 Lena Behrmann
Lena Behrmann Jasmin Wellbrock
Jasmin Wellbrock- Department of Oncology, Hematology and Bone Marrow Transplantation with Section Pneumology, Hubertus Wald University Cancer Center, University Medical Center Hamburg Eppendorf, Hamburg, Germany
The bone marrow is the home of hematopoiesis and is therefore a hotspot for the development of hematopoietic diseases. Complex interactions between the bone marrow microenvironment and hematopoietic stem cells must find a balance between proliferation, differentiation and homeostasis of the stem cell compartment. Changes in this tightly regulated network can provoke malignant transformation, leading to hematopoietic diseases. Here we focus on acute myeloid leukemia (AML), since this is the most frequent acute leukemia in adulthood with very poor overall survival rates and where relapse after chemotherapy continues to be a major challenge, driving demand for new therapeutic strategies. Current research is focusing on the identification of specific interactions between leukemic blasts and their niche components, which may be exploited as novel treatment targets along with induction chemotherapy. Significant progress has been gained over the last few years in the field of high-resolution imaging. Confocal ex vivo and intravital microscopy have revealed a detailed map of bone marrow structures and components; as well as identifying numerous alterations in the stem cell niche that correspond to disease progression. However, the underlying mechanisms are still not completely understood and due to the complexity, their elucidation remains a challenging. This review discusses the constitution of the AML niche in the bone marrow, the improvement in visualization of the complex three-dimensional niche structures and points out new therapeutic strategies to increase the overall survival of AML patients.
Introduction to Acute Myeloid Leukemia
Leukemia is characterized by infiltration of the hematopoietic organs (bone marrow, blood, spleen, and other tissues) by abnormally differentiated and nonfunctional hematopoietic blasts (1). High and uncontrolled proliferation of leukemic cells causes the expulsion of the normal hematopoietic system and the loss of their functions, leading to life-threatening symptoms such as thrombocytopenia, anemia, and immunodeficiency. Although the clinical presentation of the disease is quite uniform, the genetic and cytogenetic landscape of leukemia's is very heterogeneous (1–3). Proximal causes of leukemic development can be categorized in three major groups: (1) gene mutations and translocations, including epigenetic dysregulation, (2) immune dysregulation, and (3) changes in the bone marrow microenvironment.
Adult and elderly patients suffer mostly from the aggressive acute myeloid leukemia (AML) that shows an increasing incidence with age. Without therapy this disease leads to the death of the patient within a few weeks. Although the overall 5-year survival rate of AML patients improved by application of chemotherapy, it remains unsatisfactory. A positive outcome declines from 50% for those younger than age 65 years to only 23% for patients aged 65 years and higher (4). Although AML is known to be genetically and cytogenetically heterogeneous, the common clinical chemotherapy has not changed in the last three decades, consisting of sequential courses of cytarabine (AraC) and daunorubicine (7 + 3 regimen) (5). Particularly problematic in patients <60 years is a diminished tolerance for intensive chemotherapy with increased risk of treatment-related toxicity, leading to an estimated treatment-related mortality ranging from 10 to 30% (6). The second disadvantage of this non-specific treatment is a high rate of resistance and relapse. Disease recurrence occurs in 60–80% of AML patients within 3 years after diagnosis even in patients who achieved remission with chemotherapy.
AML is a multi-gene disease with complex subclonal populations. After chemotherapy leukemic clones are selected to those capable of activating resistance mechanisms, leading to relapsed, and refractory disease (7). Therefore, there is an urgent need for low-intensity targeted treatment to improve survival of AML patients. Over the last few years an evolving number of studies have deciphered the essential role of the microenvironment in the pathogenesis of AML. The understanding of the complex interactions between leukemia and its niche cells may reveal new drugable targets to improve AML therapy. This review will summarize the recent advances in elucidating the leukemic microenvironment in the bone marrow to decipher new perspectives for targeted therapies.
Bone Marrow Microenvironment of Leukemic Cells
To understand the reasons for the emergence of refractory AML cells under and after chemotherapy we have to understand how resistance can develop. Resistance can be divided into the categories de novo and acquired: where the latter is a sequential genetic change that finally ends in prosurvival and antiapoptotic phenotypes (8). De novo drug resistance refers to the result of environment-mediated protection from apoptosis that enables resistant AML cells to survive. The most relevant microenvironment for AML is within the bone marrow, the organ that hosts the hematopoietic stem cells (HSCs) and facilitates their differentiation into the manifold blood cell lineages (9). Within the bone marrow the HSCs are located in a tightly controlled local microenvironment, the so called niche, that regulates self-renewal, quiescence, proliferation, and differentiation of HSCs by bound or secreted molecules emanated by the surrounding cells (10). In the past decades various cell types were implicated for their roles in promoting HSC maintenance, including osteoblasts, perivascular stromal cells, endothelial cells, macrophages, CXCL12-abundant reticular cells (CAR cells), sympathetic neurons and nonmyelinating Schwann cells (11–17). Additionally there are several known soluble factors relevant for HSCs, including CXCL12, angiopoietin 1 (ANGPT1), TGF-β and signaling pathways including Notch and Wnt (11, 18–22).
Advances in bone marrow imaging technologies have improved the understanding of the physical HSC niche localization and physiological architecture but there is still the potential for discovering other undetected cell populations and factors. Despite the complexity of the bone marrow microenvironment, the HSC niche can be reduced into two physical geographies, the endosteal, and perivascular niche. The endosteum is defined by immediate proximity to trabecular or cortical bone with a high content of osteoblasts (23). The perivascular niche is located in proximity to sinusoidal and arteriolar vascular endothelium, including the surrounding supportive structures such as stromal cells and extracellular matrix (24, 25). Many publications discuss the controversial role of the distinct niches in regard to HSC dormancy and maintenance (25–27). However, the endosteal niche is consistently intertwined with vascular structures, first at the sites where arterioles enter the bone marrow via the endosteal zone, and second at the sites of sinusoids that spread as a dense network through the entire bone marrow cavity (28). In reality a separation of the two microenvironments is difficult and HSCs interact with numerous and simultaneous cell-extrinsic signaling settings (29).
Components of the AML Niche
Like the hematopoietic system, AML is depicted as a hierarchical disease based on a small subset of leukemia initiating cells (LICs). This stem cell-like compartment has alone long-term repopulating potential, the ability to propagate and maintain the AML phenotype, and is expected to be the main cause for AML relapse (30). First LICs were identified as rare CD34+CD38− events, as demonstrated by their capacity for serial transplantations in a mouse xenograft model (31, 32). As few as one in a million AML cells show this type of leukemia initiating activity, endorsing the idea of the hierarchical organization of the disease. Over the last years, an evolving number of further surface markers were defined, like CD123 or CD96, and even in the CD34− fraction LICs can reside (33–35). Hence, the heterogeneous genetic landscape of AML is recapitulated within the stem cell phenotype. Independent of their phenotype, LICs have the potential to infiltrate into the HSC niche and hijack the normal homeostatic processes, supporting their self-renewal and proliferation potential, as well as quiescence and resistance to chemotherapy. Recent findings indicate that myeloid malignancies also alter the HSC niche into a leukemia niche that becomes permissive of leukemia growth and disrupts normal hematopoiesis (36). The corrupted components of the leukemic niche cooperate with LICs to maintain their quiescence and survival. Furthermore it has been suggested that mutations in stromal cells have a primary role in AML initiation (37). However, leukemia is a very heterogeneous disease and even though we have made gains in understanding the HSC niche, not all niche related pathways are relevant for AML or might even have an inverted effect for leukemic propagation. Summarizing the AML niche needs a careful discrimination between the different concepts of niches for HSC and other leukemic subtypes. Figure 1 summarizes drugable pathways for targeted AML therapy, which will be discussed in the following paragraphs.
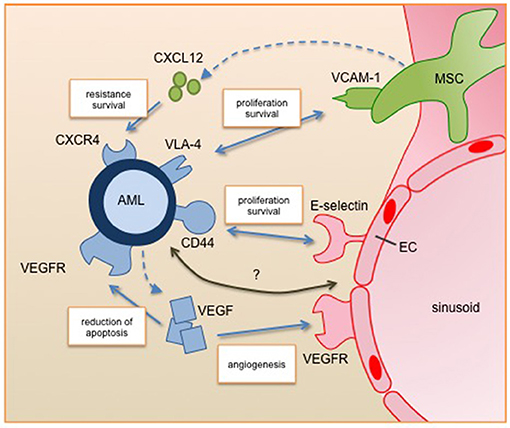
Figure 1. Leukemic blast interactions with the perivascular niche. Secreted factors and cell-cell interactions regulate the survival, proliferation and resistance of AML cells in the perivascular niche. All of them are under clinical investigation for targeted therapy. Future studies will identify further relevant pathways of the interplay between AML and its niche cells (marked with question mark). AML, acute myeloid leukemia; EC, endothelial cell; MSC, mesenchymal stromal cell; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor, CXCL12, C-X-C motif chemokine 12; CXCR4, C-X-C chemokine receptor 4; VLA-4, very late antigen 4; VCAM-1, vascular cell adhesion molecule 1.
A number of studies have confirmed that there is a tight relationship between leukemic and endothelial cells. For instance Cogle et al. presented that vascular endothelial cells are a critical component of the AML niche and leukemic cells can integrate into the vasculature (38). Furthermore, the two-way communication between AML and endothelial cells through autocrine and paracrine pathways supports AML development and progression. Here, a key player is the proangiogenic factor VEGF (vascular endothelial growth factor). VEGF has six isoforms (VEGF-A, -B, -C, -D, -E, and PLGF) that can bind to three tyrosine kinase receptors (VEGFR-1,-2, and-3) leading to a complex network of downstream signaling (39). Fiedler et al. showed that a large proportion of AML patients express high levels of VEGF, leading to both induction of angiogenesis and reduction of apoptosis in AML cells. Furthermore, VEGF induces an increase of GM-CSF secretion by endothelial cells, which is a known mitogen for AML cells (40).
Mesenchymal stromal cells (MSCs) are known to provide microenvironmental support for HSCs but also have the potential for differentiation to multiple lineages including osteoblasts, chondrocytes and adipocytes. These processes are highly regulated and disturbances are involved in oncogenic events. Battula et al. showed that AML cells induce osteoblastic but inhibit adipogenic differentiation of MSCs by bone morphogenetic protein (BMP) signaling (41). This shift leads to a pre-osteoblastic niche that enhances AML expansion. Additionally, Hanoun et al. showed that AML progression leads to a reduction of the sympathetic nervous system, which is critical for MSC quiescence osteoblast differentiation and even hematopoietic stem cells (16). This initiates an expansion of osteoblastic-primed MSCs, which can contribute to AML progression (17). Frisch et al. described that the secretion of CCL3 (also known as macrophage inflammatory protein 1α, MIP 1α) by AML cells leads to reduced levels of osteoblasts and osteocalcin in the blood. Inhibited osteoblastic cell function results in loss of mineralized bones and reduced healthy hematopoiesis (42). Taken together, these observations indicate that AML cells induce and require an osteoprogenitor-rich niche for their expansion, but block MSC differentiation to produce mature osteoblasts.
The role of adipocytes during leukemogenesis is also controversial. The existence of bone marrow adipocytes has been known for more than a century, but only recently has their function been deciphered. In contrast to the idea of silent fat storage, bone marrow adipocytes are highly active regulators of the bone marrow metabolism and the overall body homeostasis by secretion of adipokines (43). Adipocytes were identified as negative regulators for the HSC compartment (44) and Boyd et al. described the disruption of the adipocytic niche by AML (45). In contrast, Shafat et al. described a supportive effect of bone marrow adipocytes for the survival and proliferation of AML cells based on the transfer of fatty acids from adipocytes to blasts (46). In general, the amount of adipocytes in the bone marrow can fluctuate and react to several metabolic situations, such as starvation or cytotoxic stress. Since chemotherapy induces adipogenesis that leads to a different bioavailability of drugs in the bone marrow, the role of adipocytes for AML development and survival needs further investigations (47).
Although bone marrow fibroblasts are a major component of connective tissue, knowledge about the role for AML development is very limited. It is known that fibroblasts produce extracelluar matrix fibers but also regulate the microenvironment by cytokine and chemokine secretion (48). Furthermore, cancer-associated fibroblasts are considered to influence the chemo-resistance of many solid tumors. Zhai et al. presented that fibroblasts are increased in leukemic bone marrow and can provide chemo-protective elements for AML (49). Further animal experiments need to verify these findings.
The immunological microenvironment of leukemic cells plays another fundamental role for leukemic development and chemotherapy resistance (50). The normal bone marrow hosts various mature immune cell types, including T and B cells, dendritic cells, and macrophages. Usually, immune cells recognize abnormal cells on the basis of mutated proteins and are able to eliminate them (51). But AML cells can escape an immune response by creating an immunosuppressive microenvironment, where both innate and adaptive immune responses are dysregulated (50). It was shown that AML can reduce T and natural killer cell function and cytotoxicity and furthermore induce populations of regulatory T cells (Treg) through the activation of immune checkpoint markers like programmed death-1 (PD-1). These markers are crucial regulators of the immune response and play a central role for self-tolerance. Physiologically, ligands like PD-L1 are expressed on antigen-presenting cells and binding to their receptors results in the reduction of T-cell activation. Interestingly, AML cells are known to express PD-L1 to escape immune mediated eradication and PD-L1 expression levels correlate with AML progression (52). Furthermore, mesenchymal stromal cells have immune modulatory functions (53). And even though the exact mechanisms are not fully elucidated, indoleamine 2,3-dioxygenase 1 (IDO1) seems to play a key role. IDO1 expression can inhibit T-cell proliferation and modulate the function of major cell populations involved in innate and adaptive immune systems (50).
In addition to the cells of the adaptive immune system, mononuclear phagocytes contribute to the regulation of HSC populations (54). Macrophages were shown to regulate osteoblasts and the HSC mobilization in the endosteal niche (14). Interestingly, AML leads to an invasion of AML-associated macrophages into the bone marrow and spleen, supporting their progression.
One critical component for AML engraftment and progression is the adhesion to the microenvironment. Several adhesion molecules (e.g., VLA-4, E-selectin, and CD44) were deciphered as playing a relevant role for AML (29). VLA- 4 (very late antigen 4) a cell surface ligand for VCAM-1 (vascular cell adhesion molecule 1) presented on mesenchymal stromal cells is highly expressed on AML cells. Jacamo et al. showed that the interaction of VLA-4 and VCAM-1 activates prosurvival and proproliferative pathways in both the leukemia and stromal cells via the NF-κB pathway, leading to higher chemotherapy resistance (55). Interestingly, patients with VLA-4 negative AML have a generally favorable outcome (56). Mouse experiments using a VLA-4 specific antibody in combination with cytarabine therapy showed an improved outcome. The second axis of cell adhesion showing promising results for new treatment strategies is the interaction of the adhesion molecule CD44 and E-selectin. The latter is expressed by endothelial cells and is bound by CD44 that is broadly expressed on AML cells. Antagonizing E-selectin increases chemosensitivity of AML under treatment (57). Furthermore, Jin et al. showed that the activation of CD44 eradicates AML stem cells (58). Targeting the adhesion of AML cells interferes with their embedding in their protective niche and therefore influences survival.
Besides adhesion, AML cells are also regulated by soluble factors secreted by niche cells, like Notch, CCL3, TGF-β, or CXCL12 (59–61). We want to highlight CXCL12 (C-X-C motif chemokine 12), also known as stromal-cell derived factor 1 (SDF-1) (62). CXCL12 is secreted by mesenchymal stromal cells and induces chemotaxis in leukocytes by binding to its receptor CXCR4 (C-X-C chemokine receptor 4). In normal hematopoiesis this axis regulates leukocyte trafficking. Chemotherapy stress induces the expression of CXCR4 in AML cells, leading to increased resistance and survival. Inhibition of CXCR4 sensitizes AML to chemotherapy and increased therapy-related apoptosis. Another level of intercellular signaling involves the exchange of information via membranous structures, like exosomes and tunneling nanotubes (TNT). A recent publication from Kumar and colleagues presented that AML cells secrete exosomes to manipulate mesenchymal stromal cells (63). This signaling blocked osteolineage development and led to a leukemic niche that supported the leukemic development and survival. In leukemia, TNTs were first described by Polak and colleagues, showing that acute lymphoblastic leukemia cells manipulate the mesenchymal stromal cells to induce the secretion of prosurvival cytokines and chemotactic proteins, improving their capabilities for chemotherapy resistance (64).
However, since most data are based on in vitro experiments, the exact mechanisms leukemia cells use for modifying their microenvironment to generate a shelter against chemotherapy still remain largely unknown.
Animal Models for AML Niche Analysis
To understand how AML cells interact with their niches, it is crucial to know spatiotemporal cellular interactions and dynamics in the bone marrow. Quantitative analyses of bone marrow mononuclear cells by flow cytometry present a well-established technique and can deliver important hints of changes in cellularity but information about localization and cell-cell interactions get completely lost. Furthermore, extraction of structural cell types, such as endothelial or mesenchymal stromal cells for flow cytometry includes enzymatic tissue degradation steps, which can be inefficient and therefor significantly adulterate the data. Only imaging of native tissues enables the study of cellular interactions in the multicellular niche, without losing single components of the processed tissue.
One powerful model to study the development of the hematopoietic system in vivo is the transgenic zebrafish (Danio rerio) (65). Hematopoiesis is highly conserved between mammals and zebrafishes and live imaging in zebrafish embryos is a very well-established method for HSCs research. The benefits of this model are i.a. the high reproduction rates, enabling the generation of hundreds of embryos at the same time and external fertilizations, allowing the study of developing embryos without dissections. Additionally, the zebrafish embryo is small and transparent, allowing the scientist to perform live cell imaging by confocal microscopy in the complete living organism. Leonard Zon and coworkers utilized a transgenic zebrafish line to visualize the dynamic colonization of the caudal hematopoietic tissue (CHT), which is comparable to the mammalian fetal liver. Utilizing high-resolution live imaging they were able to visualize the first steps of colonization of the developing CHT. They described for the first time how endothelial cells remodel around single HSCs to form a stem cell pocket (66). However, the utility of zebrafishes for studying adult hematopoiesis and leukemogenesis is limited, since adult zebrafish hematopoiesis resides in the kidney marrow, which is structurally distinct to the bone marrow of mammals (67).
In the last decades murine models became the main mammalian model system for hematopoietic research. Their convenient maintenance and handling, as well as their small body size, short reproduction cycle, and the ability for genetic manipulations are prerequisites for a biomedical research tool. The development of immunodeficient mice strains like NSG (NOD scid gamma) enables xenografting of patient-derived cells without pre-conditioning of the mouse with radiation or chemotherapy, allowing studies about interactions between human hematopoietic cells and their microenvironment (68). One major contribution of the patient-derived xenograft (PDX) models to the AML field was the identification and characterization of leukemia initiating cells (LICs) (32). However, several signaling components of the mouse are incongruent with those of humans. In particular AML remains one of the hematologic malignancies with low engraftment performance in NSG mice (69). As a result, several attempts have been made to customize the bone marrow microenvironment to enable myeloid infiltration. One promising approach is the development of humanized mouse strains with transgenic expression of human cytokines like the NSG-SGM3 mouse strain (70). Effects on the bone marrow structure and the localization of LICs in these humanized mouse models are owing and need to be elucidated in the future.
Niche Analysis by Imaging Techniques
The first analysis of the HSC localization in murine bone marrow using confocal laser scanning microscopy was presented by Kiel et al. (24). The structural differences between the semisolid bone marrow and the hard bone surface is challenging for histological sectioning methods. Utilizing the CryoJane system they were able to transfer thin tissue sections to object slides, where histological staining can be performed. Alternatively Kawamoto developed a method for hard tissue sections, using an adhesive film to allow sectioning independent of special section equipment (71). Visualization of xenotransplanted AML stem cells in the bone marrow by confocal laser scanning microscopy revealed a preferential engraftment within the osteoblast-rich endosteal niche that was defined as a twelve cell deep zone, starting with the bone surface (72). To improve the study of cellular distribution inside two-dimensional (2D) sections the laser scanning cytometry (LSC) technology was developed. LSC is an automated cellular analyzer that allows in situ imaging and quantitative analysis of fluorescent labeled cells within thin tissue sections (73). LSC records precise spatial localization information of each fluorescent molecule within the section and generates a detailed map of the analyzed tissue. Nombela-Arrieta et al. used this technique to study the spatial distribution of HSCs in the bone marrow and defined distinct endosteal niches (25). However, these 2D histological studies of the bone marrow fail to present the complexity of the three-dimensional (3D) network of blood vessels, scaffolding structures and hematopoietic cells. The approach of serial sectioning and computational reconstruction of the tissue geometry might improve the informational outcome but remains challenging. Therefore, the establishment of protocols for 3D confocal microscopy of bone marrow samples was a crucial step for the elucidation of the HSC niche.
Kunisaki et al. were first to present a protocol for whole-mount bone marrow confocal immunofluorescence imaging, revealing new information about the regulation of quiescence in HSCs (74). A milestone for the imaging of whole-mount samples was the development of tissue clearing substances. Like most tissues, bone marrow opacity undermines high-resolution microscopy due to light scattering caused by reflection off of molecules, membranes, organelles and cells (75). During the last years, several methods for optical tissue clearing were developed, including clearing via solvent- or aqueous-based clearing solutions. The simplest passive clearing technique uses immersion in a high refractive index solution, since this procedure has no influence on fluorescent molecules or tissue structures and can easily be implemented into running staining protocols. For details, we refer the reader to the review from Richardson and Lichtman (75). Utilizing optical tissue clearing Acar et al. were able to presented a 2015 study demonstrating the distribution of dividing and non-dividing HSCs in whole-mount bone marrow samples, including sophisticated methods for image annotation and analysis (76). The combination of second harmonic generation signals to visualize the hard bone structures and 3D visualization of blood vessels revealed that dividing and non-dividing HSCs locate in a persinusoidal niche distant from the endosteum. Since then, rapidly emerging deep-tissue imaging studies are reveiling bone marrow structures and improving the map of niche formation of hematopoietic and leukemic stem cells (Figure 2) (77–80). We want to highlight the work from Coutu et al., which provide an open source for imaging data to map nonhematopoietic cells and structures in the niche (77). The highly sophisticated analysis of the physical network of cellular interactions and localization within the bone marrow presents a comprehensive atlas of the distribution of bone marrow markers. It seems like the only limitations in this field are the availability of specific antibodies or transgenic mouse lines.
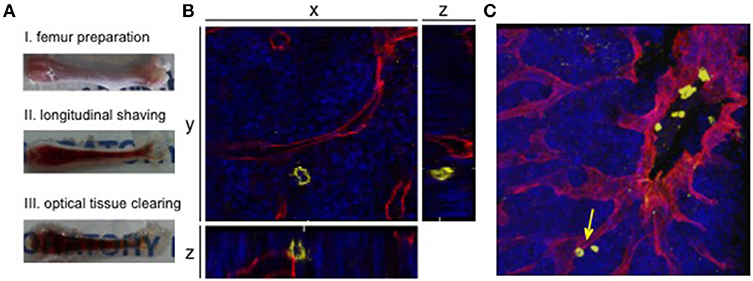
Figure 2. 3D confocal visualization of leukemic cells in the murine bone marrow. (A) Whole-mount bone preparation of fixed cryopreserved femur. (I) Bone was cleaned from tissue for fixation and cryopreservation. Next it was shaved longitudinally from both sides on a cryostat until bone marrow was fully visible (II). After optical clearing tissue appeared transparent (III). (B) Orthogonal sections of a z-stack show the relevance of 3D visualization for niche analyses. The 2D plane shows a leukemic cell (CD45, yellow) without direct contact to a sinusoidal blood vessel (endoglin, red). Orthogonal sections show an adjacent blood vessel below the AML cell. DAPI signals mark the bone marrow tissue (blue). (C) Corresponding 3D z-stack image of bone marrow vasculature with engrafted human AML cells. Cell from orthogonal section is marked with an arrow.
Scanning electron microscopy (SEM) is the most high-resolution imaging technique to visualize surfaces of cells and tissues in a high-resolution manner independent of available antibodies or transgenic mouse lines. The resolution is greater than with a light microscope but information are limited to the two-dimensional surface. However, there are several SEM techniques that can obtain 3D information about a sample, enabling the reconstruction of tissue structures. Serial block face (SBF-SEM) and focused ion beam scanning electron microscopy (FIB-SEM) both allow serial sections by ablating the specimen either with a diamond knife or a beam of ions with subsequent alignment of the data to allow reconstruction of a three-dimensional volume. Within this image individual structure can be segmented and qualitative and quantitative information can be obtained (81). FIB-SEM was used to characterize bone marrow adipocytes revealing highly dynamic cells with manifold interactions to its microenvironment (82). Identification of the cell of interest within electron microscopy is limiting this technique since it is solely dependent on visible structure hallmarks. This presents a particular challenge for xenografted AML cells, where the discrimination from to normal murine precursor cells is highly difficult.
In general, whole-mount imaging and computational modeling only allows visualization of snapshots of dynamic processes. The development of intravital microscopy solves this issue and has been used to study the behavior of HSCs and LICs in real time in the bone marrow. In 2005 Sipkins et al. presented two-photon microscopy data about the homing and engraftment of leukemic cells into the mouse calvarium bone marrow and described a specialized endothelial niche for acute lymphoblastic leukemia cells (15). This approach is minimally invasive and allows the visualization of living cells inside the bone marrow over hours. The development of a mouse calvarial window made of cover glass enables imaging even over weeks in multiple imaging sessions (83). Several groups have used and improved two-photon intravital imaging to study spatiotemporal distribution of cells in the calvarium and in the murine femur, but the latter remains highly invasive (84–89). The downside of intravital microscopy is a dependence on transplantation and transgenic models, as well as on injectable dyes (90). Since in vivo antibody staining is not sufficient for direct imaging, cells of interest have to be labeled either ex vivo before transplantation or transgenic reporter mouse strains have to be generated. In particular, ex vivo treatments can influence a broad range of cellular functions like localization, division and cell-cell interactions. It is for this reason that intravital microscopy has thusfar been unable to completely replace ex vivo confocal 3D imaging and vice versa. The future will show how well-researchers will be able to combine these techniques to visualize both a detailed and dynamic picture of the bone marrow niche of hematopoietic and leukemic stem cells.
In vitro Mimicking of the Bone Marrow Niche
Visualizing cells within their niche in vivo is a sophisticated method to study leukemia-niche interactions. However, xenograft models never fully recapitulate the physiologic niche, since human cell engraftment requires the depletion of the immune system. Mice need genetic modification to deplete T-, B- and natural killer cells, and especially for AML a humanized microenvironment is necessary for the engraftment and development of the disease in vivo. But genetic engineering of mice is time and resource consuming and alternative models to study niche related pathways are required. One possibility to generate human-mouse chimeric bone marrow tissues is the usage of ossicle systems (91). These bony structures formed in ectopic regions show formation of bone with mature, vascularized bone marrow tissue. Since scaffold materials can be seeded with mesenchymal stromal cells or/and endothelial cells before implantation, ossicles are a useful tool to test genetic modification of niche cells in regard of stem cell and leukemic engraftment. The newest developments in the field of 3D printing have improved the types of ossicles that can be generated, including variable pore size and structures, usage of biomaterials or even the incorporation of growth factors into the scaffold (92). However, the native bone marrow is a highly complex tissue and these engineered scaffolds will still represent only a model with specific limitations.
Nowadays, there is a desire to reduce mouse models and replace them by reliable in vitro assays. But classical 2D coculture models are not sufficient to mimic the in vivo situation of the bone marrow niche. In addition to solid compounds, the bone marrow is regulated by the concentration of soluble factors, oxygen levels, availability of nutrients and even the flow of blood and liquids inside the niche, which applies mechanical stress. Utilizing biomaterials or bioreactors to generate 3D cell cultures will allow modeling the niche situation more closely. Either inorganic biomaterials like hydroxyapatite or hydrogels are successfully used to culture HSCs. State-of-the-art techniques like perfused 3D bone marrow analogs by Rödling et al., or the sophisticated bone marrow-on-a-chip by Torisawa et al. are probably the most advanced in vitro models to date (93, 94). Bray et al. used a 3D tri-culture of endothelial, mesenchymal stromal and AML cells to study drug resistance in vitro. Interestingly, they were able to show a greater drug resistance in the 3D model compared to a 2D coculture, underlining the relevance of niche mimicking models (95). All of these models provide an important ex vivo alternative to mouse models. Genetic and pharmacologic manipulations, as well as stimulation with cytokines can be performed in a direct and multimodal manner, opening the possibility of personalized drug response screens for therapy prediction.
Vascularity in the Bone Marrow
The development of microscopy techniques to visualize the bone marrow niche under leukemic engraftment revealed not only changes on the molecular level but also in the overall structure, especially for the vascular system. In general, the bone marrow vascularity is responsible for the nutrient and metabolite turnover, oxygenation of the tissue and the ingress and egress of cells, like freshly differentiated immune cells. Blood vessels are made up of several different cell types. The inner layer is composed of endothelial cells (ECs), which is covered by perivascular cells (pericytes). These pericytes are embedded in the subendothelial basement membrane and connect ECs with smooth muscle cells, which generally cover large vessels, like arteries and veins (96). Remarkably, bone marrow vessels are mainly formed by a sinusoidal network consisting of a single layer of ECs (97). As in other organs, the vasculature of the bone marrow is organized in a specific order. The arteries are longitudinally aligned along the diaphysis of long bones and infiltrate into the bone marrow via branching to small arteriols and finally to a capillary network with larger diameter. To exit the bone marrow, capillaries drain together into a large central vein, which passes the hard, calcified bone matrix going back to the periphery (98).
Adams and coworkers defined two subtypes of bone capillaries, so called H and L that are distinguishable by function, surface marker expression, and structure (96, 99). Type H capillaries express high levels of CD31 and endomucin and are located in the metaphysis, which is the region of the bone containing the avascular growth plate. They are surrounded by osteoprogenitors, connected to arteriols and play an important role in angiogenesis. In contrast, type L vessels express only low levels of CD31 and endomucin and lack arteriolar connection and osteoprogenitor association but are surrounded by leptin receptor (LEPR)+ and CAR cells. They form the dense and highly branched capillary network of sinusoids in the bone marrow cavity that is known to regulate the HSC compartment (13, 18). Blood flows first through the arteries into type H capillaries before it enters the more permeable sinusoidal type L network at the interface between metaphysis and diaphysis. This leads to metabolically distinct environments, with a well-oxygenated metaphysis and a highly hypoxic diaphysis, due to the lack of direct arterial supply (99, 100).
Several studies showed that hypoxia is a key regulator of the niche-mediated dormancy and maintenance of HSCs (101, 102). Low oxygen supply has direct effects on the metabolism of a cell, changing it from oxidative phosphorylation to cytoplasmic glycolysis (103). The latter is less efficient in overall energy production and pushes cells into energy preserving dormancy. A benefit of shutting down oxidative phosphorylation are the reduced rates of intracellular reactive oxygen species (ROS) production (104). ROS management is crucial to keep HSCs in a quiescent state, since excessive levels of ROS can induce oxidative stress leading to protein, lipid, and DNA damage. Therefore, it is not surprising that high levels of ROS are linked to AML development by inducing genomic instability leading to chromosomal deletions or translocations (105). Itkins et al. showed that permeable sinusoids display higher ROS levels, lower blood flow, and shear rates compared to less permeable arterial blood vessels (106). Hematopoietic cell rolling and adhesion events as well as transendothelial migration of mature leukocytes and immature HSC occurred exclusively via sinusoids. Most recently, Passaro and colleagues showed that engraftment of patient-derived xenograft AML samples (PDXs) into mice increased the vascular permeability compared to control mice engrafted with healthy human HSCs (85). Transcriptome analysis of vascular ECs upon human AML engraftment showed that the NOX4-NOS3-axis (NADPH oxidase-nitric oxide synthase 3) is highly activated, a pathway that is known to act in response to stress induced by hypoxic conditions and to induce vascular leakiness. Accordingly, the bone marrow of xenografted mice showed increased levels of NO (nitric oxide), an inducer of vascular permeability; keeping in mind that increased vascular leakiness is associated to poor drug delivery, favoring resistance to therapy and relapse (107). Remarkably, the pharmacological inhibition of NO production in combination with cytarabine reduced AML progression and restored normal vasculature in PDX models, pointing toward a potential clinical strategy for NOS inhibition in the bone marrow vascular niche in AML. Taken together, these observations indicate that the bone marrow contains several distinct vascular sites, with defined functions and structures that regulate the fate of HSC and might have specific functions for leukemia inducing cells.
The Vessels and Angiogenesis in AML
Angiogenesis is the process of blood vessel development from pre-existing ones and is an absolute requirement for the viability and growth of solid tumors (108). The tumor-angiogenic process is mostly initiated by pro-angiogenic factors derived from tumor cells, which activate the initiation of irregular and uncontrolled vascular growth (109). Newly formed tumor vessels appear abnormal and leaky, affecting the local blood flow, metabolite exchange, and oxygenation. These aberrant structures influence drug delivery, tumor growth, metastatic potentials and ultimately the survival probability for a patient (110). Since AML is considered a liquid tumor without a compact structure, angiogenesis in AML was initially underestimated. De facto, leukemia cells, like tumor cells, are highly dependent on angiogenesis in the bone marrow and AML is associated with an increase in bone marrow microvascular density (MVD) (111, 112). More recently, clinical data has proven that bone marrow biopsies of AML patients show an increased number of sinusoidal blood vessels in comparison to healthy individuals that corrects to normal after leukemia remission. Additionally, the degree of MVD can be used as a prognostic marker, since higher MVD correlates with an increased risk of relapse and a shorter overall survival (113).
Antiangiogenic Therapy of AML
Since AML is strongly connected to the vascular niche and remodeling of bone marrow vascularization, antiangiogenic therapy seems to be a logical clinical strategy. To date, several drugs with antiangiogenic properties had been tested in clinical studies, but clinical outcomes have been disappointing (29, 114). One prominent example is bevacizumab, a monoclonal anti-VEGF antibody that has the potential to decrease VEGF levels in bone marrow. But clinical studies showed that AML progression remain unchanged (115). Only the combination of bevacizumab with standard induction chemotherapy showed a slight improvement in patient outcome (116). Since VEGF signals via receptor tyrosine kinases (RTKs) with the intracellular signaling cascade, TKIs (tyrosine kinase inhibitors) are a family of small molecules that are under clinical investigation for AML treatment with relevance to angiogenesis (e.g., semaxanib, sunitinib, or sorafenib). Interestingly, some of these drugs block other RTKs as well, broadening the spectrum of their efficacy. For example semaxanib was one of the first TKIs in a clinical trial that targets VEGFRs, but also inhibited stem cell factor receptor (KIT) and FLT3 (FMS-like tyrosine kinase 3) (117). Application of semaxanib was generally well-tolerated, but the effect on leukemic progression was absent, so further clinical development was discontinued. Comparable to semaxanib, sunitinib binds to a broad range of RTKs and is approved for the treatment of gastrointestinal stromal tumors. However, for AML treatment, sunitinib showed only modest clinical activity (118). Sorafenib is a potent inhibitor of VEGFR, KIT, PDGFR (platelet-derived growth factor receptor) and FLT3; the latter of which is mutated in 25–30% of AML cases, causing a constitutively active variant and promoting leukemogenesis (119). Clinical data showed that sorafenib reduced leukemic growth from AML patients with FLT3 mutations but not in patients without mutations. Beside its specific effect in AML carrying FLT3 mutations, clinical activity is only modest and in combination with standard chemotherapy sorafenib showed no additional benefit. Lenalidomide is another example of a VEGF inhibitor. This thalidomide derivative is an antiangiogenic and immunomodulatory drug with a decreasing effect on VEGF expression. However, clinical relevance for AML treatment is controversially discussed (120, 121).
In addition to VEGF, another major class of endothelium-specific growth factors includes the members of the angiopoietin family (Ang1, 2, 3, and 4). Interestingly, high expression of the pro-angiogenic factor Ang2 is an independent prognostic factor for overall survival in AML (122). However, clinical studies with trebananib, a neutralizing peptibody against Ang1 and Ang2 showed only a limited clinical efficacy (123). Overall, clinical data indicates that monotherapy with antiangiogenic drugs lack the efficacy to treat aggressive leukemia's, only the combination of antiangiogenic drugs with standard chemotherapy is likely to have a benefit for patient's outcome and need more clinical investigations.
An alternative approach may be to specifically harm the perivascular niche and the AML support by ECs with vascular disrupting agents (124). Combrestatins cause a breakdown of the vascular architecture by targeting the microtubule of rapidly proliferating EC. Clinical trials are ongoing to elucidate the effect of vascular disrupting agents on the progression of AML (125). In contrast, Duarte et al. found that the loss of bone marrow blood vessels can be associated with increased chemotherapy resistance of AML cells (84). In detail, AML progression leads to differential remodeling of the vasculature into a leukemia-preferential microenvironment. Dependent on their anatomical localization in the bone marrow, blood vessels were either obliterated (endosteal type H capillaries) or rescued but modified (central marrow vessels). Endosteal located AML cells are enriched in inflammatory and TNF gene signatures and express higher levels of CXCL2. Both, TNF and CXCL2 are known factors for vascular destruction and angiogenesis inhibition (126, 127). The consequences of this remodeling are the loss of HSCs specifically from endosteal areas and survival of leukemia cells under chemotherapy. Interestingly, induction of Notch signaling in ECs leads to increased numbers of endosteal vessels and furthermore to a decrease of chemotherapy resistant AML cells in the bone marrow, leading to the hypothesis that rescuing endosteal vessels before chemotherapy may actually improve the outcome and maybe the overall survival of patients, although clinical studies are needed to confirm this mechanism (84).
Other Therapeutic Targets in the Niche
Besides antiantiogenic approaches, other known niche components are under evaluation for targeted therapy. Developing CXCR4 inhibitors has made the most progress. The rational behind the blocking of this signaling pathway is to mobilize AML cells out of their protective niche to make them available for chemotherapy. Strong clinical evidence was shown in phase I studies for plerixafor (AMD3100), a small molecule inhibitor of CXCR4 that increased the rate of complete remissions in combination with cytarabine and daunorubicin but adverse events were frequent (128). Other CXCR4 inhibitors like ulocuplumab and BL-8040 present additional pro-apoptotic effects on AML cells and showed in combination with induction therapy accelerated mobilization and improved outcome for the patients (129). Finally, CX-01, a anticoagulant, binds CXCL12 with a high affinity, effectively blocking CXCL12/CXCR4 signaling. Combination with intensive therapy was well-tolerated by patients and showed enhanced treatment efficacy (130). With such promising preliminary results using CXCR4 inhibitors further clinical trials will likely follow to further elucidate the therapeutic potential of this axis.
The interaction between VLA-4 on AML cells and VCAM-1 on mesenchymal stromal cells represent another promising candidate for targeted therapy. Natalizumab was the first humanized VLA-4 monoclonal antibody tested in a xenograft AML model. Despite its beneficial effects on overall survival, its utility is limited since it can induce leucoencephalopathy (131, 132). Another pre-clinical VLA-4 inhibitor is AS101 which decreases the pro-survival PI3K/Akt/Bcl2 signaling to sensitize AML cells to chemotherapy. A mouse xenograft AML model showed that AS101 can abrogate drug resistance of leukemic cells and prolong survival in mice after chemotherapy (133).
Last, the therapeutic potential of the inhibition of E-selecting binding to CD44 should be noted. H90, an activating monoclonal antibody against CD44 and GMI-1271, a specific small molecule inhibitor of E-selectin, both showed a reduction of the leukemic burden in xenograft AML models (58, 134). These preclinical results would justify further explorations to elucidate the therapeutic potential of these inhibitors for AML treatment in combination with chemotherapy. The availability of a FDA-approved CD44 monoclonal antibody, bivatuzumab, that is currently used for clinical trials in solid tumors could potentially allow a novel clinical trial in AML patients.
Immunotherapy
The immunosuppressive role of AML cells has a strong impact on chemotherapy survival and the causative mechanisms present a promising target for therapy. One approach targets the immune checkpoint molecules expressed by AML cells. Several immune checkpoints are initiated by ligand-receptor-interactions and therefore can be blocked with antibodies or blocking fragments of the receptor or ligand (135). Unfortunately, an initial clinical trial with the monoclonal PD-1 antibody CT-011 showed disappointing results for AML patients (136). Another approach for immunotherapy is the treatment with bispecific T-cell engager (BiTE®) constructs. These recombinant antibodies include two variable domains to bind to two different antigens at the same time (137). One domain binds CD3, a component of the T-cell receptor complex, to connect T-cells to tumor cells initiating the MHC-I independent cytotoxic synapse leading to the lysis of the tumor cell. The first BiTE® for clinical application is blinatumomab, a combination of CD3 and CD19 for therapy of a specific form of acute lymphoid leukemia. For AML therapy AMG 330, a CD33/CD3-BiTE® is in a clinical phase (138). Interestingly, although being independent of MHC-I-interactions, the effectivity of AMG 330 has been shown to be dependent of costimulatory and coinhibitory T cell ligands (139). Moreover, treatment with AMG 330 led to an upregulation of the immune checkpoint molecule PD-L1 on AML cells and blocking of the PD-1/PD-L1 interaction enhanced AMG 330 mediated cell lysis (140). In this regard, the TIGIT-PVR/PVRL2 immune checkpoint axis recently emerged as a promising target for AML therapy (141). Our lab could thereby show that the blocking of the inhibitory immunoreceptor TIGIT or the corresponding ligands poliovirus receptor (PVR, CD155) and poliovirus receptor-related 2 (PVRL2, CD112) augment the antileukemic effects of AMG 330 (141).
Lastly, we want to mention the promising approaches with autologous or allogeneic T cells engineered with synthetic chimeric antigen receptors (CARs) (142). The artificial T cell receptors are designed to specifically target tumor cells, with binding inducing T-cell activation, differentiation, inflammation, and targeted cytolytic killing of the target cancer cells. For acute lymphoblastic leukemia (ALL) two CD19 CAR T cell products, tisagenlecleucel and axicabtagene ciloleucel, were approved for clinical usage in the United States. Unfortunately, AML cells lack ideal target antigens, since most surface proteins are expressed on normal myeloid precursor cells as well, and unlike B-cells patients cannot survive without functional myeloid cells. But given the remarkable cure rates for relapsed B-ALL, several preclinical trials are ongoing to identify a potent target for CAR T cell therapy of AML. Details were reviewed recently by Tasian (142). Here, we want to highlight CLL-1 (C-type lectin-like molecule-1) that is expressed on the majority of AML blasts and on leukemia initiating cells but not on hematopoietic stem cells (143). Recently Dr. Fang Liu (Chengdu Military Hospital, Chengdu, China) demonstrated that simultaneously targeting of multiple antigens like CD33 and CLL-1 is an effective strategy to eliminate AML. However, disregarding the promising results of immunotherapy for AML patients, only limited information are present about the effects of immune cells in the bone marrow. Future work will be needed to elucidate how immune cells reach the AML cells in their niches and how the microenvironment influences the outcome of the therapy. Visualization of the effects of immunotherapy to the bone marrow niche is a central task to understand and improve this approach of therapy.
Conclusions
There is an ever-growing number of targeted therapeutic strategies to treat acute myeloid leukemia, yet patient outcome remains poor and clinicians have an urgent need to receive new treatment strategies for patients. This result is likely due to the highly heterogeneous and highly polyclonal nature of AML and approaches that target specific mutations in AML cells may result in the eradication only of single subclones and therefor are inadequate to rid patients of the disease. Because complex interactions with the bone marrow microenvironment influence the survival and progression of AML, research is now focused on deciphering the bone marrow niche composition to identify new drugable targets. These interactions are less clone-specific and therefore represent a promising strategy to overcome the obstacles of individual cell heterogeneity. Furthermore, residual LICs are thought to drive disease relapse and targeting their sheltering niche might offer a means to improve eradication of AML. Unfortunately, blocking signaling pathways also harbors the risk of unpredictable side effects and toxicity. Most pre-clinical research is done in animal models to study drug effects on disease progression, and there are important limitations in translation of animal model results to human patients. Thus, researchers should seek to find good in vitro models that predict clinical drug responses. There is a strong demand for new agents with good tolerance in a broad range of AML patients, and increased efficacy relative to conventional chemotherapy. Any possibility to replace one or more components of treatment regimens in frail patients to reduce long-term effects on the maintenance of healthy HSCs would have significant clinical benefit. Technical developments of the past few years have yielded significant progress for understanding the physiological constitution of normal HSC niches and the malignant alterations that drive AML progression. However, the regulation of AML cells in their niche is highly complex and we have just started to understand the tremendous potential of targeting the bone marrow microenvironment to eradicate AML cells. Ongoing clinical studies and upcoming research results will point out new avenues of treatments, giving the hope that AML will 1 day become a treatable disease.
Author Contributions
LB wrote the manuscript. WF and JW contributed to the writing and editing of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all our collaborators for support. Furthermore we thank Scott McComb for proofreading. Schematic drawings in Figure 1 were obtained from Motifolio.
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
2. Komanduri KV, Levine RL. Diagnosis and therapy of acute myeloid leukemia in the Era of molecular risk stratification. Annu Rev Med. (2016) 67:59–72. doi: 10.1146/annurev-med-051914-021329
3. Sun Y, Chen B-R, Deshpande A. Epigenetic regulators in the development, maintenance, and therapeutic targeting of acute myeloid leukemia. Front Oncol. (2018) 8:41. doi: 10.3389/fonc.2018.00041
4. Bertoli S, Tavitian S, Huynh A, Borel C, Guenounou S, Luquet I, et al. Improved outcome for AML patients over the years 2000–2014. Blood Cancer J. (2017) 7:635. doi: 10.1038/s41408-017-0011-1
5. Büchner T, Schlenk RF, Schaich M, Döhner K, Krahl R, Krauter J, et al. Acute myeloid leukemia (AML): different treatment strategies versus a common standard arm—combined prospective analysis by the German AML intergroup. J Clin Oncol. (2012) 30:3604–10. doi: 10.1200/JCO.2012.42.2907
6. Klepin HD. Elderly acute myeloid leukemia: assessing risk. Curr Hematol Malig Rep. (2015) 10:118–25. doi: 10.1007/s11899-015-0257-2
7. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481:506–10. doi: 10.1038/nature10738
8. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer (2009) 9:665–74. doi: 10.1038/nrc2714
9. Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. (2011) 12:643–55. doi: 10.1038/nrm3184
10. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505:327–34. doi: 10.1038/nature12984
11. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature (2003) 425:841–6. doi: 10.1038/nature02040
12. Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466:829–34. doi: 10.1038/nature09262
13. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature (2012) 481:457–62. doi: 10.1038/nature10783
14. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood (2010) 116:4815–28. doi: 10.1182/blood-2009-11-253534
15. Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature (2005) 435:969–73. doi: 10.1038/nature03703
16. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell (2011) 147:1146–58. doi: 10.1016/j.cell.2011.09.053
17. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell (2014) 15:365–75. doi: 10.1016/j.stem.2014.06.020
18. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
19. Zhou BO, Ding L, Morrison SJ. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. Elife (2015) 4:e05521. doi: 10.7554/eLife.05521
20. Otten J, Bokemeyer C, Fiedler W. Tgf-Beta superfamily receptors-targets for antiangiogenic therapy? J Oncol. (2010) 2010:317068. doi: 10.1155/2010/317068
21. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood (2009) 113:1250–6. doi: 10.1182/blood-2008-04-146480
22. Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell (2008) 2:274–83. doi: 10.1016/j.stem.2008.01.003
23. Nilsson SK, Johnston HM, Coverdale JA. Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood (2001) 97:2293–9. doi: 10.1182/blood.V97.8.2293
24. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell (2005) 121:1109–21. doi: 10.1016/j.cell.2005.05.026
25. Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. quantitative imaging of hematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. (2013) 15:533–43. doi: 10.1038/ncb2730
26. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135:1118–29. doi: 10.1016/j.cell.2008.10.048
27. Ellis SL, Grassinger J, Jones A, Borg J, Camenisch T, Haylock D, et al. The relationship between bone, hemopoietic stem cells, and vasculature. Blood (2011) 118:1516–24. doi: 10.1182/blood-2010-08-303800
28. Draenert K, Draenert Y. The vascular system of bone marrow. Scan Elect. Microsc. (1980) 4:113–22.
29. Cogle CR, Bosse RC, Brewer T, Migdady Y, Shirzad R, Kampen KR, et al. Acute myeloid leukemia in the vascular niche. Cancer Lett. (2016) 380:552–60. doi: 10.1016/j.canlet.2015.05.007
30. Huntly BJP, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer (2005) 5:311–21. doi: 10.1038/nrc1592
31. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
32. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367:645–8. doi: 10.1038/367645a0
33. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia (2000) 14:1777–84. doi: 10.1038/sj.leu.2401903
34. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA. (2007) 104:11008–13. doi: 10.1073/pnas.0704271104
35. Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood (2010) 115:1976–84. doi: 10.1182/blood-2009-02-206565
36. Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood (2009) 114:1150–7. doi: 10.1182/blood-2009-01-202606
37. Blau O, Hofmann W-K, Baldus CD, Thiel G, Serbent V, Schümann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol. (2007) 35:221–9. doi: 10.1016/j.exphem.2006.10.012
38. Cogle CR, Goldman DC, Madlambayan GJ, Leon RP, Masri AA, Clark HA, et al. Functional Integration of Acute Myeloid Leukemia into the Vascular Niche. Leukemia (2014) 28:1978–87. doi: 10.1038/leu.2014.109
39. Han Y, Wang X, Wang B, Jiang G. The progress of angiogenic factors in the development of leukemia's. Intractable Rare Dis Res. (2016) 5:6–16. doi: 10.5582/irdr.2015.01048
40. Fiedler W, Graeven U, Ergün S, Verago S, Kilic N, Stockschläder M, et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood (1997) 89:1870–5.
41. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight (2017) 2:e90036. doi: 10.1172/jci.insight.90036
42. Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood (2012) 119:540–50. doi: 10.1182/blood-2011-04-348151
43. Horowitz MC, Berry R, Holtrup B, Sebo Z, Nelson T, Fretz JA, et al. Bone marrow adipocytes. Adipocyte (2017) 6:193–204. doi: 10.1080/21623945.2017.1367881
44. Naveiras O, Nardi V, Wenzel PL, Fahey F, Daley GQ. Bone marrow adipocytes as negative regulators of the hematopoietic microenvironment. Nature (2009) 460:259–63. doi: 10.1038/nature08099
45. Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. (2017) 19:1336–47. doi: 10.1038/ncb3625
46. Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood (2017) 129:1320–32. doi: 10.1182/blood-2016-08-734798
47. Sheng X, Tucci J, Parmentier J-H, Ji L, Behan JW, Heisterkamp N, et al. Adipocytes cause leukemia cell resistance to daunorubicin via oxidative stress response. Oncotarget (2016) 7:73147–59. doi: 10.18632/oncotarget.12246
48. Shirai K, Sera Y, Bulkeley W, Mehrotra M, Moussa O, LaRue AC, et al. Hematopoietic stem cell origin of human fibroblasts: cell culture studies of female recipients of gender mismatch stem cell transplantation and patients with chronic myelogenous leukemia. Exp Hematol. (2009) 37:1464–71. doi: 10.1016/j.exphem.2009.09.008
49. Zhai Y, Zhang J, Wang H, Lu W, Liu S, Yu Y, et al. Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J Exp Clin Cancer Res. (2016) 35:147. doi: 10.1186/s13046-016-0405-0
50. Isidori A, Salvestrini V, Ciciarello M, Loscocco F, Visani G, Parisi S, et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev Hematol. (2014) 7:807–18. doi: 10.1586/17474086.2014.958464
51. Janakiram M, Pareek V, Cheng H, Narasimhulu DM, Zang X. Immune checkpoint blockade in human cancer therapy: lung cancer and hematologic malignancies. Immunotherapy (2016) 8:809–19. doi: 10.2217/imt-2016-0001
52. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
53. Fibbe WE, Nauta AJ, Roelofs H. Modulation of immune responses by mesenchymal stem cells. Ann N Y Acad Sci. (2007) 1106:272–8. doi: 10.1196/annals.1392.025
54. Riether C, Schürch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ (2015) 22:187–98. doi: 10.1038/cdd.2014.89
55. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood (2014) 123:2691–702. doi: 10.1182/blood-2013-06-511527
56. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. (2003) 9:1158–65. doi: 10.1038/nm909
57. Winkler IG, Barbier V, Pattabiraman DR, Gonda TJ, Magnani JL, Levesque J-P. Vascular niche e-selectin protects acute myeloid leukaemia stem cells from chemotherapy. Blood (2014) 124:620.
58. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. (2006) 12:1167–74. doi: 10.1038/nm1483
59. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell (2013) 13:285–99. doi: 10.1016/j.stem.2013.06.009
60. Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee S-U, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell (2012) 21:577–92. doi: 10.1016/j.ccr.2012.02.018
61. Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, et al. Differential regulation of myeloid leukemia's by the bone marrow microenvironment. Nat Med. (2013) 19:1513–7. doi: 10.1038/nm.3364
62. Peled A, Tavor S. Role of CXCR4 in the pathogenesis of acute myeloid leukemia. Theranostics (2013) 3:34–9. doi: 10.7150/thno.5150
63. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia (2018) 32:575–87. doi: 10.1038/leu.2017.259
64. Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood (2015) 126:2404–14. doi: 10.1182/blood-2015-03-634238
65. Avagyan S, Zon LI. Fish to Learn: Insights into blood development and blood disorders from zebrafish hematopoiesis. Hum Gene Ther. (2016) 27:287–94. doi: 10.1089/hum.2016.024
66. Tamplin OJ, Durand EM, Carr LA, Childs SJ, Hagedorn EJ, Li P, et al. Hematopoietic stem cell arrival triggers dynamic remodeling of the perivascular niche. Cell (2015) 160:241–52. doi: 10.1016/j.cell.2014.12.032
67. Jing L, Zon LI. Zebrafish as a model for normal and malignant hematopoiesis. Dis Model Mech. (2011) 4:433–8. doi: 10.1242/dmm.006791
68. Walsh NC, Kenney LL, Jangalwe S, Aryee K-E, Greiner DL, Brehm MA, et al. Humanized Mouse Models of Clinical Disease. Ann Rev Pathol. (2017) 12:187–215. doi: 10.1146/annurev-pathol-052016-100332
69. Lysenko V, McHugh D, Behrmann L, Rochat M-A, Wilk CM, Kovtonyuk L, et al. Humanised mouse models for haematopoiesis and infectious diseases. Swiss Med Wkly. (2017) 147:w14516. doi: 10.4414/smw.2017.14516
70. Mambet C, Chivu-Economescu M, Matei L, Necula LG, Dragu DL, Bleotu C, et al. Murine models based on acute myeloid leukemia-initiating stem cells xenografting. World J Stem Cells (2018) 10:57–65. doi: 10.4252/wjsc.v10.i6.57
71. Kawamoto T, Kawamoto K. Preparation of thin frozen sections from nonfixed and undecalcified hard tissues using Kawamot's film method (2012). Methods Mol Biol. (2014) 1130:149–64. doi: 10.1007/978-1-62703-989-5_11
72. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. (2007) 25:1315–21. doi: 10.1038/nbt1350
73. Harnett MM. Laser scanning cytometry: understanding the immune system in situ. Nat Rev Immunol. (2007) 7:897. doi: 10.1038/nri2188
74. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature (2013) 502:637–43. doi: 10.1038/nature12612
75. Richardson DS, Lichtman JW. Clarifying tissue clearing. Cell (2015) 162:246–57. doi: 10.1016/j.cell.2015.06.067
76. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature (2015) 526:126–30. doi: 10.1038/nature15250
77. Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat Biotechnol. (2017) 35:1202–10. doi: 10.1038/nbt.4006
78. Stegner D, vanEeuwijk JMM, Angay O, Gorelashvili MG, Semeniak D, Pinnecker J, et al. Thrombopoiesis is spatially regulated by the bone marrow vasculature. Nat Commun. (2017) 8:127. doi: 10.1038/s41467-017-00201-7
79. Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Multicolor quantitative confocal imaging cytometry. Nat Methods (2018) 15:39–46. doi: 10.1038/nmeth.4503
80. Gomariz A, Helbling PM, Isringhausen S, Suessbier U, Becker A, Boss A, et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat Commun. (2018) 9:2532. doi: 10.1038/s41467-018-04770-z
81. Ichimura K, Miyazaki N, Sadayama S, Murata K, Koike M, Nakamura K, et al. Three-dimensional architecture of podocytes revealed by block-face scanning electron microscopy. Sci Rep. (2015) 5:8993. doi: 10.1038/srep08993
82. Robles H, Park S, Joens MS, Fitzpatrick JAJ, Craft CS, Scheller EL. Characterization of the bone marrow adipocyte niche with three-dimensional electron microscopy. Bone (2018). doi: 10.1016/j.bone.2018.01.020. [Epub ahead of print].
83. Le V-H, Lee S, Lee S, Wang T, Hyuk Jang W, Yoon Y, et al. In vivo longitudinal visualization of bone marrow engraftment process in mouse calvaria using two-photon microscopy. Sci Rep. (2017) 7:44097. doi: 10.1038/srep44097
84. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell (2018) 22:64–77.e6. doi: 10.1016/j.stem.2017.11.006
85. Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell (2017) 32:324–41.e6. doi: 10.1016/j.ccell.2017.08.001
86. Lo Celso C, Wu JW, Lin CP. In vivo imaging of hematopoietic stem cells and their microenvironment. J Biophoton. (2009) 2:619–31. doi: 10.1002/jbio.200910072
87. Lo Celso C, Lin CP, Scadden DT. In vivo imaging of transplanted hematopoietic stem and progenitor cells in mouse calvarium bone marrow. Nat Protoc. (2011) 6:1–14. doi: 10.1038/nprot.2010.168
88. Koechlein CS, Harris JR, Lee TK, Weeks J, Fox RG, Zimdahl B, et al. High-resolution imaging and computational analysis of haematopoietic cell dynamics in vivo. Nat Commun. (2016) 7:12169. doi: 10.1038/ncomms12169
89. Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science (2008) 322:1861–5. doi: 10.1126/science.1164390
90. Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell (2013) 13:520–33. doi: 10.1016/j.stem.2013.10.010
91. Abarrategi A, Mian SA, Passaro D, Rouault-Pierre K, Grey W, Bonnet D. Modeling the human bone marrow niche in mice: from host bone marrow engraftment to bioengineering approaches. J Exp Med. (2018) 215:729–43. doi: 10.1084/jem.20172139
92. Zhou X, Castro NJ, Zhu W, Cui H, Aliabouzar M, Sarkar K, et al. Improved human bone marrow mesenchymal stem cell osteogenesis in 3D bioprinted tissue scaffolds with low intensity pulsed ultrasound stimulation. Sci Rep. (2016) 6:32876. doi: 10.1038/srep32876
93. Rödling L, Schwedhelm I, Kraus S, Bieback K, Hansmann J, Lee-Thedieck C. 3D models of the hematopoietic stem cell niche under steady-state and active conditions. Sci Rep. (2017) 7:4625. doi: 10.1038/s41598-017-04808-0
94. Torisawa Y, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, et al. Bone marrow–on–a–chip replicates hematopoietic niche physiology in vitro. Nat Methods (2014) 11:663–9. doi: 10.1038/nmeth.2938
95. Bray LJ, Binner M, Körner Y, Bonin M von, Bornhäuser M, Werner C. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica (2017) 102:1215–26. doi: 10.3324/haematol.2016.157883
96. Sivaraj KK, Adams RH. Blood vessel formation and function in bone. Development (2016) 143:2706–15. doi: 10.1242/dev.136861
97. Tavassoli M. Structure and function of sinusoidal endothelium of bone marrow. Prog Clin Biol Res. (1981) 59B:249–56.
98. Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature (2016) 532:380–384. doi: 10.1038/nature17638
99. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature (2014) 507:323–8. doi: 10.1038/nature13145
100. Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature (2014) 507:376–80. doi: 10.1038/nature13146
101. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell (2010) 7:391–402. doi: 10.1016/j.stem.2010.06.020
102. Eliasson P, Jönsson J-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J Cell Physiol (2010) 222:17–22.
104. Zhang CC, Sadek HA. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid Redox Signal. (2014) 20:1891–901. doi: 10.1089/ars.2012.5019
105. Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. (2008) 270:1–9. doi: 10.1016/j.canlet.2008.03.036
106. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) 532:323–8. doi: 10.1038/nature17624
107. Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. (2013) 3:211. doi: 10.3389/fonc.2013.00211
108. Falcon BL, Chintharlapalli S, Uhlik MT, Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol Ther. (2016) 164:204–25. doi: 10.1016/j.pharmthera.2016.06.001
109. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473:298–307. doi: 10.1038/nature10144
110. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307:58–62. doi: 10.1126/science.1104819
111. Padró T, Ruiz S, Bieker R, Bürger H, Steins M, Kienast J, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood (2000) 95:2637–44.
112. Weidenaar AC, ter Elst A, Koopmans-Klein G, Rosati S, den Dunnen WFA, Meeuwsen-de Boer T, et al. High acute myeloid leukemia derived VEGFA levels are associated with a specific vascular morphology in the leukemic bone marrow. Cell Oncol. (Dordr) (2011) 34:289–96. doi: 10.1007/s13402-011-0017-9
113. Kuzu I, Beksac M, Arat M, Celebi H, Elhan AH, Erekul S. Bone marrow microvessel density (MVD) in adult acute myeloid leukemia (AML): therapy induced changes and effects on survival. Leuk Lymphoma (2004) 45:1185–90. doi: 10.1080/1042819032000159915
114. Wellbrock J, Fiedler W. Clinical experience with antiangiogenic therapy in leukemia. Curr Cancer Drug Targets (2011) 11:1053–68. doi: 10.2174/156800911798073078
115. Zahiragic L, Schliemann C, Bieker R, Thoennissen NH, Burow K, Kramer C, et al. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia (2007) 21:1310–2. doi: 10.1038/sj.leu.2404632
116. Karp JE, Gojo I, Pili R, Gocke CD, Greer J, Guo C, et al. Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemia's: therapy with sequential 1-beta-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res. (2004) 10:3577–85. doi: 10.1158/1078-0432.CCR-03-0627
117. Fiedler W, Mesters R, Tinnefeld H, Loges S, Staib P, Duhrsen U, et al. A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood (2003) 102:2763–7. doi: 10.1182/blood-2002-10-2998
118. Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, O'Farrell A-M, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood (2005) 105:986–93. doi: 10.1182/blood-2004-05-1846
119. Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene (2002) 21:2555–63. doi: 10.1038/sj.onc.1205332
120. Visani G, Ferrara F, Raimondo FD, Loscocco F, Fuligni F, Paolini S, et al. Low-dose lenalidomide plus cytarabine in very elderly, unfit acute myeloid leukemia patients: Final result of a phase II study. Leukemia Res. (2017) 62:77–83. doi: 10.1016/j.leukres.2017.09.019
121. Hütter-Krönke ML, Fiedler W, Kündgen A, Krauter J, von Lilienfeld-Toal M, Döhner H, et al. Continuous high dosing of lenalidomide in relapsed, refractory or older newly diagnosed acute myeloid leukemia patients not suitable for other treatment options - results from a phase I study. Haematologica (2018). doi: 10.3324/haematol.2018.199794. [Epub ahead of print].
122. Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U, et al. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. (2005) 23:1109–17. doi: 10.1200/JCO.2005.05.058
123. Wang ES, Fetterly GJ, Pitzonka L, Brady WE, Tan W, Greene J, et al. Phase 1 study of the angiopoietin 1/2 neutralizing peptibody, trebananib, in acute myeloid leukemia. J Clin Oncol. (2014) 32:7082. doi: 10.1200/jco.2014.32.15
124. Madlambayan GJ, Meacham AM, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood (2010) 116:1539–47. doi: 10.1182/blood-2009-06-230474
125. Turner D, Gonzalez A, Pettiford L, Meacham A, Wise E, Bosse RC, et al. A phase I study of the vascular disrupting combretastatin, oxi4503, in patients with relapsed and refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). Blood (2013) 122:1463.
126. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature (2017) 545:98–102. doi: 10.1038/nature22311
127. Cao Y, Chen C, Weatherbee JA, Tsang M, Folkman J. Gro-beta, a -C-X-C- chemokine, is an angiogenesis inhibitor that suppresses the growth of Lewis lung carcinoma in mice. J Exp Med. (1995) 182:2069–77. doi: 10.1084/jem.182.6.2069
128. Uy GL, Avigan D, Cortes JE, Becker PS, Chen RW, Liesveld JL, et al. Safety and tolerability of plerixafor in combination with cytarabine and daunorubicin in patients with newly diagnosed acute myeloid leukemia- preliminary results from a phase I study. Blood (2011) 118:82.
129. Rashidi A, DiPersio JF. Targeting the leukemia-stroma interaction in acute myeloid leukemia: rationale and latest evidence. Ther Adv Hematol. (2016) 7:40–51. doi: 10.1177/2040620715619307
130. Kovacsovics TJ, Mims AS, Salama ME, Pantin JM, Kosak KM, Rao N, et al. CX-01, a low anticoagulant heparin, may enhance count recovery and treatment efficacy in acute myeloid leukemia. Blood (2016) 128:5220.
131. Jiang E, Pham J, Kim H-N, Abdel-Azim H, Khazal S, Bug G, et al. VLA4 blockade in acute myeloid leukemia. Blood (2013) 122:3944.
132. Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. (2012) 366:1870–80. doi: 10.1056/NEJMoa1107829
133. Layani-Bazar A, Skornick I, Berrebi A, Pauker MH, Noy E, Silberman A, et al. Redox modulation of adjacent thiols in VLA-4 by AS101 converts myeloid leukemia cells from a drug-resistant to drug-sensitive state. Cancer Res. (2014) 74:3092–103. doi: 10.1158/0008-5472.CAN-13-2159
134. Chien S, Haq SU, Pawlus M, Moon RT, Estey EH, Appelbaum FR, et al. Adhesion of acute myeloid leukemia blasts to e-selectin in the vascular niche enhances their survival by mechanisms such as wnt activation. Blood (2013) 122:61.
135. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
136. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. (2008) 14:3044–51. doi: 10.1158/1078-0432.CCR-07-4079
137. Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. (2009) 69:4941–4. doi: 10.1158/0008-5472.CAN-09-0547
138. Assi R, Kantarjian H, Ravandi F, Daver N. Immune therapies in acute myeloid leukemia: a focus on monoclonal antibodies and immune checkpoint inhibitors. Curr Opin Hematol. (2018) 25:136–45. doi: 10.1097/MOH.0000000000000401
139. Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J. (2015) 5:e340. doi: 10.1038/bcj.2015.68
140. Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood (2014) 123:356–65. doi: 10.1182/blood-2013-08-523548
141. Stamm H, Klingler F, Grossjohann E-M, Muschhammer J, Vettorazzi E, Heuser M, et al. Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene (2018) 37:5269–80. doi: 10.1038/s41388-018-0288-y
142. Tasian SK. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol. (2018) 9:135–48. doi: 10.1177/2040620718774268
Keywords: acute myeloid leukemia, AML, bone marrow, angiogenesis, niche, endothelial cell, vasculature, 3D confocal microscopy
Citation: Behrmann L, Wellbrock J and Fiedler W (2018) Acute Myeloid Leukemia and the Bone Marrow Niche—Take a Closer Look. Front. Oncol. 8:444. doi: 10.3389/fonc.2018.00444
Received: 09 July 2018; Accepted: 24 September 2018;
Published: 12 October 2018.
Edited by:
Andreas Pircher, Innsbruck Medical University, AustriaReviewed by:
Nicola Giuliani, Università degli Studi di Parma, ItalyAlessandra Romano, Università degli Studi di Catania, Italy
Alessandro Isidori, Ospedali Riuniti Marche Nord, Italy
Ewa Czeslawa Izycka-Swieszewska, Gdansk Medical University, Poland
Copyright © 2018 Behrmann, Wellbrock and Fiedler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lena Behrmann, bGUuYmVocm1hbm5AdWtlLmRl