 Xuequn Xu
Xuequn Xu J. N. Rashida Gnanaprakasam
J. N. Rashida Gnanaprakasam John Sherman
John Sherman Ruoning Wang
Ruoning Wang- Center for Childhood Cancer and Blood Diseases, Hematology/Oncology & BMT, The Research Institute at Nationwide Children's Hospital, Ohio State University, Columbus, OH, United States
The adoptive transfer of T cells expressing chimeric antigen receptors (CARs) through genetic engineering is one of the most promising new therapies for treating cancer patients. A robust CAR T cell-mediated anti-tumor response requires the coordination of nutrient and energy supplies with CAR T cell expansion and function. However, the high metabolic demands of tumor cells compromise the function of CAR T cells by competing for nutrients within the tumor microenvironment (TME). To substantially improve clinical outcomes of CAR T immunotherapy while treating solid tumors, it is essential to metabolically prepare CAR T cells to overcome the metabolic barriers imposed by the TME. In this review, we discuss a potential metabolism toolbox to improve the metabolic fitness of CAR T cells and maximize the efficacy of CAR T therapy.
Cancer Cell Metabolic Program
Since cancer cells must constantly proliferate, they must also continuously generate new biomass. This in turn requires a substantially different metabolic program than that of non-proliferating somatic cells. Most non-proliferating cells utilize oxidative phosphorylation (OXPHOS) to efficiently extract ATP from pyruvate, while cancer cells reduce the majority pyruvate into lactate, a process termed aerobic glycolysis or “the Warburg Effect” (1). In 1956, Otto Warburg observed the tendency of cancer cells to metabolize glucose into lactate instead of carbon dioxide and concluded that cancer was a disease of damaged respiration (2). While not all of Warburg's conclusions have stood the test of time, it holds true that metabolism is a critical component of oncogenesis. Even if a cell has developed mutations to overcome the normal regulation of proliferation, it also requires a metabolic program that will allow the cell to synthesize all the molecules required for a new cell. Metabolism is so critical to oncogenesis that the most commonly mutated pathways, including Ras, Phosphoinositide 3-kinases (PI3K)/AKT/mammalian target of rapamycin (mTORC1), hypoxia inducible factor 1 (HIF-1), proto-oncogene MYC (c-MYC), and p53, are key metabolic regulators. HIF-1 and c-MYC in particular act in concert to express glucose and lactate transport proteins while diverting pyruvate away from OXPHOS and toward lactate production (3–5). While this method does not maximize the amount of ATP that can be extracted from glucose, it is none the less advantageous for proliferating cells. By keeping glucose derived carbon out of the TCA cycle, additional carbons are made available for lipid, protein and especially nucleotide synthesis (1, 6, 7). Sequential activation of PI3K, followed by Akt and mTORC1, are also able to aid cancer cells in capturing glucose from the environment, as well as catabolic metabolites from the mitochondria. Akt activates hexokinase and phophofructokinase-1, to retain glucose and commit it to further glycolysis, as well as ATP-citrate lyase, to convert mitochondrial citrate into cytosolic acetyl-CoA for lipid synthesis (7, 8). Finally, mTORC1 enhances mitochondrial biosynthesis to take oxaloacetate and α-ketoglutarate (α-KG) and convert them into amino acids for protein synthesis (7). Alternative sources of carbon can support growth in cell size, but glucose is required for robust DNA synthesis and concomitant cell cycle progression (9). The other major carbon and energy source is glutamine. Upon being metabolized by the cell, glutamine is shuttled into the TCA cycle to make α-KG for the production of amino acids, or citrate for the production of lipids (1, 6). Loss of p53 additionally allows malate to leave the TCA cycle to be converted into pyruvate, generating additional NADPH, another necessity for nucleotide synthesis (10). Reduction of pyruvate into lactate serves to regenerate NAD+ for further glycolysis while also conditioning the extracellular space (11).
The disorganized structure of a solid tumor means that it is not properly enervated by blood vessels. Cells located farther from the blood vessels experience hypoxia, nutrient deprivation and acidosis (3, 12). Cancer cells experiencing nutrient deprivation must rely on alternative metabolites or methods of nutrient acquisition. Ras expressing cancer cells may use macropinocytosis to scavenge nutrients from the surrounding milieu. Further, Ras driven cells utilize autophagy to degrade unnecessary cellular components into small molecule nutrients (12). Alternatively, hypoxia re-enforces glycolytic generation of lactate, while also shunting glutamine toward lipid genesis (3, 12). Finally, lactate is associated with several oncogenic processes, such as angiogenesis, cell migration and immune suppression (5). Not strictly a waste product, cells that experience chronic acidosis may take up lactate for gluconeogenesis and use in oxidative phosphorylation (11). Thus, as a tumor progresses, cancer cells condition the microenvironment, creating unique selection pressures and contributing to further heterogeneity and metabolic derangement.
T Cell Metabolic Program
T cells play a key role in mounting a robust, antigen specific adaptive immunity against invading pathogens and tumor. Upon stimulation of antigen receptors, naïve T (Tn) cells rapidly transit from a quiescent to an active state that begins with a 24 h growth phase followed by massive proliferation, differentiation, and migration. To elicit a robust immune response, T cells can differentiate into diverse functional subsets. Depending on the cytokine milieu of the microenvironment, active CD4+ T cells can differentiate into immune suppressive regulatory T (Treg) cells or inflammatory T effector cells, such as T helper TH1, TH2, TH9, TH17 and follicular helper T (Tfh). On the other hand, active CD8+ T cells mainly differentiate into CD8+ effector T (Teff) cells, also referred as cytotoxic T lymphocytes (CTL). Following pathogen clearance, the majority of the effector cells die through apoptosis while the remaining cells survive to form a population of long-lived memory T (Tmem) cells, responsible for immunity upon subsequent challenges to the same pathogen. Tmem cells are composed of distinct subsets including stem memory cells (Tscm), central memory cells (Tcm), and effector memory cells (Tem). Tscm cells exhibit a naïve like phenotype (CD44−CD62L+/CD45RA+), express interleukin-2 receptor (IL-2R) β and the chemokine C-X-C motif receptor 3 (CXCR3), representing the earliest and long-lasting developmental stage of Tmem cells. Tscm cells have a capacity to self-renew and generate the entire spectrum of more differentiated cells. Tcm cells are CD62L+, reside in lymph nodes and have limited or no effector function, but they proliferate and become effector cells upon secondary stimulation. These cells represent an intermediate population between Tscm cells and Tem cells. Tem cells are CD62L−, are the progenitor cells prone to differentiate into Teff cells upon secondary stimulation. Therefore, Tem are responsible for protective memory, and migrate into inflammatory tissues to elicit an immediate response against antigens (13).
T cell activation and differentiation are accompanied by dramatic shifts in cellular metabolic programs which fulfill their bioenergetic, biosynthetic and redox demands (14–19). Essentially, different phenotypic and functional T cell subsets are characterized by unique metabolic demands, which are tightly linked with immune modulatory signaling cascades. Specifically, quiescent Tn and Tmem cells rely on fatty acid oxidation (FAO) and OXPHOS to maintain their basic energy level, cellular function and viability (20, 21). In addition, heightened glycerol uptake and triglyceride synthesis also play an important role in promoting memory CD8+ T cells (22, 23). Overall, active T cells predominantly engage in aerobic glycolysis, the pentose phosphate pathway (PPP) and glutaminolysis to drive proliferation and subsequent effector functions (20, 24–28). However, it remains unclear whether a shift in glucose metabolism promotes activated T cells to become long-lived Tmem cells. It has recently been suggested that persistently heightened glycolysis in Teff cells compromises the formation of long-lived memory cells by driving T cells toward a terminally differentiated state, resulting in a failure to survive upon adoptive cell transfer, whereas a moderately dampened glycolysis supports the generation of long-lived memory CD8+ T cells and enhances anti-tumor immune response (29, 30). CD4+ effector T cells including TH1, TH2, TH9, TH17, and Tfh cells display heightened glycolysis, whereas FAO activity supports the differentiation and function of Treg cells (31–37). Conversely, heightened aerobic glycolysis is eventually required to drive Teff cells proliferation and differentiation into cytotoxic T cells (38).
Catabolism of glucose and glutamine generates energy, provides reducing power, and donates carbon and nitrogen to biosynthetic building blocks. During the sequential reactions of aerobic glycolysis, the breakdown of glucose into lactate generates ATP and intermediate metabolites, many of which are channeled into the biosynthesis of amino acids, lipids and nucleotide. Branching from glycolysis, the PPP starts from glucose-6-phosphate, an immediate metabolic product of glucose, and produces NADPH in the oxidative phase as well as five-carbon sugars in non-oxidative phase, the latter of which can feed back into glycolysis or provide precursors for nucleotides. Meanwhile, NADPH is required for modulating redox balance and cholesterol biosynthesis (17, 20, 39). During glutaminolysis, glutamine is converted to glutamate and subsequently to α-KG, which serves as an important anaplerotic substrate of the TCA cycle and fuels mitochondrial ATP production. As a major source of carbon and nitrogen, the catabolic products of glutamine are funneled to support the biosynthesis of amino acids, hexosamine, polyamine, lipids and nucleotides during T cell proliferation and differentiation (40–43). Similar to glucose catabolism, glutamine catabolism is composed of multiple metabolic routes branched from glutaminolysis. Glutamate is the key branch point in glutaminolysis and can be committed toward the biosynthesis of glutathione (GSH) or toward mitochondrial oxidation to produce biosynthetic precursors and ATP. Glutamate is derived from glutamine in parallel through glutaminase, in mitochondria, as well as through glutamine utilizing enzymes, including amidotransferase, largely in cytosol (44–47). As such, the subcellular compartmentalization of glutamate may represent an important mechanism that enables a fine-tuned coordination between branched metabolic routes to fulfill bioenergetics, biosynthetic and redox demand in T cells (36, 48, 49).
T cell metabolic reprogramming during activation is strictly regulated by numerous kinase signaling cascades, including mitogen-activated protein kinase (MAPK)/extra-cellular signal-regulated kinase (ERK), mTOR kinase, AMP-activated protein kinase (AMPK), and PI3K/Akt (20, 24, 50–55). T cell activation requires co stimulation of CD28 and IL-2 signaling, which activates PI3K/AKT and mTOR to promote the uptake of glucose and amino acids to support CD4 T cell proliferation and differentiation (31, 56). AMPK and mTOR coordinate metabolic status with signaling transduction in regulating T cell differentiation. TH1, TH2, and TH17 subsets, which predominantly rely on glycolysis, maintain high mTOR activity. Conversely, Treg cells, which require FAO, maintain high AMPK activity. In addition, inhibition of mTOR complex 1 (mTORC1) by rapamycin or Wnt-β-catenin signaling in T cells drives the differentiation of Tscm cells by switching T cell metabolism toward FAO and increasing the long term survival (57, 58).
Beyond these key kinase-signaling cascades, (HIF-1) and the (c-Myc), are the key transcription factors that regulate the expression of metabolic enzymes in glucose and glutamine catabolism (20, 32, 59, 60). In addition, activating enhancer binding protein 4 (AP4), Activating transcription factor 4 (ATF4), interferon regulatory factor 4 (IRF4), Bcl-6 are involved in regulating the expression of various metabolic genes to promote glycolysis and glutaminolysis as well as Teff function (61–63). On the other hand, de novo cholesterol biosynthesis is regulated by the dynamic regulation of nuclear receptor- liver X Receptor (LXR) Foxo1 protein (Foxo1) and the orphan steroid receptor, Estrogen-related receptor alpha (ERRα) (31, 33, 40, 64, 65).
Metabolic Antagonism in the TME
Emerging evidence suggests that various metabolites from various cellular compartments within the TME may serve as a complex form of intercellular communication which modulates tumor cell growth and response to therapy (66–72). T cell metabolic pathways are tightly and ubiquitously linked with T cell activation, proliferation, differentiation, and immune functions (24, 25, 27, 31, 39, 39, 51, 56, 73). Thus, the immune cells, particularly effector T cells, are intimately controlled by the metabolic communications in the TME.
Nutrients Depletion
In addition to lineage-specific metabolic requirements, which are associated with the metabolic network in the tissue-of-origin, cancer cells display a heightened ability to capture carbon and nitrogen sources from the TME and process these raw materials to meet the cell's fundamental requirements for energy, reducing power and starting materials for biosynthesis. These general metabolic features of cancer cells are required to support the needs imposed by proliferation and other neoplastic features, but at the same time often deplete the TME of nutrients (74, 75). In addition to the consumption of key carbon and nitrogen sources, glucose and glutamine, rapidly proliferating cancer cells and T effector cells have a strong demand for amino acids, some of which are not only required for protein synthesis, but are also coupled to other anabolic routes and therefore integrated into central carbon metabolism. However, both cancer and T effector cells are often dependent on the uptake of extracellular substrates from the TME, as opposed to de novo biosynthetic pathways, which are either defective or insufficient to fulfill the demands. It is well-documented that high expression of indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) by macrophages and cancer cells contributes to immune tolerance by mediating the conversion of tryptophan to kynurenine (76–79). Tryptophan depletion and kynurenine accumulation cooperatively suppress anti-tumor immunity by reciprocally impairing the growth and survival of T effector cells and enhancing the development and function of Tregs and myeloid-derived suppressor cells (MDSC) (80–85). Extracellular cysteine and arginine are also important nutritional resources, which both T and cancer cells compete over. Cysteine, alone with glycine and glutamate, are the substrates for the de novo synthesis of GSH, which is the most abundant cellular antioxidant, to ensure physiological levels of intracellular reactive oxygen species (ROS) (20, 36, 48, 49, 51, 73, 86, 87), While glucose and glutamine catabolism provide glycine, glutamate and reducing power though NADPH, proliferating cells largely obtain cysteine from the local microenvironment (20, 86, 88–101). Lack of cystathionase, the enzyme that converts methionine to cysteine, may render T cells particularly vulnerable to cysteine starvation compared to cancer cells (102). Supplementing T cells with arginine has been shown to promote the production of pro-inflammatory cytokines as well as a central memory phenotype in vitro, while also enhancing the T cell-mediated antitumor response in vivo (103–107). Conversely, the production of the arginine-degrading enzyme, arginase, in the TME has been known to causes arginine depletion and T cell anergy (104). Further, nitric oxide (NO), which is produced from arginine by nitric oxide synthases (NOS), may have cytotoxic effects on proliferating cells in the TME. However, mutated p53 may confer the cancer cells with enhanced resistance to NO-mediated cytotoxicity when compared to T effector cells (108–111).
Accumulation of Immune Suppressive Metabolic End-Products and By-Products
A fierce competition for limited carbon and nitrogen sources between tumor and T effector cells leads to the depletion of nutrients and accumulation of metabolic end-products and by-products, the latter of which also has a profound impact on T effector cells. Accumulation of lactic acid and CO2 results in the acidification of the TME, which suppresses T cell proliferation and impairs cytokine production and cytotoxic activity of T cells, while causing tumor radio resistance and promoting tumor cell migration and invasion (112–118). The acidification of the TME also profoundly impacts the cross-membrane transport of sodium ions and amino acids, as well as the pro inflammatory function of T effector cells (117–121). Additionally, tumor-derived potassium has been shown to potentially suppress T cell function (122). Stressed or damaged cells release ATP/ADP and their catabolic product adenosine into the TME, the latter of which elicits immune suppressive effects, partially through engaging cell-surface P2 purinergic receptors-mediated signaling in T cells (123–128). CD39 and CD73 are two ectonucleotidases that are widely expressed in the plasma membrane of cancer cells and cancer stromal cells and are responsible for the conversion of ADP/ATP to AMP and AMP to adenosine. As such, CD39 and CD73 play critical roles in determining the outcome of antitumor immunity through shifting ATP-driven pro-inflammatory effects to an anti-inflammatory milieu mediated by adenosine (129–131). Adenosine deaminase (ADA) converts adenosine to inosine, terminating adenosine-mediated immune suppressive effects (132). Consistent with this finding, the genetic loss of ADA results in an accumulation of adenosine, and leads to severe combined immunodeficiency (SCID) (133, 134).
The genetic context also plays a critical role in determining the composition of immunomodulatory metabolites in the TME, which differs dramatically in tumors with or without mutations in specific metabolic enzymes (135–137). R-2-hydroxyglutarate (2HG), which is enriched in tumors with gain-of-function isocitrate dehydrogenase 1/2 mutations, suppresses T cell activation and differentiation. Intriguingly, S-2HG (the other enantiomer of 2HG) supplement greatly enhances the anti-tumor capacity of adoptively transferred T cells (138). Tumors with succinate dehydrogenase (SDH) or fumarase mutations display elevated levels of succinate and fumarate, respectively. While the accumulation of intracellular 2HG, succinate and fumarate (often referred as onco-metabolites) may drive transformation in a tumor intrinsic manner, all these metabolites may indirectly contribute to tumorigenesis through their immunomodulatory effects (139, 140). A cell permeable form of fumarate, dimethyl fumarate (DMF), is the active ingredient of BG-12/TECFIDERA and FUMADERM, which have been widely used for treating autoimmune disorders (141–143). The anti-inflammatory activity of DMF has been partially attributed to its effect on suppressing T effector functions (36, 144, 145).
Depending on the nature of the metabolic stress imposed by the TME, cancer cells readily engage an array of signaling responses that are largely orchestrated by AMPK, mTOR or transcriptional factors, such as HIF1α, nuclear factor-like 2 (Nrf2), and general control non-derepressible 2 (GCN2). These stress responses are not only adaptive, but also cytoprotective and oncogenic, thus rendering cancer cells resistant to apoptosis and favoring the development of more aggressive, invasive and malignant phenotypes (146–150). Similarly, metabolic stresses favor the development of immune suppressive regulatory T (Treg) cells and tumor associated macrophages (TAM) (31, 32, 151–154). However, metabolic stresses are less tolerated in quickly proliferating T effector cells, leading to more cell death as well as less proliferation and cytokine production (14, 18, 39, 155–158). To survive, expand and exert robust anti-tumor activities in the TME, Teff cells must efficiently overcome the metabolic stress caused by the depletion of nutrients and accumulation of immune suppressive metabolites (Figure 1B). Recent research suggests that, the transfer of less differentiated Tmem subsets results in greater expansion, persistence and anti-tumor efficacy than terminally differentiated Teff cells. In particular, Tscm has a robust capacity for self-renewal and functional plasticity, though further differentiation into Tcm, Tem, and Teff subsets, thus providing a persistent anti-tumor immunity. Therefore, these cells could effectively compete with cancer stem cells over time to eradicate the tumor mass (159–161). Overall, less differentiated Tmem cells require increased mitochondrial FAO and spare respiratory capacity (SRC) for their long term survival and persistence (25, 162, 163). Clearly, the metabolic fitness of T cells is essential for successful adoptive immunotherapy.
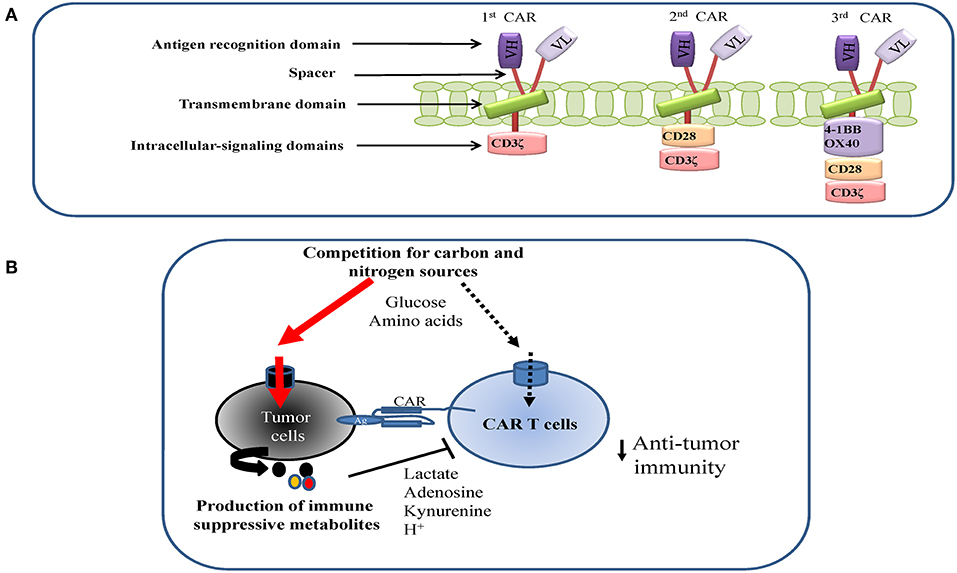
Figure 1. (A) CART design and structure. (B) Metabolic antagonism between tumor and CAR T cells in the tumor microenvironment (TME). Tumor cells may suppress CAR T cells proliferation and function by competing for key nutrients with CAR T cells and excreting immune-suppressive metabolites into the TME.
Metabolic Optimization of the Clinical Manufacture and Application of CAR T Cells
The recent breakthroughs of CD19 CAR T cell therapies to cure hematologic malignancies provide an exciting promise of extending this approach to solid tumors (164, 165). However, a critical barrier to using CAR T therapy for treating solid tumors that express appropriate antigens is the tumor's hostile microenvironment. A plethora of immunosuppressive mechanisms imposed by tumor cells suppress T cell proliferation, survival and effector function (155–169). In addition to the cytokine-mediated and cell-surface receptor-mediated signals that are essential for suppressing T cell functions and responses, the TME represents a dramatic example of metabolic derangement. Insufficient tumor vascularization due to disorganized blood vessel networks leads to hypoxia, lack of nutrients, acidosis, and the accumulation of metabolic waste and free radicals (170, 171). In addition, the increased nutrient and oxygen demands of tumor cells imposed by heightened oncogenic signaling further aggravates the metabolic stresses that suppress effector T cells function (6, 7, 46, 74, 75, 155, 169, 172, 173). As such, a rational and effective CAR T immunotherapy for solid tumors needs to integrate novel strategies which improve T cell metabolic fitness to overcome metabolic stresses imposed by the TME.
The standard manufacturing process of CAR T cells starts by obtaining the patient's peripheral blood mononuclear cells (PBMC) through leukapheresis, followed by T cell enrichment, activation and gene modification with viral or non-viral methods. These genetically modified T cells are then expanded to the required cell numbers for therapy, and then formulated and/or cryopreserved before infusion into the patient (Figure 2). The production of CAR T cells requires quality control testing throughout the entire process and is subjected to Good Manufacturing Practices (GMP) guidelines (174–177). We envision using the following metabolic strategies to optimize the manufacturing process and maximize the therapeutic efficacy of CAR T cells.
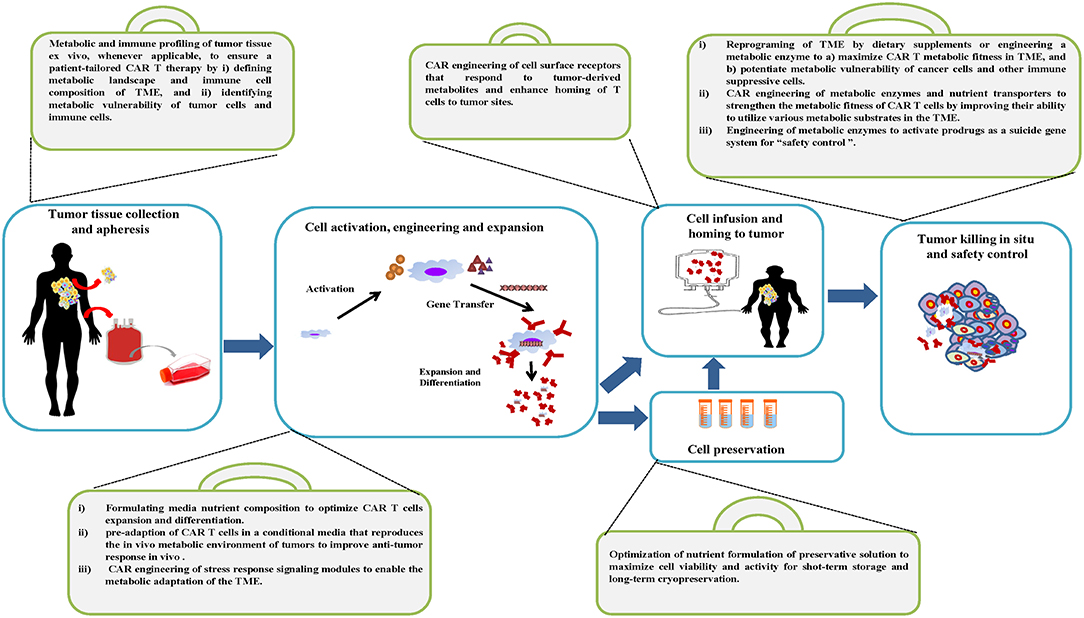
Figure 2. Metabolic tool box of the CAR T cell manufacturing process and therapy. A variety of metabolic strategies are proposed to be integrated into standard work flow to optimize the manufacturing process and maximize the therapeutic efficacy of CAR T cells.
Tumor Tissue Specimen Collection Before Leukapheresis
The personalized nature of CAR T therapy requires a patient-tailored strategy to ensure the robustness and reproducibility of personalized cell products. Whenever biopsy or surgery is applicable, a step-wise system of metabolic and analytic assessment is needed to determine the in situ immunomodulatory metabolic landscape in human tumor tissue specimens. Recently, Stable Isotope Resolved Metabolomics (SIRM) has been applied to assess the metabolic activities of thin tumor tissue slices, an adaptation of the original method of Otto Warburg's tissue slice technique (178–180). Along with conventional untargeted metabolomics, SIRM will provide complementary information, untargeted high-resolution mapping of the metabolic fate of carbon and nitrogen atoms from labeled precursors as well as quantification of nutrients and immune modulatory metabolites. In addition, the expression profile and immune modulatory impacts can be further assessed by combining RNAseq and Metabolomics-edited Transcriptomic Analysis (META) (181, 182); Using patient-derived tissue slides in studies preserves the fidelity of the original native cellular architecture and metabolic profiling on an individual patient basis. Integration of the above proposed approaches is poised to comprehensively profile the landscape of immunomodulatory metabolism in the TME, which may facilitate complementary metabolic improvements in the following steps of the manufacturing process of CAR T cells.
T Cell Activation, Genetic Modification, and Expansion
CARs are generated by combining the antigen-binding region of a monoclonal antibody with key stone intracellular-signaling domains. It consists of an extracellular targeting domain that recognizes a tumor specific antigen, which is derived from a single-chain variable fragment (scFv) of the variable heavy and variable light chains from a specific monoclonal antibody. When it is expressed on the surface of a T cell, the targeting domain allows CAR T cells to recognize and bind to the antigen that is presented by cancer cells. The intracellular signaling domain usually originates from the signal transduction subunit of co-stimulatory receptors, such as 4-1BB and CD28, which transduce extracellular ligand binding signal into CAR T cells to initiate the activation of downstream signaling cascades. CAR structure has been improved from first-generation CARs, which only had the CD3ζ signaling domain, to next generation CARs, which link the signaling endo domains of CD28, 4-1BB, and/or OX40 to provide co-stimulation signal (signal 2), which is required for optimal T cell activation (Figure 1A) (183–185). Optimal activation, gene transfer and culture conditions are essential to ensure the required cell number and quality are achieved for CAR T cell therapy. Due to the personalized nature of CAR T therapies, the degree of cell amplification, differentiation and functional activation can differ significantly from patient to patient. A patient tailored nutritional formulation for cell culture media may improve the robustness and reproducibility of cell expansion. In addition, emerging evidence suggests that the formula for commercial media does not truly reflect nutrient composition in a physiological context, which is required to ensure the metabolic fitness of cells in vivo (186–188). CAR T cells with co-stimulatory domain 4-1BB display higher mitochondrial metabolic activity than cells with a CD28 co-stimulatory domain (30). Consistent with this finding, mitochondrial characteristics including biogenesis and membrane potential have been suggested as key indicators for metabolic fitness and effector function in T cells (163, 189). Enhancing mitochondrial biogenesis through pharmacological or genetic approaches, or by enriching for cells with low mitochondrial membrane potential through cell sorting significantly improves T cell-mediated anti-tumor activities in vivo (163, 189–192). Conceivably, nutritional formulations that are tailored to meet the metabolic preferences of cells with different co-stimulatory domains may further enhance the metabolic robustness of CAR T cells.
Immune effector cells have evolved to respond to fluctuations in environmental nutrient levels and thus are able to adapt to environments with either sufficient or insufficient nutrient supply (156). We reason that the pre-adaption of CAR T cells in a conditional media that reproduces the in vivo metabolic environment of tumors may improve anti-tumor response in vivo. Given that blood flow and oxygen concentration fluctuate in solid tumors, the metabolic stress imposed on CAR T cells may also fluctuate in the TME (193, 194). As such, a fine-tuned adjustment of the severity, duration and frequency of metabolic stress may better recapitulate the metabolic conditions in the TME. Interestingly, transient glutamine restriction in vitro via either short-term nutrient starvation or metabolic inhibitor treatment enables a robust antitumor activity of adoptively transferred T effector cells in mouse models of immunotherapy (47, 195–197).
The immune cells response to the changes in the tumor's metabolic microenvironment represents a mechanism of “metabolic checkpoint” that coordinates metabolic status with cellular signaling, and in turn, determines immune function (14). Signaling kinases and transcription factors, such as AMPK and HIF1α can mediate adaptive responses that rewire T cell metabolism to determine immune function. HIF1α-dependent glycolytic pathway is preferentially enhanced in TH17 cells than Treg cells. Ablation of HIF1α or pharmacological inhibition of glycolysis reciprocally reduces TH17 cells and induce Treg cells differentiation. However, AMPK enhances Treg cells differentiation through negatively regulating OXPHOS (32, 198–205). Intrinsically edited signaling and transcriptional programs may ensure the engagement of fine-tuned metabolic programs to support T cell proliferation (associated with effector functions) or dormancy (associated with memory phenotypes) in response to the changing microenvironment (206). We reason that the engineering of the CAR by integrating a stress response signaling module may ensure a fine-tuned metabolic adaptation of T cells in the TME.
The immunosuppressive TME is one of the critical barriers for successful CAR T treatment in the solid tumor. The solid tumor is composed of lymphatic vessels, fibroblasts, infiltrated immune cells, stroma cells, and extracellular matrix (ECM) which constitute the complex TME. During tumor progression and as a result of the heightened glycolytic metabolism of cancer cells, solid tumors are subject to depleted nutrients, acidosis, and hypoxic conditions due to aberrant vascularization. Since hypoxia is a crucial aspect of the TME, targeting hypoxia could be an important strategy for promoting CAR T cell therapy in the solid tumor. Hypoxia induces the stabilization of the HIF-1α transcription factor, which regulates T cell metabolism and function. A recent study designed a novel CAR scaffold that includes the oxygen sensitive domain of HIF1α and provided a proof of concept for engineering microenvironment responsive CAR T cells. T cells which express this engineered HIF1-CAR are cable of responding to a hypoxic environment to display robust cytolytic activity in vitro (Figure 3) (207). In addition, CAR T cells that express distinct CAR co-stimulatory domains display different metabolic characters, which in turn impact memory phenotypes. 4-1BBζ CAR T cells exhibit enhanced mitochondrial SRC and mitochondrial biogenesis, which is associated with enhanced central memory phenotypes. Inclusion of CD28ζ in the CAR structure promotes effector memory differentiation and results in increased aerobic glycolysis in CAR T cells (30). In addition to the approach of engineering CAR structure, cytokine formulation can be optimized to modulate metabolic programs and promote the Tscm and Tcm phenotype that is associated with enhanced CAR T cell persistence and anti-tumor immunity in vivo (208). T cells, activated with anti-CD3/CD28 antibody, followed by expansion in the presence of IL-15 and IL-7, not only mimic a more Tscm like phenotype, but and also exhibit increased production of IFNγ, TNFα, and IL-2 as well as cytolytic activity against target cells expressing the CAR specific antigen (209, 210). It has been suggested that IL-15 promotes SRC and FAO by upregulating carnitine palmitoyl transferase, a rate-limiting metabolic enzyme that controls FAO. Since Tmem cells preferentially engage OXPHOS rather than glycolysis, IL-15 may promote Tmem cell differentiation and bioenergetic stability partially by regulating mitochondrial metabolism (162).
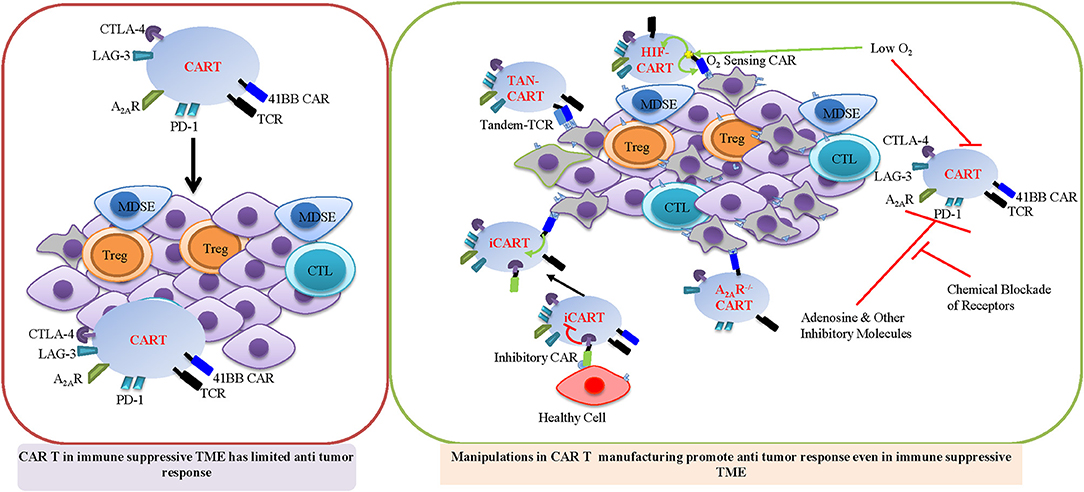
Figure 3. Important strategy for promoting CAR T cell therapy in immune suppressive TME. Manipulations during CAR T manufacturing HIF-CAR, TAN-CAR, and iCAR will promote CAR T function in immune suppressive TME. HIF-CAR T cells are activated by low oxygen concentrations. Inhibitory CAR (iCAR) limits T cell activation in the presence of off-target cells. Tandem CARs (Tan CAR) requires bispecific activation which reduces off target effects. A2AR−/− CAR T cells are resistant to adenosine mediated INF-γ suppression.
CAR T Cell Preservation
The nutritional optimization of preservative solutions is an important factor for ensuring successful reperfusion and CAR T therapy. As such, a detailed understanding of the metabolic impact on cells during short-/long-term storage or during the reperfusion period is required. The nutritional formulation requires the capability to regenerate ATP, buffer ions and scavenge free radicals in CAR T cells during the period of preservation (211–215). Finally, any inconsistency between the nutrient formulations that are utilized for preservation and those used for ex vivo cell manipulation and expansion may potentially render the cells more susceptible to preservation damage.
CAR T Cell Infusion and Homing to Tumor
Trafficking of T cells into the site of the tumor is required for achieving an optimal CAR T therapeutic response (216, 217). Novel synthetic biology approaches are needed to engineer T cells to logically respond to metabolites that are enriched in the TME, ensuring a fine-tuned tumor recognition and homing response (218, 219). For instance, adenosine is enriched in the TME and suppresses the T cell response partially by engaging cell-surface purinergic receptors-mediated signaling in T cells (220, 221). One theoretical strategy is to engineer an adenosine-responding CAR by fusing the extracellular domain of adenosine receptor to intracellular costimulatory domains, thus artificially switching a normally suppressive signal to an activating signal. We envision that a dual CAR system, with one for a tumor antigen and one for a tumor-associated metabolite, may further enhance the specificity and responsiveness of CAR T cells that may be deployed against tumors in a wide range of sites.
Tumor Killing in situ and Safety Control
The capacity for cell proliferation and persistence in the TME is the best predictor of clinical efficacy in CAR T therapy. Nutritional supplements may overcome the metabolically suppressive microenvironment and may enable CAR T cells to persist and expand, ensuring the optimal “effector vs. target” ratio for the clearance of the tumor in vivo. L-arginine is considered a conditionally essential amino acid and supplementation with L-arginine enhances the antitumor response of adoptively transferred T cells in animal models (107). Clearly, arginine is also a metabolic vulnerability of cancer cells in the TME, since circulating arginine is essential for supporting tumor growth (222, 223). Therefore, we need to understand the impact of supplemental nutrients on both T cells and tumor cells to better stratify this approach in the future.
The genetic and enzymatic approaches that enable the conversion of immune suppressive metabolites to either inert compounds or pro-inflammatory compounds are promising strategies to reprogram metabolic TME. The concentration of lactate in vertebrate plasma ranges from 1 to 30 mM under physiological and pathological conditions (224). While lactate accumulated in the TME is generally considered as metabolic “waste,” muscle cells, neurons and certain tumor cells are known to be able to take up and oxidize lactate (225–227). Consistent with these findings, emerging evidence suggests that lactate is a key carbon source in vivo and can be oxidized in the mitochondria to generate energy and feed into cataplerotic routes of the TCA cycle (228–236). Enforced expression of phosphoenolpyruvate carboxykinase 1 (PCK1), which presumably increases gluconeogenesis from lactate and thus alleviates the stress from glucose restriction in the TME, has been shown to enhance anti-tumor T cell responses in animal models (167). Also, we envision a rewired lactate flux, by enforcing LDHB expression, may render T cells capable of utilizing tumor-derived lactate, and thus may strengthen the metabolic fitness of CAR T cells in the TME.
One recent study has demonstrated that the systemic delivery of an enzyme that efficiently depletes kynurenine in the TME can enhance the T cell-mediated antitumor immunity (237). Similarly, polyethylene glycol–conjugated adenosine deaminase (PEG-ADA) and ADA gene therapies have been successfully employed to treat ADA-SCID patients (238, 239). Since only immature and transformed T cells display high ADA activity (240–242), systemic ADA treatment through PEG-ADA supplementation may be a new, complementary strategy to optimize the potency and durability of CAR T therapy. It is also conceivable that an engineered oncolytic virus or CAR T cells which express ADA may offer additional strategies to locally deliver ADA into the TME. Nucleoside transporters can rapidly remove adenosine from the extracellular nucleoside pool and direct adenosine into nucleoside salvage to support RNA/DNA synthesis. Thus, expression of high affinity nucleoside transporters may divert adenosine from eliciting an immune-suppressive purinergic signaling response into promoting T cell proliferation (243, 244). Similarly, genetic strategies which enforce the expression and affinity of transports of the key carbon and nitrogen donors (i.e., glucose and glutamine) may confer a selective advantage on T cells over tumor cells in the TME.
Another promising therapeutic strategy for remodeling the microenvironment is to modulate ion and pH balances. Reducing the potassium level through enforced expression of potassium channels in T cells can enhance their antitumor activity (122). A lactate-binding compound has been recently developed as a potent pharmaceutical approach for engineering metabolic flux of lactate and normalizing the pH in vivo (245).
The naturally occurring T cell mediated response is largely controlled through cell autonomous mechanisms. However, CAR T therapies often lead to a series of adverse effects that may be reduced through the fine-tuned regulation of the timing, location and amplitude of T cell activity. The principal of the recently developed “ON-switch” CARs can be theoretically extended by employing “metabolic switches” to control T cell activities in real-time (246, 247). In addition, the engineering of metabolic enzymes to activate prodrugs has been exploited as a suicide gene system (248–250). A similar strategy can be exploited as a “safety switch” to enable the selective ablation of CAR T cells at will by providing prodrugs and therefore limiting the on-target, but off-tumor toxicities of CAR T cells (251–253).
Future Directions
Cancer cachexia, which is responsible for the death of more than 20% of cancer patients, is characterized by dramatic body weight loss and disproportionate wasting of skeletal muscle (254–256). Supportive care including dietary treatment and physical exercise that can maintain energy balance, as well as increase insulin sensitivity, protein synthesis rate, and anti-oxidative enzyme activity, which is beneficial for relieving symptoms of cachexia (257–260). Similar strategies may be applied as systemic approaches to enhance anti-tumor immunity by fostering an optimized metabolic environment for immune cells.
The fast moving CAR T therapy field has generated tremendous excitement and will likely change the paradigm of therapeutic interventions for solid tumors. Manipulation of metabolism has been demonstrated as a potential approach to the regulation of immune responses in various preclinical settings. Beyond CAR T therapy, the above potential metabolic modulations can also be considered as adjunctive therapy together with other immunotherapies, including checkpoint blockade and cancer vaccines (261–265). TME consists of an intricate, highly complex and dynamic network of immune cell subsets. Other immune cell subsets, such as natural killer (NK) cells, TAM and MDSC are universally found in the TME and are key players in anti-tumor immunity (266, 267). We will need to continue to decipher the essential metabolic pathways by analyzing metabolic flux and assessing the consequences of metabolic intervention on these pathways in all key cellular components of the TME. Understanding how control of (or by) metabolic pathways impacts anti-tumor immune responses in these cell types is required to selectively strengthen the metabolic fitness in effector T cells and potentiate the metabolic vulnerabilities of tumor cells and immune-suppressive cells. Finally, Strategies to improve immune cell metabolic fitness may be applicable across a broad panel of cancer immunotherapies including checkpoint blockade and cancer vaccines (261–265).
Author Contributions
XX, JG, and JS wrote the manuscript and RW wrote and finalize the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors at the time of review.
Acknowledgments
This work was supported by 1UO1CA232488-01 and 1R01AI114581 from National Institute of Health, and 128436-RSG-15-180-01-LIB from the American Cancer Society (RW).
References
1. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
2. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
3. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. (2009) 29:625. doi: 10.1038/onc.2009.441
5. San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis. (2017) 38:119–33. doi: 10.1093/carcin/bgw127
6. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
7. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
8. Goncalves MD, Cantley LC. A glycolysis outsider steps into the cancer spotlight. Cell Metab. (2018) 28:3–4. doi: 10.1016/j.cmet.2018.06.017
9. Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. (2010) 24:2784–99. doi: 10.1101/gad.1985910
10. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. (2013) 493:689–93. doi: 10.1038/nature11776
11. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. (2009) 92:329–33. doi: 10.1016/j.radonc.2009.06.025
12. Boroughs LK, Deberardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. (2015) 17:351. doi: 10.1038/ncb3124
13. Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity. (2018) 48:214–26. doi: 10.1016/j.immuni.2018.02.010
14. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. (2012) 13:907–15. doi: 10.1038/ni.2386
15. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
16. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
17. O'neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
18. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
19. Ma EH, Poffenberger MC, Wong AH, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol. (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
20. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
21. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. (2015) 125:194–207. doi: 10.1172/JCI76012
22. O'sullivan D, Van Der Windt GJ, Huang SC-C, Curtis JD, Chang C-H, Buck MD, et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
23. Cui G, Staron MM, Gray SM, Ho P-C, Amezquita RA, Wu J, et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell. (2015) 161:750–61. doi: 10.1016/j.cell.2015.03.021
24. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
25. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
26. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. (2012) 209:2441–53. doi: 10.1084/jem.20112607
27. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. (2012) 33:168–73. doi: 10.1016/j.it.2012.01.010
28. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. (2016) 7:13683. doi: 10.1038/ncomms13683
29. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. (2013) 123:4479–88. doi: 10.1172/JCI69589
30. Kawalekar OU, O'connor RS, Fraietta JA, Guo L, Mcgettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
31. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, Maciver NJ, Mason EF, et al. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
32. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
33. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
34. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
35. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity. (2016) 45:540–54. doi: 10.1016/j.immuni.2016.08.017
36. Lian G, Gnanaprakasam JR, Wang T, Wu R, Chen X, Liu L, et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. eLife. (2018) 7:e36158. doi: 10.7554/eLife.36158
37. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA. (2018) 115:E6546–55. doi: 10.1073/pnas.1720113115
38. Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. (2011) 11:109–17. doi: 10.1038/nri2888
39. Wang R, Green DR. The immune diet: meeting the metabolic demands of lymphocyte activation. F1000 Biol Rep. (2012) 4:9. doi: 10.3410/B4-9
40. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. (2008) 134:97–111. doi: 10.1016/j.cell.2008.04.052
41. Igarashi K, Kashiwagi K. Modulation of cellular function by polyamines. Int J Biochem Cell Biol. (2010) 42:39–51. doi: 10.1016/j.biocel.2009.07.009
42. Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. (2010) 11:nrm2977. doi: 10.1038/nrm2977
43. Moschou PN, Roubelakis-Angelakis KA. Polyamines and programmed cell death. J Exp Bot. (2014) 65:1285–96. doi: 10.1093/jxb/ert373
44. Massiere F, Badet-Denisot M-A. The mechanism of glutamine-dependent amidotransferases. Cell Mol Life Sci. (1998) 54:205–22. doi: 10.1007/s000180050145
45. Zalkin H, Smith JL. Enzymes utilizing glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. (1998) 72:87–144. doi: 10.1002/9780470123188.ch4
46. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. (2013) 3:a014217. doi: 10.1101/cshperspect.a014217
47. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–95 e1719. doi: 10.1016/j.cell.2018.10.001
48. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity. (2017) 46:675–89. doi: 10.1016/j.immuni.2017.03.019
49. Wang R, Gnanaprakasam JNR, Wu R. Metabolic reprogramming in modulating T cell ROS generation and antioxidant capacity. Front Immunol. (2018) 9:1075. doi: 10.3389/fimmu.2018.01075
50. Douglas NC, Jacobs H, Bothwell AL, Hayday AC. Defining the specific physiological requirements for c-Myc in T cell development. Nat Immunol. (2001) 2:307–15. doi: 10.1038/86308
51. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/S1074-7613(02)00323-0
52. Grumont R, Lock P, Mollinari M, Shannon FM, Moore A, Gerondakis S. The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity. (2004) 21:19–30. doi: 10.1016/j.immuni.2004.06.004
53. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
54. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. (2012) 12:325–38. doi: 10.1038/nri3198
55. Maciver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
56. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
57. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
58. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBio Med. (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
59. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
60. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. (2010) 107:7461–6. doi: 10.1073/pnas.1002459107
61. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity–mediated metabolic programming and clonal expansion of T cells. Nat Immunol. (2013) 14:1155. doi: 10.1038/ni.2710
62. Chou C, Pinto AK, Curtis JD, Persaud SP, Cella M, Lin C-C, et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol. (2014) 15:884. doi: 10.1038/ni.2943
63. Man K, Kallies A. Bcl-6 gets T cells off the sugar. Nat Immunol. (2014) 15:904. doi: 10.1038/ni.2993
64. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
65. Newton RH, Shrestha S, Sullivan JM, Yates KB, Compeer EB, Ron-Harel N, et al. Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat Immunol. (2018) 19:838–48. doi: 10.1038/s41590-018-0157-4
66. Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. (2008) 118:3930–42. doi: 10.1172/JCI36843
67. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
68. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. (2011) 333:233–8. doi: 10.1126/science.1198973
69. Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. (2011) 7:e1002229. doi: 10.1371/journal.pgen.1002229
70. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
71. Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. (2011) 20:370–83. doi: 10.1016/j.ccr.2011.08.010
72. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. (2012) 72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949
73. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. (2001) 167:6869–76. doi: 10.4049/jimmunol.167.12.6869
74. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
75. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. doi: 10.1038/nrc2981
76. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. (2007) 117:1147–54. doi: 10.1172/JCI31178
77. Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. (2009) 9:445–52. doi: 10.1038/nrc2639
78. Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, et al. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. (2012) 72:5649–57. doi: 10.1158/0008-5472.CAN-12-0549
79. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA. (2012) 109:2497–502. doi: 10.1073/pnas.1113873109
80. Veldhoen M, Hirota K, Christensen J, O'garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. (2009) 206:43–9. doi: 10.1084/jem.20081438
81. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
82. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA. (2010) 107:19961–6. doi: 10.1073/pnas.1014465107
83. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
84. Platten M, Wick W, Van Den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
85. Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol. (2013) 25:335–43. doi: 10.1093/intimm/dxt011
86. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2011) 185:1037–44. doi: 10.4049/jimmunol.0903586
87. Dang CV. A metabolic perspective of Peto's paradox and cancer. Philos Trans R Soc B Biol Sci. (2015) 370:20140223. doi: 10.1098/rstb.2014.0223
88. Kosower NS, Kosower EM. The glutathione status of cells. Int Rev Cytol. (1978) 54:109–60. doi: 10.1016/S0074-7696(08)60166-7
89. Meister A. Metabolism and function of glutathione: an overview. Biochem Soc Trans. (1982) 10:78–9. doi: 10.1042/bst0100078
90. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci USA. (2002) 99:1491–6. doi: 10.1073/pnas.022630299
91. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation. J Exp Med. (2002) 195:59–70. doi: 10.1084/jem.20010659
92. Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. (2006) 43:143–81. doi: 10.1080/10408360500523878
93. D'autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. (2007) 8:813–24. doi: 10.1038/nrm2256
94. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. (2009) 8:579–91. doi: 10.1038/nrd2803
95. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, Von Essen MR, Woetmann A, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep. (2012) 2:266. doi: 10.1038/srep00266
96. Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
97. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. (2012) 48:158–67. doi: 10.1016/j.molcel.2012.09.025
98. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. (2013) 12:931. doi: 10.1038/nrd4002
99. Hensley CT, Wasti AT, Deberardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
100. Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci. (2015) 22:85. doi: 10.1186/s12929-015-0194-3
101. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:619–34. doi: 10.1038/nrc.2016.71
102. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. (2010) 70:68–77. doi: 10.1158/0008-5472.CAN-09-2587
103. Lind DS. Arginine and cancer. J Nutr. (2004) 134:2837S−41S; discussion 2853S. doi: 10.1093/jn/134.10.2837S
104. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
105. Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des. (2008) 14:1049–57. doi: 10.2174/138161208784246199
106. Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, Vasson MP, et al. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol. (2012) 280:182–90. doi: 10.1016/j.cellimm.2012.11.018
107. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42 e813. doi: 10.1016/j.cell.2016.09.031
108. Ambs S, Merriam WG, Ogunfusika MO, Bennett WP, Ishibe N, Hussain SP, et al. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat Med. (1998) 4:1371–6. doi: 10.1038/3957
109. Rajnakova A, Moochhala S, Goh PM, Ngoi S. Expression of nitric oxide synthase, cyclooxygenase, and p53 in different stages of human gastric cancer. Cancer Lett. (2001) 172:177–85. doi: 10.1016/S0304-3835(01)00645-0
110. Bonavida B, Baritaki S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: Downregulation of the NF-kappaB/Snail/YY1/RKIP circuitry. Nitric Oxide. (2011) 24:1–7. doi: 10.1016/j.niox.2010.10.001
111. Wongvaranon P, Pongrakhananon V, Chunhacha P, Chanvorachote P. Acquired resistance to chemotherapy in lung cancer cells mediated by prolonged nitric oxide exposure. Anticancer Res. (2013) 33:5433–44.
112. Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res. (2003) 9:1957–71.
113. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
114. Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. (2009) 182:2476–84. doi: 10.4049/jimmunol.0802059
115. Annibaldi A, Widmann C. Glucose metabolism in cancer cells. Curr Opin Clin Nutr Metab Care. (2010) 13:466–70. doi: 10.1097/MCO.0b013e32833a5577
116. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. (2010) 184:1200–9. doi: 10.4049/jimmunol.0902584
117. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
118. Bohn T, Rapp S, Luther N, Klein M, Bruehl T-J, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. (2018) 19:1319. doi: 10.1038/s41590-018-0226-8
119. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. (2013) 496:518–22. doi: 10.1038/nature11868
120. Reshkin SJ, Cardone RA, Harguindey S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat Anticancer Drug Discov. (2013) 8:85–99. doi: 10.2174/1574892811308010085
121. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. (2013) 496:513–7. doi: 10.1038/nature11984
122. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. (2016) 537:539–43. doi: 10.1038/nature19364
123. Burnstock G. (2006). Purinergic signalling–an overview. Novartis Found Symp. 276, 26–48; discussion 48–57, 275–281. doi: 10.1002/9780470032244.ch4
124. Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. (2008) 7:575–90. doi: 10.1038/nrd2605
125. Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. (2009) 32:19–29. doi: 10.1016/j.tins.2008.10.001
126. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. (2011) 11:201–12. doi: 10.1038/nri2938
127. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
128. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. (2016) 16:177–92. doi: 10.1038/nri.2016.4
129. Stagg J, Smyth M. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. (2010) 29:5346. doi: 10.1038/onc.2010.292
130. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. (2013) 19:355–67. doi: 10.1016/j.molmed.2013.03.005
131. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD 39 and CD 73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
132. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest. (1997) 100:2849–57. doi: 10.1172/JCI119833
133. Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. (1972) 2:1067–9. doi: 10.1016/S0140-6736(72)92345-8
134. Parkman R, Gelfand EW, Rosen FS, Sanderson A, Hirschhorn R. Severe combined immunodeficiency and adenosine deaminase deficiency. N Engl J Med. (1975) 292:714–9. doi: 10.1056/NEJM197504032921402
135. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. (2010) 102:932–41. doi: 10.1093/jnci/djq187
136. Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. (2011) 1807:1432–43. doi: 10.1016/j.bbabio.2011.07.003
137. Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl). (2011) 89:213–20. doi: 10.1007/s00109-011-0728-4
138. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. (2016) 540:236–41. doi: 10.1038/nature20165
139. Mills E, O'neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. (2014) 24:313–20. doi: 10.1016/j.tcb.2013.11.008
140. Sciacovelli M, Frezza C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. (2016) 100:175–81. doi: 10.1016/j.freeradbiomed.2016.04.025
141. Mrowietz U, Altmeyer P, Bieber T, Rocken M, Schopf RE, Sterry W. Treatment of psoriasis with fumaric acid esters (Fumaderm). J Dtsch Dermatol Ges. (2007) 5:716–7. doi: 10.1111/j.1610-0387.2007.06346.x
142. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. (2012) 367:1087–97. doi: 10.1056/NEJMoa1206328
143. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. (2012) 367:1098–107. doi: 10.1056/NEJMoa1114287
144. Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci USA. (2016) 113:4777–82. doi: 10.1073/pnas.1603907113
145. Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao-Draayer Y. Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J Immunol. (2017) 198:3069–80. doi: 10.4049/jimmunol.1601532
146. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. (2006) 6:729–34. doi: 10.1038/nrc1974
147. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. (2009) 9:691–700. doi: 10.1038/nrc2715
148. Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol. (2012) 4:a008763. doi: 10.1101/cshperspect.a008763
149. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. (2013) 73:2929–35. doi: 10.1158/0008-5472.CAN-12-3876
150. Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. (2006) 25:6373–83. doi: 10.1038/sj.onc.1209889
151. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
152. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. (2015) 13:e1002202. doi: 10.1371/journal.pbio.1002202
153. Angelin A, Gil-De-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93 e1287. doi: 10.1016/j.cmet.2016.12.018
154. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA. (2017) 114:580–5. doi: 10.1073/pnas.1614035114
155. Wang T, Liu G, Wang R. The intercellular metabolic interplay between tumor and immune cells. Front Immunol. (2014) 5:358. doi: 10.3389/fimmu.2014.00358
156. Slack M, Wang T, Wang R. T cell metabolic reprogramming and plasticity. Mol Immunol. (2015) 68:507–12. doi: 10.1016/j.molimm.2015.07.036
157. Gnanaprakasam JNR, Sherman JW, Wang R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. (2017) 35:63–70. doi: 10.1016/j.cytogfr.2017.03.004
158. Sugiura A, Rathmell JC. Metabolic barriers to T cell function in tumors. J Immunol. (2018) 200:400–7. doi: 10.4049/jimmunol.1701041
159. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. (2005) 115:1616–26. doi: 10.1172/JCI24480
160. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. (2005) 102:9571–6. doi: 10.1073/pnas.0503726102
161. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. (2012) 12:671–84. doi: 10.1038/nrc3322
162. Van Der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
163. Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. (2016) 23:63–76. doi: 10.1016/j.cmet.2015.11.002
164. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
165. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. (2014) 65:333–47. doi: 10.1146/annurev-med-060512-150254
166. Chang CH, Qiu J, O'sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
167. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
168. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. (2015) 162:1242–56. doi: 10.1016/j.cell.2015.08.052
169. O'sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. (2015) 36:71–80. doi: 10.1016/j.it.2014.12.004
170. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. (2011) 17:1359–70. doi: 10.1038/nm.2537
171. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
172. Denicola GM, Cantley LC. Cancer's fuel choice: new flavors for a picky eater. Mol Cell. (2015) 60:514–23. doi: 10.1016/j.molcel.2015.10.018
173. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
174. Kaiser AD, Assenmacher M, Schroder B, Meyer M, Orentas R, Bethke U, et al. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther. (2015) 22:72–8. doi: 10.1038/cgt.2014.78
175. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. (2016) 3:16015. doi: 10.1038/mto.2016.15
176. Levine BL, Miskin J, Wonnacott K, Keir C. Global Manufacturing of CAR T Cell Therapy. Mol Ther Methods Clin Dev. (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
177. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
178. Warburg O, Minami S. Versuche an überlebendem carcinom-gewebe. J Mol Med. (1923) 2:776–7. doi: 10.1007/BF01712130
179. Fan TW, Lane AN, Higashi RM. Stable isotope resolved metabolomics studies in ex vivo tissue slices. Bio Protoc. (2016) 6:e1730. doi: 10.21769/BioProtoc.1730
180. Fan TW, Warmoes MO, Sun Q, Song H, Turchan-Cholewo J, Martin JT, et al. Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator beta-glucan in a two-case ex vivo non-small-cell lung cancer study. Cold Spring Harb Mol Case Stud. (2016) 2:a000893. doi: 10.1101/mcs.a000893
181. Fan T, Bandura L, Higashi R, Lane A. Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer cells. Metabolomics. (2005) 1:325–39. doi: 10.1007/s11306-005-0012-0
182. Fan TW-M, Lane AN, Higashi RM editor. Metabolomics-edited transcriptomics analysis (META). In: The Handbook of Metabolomics: Pathway and Flux Analysis, Methods in Pharmacology and Toxicology. New York, NY: Springer Science (2012). p. 484. doi: 10.1007/978-1-61779-618-0
183. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. (2012) 1:458–66. doi: 10.4161/onci.19855
184. Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. (2016) 13:25–40. doi: 10.1038/nrclinonc.2015.187
185. Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst. (2016) 108:djv439. doi: 10.1093/jnci/djv439
186. Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. (2017) 169:258–72 e217. doi: 10.1016/j.cell.2017.03.023
187. Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. (2017) 6:e27713. doi: 10.7554/eLife.27713
188. Voorde JV, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv. (2019) 5:eaau7314. doi: 10.1126/sciadv.aau7314
189. Buck MD, O'sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
190. Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci USA. (2017) 114:E761–70. doi: 10.1073/pnas.1620433114
191. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8(+) T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res. (2018) 6:1375–87. doi: 10.1158/2326-6066.CIR-18-0095
192. Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. (2018) 562:423–8. doi: 10.1038/s41586-018-0597-x
193. Brown JM. The hypoxic cell: a target for selective cancer therapy–eighteenth Bruce F. Cain Memorial Award lecture Cancer Res. (1999) 59:5863–70.
194. Milotti E, Stella S, Chignola R. Pulsation-limited oxygen diffusion in the tumour microenvironment. Sci Rep. (2017) 7:39762. doi: 10.1038/srep39762
195. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
196. D'aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. (2018). CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 9:282. doi: 10.1038/s41419-018-0278-6
197. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer Sci. (2018) 109:3737–50. doi: 10.1111/cas.13827
198. Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. (2005) 5:712–21. doi: 10.1038/nri1685
199. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
200. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. (2009) 9:563–75. doi: 10.1038/nrc2676
201. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
202. Maciver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. (2011) 187:4187–98. doi: 10.4049/jimmunol.1100367
203. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. (2011) 13:1016–23. doi: 10.1038/ncb2329
204. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. (2012) 13:251–62. doi: 10.1038/nrm3311
205. O'neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. (2013) 493:346–55. doi: 10.1038/nature11862
206. Wang A, Luan HH, Medzhitov R. An evolutionary perspective on immunometabolism. Science. (2019) 363:eaar3932. doi: 10.1126/science.aar3932
207. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. (2017) 7:39833. doi: 10.1038/srep39833
208. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. (2015) 17:487–95. doi: 10.1016/j.jcyt.2014.12.002
209. Gomez-Eerland R, Nuijen B, Heemskerk B, Van Rooij N, Van Den Berg JH, Beijnen JH, et al. Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Hum Gene Ther Methods. (2014) 25:277–87. doi: 10.1089/hgtb.2014.004
210. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
211. Lovelock JE. The haemolysis of human red blood-cells by freezing and thawing. Biochim Biophys Acta. (1953) 10:414–26. doi: 10.1016/0006-3002(53)90273-X
212. Southard JH, Van Gulik TM, Ametani MS, Vreugdenhil PK, Lindell SL, Pienaar BL, et al. Important components of the UW solution. Transplantation. (1990) 49:251–7. doi: 10.1097/00007890-199002000-00004
213. Southard JH, Belzer FO. Organ preservation. Annu Rev Med. (1995) 46:235–47. doi: 10.1146/annurev.med.46.1.235
214. Taylor MJ, Campbell LH, Rutledge RN, Brockbank KG. Comparison of unisol with Euro-collins solution as a vehicle solution for cryoprotectants. Transplant Proc. (2001) 33:677–9. doi: 10.1016/S0041-1345(00)02198-9
215. Baust JG, Gao D, Baust JM. Cryopreservation: an emerging paradigm change. Organogenesis. (2009) 5:90–6. doi: 10.4161/org.5.3.10021
216. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
217. Van Der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, De Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. (2017) 3:797–808. doi: 10.1016/j.trecan.2017.09.006
218. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
219. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. (2018) 173:1426–38 e1411. doi: 10.1016/j.cell.2018.03.038
220. Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. (2006) 5:247–64. doi: 10.1038/nrd1983
221. Sek K, Molck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. (2018) 19:E3837. doi: 10.3390/ijms19123837
222. Patil MD, Bhaumik J, Babykutty S, Banerjee UC, Fukumura D. Arginine dependence of tumor cells: targeting a chink in cancer's armor. Oncogene. (2016) 35:4957–72. doi: 10.1038/onc.2016.37
223. Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. (2018) 563:569–73. doi: 10.1038/s41586-018-0697-7
224. Merezhinskaya N, Fishbein WN. Monocarboxylate transporters: past, present, and future. Histol Histopathol. (2009) 24:243–64. doi: 10.14670/HH-24.243
225. Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. (2004) 558:5–30. doi: 10.1113/jphysiol.2003.058701
226. Philp A, Macdonald AL, Watt PW. Lactate–a signal coordinating cell and systemic function. J Exp Biol. (2005) 208:4561–75. doi: 10.1242/jeb.01961
227. Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. (2012) 18:1319–30. doi: 10.2174/138161212799504902
228. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. (2002) 277:30409–12. doi: 10.1074/jbc.R200006200
229. Chen YJ, Mahieu NG, Huang X, Singh M, Crawford PA, Johnson SL, et al. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol. (2016) 12:937–43. doi: 10.1038/nchembio.2172
230. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell. (2017) 171:358–71 e359. doi: 10.1016/j.cell.2017.09.019
231. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. (2017) 551:115–8. doi: 10.1038/nature24057
232. Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife. (2017) 6:e28899. doi: 10.7554/eLife.28899
233. Brooks GA. The science and translation of lactate shuttle theory. Cell Metab. (2018) 27:757–85. doi: 10.1016/j.cmet.2018.03.008
234. Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z, Gladden LB. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol. (2018) 118:691–728. doi: 10.1007/s00421-017-3795-6
235. Niu X, Chen YJ, Crawford PA, Patti GJ. Transport-exclusion pharmacology to localize lactate dehydrogenase activity within cells. Cancer Metab. (2018) 6:19. doi: 10.1186/s40170-018-0192-5
236. Passarella S, Schurr A. l-Lactate transport and metabolism in mitochondria of Hep G2 cells-the cori cycle revisited. Front Oncol. (2018) 8:120. doi: 10.3389/fonc.2018.00120
237. Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol. (2018) 36:758–64. doi: 10.1038/nbt.4180
238. Hershfield MS, Buckley RH, Greenberg ML, Melton AL, Schiff R, Hatem C, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med. (1987) 316:589–96. doi: 10.1056/NEJM198703053161005
239. Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. (2009) 360:447–58. doi: 10.1056/NEJMoa0805817
240. Ben-Bassat I, Simoni F, Holtzman F, Ramot B. Adenosine deaminase activity of normal lymphocytes and leukemic cells. Isr J Med Sci. (1979) 15:925–7.
241. Minkowski MD, Castellazzi M, Buttin G. Lack of adenosine deaminase activity in cultured murine cytotoxic T lymphocytes. J Immunol. (1984) 133:52–8.
242. Minkowski MD, Bandeira A. Different functional subsets of cultured murine T cells express characteristic levels of adenosine deaminase activity. Cell Immunol. (1985) 95:380–91. doi: 10.1016/0008-8749(85)90325-9
243. Boswell-Casteel RC, Hays FA. Equilibrative nucleoside transporters–a review. Nucleosides Nucleotides Nucleic Acids. (2017) 36:7–30. doi: 10.1080/15257770.2016.1210805
244. Pastor-Anglada M, Perez-Torras S. Emerging roles of nucleoside transporters. Front Pharmacol. (2018) 9:606. doi: 10.3389/fphar.2018.00606
245. Acharya AP, Rafi M, Woods EC, Gardner AB, Murthy N. Metabolic engineering of lactate dehydrogenase rescues mice from acidosis. Sci Rep. (2014) 4:5189. doi: 10.1038/srep05189
246. Fleming AW. ‘Normal' overaction of inferior oblique muscles. Arch Ophthalmol. (1989) 107:1113. doi: 10.1001/archopht.1989.01070020179003
247. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
248. Bennett EM, Anand R, Allan PW, Hassan AE, Hong JS, Levasseur DN, et al. Designer gene therapy using an Escherichia coli purine nucleoside phosphorylase/prodrug system. Chem Biol. (2003) 10:1173–81. doi: 10.1016/j.chembiol.2003.11.008
249. Bennett EM, Li C, Allan PW, Parker WB, Ealick SE. Structural basis for substrate specificity of Escherichia coli purine nucleoside phosphorylase. J Biol Chem. (2003) 278:47110–8. doi: 10.1074/jbc.M304622200
250. Zhang Y, Parker WB, Sorscher EJ, Ealick SE. PNP anticancer gene therapy. Curr Top Med Chem. (2005) 5:1259–74. doi: 10.2174/156802605774463105
251. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
252. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
253. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol. (2014) 5:254. doi: 10.3389/fphar.2014.00254
254. Meacham LR, Mazewski C, Krawiecki N. Mechanism of transient adrenal insufficiency with megestrol acetate treatment of cachexia in children with cancer. J Pediatr Hematol Oncol. (2003) 25:414–7. doi: 10.1097/00043426-200305000-00013
255. Kumar NB, Kazi A, Smith T, Crocker T, Yu D, Reich RR, et al. Cancer cachexia: traditional therapies and novel molecular mechanism-based approaches to treatment. Curr Treat Options Oncol. (2010) 11:107–17. doi: 10.1007/s11864-010-0127-z
256. Aoyagi T, Terracina KP, Raza A, Matsubara H, Takabe K. Cancer cachexia, mechanism and treatment. World J Gastrointest Oncol. (2015) 7:17–29. doi: 10.4251/wjgo.v7.i4.17
257. Schavelzon J. On the mechanism of cancer cachexia and the possibilities of treatment. Prensa Med Argent. (1956) 43:448–50.
258. Li T, Li C. Mechanism and treatment of cancer cachexia in tumor-bearing mice. Zhonghua Zhong Liu Za Zhi. (1997)19:188–91.
259. Lenk K, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle. (2010) 1:9–21. doi: 10.1007/s13539-010-0007-1
260. De Vos-Geelen J, Fearon KC, Schols AM. The energy balance in cancer cachexia revisited. Curr Opin Clin Nutr Metab Care. (2014) 17:509–14. doi: 10.1097/MCO.0000000000000106
261. Morse MA, Chui S, Hobeika A, Lyerly HK, Clay T. Recent developments in therapeutic cancer vaccines. Nat Clin Pract Oncol. (2005) 2:108–13. doi: 10.1038/ncponc0098
262. Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol. (2012) 39:323–39. doi: 10.1053/j.seminoncol.2012.02.006
263. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
264. Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. (2018) 283:110–20. doi: 10.1111/joim.12708
265. Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. (2018) 359:1355–60. doi: 10.1126/science.aar7112
266. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
Keywords: immunotherapy, metabolism, chimeric antigen receptor (CAR), tumor microenvironment (TME), anti-tumor immune response
Citation: Xu X, Gnanaprakasam JNR, Sherman J and Wang R (2019) A Metabolism Toolbox for CAR T Therapy. Front. Oncol. 9:322. doi: 10.3389/fonc.2019.00322
Received: 24 January 2019; Accepted: 10 April 2019;
Published: 30 April 2019.
Edited by:
Prashant Trikha, Nationwide Children's Hospital, United StatesReviewed by:
Pappanaicken R. Kumaresan, University of Texas MD Anderson Cancer Center, United StatesLenka V. Hurton, University of Texas MD Anderson Cancer Center, United States
Copyright © 2019 Xu, Gnanaprakasam, Sherman and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruoning Wang, cnVvbmluZy53YW5nQG5hdGlvbndpZGVjaGlsZHJlbnMub3Jn
†These authors have contributed equally to this work