 Claudia Rossig
Claudia Rossig Sareetha Kailayangiri
Sareetha Kailayangiri Silke Jamitzky
Silke Jamitzky Bianca Altvater
Bianca Altvater- 1Department of Pediatric Hematology and Oncology, University Children's Hospital Muenster, Muenster, Germany
- 2Cells-in-Motion Cluster of Excellence (EXC 1003-CiM), University of Muenster, Muenster, Germany
Application of the CAR targeting strategy in solid tumors is challenged by the need for adequate target antigens. As a consequence of their tissue origin, embryonal cancers can aberrantly express membrane-anchored gangliosides. These are carbohydrate molecules consisting of a glycosphingolipid linked to sialic acids residues. The best-known example is the abundant expression of ganglioside GD2 on the cell surface of neuroblastomas which derive from GD2-positive neuroectoderm. Gangliosides are involved in various cellular functions, including signal transduction, cell proliferation, differentiation, adhesion and cell death. In addition, transformation of human cells to cancer cells can be associated with distinct glycosylation profiles which provide advantages for tumor growth and dissemination and can serve as immune targets. Both gangliosides and aberrant glycosylation of proteins escape the direct molecular and proteomic screening strategies currently applied to identify further immune targets in cancers. Due to their highly restricted expression and their functional roles in the malignant behavior, they are attractive targets for immune engineering strategies. GD2-redirected CAR T cells have shown activity in clinical phase I/II trials in neuroblastoma and next-generation studies are ongoing. Further carbohydrate targets for CAR T cells in preclinical development are O-acetyl-GD2, NeuGc-GM3 (N-glycolyl GM3), GD3, SSEA-4, and oncofetal glycosylation variants. This review summarizes knowledge on the role and function of some membrane-expressed non-protein antigens, including gangliosides and abnormal protein glycosylation patterns, and discusses their potential to serve as a CAR targets in pediatric solid cancers.
Introduction
The potency of T cells to control solid tumor growth is illustrated by activity of immune checkpoint inhibitors in several tumors in adults (1, 2). Most childhood tumors are not optimal candidates for this type of immunotherapy, for the following reasons [reviewed in (3)]: Checkpoint inhibitors unleash antigen-specific effector responses of T cells that were reversibly tolerized by inhibitory ligand-receptor interactions. The key prerequisite for efficacy is the presence of T cells with native specificity against tumor-associated neoantigens (4). To be recognized by T cells, the antigens must be processed and presented by the major histocompatibility complex to engage native T cell receptors (Figure 1). Tumor neoantigens are created by somatic mutations in the tumor microenvironment (5). Consequently, high non-synonymous somatic mutation frequency has emerged as one of the major prerequisites for successful T cell therapy with checkpoint inhibitors (6, 7). Most childhood tumors have very low mutational burdens, and the individual recurrent mutations typically found in childhood tumors and sarcomas are not coding for effective tumor rejection antigens. Consequently, the microenvironment of typical pediatric tumors generally is devoid of antigen-specific T cells that could potentially be reactivated by immune checkpoint inhibition (8, 9). In fact, the clinical results of early-phase trials of checkpoint inhibitors in childhood cancers are discouraging (10, 11).
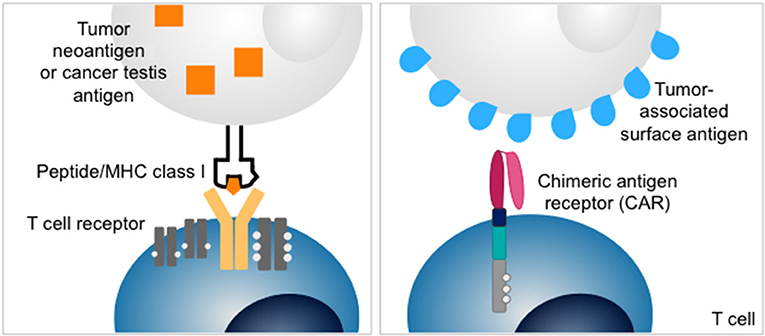
Figure 1. Structures of T cell receptor (left) and chimeric antigen receptor (CAR) (right). T cell receptors recognize intracellular antigens presented as peptides on MHC class I. Chimeric antigen receptors redirect T cells to surface antigens independent of MHC-restricted antigen presentation.
An alternative strategy to exploit T cells for cancer therapy is adoptive transfer of T cells that recognize tumor-associated surface antigens by genetic modification with chimeric antigen receptors (CARs) (12). CARs link the single-chain antigen-binding domains of a monoclonal antibody to activation domains of costimulatory receptors and the T cell receptor ζ chain (Figure 1). This allows recognition of antigens expressed on the cell surface, independent of peptide presentation on MHC. CAR T cells directed against the B lineage antigen CD19 have been highly effective to control and eliminate B cell cancers in both children and adults (13–16). Intensive efforts are on the way to extend the promise of CAR T cell therapy to solid tumors. One difficulty is the identification of adequate target antigens. Effective CAR T cell therapy requires the presence of a target antigen selectively expressed on the cell surface of tumor cells but not by essential normal tissues. Examples for protein antigens targeted by CARs in advanced preclinical or early clinical development are IL13Rα (17), HER2 (18), EGFRvIII (19), CEA (20), mesothelin (21), EphA2 (22). Overall, the range of adequate antigens beyond B lineage markers is limited.
Standard high-throughput screening tools used to identify novel cancer-associated antigens, including antigens enriched or preferentially expressed on the tumor cell membrane, analyse gene transcripts and proteins (23). The approaches do not cover the full spectrum of potential CAR targets. Unlike T cell receptor targets, antigens recognized by CARs include non-protein targets (Figure 2). These can be gangliosides (glycolipids/glycosphingolipids) or abnormally-glycosylated normal proteins (glycoforms) contributing to the outer glycan layer of the cell surface. Gangliosides are a subclass of glycosphingolipids characterized by the presence of one or more sialic acids. Aberrant glycosylation can include sialylation, fucosylation, O-glycan truncation, and N- and O-linked glycan branching. Both aberrant expression of carbohydrates and acquisition of aberrant glycosylation profiles can accompany malignant transformation. They can be unique to the malignant cell, or be restricted to immature cells but not adult somatic tissues (cancer-testis antigens), and they can contribute to tumor growth and metastasis and to immune escape. Due to their selective expression and biological role in the malignant behavior of the tumor cells, non-protein targets may be attractive target antigens. Expressed in the outer leaflet of the plasma membrane on the cell surface, they are amenable to antibody-based immunotherapeutic strategies, including antibodies (24, 25), immunotoxins, -cytokines or radioconjugates (26, 27) and more recently CAR-engineered T cells.
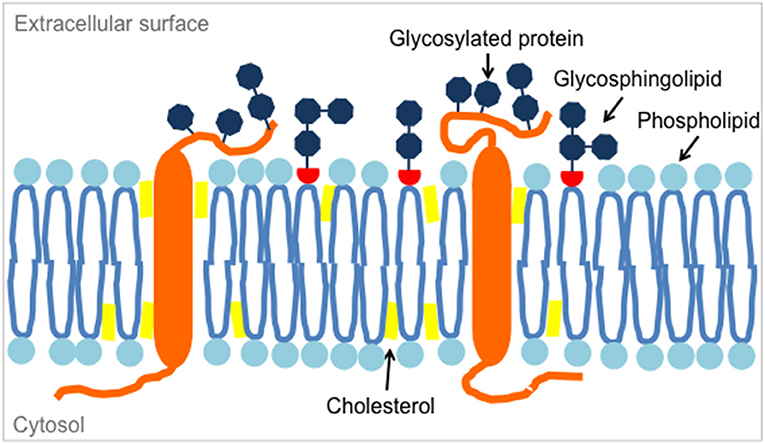
Figure 2. Glycosphingolipids are located in microdomains of the lipid bilayer of the plasma membrane, along with membrane phospholipids, cholesterol and transmembrane proteins such as glycosylated proteins.
The first to show that CARs can redirect T cells to carbohydrates and thereby extend application of targeted T cell therapy to antigens not naturally recognized by T cells was Zelig Eshhar's group (28). They generated a CAR against the difucosylated carbohydrate Lewis-Y and found that it can effectively induce T cell lysis of antigen-expressing tumor cells despite low affinity. Lewis-Y can be coupled to various proteins and lipids, including some tumor-associated antigens. It is expressed at high levels on many types of epithelial-derived cancers as well as a proportion of cells in acute myeloid leukemia (AML) and in multiple myeloma. By contrast, it is completely absent from childhood solid tumors and other cancers of neuroectodermal or mesodermal origin (29). The first carbohydrate used as a CAR target in a pediatric cancer was the ganglioside antigen GD2 (30).
This review summarizes knowledge on the current most promising carbohydrate antigens for CAR T cell therapy in pediatric cancers, including GD2 and another ganglioside, SSEA-4, as well as aberrant glycosylation motifs.
Glycolipid Targets: Gangliosides
Gangliosides were among the first antigens exploited as tumor targets for CARs. Gangliosides are complex mammalian glycolipids. They are expressed in all vertebrates and are identical in humans and in mice and other non-human species. Located in specific microdomains of the outer leaflet of the plasma membrane, they interact with membrane phospholipids and transmembrane receptors (Figure 2). As so-called glycosynapses, carbohydrate-containing microdomains are involved in cell adhesion and subsequent signal transduction. Gangliosides have been attributed various functions in cell-cell adhesion, viability, proliferation and in the modulation of cell signaling pathways (31). Ganglioside biosynthesis is performed in the Golgi apparatus by the stepwise addition of monosaccharides to ceramide by specific glycosyltransferases along complex synthesis pathways (Figure 3). As a consequence of the stepwise synthesis pathways from its precursors, individual gangliosides are often expressed in the context of their up- and downstream epitope neighbors (32).
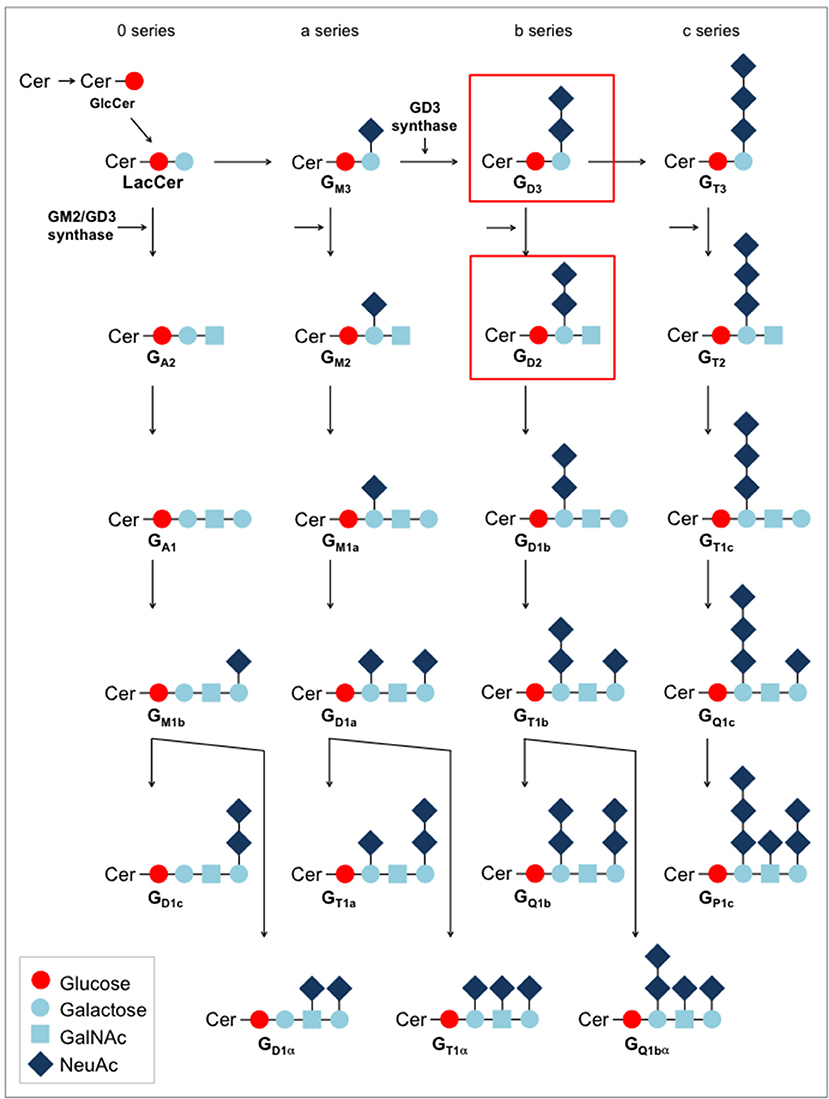
Figure 3. Biosynthesis pathways for gangliosides.
Individual gangliosides are expressed in a tissue-specific manner, with abundant expression on immature neural, sympathoadrenergic and mesenchymal cells during embryogenesis (33, 34, 35, 36). While the predominant gangliosides in early embryonic brains are simple gangliosides (GM3, GD3), later developmental stages are associated with more complex gangliosides with two or more sialic acid residues, such as GD2, GM1a, GD1a, GD1b, and GT1b (Figure 3). After birth, expression is mainly restricted to the central nervous system where complex gangliosides at low levels are found on neuronal cell bodies. The changes in the pattern of ganglioside expression during cellular differentiation suggest specific roles of individual gangliosides at different neurodevelopmental stages. Low-level expression is also found in peripheral nerves, mesenchymal stroma cells (34), and skin melanocytes (37). The pattern of expression on healthy tissues raises important safety considerations for ganglioside-targeted therapy which will be discussed in more detail. Substantial alterations in ganglioside expression have been observed in cancer cells (38).
Ganglioside GD2: Expression and Biology in Tumor Cells
Corresponding to its consistent association with immature neural crest tissue, aberrant expression of GD2 characterizes cancers of neuroectodermal origin, such as melanoma (39) and neuroblastoma (40). GD2 was further found to be expressed in several additional childhood tumors, including tumors of the CNS [retinoblastoma (41), diffuse intrinsic pons glioma (42)] and mesenchymal malignancies of the bone (Ewing sarcoma, osteosarcoma) and soft tissues (rhabdomyosarcoma, desmoplastic small round cell tumors) (43–47). Among non-melanoma cancers in adults, ganglioside GD2 was detected in a proportion of tumor cells in breast cancers (48) and in small cell lung cancer (49).
The mechanisms regulating expression of GD2 in normal human development and in cancer have not been resolved in detail. In general, expression of gangliosides during development is regulated through stage- and tissue-specific expression of ganglioside synthase genes (50). Key enzymes in GD2 synthesis are the glycosyltransferases GD3 synthase (GD3S) which synthesizes GD3 from GM3 and GM2/GD2 synthase, converting GD3 to GD2 (Figure 3). GD3S is expressed during early stages of neuronal differentiation (51). Among adult human tissues, GD3S mRNA expression is restricted to the brain. GD3S transcripts are found in GD2+ tumor cells of various histology (52), and surface expression of GD2 in breast cancer cells was found to be associated with expression of genes encoding for these enzymes, especially GD3S (53).
The activity of glycosyltransferases in normal tissues and in tumors is regulated at the transcriptional level. Studies of GD3S promoter activity in breast cancer and neuroblastoma cell lines have demonstrated a role for nuclear factor κB (NFκB) activation in transcriptional activation of the GD3S gene (54, 52). Recent studies highlight the role of epigenetic mechanisms in regulating glycosyltransferase gene expression during cellular differentiation and brain development [reviewed in (55)]. Specifically, hyperacetylation of histones was found to contribute to developmental alterations of expression of GD3S and other key enzymes of ganglioside synthesis (56, 57). Thus, epigenetic regulation of enzymes upstream of GD2 may be involved in the aberrant expression of GD2 in childhood cancers.
The biological effects of GD2 in both normal development and in tumors are not fully understood. GD2 expressed in tumor cells was found to contribute to malignant properties, including cell proliferation, invasive properties and motility (58). One potential mechanism is modulation of signal transduction along the pathways originating from GD2-containing glycosynapses. Indeed, examples for GD2-mediated activation of receptor tyrosine kinases and subsequent deregulation of critical pathways and molecules determining cell function have been reported (59, 60). Gangliosides interact with transmembrane proteins in the formation of lipid shells [reviewed in (61)]. The interaction partners of GD2 within the lipid microdomains and the structural relationships and downstream signaling of such interactions in normal cells and in cancer have not yet been systematically analyzed. In addition, interaction of cell surface GD2 with molecules in the extracellular matrix, used by neural crest cells during embryonic development to travel to distant sites, could contribute to tissue invasion and metastasis of GD2-positive cancer cells. In breast cancer, GD2 was identified as a surface marker of a subpopulation of tumor cells with specific functional properties, including self-renewal, chemoresistance, and epithelial-mesenchymal transition (EMT) (53). Inhibition of GD2 biosynthesis by genetic knockdown of GD3S in breast cancer cells hampered mammosphere formation, tumor initiation, and cell motility, as well as EMT and metastasis (53, 62). Besides cell-intrinsic functions, GD2 can contribute to immune invasion by the activation and support of immunosuppressive myeloid bystander cells in the tumor microenvironment (63). The relevance of each of these mechanisms in childhood cancers with uniform or variable GD2 expression, such as neuroblastoma and Ewing sarcoma or osteosarcoma, respectively, has not yet been elucidated.
Ganglioside GD2 as an Immunotherapeutic Target
Due to its restricted tissue expression, GD2 is a highly ranked target for immunotherapy (64). It was first evaluated as a therapeutic target in neuroblastoma, a cancer with abundant and highly consistent GD2 expression reminiscent of its origin from GD2-positive neuroectoderm (65).
Clinical development of GD2 targeting started with two murine monoclonal antibodies, IgG3 antibody 3F8 and IgG2a class-switched 14.G2a, in phase I clinical trials in neuroblastoma patients in the late 1980s (24, 25). Both antibodies had similar toxicity profiles with often severe side effects including pain, fever, hypersensitivity reactions and capillary leak syndrome. Several measures were taken to enhance the activity of the antibody by promoting antibody-dependent cell-mediated cytotoxicity (ADCC) and reduce toxic side effects, especially the pain syndrome, attributed to complement activation by the Fc domain (66). Chimeric and humanized antibodies were developed to replace murine by human Fc domains to enhance ADCC (67, 68). Subsequent clinical studies used anti-GD2 antibody to eliminate minimal residual disease after completion of intensive multimodal treatment, either alone (69), or combined with granulocyte-macrophage colony stimulating factor GM-CSF to activate granulocytes (70), or in addition with interleukin-2 to stimulate NK cells (71). In a randomized phase III trial, the combined immunotherapeutic regimen resulted in significant increases of event-free and overall survival of high-risk neuroblastoma patients at 2 years (71). The toxicities of anti-GD2 antibody therapy have remained significant but are manageable by optimized supportive care (72). Although the contribution of anti-GD2 antibody therapy to the long-term outcomes after high-risk neuroblastoma treatment has not ultimately been proven, it is now considered a standard component of state-of-the-art treatment regimens (73). Importantly, antigen escape variants by downregulation or loss of GD2 expression in response to GD2-specific targeting have been rare (71, 74, 75), supporting the value of this antigen in neuroblastoma.
Redirecting T cells to GD2 exploits the trafficking qualities and potent effector functions of T cells and thus could be a more effective strategy to target GD2-expressing tumor cells. The first CAR generated against GD2 was derived from the single-chain Fv (scFv) domains of monoclonal antibody 14.G2a (76), linked to the T cell receptor ζ chain (30). 14.G2a-redirected CAR T cells specifically and effectively lysed GD2-positive neuroblastoma cells in vitro. Since soluble GD2 is present at high concentrations in serum of advanced-stage neuroblastoma patients, competitive binding and blockade of GD2-directed CAR T cell activity had to be excluded. Soluble GD2 did not impede tumor cell lysis by 14.G2a-CAR T cells in vitro (30). Lack of a competing effect of shed antigen on the antitumor activity of CAR-redirected T cells had previously been shown for another carbohydrate target, Lewis-Y (77), and also for protein antigens shed into the blood stream by tumor cells such as CEA (78).
In an early clinical trial, treatment of neuroblastoma patients with autologous virus-specific T cells expressing the anti-GD2 CAR was safe, with some evidence of activity (79–81). Further phase I clinical studies have used signal-enhanced CARs and refined treatment regimens (82, 83). The presence of GD2 at low levels on neuronal cell bodies has caused significant safety concerns for the clinical use of GD2-specific CAR T cells. Whereas, the brain is protected from intravenous infusions of GD2 antibody by the blood-brain barrier, CAR T cells effectively penetrate into the CNS. Neither of the clinical trials performed so far has shown any evidence of neurotoxic side effects or pain (79, 80, 83), and this includes a recent trial demonstrating impressive clinical responses associated with tumor lysis syndrome and cytokine release (83). Thus, the lack of significant on-target off-tumor toxicities in the central and peripheral nervous system cannot be attributed to lack of activity. As the 14.G2a-derived GD2-specific CAR was designed to contain the isolated scFv fragment without any immunoglobulin heavy chain components to bridge it from the plasma membrane, the lack of any pain side effects is consistent with the hypothesis that activation of complement by the Fc domain of anti-GD2 antibodies is the responsible mechanism for this side effect (66). In contrast to clinical trials, neurotoxicity was reported in a mouse model following treatment with T cells expressing an affinity-enhanced 14.G2a-based CAR (84). Clinical signs of encephalopathy in this model were associated with T cell infiltration in brain regions with low-level GD2 expression. Whether the clinical picture indeed represented on-target cross-reactivities with murine brain or cytokine-mediated off-target toxicities, as well-known from the use of CD19-specific CAR T cells, remains controversial (85). In further xenograft models, potent antitumor activity of GD2-specific CAR T cells was not associated with neurotoxicities (86, 45). This encouraged the development of this therapy even for GD2-positive CNS tumors (42). In preclinical studies, GD2-specific CAR T cells were highly active against GD2-positive diffuse intrinsic pons glioma xenografts in the CNS. Neurological symptoms occurring in some of the animals were associated with tumor swelling by pseudoprogression, without histological damage of brain tissue. As long as safety concerns remain, the use of NK cells rather than long-lived T cells could be a safer alternative to targeting GD2-positive tumors (44).
Preclinical data support the value of GD2-specific CARs also for immunotherapy of GD2-expressing sarcomas, such as Ewing sarcomas (44, 87, 88) and osteosarcomas (45). In contrast to neuroblastomas, only a proportion of these tumors express significant levels of GD2, and antigen expression is often heterogeneous (45, 87). Thus, GD2-specific immunotherapy in childhood sarcomas will have to be combined with strategies eliminating also GD2low and GD2neg tumor cell subpopulations and preventing antigen-negative immune escape.
A highly related variant of GD2 is O-acetyl-GD2 (OAcGD2), characterized by a 9-O-acetyl modification on the terminal sialic acid of GD2. While tumors that express GD2 were generally found to also express the O-acetylated variant, human peripheral nerve fibers do not express OAcGD2 (89). Consequently, antibodies targeting this variant were developed to avoid the allodynic properties of GD2 (90). Whether selective recognition of the O-acetylated variant of GD2 is preferable for GD2-specific CARs is not clear.
Ganglioside GD3: An Alternative or Complementary Immune Target?
GD3 is a b-series ganglioside containing two sialic acids. It is produced from its precursor GM3 by the activity of GD3S. Physiologically it is highly expressed in embryonic neural stem cells (36). Its interest as a tumor antigen has mostly been in melanoma where GD3 is highly expressed, with no or minimal levels of GD3 on human normal melanocytes and restricted expression on other normal tissues to low levels on retinal pigment cells and in the CNS. GD3 has been investigated as a target for antibody therapy in melanoma (91, 92), and anti-GD3 CARs are in preclinical development (93, 94).
While neuroblastomas have only moderate expression of GD3 compared to GD2, an immunohistochemical analysis of various childhood sarcomas has demonstrated a high prevalence of GD3 expression especially in osteosarcomas, but also in a proportion of Ewing sarcomas and rhabdomyosarcomas (43). Moreover, malignant gliomas express high levels of GD3 as well as O-acetylated GD3 and GD3S (95, 96), and expression is associated with the degree of malignancy (96) and with neurosphere formation and clonogenic properties (97). A GD3-specific antibody, acting via complement-dependent cytotoxicity, was found to inhibit glioblastoma tumor growth in an in vivo model (97).
Thus, GD3 could be a CAR target in both CNS tumors and extracranial tumors in the pediatric population, either alone or in combination with GD2, to broaden T cell recognition in cancers with heterogeneous expression of either of the two gangliosides. However, potential on-target/off-tumor side effects by reactivity of GD3-specific CAR T cells with CNS tissues, especially with the retina, will have to be studied diligently.
Ganglioside N-Glycolyl GM3 (NeuGcGM3)
GM3 is a monosialoganglioside and the direct precursor of GD3 (Figure 3). Whereas, the acetylated form, N-acetyl GM3, is abundant in normal tissues, humans in contrast to other mammals cannot generate N-glycolylated GM3 (NeuGcGM3) due to a constitutional deletion in the gene encoding the enzyme which catalyzes the conversion of N-acetyl to N-glycolyl sialic acid (98). Expression of NeuGcGM3 was observed in human cancers and explained by expression of the sialic acid transporter under hypoxic conditions, resulting in incorporation of non-human sialic acid from dietary supplies (99). In fact, natural xeno-autoantibodies against NeuGcGM3 were found in human serum and correlated with the presence of cancers (100). Among pediatric tumors, neuroblastomas, Wilms tumors, Ewing sarcomas and retinoblastomas were reported to express NeuGcGM3 (101) whereas various normal tissues were negative [summarized in (102)]. An anti-idiotype vaccine, racotumumab, was able to induce antibody responses to the target in a phase I clinical trial in refractory childhood cancers, without any evidence for off-tumor toxicities (103). Direct antibody or CAR targeting of this antigen has not yet reached clinical translation. Again, although immunoreactivity of the anti-NeuGcGM3 antibody has so far been restricted to tumor tissues, interactions with healthy tissues, with the potential to cause limiting toxicities, cannot be excluded.
Stage-Specific Embryonic Antigen-4 (SSEA-4): A Marker for Embryonic Stem Cells
SSEA-4 is a globo-series ganglioside synthesized from SSEA-3 by the enzyme ST3 beta-galactoside alpha-2,3-sialyltransferase 2 (ST3GAL2) (104) (Figure 4). As other glycosphingolipids, SSEA-4 is a component of glycosynapses of the plasma membrane (Figure 2). Due to its highly restricted expression in pluripotent human embryonic stem cells, SSEA-4 conceptually is an attractive target for CAR T cell therapy. During human preimplantation development, SSEA-4 is first observed on the pluripotent cells of the inner cell mass and lost upon differentiation (105). After birth, human germ stem cells in the testis and ovary (106, 107) as well as mesenchymal stem cells (108) express SSEA-4. Its biological function has not yet been resolved in detail. In human tumors, SSEA-4 was first identified in a teratocarcinoma cell line (109). It was further found to be overexpressed in osteosarcoma (110), prostate cancer (111), breast cancer (112), and glioblastoma (113).
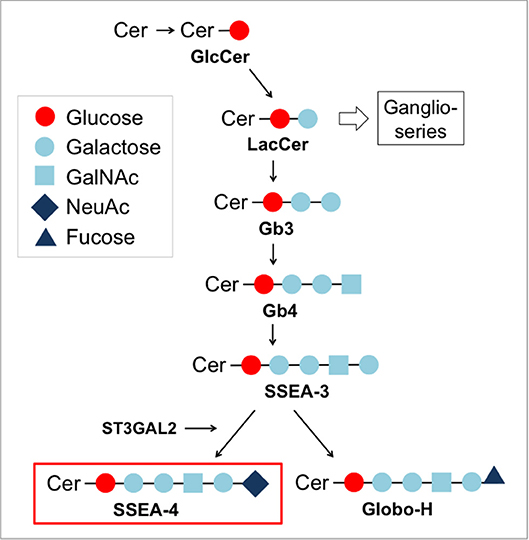
Figure 4. Biosynthesis pathway for globo-series gangliosides.
Several studies suggest that SSEA-4 expression in cancer marks subpopulations with specific biological properties within the tumor bulk. Among osteosarcoma cells, SSEA-4-positive subpopulations, but not SSEA-4-negative tumor cells, reliably established xenografts in mice, and in patients with this cancer, the frequency of SSEA-4 expressing tumor cells was inversely correlated with overall survival (110). Moreover, tumor tissue extracted after chemotherapy showed a higher number of SSEA-4-positive tumor cells, suggesting selection by chemotherapy, and/or an upregulation of the antigen by cytotoxic agents (110). In prostate cancer, SSEA-4 expressing cell subpopulations were highly tumorigenic, had higher cellular adhesion and a migratory phenotype, indicating a role of SSEA-4 in cancer invasion (111). SSEA-4 in breast cancer cells was found to be highly expressed in a subpopulation of chemotherapy-resistant tumor cells, and expression of enzyme ST3GAL2 was a predictive marker for poor outcome (112). In astrocytomas, SSEA-4 expression was associated with higher tumor grades (113).
Selective presence in cancer cells and association with self-renewing and migratory properties makes SSEA-4 a candidate target for antibody-based immunotherapies. In preclinical experiments, the SSEA-4 specific monoclonal antibody MC 813-70 was indeed active to inhibit the growth of glioblastomas in nude mice (113). Apparent toxicities were not described in this model (113). By contrast, SSEA-4-specific CAR T cells were found to cause major alterations in the composition of the hematopoietic compartment in a preclinical mouse model, suggestive of on-target/off-tumor toxicity (114). Whether SSEA-4 expression in humans after birth is sufficiently restricted to cancer cells to allow for safe targeting of cancer with CAR-engineered T cells remains an open question.
Carbohydrate Modifications of Cell Surface Proteins
Besides membrane glycolipids, aberrant glycosylation of cell surface proteins in cancers leads to expression of distinct glycoproteins which could allow for selective CAR targeting. Cancer-associated cell surface glycosylation was shown to be directly involved in malignant transformation and metastasis [reviewed in (115)], supporting the potential of glyco-epitopes as immune targets. The most prevalent glyco-epitopes in carcinomas are Tn (GalNAcα1-Ser/Thr) and sialyl-Tn (STn, NeuAcα2,6GalNAc-Ser/Thr). Tn and STn are present on many glycoproteins expressed in epithelial cancers, including MUC1 (116, 117). MUC1 carring such variations was associated with poor prognosis, reduced chemosensitivity and with immune-inhibitory properties in breast cancer (116).
The first CARs against MUC1 were directed against the protein core. Their tumor selectivity was mediated by MUC1 hypoglycosylation which enabled the scFv to access the protein (118). More recently, CARs were directed to specific truncated O-glycopeptide epitopes not expressed on normal tissues and shown to differentiate between wild-type and Tn-glycoforms of MUC1 (117). CAR T cells against Tn-MUC1 effectively controlled tumor growth in murine xenograft models. Thus, aberrant glycosylation can turn a self-antigen into a neoantigen which can serve as target for tumor rejection by CAR T cells.
Proteins that recognize glycans, so-called lectins, could be alternative recognition domains for CARs targeting aberrant glycosylation. In a recent report, lectins were identified that specifically bind to a fucosylated glycan epitope on the surface of pancreatic cancer cell lines (119).
In pediatric cancers, oncofetal chondrosulfate (ofCS) modifications of glycosaminoglycans attached to proteoglycans on the cell surface have potential to serve as CAR targets (120). Chondroitin sulfate chains normally restricted to the placenta are found on many cancer-associated proteoglycans, including CSPG4, syndecan-1,−4, CD44, and glypican-1, with widespread expression also in pediatric tumors (120). Placental ofCS is detected by malaria protein VAR2CSA, suggesting a role in pregnancy-associated malaria (120). Studies using VAR2CSA as a model ligand for ofCS demonstrated a key role of this modification in the malignant phenotype, especially in tumor cell motility and metastatic potential (121). The ligand was further found to be able to isolate circulating tumor cells from peripheral blood (122). Thus, ofCS could be both a marker and a therapeutic target of the highly metastatic subpopulation of tumor cells across various types of cancer, including childhood solid tumors. A concern is the variability of protein glycosylation, which despite the biological function of the oncofetal modification could allow easy escape from immune targeting. Moreover, lack of expression on normal tissues besides placenta will have to be unequivocally demonstrated.
Outlook
The feasibility of redirecting T cells against carbohydrate antigens which they do not normally recognize using CARs has been well demonstrated in both preclinical and clinical studies. Still, among the large numbers of CAR targets currently explored, relatively few are non-protein antigens. Carbohydrate antigens may be under-studied for technical reasons. Immunohistochemistry detection of carbohydrates is often limited to cryopreserved material not generally available from biopsies of pediatric tumors. Gangliosides and other glycolipids do not appear in gene and protein expression analyses, and genes encoding for the enzymes necessary for ganglioside synthesis are not useful surrogates to predict expression of specific gangliosides since these are products of numerous glycoslytransferases. A major challenge to the identification of cancer-associated glycosylation variants is their diversity and heterogeneity. Known carbohydrate targets in cancer were identified by their interaction with monoclonal anti-tumor antibodies followed by analysis of the chemical structure of the antibody target. Novel mass spectrometry-based technologies in modern glycomics now allow systematic and high-throughput comparative analysis of the patterns of gangliosides and oligosaccharides in tissues, followed by purification and structural elucidation (123–125). Glycomic screening of the cancer cell surface in pediatric cancers could be a useful next step to identify novel carbohydrate targets for CAR gene-modified T cells (126).
Many questions with regard to the tissue specificity and biology of non-protein antigens are still unanswered. Many gangliosides are physiologically expressed in a stage- and tissue-specific manner in human embryogenesis and could be present on neuronal, mesenchymal or hematopoietic stem cells after birth, raising important safety concerns. The mechanisms regulating tissue-specific expression of individual gangliosides are often unknown but highly relevant to better understand their biological role. This knowledge may also serve to find interventions that upregulate presence of the antigen on cancer cells with heterogeneous antigen expression, a major limitation for effective tumor targeting by CAR T cells.
While it is unlikely that carbohydrate targets with restricted expression on tumor cells will be found in the majority of childhood cancers, novel T cell engineering strategies may allow safe targeting of antigens despite low level expression in normal tissues. One approach is combinatorial antigen recognition, resulting in full T cell activation responses only in the presence of two or more target antigens. For this purpose, T cells can be cotransduced with a CAR that provides suboptimal activation in response to antigen, along with a chimeric costimulatory receptor that recognizes a second antigen (127). Alternatively, modular synthetic receptors were designed on the basis of the capacity of Notch receptors to perform transcriptional switches (128). Engagement of a first CAR by its target antigen releases an intracellular synthetic transcription factor that selectively induces gene expression of a second CAR. In a mouse model, this resulted in selective clearance of tumors coexpressing both antigens. An alternative strategy to protect off-target tissues against CAR T cells is cotransduction with an activating CAR and a CAR that delivers inhibitory signaling in response to a second antigen expressed exclusively on normal cells (129).
Moreover, control systems have been developed that allow to remove T cells on demand in cases of on- or off-target toxicities. Such suicide switches rely on genes that render the cells sensitive to prodrugs (130, 131) or proapoptotic genes activated by dimerization(132), or on surface markers for antibody-mediated depletion, e.g., truncated epithelial growth factor receptor (tEGFR) (133) or rituximab(134). An even safer solution could be “on-switch” CARs. Separation of intracellular or costimulatory signals from extracellular antigen recognition in inducible gene expression systems allows to turn on the respective signaling pathway by small molecules on demand (135, 136). In the view of the rapidly expanding toolbox for tuning and controlling the in vivo activity and persistence of CAR T cells, carbohydrate antigens as well as protein antigens with and without carbohydrate modifications, despite low-level coexpression on healthy tissues, deserve investigation as potential targets for future generations of CARs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
2. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
3. Majzner RG, Heitzeneder S, Mackall CL. Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell (2017) 31:476–85. doi: 10.1016/j.ccell.2017.03.002
4. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
5. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
6. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi: 10.1038/nature13988
7. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell (2017) 171:934–49.e15. doi: 10.1016/j.cell.2017.09.028
8. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555:321–7. doi: 10.1038/nature25480
9. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature (2018) 555:371–6. doi: 10.1038/nature25795
10. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res. (2016) 22:1364–70. doi: 10.1158/1078-0432.CCR-15-0491
11. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. (2017) 18:1493–501. doi: 10.1016/S1470-2045(17)30624-1
12. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma-subunit or zeta-subunit of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4.
13. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. (2017) 35:1803–13. doi: 10.1200/JCO.2016.71.3024
14. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
15. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
16. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
17. Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. (2017) 9:eaag2196. doi: 10.1126/scitranslmed.aag2196
18. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific t cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
19. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
20. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
21. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
22. Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. (2018) 9:70–80. doi: 10.1016/j.omtm.2018.01.009
23. Roesli C, Borgia B, Schliemann C, Gunthert M, Wunderli-Allenspach H, Giavazzi R, et al. Comparative analysis of the membrane proteome of closely related metastatic and nonmetastatic tumor cells. Cancer Res. (2009) 69:5406–14. doi: 10.1158/0008-5472.CAN-08-0999
24. Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, et al. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol. (1987) 5:1430–40. doi: 10.1200/JCO.1987.5.9.1430
25. Handgretinger R, Baader P, Dopfer R, Klingebiel T, Reuland P, Treuner J, et al. A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14.G2a. Cancer Immunol Immunother. (1992) 35, 199–204.
26. Lee FT, Rigopoulos A, Hall C, Clarke K, Cody SH, Smyth FE, et al. Specific localization, gamma camera imaging, and intracellular trafficking of radiolabelled chimeric anti-G(D3) ganglioside monoclonal antibody KM871 in SK-MEL-28 melanoma xenografts. Cancer Res. (2001) 61:4474–82.
27. Yang RK, Kalogriopoulos NA, Rakhmilevich AL, Ranheim EA, Seo S, Kim K, et al. Intratumoral hu14.18-IL-2 (IC) induces local and systemic antitumor effects that involve both activated T and NK cells as well as enhanced IC retention. J Immunol. (2012) 189:2656–64. doi: 10.4049/jimmunol.1200934
28. Mezzanzanica D, Canevari S, Mazzoni A, Figini M, Colnaghi MI, Waks T, et al. Transfer of chimeric receptor gene made of variable regions of tumor-specific antibody confers anticarbohydrate specificity on T cells. Cancer Gene Ther. (1998) 5:401–7.
29. Zhang S, Zhang HS, Cordon-Cardo C, Reuter VE, Singhal AK, Lloyd KO, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group-related antigens. Int J Cancer (1997) 73:50–6.
30. Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of GD2-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer (2001b) 94:228–36. doi: 10.1002/ijc.1457
31. Todeschini AR, Dos Santos JN, Handa K, Hakomori SI. Ganglioside GM2/GM3 complex affixed on silica nanospheres strongly inhibits cell motility through CD82/cMet-mediated pathway. Proc Natl Acad Sci USA. (2008) 105:1925–30. doi: 10.1073/pnas.0709619104
32. Hu J, Huang X, Ling CC, Bundle DR, Cheung NK. Reducing epitope spread during affinity maturation of an anti-ganglioside GD2 antibody. J Immunol. (2009) 183:5748–55. doi: 10.4049/jimmunol.0901409
33. Draper JS, Pigott C, Thomson JA, Andrews PW. Surface antigens of human embryonic stem cells: changes upon differentiation in culture. J Anat. (2002) 200(Pt 3):249–58. doi: 10.1046/j.1469-7580.2002.00030.x
34. Martinez C, Hofmann TJ, Marino R, Dominici M, Horwitz EM. Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood (2007) 109:4245–8. doi: 10.1182/blood-2006-08-039347
35. Saxena S, Wahl J, Huber-Lang MS, Stadel D, Braubach P, Debatin KM, et al. Generation of murine sympathoadrenergic progenitor-like cells from embryonic stem cells and postnatal adrenal glands. PLoS ONE (2013) 8:e64454. doi: 10.1371/journal.pone.0064454
36. Yanagisawa M, Yoshimura S, Yu RK. Expression of GD2 and GD3 gangliosides in human embryonic neural stem cells. ASN Neuro (2011). 3:e00054. doi: 10.1042/AN20110006
37. Hersey P, Jamal O, Henderson C, Zardawi I, D'Alessandro G. Expression of the gangliosides GM3, GD3 and GD2 in tissue sections of normal skin, naevi, primary and metastatic melanoma. Int J Cancer (1988) 41:336–43.
38. Krug LM, Ragupathi G, Hood C, Kris MG, Miller VA, Allen JR, et al. Vaccination of patients with small-cell lung cancer with synthetic fucosyl GM-1 conjugated to keyhole limpet hemocyanin. Clin Cancer Res. (2004) 10(18 Pt 1):6094–100. doi: 10.1158/1078-0432.CCR-04-0482
39. Cahan LD, Irie RF, Singh R, Cassidenti A, Paulson JC. Identification of a human neuroectodermal tumor antigen (OFA-I-2) as ganglioside GD2. Proc Natl Acad Sci USA. (1982) 79:7629–33.
40. Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. (1984) 44:5914–20.
41. Portoukalian J, David MJ, Gain P, Richard M. Shedding of GD2 ganglioside in patients with retinoblastoma. Int J Cancer (1993) 53:948–51.
42. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
43. Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr Blood Cancer (2016) 63:1780–5. doi: 10.1002/pbc.26097
44. Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology (2017) 6:e1250050. doi: 10.1080/2162402X.2016.1250050
45. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
46. Modak S, Gerald W, Cheung NK. Disialoganglioside GD2 and a novel tumor antigen: potential targets for immunotherapy of desmoplastic small round cell tumor. Med Pediatr Oncol. (2002) 39:547–51. doi: 10.1002/mpo.10151
47. Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer (2014) 120:548–54. doi: 10.1002/cncr.28461
48. Orsi G, Barbolini M, Ficarra G, Tazzioli G, Manni P, Petrachi T, et al. GD2 expression in breast cancer. Oncotarget (2017) 8:31592–600. doi: 10.18632/oncotarget.16363
49. Yoshida S, Fukumoto S, Kawaguchi H, Sato S, Ueda R, Furukawa K. Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res (2001) 61:4244–52.
50. Ngamukote S, Yanagisawa M, Ariga T, Ando S, Yu RK. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem. (2007) 103:2327–41. doi: 10.1111/j.1471-4159.2007.04910.x
51. Yamamoto A, Haraguchi M, Yamashiro S, Fukumoto S, Furukawa K, Takamiya K, et al. Heterogeneity in the expression pattern of two ganglioside synthase genes during mouse brain development. J Neurochem. (1996) 66:26–34.
52. Kwon HY, Dae HM, Song NR, Kim KS, Kim CH, Lee YC. Valproic acid induces transcriptional activation of human GD3 synthase (hST8Sia I) in SK-N-BE(2)-C human neuroblastoma cells. Mol Cells (2009) 27:113–8. doi: 10.1007/s10059-009-0012-4
53. Battula VL, Shi YX, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. (2012) 122:2066–78. doi: 10.1172/JCI59735
54. Bobowski M, Vincent A, Steenackers A, Colomb F, Van Seuningen I, Julien S, et al. Estradiol represses the G(D3) synthase gene ST8SIA1 expression in human breast cancer cells by preventing NFkappaB binding to ST8SIA1 promoter. PLoS ONE (2013) 8:e62559. doi: 10.1371/journal.pone.0062559
55. Itokazu Y, Tsai YT, Yu RK. Epigenetic regulation of ganglioside expression in neural stem cells and neuronal cells. Glycoconj J. (2017) 34:749–56. doi: 10.1007/s10719-016-9719-6
56. Tsai YT, Yu RK. Epigenetic activation of mouse ganglioside synthase genes: implications for neurogenesis. J Neurochem. (2014) 128:101–10. doi: 10.1111/jnc.12456
57. Suzuki Y, Yanagisawa M, Ariga T, Yu RK. Histone acetylation-mediated glycosyltransferase gene regulation in mouse brain during development. J Neurochem. (2011) 116:874–80. doi: 10.1111/j.1471-4159.2010.07042.x
58. Furukawa K, Hamamura K, Ohkawa Y, Ohmi Y, Furukawa K. Disialyl gangliosides enhance tumor phenotypes with differential modalities. Glycoconj J. (2012) 29:579–84. doi: 10.1007/s10719-012-9423-0
59. Probstmeier R, Pesheva P. Tenascin-C inhibits beta1 integrin-dependent cell adhesion and neurite outgrowth on fibronectin by a disialoganglioside-mediated signaling mechanism. Glycobiology (1999) 9:101–14.
60. Wu L, Bernard-Trifilo JA, Lim Y, Lim ST, Mitra SK, Uryu S, et al. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene (2008) 27:1439–48. doi: 10.1038/sj.onc.1210770
61. Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. (2010) 584:1748–59. doi: 10.1016/j.febslet.2009.12.010
62. Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Pena EQ, Taube JH, et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene (2014) 34:2958–67. doi: 10.1038/onc.2014.245
63. Wondimu A, Liu Y, Su Y, Bobb D, Ma JS, Chakrabarti L, et al. Gangliosides drive the tumor infiltration and function of myeloid-derived suppressor cells. Cancer Res. (2014) 74:5449–57. doi: 10.1158/0008-5472.CAN-14-0927
64. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. (2009) 15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737
65. Wu ZL, Schwartz E, Seeger R, Ladisch S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. (1986) 46:440–3.
66. Sorkin LS, Otto M, Baldwin WM III, Vail E, Gillies SD, Handgretinger R, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain (2010) 149:135–42. doi: 10.1016/j.pain.2010.01.024
67. Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology (2012b) 1:477–86. doi: 10.4161/onci.19864
68. Mueller BM, Romerdahl CA, Gillies SD, Reisfeld RA. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol. (1990) 144:1382–6.
69. Simon T, Hero B, Faldum A, Handgretinger R, Schrappe M, Niethammer D, et al. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol. (2004) 22:3549–57. doi: 10.1200/JCO.2004.08.143
70. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. (2012) 30:3264–70. doi: 10.1200/JCO.2011.41.3807
71. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
72. Ozkaynak MF, Gilman AL, London WB, Naranjo A, Diccianni MB, Tenney SC, et al. A Comprehensive safety trial of chimeric antibody 14.18 with GM-CSF, IL-2, and isotretinoin in high-risk neuroblastoma patients following myeloablative therapy: children's oncology group study ANBL0931. Front Immunol. (2018) 9:1355. doi: 10.3389/fimmu.2018.01355
73. Simon T, Hero B, Schulte JH, Deubzer H, Hundsdoerfer P, von Schweinitz D, et al. GPOH guidelines for diagnosis and treatment of patients with neuroblastic tumors. Klin Padiatr. (2017) 229:147–67. doi: 10.1055/s-0043-103086
74. Kramer K, Gerald WL, Kushner BH, Larson SM, Hameed M, Cheung NK. Disaloganglioside GD2 loss following monoclonal antibody therapy is rare in neuroblastoma. Med Pediatr Oncol. (2001) 36:194–6. doi: 10.1002/1096-911X(20010101)36:1 < 194::AID-MPO1046>3.0.CO;2-B
75. Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol. (2001) 19:4189–94. doi: 10.1200/JCO.2001.19.22.4189
76. Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, et al. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. (1989) 49:2857–61.
77. Westwood JA, Murray WK, Trivett M, Haynes NM, Solomon B, Mileshkin L, et al. The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J Immunother. (2009) 32:292–301. doi: 10.1097/CJI.0b013e31819b7c8e
78. Hombach A, Koch D, Sircar R, Heuser C, Diehl V, Kruis W, et al. A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Ther. (1999) 6:300–4. doi: 10.1038/sj.gt.3300813
79. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
80. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
81. Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood (2002) 99:2009–16. doi: 10.1182/blood-2006-11-059139
82. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
83. Straathof KFB, Wallace R, Thomas S, Cheung G, Collura A, Gileadi T, et al. A Cancer Research UK Phase I Trial of Anti-GD2 Chimeric Antigen Receptor (CAR) Transduced T-Cells (1RG-CART) in Patients With Relapsed or Refractory Neuroblastoma. Chicago, IL: Paper presented at the Annual Meeting of the American Association for Cancer Research (2018).
84. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
85. Majzner RG, Weber EW, Lynn RC, Xu P, Mackall CL. Neurotoxicity associated with a high-affinity GD2 CAR-letter. Cancer Immunol Res. (2018) 6:494–5. doi: 10.1158/2326-6066.CIR-18-0089
86. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
87. Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer (2012) 106:1123–33. doi: 10.1038/bjc.2012.57
88. Liebsch L, Kailayangiri S, Beck L, Altvater B, Koch R, Dierkes C, et al. Ewing sarcoma dissemination and response to T-cell therapy in mice assessed by whole-body magnetic resonance imaging. Br J Cancer (2013) 109:658–66. doi: 10.1038/bjc.2013.356
89. Alvarez-Rueda N, Desselle A, Cochonneau D, Chaumette T, Clemenceau B, Leprieur S, et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS ONE (2011) 6:e25220. doi: 10.1371/journal.pone.0025220
90. Cochonneau D, Terme M, Michaud A, Dorvillius M, Gautier N, Frikeche J, et al. Cell cycle arrest and apoptosis induced by O-acetyl-GD2-specific monoclonal antibody 8B6 inhibits tumor growth in vitro and in vivo. Cancer Lett. (2013) 333:194–204. doi: 10.1016/j.canlet.2013.01.032
91. Forero A, Shah J, Carlisle R, Triozzi PL, LoBuglio AF, Wang WQ, et al. A phase I study of an anti-GD3 monoclonal antibody, KW-2871, in patients with metastatic melanoma. Cancer Biother Radiopharm. (2006) 21:561–8. doi: 10.1089/cbr.2006.21.561
92. Tarhini AA, Moschos SJ, Lin Y, Lin HM, Sander C, Yin Y, et al. Safety and efficacy of the antiganglioside GD3 antibody ecromeximab (KW2871) combined with high-dose interferon-alpha2b in patients with metastatic melanoma. Melanoma Res. (2017) 27:342–50. doi: 10.1097/CMR.0000000000000353
93. Lo AS, Ma Q, Liu DL, Junghans RP. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res. (2010) 16:2769–80. doi: 10.1158/1078-0432.CCR-10-0043
94. Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans RP. Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia (2000) 2:449–59. doi: 10.1038/sj.neo.7900108
95. Fabris D, Rozman M, Sajko T, Vukelic Z. Aberrant ganglioside composition in glioblastoma multiforme and peritumoral tissue: a mass spectrometry characterization. Biochimie (2017) 137:56–68. doi: 10.1016/j.biochi.2017.03.001
96. Gaini SM, Riboni L, Cerri C, Grimoldi N, Sganzerla EP, Berra B. Ganglioside content and composition in human gliomas. Acta Neurochir Suppl. (1988) 43:126–9.
97. Yeh SC, Wang PY, Lou YW, Khoo KH, Hsiao M, Hsu TL, et al. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc Natl Acad Sci USA. (2016) 113:5592–7. doi: 10.1073/pnas.1604721113
98. Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J Biol Chem. (1998) 273:15866–71.
99. Yin J, Hashimoto A, Izawa M, Miyazaki K, Chen GY, Takematsu H, et al. Hypoxic culture induces expression of sialin, a sialic acid transporter, and cancer-associated gangliosides containing non-human sialic acid on human cancer cells. Cancer Res. (2006) 66:2937–45. doi: 10.1158/0008-5472.CAN-05-2615
100. Padler-Karavani V, Hurtado-Ziola N, Pu M, Yu H, Huang S, Muthana S, et al. Human xeno-autoantibodies against a non-human sialic acid serve as novel serum biomarkers and immunotherapeutics in cancer. Cancer Res. (2011) 71:3352–63. doi: 10.1158/0008-5472.CAN-10-4102
101. Scursoni AM, Galluzzo L, Camarero S, Lopez J, Lubieniecki F, Sampor C, et al. Detection of N-glycolyl GM3 ganglioside in neuroectodermal tumors by immunohistochemistry: an attractive vaccine target for aggressive pediatric cancer. Clin Dev Immunol. (2011) 2011:245181. doi: 10.1155/2011/245181
102. Torbidoni AV, Scursoni A, Camarero S, Segatori V, Gabri M, Alonso D, et al. Immunoreactivity of the 14F7 Mab raised against N-Glycolyl GM3 Ganglioside in retinoblastoma tumours. Acta Ophthalmol. (2015) 93:e294–300. doi: 10.1111/aos.12578
103. Cacciavillano W, Sampor C, Venier C, Gabri MR, de Davila MT, Galluzzo ML, et al. A Phase I study of the anti-idiotype vaccine racotumomab in neuroblastoma and other pediatric refractory malignancies. Pediatr Blood Cancer (2015) 62:2120–4. doi: 10.1002/pbc.25631
104. Saito S, Aoki H, Ito A, Ueno S, Wada T, Mitsuzuka K, et al. Human alpha2,3-sialyltransferase (ST3Gal II) is a stage-specific embryonic antigen-4 synthase. J Biol Chem. (2003) 278:26474–9. doi: 10.1074/jbc.M213223200
105. Tondeur S, Assou S, Nadal L, Hamamah S, De Vos J. Biology and potential of human embryonic stem cells. Ann Biol Clin. (2008) 66:241–7. doi: 10.1684/abc.2008.0224
106. Harichandan A, Sivasubramaniyan K, Bühring HJ. Prospective isolation and characterization of human bone marrow-derived MSCs. Adv Biochem Eng Biotechnol. (2013) 129:1–17. doi: 10.1007/10_2012_147
107. Virant-Klun I, Skutella T, Hren M, Gruden K, Cvjeticanin B, Vogler A, et al. Isolation of small SSEA-4-positive putative stem cells from the ovarian surface epithelium of adult human ovaries by two different methods. Biomed Res Int. (2013) 2013:690415. doi: 10.1155/2013/690415
108. Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood (2007) 109:1743–51. doi: 10.1182/blood-2005-11-010504
109. Kannagi R, Cochran NA, Ishigami F, Hakomori S, Andrews PW, Knowles BB, et al. Stage-specific embryonic antigens (SSEA-3 and−4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. (1983) 2:2355–61.
110. Zhang W, Ding ML, Zhang JN, Qiu JR, Shen YH, Ding XY, et al. mTORC1 maintains the tumorigenicity of SSEA-4(+) high-grade osteosarcoma. Sci Rep. (2015) 5:9604. doi: 10.1038/srep09604
111. Sivasubramaniyan K, Harichandan A, Schilbach K, Mack AF, Bedke J, Stenzl A, et al. Expression of stage-specific embryonic antigen-4 (SSEA-4) defines spontaneous loss of epithelial phenotype in human solid tumor cells. Glycobiology (2015) 25:902–17. doi: 10.1093/glycob/cwv032
112. Aloia A, Petrova E, Tomiuk S, Bissels U, Deas O, Saini M, et al. The sialyl-glycolipid stage-specific embryonic antigen 4 marks a subpopulation of chemotherapy-resistant breast cancer cells with mesenchymal features. Breast Cancer Res. (2015) 17:146. doi: 10.1186/s13058-015-0652-6
113. Lou YW, Wang PY, Yeh SC, Chuang PK, Li ST, Wu CY, et al. Stage-specific embryonic antigen-4 as a potential therapeutic target in glioblastoma multiforme and other cancers. Proc Natl Acad Sci USA. (2014) 111:2482–7. doi: 10.1073/pnas.1400283111
114. Pfeifer R, Lock D, Aloia A, Tomiuk S, Hellmer J, Assenmacher M, et al. Development of a CAR T cell therapy for the treatment of chemoresistant subpopulations in solid tumors (Abstract). Hum Gene Ther. (2016) 27:A58–9. doi: 10.3389/fimmu.2018.01740
115. Ohtsubo K, Chen MZ, Olefsky JM, Marth JD. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med. (2011) 17:1067–75. doi: 10.1038/nm.2414
116. Beatson R, Maurstad G, Picco G, Arulappu A, Coleman J, Wandell HH, et al. The breast cancer-associated glycoforms of MUC1, MUC1-Tn and sialyl-Tn, are expressed in COSMC wild-type cells and bind the C-type lectin MGL. PLoS ONE (2015) 10:e0125994. doi: 10.1371/journal.pone.0125994
117. Posey ADJr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
118. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. (2008) 180:4901–9. doi: 10.4049/jimmunol.180.7.4901
119. Shimomura O, Oda T, Tateno H, Ozawa Y, Kimura S, Sakashita S, et al. A novel therapeutic strategy for pancreatic cancer: targeting cell surface glycan using rBC2LC-N Lectin-Drug Conjugate (LDC). Mol Cancer Ther. (2018) 17:183–95. doi: 10.1158/1535-7163.MCT-17-0232
120. Salanti A, Clausen TM, Agerbaek MO, Al Nakouzi N, Dahlback M, Oo HZ, et al. Targeting human cancer by a glycosaminoglycan binding malaria protein. Cancer Cell (2015) 28:500–14. doi: 10.1016/j.ccell.2015.09.003
121. Clausen TM, Pereira MA, Al Nakouzi N, Oo HZ, Agerbaek MO, Lee S, et al. Oncofetal chondroitin sulfate glycosaminoglycans are key players in integrin signaling and tumor cell motility. Mol Cancer Res. (2016) 14:1288–99. doi: 10.1158/1541-7786.MCR-16-0103
122. Agerbaek MO, Bang-Christensen SR, Yang MH, Clausen TM, Pereira MA, Sharma S, et al. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat Commun. (2018) 9:3279. doi: 10.1038/s41467-018-05793-2
123. Fukui S, Feizi T, Galustian C, Lawson AM, Chai W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat Biotechnol. (2002) 20:1011–7. doi: 10.1038/nbt735
124. Hancock SM, Tarling CA, Withers SG. High-throughput screening of cell lysates for ganglioside synthesis. Anal Biochem. (2008) 382:48–54. doi: 10.1016/j.ab.2008.07.023
125. Serb A, Schiopu C, Flangea C, Vukelic Z, Sisu E, Zagrean L, et al. High-throughput analysis of gangliosides in defined regions of fetal brain by fully automated chip-based nanoelectrospray ionization multi-stage mass spectrometry. Eur J Mass Spectrom. (2009) 15:541–53. doi: 10.1255/ejms.1009
126. Cho HC, Liao CH, Yu AL, Yu J. Surface markers in stem cells and cancer from the perspective of glycomic analysis. Int J Biol Markers (2012) 27:e344–352. doi: 10.5301/JBM.2012.10361
127. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
128. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T Cells with combinatorial antigen-sensing circuits. Cell (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
129. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
130. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. (2009) 10:489–500. doi: 10.1016/S1470-2045(09)70074-9
131. Roellecke K, Virts EL, Einholz R, Edson KZ, Altvater B, Rossig C, et al. Optimized human CYP4B1 in combination with the alkylator prodrug 4-ipomeanol serves as a novel suicide gene system for adoptive T-cell therapies. Gene Ther. (2016) 23:615–26. doi: 10.1038/gt.2016.38
132. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood (2015) 125:4103–13. doi: 10.1182/blood-2015-02-628354
133. Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. (2016) 126:4262–72. doi: 10.1172/JCI84813
134. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
135. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. (2017) 7:1306–19. doi: 10.1158/2159-8290.CD-17-0263
Keywords: CAR T cells, childhood cancer, sarcomas, gangliosides, glycosylation
Citation: Rossig C, Kailayangiri S, Jamitzky S and Altvater B (2018) Carbohydrate Targets for CAR T Cells in Solid Childhood Cancers. Front. Oncol. 8:513. doi: 10.3389/fonc.2018.00513
Received: 04 September 2018; Accepted: 22 October 2018;
Published: 12 November 2018.
Edited by:
Rimas J. Orentas, Seattle Children's Research Institute, United StatesReviewed by:
Cynthia Hawkins, Hospital for Sick Children, CanadaDina Schneider, Lentigen Technology, United States
Copyright © 2018 Rossig, Kailayangiri, Jamitzky and Altvater. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Rossig, cm9zc2lnQHVuaS1tdWVuc3Rlci5kZQ==