200/JCO.2018.36.15_suppl.7042
95. Burris H, Mellinghoff I, Maher E, Wen P, Beeram M, Touat M, et al. Abstract PL04–05: the first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol Cancer Ther. (2015) 14(suppl. 2):PL04–05. doi: 10.1158/1535-7163.TARG-15-PL04-05
96. Mellinghoff IK, Touat M, Maher E, DeLaFuente M, Cloughesy TF, Holdhoff M, et al. ACTR-46. AG120, A first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: results from the Phase 1 glioma expansion cohorts. Neuro Oncol. (2016) 18(suppl. 6):vi12. doi: 10.1093/neuonc/now212.044
97. Fan B, Mellinghoff IK, Wen PY, Lowery MA, Goyal L, Tap WD, et al. Pharmacokinetics/pharmacodynamics (PK/PD) of ivosidenib in patients with IDH1-mutant advanced solid tumors from a phase 1 study. J Clin Oncol. (2018) 36(suppl. 15):2577. doi: 10.1200/JCO.2018.36.15_suppl.2577
98. Mellinghoff IK, Penas-Prado M, Peters KB, Cloughesy TF, Burris HA, Burris EA, et al. Phase 1 study of AG-881, an inhibitor of mutant IDH1/IDH2, in patients with advanced IDH-mutant solid tumors, including glioma. J Clin Oncol. (2018) 36(suppl. 15):2002. doi: 10.1200/JCO.2018.36.15_suppl.2002
99. Harding JJ, Lowery MA, Shih AH, Schvartzman JM, Hou S, Famulare C, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. (2018) 8:1540–7. doi: 10.1158/2159-8290.CD-18-0877
100. Lowery MA, Abou-Alfa GK, Valle JW, Kelley RK, Goyal L, Shroff RT, et al. ClarIDHy: a phase 3, multicenter, randomized, double-blind study of AG-120 vs placebo in patients with an advanced cholangiocarcinoma with an IDH1 mutation. J Clin Oncol. (2017) 35(suppl. 15):TPS4142. doi: 10.1200/JCO.2017.35.15_suppl.TPS4142
101. Mellinghoff I, Le K, Young R, Maher E, Wen P, Cloughesy T, et al. RBTT-03. A phase 1, multicenter, randomized, open-label, perioperative study of AG-120 (ivosidenib) and AG-881 in patients with recurrent, nonenhancing, IDH1-mutant, low-grade glioma. Neuro Oncol. (2018) 20(suppl. 6):vi234. doi: 10.1093/neuonc/noy148.973
102. Lowery MA, Abou-Alfa GK, Burris HA, Janku F, Shroff RT, Cleary JM, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. (2017) 35(15_suppl):4015. doi: 10.1200/JCO.2017.35.15_suppl.4015
103. Frankel SR, Eardley A, Lauwers G, Weiss M, Warrell RP Jr. The “retinoic acid syndrome” in acute promyelocytic leukemia. Ann Intern Med. (1992) 117:292–6. doi: 10.7326/0003-4819-117-4-292
104. Au WY, Kwong YL. Arsenic trioxide: safety issues and their management. Acta Pharmacol Sin. (2008) 29:296–304. doi: 10.1111/j.1745-7254.2008.00771.x
105. Fathi AT, DiNardo CD, Kline I, Kenvin L, Gupta I, Attar EC, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. (2018) 4:1106–10. doi: 10.1001/jamaoncol.2017.4695
106. Birendra KC, DiNardo CD. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant IDH1 inhibitor, AG-120. Clin Lymphoma Myeloma Leuk. (2016) 16:460–5. doi: 10.1016/j.clml.2016.04.006
107. Mascarenhas J. A concise update on risk factors, therapy, and outcome of leukemic transformation of myeloproliferative neoplasms. Clin Lymphoma Myeloma Leuk. (2016) 16:S124–9. doi: 10.1016/j.clml.2016.02.016
108. Yogarajah M, Tefferi A. Leukemic transformation in myeloproliferative neoplasms: a literature review on risk, characteristics, and outcome. Mayo Clin Proc. (2017) 92:1118–28. doi: 10.1016/j.mayocp.2017.05.010
109. Daly PA, Schiffer CA, Wiernik PH. Acute promyelocytic leukemia–clinical management of 15 patients. Am J Hematol. (1980) 8:347–59. doi: 10.1002/ajh.2830080403
110. Leblebjian H, DeAngelo DJ, Skirvin JA, Stone RM, Wadleigh M, Werner L, et al. Predictive factors for all-trans retinoic acid-related differentiation syndrome in patients with acute promyelocytic leukemia. Leuk Res. (2013) 37:747–51. doi: 10.1016/j.leukres.2013.04.011
111. Jeddi R, Ghedira H, Mnif S, Gouider E, Fenaux P, Meddeb B. High body mass index is an independent predictor of differentiation syndrome in patients with acute promyelocytic leukemia. Leuk Res. (2010) 34:545–7. doi: 10.1016/j.leukres.2009.09.017
112. Montesinos P, Bergua JM, Vellenga E, Rayón C, Parody R, de la Serna J, et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood. (2009) 113:775–83. doi: 10.1182/blood-2008-07-168617
113. Elemam O, Abdelmoety D. Acute promyelocytic leukemia, study of predictive factors for differentiation syndrome, single center experience. J Egypt Natl Cancer Inst. (2013) 25:13–9. doi: 10.1016/j.jnci.2012.10.004
114. Cardinale L, Asteggiano F, Moretti F, Torre F, Ulisciani S, Fava C, et al. Pathophysiology, clinical features and radiological findings of differentiation syndrome/all-trans-retinoic acid syndrome. World J Radiol. (2014) 6:583–8. doi: 10.4329/wjr.v6.i8.583
115. Sanz MA, Fenaux P, Tallman MS, Estey EH, Löwenberg B, Naoe T, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. (2019) 133:1630–43. doi: 10.1182/blood-2019-01-894980
116. Montesinos P, Sanz MA. The differentiation syndrome in patients with acute promyelocytic leukemia: experience of the pethema group and review of the literature. Mediterr J Hematol Infect Dis. (2011) 3:e2011059. doi: 10.4084/mjhid.2011.059
117. Luesink M, Jansen JH. Advances in understanding the pulmonary infiltration in acute promyelocytic leukaemia. Br J Haematol. (2010) 151:209–20. doi: 10.1111/j.1365-2141.2010.08325.x
118. De Botton S, Dombret H, Sanz M, Miguel JS, Caillot D, Zittoun R, et al. Incidence, clinical features, and outcome of all trans-retinoic acid syndrome in 413 cases of newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. (1998) 92:2712–8.
119. Tang L, Chai W, Ye F, Yu Y, Cao L, Yang M, et al. HMGB1 promotes differentiation syndrome by inducing hyperinflammation via MEK/ERK signaling in acute promyelocytic leukemia cells. Oncotarget. (2017) 8:27314–27. doi: 10.18632/oncotarget.15432
120. Emadi A, Faramand R, Carter-Cooper B, Tolu S, Ford LA, Lapidus RG, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. (2015) 90:E77–9. doi: 10.1002/ajh.23965
121. Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. (2013) 4:1729–36. doi: 10.18632/oncotarget.1412
122. Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. (2013) 4:1737–47. doi: 10.18632/oncotarget.1408
123. Yamashita AS, da Costa Rosa M, Borodovsky A, Festuccia WT, Chan T, Riggins GJ. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. (2018) 21:189–200. doi: 10.1093/neuonc/noy146
124. Dixit D, Xie Q, Rich JN, Zhao JC. Messenger RNA methylation regulates glioblastoma tumorigenesis. Cancer Cell. (2017) 31:474–5. doi: 10.1016/j.ccell.2017.03.010
125. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
126. Williams MJ, Singleton WG, Lowis SP, Malik K, Kurian KM. Therapeutic targeting of histone modifications in adult and pediatric high-grade glioma. Front Oncol. (2017) 7:45. doi: 10.3389/fonc.2017.00045
127. Lu R, Wang GG. Pharmacologic targeting of chromatin modulators as therapeutics of acute myeloid leukemia. Front Oncol. (2017) 7:241. doi: 10.3389/fonc.2017.00241
128. Ungerstedt JS. Epigenetic modifiers in myeloid malignancies: the role of histone deacetylase inhibitors. Int J Mol Sci. (2018) 19:E3091. doi: 10.3390/ijms19103091
129. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res. (2016) 22:291–300. doi: 10.1158/1078-0432.CCR-14-3296
130. Fujiwara H, Tateishi K, Kato H, Nakatsuka T, Yamamoto K, Tanaka Y, et al. Isocitrate dehydrogenase 1 mutation sensitizes intrahepatic cholangiocarcinoma to the BET inhibitor JQ1. Cancer Sci. (2018) 109:3602–10. doi: 10.1111/cas.13784
131. Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell. (2016) 30:337–48. doi: 10.1016/j.ccell.2016.05.018
Keywords: acute myeloid leukemia, enasidenib, glioma, IDH, isocitrate dehydrogenase, ivosidenib
Citation: Golub D, Iyengar N, Dogra S, Wong T, Bready D, Tang K, Modrek AS and Placantonakis DG (2019) Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 9:417. doi: 10.3389/fonc.2019.00417
Received: 24 March 2019; Accepted: 02 May 2019;
Published: 17 May 2019.
Edited by:
Tomofusa Fukuyama, The University of Tokyo, JapanReviewed by:
Wu Xu, University of Louisiana at Lafayette, United StatesCopyright © 2019 Golub, Iyengar, Dogra, Wong, Bready, Tang, Modrek and Placantonakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danielle Golub, ZGFuaWVsbGUuZ29sdWJAbnl1bGFuZ29uZS5vcmc=
Dimitris G. Placantonakis, ZGltaXRyaXMucGxhY2FudG9uYWtpc0BueXVsYW5nb25lLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.