 Yimei Feng
Yimei Feng Xiaoping Li
Xiaoping Li Kaniel Cassady
Kaniel Cassady Zhongmin Zou5
Zhongmin Zou5 Xi Zhang
Xi Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 02 April 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00210
This article is part of the Research Topic Molecular and Immunological Advances in Hematological Malignancies View all 11 articles
Over the last decade, investigation of Ten-Eleven Translocation 2 (TET2) gene function and TET2 mutation have become of increasing interest in the field of hematology. This heightened interest was sparked by the seminal discoveries that (1) TET2 mutation is associated with development of hematological malignancies and that (2) the TET family of proteins is critical in promoting DNA demethylation and immune homeostasis. Since then, additional studies have begun to unravel the question “Does TET2 have additional biological functions in the regulation of hematopoiesis?” Here, we present a mini-review focused on the current understanding of TET2 in hematopoiesis, hematological malignancies, and immune regulation. Importantly, we highlight the critical function that TET2 facilitates in maintaining the stability of the genome. Based on our review of the literature, we provide a new hypothesis that loss of TET2 may lead to dysregulation of the DNA repair response, augment genome instability, and subsequently sensitize myeloid leukemia cells to PARP inhibitor treatment.
In 2009, expression of the Ten-Eleven Translocation-2 (TET2) gene and its variants was demonstrated in myeloid malignancies (1). Followup studies demonstrating that proteins of the TET family play a key role in DNA hydroxymethylation further fueled interest in investigation of TET2 function in both basic and clinical research (1, 2). Here we present a review on the current understanding of TET2 in hematopoiesis and hematopoietic malignancies, as well as discuss the disputed clinical prognosis of TET2 mutation. Moreover, we highlight the role of TET2 in DNA damage and repair, and provide evidence to support the hypothesis that TET2 interacts with other DNA repair-related proteins to maintain genome stability. We further propose that loss of TET2 may sensitize myeloid leukemia cells to DNA repair stress, such as is induced by treatment with PARP inhibitors.
TET2 gene, a member of the TET family of enzymes, is located on chromosome 4q24, and its protein product TET2 modulates DNA hydroxymethylation by converting 5-methylcytosine (5 mC) to 5-hydroxymethylcytosine (5 hmC) to promote DNA demethylation (3). TET enzymes can modify 5 mC through oxidation, a phenomenon which revealed an alternative pathway for DNA demethylation mechanisms. It is noted that TET2 mutations are consistently associated with a decrease in 5hmC, which has been suggested as a potential diagnostic and prognostic biomarker for hematopoietic malignancies, especially myeloid malignancies (4).
The TET2 functional domain is at the C-terminus, consisting of a cysteine (Cys)-rich domain and a double-stranded β-helix fold (DSβH) domain (Figure 1). The DSβH domain contains the important metal-binding residues for Fe (II)/α-KG (also known as 2-oxoglutarate). TET dioxygenases require oxygen, α-KG and Fe (II) for their activity (5). TET uses molecular oxygen as a substrate to catalyze the oxidative decarboxylation of α-KG, which generates enzyme-bound Fe (IV)-oxo, and subsequently converts 5 mC to 5 hmC (6, 7).
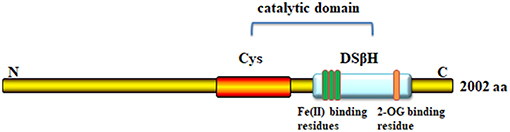
Figure 1. Domain structure of TET2 protein. The C-terminal core catalytic domain consists of a cysteine (Cys)-rich domain, and double-stranded β-helix fold domain (DSβH domain), each of which contains three Fe-binding domains and one site for 2-oxoglutarate binding.
Since the discovery of TET2, numerous studies have focused attention on the enzymatic effects of the TET2 protein, presumably through its function in DNA demethylation. TET2-interacting proteins have been identified and analyzed by expressing tagged-TET2, followed by affinity purification coupled with mass spectrometry. Known TET2 binding partners include small molecules, micro-RNAs, and some transcription factors (8). TET2 can recruit O-linked β-D-N-acetylglucosamine (O-GlcNAc) transferase (OGT) to chromatin, an effect that is independent of its enzymatic activity. TET2 and OGT colocalize on chromatin, increase H3K4me3 level, and regulate gene transcription (9, 10). Additionally, multiple studies have confirmed that microRNAs (miR-7, miR-22, miR-26, miR-29, miR-101, miR-125) (11) negatively regulate TET2 gene expression at the pre-mRNA level (8, 12). Vitamin C can restore and enhance TET2 enzymatic activity to suppress leukemia (13, 14). Furthermore, several proteins, such as WT1 (15, 16), VprBP (17), EBF1 (18), IDAX (19), and UHRF2 (20), are reported to exert their biological functions in combination with TET2 (5). Sun et al. recently reported sirtuin 1 (SIRT1) deacetylates TET2 in myelodysplastic syndrome stem and progenitor cells; moreover, SIRT1-deficient MDS HSPCs (CD34+ cells) exhibit enhanced cell growth and self-renewal (21). In summary, TET2 expression is tightly regulated at the pre-translational level and the TET2 protein is believed to exert its function via TET2-containing protein complexes (Figure 2). However, additional TET2 partners remain to be identified.
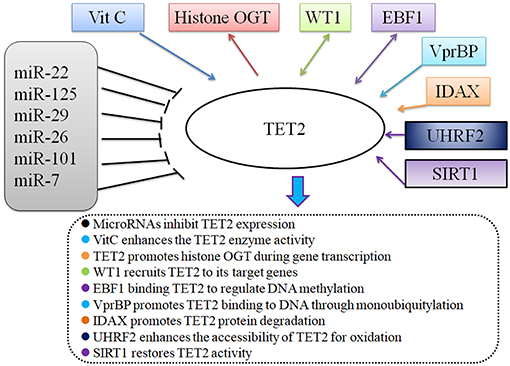
Figure 2. TET2 and its partners.
TET2 plays important roles in hematopoiesis, including promoting the self-renewal of stem cells and lineage commitment and terminal differentiation of monocytes (5). TET2 is highly expressed in hematopoietic stem/progenitor cells, and the deletion of TET2 in primary bone marrow cells leads to the increase of the percentage of immature c-Kit+Lin− cells, suggesting that loss of TET2 may affect stem/progenitor cell differentiation (4, 22). TET2 deletion in CD34+CD38+ cells can promote monocyte expansion, indicating a regulatory role of TET2 in lineage commitment (23, 24).
TET2 has been widely recognized as a tumor-suppressor gene. TET2 deletion is sufficient to cause myeloid and lymphoid malignancies in mice (25). Homozygous and heterozygous mutations in the TET2 gene are recurrent events in human hematopoietic malignancies. In a range of myeloid and lymphoid neoplasms, the frequencies of TET2 mutations are 20–35% in myelodysplastic syndrome (MDS) (26), 30–60% in chronic myelomonocytic leukemia (CMML) (27), 12–34% in acute myeloid leukemia (AML) (28) and 2–33% in lymphoid malignancies (5) (Figure 3).
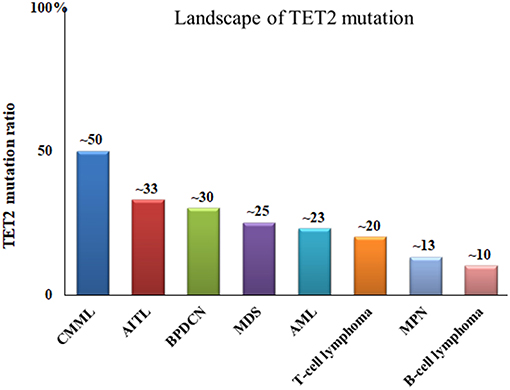
Figure 3. Frequencies of TET2 gene mutations in malignant blood disease. CMML, chronic myelomonocytic leukemia; AITL, angioimmunoblastic T-cell lymphoma; BPDCN, blastic plasmacytoid dendritic cell neoplasm; MDS, myelodysplastic syndromes; AML, acute myelogenous leukemia; MPN, myeloproliferative neoplasms.
TET2 gene mutations include frame shift, generated stop codons, in-frame deletion, and amino acid substitutions of highly conserved residues. As for the incidence of homozygous or heterozygous mutations in given disease patterns, such as AML, CMML and MDS, there is no specific genotypic pattern. Viguie et al. (29) analyzed six patients with deletions or mutations of TET2 in myeloid cancers, and reported that five patients had a heterozygous loss, and one had a deletion of both TET2 copies (1). Solary et al. (5) reported the highest rate of TET2 genetic alterations was observed in CMML in which a heterozygous or homozygous mutation was identified in 50–60% of this overlapping MDS/MPN; however prognosis associated with prevalence of heterozygous vs. homozygous mutation was not reported.
Genomic sequencing of all coding TET2 exons revealed those homozygous mutations tend to be associated with uniparental disomy 4q, and the heterozygous genotype is associated with lack of chromosomal lesions (30). Heterozygous TET2 knockout in mice, which led to ~50% loss of TET2 gene expression, resulted in significant but slower and less frequent malignant transformation than double-allele knockout (25). The 5-hydroxymethylcytosine level in DNA was reduced dramatically in homozygous TET2-mutant mice compared to heterozygous TET2-mutant mice. Approximately 33% of homozygous TET2-mutant and 8% of heterozygous TET2-mutant mice developed lethal myeloid malignancies in the first year of life (25). However, heterozygous and homozygous mutations of TET2 were found in patients with similar clinical manifestations and disease phenotype, and Kaplan-Meier curves suggested there was also no difference on Overall Survival (OS) between the homo/hemizygous and heterozygous cases (30). On the contrary, patients with homozygous TET2 mutation showed significantly inferior Event-Free Survival (EFS) and a higher relapse rate compared with those with heterozygous TET2 mutation (31). Thus, while homozygous TET2 deletion in murine models conveys increased susceptibility to malignancies, TET2 genotype relative to patient outcome in human malignancies is less clear (see below).
When it comes to prognosis of myeloid malignancies with TET2 mutations, the effect of TET2 mutations remains controversial (Table 1) (39). Those mutations include missense, non-sense, and frame shift mutations spanning the entire TET2 coding sequences (28). Large cohort studies showed that TET2 mutations did not impact the overall survivals in AML (32, 33) and myeloproliferative neoplasma (MPN) patients (34). On the other hand, some reports found that TET2 mutant-CMML (35) and -AML (36, 37, 40) patients had poorer outcomes compared with patients without TET2 mutations. In contrast, TET2 mutations have been shown to confer superior survival in MDS (26, 38). Furthermore, TET2 mutations may predict a more favorable response to hypomethylating agents (HMAs) in high-risk patients (41–43). Two meta-analyses (44, 45) suggest that TET2 mutations have no prognosis impact on OS of patients with MDS. However, low expression of TET2 is clearly associated with an unfavorable prognosis in MDS patients. Because these reports are clinical retrospective studies lacking the support of basic, preclinical scientific research, the possible underlying mechanisms that account for the prognosis of TET2 mutations in different malignancies are unknown. Whether TET2 mutations dysregulate pathways already known to contribute to hematopoietic transformation, or represent a novel pathway to transformation, remains to be elucidated (36).
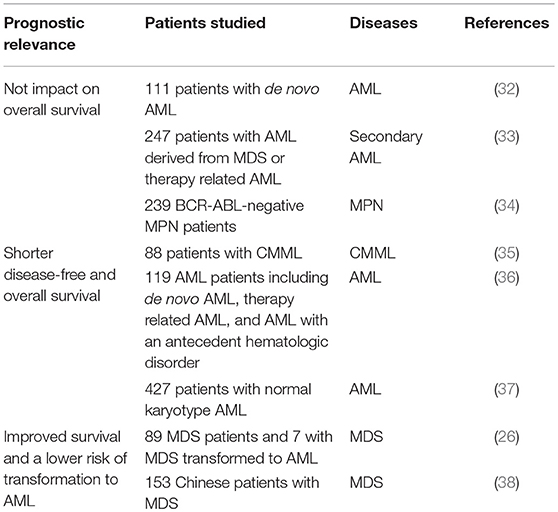
Table 1. Prognosis of TET2 mutations in myeloid malignancies.
TET2 mutations, as an early event in pathogenesis, may cooperate with other gene mutations, deemed background mutations, to promote various hematological malignancies. For example, TET2 mutations, when harboring FLT3-ITD mutation, induced AML (46); TET2 mutations, if combined with JAK2 and ASXL1 mutation, generated MPN, such as polycythemia vera (PV) and secondary myelofibrosis (MF) (47–50); finally, TET2 mutations, together with mutations in SRSF2 and KRAS, were associated with CMML (23) (Figure 4). It is obvious that different combinations of TET2 mutations along with other gene mutations will inevitably predict different prognoses. With the use of new drugs, such as HMAs, the clinical prognosis of TET2 mutation may also be improved.
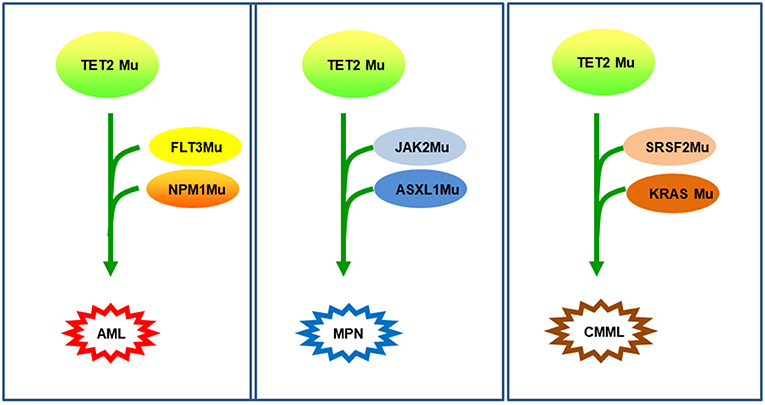
Figure 4. TET2 mutations as background mutations. Thanks to TET2 mutations as background mutations, the disease will occur and develop by the accumulation of additional mutations. In the first example (42), TET2-D1384V in HSCs, then in NPM1 and FLT3 mutation, induced an AML. In the second example (31, 44), TET2 mutation with JAK2V617F-positive generated thrombocythemia, which can transform into secondary myelofibrosis due to an additional ASXL1 mutation. In the third example (23), mutations in SRSF2, then in KRAS mutation developed a chronic myelomonocytic leukemia (CMML).
Recently, it was reported that TET2 also participated in regulation of the immune system (51). TET2 is highly expressed in each T helper cell (Th cell) subset compared with the other TET family genes. TET2−/− T cells showed a marked reduction in IFN-γ and IL-10 production at both the mRNA and protein level, which was associated with reduced p300 recruitment, suggesting TET2 may regulates Th1 and Th17 cell differentiation (52). In a murine model of multiple sclerosis, (autoimmune encephalomyelitis-EAE) model, upregulation of TET2 inhibited naive CD4+ T cell proliferation and differentiation into Th1 and Th17 cells and was largely protective against development of EAE (53). In another study, it was shown that TET2 mutation, cooperating closely with RhoA, leads to abnormal CD4+ T cell proliferation and disruption of T cell homeostasis (54). TET2 also can mediate Foxp3 demethylation to drive T regulatory cell (Treg) differentiation and maintain immune homeostasis (55). TET2 also plays a role in the regulation of the innate immune system and was found to be highly expressed during murine macrophage (MΦ) differentiation. TET2 loss increased IL-1b, IL-6, and Arginase1 mRNA expression, indicating that TET2 can restrain inflammation mediated by murine MΦs (56) (Figure 5). TET2/TET3 conditional knockout at early stages of B-cell development largely prevents lineage-specific programmed demethylation events, thus causing defects in B-cell differentiation and function (57). TET2 deficiency results in germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis (58). Interestingly, introduction of anti-CD19 chimeric antigen receptors (CARs) occasionally disrupts the TET2 base sequence (59). Consequently, TET2-disrupted CAR T cells were shown to display a central memory phenotype and induced long-term leukemia remission (59). TET2-deficient macrophages altered the tumor microenvironment to reduce tumor burden during melanoma progression (60). In a separate study, TET2-deficient CD8+ tumor infiltrating lymphocytes (TILs) also displayed increased anti-tumor efficiency in a mouse model of melanoma (61). In summary, TET2 plays a critical role in maintaining and/or establishing immune tolerance. Thus, manipulation of TET2 function may be possible for the promotion of tolerance (such as during autoimmunity) or in the promotion of antitumor immunity.
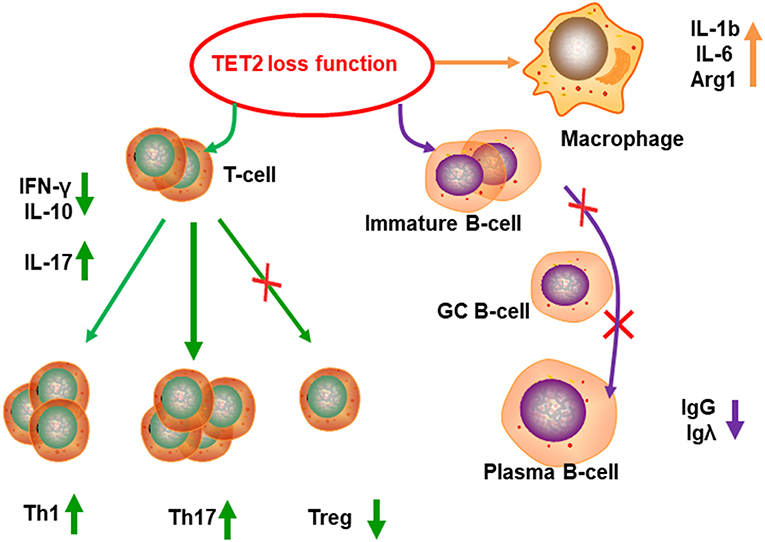
Figure 5. TET2 functions in immune regulation. TET2 can regulate the T cell differentiation. Because of TET2 loss of function, population of Treg cells decreased, Th1 and Th17 subsets increased, cytokines secretion changed, such as increased IL-17, and reduction of IL-10 and IFN-γ. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, restrains IgG and Igλexpression, and promotes B-cell lymphomagenesis. TET2 can inhibit inflammation in MΦs, TET2−/− MΦs produce more inflammatory factor, such as IL-1b, IL-6, and arginase1.
DNA strand damage is primarily sub-divided into double-strand breaks (DSB) and single strand breaks (SSB). DSBs are the most hazardous of all types of DNA damage; when unrepaired, DSBs can be lethal and trigger cellular apoptosis. There are two major pathways for DSB repair, non-homologous end joining (NHEJ) and homologous recombination (HR). Ku70, Ku80, and Lig4 are involved in NHEJ, while BRCA1/BRCA2, RAD51 are involved in the HR pathway. PARP1/PARP2 participates in SSB repair, and PARP (poly(ADP-ribose) polymerase) inhibition causes PARP-1 to be trapped onto DNA repair intermediates, especially during base excision repair of SSB (Figure 6).
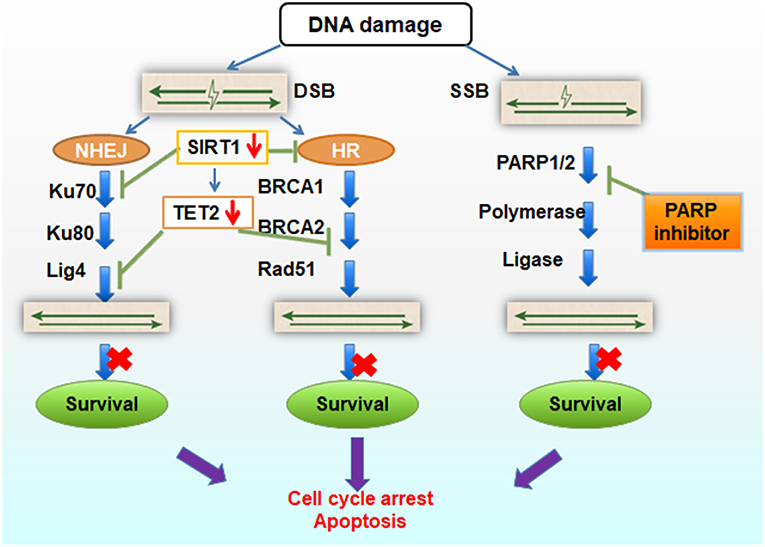
Figure 6. Schematic diagram of TET2 function in DNA repair. TET2 knock-down results in lower expression of Lig4 and BRCA2 expression, and SIRT1 deficiency results in Ku70 and TET2 inactivation, blocking the NHEJ and HR repair. If combined with inhibition of PARP inhibitor in SSB repair, the whole DNA repair pathways are harmed, cell survival gets impeded, and cell apoptosis is elevated.
In fact, the Tet family (Tet1/2/3) is reported to be involved in the DNA damage and repair response pathways. TET1 deficient cells harbored significantly more DNA strand breaks even in the absence of exogenous DNA damaging agents (62). TET1 may be involved in protecting cells against more severe injuries, such as DNA strand breaks. TET1 regulates several important DNA repair genes via modulating H4K16ac at the promoter regions of DNA repair genes including RAD50, BRCA1, RAD51, and TP53BP1 (63). Loss of TET1 leads to DNA instability and resulted in the development of myeloid malignancy in mouse models (64, 65). Additionally, Tet3-mediated conversion of 5 mC to 5 hmC promotes ATR-dependent DNA damage response and regulates DNA repair (66). Although the biochemical properties of TET2 have been extensively studied, the effect of TET2 loss-of-function in DNA damage repair is less understood. Recently, Kafer et al. reported that 5 hmC is actively enriched at endogenous DNA damage sites in cancer cell lines, precisely where TET2 creates damage-associated 5 hmC foci (67). Deficiency of TET2 elicits chromosome segregation defects in response to DNA replication stress. It has also been reported that TET2 knockout can decrease BRCA2 mRNA expression (67). As previously mentioned, BRCA1/BRCA2 proteins are important in maintaining genomic stability, especially in HR during the DNA damage response. It is reported that wild-type TET2 induction resulted in an increased level of 5-hmC and a cell cycle defect in S phase associated with higher level of phosphorylated P53, a process which is tightly controlled to avoid genetic and chromosomal instabilities (68). Taken together, it is reasonable to suppose that TET2 can maintain genomic stability via promotion of the DNA damage repair.
Based on our review of the literature, we hypothesize that loss of TET2 might sensitize myeloid leukemia cells to poly (ADP-ribose) polymerase inhibitors (PARPis). PARPis have become a mainstay for treatment of certain malignancies, especially breast and ovarian cancers (69), because they can selectively target tumor cells with BRCA1/2 mutation or HR deficiency (70). In CML (71), fanconi anemia (FA) (72) and some therapy-related AML samples (73, 74), BRCA1 or/and BRCA2 mutations have been found and suggested to be pathogenic genotypes, which may serve as models for PARPis application to leukemia therapy. Moreover, several clinical studies have reported that PARPis are effective in leukemia treatment (75, 76), but the detailed mechanism underlying their effectiveness needs to be fully explored. As mentioned earlier, TET2 deletion also leads to downregulation of BRCA2 gene expression—a phenomenon which directly supports the underlying principle of our hypothesis. It is important to highlight that reduction of BRCA function as a result of TET2 deletion may not be equivalent to loss-of-function resulting from BRCA mutation. However, it is reported that BRCA reduction can also sensitize tumor cells to PARPis. For example, miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARPis (77). MiR-9 mediates the downregulation of BRCA1, impedes DNA damage repair in ovarian cancer and improves chemotherapeutic efficacy by increasing the sensitivity of cancer cells to DNA damage (78). PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition (79). Additionally, BRCA-deficient tumors are hypersensitive to DNA damaging chemotherapeutic agents such as cisplatin, mitomycin C, etc. (80, 81). Accordingly, loss of TET2 results in BRCAs reduction, impairs homologous recombination in DNA repair, and, if combined with DNA damaging chemotherapeutic agents, might sensitize tumor cells to PARPis.
Secondly, TET2 plays a particular role in the NHEJ pathway of DNA repair. Wang et al. (15) over-expressed TET2 in HL60 cells, and subsequently found that expression of Lig4 mRNA increased 4-fold. Because Lig4 is critical for DNA repair through NHEJ pathway (82), loss of TET2 might decrease the Lig4 recruitment, result in DNA repair failure, and cell death. In our own studies, we found that SIRT1 can bind to TET2 and restore its activity (21). Overexpressing SIRT is reported to activate TET2 function, which suggests that SIRT1 may provide a novel therapeutic target in MDS (83, 84). These published data suggest that SIRT1 might interact with TET2 to promote DNA stability. It is reported that active SIRT1 promotes genomic stability and that SIRT1 specifically activates Rad51-independent HR (85). On the other hand, SIRT1 promotes DNA repair activity in NHEJ by the deacetylation of Ku70 (86). Thus, SIRT1 insufficiency could feasibly restrain function of TET2, and disable the DNA DSB repair. BRCAs, Lig4 and SIRT1, as partner proteins of TET2, are closely related to NHEJ and HR pathway of DNA repair.
Taken together, we propose that loss of TET2 can inhibit its partner proteins function (such as BRCAs, Lig4, SIRT1) in DSB DNA repair. If combined with PARPis to block the SSB repair pathway, cells are prone to cell cycle arrest and apoptosis (Figure 6). The experiments required to test our hypothesis is relatively straight-forward. For example, our hypothesis can be tested by treating TET2KO cells with PARPis, such as AZD2281 (Olaparib) and MK4827 (Niraparib), two selective inhibitors of PARP1/2, and analyzing cellular function/viability through cytotoxicity assays and apoptosis detection. On the other hand, rescue experiments, knocking in and overexpressing TET2, can further test whether TET2 overexpression provides protection from PARPis. Finally TET2−/− AML mice model and/or clinical patients‘ samples with TET2 mutation also can be treated with PARPis to observe the treatment's effect and further explore the mechanism of TET2 in regulating genomic stability.
TET2 plays important roles in epigenetic regulation, stem cell differentiation, and development of hematopoietic malignancies. Loss of TET2 function leads to DNA hypermethylation and subsequent dysregulated gene expression in hematopoietic stem cells, and has been considered as an initial step of myeloid malignant transformation including MDS and AML. While enzymatic activity of TET2 is well studied, little is known about other biological functions of TET2, for example, as a scaffold protein to recruit partners in DNA damage repair. Furthermore, the prognosis of TET2 mutation in hematologic malignancies has been controversial and the detailed mechanism of TET2 in promotion of malignancy needs to be further explored. In general, malignancies expressing the TET2 mutation are sensitive to treatment with HMAs. However, as some malignancies are resistant to HMAs, investigating additional drugs for the treatment of patients with TET2 mutation expressing malignancies is of dire importance. Due to the role of TET2 in promoting DNA stability, PARPis represent a class of drugs that offer potential effectiveness in treating HMA-resistant myeloid neoplasms. Additional studies are required to shed light on the potential therapeutic role of disrupting TET2 pathway to augment genome instability for the treatment of cancer.
XL, KC, XZ, ZZ, and YF: conceptualization; XZ and YF: funding acquisition; XL, XZ, ZZ, and YF: methodology; YF: wrote original draft and revised the manuscript; YF, KC, ZZ, and XZ: writing-review and editing.
This study was supported by the National Natural Youth Science Foundation of China (No. 81400081) and National Natural Science Foundation of China (Nos. 81873424, 8197011220), and the Natural Science Foundation of Chongqing (cstc2019jcyj-msxm1114).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Jie Sun, Haojie Dong, Yinghui Zhu, and Ling Li (Department of Hematological Malignancies Translational Science, City of Hope Medical Center, Duarte, USA), who help us in manuscript design and experimental proceeding.
TET2, Ten-Eleven Translocation 2; HMAs, hypomethylating agents; SIRT1, sirtuin 1; MDS, myelodysplastic syndrome; OS, overall survivals; PARP, poly (ADP-ribose) polymerase; BRCA, breast cancer susceptibility (BRCA) genes; HR, homologous recombination; NHEJ, non-homologous end-joining.
1. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
2. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–849 e835. doi: 10.1016/j.stem.2018.10.013
3. Fan S, Twu NF, Wang JA, Yuan RQ, Andres J, Goldberg ID, et al. Down-regulation of BRCA1 and BRCA2 in human ovarian cancer cells exposed to adriamycin and ultraviolet radiation. Int J Cancer. (1998) 77:600–9.
4. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. (2011) 20:11–24. doi: 10.1016/j.ccr.2011.06.001
5. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The ten-eleven translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. (2014) 28:485–96. doi: 10.1038/leu.2013.337
6. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. (2013) 502:472–9. doi: 10.1038/nature12750
7. Mohr F, Dohner K, Buske C, Rawat VP. TET genes: new players in DNA demethylation and important determinants for stemness. Exp Hematol. (2011) 39:272–81. doi: 10.1016/j.exphem.2010.12.004
8. Pan F, Weeks O, Yang FC, Xu M. The TET2 interactors and their links to hematological malignancies. IUBMB Life. (2015) 67:438–45. doi: 10.1002/iub.1389
9. Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. (2013) 493:561–4. doi: 10.1038/nature11742
10. Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. (2013) 32:645–55. doi: 10.1038/emboj.2012.357
11. Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D'Alessio AC, et al. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep. (2013) 5:471–81. doi: 10.1016/j.celrep.2013.08.050
12. Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. (2013) 13:87–101. doi: 10.1016/j.stem.2013.06.003
13. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. (2017) 170:1079–95 e1020. doi: 10.1016/j.cell.2017.07.032
15. Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. (2015) 57:662–73. doi: 10.1016/j.molcel.2014.12.023
16. Zhang X, Yang C, Peng X, Chen X, Feng Y. Acute WT1-positive promyelocytic leukemia with hypogranular variant morphology, bcr-3 isoform of PML-RARalpha and Flt3-ITD mutation: a rare case report. Sao Paulo Med J. (2017) 135:179–84. doi: 10.1590/1516-3180.2016.020104102016
17. Nakagawa T, Lv L, Nakagawa M, Yu Y, Yu C, D'Alessio AC, et al. CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol Cell. (2015) 57:247–60. doi: 10.1016/j.molcel.2015.08.021
18. Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. (2013) 4:2166. doi: 10.1038/ncomms3166
19. Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. (2013) 497:122–6. doi: 10.1038/nature12052
20. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. (2013) 152:1146–59. doi: 10.1016/j.cell.2013.02.004
21. Sun J, Zhu Y, He X, Ding Z, Dong H, Feng Y, et al. Activation of SIRT1 deacetylase as a therapeutic approach for myelodysplastic syndromes by restoring TET2 function. Blood. (2017) 130:2497. Available online at: http://www.bloodjournal.org/content/130/Suppl_1/2497
22. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. (2010) 17:13–27. doi: 10.1016/j.ccr.2009.11.020
23. Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. (2013) 121:2186–98. doi: 10.1182/blood-2012-06-440347
24. Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. (2013) 27:1441–50. doi: 10.1038/leu.2013.100
25. Li Z, Cai XQ, Cai CL, Wang JP, Zhang WY, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. (2011) 118:4509–18. doi: 10.1182/blood-2010-12-325241
26. Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood. (2009) 114:3285–291. doi: 10.1182/blood-2009-04-215814
27. Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, et al. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. (2011) 25:877–9. doi: 10.1038/leu.2011.10
28. Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. (2012) 26:934–42. doi: 10.1038/leu.2011.326
29. Viguie F, Aboura A, Bouscary D, Ramond S, Delmer A, Tachdjian G, et al. Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia. (2005) 19:1411–5. doi: 10.1038/sj.leu.2403818
30. Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. (2009) 113:6403–10. doi: 10.1182/blood-2009-02-205690
31. Ahn JS, Kim HJ, Kim YK, Jung SH, Yang DH, Lee JJ, et al. Adverse prognostic effect of homozygous TET2 mutation on the relapse risk of acute myeloid leukemia in patients of normal karyotype. Haematologica. (2015) 100:e351–353. doi: 10.3324/haematol.2015.126227
32. Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood. (2010) 116:1132–5. doi: 10.1182/blood-2009-07-234484
33. Kosmider O, Delabesse E, de Mas VM, Cornillet-Lefebvre P, Blanchet O, Delmer A, et al. TET2 mutations in secondary acute myeloid leukemias: a French retrospective study. Haematologica. (2011) 96:1059–63. doi: 10.3324/haematol.2011.040840
34. Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. (2009) 23:905–11. doi: 10.1038/leu.2009.47
35. Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. (2009) 94:1676–81. doi: 10.3324/haematol.2009.011205
36. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. (2009) 114:144–7. doi: 10.1182/blood-2009-03-210039
37. Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European leukemianet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. (2011) 29:1373–81. doi: 10.1200/JCO.2010.32.7742
38. Wang J, Ai X, Gale RP, Xu Z, Qin T, Fang L, et al. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res. (2013) 37:305–11. doi: 10.1016/j.leukres.2012.10.004
39. Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. (2011) 9:193–204. doi: 10.1016/j.stem.2011.08.007
40. Liu WJ, Tan XH, Luo XP, Guo BP, Wei ZJ, Ke Q, et al. Prognostic significance of Tet methylcytosine dioxygenase 2 (TET2) gene mutations in adult patients with acute myeloid leukemia: a meta-analysis. Leuk Lymphoma. (2014) 55:2691–8. doi: 10.3109/10428194.2014.893308
41. Flach J, Dicker F, Schnittger S, Kohlmann A, Haferlach T, Haferlach C. Mutations of JAK2 and TET2, but not CBL are detectable in a high portion of patients with refractory anemia with ring sideroblasts and thrombocytosis. Haematologica. (2010) 95:518–9. doi: 10.3324/haematol.2009.013631
42. Couronne L, Lippert E, Andrieux J, Kosmider O, Radford-Weiss I, Penther D, et al. Analyses of TET2 mutations in post-myeloproliferative neoplasm acute myeloid leukemias. Leukemia. (2010) 24:201–3. doi: 10.1038/leu.2009.169
43. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. (2009) 41:838–42. doi: 10.1038/ng.391
44. Guo Z, Zhang SK, Zou Z, Fan RH, Lyu XD. Prognostic significance of TET2 mutations in myelodysplastic syndromes: A meta-analysis. Leuk Res. (2017) 58:102–7. doi: 10.1016/j.leukres.2017.03.013
45. Lin Y, Lin Z, Cheng K, Fang Z, Li Z, Luo Y, et al. Prognostic role of TET2 deficiency in myelodysplastic syndromes: A meta-analysis. Oncotarget. (2017) 8:43295–305. doi: 10.18632/oncotarget.17177
46. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. (2012) 4:149ra118. doi: 10.1126/scitranslmed.3004315
47. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. (2012) 44:1179–81. doi: 10.1038/ng.2413
48. Beer PA, Delhommeau F, LeCouedic JP, Dawson MA, Chen E, Bareford D, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. (2010) 115:2891–900. doi: 10.1182/blood-2009-08-236596
49. Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. (2012) 5:12. doi: 10.1186/1756-8722-5-12
50. Verstovsek S, Odenike O, Singer JW, Granston T, Al-Fayoumi S, Deeg HJ. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J Hematol Oncol. (2016) 9:137. doi: 10.1186/s13045-016-0367-x
51. Tsagaratou A, Rao A. TET proteins and 5-methylcytosine oxidation in the immune system. Cold Spring Harb Symp Quant Biol. (2013) 78:1–10. doi: 10.1101/sqb.2013.78.020248
52. Ichiyama K, Chen TT, Wang XH, Yan XW, Kim BS, Tanaka S, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. (2015) 42:613–26. doi: 10.1016/j.immuni.2015.06.005
53. Wang X, Wang J, Yu Y, Ma T, Chen P, Zhou B, et al. Decitabine inhibits T cell proliferation via a novel TET2-dependent mechanism and exerts potent protective effect in mouse auto- and allo-immunity models. Oncotarget. (2017) 8:56802–15. doi: 10.18632/oncotarget.18063
54. Zang S, Li J, Yang H, Zeng H, Han W, Zhang J, et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest. (2017) 127:2998–3012. doi: 10.1172/JCI92026
55. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity. (2015) 43:251–63. doi: 10.1016/j.immuni.2015.07.017
56. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. (2017) 55:56–70 e13. doi: 10.1016/j.exphem.2017.08.001
57. Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci USA. (2016) 113:5018–23. doi: 10.1073/pnas.1604365113
58. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, et al. TET2 Deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell Lymphomagenesis. Cancer Discov. (2018) 8:1632–53. doi: 10.1158/2159-8290.CD-18-0657
59. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
60. Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, et al. The DNA Methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity. (2017) 47:284–297 e285. doi: 10.1016/j.immuni.2017.07.020
61. Minjung Lee, Hongxiang Zeng, Jia Li, Wei Han, Deqiang Sun, Huang. Y. Disruption of TET2 dioxygenase enhances antitumor efficiency in CD8+ tumor infiltrating lymphocytes. Blood. (2018):132:860. doi: 10.1182/blood-2018-99-115405
62. Coulter JB, Lopez-Bertoni H, Kuhns KJ, Lee RS, Laterra J, Bressler JP. TET1 deficiency attenuates the DNA damage response and promotes resistance to DNA damaging agents. Epigenetics. (2017) 12:854–64. doi: 10.1080/15592294.2017.1359452
63. Zhong JN, Li XF, Cai WS, Wang Y, Dong SS, Yang J, et al. TET1 modulates H4K16 acetylation by controlling auto-acetylation of hMOF to affect gene regulation and DNA repair function. Nucleic Acids Res. (2017) 45:672–84. doi: 10.1093/nar/gkw919
64. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu YT, et al. TET1 is a tumor suppressor of hematopoietic malignancy Nat Immunol. (2015) 16:653–62. doi: 10.1038/ni.3148
65. An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun. (2015) 6:1–14. doi: 10.1038/ncomms10071
66. Jiang DW, Wei S, Chen F, Zhang Y, Li JL. TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep. (2017) 18:781–96. doi: 10.15252/embr.201643179
67. Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, et al. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep. (2016) 14:1283–92. doi: 10.1016/j.celrep.2016.01.035
68. Mahfoudhi E, Talhaoui I, Cabagnols X, Della Valle V, Secardin L, Rameau P, et al. TET2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Repair. (2016) 43:78–88. doi: 10.1016/j.dnarep.2016.05.031
69. Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. (2016) 108:73–85. doi: 10.1016/j.critrevonc.2016.10.010
70. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. (2010) 376:235–44. doi: 10.1016/S0140-6736(10)60892-6
71. Deutsch E, Jarrousse S, Buet D, Dugray A, Bonnet ML, Vozenin-Brotons MC, et al. Down-regulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood. (2003) 101:4583–8. doi: 10.1182/blood-2002-10-3011
72. Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, et al. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. (2004) 103:3226–9. doi: 10.1182/blood-2003-09-3138
73. Rashidi A, Amarillo I, Fisher SI. BRCA2-associated therapy-related acute myeloid leukemia. Med Oncol. (2015) 32:371. doi: 10.1007/s12032-014-0371-3
74. Scardocci A, Guidi F, D'Alo F, Gumiero D, Fabiani E, Diruscio A, et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br J Cancer. (2006) 95:1108–3. doi: 10.1038/sj.bjc.6603392
75. Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, et al. A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin Cancer Res. (2017) 23:693–706. doi: 10.1158/1078-0432.CCR-16-0984
76. Zhao L, So CW. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Exp Hematol. (2016) 44:902–7. doi: 10.1016/j.exphem.2016.07.007
77. Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. (2011) 41:210–20. doi: 10.1016/j.molcel.2010.12.005
78. Sun C, Li N, Yang Z, Zhou B, He Y, Weng D, et al. miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J Natl Cancer Inst. (2013) 105:1750–8. doi: 10.1093/jnci/djt302
79. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. (2012) 2:1036–47. doi: 10.1158/2159-8290.CD-11-0348
80. Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. (2000) 275:23899–903. doi: 10.1074/jbc.C000276200
81. Fedier A, Steiner RA, Schwarz VA, Lenherr L, Haller U, Fink D. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53-deficient cells. Int J Oncol. (2003) 22:1169–73. doi: 10.3892/ijo.22.5.1169
82. Liu Y, Zhou K, Zhang H, Shugart YY, Chen L, Xu Z, et al. Polymorphisms of LIG4 and XRCC4 involved in the NHEJ pathway interact to modify risk of glioma. Hum Mutat. (2008) 29:381–9. doi: 10.1002/humu.20645
83. Sun J, He X, Zhu Y, Ding Z, Dong H, Feng Y, et al. SIRT1 Activation Disrupts maintenance of myelodysplastic syndrome stem and progenitor cells by restoring TET2 function. Cell Stem Cell. (2018) 23:355–369 e359. doi: 10.1016/j.stem.2018.07.018
84. Simeoni F, Somervaille TCP. Revert the SIRT: Normalizing SIRT1 Activity in Myelodysplastic Stem Cells. Cell Stem Cell. (2018) 23:315–7. doi: 10.1016/j.stem.2018.08.003
85. Uhl M, Csernok A, Aydin S, Kreienberg R, Wiesmuller L, Gatz SA. Role of SIRT1 in homologous recombination. DNA Repair. (2010) 9:383–93. doi: 10.1016/j.dnarep.2009.12.020
Keywords: TET2, SIRT1, DNA demethylation, DNA repair, DNA stability
Citation: Feng Y, Li X, Cassady K, Zou Z and Zhang X (2019) TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 9:210. doi: 10.3389/fonc.2019.00210
Received: 25 December 2018; Accepted: 11 March 2019;
Published: 02 April 2019.
Edited by:
Ritu Gupta, All India Institute of Medical Sciences, IndiaReviewed by:
Liren Qian, Sixth Medical Center of People's Liberation Army General Hospital, ChinaCopyright © 2019 Feng, Li, Cassady, Zou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yimei Feng, eWltZWlmZW5nQDE2My5jb20=
Xi Zhang, emhhbmd4eGlAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.