
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 12 February 2019
Sec. Cancer Immunity and Immunotherapy
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00056
This article is part of the Research Topic Current Perspectives, Challenges and Advances in Cell Based Therapies View all 14 articles
 Aesha I. Ali1,2
Aesha I. Ali1,2 Amanda J. Oliver1,2
Amanda J. Oliver1,2 Tinaz Samiei1,2
Tinaz Samiei1,2 Jack D. Chan1,2
Jack D. Chan1,2 Michael H. Kershaw1,2*
Michael H. Kershaw1,2* Clare Y. Slaney1,2*
Clare Y. Slaney1,2*Conventional treatments for pancreatic cancer are largely ineffective, and the prognosis for the vast majority of patients is poor. Clearly, new treatment options are desperately needed. Immunotherapy offers hope for the development of treatments for pancreatic cancer. A central requirement for the efficacy of this approach is the existence of cancer antigen-specific T cells, but these are often not present or difficult to isolate for most pancreatic tumors. Nevertheless, specific T cells can be generated using genetic modification to express chimeric antigen receptors (CAR), which can enable T cell responses against pancreatic tumor cells. CAR T cells can be produced ex vivo and expanded in vitro for infusion into patients. Remarkable responses have been documented using CAR T cells against several malignancies, including leukemias and lymphomas. Based on these successes, the extension of CAR T cell therapy for pancreatic cancer holds great promise. However, there are a number of challenges that limit the full potential of CAR T cell therapies for pancreatic cancer, including the highly immunosuppressive tumor microenvironment (TME). In this article, we will review the recent progress in using CAR T cells in pancreatic cancer preclinical and clinical settings, discuss hurdles for utilizing the full potential of CAR T cell therapy and propose research strategies and future perspectives. Research into the use of CAR T cell therapy in pancreatic cancer setting is rapidly gaining momentum and understanding strategies to overcome the current challenges in the pancreatic cancer setting will allow the development of effective CAR T cell therapies, either alone or in combination with other treatments to benefit pancreatic cancer patients.
Pancreatic cancer presents a major challenge in the clinic and is one of the most aggressive tumor types. Pancreatic cancer is the fourth most common cause of cancer death (1, 2) and it is on its way to be the second most common cause of cancer-related deaths by 2030 (3). Patients with pancreatic cancers have a median survival rate of 5 months after diagnosis and the overall 5-years survival rate is <5% (4).
The most common clinical therapeutic approaches against pancreatic cancer include surgical resection, radiotherapy, chemotherapy, and combination of these treatments (5). Surgical resection may lead to longer-term survival, but only a small number of the patients are considered resectable (6–8) because most patients that present to the clinic are with advanced or metastatic disease (9). In addition, the removal of part or the full pancreas is technically difficult, and even if the operation is successful, the 10-years survival rate is still <10% (10). Radiotherapy is usually not curative on its own and is used to alleviate symptoms. Although in the last two decades, chemotherapy and targeted therapy (such as Erlotinib targeting epidermal growth factor receptor, Sunitinib targeting multiple receptor tyrosine kinases, and Everolimus targeting mTOR kinase) have been used for patients with unresectable locally advanced or metastatic pancreatic cancer (11), these have only generated modest improvements in survival (12, 13). Therefore, the need to develop alternative effective therapies for pancreatic cancer is urgent.
Currently, immunotherapy has been offered as an important cancer treatment for a number of cancer types (14, 15) and recent preclinical and clinical evidence suggest that therapies utilizing the immunity could potentially be effective against this devastating disease (16). However, there has been limited success in the use of checkpoint blockade immunotherapies such as PD1/CTLA4 antibodies or vaccines in the treatment of pancreatic cancer (17).
Adoptive cellular therapies involving an infusion of effector immune cells into patients have generated remarkable responses in some cancers (18) and chimeric antigen receptor (CAR) T cell therapy represents a promising therapeutic modality for some difficult cancers including pancreatic cancers. This review aims to summarize the recent development of cellular therapies and clinical data in CAR T cell trials for pancreatic cancer. As pancreatic ductal adenocarcinoma (PDAC) is the most common malignancy of the pancreas and represents the vast majority of pancreatic cancer deaths, we will emphasize CAR T cell work on PDAC in this article.
CAR T cells present an exciting opportunity for cancer immunotherapy. As a form of adoptive immunotherapy, CAR T cells are generated from patient autologous T cells isolated from peripheral blood. Patient T cells are transduced ex vivo to express a CAR specific for a tumor antigen of choice and adoptively transferred into the patient to treat established cancers (19). CARs are composed of an antibody single-chain variable fragment (scFv) conjugated to intracellular signaling domains containing CD3-ζ chain and one or more co-stimulatory domains such as CD28 and CD137 (18, 20–22) (Figure 1). The CAR scFv confers the ability to T cells to directly recognize cancer antigens independent of MHC antigen presentation, and CAR specific recognition/binding to tumor antigen drives CAR T cell activation and tumor cell killing (23, 24). The first generation of CARs that was designed to contain CD3ζ or FcRγ signaling domains was limited by the lack of costimulatory signaling. The subsequent second generation of CARs has been designed to incorporate CD28 or CD137 cytoplasmic co-stimulatory domains. The third generation of CARs contains additional signaling domains (CD137, CD28, and/or OX40) (18, 20). The latter generations of CAR T cells are better equipped to overcome the immunosuppressive tumor microenvironment (TME), however, it remains unclear what combination of signaling domains is necessary for maximal anti-tumor response.
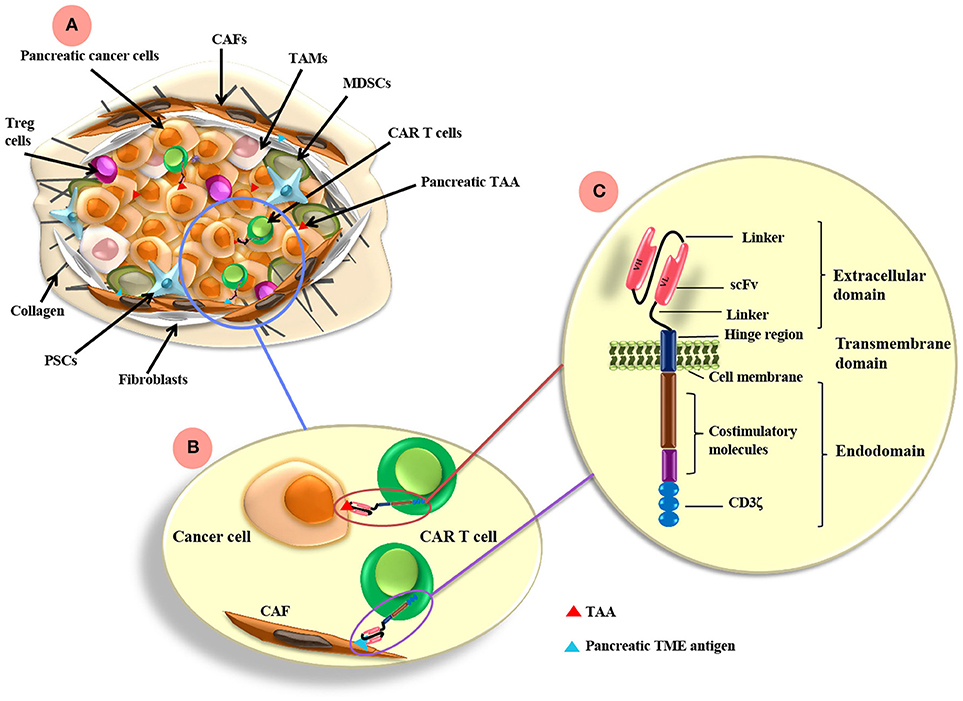
Figure 1. CAR T cell antigen-targeting strategies and pancreatic cancer TME. (A) The pancreatic TME consists of tumor cells as well as many immunosuppressive cells, such as CAFs, TAMs, MDSCs, PSCs, and Treg cells. (B) CAR T cells can be directed to the TAA expressed on pancreatic cancer cells and/or other antigens targeting the TME components, such as FAP on CAFs. (C) CARs are composed of extracellular, transmemebrane and endo-domains. The extracellular domain consists of an antibody variable heavy chain (VH) and a light chain (VL) domain, which are derived from an scFv from an antibody specific for a TAA. A flexible hinge region links the extracellular domain to a transmembrane and endodomain. The endodomain has cytoplasmic signaling regions derived from CD3ζ and costimulatory signaling domains. TAMs, tumor-associated macrophages; CAFs, cancer associated fibroblasts; MDSCs, myeloid-derived suppressor cells; Tregs, regulatory T cells; PSCs, pancreatic stellate cells; FAP, fibroblast activation protein; scFv, single chain variable fragment. TAA, tumor associated antigen; TME, tumor microenvironment.
The use of CAR T cells for the treatment of B cell malignancies demonstrated significant responses in patients (25, 26). Given the success in clinical trials, the use of CD19-targeted CAR T cell therapies was approved by the FDA in 2017. Approved CAR T cell therapies include tisagenlecleucel (Kymriah) for the treatment of children and adolescents with refractory/relapsed B-cell acute lymphoblastic leukemia (B-ALL), and axicabtagene ciloleucel (Yescarta) for adult relapsed-refractory large B-cell lymphoma patients. However, despite the successes in hematological cancers, clinical trials targeting solid tumors have demonstrated only moderate efficacy. This is largely attributed to the immunosuppressive TME, limited activation and trafficking of CAR T cells to the tumor site, heterogeneous antigen expression/distribution in some solid tumors and availability of validated antibodies that could be utilized in the CAR constructs (27–29).
A range of approaches aimed at enhancing CAR T cell efficacy is currently undergoing investigation. A notable strategy that has demonstrated promising effects in vivo is the use of dual-specific T cells. Dual-specific T cells co-express a CAR against a tumor antigen and a TCR against a strong immunogen (30). Through vaccination, dual-specific T cells can engage the cognate immunogen of the chosen TCR presented by antigen presenting cells (APCs) on MHC molecules. A recent study using the “adoptive cell transfer incorporating vaccination” (ACTIV) therapy regimen for dual-specific T cell treatment has demonstrated durable responses in a range of solid tumors in vivo (31, 32). Use of the specialized “CARaMEL” dual-specific T cells, expressing a CAR against HER2 and TCR specific for the melanocyte protein gp100 (also known as pMEL), drove dramatic T cell expansion and tumor regression in a number of solid tumor models. Moreover, surviving mice that received ACTIV therapy developed potent immune memory responses against pre-existing tumor cells. These results provide encouraging evidence for the investigation and development of dual-specific T cells for the treatment of difficult cancers including pancreatic cancer.
In recent years, CAR T cell therapies have been tested in both preclinical and clinical settings for treating pancreatic cancers. However, a focus for the field remains the discovery and validation of pancreatic cancer-specific antigens.
Mesothelin (MSLN) is a glycoprotein present mainly in mesothelial cells and overexpressed in a variety of human cancers including malignant pleural mesothelioma, ovarian, lung and pancreatic cancers (33). MSLN has been reported to be expressed by >80% of PDACs and its expression correlated with poor prognosis (34). Although MSLN is believed to play a role in cell adhesion and positively regulates tumor invasion and growth, its biological function is unclear (34). Because MSLN is expressed only on non-crucial tissues, it is an attractive target for CAR T cell based immunotherapy.
Preclinical studies have demonstrated that CAR T cells against MSLNs could potentially be effective against PDAC. When MSLN-CAR T cells were adoptively transferred intratumorally or intravenously (i.v.) into NSG mice bearing pre-established subcutaneous patient-derived mesothelioma, the tumor burden was greatly reduced in size and some tumors were completely eradicated, demonstrating the potential of targeting this antigen (35). Due to the concerns for potential on-target/off-tumor toxicity, phase I clinical studies used mRNA-based methods to generate CAR T cells that express CARs transiently to limit the duration of toxicity. Given the short-term expression of CARs, multiple injections were required. In a study carried out by Beatty et al (36), a patient with metastatic PDAC was given eight doses of MSLN-CAR T cells by i.v. infusion and two doses via intratumoral injections. The CAR T cells were detected in the extravascular tumor compartments 3 days after the initial i.v. infusion. The patient demonstrated stable disease 3 weeks post MSLN-CAR T cell administration, without overt evidence of toxicity against normal tissues. An anti-tumor effect was observed based on the development of novel humoral immune responses post the cell infusion. In another phase I trial carried out by the same group, six metastatic PDAC patients were treated with mRNA-based MSLN-CAR T cells three times per week for three consecutive weeks. The treatment was well-tolerated and the disease was stabilized in two of the treated patients. Importantly, one patient had total metabolic active volume decreased by 69.2%, although there was no detected effect on the primary tumor (37). Given the promising results, trials of MSLN-CAR T cells engineered by traditional viral transduction methods have also been initiated (Table 1).
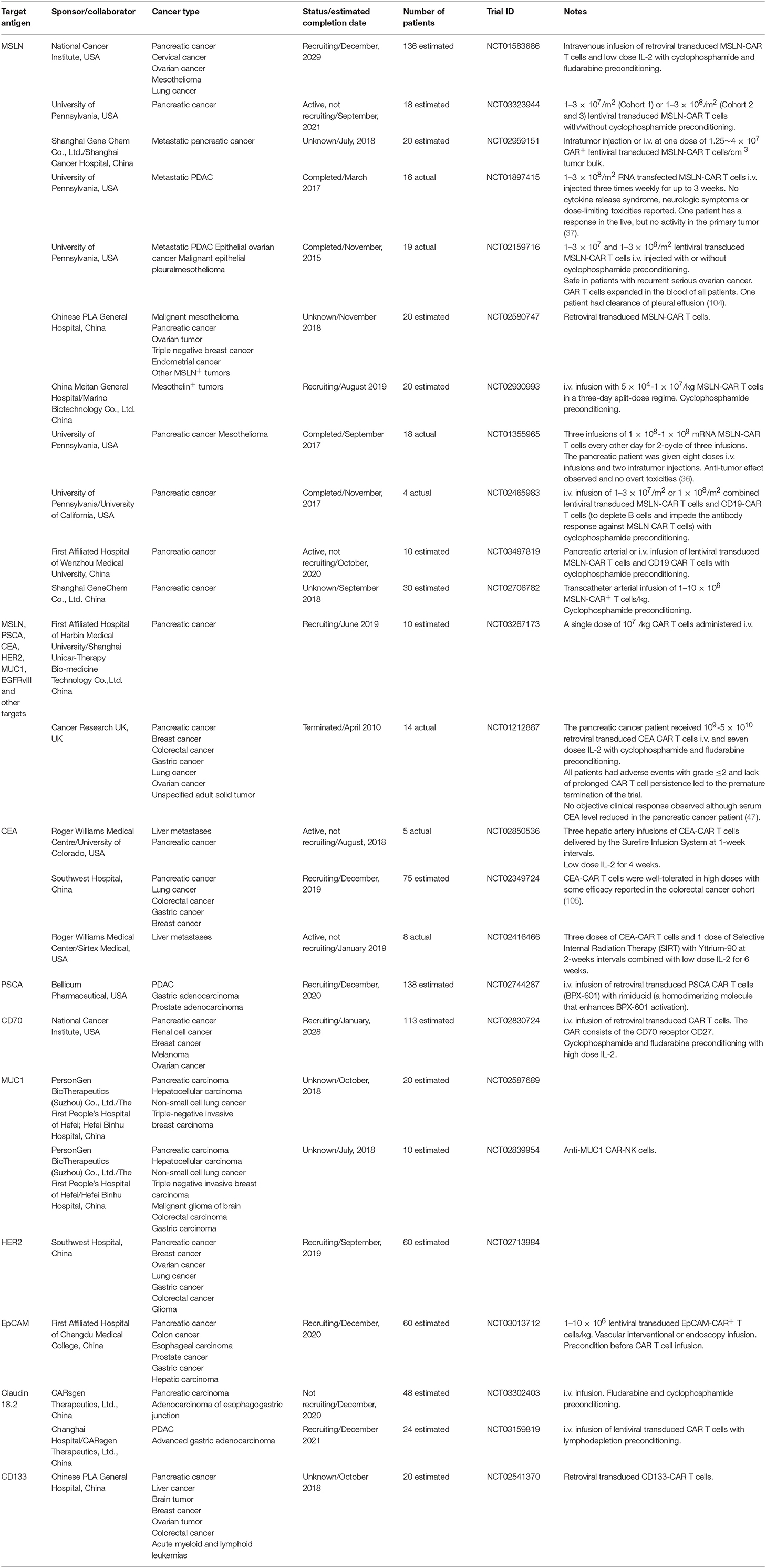
Table 1. Clinical trials involving CAR-based immunotherapy in pancreatic cancer from www.clinicaltrials.gov.
Prostate stem cell antigen (PSCA) is a glycosylphosphatidylinositol-anchored cell surface protein involved in intracellular signaling, although much of its function remains unclear. PSCA is expressed in the epithelial cells including that of prostate, kidney, skin, stomach, urinary bladder, esophagus and placenta, and also expressed in differentiating cells such as the ones of prostate and gastric epithelial cells. PSCA has also been detected in several cancer types including prostate, urinary bladder and pancreatic cancers (38). Aberrant overexpression of PSCA is detected in nearly 60% of the primary PDACs, while the gene expression is not detected in normal pancreatic duct (39). Therefore, PSCA has been proposed as a specific biomarker for PDAC patients and a promising target for CAR T cell therapies in treating PDAC. An advantage of targeting PSCA is that it is upregulated in pancreatic cancer cells from early stages of malignant transformation (40), including premalignant pancreatic intraepithelial neoplasias. PSCA may therefore serve as a useful target of immunotherapy that could eliminate malignant cells at all stages of PDAC.
Strategies using CARs against PSCA have been tested in preclinical settings. The 1st generation CAR T cells specifically killed PSCA+ pancreatic cancer cell lines without lysing PSCA− target cell in vitro (8). In a more recent study, multiple CAR constructs were compared. Adoptive transfer of these human CAR T cells in this study demonstrated significant antitumor activity in a murine model of human pancreatic cancer. Interestingly, although the third-generation CAR containing CD28 and CD137 costimulatory domains induced greater persistence of CAR T cells in vivo, the second generation CAR that does not contain CD137 domain, induced a better antitumor effect, with 40% of mice demonstrating tumor eradication (40). The efficacy between second- and third- generation CARs have been compared by various studies and the discrepancies between these reports indicate that the optimal CAR design needs to be empirically determined for disease and antigen targeted (41, 42). Being encouraged by the preclinical success, a trial using CAR T cells against PSCA has been initiated and is currently recruiting patients (NCT02744287).
Carcinoembryonic antigen (CEA) is a cell surface glycoprotein belonging to the immunoglobulin (Ig) superfamily and plays a role in cell adhesion. CEA is one of the “oncofetal” antigens, typically produced in the gastrointestinal tissue during fetal development (43, 44). Although CEA is expressed in various healthy epithelia of pulmonary and gastrointestinal tracts, its distribution is often limited to the luminal surface, thus difficult for CAR T cells to access (44). After neoplastic transformation, luminal epithelia cells lose the apical polarity of CEA expression and CEA becomes accessible to immune cells. Some CEA is released to the serum and soluble CEA in the circulation of patients is used as a marker for cancer progression. CEA is highly expressed on the surface of the majority of PDAC cells (45). Together with its restricted expression in normal tissues, CEA is an attractive target for CAR T cell treatment in PDAC. A few versions of anti-CEA CARs have been developed in the past few years, each targeting different CEA epitopes.
CEA-CAR T cells exhibited cytotoxicity against CEA expressing cancer cells in vitro and demonstrated anti-tumor effect in vivo in a clinically relevant orthotopic CEA+ murine model. The recipient CEA transgenic mice express CEA in their intestinal and pulmonary tracts. Ten days post intrapancreatic injections of Panc02-CEA+ cells, the recipient mice received CEA-CAR T cells. Interestingly, the injection of the CAR T cells eradicated tumors without any damage to normal tissues that are CEAlow (46).
However, a recent clinical trial using CEA-CAR T cells treating patients with advanced CEA+ cancers demonstrated acute respiratory toxicity, which resulted in the premature closure of the trial (47). The expression of CEA on lung epithelium was considered to result in the toxicity, which was associated with pre-conditioning. In a recent clinical trial using T cells modified to express an anti-CEA TCR for treating CEA+ metastatic colorectal cancer, severe autoimmune colitis and pneumonia was observed in all of the three patients and led to the halt of the trial (48). This was likely due to the ability of TCR-redirected T cells to engage CEA presented by MHC.
Mucins are high-molecular-weight glycoproteins with the presence of a heavily O-glycosylated tandem repeat region that is rich in proline, threonine and serine residues. The large gel-forming mucins are an extracellular secretion of goblet cells and their functions include lubrication of the epithelial surfaces and protection from physical and chemical insult. The epithelial membrane-tethered mucins are distinct from the conventional secreted mucins and are transmembrane molecules expressed by most glandular and ductal epithelial cells. It is widely accepted that the transmembrane mucin 1 (MUC1) is overexpressed in multiple epithelial adenocarcinomas, such as that of breast, colon, and pancreatic cancers. Importantly, under normal conditions, MUC1 is heavily glycosylated and expressed on the apical surface of epithelial cells, but in tumor cells MUC1 is aberrantly glycosylated. This modification of the MUC1 antigen reveals epitopes associated with the core protein, which is usually masked by oligosaccharides. Overexpression of aberrantly glycosylated MUC1 is associated with multiple metastatic cancers. In particular, MUC1 is aberrantly expressed in 60% of pancreatic cancers and is correlated with poor prognosis, enhanced metastasis, and chemoresistance (49, 50).
Strategies using CAR T cells targeting aberrantly expressed MUC1 have generated exciting results in preclinical studies. Posey et al. (51) generated a CAR that recognizes the aberrant glycoform Tn (GalNAca1-O-Ser/Thr) antigen on MUC1. These CAR T cells are able to recognize multiple types of tumors in vitro and exhibited superior tumor rejection and prolonged survival against disseminated pancreatic cancers in a xenograft model. This elegant study highlighted the potential for protein modifications as a target. As one of the most characteristic features of cancer cells is altered glycosylation, changes in glycosylation may expose a range of different cancer-associated epitopes and can serve as a target for CAR T cells. Given the promising outcomes from preclinical studies, multiple early phase trials have been planned and are active for using MUC1-CAR T cells in treating PDAC (Table 1).
CD47 is a transmembrane protein known to mediate a “do not eat me” signal. Structurally, CD47 contains an extracellular N-terminal hydrophilic Ig superfamily domain and an intracellular hydrophobic domain. Signal regulatory protein alpha (SIRPα) has been identified as the receptor to CD47. The binding of tumor-expressing CD47 to SIRPα on immune cells leads to the activation of the downstream signaling pathway in immune cells and inhibits the immune phagocyte-dependent clearance of tumors. CD47 has been identified in several types of hematological and solid cancers including pancreatic cancer (52, 53). In addition, CD47 is expressed on high levels on cancer stem cells (CSCs), but not on normal cells in the pancreas (52). Therefore, targeting CD47 has been a subject of intense interest in recent years.
CD47-specific CAR T cells were recently developed by ProMab Biotechnologies. These CAR T cells demonstrated high cytotoxicity against a few types of cancer cells including pancreatic cancer cells. Importantly, intratumoral injection of these CAR T cells significantly decreased pancreatic xenograft tumor growth. The same research group also developed humanized CD47-CAR T cells that contain humanized CD47 scFv. These humanized CD47-CAR T cells demonstrated specific killing of CD47+ cancer cells in vitro and it will be interesting to assess their efficacy in clinical trials (54).
The tyrosine kinase growth factor receptors are transmembrane proteins that play a key role in medicating intracellular signal transduction cascade for cell proliferation and differentiation. Up-regulation of some of the receptors are mechanisms of cancer development and progression, and the overexpression of a number of these receptors have been identified in many types of cancers (55). Therefore, CARs designed to target tyrosine kinase growth factor receptors have been developed and clinical trials are under way to test the safety and efficacy of many of these targets.
Human epidermal growth factor receptor 2 (HER2, also known as ERBB2) is a transmembrane glycoprotein belonging to the epidermal growth factor receptor (EGFR) family. The binding of HER2 to its ligand induces heterodimerization of the receptors, which mediates the activation of intracellular tyrosine kinase signaling cascades and leads to cell proliferation and differentiation. Overexpression of HER2 induces dimerization of HER2 and initiates signal transduction without ligand binding. Overexpression of HER2 has been reported in multiple cancer types making it an attractive target for CAR T cell treatment (20). However, an early HER2-CAR T cell trial, utilizing a third generation CAR, reported a patient death post the CAR T cell treatment (56). This patient was diagnosed with colon cancer metastatic to the lung and liver. The patient was preconditioned using cyclophosphamide (CY) and flurodarabine. Following intravenous infusion of 1010 HER2-CAR cells in 30 min, the patient experienced acute respiratory distress and subsequent death. At autopsy, multiple organs including the lung showed signs of ischemia and injury, and her serum samples post infusion showed significant increased levels of multiple cytokines, including IFN-γ, IL-6, and TNF-α. This report raised safety concerns on targeting HER2, but a more recent study using lower numbers of HER2-CAR T cells, and a second generation CAR in treating 19 patients with HER2+ sarcoma, demonstrated safety (57).
HER2 expression is observed in 20–60% of pancreatic cancer cases and therefore, it is a potential target for CAR T cell treatment (58–61). Recently, a Phase I clinical trial used HER2-CAR T cells to treat two pancreatic cancer patients. Although the trial demonstrated safety, only moderate responses were achieved (62). In contrast, results from preclinical studies have been promising. A recent study used HER2-CAR T cells to treat mice bearing xenografts derived from stage IV PDAC patients and achieved complete remission in both local and disseminated disease settings. In addition, in this study, the authors used an antibody-based switchable CAR system. These switchable CAR T cells bind to a specific peptide that is genetically engrafted onto a tumor-binding Fab molecule. The switch acts as a bridge between the tumor cells and CAR T cells and has a short half-life and thus, limits potential immunogenicity. The comparison between the switchable and conventional CAR T cells demonstrated that the anti-tumor efficacy of these switchable CAR T system was not compromised (63). The discrepancy in efficacy results from clinical and preclinical studies may be due to the different levels of HER2 expression in patients and xenografts, and the immunosuppressive TME in the patients.
Other putative growth factor receptors that could be targeted against pancreatic cancer using CARs include insulin-like growth factor receptor-1 (IGF1R), EGFR, vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet-derived growth factors (PDGFRs) that are expressed at elevated levels in pancreatic cancers and contribute the cancer's malignant phenotype (55). IGF1R is expressed in a variety of cancers and blocking IGF1R expression enhances apoptosis and suppresses metastasis in pancreatic cancer cells (64). IGF1R-CAR cells have demonstrated efficacy in murine models of sarcoma xenograft (65) and could be potentially useful in the treatment of PDAC. EGFR is a surface glycoprotein that belongs to the EGFR family of tyrosine kinase receptors. EGFR is aberrantly activated in a number of epithelial tumors and its overexpression has been detected in up to 90% of pancreatic tumors (66, 67). The value of EGFR-CAR T cells in treating solid cancers has been demonstrated in both preclinical (68) and clinical settings (69) in a few different cancer types but its potential in treating PDAC is yet to be tested.
CD24 is a mucin-like protein. It was originally discovered as the ligand for P-selectin and involved in signal transduction mediated by the members of the protein tyrosine kinase family. CD24 expression is observed in over 70% of PDAC tumors and in putative PDAC cancer stem cells (CSC). CSC can also express CD44 and epithelial specific antigen (ESA) or CD133. Given the prevalence of early-disseminated metastases, CSCs are believed to play a key role in the pancreatic cancer development and progression. Among these PDAC CSC protein markers, CD24 has the lowest expression in normal tissue and thus is proposed as a suitable target antigen for immunotherapy (70, 71). A study carried out by Maliar et al. examined the therapeutic efficacy of both HER2-CAR and CD24-CAR in treating PDACs in murine models. In mice bearing subcutaneous human PDAC cell line Capan-1 (positive for both HER2 and CD24), intratumor injection of the CAR T cells targeting either HER2 or CD24 greatly inhibited tumor growth, in some cases, eradicated tumors. In mice bearing orthotopic Capan-1 tumors, i.v. injections of these two different CAR T cells also demonstrated anti-tumor effects to both primary and metastatic tumors at the liver and lymph nodes. Interestingly, the cells that were dissociated from patient pancreatic cancers for xenograft in this study, displayed a heterogeneous expression pattern of antigens, including both HER2 and CD24. The adoptive transfer of HER2-CAR or CD24-CAR to mice bearing these xenografts significantly arrested the tumor growth, but to a different degree, dependent on the CAR-targeted antigen specificity. The results from this study highlight the antigen heterogeneity nature of human pancreatic tumors (72). It remains to be seen if the administration of CAR T cells targeting both antigens will enhance efficacy. This study highlights potential therapeutic limitations using CARs targeting a single tumor antigen in treating complex cancers that are heterogeneous in antigen expression patterns and distributions.
Fibroblast activation protein (FAP) is a type II integral membrane serine protease. Healthy adult tissues have no detectable FAP expression. However, under certain biological circumstances, such as remodeling, wound healing, and embryogenesis, FAP expression has been observed. FAP is also present in a large proportion of tumor stromal fibroblasts in the majority of epithelial carcinomas including pancreatic cancers (73) and its expression correlates with poor prognosis in pancreatic cancer patients (74). The carcinoma-associated fibroblasts (CAFs) are a central player in tumorigenesis and metastasis and the key characteristics of CAFs is the expression of FAP (75). Due to its high expression in CAFs, FAP has been tested as a CAR target.
Tran et al. investigated the use of FAP-CAR T cells targeting tumor stromal fibroblasts in a number of mouse tumor models and human pancreatic cancer xenografts. Despite in vitro activity observed, the injection of the FAP-CAR T cells only elicited limited in vivo anti-tumor effect. Unexpected side effects such as cachexia and lethal bone toxicities were observed. The off-target effect was due to the expression of FAP on murine bone marrow stromal cells (BMSCs). In this study, human BMSCs were also identified in expressing FAP and could be recognized by FAP-CAR T cells (76). The finding that FAP is expressed by BMSCs raised safety concerns for therapies targeting FAP.
Interestingly, in separate studies carried out by Wang et al. (77) and Kakarla et al. (78) using FAP-CAR T cells, no toxicity was observed. The reason may be due to the different scFvs used. The scFv used by Wang et al. targets a different FAP epitope and only eliminate FAPhi cells, while sparing FAPlow cells including BMSCs.
A substantial number of clinical trials involving the use of CAR technology have recently been undertaken on pancreatic cancer patients in an attempt to investigate the potential safety, effectiveness and feasibility of the approach. A variety of tumor associated antigens (TAA) expressed on pancreatic cancer cells have been targeted in various clinical trials to redirect CAR T cells against pancreatic cancer including MSLN, CEA, PSCA, MUC1, and HER2 (Table 1).
A range of CAR formats are used in these clinical studies, although chiefly second-generation formats, with either CD28 or CD137 cytoplasmic domains. Transduction methods also vary, including electroporation with CAR-encoding RNA, but viral vectors, either retroviral or lentiviral, are the main method of CAR gene delivery (Table 1). Typically, dose escalation is used in these trials, which aim to determine safety, with doses ranging from 1 × 107 to 3 × 108 per m2. Higher doses of RNA transfected T cells are used, since they present less of a risk due to their limited duration of CAR expression. The majority of studies involve a single dose of CAR T cells; some trials use multiple doses (Table 1).
There is substantial variation in the use of preconditioning, although some level of lymphodepletion is proposed for most studies. The combination of CY and fludarabine is often preferred to induce a deep level of lymphodepletion to enhance engraftment of transferred CAR T cells. While most of the studies detailed in Table 1 involve the transfer of CAR T cells alone, some propose to use additional drugs to enhance CAR T cell activity. Thus, IL-2 is proposed for some studies to provide T cells with a supporting growth factor.
To enhance the effectiveness and the safety of CAR T cell therapy, some clinical trials use reagents that activate CAR T cells. For example, in trial NCT02744287 targeting PSCA, a dimerizer agent, Rimiducid (AP1903) is used. Rimiducid is administered with PSCA-specific CAR T cells that contain an inducible MyD88/CD40 (iMC) costimulatory domain. In another trial (NCT02416466), radiation is delivered using Yttrium-90 microspheres to maximize the tumoricidal effects of CAR T cells and minimize the effects on healthy liver parenchyma in patients with liver metastases.
The majority of clinical trials of CAR T cells in pancreatic cancer are in early stages of recruitment, but some have been completed and initial reports are available. CAR T cells are generally well-tolerated in pancreatic cancer patients, although some toxicity was reported when targeting CEA (47). Despite the early nature of most trials, there are some reports of CAR T cell efficacy. The CARsgen trial reported their early results at the 2018 CAR-TCR Summit in Boston that using an anti-Claudin-18.2 CAR treating pancreatic and gastric cancer patients resulted in some objective responses without overt toxicities (79, 80). However, there are no descriptions of significant responses in most trials to date, some unique challenges presented by pancreatic cancer may have to be overcome to maximize responses.
A major obstacle for immunotherapies, in particular CAR T cell therapies, in solid tumors is the immunosuppressive tumor microenvironment (TME) (75). The TME impacts on the efficacy of CAR T cells both by limiting their infiltration and suppressing their function within the tumor (29, 81).
A unique feature of the pancreatic TME is the desmoplastic stromal reaction, which promotes tumor growth and provides a physical barrier for therapeutic drugs and T cell infiltration. In fact, PDAC is one of the most stroma-rich cancers and in some cases, the stromal components precede pancreatic cancer cells. In normal pancreas, the pancreatic stellate cells (PSCs) are a rare population and function to store retinoids in a form of lipid droplets in the cytosol. During pancreatic cancer progression, the pancreatic stellate cells (PSCs) become activated by tumor-secreted cytokines, lose retinoid droplets and transform into a myofibroblast phenotype. The activated PSCs secrete extracellular matrix (ECM) proteins and deposit collagens to form a dense fibrotic cancer stroma (82). In addition, activated PSCs secrete cytokines (such as IL-6 and IL-11) and chemokines (such as CXCL12, CCL5, CCL2, and CCL17) that recruit immunosuppressive leukocyte subsets (82–85). Indeed, as discussed above, CAR T cells that target the highly expressed CAF protein, FAP, are considered in pancreatic cancer due to the significant role these cells play in tumorigenesis (77, 86, 87) and in addition, FAP is positive in PDAC-derived ECMs but negative in normal PSCs (88). The particularly harsh TME is a major barrier to CAR T cell efficacy in pancreatic cancer and thus additional modulators will be required for durable responses (Figure 1).
Approaches that enable CAR T cells to sustain and function in the TMEs have been investigated in a large number of preclinical and clinical studies in a range of malignancies, and the results have revealed various potential strategies against pancreatic cancers. For example, depleting immunosuppressive cell subsets such as Tregs, MDSCs and TAMs has demonstrated enhanced efficacy of CAR T cell therapies (29). These suppressive immune cell types are enriched in pancreatic cancer and are associated with increased tumor growth and poorer prognosis (89–91). Various reagents that have shown to eliminate these suppressive immune cells or modulate their functions have been tested in murine models in recent years, such as IL-2 toxin and anti-CD25 (for Treg depletion), CSF-1R inhibitor and CCR2 toxin (for MDSCs/TAMs) (92–95). A few anti-TAM drugs such as antibodies against CCL2, CSF1R that inhibit the recruitment and survival of TAMs and MDSCs are currently under clinical investigation (96). Strategies of using CARs against these suppressive cells are emerging. For examples, inserting CSF1R-CARs into NK and T cells for killing TAMs have demonstrated promising outcomes in vitro (97). In our laboratory, using ACTIV therapy that involved injecting dual-specific CAR T cells and a vaccine, TAMs decreased significantly post the treatment, coinciding with tumor regression (32). In humans, the engraftment of T cells is enhanced with myeloablative preconditioning regimes and thus specific depletion methods could enhance CAR T cell efficacy in pancreatic cancer patients (98, 99).
Another strategy for enhancing CAR T cell efficacy in solid tumors is blocking the inhibitory signals received by the T cells from immunosuppressive populations in the TME. Checkpoint inhibitors, against molecules such as PD-1, CTLA4, TIM-3, and LAG-3 have shown promise as single agents in a number of cancer types (100) but unfortunately, none of these treatment as a single therapy has generated significant clinical benefit in pancreatic cancers. CAR T cells can express high amounts of these checkpoint molecules, which can lead to apoptosis and hypo-function. Results from preclinical studies clearly demonstrated that CAR T cell treatments could benefit from the addition of checkpoint inhibitors (101, 102). Some early clinical data also support the use of a combination of the checkpoint inhibitors with CAR T cell therapy in treating difficult cancers. A study carried out by Chong et al. reported that in a diffuse large B cell lymphoma patient refractory to CART19, after PD-1 blockade, the patient had an expansion of the CAR T cells and clinically significant antitumor response (103).
In addition, a number of strategies have been tested to enhance CAR cells to sustain in a suppressive cytokine milieu. Methods such as introducing the CD137 signaling domain within the CAR intracellular domain to increase mitochondrial biogenesis (104), expressing regulatory subunit I anchoring disruptor (RIAD) peptide to CAR T cells to disrupt protein kinase A (PKA) activation (105), and constitutively expressing CD40L by CAR T cells (106) have all demonstrated potential in enhancing CAR T cell treatment efficacy in solid cancers. A recent elegant study combined MSLN-CAR T cells with an oncolytic adenovirus expressing TNF-α and IL-2 to treat human PDAC xenograft models and a sygeneic mouse tumor model. This strategy significantly enhanced CAR T cell anti-tumor efficacy. The anti-tumor effect was linked to increased tumor-infiltrating lymphocytes and altered TME including altered polarization of macrophages and maturation of dendritic cells (107).
Given the limited options for efficacious pancreatic cancer treatment and the success of CAR T cells in hematological malignancies, in which TME differs in its degree of immunosuppression, overcoming this obstacle in pancreatic cancer will be an important consideration for future research.
Pancreatic cancer is the most lethal cancer and new therapies are urgently needed. CAR T cell therapy represents a revolutionary treatment for cancers and has generated remarkable responses in hematological malignancies. The extension of CAR T cell therapy into pancreatic cancer recently started and this field is moving forward rapidly. Due to its unique immunosuppressive TME and antigen complexity and heterogeneity (108), pancreatic cancer presents one of the most difficult cancers for immunotherapies. Understanding the pancreatic cancer TME and how this TME affects CAR T cell efficacy is key in designing effective CAR T cell treatments. In addition, besides expanding the CAR-antigen landscape, targeting multiple antigens simultaneously (109) and using strategies targeting neoantigens (16) could provide significant opportunities for treating pancreatic cancer.
AA, MK, and CS: conception and design; AA, AO, TS, JC, MK, and CS: write, review, and revision of the manuscript; MK and CS: supervision.
This work was supported by grants from the Peter MacCallum Cancer Center Foundation, the National Health and Medical Research Council (NHMRC) of Australia (1103352 and 1132373), the National Breast Cancer Foundation (NBCF) of Australia (IIRS-18-064) and Susan G. Komen Breast Cancer Foundation (16376637). CS was supported by a Fellowship from the NBCF. AA was supported by a University of Melbourne International Research scholarship, AO by an Australian Postgraduate Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bittoni A, Andrikou K, Lanese A, Santoni M, Pellei C, Faloppi L, et al. Treatment of pancreatic cancer. Actuality and perspective. Recent Prog Med. (2015) 106:208–16. doi: 10.1701/1868.20404
2. Teague A, Lim KH, Wang-Gillam A. Advanced pancreatic adenocarcinoma: a review of current treatment strategies and developing therapies. Ther Adv Med Oncol. (2015) 7:68–84. doi: 10.1177/1758834014564775
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
4. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. (2010) 60:277–300. doi: 10.3322/caac.20073
5. Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, et al. Current therapeutic strategies for advanced pancreatic cancer: a review for clinicians. World J Clin Oncol. (2016) 7:27–43. doi: 10.5306/wjco.v7.i1.27
6. Williamson RC. Pancreatic cancer: the greatest oncological challenge. Br Med J. (1988) 296:445–6. doi: 10.1136/bmj.296.6620.445
7. Nitecki SS, Sarr MG, Colby TV, van Heerden JA. Long-term survival after resection for ductal adenocarcinoma of the pancreas. Is it really improving? Ann Surg. (1995) 221:59–66. doi: 10.1097/00000658-199501000-00007
8. Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, et al. Engineered T cells for pancreatic cancer treatment. HPB (2011) 13:643–50. doi: 10.1111/j.1477-2574.2011.00344.x
9. Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets (2011) 15:817–28. doi: 10.1517/14728222.2011.566216
10. Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. J Gastrointest Surg. (2006) 10:1199–210; discussion: 210–1. doi: 10.1016/j.gassur.2006.08.018
11. Burris HA, III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. (1997) 15:2403–13. doi: 10.1200/JCO.1997.15.6.2403
12. Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. (2004) 350:1200–10. doi: 10.1056/NEJMoa032295
13. Valle JW, Palmer D, Jackson R, Cox T, Neoptolemos JP, Ghaneh P, et al. Optimal duration and timing of adjuvant chemotherapy after definitive surgery for ductal adenocarcinoma of the pancreas: ongoing lessons from the ESPAC-3 study. J Clin Oncol. (2014) 32:504–12. doi: 10.1200/JCO.2013.50.7657
14. Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science (2004) 305:200–5. doi: 10.1126/science.1100369
15. Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother Cancer (2016) 4:14. doi: 10.1186/s40425-016-0119-z
16. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551:512–6. doi: 10.1038/nature24462
17. Torphy RJ, Zhu Y, Schulick RD. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann Gastroenterol Surg. (2018) 2:274–81. doi: 10.1002/ags3.12176
18. Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. (2018) 8:924–34. doi: 10.1158/2159-8290.CD-18-0297
19. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
20. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13:525–41. doi: 10.1038/nrc3565
21. Duong CP, Yong CS, Kershaw MH, Slaney CY, Darcy PK. Cancer immunotherapy utilizing gene-modified T cells: from the bench to the clinic. Mol Immunol. (2015) 67(2 Pt A):46–57. doi: 10.1016/j.molimm.2014.12.009
22. Kershaw MH, Westwood JA, Slaney CY, Darcy PK. Clinical application of genetically modified T cells in cancer therapy. Clin Transl Immunol. (2014) 3:e16. doi: 10.1038/cti.2014.7
23. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
24. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
25. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
26. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
27. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol. (2017) 95:356–63. doi: 10.1038/icb.2016.128
28. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
29. Beavis PA, Slaney CY, Kershaw MH, Gyorki D, Neeson PJ, Darcy PK. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin Immunol. (2016) 28:64–72. doi: 10.1016/j.smim.2015.11.003
30. Kershaw MH, Westwood JA, Hwu P. Dual-specific T cells combine proliferation and antitumor activity. Nat Biotechnol. (2002) 20:1221–7. doi: 10.1038/nbt756
31. Slaney CY, von Scheidt B, Davenport AJ, Beavis PA, Westwood JA, Mardiana S, et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res. (2017) 23:2478–90. doi: 10.1158/1078-0432.CCR-16-1860
32. Westwood JA, Ellis S, Danne J, Johnson C, Oorschot V, Ramm G, et al. An ultrastructural investigation of tumors undergoing regression mediated by immunotherapy. Oncotarget (2017) 8:115215–29. doi: 10.18632/oncotarget.23215
33. Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. (1996) 93:136–40. doi: 10.1073/pnas.93.1.136
34. Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol. (2016) 34:4171–9. doi: 10.1200/JCO.2016.68.3672
35. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
36. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
37. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
38. Saeki N, Gu J, Yoshida T, Wu X. Prostate stem cell antigen: a Jekyll and Hyde molecule? Clin Cancer Res. (2010) 16:3533–8. doi: 10.1158/1078-0432.CCR-09-3169
39. Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. (2001) 61:4320–4. Available online at: http://cancerres.aacrjournals.org/content/61/11/4320
40. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. (2014) 25:1003–12. doi: 10.1089/hum.2013.209
41. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4–1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. (2007) 179:4910–8. doi: 10.4049/jimmunol.179.7.4910
42. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T Cells in B cell non-Hodgkin's Lymphomas. Mol Ther. (2018). 26:2727–37. doi: 10.1016/j.ymthe.2018.09.009.
43. Jessup JM, Petrick AT, Toth CA, Ford R, Meterissian S, O'Hara CJ, et al. Carcinoembryonic antigen: enhancement of liver colonisation through retention of human colorectal carcinoma cells. Br J Cancer (1993) 67:464–70. doi: 10.1038/bjc.1993.88
44. Nap M, Mollgard K, Burtin P, Fleuren GJ. Immunohistochemistry of carcino-embryonic antigen in the embryo, fetus and adult. Tumour Biol. (1988) 9:145–53. doi: 10.1159/000217555
45. Yamaguchi K, Enjoji M, Tsuneyoshi M. Pancreatoduodenal carcinoma: a clinicopathologic study of 304 patients and immunohistochemical observation for CEA and CA19–9. J Surg Oncol. (1991) 47:148–54. doi: 10.1002/jso.2930470303
46. Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, et al. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology (2012) 143:1095–107.e2. doi: 10.1053/j.gastro.2012.06.037
47. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
48. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
49. Qu CF, Li Y, Song YJ, Rizvi SM, Raja C, Zhang D, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br J Cancer (2004) 91:2086–93. doi: 10.1038/sj.bjc.6602232
50. Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. (2013) 10:607–20. doi: 10.1038/nrgastro.2013.120
51. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-Glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
52. Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. (2015) 21:2325–37. doi: 10.1158/1078-0432.CCR-14-1399
53. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. (2001) 11:130–5. doi: 10.1016/S0962-8924(00)01906-1
54. Golubovskaya V, Berahovich R, Zhou H, Xu S, Harto H, Li L, et al. CD47-CAR-T cells effectively kill target cancer cells and block pancreatic tumor growth. Cancers (2017) 9:E139. doi: 10.3390/cancers9100139
55. Ozawa F, Friess H, Tempia-Caliera A, Kleeff J, Buchler MW. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen. (2001) 21:27–44. doi: 10.1002/1520-6866(2001)21:1<27::AID-TCM4>3.0.CO;2-9
56. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
57. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
58. Yamanaka Y, Friess H, Kobrin MS, Buchler M, Kunz J, Beger HG, et al. Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Human Pathol. (1993) 24:1127–34. doi: 10.1016/0046-8177(93)90194-L
59. te Velde EA, Franke AC, van Hillegersberg R, Elshof SM, de Weger RW, Borel Rinkes IH, et al. HER-family gene amplification and expression in resected pancreatic cancer. Eur J Surg Oncol. (2009) 35:1098–104. doi: 10.1016/j.ejso.2009.02.013
60. Safran H, Steinhoff M, Mangray S, Rathore R, King TC, Chai L, et al. Overexpression of the HER-2/neu oncogene in pancreatic adenocarcinoma. Am J Clin Oncol. (2001) 24:496–9. doi: 10.1097/00000421-200110000-00016
61. Komoto M, Nakata B, Amano R, Yamada N, Yashiro M, Ohira M, et al. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. (2009) 100:1243–7. doi: 10.1111/j.1349-7006.2009.01176.x
62. Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell (2018) 9:838–47. doi: 10.1007/s13238-017-0440-4
63. Raj D, Yang MH, Rodgers D, Hampton EN, Begum J, Mustafa A, et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut (2018). doi: 10.1136/gutjnl-2018-316595 Available online at: https://gut.bmj.com/content/gutjnl/early/2018/08/17/gutjnl-2018-316595.full.pdf
64. Subramani R, Lopez-Valdez R, Arumugam A, Nandy S, Boopalan T, Lakshmanaswamy R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS ONE (2014) 9:e97016. doi: 10.1371/journal.pone.0097016
65. Huang X, Park H, Greene J, Pao J, Mulvey E, Zhou SX, et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE (2015) 10:e0133152. doi: 10.1371/journal.pone.0133152
66. Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, et al. Targeting EGFR in pancreatic cancer treatment. Curr drug Targets (2012) 13:802–10. doi: 10.2174/138945012800564158
67. Li H, Huang Y, Jiang DQ, Cui LZ, He Z, Wang C, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. (2018) 9:177. doi: 10.1038/s41419-017-0238-6
68. Zhou X, Li J, Wang Z, Chen Z, Qiu J, Zhang Y, et al. Cellular immunotherapy for carcinoma using genetically modified EGFR-specific T lymphocytes. Neoplasia (2013) 15:544–53. doi: 10.1593/neo.13168
69. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. (2016) 59:468–79. doi: 10.1007/s11427-016-5023-8
70. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. (2007) 67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030
71. Kristiansen G, Sammar M, Altevogt P. Tumour biological aspects of CD24, a mucin-like adhesion molecule. J Mol Histol. (2004) 35:255–62. doi: 10.1023/B:HIJO.0000032357.16261.c5
72. Maliar A, Servais C, Waks T, Chmielewski M, Lavy R, Altevogt P, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology (2012) 143:1375–84.e1–5. doi: 10.1053/j.gastro.2012.07.017
73. Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci USA. (1990) 87:7235–9. doi: 10.1073/pnas.87.18.7235
74. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z, et al. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas (2008) 37:154–8. doi: 10.1097/MPA.0b013e31816618ce
75. Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH, et al. Tissue-dependent tumor microenvironments and their impact on immunotherapy responses. Front Immunol. (2018) 9:70. doi: 10.3389/fimmu.2018.00070
76. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. (2013) 210:1125–35. doi: 10.1084/jem.20130110
77. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
78. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
79. Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, et al. Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. J Natl Cancer Inst. (2018). doi: 10.1093/jnci/djy134 [Epub ahead of print].
80. NewsFeed. CAR-TCR SUMMIT 2018: Preliminary First-in-Human Data with CAR-CLAUDIN18.2-T in Gastric and Pancreatic Cancer. Medipaper Medical Communications Ltd. (2018). Available online at: https://medi-paper.com/car-tcr-summit-2018-preliminary-first-in-human-data-with-car-claudin18–2-t-in-gastric-and-pancreatic-cancer/
81. Scarfo I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer (2017) 5:28. doi: 10.1186/s40425-017-0230-9
82. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
83. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. (2015) 47:1168–78. doi: 10.1038/ng.3398
84. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. (2008) 68:918–26. doi: 10.1158/0008-5472.CAN-07-5714
85. Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology (2005) 128:907–21. doi: 10.1053/j.gastro.2004.12.036
86. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. (2015) 75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041
87. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
88. Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer (2011) 11:245. doi: 10.1186/1471-2407-11-245
89. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369
90. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
91. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE (2014) 9:e91551. doi: 10.1371/journal.pone.0091551
92. Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood (2009) 114:3793–802. doi: 10.1182/blood-2009-03-208181
93. Baba J, Watanabe S, Saida Y, Tanaka T, Miyabayashi T, Koshio J, et al. Depletion of radio-resistant regulatory T cells enhances antitumor immunity during recovery from lymphopenia. Blood (2012) 120:2417–27. doi: 10.1182/blood-2012-02-411124
94. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
95. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. (2012) 72:876–86. doi: 10.1158/0008-5472.CAN-11-1792
96. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
97. Zhang P, Zhao S, Wu C, Li J, Li Z, Wen C, et al. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy (2018) 10:935–49. doi: 10.2217/imt-2018-0012
98. Seyedin SN, Schoenhals JE, Lee DA, Cortez MA, Wang X, Niknam S, et al. Strategies for combining immunotherapy with radiation for anticancer therapy. Immunotherapy (2015) 7:967–80. doi: 10.2217/imt.15.65
99. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
100. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
101. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
102. Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O'Brien S, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res. (2016) 22:436–47. doi: 10.1158/1078-0432.CCR-15-1070
103. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
104. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
105. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. (2016) 4:541–51. doi: 10.1158/2326-6066.CIR-15-0263
106. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. (2015) 23:769–78. doi: 10.1038/mt.2015.4
107. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:99573. doi: 10.1172/jci.insight.99573
108. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (2008) 321:1801–6. doi: 10.1126/science.1164368
Keywords: chimeric antigen receptor, pancreatic cancer, tumor microenvironment, pancreatic ductal adenocarcinoma, adoptive cell transfer, immunotherapy
Citation: Ali AI, Oliver AJ, Samiei T, Chan JD, Kershaw MH and Slaney CY (2019) Genetic Redirection of T Cells for the Treatment of Pancreatic Cancer. Front. Oncol. 9:56. doi: 10.3389/fonc.2019.00056
Received: 16 November 2018; Accepted: 21 January 2019;
Published: 12 February 2019.
Edited by:
Catherine Sautes-Fridman, INSERM U1138 Centre de Recherche des Cordeliers, FranceReviewed by:
Bipulendu Jena, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2019 Ali, Oliver, Samiei, Chan, Kershaw and Slaney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael H. Kershaw, bWljaGFlbC5rZXJzaGF3QHBldGVybWFjLm9yZw==
Clare Y. Slaney, Y2xhcmUuc2xhbmV5QHBldGVybWFjLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.