 Carolina Salazar
Carolina Salazar Ian G. Campbell
Ian G. Campbell Kylie L. Gorringe
Kylie L. Gorringe- 1Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 2Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, VIC, Australia
The dualistic classification of epithelial ovarian cancer (EOC) into “type I” and “type II” is widely applied in the research setting; it is used as a convenient way of conceptualizing different mechanisms of tumorigenesis. However, this classification conflicts with recent molecular insights of the etiology of EOC. Molecular and cell of origin studies indicate that while type II tumors could be classed together, type I tumors are not homogenous, even within the histological types, and can have poor clinical outcomes. Type II high grade serous carcinoma and type I low grade serous carcinomas best fit the description of the dualistic model, with different precursors, and distinct molecular profiles. However, endometriosis-associated cancers should be considered a separate group, without assuming an indolent course or type I genetic profiles. Furthermore, the very clear differences between mucinous ovarian carcinomas and other type I tumors, including an uncertain origin, and heterogeneous mutational spectrum and clinical behavior, indicate a non-type I classification for this entity. The impression that only type II carcinomas are aggressive, have poor prognosis, and carry TP53 mutations is an unhelpful misinterpretation of the dualistic classification. In this review, we revisit the history of EOC classification, and discuss the misunderstanding of the dualistic model by comparing the clinical and molecular heterogeneity of EOC types. We also emphasize that all EOC research, both basic and clinical, should consider the subtypes as different diseases beyond the type I/type II model, and base novel therapies on the molecular characteristics of each tumor.
Introduction
Epithelial Ovarian cancer (EOC) is one of the four malignancies of the female genital tract (1) and ranks fifth in deaths caused by cancer among women (2–4). The median age of EOC patients is 63 years (5) and persistent pelvic, abdominal and back pain, unusual bloating, frequent urination and lack of energy are all EOC symptoms related to everyday conditions for women in this age group (6, 7). The non-specific symptoms of EOC is one of the explanations for generally advanced-stage disease at diagnosis (8) which is associated with less favorable prognosis, abdominal metastasis and recurrence within 18 months for most patients with advanced disease (5).
EOC comprise diverse types of histology, and over the years a number of classification systems have been proposed. One system that is widely applied in the research setting is a dualistic classification into “type I” and “type II” cancers (9). However, this system was devised prior to the availability of very large data sets of base-pair level genomic information for EOC, and seems incompatible with the latest molecular insights of the origin and relationships of the various forms of EOC. In this review, we argue that this dualistic model is widely misinterpreted when applied across the full spectrum of EOC and fails to incorporate the clinical and molecular heterogeneity of this disease. Application of this framework to study design and interpretation has led to many studies in contexts as varied as transgenic mouse models (10), biomarker studies (11), clinical research (11, 12), and biological analysis (13). By using the dualistic model, these studies have likely missed true biological signals by merging at least four distinct subtypes into a single Type I grouping.
History of EOC Classification
As noted by Peaslee in 1873, for more than 200 years ovarian cancer (OC) was classified according to tumor size and character into cystic and solid tumors (14, 15). In the late nineteenth century, improvements in pathological techniques led von Waldeyer-Hartz (16) to the first classification based on histogenesis of ovarian tumors; differentiating epithelial and teratoid tumors from connective tissue growths. Subsequently, others have proposed several histogenesis-based classifications (14) (Figure 1).
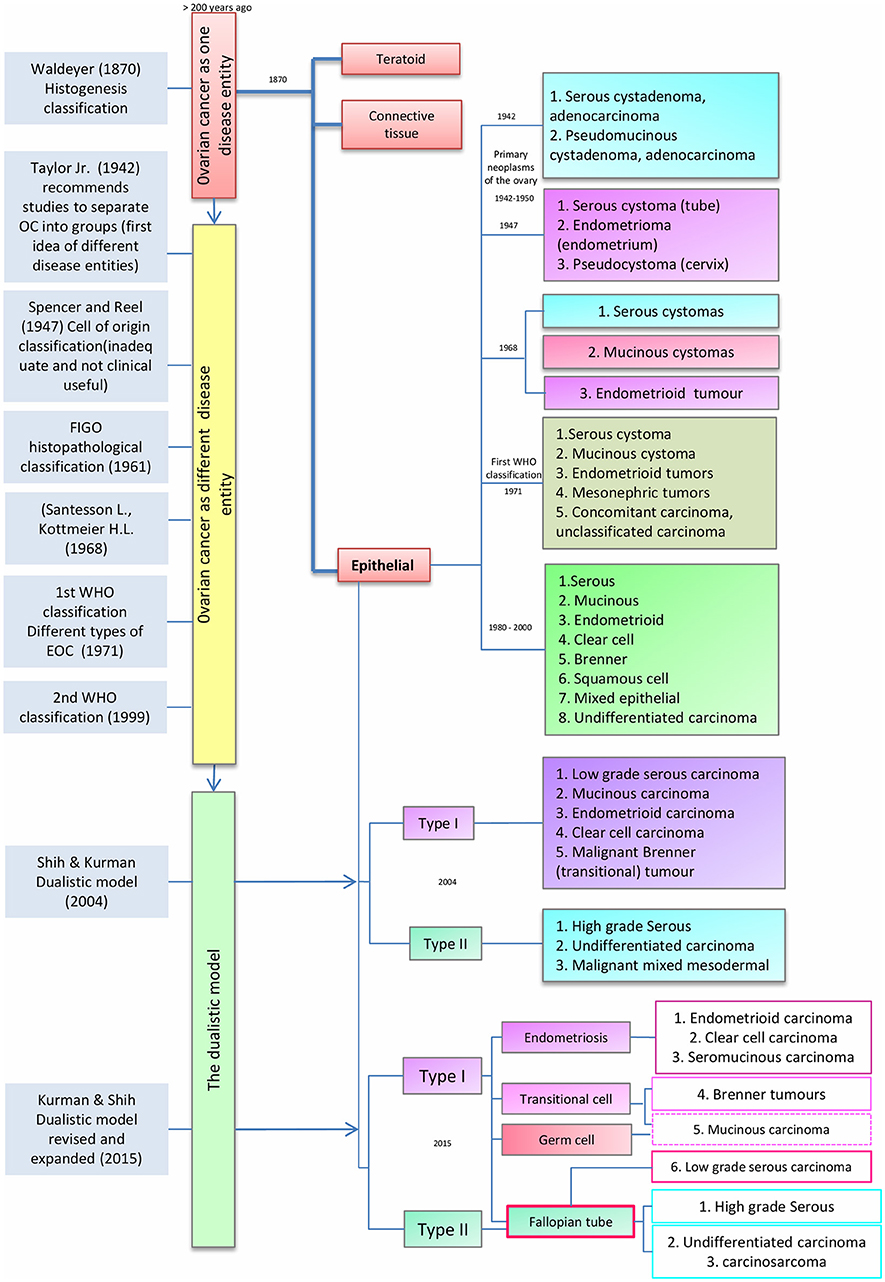
Figure 1. Timeline classification of epithelial ovarian cancer. Blue boxes on the left show author contributions to ovarian cancer (OC) classification over time. Red, yellow, and green boxes represent the evolution of OC classification from a single disease entity to a binary model. Finally, the different sub classifications of epithelial ovarian cancer (center red box), are presented from 1942 to the latest model in 2015.
In 1942, Taylor Jr. noted that research studies and clinicopathological reports had to take account of the specific type of OC. This idea was supported by Marchetti, who in 1950 recommended a standardized system to classify OC whereby epithelial tumors were part of the “primary neoplasm of the ovary” and divided into two groups: (1) serous cystadenoma and cystadenocarcinoma and (2) Pseudomucinous cystadenoma and cystadenocarcinoma. Subsequently, in 1947, Spencer and Reel proposed a modification of the classification described by Schiller (1940), in which EOC was classified on the origin of tumors into three groups: (1) serous cystomas (tubal origin), (2) endometrioma (endometrial origin) and (3) pseudocystoma (cervical origin) (14, 17). This classification system was ahead of its time given the limited data that was available about the cellular origins of OC. In 1961 this model was supplanted by an ovarian-centric histopathological classification as proposed by the Cancer Committee of the International Federation of Gynecology and Obstetrics (FIGO). The FIGO model mandated that all cases of neoplasms producing hormones, germ cell tumors and metastatic carcinomas from non-ovarian primaries should be excluded from therapeutic statistics on ovarian epithelial tumors (18). In 1968, Santesson and Kottmeir proposed EOC as a group of diseases instead of one entity and stated that treatments should be based on homogeneous groups of tumors. From that time, EOC primary classification consisted of serous, mucinous and endometrioid tumors with each type subclassified into benign, borderline, or malignant (19).
The first widely used clinical histological classification of the common primary epithelial tumors of the ovary was developed by the World Health Organization (WHO) in 1971 and divided EOCs into serous cystomas, mucinous cystomas, endometrioid tumors, mesonephric tumors, concomitant carcinoma, and unclassified carcinoma (18). This classification was endorsed without modifications until 1999 when the WHO published a new classification in which EOCs were grouped into serous, mucinous, endometrioid, clear cell, Brenner, squamous cell, mixed epithelial tumors and undifferentiated carcinomas (20). These subtypes are defined according to the histological appearance of the epithelium; i.e., serous carcinomas resemble the serous epithelium of the fallopian tube, mucinous carcinomas have mucin-containing cells, clear cell tumors have cells with a clear cytoplasm and “hobnail” cells, endometrioid carcinomas have epithelium similar to the glands of the endometrium, and Brenner tumors have transitional cell aggregates. Undifferentiated carcinomas generally lack these defining cell types and are extensively atypical and highly proliferative (21, 22). However, the absence of reliable knowledge about the origin and model of progression of EOC meant that the etiology of EOC was not taken into account in the revised WHO classification.
The Dualistic Model
Since 1942, the idea that OC does not represent a single disease and should be classified according to clinicopathologic types and groups became increasingly clear. Fukunaga et al. suggested that EOC development pathways are different for each subtype. For example, endometrioid ovarian carcinomas were shown to have an origin in endometriosis, instead of the ovarian surface epithelium (23). This concept was supported by Obata et al. who demonstrated that PTEN alterations are common in endometrioid carcinomas, contrary to serous or mucinous carcinomas (24). Shih and Kurman (9) proposed a dualistic model, using molecular and morphological features rather than predominantly histological appearance. They noted that EOC origin and pathogenesis was poorly understood and specifically its classification was inconsistent regarding the relationship between borderline tumors and invasive carcinomas. Under the dualistic model, the third WHO EOC classification (2000-2005) instead grouped EOC into two broad categories: type I and type II tumors, corresponding to tumorigenic pathways and not to specific histopathology. Type I tumors included low grade serous (LGSC), mucinous (MOC), endometrioid (ENOC), clear cell carcinomas (CCOC), and malignant Brenner (transitional) tumors, while type II included high grade serous (HGSC), undifferentiated carcinomas and malignant mixed mesodermal tumors. Type I tumors appeared to have clearly defined precursor lesions, cystadenomas, atypical proliferative tumors, and non-invasive carcinoma, and evolved along a pathway that resembles the adenoma-carcinoma sequence described for colorectal cancer. The evolution of type I tumors was described as slow and associated with molecular changes in BRAF, KRAS, and PTEN that were not found in type II. In contrast, type II tumors were defined as rapidly evolving from the ovarian surface epithelium or inclusion cysts but lacking morphological precursors, carrying TP53 mutations and with early metastatic spread (9, 25).
The dualistic model is not used clinically in real-world settings, whereas WHO and FIGO classifications are important criteria for interpretation of clinical data and deciding EOC therapy (21, 22, 26). In 2014, the WHO updated the classification of tumors of the female reproductive organs based on the newly understood biology of ovarian tumors. The idea that OC is a highly heterogeneous disease in terms of etiology, histological, epidemiological, clinical and molecular features is no longer controversial and this heterogeneity suggested a classification according to the different epithelial cell types present in the reproductive female tract. In the previous WHO classification, the origin of all EOC was considered to be the mesothelial surface of the ovary but in the new classification tubal carcinogenesis is cited as the point of origin for high-grade serous carcinomas. Although the origin of mucinous and some advanced serous carcinomas is still unclear, the new classification is more consistent with our current understanding of the disease. The new classification removes transitional cell tumors (21, 22) but includes seromucinous tumors as a new type [ Kurman et al. (27) designated them as mixed Müllerian tumors]. The intermediate step from benign to invasive lesions remains as borderline tumors.
Recent genetic and histopathological studies, as well as the new WHO classification for EOC, prompted an update of Kurman and Shih's dualistic model. Type I tumors now include MOC, LGSC, seromucinous, ENOC, CCOC, and Brenner (transitional) tumors (Figure 1), while type II tumors include HGSC, carcinosarcomas, and undifferentiated carcinomas (27). Genetic stability and mutational profiles are key molecular factors distinguishing type I and II tumors: type II have high chromosome instability and carry TP53 mutations. Somatic mutations in PIK3CA, PTEN, CTNNB1, KRAS, BRAF, and ARID1A are frequent in type I tumors, whereas chromosomal instability affecting genes like CCNE1, RB1, and NF1 are common in type II (27) (Figure 2).
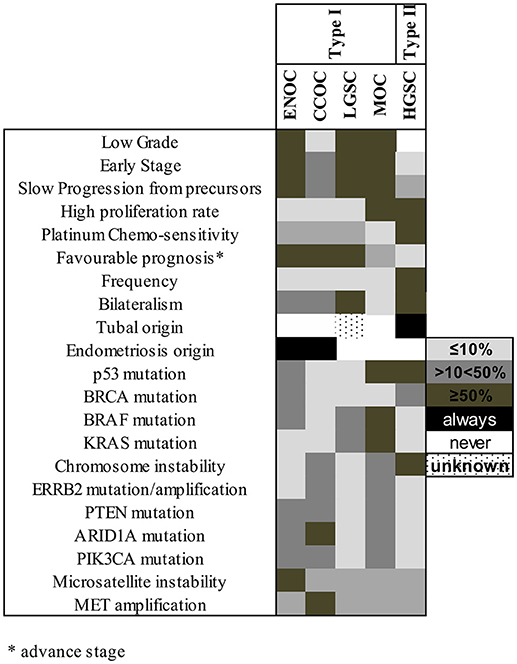
Figure 2. Molecular factors and characteristics distinguishing type I and II tumors. The shade of each box represents the frequency of the characteristics listed at left in each subtype, grouped according to their previous type I/type II assignment. ENOC, endometrioid ovarian carcinoma; CCOC, clear cell ovarian carcinoma; LGSC, low grade serous carcinoma; MOC, mucinous ovarian carcinoma; HGSC, high grade serous carcinoma.
The dualistic model is conceptually attractive and a convenient way of describing different mechanisms of disease initiation and progression. However, this model has led to the inaccurate impression that only HGSC is aggressive, has poor prognosis and carries TP53 mutations (5, 25, 28) while type I tumors are relatively homogenous and less aggressive. It is now clear that, particularly for non-HGSC, these groups are far from homogenous, even within their histological types, and many can be clinically very aggressive as described below.
Type I Tumors and MOC
Type I tumors are currently thought to arise from benign precursor lesions of the ovarian surface epithelium (29, 30) or the fallopian tube (31, 32) (LGSC), or endometriotic implants in the pelvis (CCOC, ENOC) (33–35). The exact cell of origin of LGSC is still unclear and more studies are needed to determine whether the ovary or fallopian tube or potentially both are the source. Clear cell and endometrioid carcinomas share similar mutational profiles however; the histological differences suggest that the cells of origin could be different. Cochrane et al. showed that clear cell carcinoma and endometrioid carcinoma arises from different types of cells, ciliated and secretory cells, respectively, (36). Seromucinous tumors are also thought to be derived from endometriosis at a similar frequency as endometrioid and clear cell tumors (37). Type I tumors are best exemplified by LGSC, which indeed fit many of the characteristics, with clearly defined benign and borderline precursor tumors, mutations in KRAS or BRAF but not TP53, stable chromosomal profiles, and low proliferation rates (38). However, features of the other type I tumors vary widely.
Comprehensive genome-wide analyses have shown that inactivating mutations in the ARID1A gene occur at high frequency in ENOC and CCOC (30 and 57%, respectively) (39). TP53 mutations are less frequent, but not absent, in CCOC (~5–10% in recent studies) (40–43) while PIK3CA mutations are common. ENOC have a similar pattern of genetic aberrations to CCOC, with prevalent ARID1A, PIK3CA, and CTNNB1 mutations and a low rate of TP53 mutations (44, 45). A recent whole genome analysis identified TP53 mutations in 28% (8/29) of ENOC (43). Interestingly, ENOC and CCOC can present with microsatellite instability, rarely detected in other ovarian cancers (46, 47). Copy number data from ENOC are limited, with a substantial percentage of high-grade ENOC cases reported in older studies likely to be misdiagnosed HGSC (48). CCOC have more copy number events than serous borderline tumors (49) including frequent gain of 8q (50), although less than HGSC. Some studies have shown that a subset of CCOC has a high level of chromosome instability, and this profile correlates with a worse prognosis (51, 52). Therefore, the endometriosis-related ovarian carcinomas may themselves be heterogeneous and subsets may carry type II characteristics.
MOC is the clear outlier in the type I group: its origin remains unclear and it behaves differently to the other histologic subtypes of EOC. Epidemiologic studies have suggested ovulation as a factor for developing EOC (53), whereby oral contraceptives reduce the risk of EOC (54) and menarchal age, nulliparity (55), estrogen exposure, lack of breastfeeding and tubal sterilization are all associated with ovarian cancer risk. In contrast, these factors are only weakly, if at all, associated with MOC, whereas smoking is a risk factor (25, 56–60). Primary MOC commonly express CK7, CEA, CDX2, and CA 19.9, however CK20 positivity is sometimes observed, unlike other type I tumors (56). Most MOC do not express ER or PR, consistent with a non-Müllerian origin (27), but positivity is seen in a subset (56, 61), along with the Mullerian marker PAX8 in 50% of MOCs (62, 63). MOC tumors predominantly have gastrointestinal-type differentiation but the endo-cervical type is sometimes seen (64). They can also present with a range of histopathological grades, from mostly borderline with small foci of invasion, to tumors with large areas of high architectural complexity through to a loss of differentiation, infiltrative growth and extensive nuclear atypia (grade 3). In keeping with a type I origin, KRAS-activating mutations are present in ~65% of MOC, representing the most common molecular genetic alteration (65–67). Additionally, RAS/MEK pathway activation is frequent in this cancer, since >90% of MOCs have ERRB2 amplification and/or KRAS or BRAF mutations (68). Notably different from other type I tumors, 50% of MOC carry a TP53 mutation (27, 68) which is in stark disagreement with a type I model for MOC, and it is unclear why these events have not been commonly reported in the previous literature. Other genetic events, such as RNF43, PIK3CA, or ARID1A mutation, are present in < 20% of cases (66, 68).
Inter-tumoral heterogeneity is almost a hallmark of MOC and is reflected in the multiple hypotheses for its origin. One suggestion is that these tumors are only ever metastases derived from occult extra-ovarian carcinomas, however, the existence of mucinous benign and borderline precursors as well as early stage tumors with excellent outcomes suggests this is cannot commonly be the case. MOC has been shown to share key genetic events with benign and borderline tumors (66, 68) but the cell of origin of these is not known, with suggested origins including the ovarian surface epithelium, metaplastic transformation of ovarian inclusion cyst epithelia (69), paraovarian epithelial nests of the tuboperitoneal junction as well as development from Brenner tumors or teratomas (25, 34). Mucinous cystoadenomas have been shown to be potential precursor lesions for MOC; studies revealed identical KRAS mutation at codons 12 and 13 in cystadenoma, atypical proliferative and mucinous carcinoma (65, 70). MOC and Brenner tumors share a 12q14-21 amplification and a possible tuboperitoneal junction origin (71); this could be the reason why small MOC tumors can be misdiagnosed as Brenner tumors. However, Brenner tumors could also derive from cystadenomas (27). Recent studies showed that the mucinous epithelial expansion in Brenner tumors leads to the development of cystadenomas and there is a clonal relationship between the mucinous and Brenner components (72, 73). Atypical Brenner tumors also have frequent deletion of CDKN2A, a genetic feature characteristic of MOC (74). Similarly, teratomas have also been shown to share a clonal origin with co-existing MOC (72, 75). Thus, genetic and other evidence suggests multiple potential origins for MOC: perhaps the heterogeneity of the disease is reflective of different sources, some of which may remain to be identified.
Type II Tumors
Recent studies where the entire fallopian tube and fimbria were closely examined have provided strong evidence that HGSC develops from an intraepithelial carcinoma in the fallopian tube fimbria (STIC). Sixty percent of women diagnosed with sporadic HGSC also had STIC (27, 76–78). Moreover, this tubal origin was supported by epidemiological studies were women with intact fallopian tubes have a higher risk of developing HGSC compared to women who had a previous salpingectomy (79, 80). Further evidence has been obtained from genome-wide mutation and gene expression data, clearly supporting a tubal origin for HGSC (81–83). A small subset of HGSC may develop from borderline or LGSC precursors (31, 84–86), but this is rare. The origin of the other variant of type II tumors, solid pseudoendometrioid transition tumors (SET), is still unclear but may develop from either STIC or another tubal precursor (87, 88).
HGSC tumors are frequency diagnosed at an advanced stage, and almost 100% of cases show a TP53 gene mutation (89, 90). Recent studies have shown that somatic and germline BRCA mutations are common in HGSC (83, 88) and The Cancer Genome Atlas study has shown that CCNE1 amplification, and aberrant NOTCH3 and FOXM signaling are also frequent (91). SET type tumors, which are reported in younger women, have a higher mitotic index and number of tumor-infiltrating lymphocytes than HGSC. Moreover, SET tumors have a yet higher BRCA1 mutation rate (~17%) and have a better outcome due to greater chemosensitivity (27, 88, 92, 93). Undifferentiated ovarian carcinomas are uncommon and thought to be poorly differentiated HGSC or high grade ENOC although no substantive molecular genetic studies have been reported on this type (34). Undifferentiated carcinomas share the same molecular profile as HGSOC including overexpression of p53, indicating that most probably correspond to HGSC (94–96). Carcinosarcomas (Malignant Mixed Müllerian Tumor) which are biphasic tumors with a carcinoma and a sarcoma component, show frequent CDKN2A overexpression and TP53 mutations (>90%)(9).
The Dualistic Model Is Not Informative
Independent retrospective studies that have evaluated the prognostic and clinical value of the dualistic model showed that this classification does not correlate with patients' prognosis (94–97). Panici et al. demonstrated that type II EOC present more frequently at an advanced stage than type I tumors and require more aggressive surgery (96). However, multiple studies have shown that the survival rate of patients diagnosed with type I and type II tumors is not significantly different when factors like stage and age are taken into account (96). For example, while over 80% of MOCs are diagnosed at early stage and have a better prognosis than HGSC, when detected at Stage III they have a worse prognosis due to intrinsic chemoresistance (98, 99). Similarly, Ki67 staining has found that while indeed LGSC and HGSC are markedly different, CCOC, ENOC, and especially MOC can have high proliferation rates (100).
Molecular genetics and cell of origin studies have illustrated that while type II tumors can perhaps be considered together, type I tumors are too heterogeneous and instead should be considered as different diseases. The only context where the dualistic model has relevance is for serous carcinoma of the ovary since the type II HGSC and type I LGSC have distinct carcinogenesis, different precursors, well defined molecular profiles and different morphology and prognosis that fit within the description of the model. In contrast, the cancers with an origin in endometriosis should be considered a distinct group, with a shared origin but not necessarily an indolent course or with type I genetic profiles. The very clear differences between MOC and other type I tumors, the uncertainty about the origin of MOC and the heterogeneity of its mutational spectrum, suggests a separate classification for this entity. The wide variety in clinical behavior for MOC also suggests it should never be considered as an indolent “type I” tumor, although within MOC there may be indolent subgroups. Therefore, future studies for the diagnosis and treatment of EOC should consider the subtypes as different diseases, and novel therapies should also stratify EOC on the molecular characteristics of each tumor. Molecular profiling may suggest further stratification within histological subtypes, such as the already clinically relevant separation of HGSC and LGSC.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was funded by the National Health and Medical Research Council of Australia APP1045783. CS was supported by a Melbourne University International Research Scholarship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Berek JS, Bast RC Jr. Epithelial Ovarian Cancer. In: Kufe DW, Pollock RE, Weichselbaum RR editors. Hamilton, ON (2003). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK12433/
2. Dai J, Shen W, Wen W, Chang J, Wang T, Chen H, et al. Estimation of heritability for nine common cancers using data from genome-wide association studies in Chinese population. Int J Cancer (2017) 140:329–36. doi: 10.1002/ijc.30447
3. Matz M, Coleman MP, Sant M, Chirlaque MD, Visser O, Gore M, et al. The histology of ovarian cancer: worldwide distribution and implications for international survival comparisons (CONCORD-2). Gynecol Oncol. (2017) 144:405–13. doi: 10.1016/j.ygyno.2016.10.019
4. Oronsky B, Ray CM, Spira AI, Trepel JB, Carter CA, Cottrill HM. A brief review of the management of platinum-resistant–platinum-refractory ovarian cancer. Med Oncol. (2017) 34:103. doi: 10.1007/s12032-017-0960-z
5. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet (2014) 384:1376–88. doi: 10.1016/S0140-6736(13)62146-7
6. Olson SH, Mignone L, Nakraseive C, Caputo TA, Barakat RR, Harlap S. Symptoms of ovarian cancer. Obstet Gynecol. (2001) 98:212–7. doi: 10.1097/00006250-200108000-00006
7. Goldstein CL, Susman EP, Lockwood S, Medlin EE, Behbakht K. Awareness of symptoms and risk factors of ovarian cancer in a population of women and healthcare providers. Clin J Oncol Nurs. (2015) 19:206–12. doi: 10.1188/15.CJON.206-212
8. Matz M, Coleman MP, Carreira H, Salmerón D, Chirlaque MD, Allemani C. Worldwide comparison of ovarian cancer survival: histological group and stage at diagnosis (CONCORD-2). Gynecol Oncol. (2017) 144:396–404. doi: 10.1016/j.ygyno.2016.11.019
9. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. (2004) 164:1511–8. doi: 10.1016/S0002-9440(10)63708-X
10. Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER, Cho KR. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. Am J Pathol. (2013) 182:1391–9. doi: 10.1016/j.ajpath.2012.12.031
11. Skirnisdottir I, Seidal T, Åkerud H. Differences in clinical and biological features between type I and type II tumors in FIGO stages I-II epithelial ovarian carcinoma. Int J Gynecol Cancer (2015) 25:1239–47. doi: 10.1097/IGC.0000000000000484
12. Lim AWW, Mesher D, Gentry-Maharaj A, Balogun N, Widschwendter M, Jacobs I, et al. Time to diagnosis of Type I or II invasive epithelial ovarian cancers: a multicentre observational study using patient questionnaire and primary care records. BJOG (2016) 123:1012–20. doi: 10.1111/1471-0528.13447
13. Schwede M, Spentzos D, Bentink S, Hofmann O, Haibe-Kains B, Harrington D, et al. Stem cell-like gene expression in ovarian cancer predicts type II subtype and prognosis. PLoS ONE (2013) 8:e57799. doi: 10.1371/journal.pone.0057799
14. Kottmeier HL. The classification and treatment of ovarian tumours. Acta Obstet Gynecol Scand. (1952) 31:313–63. doi: 10.3109/00016345209154959
15. Randolph PE. Ovarian Tumors: Their Pathology, Diagnosis, and Treatment, Especially by Ovariotomy. New York, NY: Appleton & Company (1872).
16. von Waldeyer-Hartz W. Eierstock und Ei: ein Beitrag zur Anatomie und Entwicklungeschichte der Sexualorgane. Leipzig: Verlag von Wilhelm Engelmann (1870).
17. Spencer JA, Reel PJ. A classification of ovarian tumors based upon histogenesis. Am J Obstet Gynecol. (1947) 54:273–80. doi: 10.1016/S0002-9378(16)39534-5
18. SO AOGS. Classification and staging of malignant tumours in the female pelvis. Acta Obstet Gynec Scand. (1971) 50:1–7. doi: 10.3109/00016347109157278
19. Santesson L, Kottmeier H. General Classification of Ovarian Tumours. Ovarian cancer Berlin; Heidelberg: Springer (1968). p. 1–8.
20. Scully RE. Histological Classification of Ovarian Tumours. Histological Typing of Ovarian Tumours. Berlin; Heidelberg: Springer (1999). p. 3–9. doi: 10.1007/978-3-642-58564-7_2
21. Kurman RJ. WHO Classification of Tumours of Female Reproductive Organs, 4th Edn. Lyon: International Agency for Research on Cancer (2014).
22. Meinhold-Heerlein I, Fotopoulou C, Harter P, Kurzeder C, Mustea A, Wimberger P, et al. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch Gynecol Obstet. (2016) 2016:695. doi: 10.1007/s00404-016-4035-8
23. Fukunaga M, Nomura K, Ishikawa E, Ushigome S. Ovarian atypical endometriosis: its close association with malignant epithelial tumours. Histopathology (1997) 30:249–55. doi: 10.1046/j.1365-2559.1997.d01-592.x
24. Obata K, Morland SJ, Watson RH, Hitchcock A, Chenevix-Trench G, Thomas EJ, et al. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. (1998) 58:2095–7.
25. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. (2014) 2014:934261. doi: 10.1155/2014/934261
26. Meinhold-Heerlein I, Hauptmann S. The heterogeneity of ovarian cancer. Arch Gynecol Obstet. (2014) 289:237–9. doi: 10.1007/s00404-013-3114-3
27. Kurman RJ, Shih Ie M. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am J Pathol. (2016) 186:733–47. doi: 10.1016/j.ajpath.2015.11.011
28. Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. (2013) 19:961–8. doi: 10.1158/1078-0432.CCR-12-2243
29. Bell DA. Origins and molecular pathology of ovarian cancer. Mod Pathol. (2005) 18:S19. doi: 10.1038/modpathol.3800306
30. Hunter SM, Anglesio MS, Sharma R, Gilks B, Melnyk N, Chiew Y-E, et al. Copy number aberrations in benign serous ovarian tumors: a case for reclassification? Clin Cancer Res. (2011) 17:7273–82. doi: 10.1158/1078-0432.CCR-11-2080
31. Nik NN, Vang R, Shih I-M, Kurman RJ. Origin and pathogenesis of pelvic (ovarian, tubal, and primary peritoneal) serous carcinoma. Ann Rev Pathol. (2014) 9:27–45. doi: 10.1146/annurev-pathol-020712-163949
32. Qiu C, Lu N, Wang X, Zhang Q, Yuan C, Yan S, et al. Gene expression profiles of ovarian low-grade serous carcinoma resemble those of fallopian tube epithelium. Gynecol Oncol. (2017) 147:634–41. doi: 10.1016/j.ygyno.2017.09.029
33. Jiang X, Morland SJ, Hitchcock A, Thomas EJ, Campbell IG. Allelotyping of endometriosis with adjacent ovarian carcinoma reveals evidence of a common lineage. Cancer Res. (1998) 58:1707–12.
34. Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer–shifting the paradigm. Hum Pathol. (2011) 42:918–31. doi: 10.1016/j.humpath.2011.03.003
35. Veras E, Mao T-L, Ayhan A, Ueda S, Lai H, Hayran M, et al. Cystic and adenofibromatous clear cell carcinomas of the ovary: distinctive tumors that differ in their pathogenesis and behavior: a clinicopathologic analysis of 122 cases. Am J Surg Pathol. (2009) 33:844–53. doi: 10.1097/PAS.0b013e31819c4271
36. Cochrane DR, Tessier-Cloutier B, Lawrence KM, Nazeran T, Karnezis AN, Salamanca C, et al. Clear cell and endometrioid carcinomas: are their differences attributable to distinct cells of origin? J Pathol. (2017) 243:26–36. doi: 10.1002/path.4934
37. Shappell HW, Riopel MA, Sehdev AES, Ronnett BM, Kurman RJ. Diagnostic criteria and behavior of ovarian seromucinous (endocervical-type mucinous and mixed cell-type) tumors: atypical proliferative (borderline) tumors, intraepithelial, microinvasive, and invasive carcinomas. Am J Surg Pathol. (2002) 26:1529–41. doi: 10.1097/00000478-200212000-00001
38. Gershenson DM. Low-grade serous carcinoma of the ovary or peritoneum. Ann Oncol. (2016) 27(Suppl. 1):i45–i9. doi: 10.1093/annonc/mdw085
39. Takeda T, Banno K, Okawa R, Yanokura M, Iijima M, Irie-Kunitomi H, et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol Rep. (2016) 35:607–13. doi: 10.3892/or.2015.4421
40. Murakami R, Matsumura N, Brown JB, Higasa K, Tsutsumi T, Kamada M, et al. Exome sequencing landscape analysis in ovarian clear cell carcinoma shed light on key chromosomal regions and mutation gene networks. Am J Pathol. (2017) 187:2246–58. doi: 10.1016/j.ajpath.2017.06.012
41. Maru Y, Tanaka N, Ohira M, Itami M, Hippo Y, Nagase H. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol Oncol. (2017) 144:377–83. doi: 10.1016/j.ygyno.2016.11.045
42. Shibuya Y, Tokunaga H, Saito S, Shimokawa K, Katsuoka F, Bin L, et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer. (2018) 57:51–60. doi: 10.1002/gcc.22507
43. Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet. (2017) 49:856–65. doi: 10.1038/ng.3849
44. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. (2010) 363:1532–43. doi: 10.1056/NEJMoa1008433
45. Kuo K-T, Mao T-L, Jones S, Veras E, Ayhan A, Wang T-L, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. (2009) 174:1597–601. doi: 10.2353/ajpath.2009.081000
46. Fujita M, Enomoto T, Yoshino K, Nomura T, Buzard G, Inoue M, et al. Microsatellite instability and alterations in the hMSH2 gene in human ovarian cancer. Int J Cancer (1995) 64:361–6. doi: 10.1002/ijc.2910640602
47. Gras E, Catasus L, Argüelles R, Moreno-Bueno G, Palacios J, Gamallo C, et al. Microsatellite instability, MLH-1 promoter hypermethylation, and frameshift mutations at coding mononucleotide repeat microsatellites in ovarian tumors. Cancer (2001) 92:2829–36. doi: 10.1002/1097-0142(20011201)92:11<2829::AID-CNCR10094>3.0.CO;2-3
48. Köbel M, Bak J, Bertelsen BI, Carpen O, Grove A, Hansen ES, et al. Ovarian carcinoma histotype determination is highly reproducible, and is improved through the use of immunohistochemistry. Histopathology (2014) 64:1004–13. doi: 10.1111/his.12349
49. Kuo KT, Mao TL, Chen X, Feng Y, Nakayama K, Wang Y, et al. DNA copy numbers profiles in affinity-purified ovarian clear cell carcinoma. Clin Cancer Res. (2010) 16:1997–2008. doi: 10.1158/1078-0432.CCR-09-2105
50. Anglesio MS, George J, Kulbe H, Friedlander M, Rischin D, Lemech C, et al. IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res. (2011) 17:2538–48. doi: 10.1158/1078-0432.CCR-10-3314
51. Uehara Y, Oda K, Ikeda Y, Koso T, Tsuji S, Yamamoto S, et al. Integrated copy number and expression analysis identifies profiles of whole-arm chromosomal alterations and subgroups with favorable outcome in ovarian clear cell carcinomas. PLoS ONE (2015) 10:e0128066. doi: 10.1371/journal.pone.0128066
52. Tan DS, Iravani M, McCluggage WG, Lambros MB, Milanezi F, Mackay A, et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin Cancer Res. (2011) 17:1521–34. doi: 10.1158/1078-0432.CCR-10-1688
53. Purdie DM, Bain CJ, Siskind V, Webb PM, Green AC. Ovulation and risk of epithelial ovarian cancer. Int J Cancer (2003) 104:228–32. doi: 10.1002/ijc.10927
54. Risch HA, Marrett LD, Jain M, Howe GR. Differences in risk factors for epithelial ovarian cancer by histologic type: results of a case-control study. Am J Epidemiol. (1996) 144:363–72. doi: 10.1093/oxfordjournals.aje.a008937
55. Hankinson SE, Colditz GA, Hunter DJ, Willett WC, Stampfer MJ, Rosner B, et al. A prospective study of reproductive factors and risk of epithelial ovarian cancer. Cancer (1995) 76:284–90. doi: 10.1002/1097-0142(19950715)76:2<284::AID-CNCR2820760219>3.0.CO;2-5
56. Xu W, Rush J, Rickett K, Coward JI. Mucinous ovarian cancer: a therapeutic review. Crit Rev Oncol Hematol. (2016) 102:26–36. doi: 10.1016/j.critrevonc.2016.03.015
57. Gates MA, Rosner BA, Hecht JL, Tworoger SS. Risk factors for epithelial ovarian cancer by histologic subtype. Am J Epidemiol. (2009) 171:45–53. doi: 10.1093/aje/kwp314
58. Jha P, Ranson MK, Nguyen SN, Yach D. Estimates of global and regional smoking prevalence in 1995, by age and sex. Am J Public Health. (2002) 92:1002–6. doi: 10.2105/AJPH.92.6.1002
59. Benito V, Lubrano A, Arencibia O, Medina N, Eva EÁ, Andújar M, et al. Serous and mucinous borderline ovarian tumors: are there real differences between these two entities? Eur J Obst Gynecol Reproduct Biol. (2010) 153:188–92. doi: 10.1016/j.ejogrb.2010.07.024
60. Jordan SJ, Green AC, Whiteman DC, Webb PM, Study AC, Group AOCS. Risk factors for benign, borderline and invasive mucinous ovarian tumors: epidemiological evidence of a neoplastic continuum? Gynecol Oncol. (2007) 107:223–30. doi: 10.1016/j.ygyno.2007.06.006
61. Tkalia I, Vorobyova L, Svintsitsky V, Nespryadko S, Goncharuk I, Lukyanova NY, et al. Clinical significance of hormonal receptor status of malignant ovarian tumors. Exp Oncol. (2014) 36:125–33.
62. Adler E, Mhawech-Fauceglia P, Gayther SA, Lawrenson K. PAX8 expression in ovarian surface epithelial cells. Hum Pathol. (2015) 46:948–56. doi: 10.1016/j.humpath.2015.03.017
63. Auersperg N. The origin of ovarian carcinomas: a unifying hypothesis. Int J Gynecol Pathol. (2011) 30:12–21. doi: 10.1097/PGP.0b013e3181f45f3e
64. Lin X, Lindner JL, Silverman JF, Liu Y. Intestinal type and endocervical-like ovarian mucinous neoplasms are immunophenotypically distinct entities. Appl Immunohistochem Mol Morphol. (2008) 16:453–8. doi: 10.1097/PAI.0b013e3181672574
65. Cuatrecasas M, Villanueva A, Matias-Guiu X, Prat J. K-ras mutations in mucinous ovarian tumors. Cancer (1997) 79:1581–6. doi: 10.1002/(SICI)1097-0142(19970415)79:8&1581::AID-CNCR21&3.0.CO;2-T
66. Ryland GL, Hunter SM, Doyle MA, Caramia F, Li J, Rowley SM, et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. (2015) 7:87. doi: 10.1186/s13073-015-0210-y
67. Mackenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer (2015) 15:415. doi: 10.1186/s12885-015-1421-8
68. Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J Pathol. (2013) 229:469–76. doi: 10.1002/path.4134
69. Cuellar-Partida G, Lu Y, Dixon SC, Australian Ovarian Cancer S, Fasching PA, Hein A, et al. Assessing the genetic architecture of epithelial ovarian cancer histological subtypes. Hum Genet. (2016) 135:741–56. doi: 10.1007/s00439-016-1663-9
70. Mok SC-H, Bell DA, Knapp RC, Fishbaugh PM, Welch WR, Muto MG, et al. Mutation of K-ras protooncogene in human ovarian epithelial tumors of borderline malignancy. Cancer Res. (1993) 53:1489–92.
71. Pejovic T, Bürki N, Odunsi K, Fiedler P, Achong N, Schwartz PE, et al. Well-differentiated mucinous carcinoma of the ovary and a coexisting Brenner tumor both exhibit amplification of 12q14–21 by comparative genomic hybridization. Gynecol Oncol. (1999) 74:134–7. doi: 10.1006/gyno.1999.5402
72. Wang Y, Wu Rc, Shwartz LE, Haley L, Lin Mt, Shih Im, et al. Clonality analysis of combined Brenner and mucinous tumours of the ovary reveals their monoclonal origin. J Pathol. (2015) 237:146–51. doi: 10.1002/path.4572
73. Tafe LJ, Muller KE, Ananda G, Mitchell T, Spotlow V, Patterson SE, et al. Molecular genetic analysis of ovarian Brenner tumors and associated mucinous epithelial neoplasms: high variant concordance and identification of mutually exclusive RAS driver mutations and MYC amplification. Am J Pathol. (2016) 186:671–7. doi: 10.1016/j.ajpath.2015.11.008
74. Kuhn E, Ayhan A, Shih I-M, Seidman JD, Kurman RJ. The pathogenesis of atypical proliferative Brenner tumor: an immunohistochemical and molecular genetic analysis. Mod Pathol. (2014) 27:231–7. doi: 10.1038/modpathol.2013.142
75. Kerr SE, Flotte AB, McFalls MJ, Vrana JA, Halling KC, Bell DA. Matching maternal isodisomy in mucinous carcinomas and associated ovarian teratomas provides evidence of germ cell derivation for some mucinous ovarian tumors. Am J Surg Pathol. (2013) 37:1229–35. doi: 10.1097/PAS.0b013e31828f9ecb
76. Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. (2001) 195:451–6. doi: 10.1002/path.1000
77. Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol. (2006) 30:230–6. doi: 10.1097/01.pas.0000180854.28831.77
78. Przybycin CG, Kurman RJ, Ronnett BM, Shih I-M, Vang R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol. (2010) 34:1407–16. doi: 10.1097/PAS.0b013e3181ef7b16
79. McAlpine JN, Hanley GE, Woo MM, Tone AA, Rozenberg N, Swenerton KD, et al. Opportunistic salpingectomy: uptake, risks, and complications of a regional initiative for ovarian cancer prevention. Am J Obstet Gynecol. (2014) 210:471. e1–e11. doi: 10.1016/j.ajog.2014.01.003
80. Falconer H, Yin L, Grönberg H, Altman D. Ovarian cancer risk after salpingectomy: a nationwide population-based study. J Natl Cancer Inst. (2015) 107:dju410. doi: 10.1093/jnci/dju410
81. Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun. (2017) 8:990. doi: 10.1038/s41467-017-01217-9
82. Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. (2017) 8:1093. doi: 10.1038/s41467-017-00962-1
83. Tone AA, Begley H, Sharma M, Murphy J, Rosen B, Brown TJ, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin Cancer Res. (2008) 14:4067–78. doi: 10.1158/1078-0432.CCR-07-4959
84. Emmanuel C, Chiew YE, George J, Etemadmoghadam D, Anglesio MS, Sharma R, et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin Cancer Res. (2014) 20:6618–30. doi: 10.1158/1078-0432.CCR-14-1292
85. Boyd C, McCluggage WG. Low-grade ovarian serous neoplasms (low-grade serous carcinoma and serous borderline tumor) associated with high-grade serous carcinoma or undifferentiated carcinoma: report of a series of cases of an unusual phenomenon. Am J Surg Pathol. (2012) 36:368–75. doi: 10.1097/PAS.0b013e31823732a9
86. Dehari R, Kurman RJ, Logani S, Shih I-M. The development of high-grade serous carcinoma from atypical proliferative (borderline) serous tumors and low-grade micropapillary serous carcinoma: a morphologic and molecular genetic analysis. Am J Surg Pathol. (2007) 31:1007–12. doi: 10.1097/PAS.0b013e31802cbbe9
87. Soslow RA, Han G, Park KJ, Garg K, Olvera N, Spriggs DR, et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol. (2012) 25:625–36. doi: 10.1038/modpathol.2011.183
88. Howitt BE, Hanamornroongruang S, Lin DI, Conner JE, Schulte S, Horowitz N, et al. Evidence for a dualistic model of high-grade serous carcinoma: BRCA mutation status, histology, and tubal intraepithelial carcinoma. Am J Surg Pathol. (2015) 39:287–93. doi: 10.1097/PAS.0000000000000369
89. Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. (2010) 221:49–56. doi: 10.1002/path.2696
90. Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474:609–15. doi: 10.1038/nature10166
91. Patch A-M, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature (2015) 521:489–94. doi: 10.1038/nature14410
92. Köbel M, Kalloger SE, Lee S, Duggan MA, Kelemen LE, Prentice L, et al. Biomarker-based ovarian carcinoma typing: a histologic investigation in the ovarian tumor tissue analysis consortium. Cancer Epidemiol Prevent Biomark. (2013) 22:1677–86. doi: 10.1158/1055-9965.EPI-13-0391
93. Silva EG, Robey-Cafferty SS, Smith TL, Gershenson DM. Ovarian carcinomas with transitional cell carcinoma patter. Am J Clin Pathol. (1990) 93:457–65. doi: 10.1093/ajcp/93.4.457
94. Braicu E, Sehouli J, Richter R, Pietzner K, Denkert C, Fotopoulou C. Role of histological type on surgical outcome and survival following radical primary tumour debulking of epithelial ovarian, fallopian tube and peritoneal cancers. Br J Cancer (2011) 105:1818–24. doi: 10.1038/bjc.2011.455
95. Bamias A, Sotiropoulou M, Zagouri F, Trachana P, Sakellariou K, Kostouros E, et al. Prognostic evaluation of tumour type and other histopathological characteristics in advanced epithelial ovarian cancer, treated with surgery and paclitaxel/carboplatin chemotherapy: cell type is the most useful prognostic factor. Eur J Cancer. (2012) 48:1476–83. doi: 10.1016/j.ejca.2011.09.023
96. Panici PB, Marchetti C, Salerno L, Musella A, Vertechy L, Palaia I, et al. Dualistic classification of epithelial ovarian cancer: surgical and survival outcomes in a large retrospective series. Ann Surg Oncol. (2014) 21:3036–41. doi: 10.1245/s10434-014-3714-6
97. Lopez-Garcia M-A, Palacios J. Pathologic and molecular features of uterine carcinosarcomas. Semin Diagn Pathol. (2010) 27:274–86. doi: 10.1053/j.semdp.2010.09.005
98. Hess V, A'hern R, Nasiri N, King DM, Blake PR, Barton DP, et al. Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol. (2004) 22:1040–4. doi: 10.1200/JCO.2004.08.078
99. Zaino RJ, Brady MF, Lele SM, Michael H, Greer B, Bookman MA. Advanced stage mucinous adenocarcinoma of the ovary is both rare and highly lethal. Cancer (2011) 117:554–62. doi: 10.1002/cncr.25460
Keywords: epithelial ovarian cancer, type I-II tumors, dualistic model, classification, ovarian carcinoma
Citation: Salazar C, Campbell IG and Gorringe KL (2018) When Is “Type I” Ovarian Cancer Not “Type I”? Indications of an Out-Dated Dichotomy. Front. Oncol. 8:654. doi: 10.3389/fonc.2018.00654
Received: 10 October 2018; Accepted: 10 December 2018;
Published: 21 December 2018.
Edited by:
Sarah M. Temkin, Virginia Commonwealth University, United StatesReviewed by:
Sophia H. L. George, University of Miami, United StatesEmily K. Colvin, University of Sydney, Australia
Copyright © 2018 Salazar, Campbell and Gorringe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kylie L. Gorringe, a3lsaWUuZ29ycmluZ2VAcGV0ZXJtYWMub3Jn
†These authors have contributed equally to this work