
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 25 October 2018
Sec. Molecular and Cellular Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00480
This article is part of the Research TopicAccomplishments, Collaborative Projects and Future Initiatives in Breast Cancer Genetic PredispositionView all 13 articles
 Laura Caleca1
Laura Caleca1 Irene Catucci2
Irene Catucci2 Gisella Figlioli2Loris De Cecco3
Gisella Figlioli2Loris De Cecco3 Tina Pesaran4
Tina Pesaran4 Maggie Ward5Sara Volorio6,7
Maggie Ward5Sara Volorio6,7 Anna Falanga8Marina Marchetti7Maria Iascone9Carlo Tondini10Alberto Zambelli10Jacopo Azzollini11Siranoush Manoukian11
Anna Falanga8Marina Marchetti7Maria Iascone9Carlo Tondini10Alberto Zambelli10Jacopo Azzollini11Siranoush Manoukian11 Paolo Radice1
Paolo Radice1 Paolo Peterlongo2*
Paolo Peterlongo2*PALB2 (partner and localizer of BRCA2) was initially identified as a binding partner of BRCA2. It interacts also with BRCA1 forming a complex promoting DNA repair by homologous recombination. Germline pathogenic variants in BRCA1, BRCA2 and PALB2 DNA repair genes are associated with high risk of developing breast cancer. Mutation screening in these breast cancer predisposition genes is routinely performed and allows the identification of individuals who carry pathogenic variants and are at risk of developing the disease. However, variants of uncertain significance (VUSs) are often detected and establishing their pathogenicity and clinical relevance remains a central challenge for the risk assessment of the carriers and the clinical decision-making process. Many of these VUSs are missense variants leading to single amino acid substitutions, whose impact on protein function is uncertain. Typically, VUSs are rare and due to the limited genetic, clinical, and pathological data the multifactorial approaches used for classification cannot be applied. Thus, these variants can only be characterized through functional analyses comparing their effect with that of normal and mutant gene products used as positive and negative controls. The two missense variants BRCA2:c.91T >G (p.Trp31Gly) and PALB2:c.3262C >T (p.Pro1088Ser) were detected in two breast cancer probands originally ascertained at Breast Cancer Units of Institutes located in Milan and Bergamo (Northern Italy), respectively. These variants were located in the BRCA2-PALB2 interacting domains, were predicted to be deleterious by in silico analyses, and were very rare and clinically not classified. Therefore, we initiate to study their functional effect by exploiting a green fluorescent protein (GFP)-reassembly in vitro assay specifically designed to test the BRCA2-PALB2 interaction. This functional assay proved to be easy to develop, robust and reliable. It also allows testing variants located in different genes. Results from these functional analyses showed that the BRCA2:p.Trp31Gly and the PALB2:p.Pro1088Ser prevented the BRCA2-PALB2 binding. While caution is warranted when the interpretation of the clinical significance of rare VUSs is based on functional studies only, our data provide initial evidences in favor of the possibility that these variants are pathogenic.
Approximately 20% of the familial aggregation of breast cancer is related to the presence of germline pathogenic variants in the tumor suppressor high-risk genes BRCA1 (MIM#113705) and BRCA2 (MIM#600185) [reviewed in (1)]. Additional germline variants in several other genes, including PALB2 (partner and localizer of BRCA2) (MIM#610355) have also been implicated in increased predisposition to breast cancer (2, 3). Estimated cumulative breast cancer risk by age of 70 conferred by pathogenic variants in BRCA1 and BRCA2 is approximately 60 and 50%, respectively (4, 5). Loss of function PALB2 pathogenic variants confer a breast cancer risk of 35% by age of 70, that is comparable to that conferred by BRCA2 pathogenic variants (6). Sequencing of these genes has become a key step of the clinical management of breast cancer families as the carriers of a pathogenic variants may be offered appropriate surveillance programs or risk reducing options, whereas the non-carriers may be advised to follow the same recommendations offered to the general population (7).
The clinical utility and efficacy of genetic testing rely on the possibility to establish a correlation between the detected genetic variant and its protein functional effect. As an example, pathogenicity is generally inferred for variants introducing premature termination codons (PTCs), or affecting mRNA integrity and/or stability that give rise to functionally compromised proteins. However, the assessment of the clinical relevance of other variants, especially those that are rare, may not be equally straightforward. These are referred to as variants of uncertain significance (VUSs) and typically include missense variants, small in-frame deletions or insertions, exonic and intronic alterations potentially affecting the mRNA splicing, and variants in regulatory sequences (4, 8). Many of such variants located in the BRCA1, BRCA2, and PALB2 genes have been deposited as “unclassified” in publicly available databases. The current approach to clinically classify a VUS is the multifactorial likelihood prediction model in which, data from epidemiological, genetic, pathological and clinical analyses are combined in order to derive a posterior likelihood of pathogenicity. However, reaching odds ratios in favor of or against causality requires such analyses to be based on several independent observations or to be carried out in large sample series which are usually difficult to obtain if a variant is rare (9, 10). This provides a compelling rationale to the inclusion in the multifactorial model of additional experimental evidences. As a possibility, VUSs —especially those located in the coding regions—can be studied using in vitro and functional assays that compare the effect of normal and mutant gene products.
At the molecular level, PALB2 was identified as a binding partner of BRCA2 and was subsequently shown to bridge, via direct protein-protein interaction, BRCA1 and BRCA2 at sites of DNA damage (11–13). Here, this complex promotes the repair by homologous recombination (HR) of the highly genotoxic DNA lesions, such as double-strand breaks (DSBs) or inter-strand crosslinks (ICLs) (14, 15). These BRCA1-PALB2-BRCA2 interactions are mediated via the coiled-coil domains located at the N-terminus of PALB2 (amino acids 9-44) and at the C-terminus of BRCA1 (amino acids 1,393–1,424), and by the seven-bladed β-propeller WD40 (tryptophan-aspartic acid rich) domain of the C-terminal end of PALB2 (amino acids 836–1,186) binding a domain in the N-terminal end of the BRCA2 (amino acids 21–39) (16, 17). Functional assays based on these domain bindings were used to study patient-derived missense variants in BRCA1 and BRCA2 to provide evidence in favor of or against pathogenicity. Three BRCA2 missense variants, the c.73G>A (p.Gly25Arg), c.91T>C (p.Trp31Arg), and c.93G>T (p.Trp31Cys) were found to disrupt the BRCA2-PALB2 interaction, causing deficiencies in BRCA2 localization to the nucleus and in HR mediated DSB repair (16). Similarly, three BRCA1 missense variants, the c.4198A>G (p.Met1400Val), c.4220T>C (p.Leu1407Pro), and c.4232T>C (p.Met1411Thr) abrogated or moderately impaired the BRCA1-PALB2 binding, causing reduced HR activity (17, 18). To date, only few patient-derived missense variants in the PALB2 gene have been investigated for pathogenicity. Among these, the PALB2:c.104T>C (p.Leu35Pro), located in the coiled-coil domain, was found to co-segregate with two breast cancer cases in a family with a strong history for the disease, and was shown to abrogate the BRCA1-PALB2 binding and to completely prevent HR and resistance to DNA damaging agents. As a result, the p.Leu35Pro was suggested to be a pathogenic variant (19) and is to our knowledge the sole variant in PALB2 to date suggested to be pathogenic. All these findings emphasize that functional assays on VUS located in the BRCA1-PALB2-BRCA2 interaction domains may provide clues on their pathogenicity and that other variants affecting such interactions may be associated with breast cancer susceptibility.
In the current study, we aimed to characterize functionally the two rare missense variants, PALB2:c.3262C>T (p.Pro1088Ser) and BRCA2:c.91T>G (p.Trp31Gly), that were initially identified in breast cancer families and that are located in the protein interaction domains. These two variants were tested for pathogenicity using the green fluorescent protein (GFP)-reassembly in vitro assay that was recently developed for the study of protein-protein interactions (20, 21).
The two female Italian breast cancer probands included in this study were originally considered eligible for clinical genetic testing in breast cancer genes, based on criteria including age of onset for breast cancer and family history for the disease. One proband, recruited at the Genetics Unit of Fondazione IRCCS Istituto Nazionale dei Tumori in Milan (INT), was tested for mutations in the coding regions of BRCA1 and BRCA2 by massively parallel sequencing, using TruSeq Custom Amplicon v.1.2 (Illumina), and multiplex ligation-dependent probe amplification (MLPA) resulting carrier of the BRCA2:p.Trp31Gly. These tests were performed at Cogentech Cancer Genetic Test Laboratory (CGT Lab). The other proband, recruited at the Unit of Medical Oncology of the Ospedale Papa Giovanni XXIII in Bergamo (HPG23), was tested for mutations in the coding regions of BRCA1 and BRCA2 by Sanger sequencing and MLPA at Cogentech CGT Lab. No BRCA1 or BRCA2 mutations were detected, and so this probands was tested at Laboratorio Genetica Medica, HPG23 by massively parallel sequencing using TruSight Cancer assay (Illumina). No pathogenic or likely pathogenic variants were found and the only deleterious variant detected was the missense PALB2:p.Pro1088Ser variant.
Genotyping of the PALB2:p.Pro1088Ser was performed using a custom TaqMan assay (probes and experimental conditions are available upon request). This variant was tested in familial and consecutive breast cancer cases ascertained at HPG23, and in female blood donors used as controls recruited at the AVIS Bergamo.
All individuals included in this study and herein described signed an informed consent to the use of their biological samples and clinical data for research project. This study was approved by Ethical Committee of Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, and Ethical Committee of the Province of Bergamo.
Six BRCA2 or PALB2 variants located in the protein interaction domain were included as positive and negative controls. The PALB2:c.2816T>G (p.Leu939Trp) variant was reported to be not associated with breast cancer risk and to not alter the protein DNA repair activity by HR (22, 23). The BRCA2:c.79A>G (p.Ile27Val) and PALB2:c.3064AT>GC (p.Met1022Ala) missense variants were functionally tested and resulted not disrupting the BRCA2-PALB2 interaction (16, 24). These three variants were used as positive controls. The three missense variants BRCA2:c.93G>T (p.Trp31Cys), BRCA2:c.91T>C (p.Trp31Arg) and PALB2:c. 3073G>A (p.Ala1025Arg) were reported to functionally prevent the BRCA2-PALB2 binding and were used as negative controls (16, 24). All of these variants were patient-derived with the exception of the PALB2:p.Met1022Ala and PALB2:p.Ala1025Arg that were synthetically designed based on crystallography analyses.
The pET11a-NfrGFP-Z and pMRBAD-Z-CfrGFP expression vectors, encoding anti-parallel leucine zipper motifs (Z) fused to the N-terminal or C-terminal fragment of the GFP Protein (NfrGFP and CfrGFP, respectively) (20) were kindly donated by TJ Magliery from the Ohio State University in Columbus (OH, USA). The DNA fragments encoding the N-terminal end of BRCA2 (amino acids 10–40) and the WD40 domain of PALB2 (amino acids 836-1186) were amplified from the cDNA of the 293T cells by PCR. The purified PCR products were subcloned into pET11a-NfrGFP between XhoI and BamHI restriction sites and pMRBAD-Z-CfrGFP between NcoI and AatII restriction sites, replacing the fragments encoding Z motifs. The BRCA2 c.79A>G (p.Ile27Val), c.91T>C (p.Trp31Arg), c.91T>G (p.Trp31Gly), c.93G>T (p.Trp31Cys) and the PALB2 c.2816T>G (p.Leu939Trp), c.3073G>A (p.Ala1025Arg), c.3266C>T (p.Pro1088Ser) variants were obtained by direct mutagenesis of pET11a-NfrGFP-BRCA2 and of pMRBAD-PALB2-CfrGFP using the QuickChange XL Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer's instruction. The PALB2 c.3064AT>GC (p.Met1022Ala) was obtained by the overlap extension PCR mutagenesis method (25). The presence of variants in recombinant clones was verified by DNA sequencing (Eurofins Genomics).
Compatible pairs of plasmids (pET11a-NfrGFP-BRCA2 and pMRBAD-PALB2-CfrGFP, both as wild-type and mutant forms) were co-transformed into BL21 (DE3) E. coli competent cells by electroporation. Single colonies were then picked and used to inoculate 2 ml of LB broth medium containing ampicillin (100 μg/ml) and kanamycin (35 μg/ml). Following overnight incubation at 37°C, the cultured cells were diluted 1:1,000 and 100 μl were plated on inducing LB agar (LBA) plates supplemented with 20 μM Isopropyl β-D-1-tiogalattopiranoside (IPTG) and 0.2% L-arabinose, to promote the expression of recombinant proteins. The plates were incubated at 30°C for 24 h and then 3 days at room temperature (RT). Fluorescence was observed after excitation with long-wave (365 nm) UV light in combination with the short pass (SP) emission filter using a Syngene image capture system (SYNGENE) as specified by the manufacturer.
The pET11a-NfrGFP-BRCA2 (both wt and mutant forms) also encode a hexa histidine (H6)-tag at the N-terminus of the NfrGFP useful for rapid purification by Immobilized metal affinity chromatography (IMAC) method of the H6-tagged proteins. This method exploits the strong binding of H6-tagged protein to metal ions as nickel, allowing them to be separated from other proteins that have lower or no affinity.
Co-transformed bacterial cells were recovered from inducing LBA media using a plate spreader and resuspended in two 1 ml-aliquots of 1X phosphate buffered saline (PBS). After centrifugation, each pellet was resuspended in 50 μl of 1xSDS loading buffer (whole cell extracts), or in 1 ml of lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 0.1% v/v Triton X-100, 100 μM EDTA pH8.0, 0.5 mg/ml lysozime, 20 mM Imidazole, protease inhibitors, 5 μg/ml DNase and RNase) for IMAC purification using the nickel nitrilotriacetic (Ni-NTA) agarose resin (QIAGEN), following the manufacturer's instructions. The purified protein complexes were subjected to 13% SDS-PAGE and visualized by Western blotting using a polyclonal anti-GFP antibody (#600-101-215; Rockland). Whole cell extracts were similarly resolved and visualized, to detect expression levels of the all NfrGFP-BRCA2 and CfrGFP-PALB2 fusion peptides.
Part of our research activity stems from the collaboration with several Breast Cancer Units in which clinical genetic testing is routinely performed. One of our major interest is to functionally study and characterize VUSs in breast cancer genes. In this study, we report the identification and describe the initial functional analyses of the BRCA2:p.Trp31Gly and the PALB2:p.Pro1088Ser variants. The BRCA2:p.Trp31Gly and the PALB2:p.Pro1088Ser variants were originally identified in two different Italian breast cancer probands born in Milano and Bergamo, respectively and are located in the BRCA2-PALB2 interacting domains. Both these probands developed breast cancer at a young age and reported a close relative affected with early onset breast cancer (≤ 40 years). Unfortunately, we were not able to ascertain other family members to be genotyped in order to attempt co-segregation analyses (Figure 1). None of these two variants were reported in public databases such as GnomAD and 1000 genomes. However, the BRCA2: < underline >p.Trp31Gly was annotated in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) in a single individual submitted by Ambry Genetics and classified as a VUS. On the contrary, the PALB2:p.Pro1088Ser was not found in any of the clinical databases we searched. However, during the last annual meeting of the PALB2 Interest Group (PIG; http://www.palb2.org/) colleagues from Ambry Genetics reported the finding of an additional carrier of the PALB2:p.Pro1088Ser variant. To our knowledge, to date, this is only the second proband found to carry this variant. For this reason, we report here his clinical phenotype and family cancer history. This proband was a male affected with colorectal polyps, type unknown, and from a family with cases of melanoma and pancreatic cancer but not breast cancers (Figure 2). Unfortunately, also in this case, no other samples were available for genotyping. We previously reported two different founder mutations, the PALB2:c.1027C>T (p.Gln343*) and the BRCA2:c.190T>C (p.Cys64Arg), originally identified in the Bergamo province where they have a carrier frequency approximately 10-fold higher than that of the Italian population (21, 26, 27). Hence, we genotyped the PALB2:p.Pro1088Ser in 126 familial and 477 consecutive breast cancer cases, and 1,074 controls all born in the province of Bergamo but no additional carriers were found.
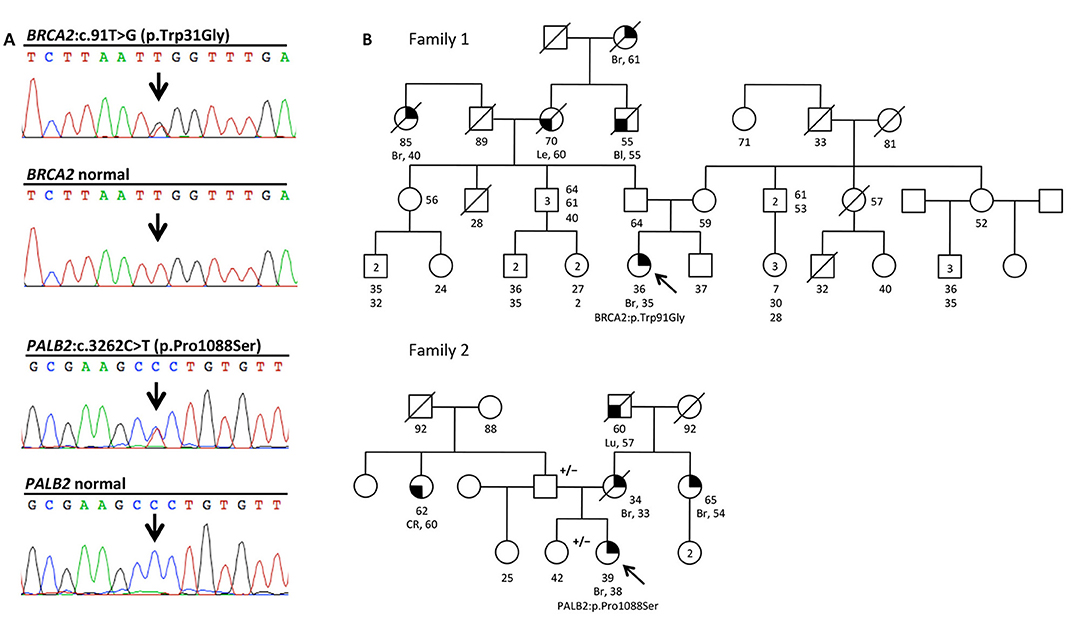
Figure 1. The BRCA2:c.91T>G (p.Trp31Gly) and the PALB2:c.3262C>T (p.Pro1088Ser) variants, and pedigrees of the two mutation carriers. (A) Electropherograms showing the BRCA2 and PALB2 sequence of the individuals carrying the two variants, and normal controls. (B) Family pedigrees 1 and 2 of the two Italians probands carrying the BRCA2:c.91T>G (p.Trp31Gly;) and the PALB2:c.3262C>T (p.Pro1088Ser) variants, respectively. Probands are indicated by arrow. Cancer type, age at diagnosis and age of death are reported when known. Age of healthy individuals, if known, was annotated at date of genetic counseling. Events occurred after genetic counseling, if known, are annotated. Additional relatives carrying the variants are indicated by +/–. Cancer type is reported as follows: Bl, bladder cancer; Br, breast cancer; CR, colorectal cancer; Le, leukemia; Lu, lung cancer.
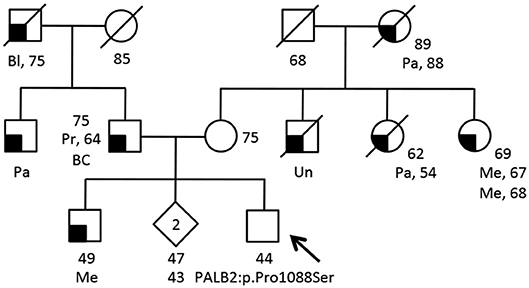
Figure 2. Family pedigree of the additional proband carrying the PALB2:c.3262C>T (p.Pro1088Ser) variant. The proband had large multi gene panel test (67 genes) due to a family history of cancer. The analysis was performed at the Ambry Genetics using Next Generation Sequencing and Array CGA (https://www.ambrygen.com/clinician/genetic-testing/28/oncology/cancernext-expanded). Proband is indicated by arrow. Cancer type, age at diagnosis and age of death are reported when known. Age of healthy individuals, if known, was annotated at date of genetic counseling. Events occurred after genetic counseling, if known, are annotated. Cancer type is reported as follows: BC, basal cells cancer; Bl, bladder cancer; Me, melanoma; Pa, pancreatic cancer; Pr, prostate cancer; Un, unknown.
The BRCA2:c.91T>G and the PALB2:c.3262C>T variants were located within the protein domains mediating the BRCA2-PALB2 interaction (Figure 3A). To evaluate the effect of these variants on the BRCA2-PALB2 interaction, we exploited a bimolecular fluorescence complementation-based assay, the GFP-reassembly in vitro assay. In this assay, the GFP is dissected into two fragments, the N-terminal, NfrGFP and the C-terminal, CfrGFP which are fused to the N-terminal end of BRCA2 (amino acids 10-40) and the WD40 domain of PALB2 (amino acids 836-1186), respectively. These two plasmids are co-expressed in BL21 (DE3) E. coli cells and only if BRCA2-PALB2 interaction occurs, the GFP reassemble emitting cellular fluorescence after ultraviolet (UV) irradiation.
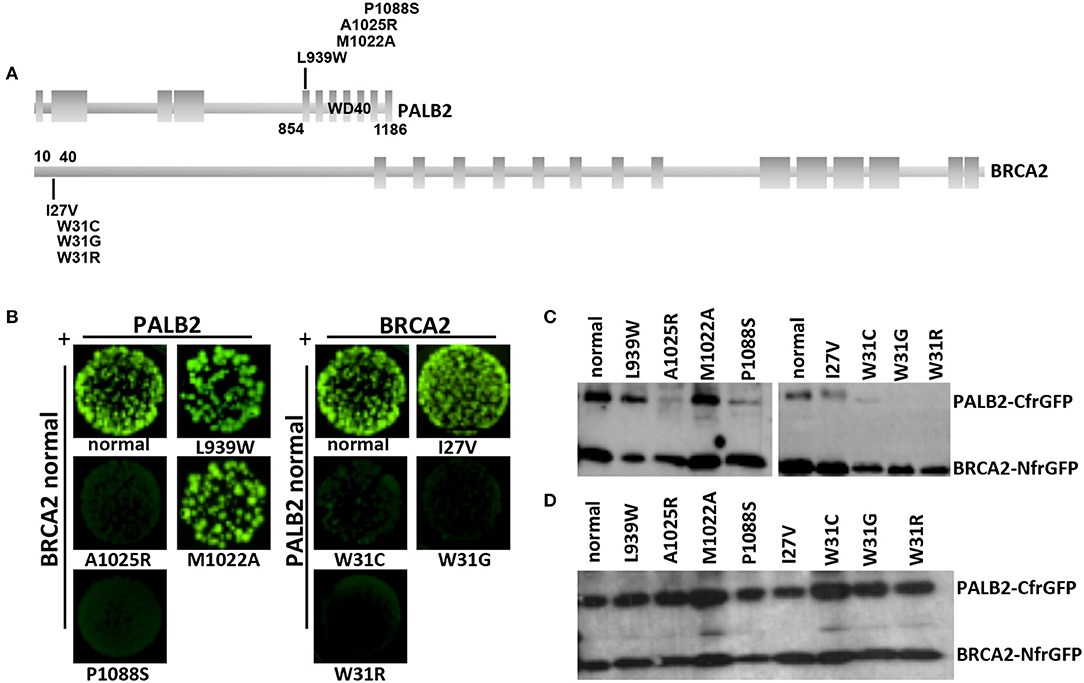
Figure 3. Detection of the BRCA2-PALB2 interaction. (A) Schematic representation of the specific domains mediating the BRCA2-PALB2 binding and of the location of variants analyzed in the present study. (B) GFP-reassembly in vitro assay. Fluorescence was recovered, under long-wave UV light (365 nm), after 24 h of growth at 30°C followed by 3 days of incubation at room temperature. The variants PALB2 L939W or M1022A and BRCA2 I27V were included as positive controls. The variants PALB2 A1025R and BRCA2 W31C or W31R were included as negative controls. (C) IMAC purification. The BRCA2-PALB2 reassembled complexes were purified from the co-transformed E. coli BL21 (DE3) cells, subjected to SDS-PAGE and visualized by Western blotting using a polyclonal anti-GFP antibody. (D) Analysis of expression of BRCA2-NfrGFP and PALB2-CfrGFP normal and mutant forms. Whole cell extracts from the co-transformed E. coli BL21 (DE3) cells were subjected to SDS-PAGE and visualized by Western blotting using a polyclonal anti-GFP antibody. Images of (C, D) were selected from original autoradiographic films shown in Supplementary Figure 1.
Bright GFP fluorescence was observed in bacterial cells co-expressing NfrGFP fused with normal BRCA2 and CfrGFP fused with either normal PALB2, the clinically neutral variant PALB2:p.Leu939Trp or PALB2:p.Met1022Ala (positive controls). Similar results were observed in bacterial cells co-expressing CfrGFP fused with normal PALB2 and NfrGFP fused with BRCA2:p.Ile27Val (positive control). On the contrary, no fluorescence was observed in bacterial cells co-expressing NfrGFP fused with normal BRCA2 and CfrGFP fused with either PALB2:p.Pro1088Ser, or PALB2:p.Ala1025Arg (negative control). Similar results were observed in bacterial cells co-expressing CfrGFP fused with normal PALB2 and NfrGFP fused with either BRCA2:p.Trp31Gly, or the negative controls BRCA2:p.Trp31Cys, or BRCA2:p.Trp31Arg (Figure 3B).
To confirm that GFP-reassembly was effectively due to the BRCA2-PALB2 interaction, the IMAC purified reassembled complexes were analyzed by Western blotting using a polyclonal anti-GFP antibody. Two bands corresponding to the components of the GFP reassembled complexes were detected in lysates of the bacterial cells that resulted fluorescent in the GFP-reassembly screening. On the contrary, no or low intensity bands corresponding to the PALB2-CfrGFP fused domains were observed in lysates from bacterial cells for whom no fluorescence was detected (Figure 3C). In general, any mutations can cause the decrease or the complete loss of expression of the encoded GFP fused peptides. Thus, we wanted to confirm that the lack or the low intensity of the PALB2-CfrGFP bands was not due to loss of expression. To this aim, the whole cell extracts were analyzed by Western blotting using a polyclonal anti-GFP antibody as previously described. In this experiment, we showed that normal and mutated fusion peptides were expressed to a similar extent indicating that the loss of fluorescence observed in the GFP-reassembly in vitro assay, was attributable to the lack of binding between the proteins and not to poor expression of the mutants (Figure 3D). All these results provided experimental evidence that both the BRCA2:p.Trp31Gly and the PALB2:p.Pro1088Ser variants abrogate the BRCA2-PALB2 binding.
In clinical settings, VUSs in breast cancer genes represent a serious issue in the process of disease risk assessment in carriers. Typically, results from different sources such as epidemiological, genetic, and clinical analyses are combined together in order to derive a posterior likelihood of pathogenicity used to classify a VUS. While this multifactorial approach is successful to classify common VUSs, variants that are rare or unique can only be studied through functional analyses.
In the present study, we investigated the pathogenicity of the two BRCA2:p.Trp31Gly and PALB2:p.Pro1088Ser variants performing functional analyses. The BRCA2:p.Trp31Gly was previously reported in a single proband and annotated as VUS. To our knowledge, the PALB2:p.Pro1088Ser was never detected before. Both variants are located in the interaction domains of BRCA2 and PALB2. Large part of the BRCA2 functions in the repair of the DNA double strand breaks and inter-strand crosslinks by HR depends from its interaction with PALB2. Thus, we developed a GFP-reassembly assay based on the testing of this interaction speculating that this binding assay would be a predictor of the effect of the variants on the BRCA2 integrity.
In the GFP-reassembly assay, we used six different BRCA2 and PALB2 missense variants as controls. Two patient-derived BRCA2 variants, the p.Trp31Arg and p.Trp31Cys, and one synthetic PALB2 variant, the p.Ala1025Arg, were known to prevent the BRCA2-PALB2 interaction. The patient derived BRCA2:p.Ile27Val and PALB2:Leu939Trp, and the synthetic PALB2:p.Met1022Ala were expected to not alter the binding of these proteins. For all of these variants, the results were concordant with their expected effect on the BRCA2-PALB2 binding.
The GFP-reassembly assay results indicated that both the BRCA2:p.Trp31Gly and PALB2:p.Pro1088Ser prevented the BRCA2-PALB2 interaction suggesting that in physiological conditions these alleles encode proteins that might be unable to interact with PALB2 and BRCA2, respectively. To our knowledge, the PALB2:p.Pro1088Ser is the first missense variant in the gene that was functionally shown to abrogate the binding with BRCA2. As the correct formation of the BRCA1-PALB2-BRCA2 complex is necessary for DNA repair by HR, our results provide evidences in favor of the hypothesis that the BRCA2:p.Trp31Gly and PALB2:p.Pro1088Ser are pathogenic variants. While this assumption is at present most likely—in example vs. the possibility that the variants are neutral—other aspects need to be considered for a clearer picture of the effect of these variants on breast cancer risk. Firstly, Foo and colleagues showed that both the PALB2:p.Leu35Pro and p.Tyr28Cys caused the loss of the interaction with BRCA1; however, only the p.Leu35Pro completely abrogated the HR activity and the p.Tyr28Cys caused a loss of approximately 65% of the HR activity (19). Hence, PALB2 missense variants causing the loss of the binding with BRCA1 might confer different risk magnitude for breast cancer. To be conservative, we cannot exclude that this might be true as well for the PALB2 variants abrogating the binding with BRCA2. As a second point, caution should be taken when inferring on the nature of a missense variant on the bases of functional studies only. Park and colleagues reported that the PALB2:p.Leu939Trp variant might be pathogenic based on the fact that it resulted in altered BRCA2-PALB2 binding, decreased HR capacity, and increased sensitivity to ionizing radiation (28). However, we provided strong evidences deriving from additional functional studies and very large case-control studies that the PALB2:p.Leu939Trp is a neutral variant (23). As a final consideration, it should be noted that of the many missense variants that were functionally proved to prevent the BRCA1-PALB2-BRCA2 complex formation (16, 19, 24), all, with the only exception of the PALB2:p.Leu939Trp that is consider benign or likely benign, are annotated or should be treated clinically as VUS.
In conclusion, we report here results from functional studies indicating that the BRCA2:c.91T>G (p.Trp31Gly) and the PALB2:c.3262C>T (p.Pro1088Ser) missense variants abrogate the BRCA2-PALB2 protein binding. These data provide initial evidences corroborating the hypothesis that these variants are pathogenic. Importantly, novel data are warranted to progress in the clinical classification of these variants. The search for additional variant carriers and collection of their family members is crucial to provide genetic or pathological data; however, as the variants in study are very rare, we expect that not many variant carriers will be found in the near future. On the contrary, additional functional studies (i.e. testing specific protein functions in eukaryotic cells) could be immediately performed. While caution is warranted when clinical classification of a VUS is based on in vitro assays only, these results will provide additional evidences to better clarify the functional effect of the variants in study.
PP and PR designed and supervised the study. TP, MW, SV, AF, MM, MI, CT, AZ, JA, and SM provided samples and data. LC, IC, and LD performed experiments. LC, IC, GF, PP, and PR analyzed data. LC, IC, PP, and PR wrote the manuscript. All authors contributing to, critically revised and approved the manuscript.
This work was partially supported by the following entities. Ministero della Salute, Italy Ricerca Finalizzata–Bando 2010 to PP; AIRC (Associazione Italiana per la Ricerca sul Cancro) to PP (IG 16732), PR (IG 15547) and AF (5 × 1,000 n. 12237); Fondazione Umberto Veronesi (FUV-Post-doctoral Fellowships−2016) to IC; the Italian citizens who allocated the 5 × 1,000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale dei Tumori, according to the Italian laws, to SM.
TP is a full time paid employee of Ambry Genetics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are particularly grateful to individuals participating in this study and their families. We also thank the Real Time PCR and the DNA Sequencing Service of Cogentech, Milan, Dr. Thomas J. Magliery from the Ohio State University in Columbus (OH, USA) for kindly providing the pET11a-NfrGFP-Z and pMRBAD-Z-CfrGFP plasmids necessary for the GFP-reassembly assay, Dr. Maria Teresa Radice of Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, for technical assistance with plasmid construction and Drs. Cristina Zanzottera and Roberta Villa of Fondazione IRCCS Istituto Nazionale dei Tumori, Milan.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00480/full#supplementary-material
1. Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. (2010) 4:174–91. doi: 10.1016/j.molonc.2010.04.011
2. Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. (2015) 372:2243–57. doi: 10.1056/NEJMsr1501341
3. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. (2017) 3:1190–6. doi: 10.1001/jamaoncol.2017.0424
4. Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science (2014) 343:1466. doi: 10.1126/science.1251827
5. Foulkes WD. BRCA1 and BRCA2—Update and implications on the genetics of breast cancer: a clinical perspective. Clin Genet. (2014) 85:1–4. doi: 10.1111/cge.12291
6. Antoniou AC, Foulkes WD, Tischkowitz M. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. (2014) 371:1651–2. doi: 10.1056/NEJMc1410673
7. Girardi F, Barnes DR, Barrowdale D, Frost D, Brady AF, Miller C, et al. Risks of breast or ovarian cancer in BRCA1 or BRCA2 predictive test negatives: findings from the EMBRACE study. Genet Med. (2018). doi: 10.1038/gim.2018.44
8. Radice P, De Summa S, Caleca L, Tommasi S. Unclassified variants in BRCA genes: guidelines for interpretation. Ann Oncol. (2011) 22(Suppl 1):i18–23. doi: 10.1093/annonc/mdq661
9. Spurdle AB. Clinical relevance of rare germline sequence variants in cancer genes: evolution and application of classification models. Curr Opin Genet Dev. (2010) 20:315–23. doi: 10.1016/j.gde.2010.03.009
10. Lindor NM, Guidugli L, Wang X, Vallée MP, Monteiro AN, Tavtigian S, et al. A review of a multifactorial probability based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. (2012) 33:8–21. doi: 10.1002/humu.21627
11. Pauty J, Rodrigue A, Couturier A, Buisson R, Masson JY. Exploring the roles of PALB2 at the crossroads of DNA repair and cancer. Biochem J. (2014) 460:331–42. doi: 10.1042/BJ20140208
12. Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. (2009) 7:1110–8. doi: 10.1158/1541-7786.MCR-09-0123
13. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. (2009) 19:524–9. doi: 10.1016/j.cub.2009.02.018
14. Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. (2016) 17:337–49. doi: 10.1038/nrm.2016.48
15. Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. (2015) 7:a016600. doi: 10.1101/cshperspect.a016600
16. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell (2006) 22:719–29. doi: 10.1016/j.molcel.2006.05.022
17. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. (2009) 106:7155–60. doi: 10.1073/pnas.0811159106
18. Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife (2017) 6:e21350. doi: 10.7554/eLife.21350
19. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene (2017) 36:4161–70. doi: 10.1038/onc.2017.46
20. Sarkar M, Magliery TJ. Re-engineering a split-GFP reassembly screen to examine RING-domain interactions between BARD1 and BRCA1 mutants observed in cancer patients. Mol Biosyst. (2008) 4:599–605. doi: 10.1039/b802481b
21. Caleca L, Putignano AL, Colombo M, Congregati C, Sarkar M, Magliery TJ, et al. Characterization of an Italian founder mutation in the RING-finger domain of BRCA1. PLoS ONE (2014) 9:e86924. doi: 10.1371/journal.pone.0086924
22. Southey MC, Winship I, Nguyen-Dumont T. PALB2: research reaching to clinical outcomes for women with breast cancer. Hered Cancer Clin Pract. (2016) 14:9. doi: 10.1186/s13053-016-0049-2
23. Catucci I, Radice P, Milne RL, Couch FJ, Southey MC, Peterlongo P. The PALB2 p.Leu939Trp mutation is not associated with breast cancer risk. Breast Cancer Res. (2016) 18:111. doi: 10.1186/s13058-016-0762-9
24. Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. (2009) 10:990–6. doi: 10.1038/embor.2009.126
25. Hussain H, Chong NF. Combined overlap extension PCR method for improved site directed mutagenesis. Biomed Res Int. (2016) 2016:8041532. doi: 10.1155/2016/8041532
26. Catucci I, Peterlongo P, Ciceri S, Colombo M, Pasquini G, Barile M, et al. PALB2 sequencing in Italian familial breast cancer cases reveals a high-risk mutation recurrent in the province of Bergamo. Genet Med. (2014) 16:688–94. doi: 10.1038/gim.2014.13
27. Catucci I, Casadei S, Ding YC, Volorio S, Ficarazzi F, Falanga A, et al. Haplotype analyses of the c.1027C>T and c.2167_2168delAT recurrent truncating mutations in the breast cancer-predisposing gene PALB2. Breast Cancer Res Treat. (2016) 160:121–9. doi: 10.1007/s10549-016-3981-y
Keywords: breast cancer, breast cancer predisposition genes, PALB2, BRCA2, VUS, functional analyses, PALB2-BRCA2 interacting domain
Citation: Caleca L, Catucci I, Figlioli G, De Cecco L, Pesaran T, Ward M, Volorio S, Falanga A, Marchetti M, Iascone M, Tondini C, Zambelli A, Azzollini J, Manoukian S, Radice P and Peterlongo P (2018) Two Missense Variants Detected in Breast Cancer Probands Preventing BRCA2-PALB2 Protein Interaction. Front. Oncol. 8:480. doi: 10.3389/fonc.2018.00480
Received: 03 August 2018; Accepted: 08 October 2018;
Published: 25 October 2018.
Edited by:
Haining Yang, University of Hawaii Cancer Center, United StatesReviewed by:
Adriana De Siervi, Instituto de Biología y Medicina Experimental (IBYME), ArgentinaCopyright © 2018 Caleca, Catucci, Figlioli, De Cecco, Pesaran, Ward, Volorio, Falanga, Marchetti, Iascone, Tondini, Zambelli, Azzollini, Manoukian, Radice and Peterlongo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Peterlongo, cGFvbG8ucGV0ZXJsb25nb0BpZm9tLmV1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.