 Patrick Weyerhäuser
Patrick Weyerhäuser Sven R. Kantelhardt
Sven R. Kantelhardt Ella L. Kim
Ella L. Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 27 August 2018
Sec. Cancer Molecular Targets and Therapeutics
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00335
This article is part of the Research TopicTargeted Therapies for Glioblastoma: A Critical AppraisalView all 16 articles
There is a growing evidence that antimalarial chloroquine could be re-purposed for cancer treatment. A dozen of clinical trials have been initiated within the past 10 years to test the potential of chloroquine as an adjuvant treatment for therapy–refractory cancers including glioblastoma, one of the most aggressive human cancers. While there is considerable evidence for the efficacy and safety of chloroquine the mechanisms underlying the tumor suppressive actions of this drug remain elusive. Up until recently, inhibition of the late stage of autophagy was thought to be the major mechanism of chloroquine-mediated cancer cells death. However, recent research provided compelling evidence that autophagy-inhibiting activities of chloroquine are dispensable for its ability to suppress tumor cells growth. These unexpected findings necessitate a further elucidation of the molecular mechanisms that are essential for anti-cancer activities of CHQ. This review discusses the versatile actions of chloroquine in cancer cells with particular focus on glioma cells.
Glioblastoma (GB) is one of the most lethal human cancers (1). Despite its rarity, GB is among the top priorities in clinical oncology due to its extremely aggressive pattern, high mortality rate and unsatisfactory efficacy of current treatments. An eventual mortality rate close to 100%, 5–years survival rate of <10%, and a median survival of only 15 months remain unimproved since the establishment of standard frontline therapy for GB in 2005 (2, 3). The current standard of care for GB is based on the “one-treatment-for-all” principle and consists of a surgical resection as complete as feasible, followed by combined treatment with hypofractionated radiation therapy and non-selective chemotherapy with DNA alkylating agent temozolomide (TMZ) followed by six cycles of chemotherapy alone (3). However, the clinical effectiveness of TMZ is rather moderate (survival benefit of 2 months compared with radiotherapy alone) and restricted to a subset of GBs (~50%) lacking methyl-guanine-methyl-transferase (MGMT), an enzyme that removes the alkyl group from TMZ-induced O6-methylguanine DNA adducts (4). GBs re-grow inevitably after (or under) radio-chemotherapy. For recurrent GBs, there is no generally accepted standard therapy. None of the experimentally tested therapeutic options led to significant survival benefit (5). Post-treatment recurrence due to intrinsic and acquired resistance to cytotoxic treatments pose the major challenge to effective treatments of GB. The hallmark of GB's genetic landscape is the co-occurrence of multiple defects in key cancer-related pathways that use distinct mechanisms yet have partially overlapping functions. RTK, pRb and p53 have been identified as core pathways impaired nearly universally in the majority of GBs (6). Multiplicity of genetic aberrations affecting different pathways in conjunction with the functional redundancy of affected pathways poses a challenge for mono-targeted therapies for GB. Adding a further level of complexity, there is considerable heterogeneity of cell types constituting GBs.
Development of multi-targeted therapeutic approaches using a combination of drugs or a drug with a broad spectrum of targets might provide the solution to overcome intrinsic and acquired resistance of GBs to cytotoxic treatments.
Chloroquine (CHQ) is a well-known antimalarial that has recently attracted considerable attention for its anti-neoplastic activites. Application of CHQ for cancer treatment is an example of drug re-purposing, a strategy for identifying new therapeutic indications for drugs that have initially been developed for different medical applications (7). Synthetized at I.G. Farbenindustrie Bayer A.G. Laboratories (Elberfeld, Germany) in 1934, CHQ has been the drug of choice for malaria treatment for several decades till its role as anti–malarial has diminished due to the emergence of CHQ–resistant strains of the malaria parasite. One of the early encounters of anti-neoplastic effects of CHQ have been made during an anti-malaria trial launched by WHO in North Africa in the 1970's. It was noticed that the incidence of Burkitt's lymphoma dropped profoundly in the CHQ-treated population during the trial but returned to the basal level after the trial has been discontinued (8). This unexpected observation has remained unfollowed until a series of experimental studies reported on anti–neoplastic effect of CHQ in different types of cancer cells (9). In particular, the potential of CHQ to sensitize neoplastic cells to radiation and some other types of chemotherapy has been emerging as an approach to target treatment-refractory cancers including GBs. Currently, 17 clinical studies have been initiated to test the effects of CHQ as adjuvant treatment for different types of cancer including GB (Table 1) (12). Interest to CHQ as an adjuvant treatment for GB was sparked by the initial observation that addition of CHQ to standard therapy leads to a significant prolongation of survival in patients with GB (17) (10). After the initial demonstration that CHQ potentiates therapeutic effects of standard therapy in a double-blinded clinical trial (Phase III) involving a cohort of 30 patients with newly diagnosed GB, (10) further encouraging results have been reported in a case study with 5 patients suffering from recGB treated with CHQ and re-irradiation (18). These observations are coherent with the results from experimental studies indicating that chloroquine can potentiate cytotoxicity of TMZ and ionizing radiation in glioma cells (19–22).
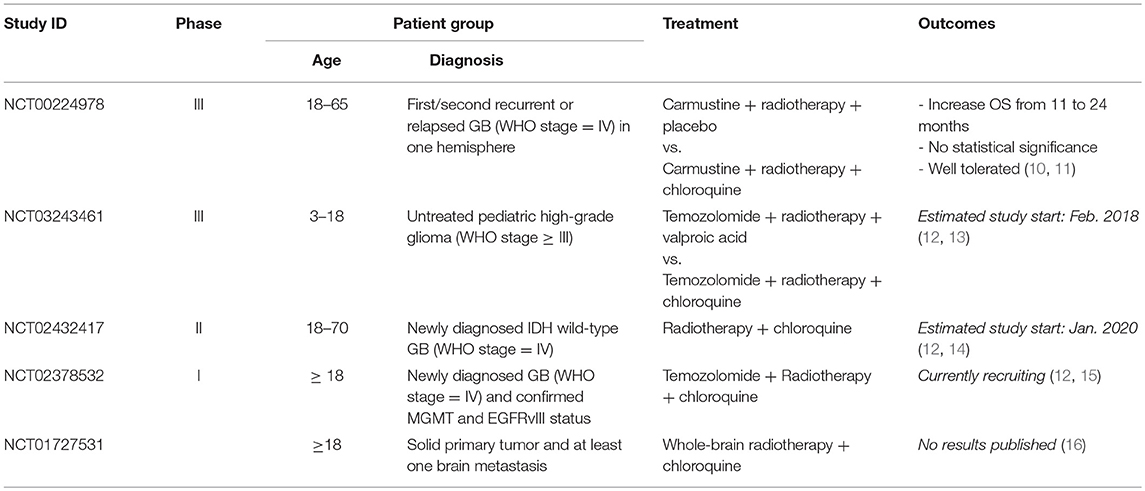
Table 1. Summary of clinical trials testing chloroquine in GBs.
CHQ (7-chloro-4-(4-diethylamino-1-methylbutylamino)-quinoline) is a small, lipophilic, amphiphilic and weakly basic tertiary amine with pKas of 8.4 and 10.2 (12, 23). At the physiological pH of 7.4, CHQ is unprotonated and highly membrane-penetrating (12). Once inside the cell, CHQ accumulates in acidic compartments and becomes protonated. As a consequence, it raises the intra–organellar pH and affects the activity of endosomes, lysosomes, autophagosomes, and autophagolysosomes (23). Owing to its lysosomotropic properties, CHQ accumulates primarily in the lysosome, where the increase of the lysosomal pH leads to a blockage of the lysosome-autophagosome fusion, a critical event during the late stage of autophagy (24). Good solubility and rapid absorption are attractive pharmacological properties of CHQ. It is rapidly absorbed when administered orally, but sub cutaneous, intra muscular, and rectal administrations are likewise possible (25).
CHQ can elicit an array of distinct biological responses in the CNS, depending on the dose and cell type. The lowest threshold of CHQ concentrations to induce neuronal death in vitro is around 20 μM (26, 27). Similar values for cytotoxic concentrations of CHQ were found in normal astrocytes (28) or neoplastic cells derived from astrocytic tumors (29, 30). However, at concentrations of 10 μM or lower, CHQ elicits neuroprotective effects in the context of oxidative damage (31). Thus, various functional outcomes can be elicited by CHQ depending on the cell type, particular pathophysiological condition, dose of the drug and treatment context. While there is an abundance of information about safety and tolerability profiles of CHQ in the context of non–cancer pathologies, CHQ application for cancer treatment will require establishing tolerability ranges in cancer patients and at cancer-relevant doses. This consideration is of special importance in the context of brain tumors, which are protected by the blood brain barrier. A phase I/II trial addressing the effects and feasibility of escalating CHQ doses for GB treatment found that CHQ doses used for treating rheumatoid arthritis may not be sufficient to effectively inhibit autophagy when used in combination with TMZ and radiation in patients with GB (32).
The mechanisms of radio- or chemo sensitization mediated by CHQ in glioma cells are not entirely understood. Modulation of the autophagic response is by far the most intensively investigated mechanism of CHQ in non-neoplastic and cancer cells. Until recently, the generally accepted view was that inhibition of autophagy is the major route of cancer cell death induced by CHQ (33). Indeed, several lines of experimental evidence suggest the importance of autophagic inhibition as the underlying mechanisms of radio-sensitization by CHQ. Knock down of beclin-1 or pharmacological inhibition of autophagy by 3-methyladenine or interference with autophagy-promoting signaling mediated through the PI3K/Akt (20) or EGFR signaling (34) have been shown to impair the radio/chemo-sensitizing ability of CHQ in glioma cells. However, the seemingly well delineated causative relationship between CHQ effects on autophagy and tumor suppression has recently been challenged by some very surprising findings coming from the pharmaceutical oncology field. Nearly simultaneously, research teams from AstraZeneka, Novartis and Pfizer have provided compelling evidence that tumor–suppressing effects of CHQ are independent from its autophagy-inhibiting activities (35, 36). Intriguingly, CHQ-induced cell death was found to be related with the inhibition of cholesterol biosynthesis by autophagy-related pathways but not with autophagy inhibition per se (36). These findings prompt to hypothesize that modulation of the cell metabolism might be one of the mechanisms underlying the anti-neoplastic efficacy of CHQ, which affects a range of metabolic processes including the amino acid metabolism, (37) glucose metabolism (38) and mitochondrial metabolism (39). Interestingly, CHQ potently inhibits glyconeogenesis, (40) which is a compensatory mechanism supporting the survival of cancer cells bearing mutations in the isocitrate dehydrogenase (IDH) gene. IDH1/2 genes code for metabolic enzymes that interconvert isocitrate and α-ketoglutarate. Loss of catalytic activity caused by point mutations in IDH1/2 genes leads to a decrease in α-ketoglutarate and increased production of D–2–hydroxyglutarate (41, 42). In glial tumors, IDH1/2 mutational status is regarded as one of the most important diagnostic and prognostic biomarkers (43, 44). Point mutations in IDH1/2 associate with longer survival and are found in about 80% of anaplastic astrocytoma (WHO Grade III) and secondary GBs (GBs that progress from lower grade gliomas), but only rarely (< 10%) in primary GBs (GBs that occur without precursor lesions). Although the relationship between IDH1/2 mutational status and sensitivity to CHQ in gliomas remains to be established, the recently proposed hypothesis that IDH1/2 mutations might be predictive of the efficacy of CHQ in gliomas seems plausible (42). Recently launched clinical studies aiming to validate the association between IDH1/2-mutated molecular subtype and sensitivity to CHQ will test this hypothesis (45).
The diversity of CHQ effects reflects the functional pleiotropy of its molecular targets, which include multi-functional factors as transcription factor NF-κB, (46) or DNA damage-inducible factors like the ataxia telangiectasia mutated (ATM) kinase (47) and its downstream target tumor suppressor p53 (48). A broad versatility of responses that can be mediated by CHQ can be exemplified by its effects on p53 whose functional status is an important factor determining the ultimate outcome from CHQ treatment in cancer cells. This, in fact, is not surprising considering the nodal position of p53 in several regulatory hubs that govern diverse cellular responses to different types of stress (49, 50). The ability to trigger distinct effects such as cell survival or cell death is the key fundamental of p53 function as the “guardian of the genome” (51). Amidst a great multitude of factors influencing the choice between pro-survival and death-promoting activities of p53, (52) the ability to repair DNA damages is essential for promoting cell survival after cell injury. Activation of p53 signaling upon DNA damage can lead to a transient arrest of the cell cycle, enabling DNA repair, or cell death, if the extent of DNA damage exceeds the repair capacity of the cell. Whereas the ability of CHQ to induce p53-dependent apoptosis has been well-documented (22, 27, 29), the mechanism of p53 activation by CHQ remains elusive. In the canonical DNA damage response (DDR), activation of the ATM/Chk1/p53 signaling is the initial event in a signaling cascade triggered by DNA-double strand breaks (53). However, CHQ does not cause direct DNA damage. It has been proposed that topological perturbations in the chromatin structure caused by CHQ intercalation into the DNA helix (54–57) may be sensed by ATM leading to its activation by autophosphorylation (47). Alternatively (or in addition) to its direct effects on DNA topology, CHQ can cause DNA breakage through an indirect mechanism involving mitochondrial damage (58). Considering that both ATM and p53 are sensitive to oxidative stress, (59, 60) these findings indicate that activation of the ATM-p53 signaling by CHQ might be triggered by oxidative DNA damage. Interestingly, while activating key mediators of DDR, CHQ has an intrinsic repair-inhibiting activity manifest in different types of normal and neoplastic cells in vitro (30, 58) and in vivo (61). Although the exact mechanisms of CHQ–mediated inhibition of DNA repair remain unknown, they are likely to reflect the causative relationship between impaired autophagy and deficient DNA repair (62). It is tempting to hypothesize that conflicting signals generated through the dual ability of CHQ to activate key mediators of DDR and to suppress DNA repair, play a role in shifting the balance in favor of cell death. Potentially conflicting signals can also emanate from the p53 transcriptional response induced by CHQ. p53 activation leads to transcriptional up-regulation of Bax1, which is indispensable for CHQ–induced apoptosis, (27) but also induces a battery of genes that promote cell survival through the activation of autophagic response (52).
The concurrent activation of cell death and pro-survival pathways through the modulation of autophagy might represent yet another death-survival axis regulated by CHQ: On the one hand, CHQ can activate cell death through the lysosome-initiated apoptosis via cathepsin signaling (63, 64). On the other hand, CHQ leads to the accumulation of a multifunctional protein chaperone p62 (also known as sequestome-1, SQSTM-1), whose expression is associated with increased cell proliferation, tumor growth and cytotoxic resistance in different types of human cancers (65). In gliomas, p62 expression correlates with the tumor grade and shorter survival (66, 67). As p62 is an autophagy adaptor targeted for degradation through autophagic clearance, autophagy inhibition by CHQ leads to the increase of the p62 protein levels (68). One of the mechanisms underlying pro-tumor activities of p62 relies on its ability to activate NF-κB, a key pathway regulating cell survival and proliferation. Augmented NF-κB signaling is linked to poor prognosis and treatment resistance in gliomas (69, 70). Moreover, there is evidence that activation of the p62/NF-κB signaling by CHQ may be further amplified through a positive feedback loop whereby CHQ-induced p62 activates NF-κB, which in turn activates the expression of p62 (71). Thus, inhibition of autophagy by CHQ can activate not only the lysosome-mitochondria death pathway, (63, 64) but also survival–promoting signaling mediated through the p62/NF-κB feedback loop (71). Considering that ATM is essential for the function of both p53 and NF-kB proteins, which often act in an antagonistic way in the regulation of cell survival, (72) and that CHQ modulates activities of all three factors (Figure 1), it is conceivable that p53 status is an important factor in determining cell fate in response to CHQ treatment.
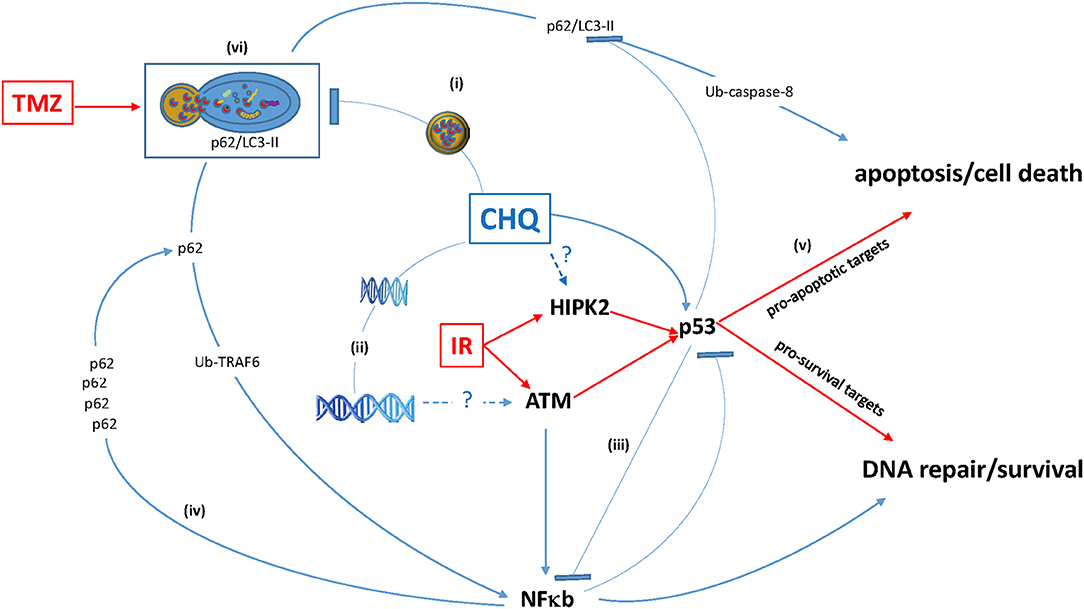
Figure 1. Antagonistic pleiotropy of multifunctional hub proteins modulated by CHQ. (i) CHQ accumulation in the lysosome inhibits the lysosome-autophagosome fusion and impairs degradation of proteins including the ubiquitin (Ub)-binding protein p62 and its binding partner pro-apoptotic LC3-II. (ii) CHQ intercalates into the DNA helix and cause relaxation of chromatin structure, which may be the mechanism of ClQ-mediated activation of a DNA damage-inducible kinase ATM. (iii) CHQ modulates activities of pleiotropic transcription factors p53 and NF-κB and may influence cross-talk between these pathways. (iv) p62 functional duality and positive p62 / NF-κB feedback loop. (v) Augmentation of pro-apoptotic activities of p53 may be a possible mechanism of CHQ-mediated radiosensitization. (vi) Autophagy inhibition by ClQ may counteract autophagic activation by TMZ and thereby sensitize glioma cells to chemotherapy.
The dichotomy of cellular responses elicited by CHQ is also manifest in its inhibitory effects on the inflammatory response which might be particularly important considering the tumor microenvironment. Normalization of the tumor vasculature has been implicated as a potential mechanism underlying the ability of CHQ to increase the efficacy of chemotherapeutic drugs, by facilitating their delivery to the tumor mass (73, 74). Indeed, there is evidence that CHQ normalizes the tumor vasculature through the reduction of vessel density, improvement of cell alignment, formation of tight junctions and promotion of quiescent phenotype of endothelial cells (73, 74). However, CHQ has also been shown to have pro-inflammatory effects in some types of cells. Within the CNS, CHQ inhibits pro–inflammatory cytokines in microglial cells, but not in astrocytes, in which it induces inflammatory cytokines through the activation of NF-κB signaling (46). Considering that GBs are tumors of astrocytic origin, their responses to CHQ may resemble those observed in astrocytes.
Thus, the ultimate outcome of CHQ treatment is likely to be determined by the intricate balance between activities of pleiotropic pathways involved in the regulation of autophagy, DDR and apoptosis/cell death (Figure 1).
Despite recent advances in the understanding of molecular mechanisms of anti-tumor effects of CHQ, a number of issues remain unsolved. One confounding factor is that experimental models used for investigating the effects of CHQ may not fully recapitulate distinctive characteristics of treatment-resistant GB. The current paradigm of therapeutic resistance in GB is centered on so-called glioma stem-like cells (GSCs). GSCs are considered the most clinically relevant type of glioma cells driving GBs propagation before and after therapy (75). It has been shown that GSCs possess an augmented DNA damage response (DDR), (76) which renders them capable of surviving cytotoxic treatments that are otherwise effective in killing non-stem glioma cells (76–78). In conjunction with augmented DDR, radiation-induced activation of anti-death and autophagic responses make important contributions to GSCs ability to escape from the cytotoxic effect of radiation (79, 80). Most of the existing studies addressing the effects of CHQ in glioma cells have used conventional serum-dependent cell lines that lack stemness properties and/or poorly recapitulate characteristic features of human GBs. For example, the human glioma cell line U87MG, which has been widely used as an experimental model for investigating biological responses mediated by CHQ (21, 22, 29, 30, 81, 82) does not reproduce certain characteristic traits of GBs such as an invasive tumor phenotype, intra-tumoral heterogeneity and high degree of intrinsic radio-resistance. Considering that GSCs are fundamentally distinct from non-stem glioma cells, it is conceivable that their responses to CHQ might also differ from those operating in the latter ones. Further underscoring this notion, activation of the p53 signaling by CHQ seems to lead to different outcomes in GSCs or non–stem glioma cells. In non-stem glioma cells with wtp53, p53-dependent apoptosis is a profound response to high concentrations of CHQ (≥20 μM) either applied alone or combined with other treatments (22, 29, 30). In contrast, GSCs with functional p53 do not activate apoptosis, but undergo predominantly a G1 arrest in response to similar CHQ concentrations (20).
Furthermore, the threshold of CHQ concentrations required for inducing cell death in the experimental setting (~20 μM) is considerably higher than clinically acceptable doses of CHQ (~5 μM). Therefore, the potential therapeutic value of clinically acceptable doses of CHQ for GB treatment requires further validation. Clarifying this question is particularly important considering the results of a phase I/II trial addressing the feasibility of dose escalation for CHQ treatment of GB (32). It was found that CHQ doses used for treating rheumatoid arthritis may not be sufficient to effectively inhibit autophagy when used in combination with TMZ and radiation in patients with GB (32). Likewise, CHQ potential in sensitizing glioma cells to radiation, observed under experimental conditions (single treatment with 10 Gy) (20) needs to be reproduced under clinically relevant conditions, applying hypofractionated radiation (multiple fractions of 2.0–2.5 Gy).
The chemo- and radio-sensitizing effects of CHQ observed under experimental conditions warrant further explorations of CHQ potential as an adjuvant treatment for GB. In order to better define the potential benefits of using this drug as an adjuvant treatment for GB, the remarkable diversity of outcomes that can be elicited by CHQ need to be considered.
PW: literature analysis, manuscript writing and preparation. SK: manuscript revision and preparation. EK: concept, literature analysis, manuscript writing, preparation and final approval.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KR and the handling Editor declared their shared affiliation.
We thank the JGU Mainz Library's Open Access Funds for funding this publication.
1. Anjum K, Shagufta BI, Abbas SQ, Patel S, Khan I, Shah SAA, et al. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: a review. Biomed Pharmacother. (2017) 92:681–89. doi: 10.1016/j.biopha.2017.05.125
2. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. (2016) 20:S2–8. doi: 10.1188/16.CJON.S1.2-8
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
4. Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT Gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
5. Weller MP, Pfister SMMD, Wick WP, Hegi MEP, Reifenberger GP, Stupp RP. Molecular neuro-oncology in clinical practice: a new horizon. Lancet Oncol. (2013) 14:e370-e79. doi: 10.1016/S1470-2045(13)70168-2
6. Chin L, Meyerson M, Aldape K, Bigner D, Mikkelsen T. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–68. doi: 10.1038/nature07385
8. Geser A, Brubaker G, Draper CC. Effect of a malaria suppression program on the incidence of Afrikan Burkitt's lymphoma. Am J Epidemiol. (1989) 129:740–52. doi: 10.1093/oxfordjournals.aje.a115189
9. Al-Bari MAA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. (2015) 70:1608. doi: 10.1093/jac/dkv018
10. Sotelo J, Brice-o E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Int Med. (2006) 144:337. doi: 10.7326/0003-4819-144-5-200603070-00008
11. Sotelo J. Chloroquine for Treatment of Glioblastoma Multiforme U.S. National Library of Medicine (2009). Available online at: https://clinicaltrials.gov/ct2/show/NCT00224978 (Accessed April 18, 2018).
12. Verbaanderd C, Maes H, Schaaf M, Sukhatme VP, Pantziarka P, Sukhatme V, et al. Repurposing Drugs in Oncology (ReDO) - chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience (2017) 11:781. doi: 10.3332/ecancer.2017.781
13. Kramm C. International Cooperative Phase III Trial of the HIT-HGG Study Group (HIT-HGG-2013) (HIT-HGG-2013) U.S. National Library of Medicine (2018) Available online at: https://clinicaltrials.gov/ct2/show/NCT03243461 (Accessed April 18, 2018).
14. Lambin P, Compter I, Eekers D. The Addition of Chloroquine to Chemoradiation for Glioblastoma U.S. National Library of Medicine (2018). Available online at: https://clinicaltrials.gov/ct2/show/NCT02432417 (Accessed April 18, 2018).
15. De Ruysscher D, Compter I, Eekers D. The Addition of Chloroquine to Chemoradiation for Glioblastoma (CHLOROBRAIN) U.S. National Library of Medicine (2018). Available online at: https://clinicaltrials.gov/ct2/show/NCT02378532 (Accessed April 18, 2018).
16. DeNittis A. IDO2 Genetic Status Informs the Neoadjuvant Efficacy of Chloroquine (CQ) in Brain Metastasis Radiotherapy U.S. National Library of Medicine (2015). Available online at: https://clinicaltrials.gov/ct2/show/study/NCT01727531 (Accessed April 18, 2018).
17. Brice-o E, Reyes S, Sotelo J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus (2003) 14:1–6. doi: 10.3171/foc.2003.14.2.4
18. Bilger A, Bittner M-I, Grosu A-L, Wiedenmann N, Meyer PT, Firat E, et al. FET-PET-based reirradiation and chloroquine in patients with recurrent glioblastoma: first tolerability and feasibility results. Strahlenther und Onkol. (2014) 190:957–61. doi: 10.1007/s00066-014-0693-2
19. Djordevic B, Lange CS, Rotman M. Potentiation of radiation lethality in mouse melanoma cells by mild hyperthermia and chloroquine. Melanoma Res. (1993) 2:321–26. doi: 10.1097/00008390-199212000-00005
20. Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote γ-irradiation-induced cell death in primary stem-like glioma cells. PLoS ONE (2012) 7:e47357. doi: 10.1371/journal.pone.0047357
21. Hori YS, Hosoda R, Akiyama Y, Sebori R, Wanibuchi M, Mikami T, et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J Neuro Oncol. (2015) 122:11. doi: 10.1007/s11060-014-1686-9
22. Lee SW, Kim HK, Lee NH, Yi HY, Kim HS, Hong SH, et al. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. (2015) 360:195–204. doi: 10.1016/j.canlet.2015.02.012
23. Park J, Choi K, Jeong E, Kwon D, Benveniste EN, Choi C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia (2004) 47:9–20. doi: 10.1002/glia.20017
24. Thomé R, Lopes SCP, Costa FTM, Verinaud L. Chloroquine: modes of action of an undervalued drug. Immunol Lett. (2013) 153:50–7. doi: 10.1016/j.imlet.2013.07.004
25. Cooper RG, Magwere T. Chloroquine: novel uses & manifestations. Indian J Med Res. (2008) 127:305–16.
26. Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. (2006) 69:1125–36. doi: 10.1124/mol.105.018408
27. Zaidi AU, McDonough JS, Klocke BJ, Latham CB, Korsmeyer SJ, Flavell RA, et al. Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J Neuropathol ExpNeurol. (2001) 60:937–45. doi: 10.1093/jnen/60.10.937
28. Vijaykumar TS, Nath A, Chauhan A. Chloroquine mediated molecular tuning of astrocytes for enhanced permissiveness to HIV infection. Virology (2008) 381:1–5. doi: 10.1016/j.virol.2008.07.039
29. Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker E-M, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. (2010) 12:389–400. doi: 10.1093/neuonc/nop046
30. Ye H, Chen M, Cao F, Huang H, Zhan R, Zheng X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. (2016) 16:178. doi: 10.1186/s12883-016-0700-6
31. Yoko H, Hideko Y, Moursy AMS, Shawky M, Kentaro O, Kazutoshi K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J Neurochem. (2011) 119:839–47. doi: 10.1111/j.1471-4159.2011.07464.x
32. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy (2014) 10:1359–68. doi: 10.4161/auto.28984
33. Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. (2013) 73:3–7. doi: 10.1158/0008-5472.CAN-12-2464
34. Jutten B, Keulers TG, Schaaf MBE, Savelkouls K, Theys J, Span PN, et al. EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival. Radiother Oncol. (2013) 108:479–83. doi: 10.1016/j.radonc.2013.06.033
35. Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA. (2016) 113:182–87. doi: 10.1073/pnas.1515617113
36. King MA, Ganley IG, Flemington V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene (2016) 35:4518. doi: 10.1038/onc.2015.511
37. Choi MM, Kim EA, Choi SY, Kim TU, Cho SW, Yang SJ. Inhibitory properties of nerve-specific human glutamate dehydrogenase isozyme by chloroquine. J Biochem Mol Biol. (2007) 40:1077–82. doi: 10.5483/BMBRep.2007.40.6.1077
38. Sutorius AHM, Cotton DWK. Inhibiting effect of some antimalarial substances on glucose-6-phosphate dehydrogenase. Nature (1971) 233:197–97. doi: 10.1038/233197a0
39. Putchen D, Kumaravelu P, Shanthi S, Devaraj N. Effect of chloroquine on rat liver mitochondria. Indian J Exp Biol. (1994) 32:797–9.
40. Jarzyna R, Kiersztan A, Lisowa O, Bryla J. The inhibition of gluconeogenesis by chloroquine contributes to its hypoglycaemic action. Eur J Pharmacol. (2001) 428:381–8. doi: 10.1016/S0014-2999(01)01221-3
41. Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci USA. (2014) 111:14217. doi: 10.1073/pnas.1409653111
42. Molenaar RJ, Coelen RJ, Khurshed M, Roos E, Caan MW, van Linde ME, et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open (2017) 7:e014961. doi: 10.1136/bmjopen-2016-014961
43. Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. (2010) 120:707–18. doi: 10.1007/s00401-010-0781-z
44. Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. (2014) 15:e395–403. doi: 10.1016/S1470-2045(14)70011-7
45. Wilmink JW. Metformin And Chloroquine in IDH1/2-mutated Solid Tumors (MACIST) U.S. National Library of Medicine (2015). Available online at: https://clinicaltrials.gov/ct2/show/NCT02496741 (Accessed April 18, 2018).
46. Park J, Kwon D, Choi C, Oh J-W, Benveniste EN. Chloroquine induces activation of nuclear factor-kappaB and subsequent expression of pro-inflammatory cytokines by human astroglial cells. J Neurochem. (2003) 84:1266. doi: 10.1046/j.1471-4159.2003.01623.x
47. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature (2003) 421:499–506. doi: 10.1038/nature01368
48. Loehberg CR, Thompson T, Kastan MB, Maclean KH, Edwards DG, Kittrell FS, et al. Ataxia telangiectasia-mutated and p53 are potential mediators of chloroquine-induced resistance to mammary carcinogenesis. Cancer Res. (2007) 67:12026–33. doi: 10.1158/0008-5472.CAN-07-3058
49. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell (2017) 170:1062. doi: 10.1016/j.cell.2017.08.028
50. Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. (2018) 25:93. doi: 10.1038/cdd.2017.171
52. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nature reviews. Mol Cell Biol. (2015) 16:393. doi: 10.1038/nrm4007
53. Stracker TH, Roig I, Knobel PA, Marjanović M. The ATM signaling network in development and disease. Front Genet (2013) 4:37. doi: 10.3389/fgene.2013.00037
54. O'Brien RL, Olenick JG, Hahn FE. Reactions of quinine, chloroquine, and quinacrine with DNA and their effects on the DNA and RNA polymerase reactions. Proc Natl Acad Sci USA. (1966) 55:1511–7. doi: 10.1073/pnas.55.6.1511
55. Li G-D. Nucleus may be the key site of chloroquine antimalarial action and resistance development. Medi Hypotheses (2006) 67:323–26. doi: 10.1016/j.mehy.2006.02.008
56. Yin F, Guo M, Yao S. Kinetics of DNA binding with chloroquine phosphate using capacitive sensing method. Biosens Bioelectron. (2003) 19:297–304. doi: 10.1016/S0956-5663(03)00197-0
57. Cheng J, Zeidan R, Mishra S, Liu A, Pun SH, Kulkarni RP, et al. Structure-function correlation of chloroquine and analogues as transgene expression enhancers in nonviral gene delivery. J Med Chem. (2006) 49:6522-31. doi: 10.1021/jm060736s
58. Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. (2016) 376:249–58. doi: 10.1016/j.canlet.2016.04.002
59. Gambino V, De Michele G, Venezia O, Migliaccio P, Dall'Olio V, Bernard L, et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell (2013) 12:435–45. doi: 10.1111/acel.12060
60. Maciejczyk M, Mikoluc B, Pietrucha B, Heropolitanska-Pliszka E, Pac M, Motkowski R, et al. Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. (2017) 11:375–83. doi: 10.1016/j.redox.2016.12.030
61. Giovanella F, Ferreira GK, de Prá SDT, Carvalho-Silva M, Gomes LM, Scaini G, et al. Effects of primaquine and chloroquine on oxidative stress parameters in rats. Anais da Academia Brasileira de Ciências (2015) 87:1487–96. doi: 10.1590/0001-3765201520140637
62. Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci USA. (2015) 112:773–78. doi: 10.1073/pnas.1409563112
63. Boya P, Gonzalez-Polo R-A, Poncet D, Andreau K, Vieira HLA, Roumier T, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene (2003) 22:3927–36. doi: 10.1038/sj.onc.1206622
64. Seitz C, Hugle M, Cristofanon S, Tchoghandjian A, Fulda S. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int J Cancer (2013) 132:2682–93. doi: 10.1002/ijc.27935
65. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. (2015) 282:4672–78. doi: 10.1111/febs.13540
66. Zhao M, Xu H, Zhang B, Hong B, Yan W, Zhang J. Impact of nuclear factor erythroid-derived 2–like 2 and p62/sequestosome expression on prognosis of patients with gliomas. Hum Pathol. (2015) 46:843–49. doi: 10.1016/j.humpath.2015.02.009
67. Xu H, Sun L, Zheng Y, Yu S, Ou-yang J, Han H, et al. GBP3 promotes glioma cell proliferation via SQSTM1/p62-ERK1/2 axis. Biochem Biophys Res Commun. (2018) 495:446–53. doi: 10.1016/j.bbrc.2017.11.050
68. Liu EY, Ryan KM. Autophagy and cancer - issues we need to digest. J Cell Sci. (2012) 125(Pt 10):2349–58. doi: 10.1242/jcs.093708
69. Puliyappadamba VT, Hatanpaa KJ, Chakraborty S, Habib AA. The role of NF-κB in the pathogenesis of glioma. Mol Cell Oncol. (2014) 1:e963478. doi: 10.4161/23723548.2014.963478
70. Cahill KE, Morshed RA, Yamini B. Nuclear factor-κB in glioblastoma: insights into regulators and targeted therapy. Neuro Oncol. (2016) 18:329–39. doi: 10.1093/neuonc/nov265
71. Yang S, Qiang L, Sample A, Shah P, He Y-Y. NF-κB Signaling activation induced by chloroquine requires autophagosome, p62 protein, and c-Jun N-terminal Kinase (JNK) signaling and promotes tumor cell resistance. J Biol Chem. (2017) 292:3379–88. doi: 10.1074/jbc.M116.756536
72. Ahmed KM, Li JJ. ATM-NF-kappaB connection as a target for tumor radiosensitization. Curr Cancer Drug Targets (2007) 7:335–42. doi: 10.2174/156800907780809769
73. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell (2014) 26:190–206. doi: 10.1016/j.ccr.2014.06.025
74. Maes H, Kuchnio A, Carmeliet P, Agostinis P. How to teach an old dog new tricks: autophagy-independent action of chloroquine on the tumor vasculature. Autophagy (2014) 10:2082–4. doi: 10.4161/auto.36259
75. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. (2015) 29:1203. doi: 10.1101/gad.261982.115
76. McLendon RE, Hjelmeland AB, Dewhirst MW, Shi Q, Rich JN, Wu Q, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi: 10.1038/nature05236
77. Wang J, Ma Y, Cooper MK. Cancer stem cells in glioma: challenges and opportunities. Transl Cancer Res. (2013) 2:429–41. doi: 10.3978/j.issn.2218-676X.2013.08.01
78. Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. (2015) 75:4416–28. doi: 10.1158/0008-5472.CAN-14-3790
79. Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer (2009) 125:717–22. doi: 10.1002/ijc.24402
80. Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. (2015) 4:226–38. doi: 10.1016/j.stemcr.2014.12.006
81. Li C, Liu Y, Liu H, Zhang W, Shen C, Cho K, et al. Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol Biochem. (2015) 35:1303–16. doi: 10.1159/000373952
Keywords: glioblastoma, chloroquine, radio-sensitization, autophagy, glioma stem-like cells
Citation: Weyerhäuser P, Kantelhardt SR and Kim EL (2018) Re-purposing Chloroquine for Glioblastoma: Potential Merits and Confounding Variables. Front. Oncol. 8:335. doi: 10.3389/fonc.2018.00335
Received: 18 June 2018; Accepted: 02 August 2018;
Published: 27 August 2018.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
Yun Dai, Virginia Commonwealth University, United StatesCopyright © 2018 Weyerhäuser, Kantelhardt and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Correspondence: Ella L. Kim, ZWxsYS5raW1AdW5pbWVkaXppbi1tYWluei5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.