 Emira Bousoik1,2
Emira Bousoik1,2 Hamidreza Montazeri Aliabadi1*
Hamidreza Montazeri Aliabadi1*- 1Department of Biomedical and Pharmaceutical Sciences, Center for Targeted Drug Delivery, School of Pharmacy, Chapman University, Irvine, CA, United States
- 2School of Pharmacy, Omar Al-Mukhtar University, Dèrna, Libya
Janus tyrosine kinase (JAK) family of proteins have been identified as crucial proteins in signal transduction initiated by a wide range of membrane receptors. Among the proteins in this family JAK2 has been associated with important downstream proteins, including signal transducers and activators of transcription (STATs), which in turn regulate the expression of a variety of proteins involved in induction or prevention of apoptosis. Therefore, the JAK/STAT signaling axis plays a major role in the proliferation and survival of different cancer cells, and may even be involved in resistance mechanisms against molecularly targeted drugs. Despite extensive research focused on the protein structure and mechanisms of activation of JAKs, and signal transduction through these proteins, their importance in cancer initiation and progression seem to be underestimated. This manuscript is an attempt to highlight the role of JAK proteins in cancer biology, the most recent developments in targeting JAKs, and the central role they play in intracellular cross-talks with other signaling cascades.
Introduction
The fate of our cells is mainly decided by the intracellular signaling pathways that control mechanisms involved in phenotypical modifications. This crucial role becomes even more significant in cancer cells that rely upon a vast, complicated, and inter-connected network of signaling pathways for their survival and proliferation. Signaling pathways are mostly activated through cell membrane receptors that are triggered by different ligands, which initiate the mechanisms responsible for controlling phenotypical outcomes, e.g., proliferation, or apoptosis. For instance, Receptor tyrosine kinases (RTKs, including epidermal growth factor receptor or EGFR, and human epidermal growth factor receptor 2 or HER2) and cytokine receptors are among the most important cell surface receptors that activate these signaling cascades. It is well-established that cancer is a heterogeneous disease (1–5). In 2015, Sottoriva et al. proposed a “Big Bang” model of tumor initiation in colorectal cancer that suggests after initial oncogenic mutation, future generations acquire further mutations, which are present in discrete populations of cells that then expand as the tumor grows, leading to spatial heterogeneity (6). A similar and even more diverse pattern has been reported for other types of cancer. Amir et al. studied two human acute lymphoblastic leukemia samples with viSNE technology, and reported a large, irregular mass of abnormal cells that were more different than similar (7). The sub-population of a sample with intrinsic resistance to a therapeutic assault (due to different mutations in the target protein, and/or reliance on an alternative mechanism) would survive and outgrow other cells due to the selection pressure, and promotes relapse after therapy, which results in abundance of cells that were once minority (8). This “Darwinian clone selection” has been well-documented in different types of cancer cells in respond to a variety of molecularly targeted drugs (9). This inter- and intra-tumor heterogeneity means that each cancer cell potentially has access to a wide variety of mechanisms to arrive at the same phenotypical outcome.
In addition to the diversity of the signaling pathways, which provides ample opportunities for cancer cells to “switch” pathways as a response to molecularly targeted drugs, and to make the matters even more complicated, these pathways are also not completely independent, and are engaged in signaling cascade “cross-talk.” Recent findings have revealed extensive interactions between traditionally categorized cascades, which has blurred the line between parallel pathways. Many of the effector proteins seem to multi-task and be involved in different mechanisms. Activation of HER2, a tyrosine kinase membrane receptor specifically expressed in breast cancer cell membrane, triggers phosphorylation of RAF and Ras, which results in over-expression of Bcl-2 family proteins (10). Another widely inter-connected protein is MUC1, which is over-expressed in different types of carcinomas and is correlated with higher risk of metastasis and poor survival rate (10). MUC1 interacts with several cytoplasmic proteins and Ras-Raf-MEK-ERK signaling pathway (11), STAT3 (mediated by Src) (12), and proteins known to be activated by EGFR (13). Studies also indicate that crosstalk among signaling pathways contributes to a deregulation of PI3K–PTEN signaling that can lead to tumorigenesis (14). This could at least partially explain the growing number of preclinical data that indicate a failure to induce apoptosis despite effective inhibition of PI3K-Akt components (15, 16).
Janus tyrosine kinase (JAK) and signal transducers and activators of transcription (STATs) are among the major proteins involved in this inter-pathway crosstalk, and latest reports have led to the elucidation of a key role of JAK/STAT signaling pathway in development, proliferation, differentiation, and survival of cancer cell, and in fact, Vogelstein et al. have included JAK/STAT pathway among 12 core cancer pathways (17). The effect of STAT3 activation on Ras and PI3K/Akt pathways (18), and the connections of JAK2 to PI3K and ERK pathways (19, 20) are examples of these inter-pathway cross-talks. JAKs are a family of proteins that belong to a category of intracellular non-receptor tyrosine kinases. In mammals, the JAK family contains four members: JAK1, JAK2, JAK3, and TYK2. STAT family is composed of seven members STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6, which mainly act as transcription factors (21, 22). Compared to other major cell signaling pathways, JAK/STAT pathway seems relatively simple, with few components. Multiple review articles have focused on this important pathway and its role in cancer cells; however, this manuscript will try to take a closer look at the versatile mechanisms involved in this seemingly simple and straightworward cascade, and to analyze the efforts that have been made in altering its activity as a therapeutic strategy. Figure 1 summarizes the intracellular signaling cascades involving activation of JAK proteins.
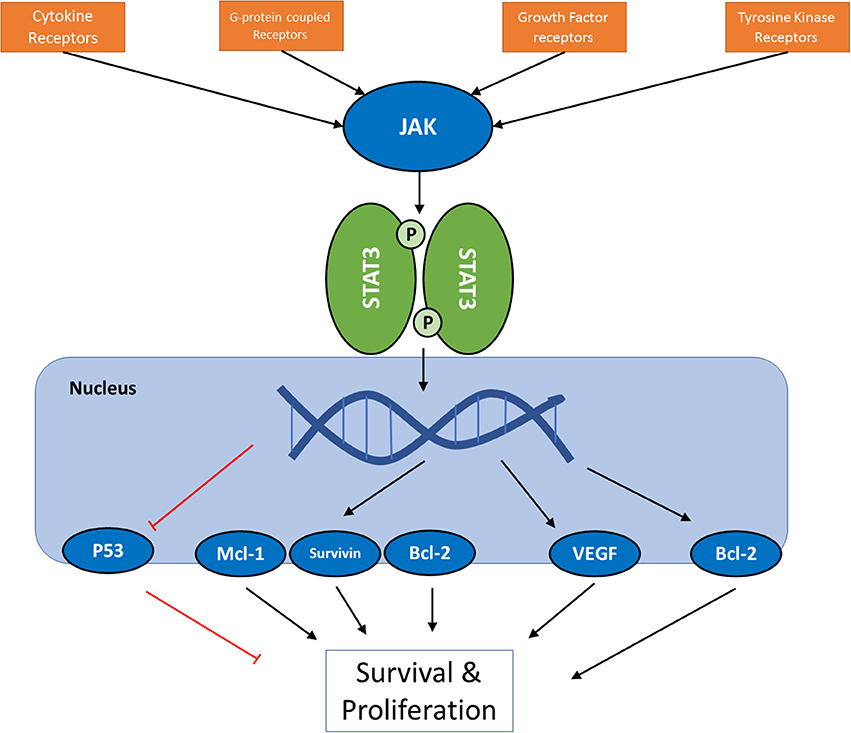
Figure 1. A schematic presentation of JAK/STAT pathway in cancer cells. For the full list of proteins regulated by the pathway via different STAT proteins, see Table 3.
A Brief History
JAK/STAT signaling cascade is among the highly conserved metazoan pathways observed in a wide range of species (23), which is involved in an array of cytokines and growth factors (22). The “story” of this silencing pathway has been previously reviewed in detail (24), where the authors track the origins of the discovery to 1950s and interferons. Our knowledge of existence of JAK/STAT pathway, however, is < 3 decades old. JAK1and JAK2 were identified in 1989, and the first member off JAK family of proteins was cloned and synthesized in 1990 (25). The gene was named Tyk2, and attracted attention due to a “kinase-like” domain next to the well-known and conserved protein tyrosine kinase domain (26). JAK name was taken from two-faced Roman God of doorways, Janus because the way that JAK possesses two near-identical phosphate-transferring domains: One domain exhibits the kinase activity while the other negatively regulates the kinase activity of the first (24, 27). Wilks et al. cloned and published the complementary DNA sequence for JAK2 in 1992, and demonstrated the same kinase/“pseudo-kinase” sections in the protein structure (28). In 1992 Shuai et al reported sequencing cDNA clones that were later called signal transducer and activator of transcription (STAT1; a and b) and STAT2 (29, 30). Later on, graduate students in Darnell lab cloned STAT3 and 4 from a lymphocyte cDNA library, establishing their membership in STATs family, and demonstrating that IL6 and EGF triggered phosphorylation of STAT3 (31, 32). The gene encoding STAT5 (initially named mammary gland factor; MGF) was cloned and sequenced in Groner's lab in 1994 (33). It was not until twenty first century that initial reports were published to link mutations in JAKs and STATs (resulting in persistent activation of the pathway) to several disorders (24).
Mutations
Predictably, mutations that affect JAK/STAT pathway activity impact multiple cellular events (34, 35). The first report on mutation in this pathway surfaced in 1997, which explains the role of E695K mutation in the JH2 domain of JAK2 in murine cells that results in increased autophosphrylation and increased activation of STAT5 (36). First patient with confirmed mutation in the JAK/STAT pathway was reported in 2003 (37). Classical cases of growth hormone insensitivity (GHI) are usually due to mutations in GH receptor (GHR), which is a member of cytokine-hematopoietin family of receptors that do not show any intrinsic kinase activity (38). Dimerization of GHR leads to activation of JAK2, and in turn, activation of STATs. In 2003, a 16-years old Argentinian girl was diagnosed with severe growth retardation and immunodeficiency, which suggested involvement a pathway affecting both GH and cytokines (37). Immunoblotting showed absence of STAT5b (despite normal levels of total STAT5), and RT-PCR confirmed homozygous missense mutation in codon 630, resulting in substituting alanine with proline (39).
In 2005, four separate groups working on tyrosine kinase signal transduction reported a valine-to-phenylalanine mutation at position 617 in the JH2 domain of JAK2 that causes “gain-of-function” (40–43). To this day, V617F remains the most studied mutation in JAK family of proteins, and seems to be present in the majority of patient with Polycythemia Vera. This type of mutation is also frequently observed in other myeloproliferative neoplasms (44), and has been previously reviewed, extensively (45–48). Table 1 summarizes selected mutations (based on the significance and frequency of reports) in components of JAK/STAT signaling pathway. The list of all mutations reported in literature is out of the scope of this manuscript.
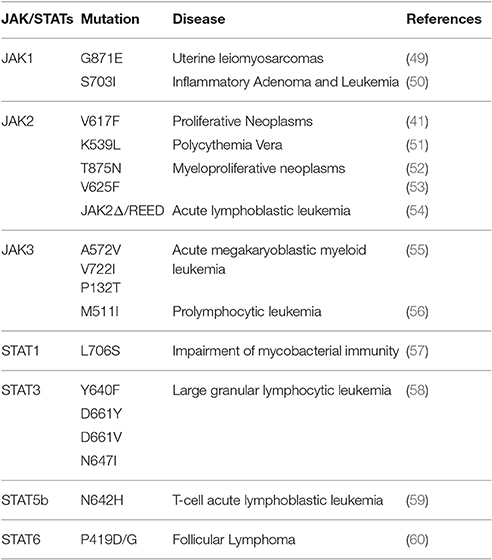
Table 1. Selected JAK/STAT mutations and resulting disorders.
Triggering the Signal: Receptors
JAK/STAT signaling begins with the activation of JAK by binding of a ligand such as growth factors, interferons, or interleukins to specific transmembrane receptors. A wide array of receptors has been associated with JAK/STAT pathway activation, which are summarized in Table 2. The cytokine receptors are probably the most commonly known family of transmembrane receptors associated with JAK activation. Generation of knockout mice for JAK family members, and evaluation of response to cytokine stimuli has contributed significantly to our understanding of the relationships between JAKs and cytokine receptors, which has led to the belief that cytokine receptors each prefer specific member(s) of JAK family for the signal transduction effector (109, 110). However, homozygous deletion of JAK1 and JAK2 causes lethality in mice due to disruption of neuronal development (111) and definitive hematopoiesis (110), respectively.
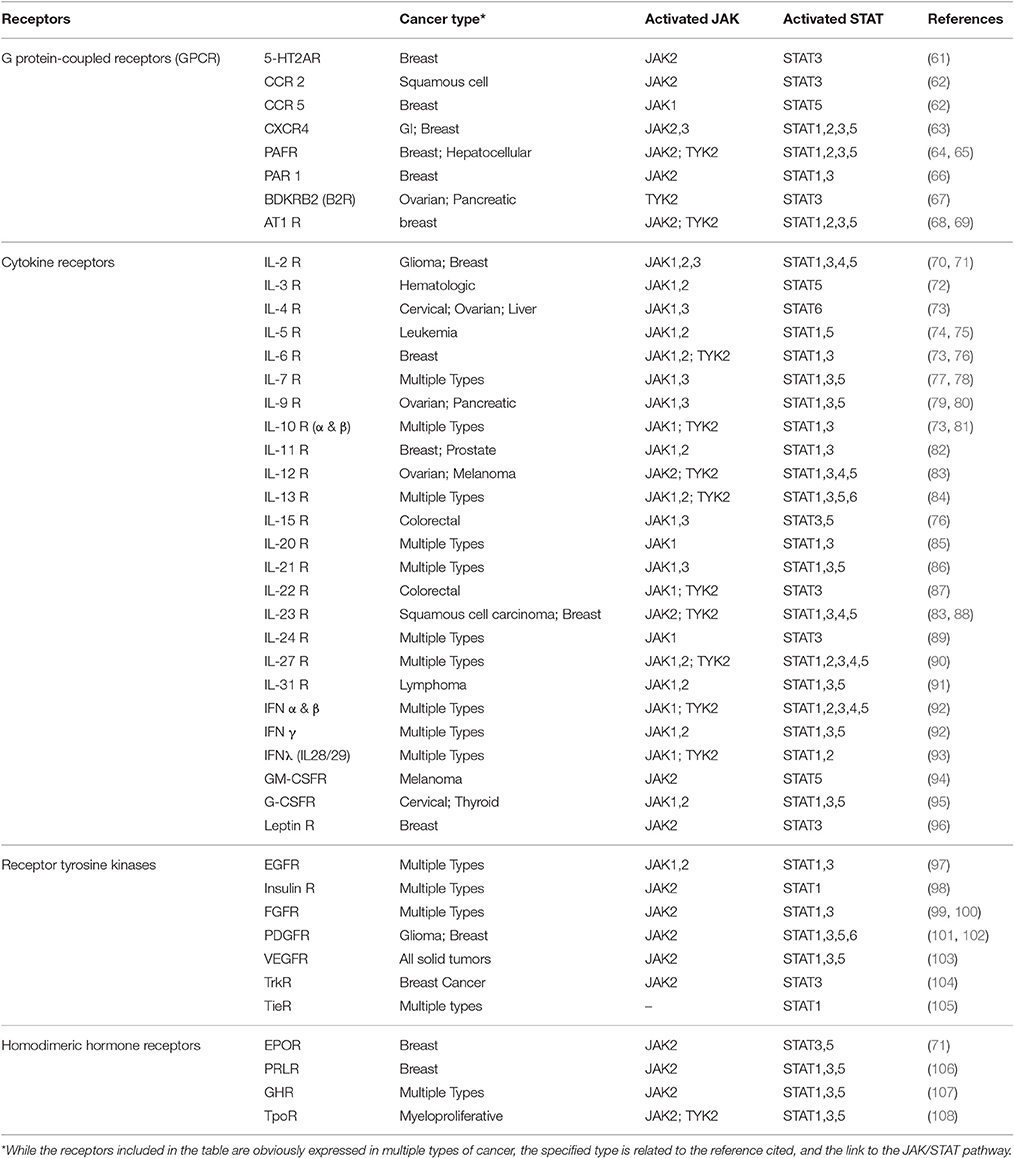
Table 2. A summary of the receptors involved in activating JAK/STAT pathway in cancer.
Cytokine receptors activate JAK/STAT pathway through a variety of combinations of different JAK and STAT family members, which highlights the versatile nature of this pathway. The receptors in this family that are linked to JAK activation could be categorized as interleukin (IL) receptors, interferon (IFN) receptors, and colony stimulating factor receptors (CSFRs). Among IL receptors, gp130 subunit and receptors for IL-2, IL-3, IL-4, IL-6, IL-7, IL-9, IL-10, IL-11, IL-12, IL-13, IL-15, IL-20, IL-21, IL-22, IL-23, IL-27, IL-31, and Leptin have been reported to trigger activation of specific members of JAK family of proteins; however, while JAK1 seems to be a common factor among them (except for IL-12 and IL-23 receptors), a wide range of combinations of downstream effectors are observed. For example, heterodimerization of the IL-2Rβ and γc cytoplasmic domains has been reported to activate JAK1 and JAK3, with JAK1 associating with IL-2Rβ and JAK3 with γc (70). Interaction of IL-2 and the receptor, mostly results in activation of STAT5; however, STAT3 and STAT1 are also activated to lower degrees (112). Crucial role of IL-2 and its receptor in development of breast tumors, and a correlation between the malignancy of the tumor and expression of these receptors has been reported (113). The erythropoietin receptor (EPOR) is a hormone receptor that shares extra-cytoplasmic structural characteristics with cytokine receptor family [EPOR and IL-2Rβ share 45% amino acid identity in box 1 and box 2 cytoplasmic regions (114)]. In 1994, D'Andrea and Barber reported that EPOR stimulation results in rapid and dose-dependent JAK2 phosphorylation (71). On the other hand, simulation of IL-3 receptor via treatment with proteasome inhibitor, N-acetyl-L-leucinyl-L-leucinyl-norleucinal (LLnL) has resulted in prolonged activation of JAK 1 and 2, and stable phosphorylation of STAT5 (72).
It has been suggested that different cytokine receptors preferentially use one of the members of JAK family of proteins, or a specific combination (115). However, the mechanism of this selectivity is unclear. Interestingly, IL-4R (73) and IL-13R (84) are the only cytokine receptors that transduce the signal to STAT6. STAT6 has different functions in different cell types, and activates the transcription of a different set of proteins in T cells compared to non-lymphocyte cells (116). IL-13R is expressed in different tumor types, and although breast cancer cells are not among those, in a recent study Kawakami et al. reported targeting breast cancer cells by IL-13, after transfecting the cells with IL-13Rα2 plasmids (117). The receptor for IL-5 is a crucial factor in the physiology of eosinophils, multifunctional granulocytes that play a role in immune system, and are associated with the pathology of asthma and inflammation (118). STAT1 and STAT5 are activated through the signaling triggered by this receptor (Table 2).
IL-6 and IL-10 predominantly activate STAT3 with sometimes diverse outcomes (73, 76). Both STAT1 and STAT3 have been reported to be activated via IL-6R signal; however, different cell types show strong preference toward one STAT. SOCS3 is a protein that is induced via STAT signaling from different cytokine receptors, and acts as a feedback inhibitor on the expression of IL-6R (among other receptors). In the absence of SOCS3, STAT3 activation is significantly increased (119). However, STAT1 activation is not affected similarly, and therefore, in presence of SOCS3 the path activated by IL-6R switches from STAT3 to STAT1 to some extent (120). Even though IL-10R signaling resembles IL-6R pathway closely, the STAT3 activation of IL-10 induces transcription of a different set of proteins that are mostly involved in inhibition of inflammatory responses (121). IL-12R and IL-23R are structurally related, use the same signaling pathway, and are among the cytokine receptors that require TYK2 for their signal transduction (73). In T-cells, IL-12R activation results in STAT4 stimulation, which induces IFN-γ expression. IL-11 and its receptor have been indicated in breast cancer development and progression, and in 2006 IL-11 was reported as a predictor of poor prognosis in this type of cancer (122). IL-31, mainly produced by CD4(+) T cells and is a member of the gp130/IL-6 cytokine family. IL-31R activates JAK/STAT, PI3K/AKT, and MAPK pathways and acts on a broad range of cells (91). While other IL receptors [e.g., IL-19 (123) and IL-35 receptors (124)] have been reported to activate JAK/STAT pathway, their role in cancer cells is unclear.
TYK2 is the main difference between the pathways activated by type I (IFNα and β) and type II (IFNγ) IFN receptors. It has been reported that IFNαR1 and R2 (β) are associated with TYK2 and JAK2, respectively, while IFNγR1 and R2 activate JAK1 and JAK2 respectively (92). Briscoe et al. reported that JAK1 negative U4A cells demonstrate a partial response to IFNγ; however, the JAK2 negative γ2A cells did not response to IFNγ at all (125). IFNγ predominantly triggers activation of STAT1. There is evidence that IFN receptor activation triggers other intracellular proteins involved in other signaling pathways, including MAP kinase, PI3-K, CaMKII and NF-κB (126). Granulocyte Colony Stimulating Factor (G-CSF) and Granulocyte/Macrophage Colony Stimulating Factor (GM-CSF) are among other cytokines that have been linked to JAK/STAT pathway activation. G-CSFR is reported to mainly activate JAK2 and STAT3, and is expressed in several normal and malignant tissue (95). GM-CSF receptors have been identified on most types of myeloid progenitors, mature monocytes, neutrophils, eosinophils, basophils, and dendritic cells and mainly contribute to defense mechanisms against bacterial infections (127). GM-CSFR is also reported to activate JAK2; however, STAT5 is reported to be the main member of STAT family of protein to be activated via this pathway (94). Leptin is a cytokine normally secreted from adipose tissue, and is involved in regulating energy consumption and appetite (128). Interaction of leptin with the leptin receptor (which is categorized as a type I cytokine receptor) initiates the signaling cascade by phosphorylating associated JAK2. This, in turn activates STAT3, and MAPK extracellular signal-activated kinase 1/2 (ERK1/2) (128). Leptin signaling pathway has been reported to play a role in the proliferation of breast cancer cells via JAK/STAT, ERK1/2, PI3K-Akt pathways, and by enhancing angiogenesis through up-regulating vascular endothelial growth factor (VEGF) (96).
While JAK/STAT pathway was originally identified as a pathway activated by IFN signaling cascades, it has been recently reported that JAK proteins can be activated by other types of receptors to widen the array of the signals that could trigger this signaling pathway. G protein-coupled receptors (GPCR) are one of the categories of receptors that have shown capability to activate JAK (129). Among GPCRs, CXCR4 has been indicated to play a role in breast cancer cell growth. This receptor, activated by chemokine stromal cell-derived factor (SDF-1alpha), becomes tyrosine phosphorylated through activation and association with the receptor of JAK2 and JAK3 kinases, which in turn recruit and tyrosine phosphorylate multiple STAT family members (63). In this category of receptors, platelet-activating factor receptor (PAFR), bradykinin B2 receptor (B2R), and angiotensin II receptor type 1 (AT1R) all activate TYK2 (along with JAK2 for PAFR and AT1R) to trigger the JAK/STAT pathway. Among GPCRs, 5-HT2A receptor (5-HT2AR) has been identified as the main receptor to mediate the cell growth enhancing effect of serotonin (5-hydroxytryp-tamine; 5-HT) in different tissue, including MCF-7 breast cancer cells (130). The signal transduced through this membrane is known to activate both Ras/Raf and JAK/STAT pathways, and a recent study confirmed activation of JAK2/STAT3 combination by this receptor in JEG-3 human trophoblast choriocarcinoma cells (61). Receptors to both families of chemokines (CC and CXC) are also known to activate JAK/STAT signaling pathway. In a 2001 manuscript, Mellado et al. identified JAK1 (but not JAK2 or 3) to be associated with CCR5, while CCR2 promoted JAK2 activation in HEK-293 cells transfected with CCR5 and CCR2, respectively (62). On the other hand, CXCR4 has been found to be a prognostic marker in a variety of cancers, including breast cancer (131). Activation of CXCR4 receptor by chemokine stromal cell-derived factor (SDF-1α) has been reported to activate JAK2 and JAK3 independent of Gαi-1, and in turn recruit several members of STAT family (63). Angiotensin II also activates JAK/STAT pathway via AT1 receptor (AT1R) (68). In 2000, Ali et al. reported activation of JAK2/STAT1 combination that was independent of the tyrosine residues of the receptor, but completely dependent on the catalytic activity of JAK2 (69).
Activation of STAT family of proteins by RTKs have been long speculated; however, involvement of JAK proteins in the process has been a topic of debate. A possible link between signals transduced by epithelial growth factor receptor (EGFR) and STAT family activation has long been identified (132). However, the exact mechanism was not clear. In 2004, Andl et al. reported a JAK-dependent activation of STAT1 and 3, using a specific JAK inhibitor (AG-490), and suggested that EGFR induces the phosphorylation of STAT1, which triggers complex formation of STAT1 and 3 with JAK1 and 2 (97). Other reports since then have confirmed this link, and indicated the regulation of PD-L1 expression, among other intracellular roles, via this link (133–135). Among other growth factor receptors, fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor receptor (VEGFR) have also been linked to this cascade. FGFR has been reported to stimulate STAT1 and 3 through JAK2 (among other downstream proteins) (100). It has been reported that tyrosine phosphorylation of STAT3 via this receptor is JAK-dependent, relying on formation of a complex by JAK2 and Src with FGFR1 (99). On the other hand, a recent study has indicated VEGFR-2 to activate the JAK2/STAT3 signaling axis by recruiting JAK2 and STAT3, which results in over-expression of MYC and SOX2 (136).
There are a few reports that claim toll-like receptors (TLRs) could also activate STAT3, which is one of the pathways for this family of receptors to play a role in tumor development (137); however, their involvement is contentious. TLR4 and TLR9 are among the receptors that have shown the most significant correlation with STAT3 activation. It has been reported that TLR4 overexpression could lead to STAT3 activation in intestinal epithelial cells, which also correlates with the clinical outcomes of colon adenocarcinoma (138). Upregulation of IL-6 by TLR4 is also reported in lymphoma (139), as a possible mechanism in the carcinogenesis. TLR9 is overexpressed in glioma stem cells, and a correlation between the expression level of TLR9 and survival rate in glioblastoma has been reported (140). It has been speculated that TLR9 activates JAK2 via Frizzled 4, which results in phosphorylation of STAT3 (141).
Hormone receptors are another family of receptors that have been associated with JAK/STAT pathway. In addition to EPOR, prolactin receptor (PRLR) has also been indicated in the activation of this signaling cascade. In 1997, Pezet et al. reported that binding of prolactin to its receptor results in dimerization of JAK2, which is “constitutively” associated with this receptor (142). It has been speculated that JAK/STAT is the principal signaling cascade activated by PRLR (106). Although considered an “archetypal” cytokine receptor, growth hormone receptor (GHR) is also associated with JAK2 activation and triggers JAK/STAT pathway (107).
JAK Activation
Unlike RTKs, cytokine receptors do not possess intrinsic kinase domain, and therefore, rely on JAK family to transfer the signal to the cytoplasmic components of the cascade (143). JAK is associated with cytoplasmic domains of cytokine receptors via JAK binding sites that are located close to the membrane and forms a complex that is equivalent in function to RTKs (144). However, the where and when of this association has been a topic of discussion (73). Members of JAK family consist of seven different JAK homology (JH) domains that include a four-point-one, ezrin, radixin, moesin (FERM) domain (JH5, 6, and 7) and a Src homology 2 (SH2) domain (JH3, and 4). JH1 and JH2 form the kinase and pseudokinase domains, respectively (145). The N-terminus half of all four members comprises FERM and SH2 domains that associate JAK with the cytoplasmic tail of the cytokine receptors (146). Binding of the ligand to cytokine receptor reorients the receptor/JAK dimers, which brings the JAKs close enough to transphosphorylate the partner JAK in the dimer at JH1. The activity of JH2 domain has only been reported for JAK2, as 10% catalytic activity compared to JH1 (133), and had been speculated to play an auto-inhibitory role, since the loss of JH2 leads to constant activity (147). Activated JAKs in turn, phosphorylate the residues on the cytoplasmic tail of the cytokine receptor to create “docking sites” for recruitment of downstream proteins with SH2 domains, e.g., STAT family of proteins (145). It is evident that different receptors have specific preferences for the JAK family protein they use as signaling effector, which means there is an obligate relationship between the receptor and the specific JAK protein(s) activated (143). However, in many cases, it has been shown that in the absence of the specific JAK family member, other proteins in the family have taken the responsibility and transferred the signal.
Recruitement of Stats and Transport to Nucleus
The next step in this signaling cascade is the recruitment of members of STAT family of proteins. Inactivated (or “latent”) STATs are found in cytoplasm [although, non-canonical mechanism of activation indicates presence of non-phosphorylated STATs in nucleus (148)]. As their name indicates, STATs act both as signal transducer and transcription factor; however, two structural components make them unique among transcription factors: an SH2 domain and a highly conserved C-terminal tyrosine residue (149). It is this tyrosine residue that is phosphorylated by activated JAKs. After phosphorylation, STATs form stable homodimers or heterodimers with other STAT proteins via SH2 domain interactions (150). A similar specificity observed with JAKs is seen here as well, where specific members of STAT family respond to a defined set of stimuli and receptors. Among STATs, STAT3 has been shown to be activated through other pathways, most importantly via EGFR and SRC (31, 151). The JAK/STAT activation could also be inactivated by negative regulators, e.g., SH2-containing protein tyrosine phosphatase (SHP) and suppressor of cytokine signaling (SOCS) proteins (152).
After activation by tyrosine phosphorylation, STATs become dimerized and translocate into the nucleus, where they act as transcription factors. Most STATs form homodimers; however, heterodimer formations (including STAT1/2, STAT1/3, and STAT5a/b) have also been reported (153). STAT1 has been reported to exist as pre-formed homodimers, and phosphorylation induces reorientation (anti-parallel to parallel conformation), which presumably could be true for other STATs as well (154). While translocation between cytoplasm and nucleus is a regular occurrence, the nuclear envelope provides a barrier that prevents free diffusion of large molecules (more than 40–60 kilo Dalton in molecular weight). These large molecules, including STATs usually require a specific transport receptor for facilitated transport (155). The receptors involved in importing molecules into nucleus are commonly known as importins, which consist of α and β subunits, known as importin α and β, respectively (156). The protein to be imported into the nucleus binds to the importin α via a specific motif on the protein called nuclear localization sequence (NLS) (157). After binding to the protein, importin α interacts with importin β, which docks the protein/importin complex at nuclear core complexes (NPCs). The translocation process is an active transport that requires energy, which is provided by NPC-associated GTPase, known as Ran (156). The translocation of STATs into nucleus via importins is a subject of discussion. For example, there are six human importin αs reported in literature, which show some similar structural characteristics, but binding specificity as well. While it seems accepted that STAT1 and STAT5 are transported into nucleus by importin α5 and α3, respectively, the same certainty does not seem to exist for STAT3. Different reports indicate involvement of importin α5 and α7 (158), importin α3 and α6 (limited to testis) (159), or a variety of importin αs (160). On the other hand, there are speculations that STATs do not contain functional NLS altogether, and therefore, NLS-containing chaperons are required to associate with STATs to facilitated binding to importins (153). Other reports indicate a binding site on STAT1 and STAT3 slightly different than the NLS binding site observed on other proteins (161). STAT3, 5, and 6 could be translocated into nucleus in the un-phosphorylated form as well (162).
Stats as Transcription Factors
STATs have demonstrated the capability to activate the transcription of non-active genes in a few minutes (163). STAT family of proteins play multiple roles in cancer cells, and specifically, STAT3 has been shown to enhance cancer cell proliferation, migration, and survival, in addition to suppression of antitumor immune response (137). JAK activation is not the only mechanism of the activation of STATs and their migration into nucleus. For instance, a link between STAT activation and Src family of kinases has been proposed by researchers, which will promote the transcription of proteins such as VEGF and IL-8 (164). This ability of STATs to integrate the signal from a wide variety of signaling cascades indicates the possibility of regulation of a variety of genes through this family of transcription factors that serve different mechanisms involved in growth, differentiation, and survival. Among STATs, STAT1 and STAT2 are known as the targets of interferon activation (165). However, activation of STAT1 via PDGF has also been reported (166). STAT1 forms a heterodimer with phosphorylated STAT2, and IFN-regulatory factor 9, and is transported into nucleus to bind to IFN-stimulated response element (ISRE) in promoters of the responsive genes (167, 168). STAT1 homodimers are also formed. Both dimers seem to promote the expression of genes that enhance growth arrest and apoptosis (Table 3). For instance, STAT1 is involved in expression of several caspases, as executives of apoptosis (169). Based on the downregulation and activation pattern of downstream proteins, STAT1 seems to be involved in controlling the cell growth, enhancing vascularization, and inducing cell death, which are all characteristics that inhibit tumor growth.
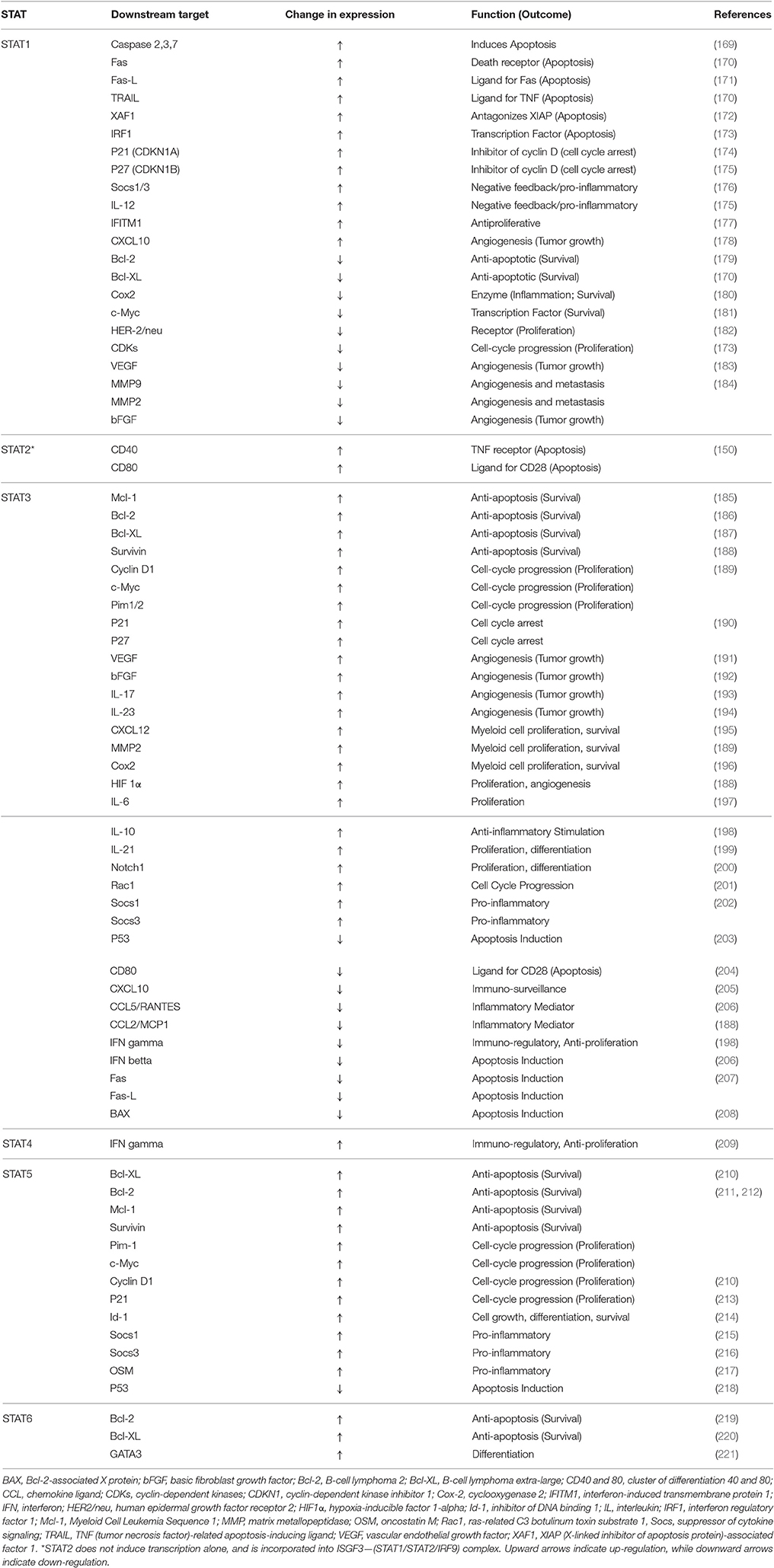
Table 3. Selected survival-related genes regulated by members of STAT family of proteins.
STAT2 was also initially identified as a component of the STAT1/STAT2 heterodimer and IFN-regulatory factor 9, and is the largest molecule among the proteins in this family. It has been reported that tyrosine phosphorylation and heterodimerization with STAT1 are necessary for STAT2 transportation into nucleus (167). However, non-phosphorylated STAT2 is also translocated into nucleus without interferon receptor signaling, as a result of interaction with a non-STAT transcription factor called IRF9 (222). There is little information available about the formation of STAT2 homodimers, and the role of STAT2 as an independent transcription factor is largely unknown and controversial (175). STAT2 activation has been linked to increased expression level of Cluster of differentiation (CD) 40 and CD80 (150), receptors involved in a multiple-step T-cell activation model (223). Similar to STAT1, impaired response to interferons observed subsequent to STAT2 knockdown in animal models has resulted in viral infections (224).
STAT3 is by far the most studied and best-known member of STAT family of proteins, and along with STAT5 have been extensively investigated in cancer biology. The outcome of STAT3 activation, however, is almost the exact opposite of STAT1 (despite almost 70% sequence homology, and similar crystal structure as tyrosine phosphorylated dimers) and contributes to carcinogenic processes and cancer progression (150, 225). It has been interconnected with nuclear factor-κB (NF-κB) signaling, and they seem to co-regulate a variety of oncogenic and inflammatory genes (226). It also seems to play a crucial role in development, as knocking down STAT3 in mice has been proven to be lethal to the embryo (227). STAT3 could be transported into nucleus both as tyrosine phosphorylated and non-phosphorylated, which is mediated by importin α3 (159) (silencing importin α3 using RNA interference approaches has shown to induce STAT3 accumulation in cytoplasm), while the main transporter for STAT1 is importin α5 (162). It has been reported that non-phosphorylated STAT3 present in nucleus could also affect the expression of many oncogenic proteins, independently, or after forming complexes with other transcription factors, e.g., JUN (228). Among well-known proteins that are overexpressed by STAT3 activation, Mcl-1, Bcl-2, Bcl-XL, and survivin are anti-apoptotic proteins that play a crucial role in cancer cell survival (186, 187, 189), cyclin D1 and c-Myc enhance proliferation (189), and VEGF promotes angiogenesis, which is required for tumor growth (229). On the other hand, STAT3 is reported to downregulate expression of important proteins involved in apoptosis induction or mechanism, including P53 (203), interferon β (206), Fas and its ligand, and BAX [(207, 208); Table 3).
STAT1/STAT2, STAT1/STAT3, (163), and STAT1/STAT4 (230) are the only heterodimers reported. Therefore, STAT4 is known to form homodimers, and the activation of this member of STAT family is triggered by IL-12 receptor, and is only linked directly to enhanced expression of interferon γ as a result (209), which is crucial in differentiation of T-helper cells 1 (231). On the other hand, STAT5 is the other member of the family usually associated with cancer. Two versions of this protein, known as STAT5A and 5B, are identified, which share a similar protein structure (more than 95% identical), and are reportedly involved in development and hematopoiesis, since impaired T-cell proliferation and severe anemia are reported in STAT5A/5B double-knockdown mice (232). STAT5B transport into nucleus is similar to STAT3, and can be transported in and out of nucleus in non-phosphorylated form as well (233). Also, similar to STAT3, STAT5 is also overactive in many invasive human cancers (163). STAT5 is involved in expression of many proteins that are linked to STAT3 as well, and therefore seems to contribute to similar outcomes (cell survival and enhanced proliferation). However, the expression of inhibitor of DNA binding 1 (Id-1) seems to be exclusively linked to activation of STAT5 (214). Id-1 is a protein involved in cancer progression, angiogenesis, and cell survival (234). STAT5 and STAT6 are both reportedly overactive in hematopoietic malignancies (226, 235). STAT6 activation seems to be triggered mainly by IL-4 and IL-13, and the loss of these cascades has been reported to impaired T-helper 2 cell differentiation (236) and development of certain types of leukemias and lymphomas (235), respectively. Majoros et al. have reviewed reports on “non-canonical” mechanisms of activation (including kinase-independent JAK functions and activity of non-phosphorylated STATs) recently (237).
MicroRNAs (miRNAs) are part of cellular gene expression regulators that can significantly change the phenotypic characteristics of the cell. They are expressed as hairpin structures, transformed in a multi-step process to a single strand RNA, and are incorporated into the RNA-induced silencing complex (RISC) to identify and bind to a partial or perfect complementary match on targeted mRNAs (238). Not only are miRNAs involved in the regulation of proteins involved in JAK/STAT pathway (similar to the majority of other cellular proteins), STAT family of proteins are also reported to regulate miRNA expression levels. For instance, it has been shown that STAT3 directly activates miRNA-21, which in turn, downregulate PTEN, among other proteins, which is a well-known tumor suppressor gene (239, 240). The interactions between STAT proteins and miRNAs have been previously reviewed (241).
Targeting JAK/STAT Pathway
Targeting members of JAK and STAT families of proteins with molecularly targeted drugs, and/or RNA interference (RNAi) approaches has been extensively studied, with many of them already in clinics or clinical trials. It has been hypothesized that blocking this signaling pathway could inhibit cancer progression as a single therapy, or in combination with other anticancer agents. The small molecule drugs targeting these proteins in clinical trials or used in clinics are summarized in Table 4. A quick look at the table reveals a few facts:
a. Members of JAK family have been a more popular target of molecularly targeted drugs than STATs. JAK is an upstream protein to STAT, which means it has to be activated in order to activate STATs, and this might be a hypothetical explanation for this exaggerated focus. However, STATs have been reported to be activated by other signaling mechanisms, independent of JAKs. The other explanation could be based on the hypothesis that upstream proteins might be involved in cross-talk with other signaling cascades, and therefore, by targeting JAKs we could also interfere with other mechanism involved in cancer progression. The emphasis on JAKs is also apparent in number of drugs in clinics and clinical trials compared to drugs targeting STATs (which are mostly still in pre-clinical stages);
b. While there is a variety of JAK proteins that have been targeted by small molecule drugs (including TYK2), the only member of STAT family that has been the focus of therapeutic attempts is STAT3 (with the exception of fludarabine that targets STAT1). This is mostly due to the fact that STAT3 has been one of most promising targets for molecularly targeted treatment. This also indicates less specificity seen in JAK inhibitors (especially pan-JAK inhibitors, e.g. ruxolitinib), rather than intentional targeting more than one JAK at a time;
c. JAK2/STAT3 seem to be the most popular targets in cancer treatment. In fact, cancer seems to be the dominant target for these therapeutic approaches. Autoimmune diseases (e.g., rheumatoid arthritis and psoriasis) are the second focus of attention.
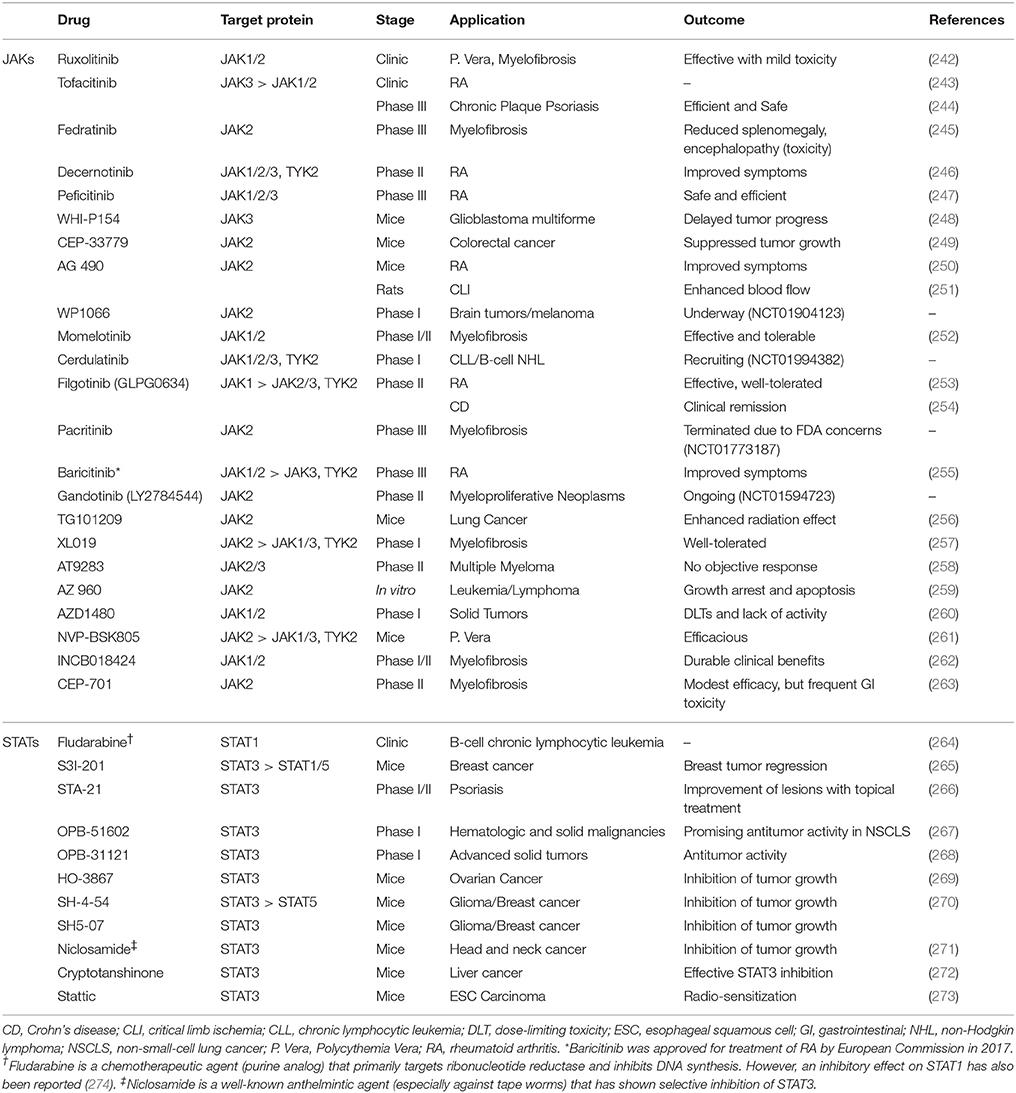
Table 4. Selected small molecule drugs targeting JAK or STAT proteins used in clinical setting or in different stages of clinical trials.
Ruxolitinib, tofacitinib, and fludarabine are the only molecularly targeted drugs against JAK/STAT pathway used in clinics. Fedratinib reached Phase III clinical trials; however, a report published in 2015 indicates that the clinical development has been discontinued due to toxic effects in some patients (most importantly encephalopathy), despite significant reduction of splenomegaly and symptom of myelofibrosis (245). Another interrupted development was recently reported for Pacritinib (a specific JAK2 inhibitor) in Phase III clinical trial, due to patient deaths, despite previous reports on its efficacy and safety in myelofibrosis (275). However, there are still four active Phase 1 and/or 2 trials that seem to continue on this drug. Peficitinib is another pan-JAK inhibitor in Phase III, which was recently reported efficacious in treatment moderate to severe rheumatoid arthritis (RA) with “acceptable safety profile” in a double-blind 12-wwek study in Japan (247). Recent reports also indicate development of TYK2-specific inhibitors, including NDI-031301 which has shown promising results in acute lymphoblastic leukemia (276). In addition to the small molecules included in Table 4, there are numerous new inhibitors of JAKs and STATs. A comprehensive review on investigational JAK inhibitors was recently published by Musumeci et al. (277).
An alternative approach in blocking signaling pathways involved in cancer progression is RNAi approaches that rely on temporary or permanent “silencing” of the targeted protein by targeting the mRNA responsible for the expression of the targeted protein. Due to a wider range of targets for these approaches, a larger number of effectors have been silenced via RNAi-based attempts, which include the downstream proteins activated by this pathway, and have been reviewed previously (278). Antisense oligonucleotides (ASOs) have also been studied for silencing proteins involved in this pathway. Recently, Hong et al. reported preclinical and initial clinical evaluation of methyl-modified ASOs (AZD9150) targeting STAT3 in patient-derived xenograft models and highly treatment-refractory lymphoma and non-small cell lung cancer patients (279). Another approach to this type of expression inhibition is known as “decoys.” Decoys targeting transcription factors, specifically, consist of nucleotide sequence derived from conserved regulatory elements, and block binding of transcription factor to genomic DNA by competitive inhibition. Sen et al. reported using cyclic decoys (by linking oligonucleotide strands using hexaethylene glycol spacers) in a “phase 0” study to target STAT3 in head and neck cancer patients (280). The newest strategy in silencing, the Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein (Cas)9 gene editing system, has been recently used to silence components of JAK/STAT pathway, mostly for investigational purposes. Quick et al. reported targeting JAK1 or STAT3 using CRISPR, which significantly reduced oncogene ubiquitin-specific protease 6 (USP6)/TRE17 in bone and soft tissue tumors, which indicates possibility of treatment of this type of malignancy by inhibition of JAK/STAT pathway (281).
Role of JAKs in Intracellular Crosstalks
The pivotal role of JAKs in intracellular signaling is not limited in the JAK/STAT axis. Crosstalk between JAK and other well-known signaling pathways has been documented in recent years. In 2007, Levine et al. reviewed the role of JAK2 in myeloproliferative disorders, and reported activation of two other major signaling pathways (PI3K/Akt and Ras/Raf/MAPK/ERK) through JAK2 (282), which was later reported by Birzniece et al. (19) as part of growth factor signaling, and Chiba et al. in Alzheimer's disease (20). It has been suggested that JAK2-mediated ERK activation is conducted through Ras, and via SH2-domain containing transforming protein (SHC), growth factor receptor-bond protein (GRB), and son of sevenless (SOS) (283, 284). Activation of PI3K has been proposed to be via phosphorylation of IRS1/2 (285). In a review of IFN-mediated signaling, Platanias has reported the activation of the catalytic subunit (p110) of PI3K, and MAPKs via phosphorylation of VAV or other guanine-nucleotide-exchange factors (GEFs), as a result of activation of members of JAK family (286). Direct activation of FAK via JAK2 has also been reported in multiple studies (287–289). Figure 2 illustrates the central role of JAK protein in activation of these three major pathways. Additionally, there is ample evidence in literature for JAK-independent activation of STAT3 via Src (290, 291).
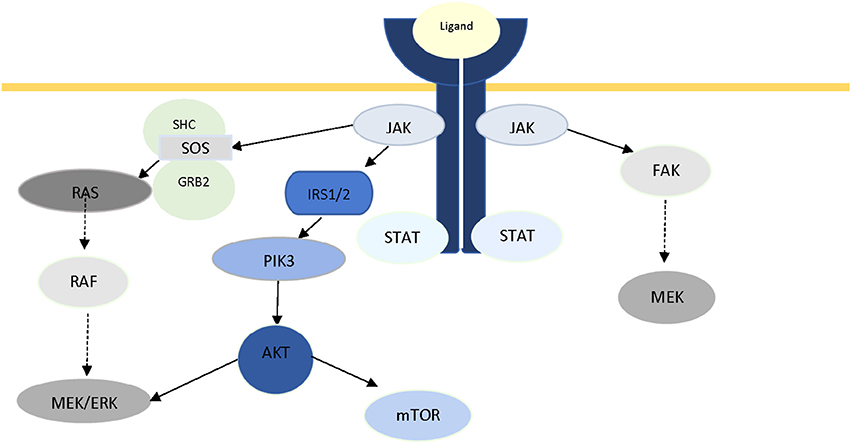
Figure 2. The role of JAK2 protein in intracellular crosstalk.
Conclusion
JAK/STAT is a major and versatile signaling pathway that has been extensively studied in the past two decades for crucial roles in cancer and inflammation. The variety of the receptors triggering this pathway is unmatched among known signaling cascades, and the wide range of downstream proteins indicate the importance of JAK2/STAT3 axis in cancer progression. Despite promising tumor suppression in animal studies as a result of inhibition of this pathway, however, the safety issues have marred the success of this therapeutic approach in clinical settings to some extent. Also, due to the versatile nature of the pathway, and potential crosstalks with multiple alternative pathways, a monotherapy-based approach might not create reliable results on the long term. A more systematic exploration of intra- and inter-pathway connections would be helpful in understanding the molecular mechanisms of the signal transduction in this cascade, as well, as identification of novel targets in cancer therapy.
Author Contributions
Both authors made an intellectual contribution to this work. Literature search, tables and figures are mostly done by EB. The outline, the text and final editing was done by HM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature (2013) 501:338–45. doi: 10.1038/nature12625
2. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature (2013) 501:328–37. doi: 10.1038/nature12624
3. Bertucci F, Birnbaum D. Reasons for breast cancer heterogeneity. J Biol. (2008) 7:6. doi: 10.1186/jbiol67
4. Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle (2007) 6:2332–8. doi: 10.4161/cc.6.19.4914
6. Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. (2015) 47:209–16. doi: 10.1038/ng.3214
7. Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. (2013) 31:545–52. doi: 10.1038/nbt.2594
8. Alderton GK. Tumour heterogeneity: the rise of the minority. Nat Rev Cancer (2013) 13:225. doi: 10.1038/nrc3499
9. Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer (2013) 13:365–76. doi: 10.1038/nrc3498
10. Zhou X, Agazie YM. Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J Biol Chem. (2009) 284:12226–34. doi: 10.1074/jbc.M900020200
11. Senapati S, Das S, Batra SK. Mucin-interacting proteins: from function to therapeutics. Trends Biochem Sci. (2010) 35:236–45. doi: 10.1016/j.tibs.2009.10.003
12. Al Masri A, Gendler SJ. Muc1 affects c-Src signaling in PyV MT-induced mammary tumorigenesis. Oncogene (2005) 24:5799–808. doi: 10.1038/sj.onc.1208738
13. Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene (2013) 32:1073–81. doi: 10.1038/onc.2012.158
14. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer (2006) 6:184–92. doi: 10.1038/nrc1819
15. Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. (2007) 67:7960–5. doi: 10.1158/0008-5472.CAN-07-2154
16. Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. (2007) 67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615
17. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi: 10.1126/science.1235122
18. Bruno B, Giaccone L, Rotta M, Anderson K, Boccadoro M. Novel targeted drugs for the treatment of multiple myeloma: from bench to bedside. Leukemia (2005) 19:1729–38. doi: 10.1038/sj.leu.2403905
19. Birzniece V, Sata A, Ho KK. Growth hormone receptor modulators. Rev Endocr Metab Disord. (2009) 10:145–56. doi: 10.1007/s11154-008-9089-x
20. Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer's disease. Expert Opin Ther Targets (2009) 13:1155–67. doi: 10.1517/14728220903213426
21. Aittomaki S, Pesu M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol. (2014) 114:18–23. doi: 10.1111/bcpt.12164
22. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. (2004) 117(Pt 8):1281–3. doi: 10.1242/jcs.00963
23. Harrison DA. The Jak/STAT pathway. Cold Spring Harb Perspect Biol. (2012) 4:a011205. doi: 10.1101/cshperspect.a011205
24. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
25. Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R, Krolewski JJ. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene (1990) 5:1329–36.
26. Sandberg EM, Wallace TA, Godeny MD, VonDerLinden D, Sayeski PP. Jak2 tyrosine kinase: a true jak of all trades? Cell Biochem Biophys. (2004) 41:207–32. doi: 10.1385/CBB:41:2:207
27. Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene (2000) 19:5662–79. doi: 10.1038/sj.onc.1203925
28. Harpur AG, Andres AC, Ziemiecki A, Aston RR, Wilks AF. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene (1992) 7:1347–53.
29. Shuai K, Schindler C, Prezioso VR, Darnell JE Jr. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science (1992) 258:1808–12.
30. Shuai K, Stark GR, Kerr IM, Darnell JE Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science (1993) 261:1744–6.
31. Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science (1994) 264:95–8.
32. Zhong Z, Wen Z, Darnell JE. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci USA. (1994) 91:4806–10.
33. Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. (1994) 13:2182–91.
34. Jatiani SS, Baker SJ, Silverman LR, Reddy EP. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer (2010) 1:979–93. doi: 10.1177/1947601910397187
35. Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood (2000) 95:19–29.
36. Luo H, Rose P, Barber D, Hanratty WP, Lee S, Roberts TM, et al. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. (1997) 17:1562–71.
37. Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. (2003) 349:1139–47. doi: 10.1056/NEJMoa022926
38. Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab. (2001) 12:252–7. doi: 10.1016/S1043-2760(01)00423-4
39. Rosenfeld RG, Kofoed E, Buckway C, Little B, Woods KA, Tsubaki J, et al. Identification of the first patient with a confirmed mutation of the JAK-STAT system. Pediatr Nephrol. (2005) 20:303–5. doi: 10.1007/s00467-004-1678-7
40. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (2005) 365:1054–61. doi: 10.1016/S0140-6736(05)71142-9
41. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434:1144–8. doi: 10.1038/nature03546
42. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
43. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell (2005) 7:387–97. doi: 10.1016/j.ccr.2005.03.023
44. Waterhouse M, Follo M, Pfeifer D, von Bubnoff N, Duyster J, Bertz H, et al. Sensitive and accurate quantification of JAK2 V617F mutation in chronic myeloproliferative neoplasms by droplet digital PCR. Ann Hematol. (2016) 95:739–44. doi: 10.1007/s00277-016-2623-0
45. Langabeer SE, Haslam K, O'Brien D, Enright H, Leahy M. Transient JAK2 V617F mutation in an aplastic anaemia patient with a paroxysmal nocturnal haemoglobinuria clone. Br J Haematol. (2013) 161:297–8. doi: 10.1111/bjh.12224
46. Oh ST, Gotlib J. JAK2 V617F and beyond: role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert Rev Hematol. (2010) 3:323–37. doi: 10.1586/ehm.10.28
47. Saki N, Shirzad R, Rahim F, Saki Malehi. A Estimation of diagnosis and prognosis in ET by assessment of CALR and JAK2(V617F) mutations and laboratory findings: a meta-analysis. Clin Transl Oncol. (2017) 19:874–883. doi: 10.1007/s12094-017-1618-1
48. Stein BL, Oh ST, Berenzon D, Hobbs GS, Kremyanskaya M, Rampal RK, et al. Polycythemia Vera: an appraisal of the biology and management 10 years after the discovery of JAK2 V617F. J Clin Oncol. (2015) 33:3953–60. doi: 10.1200/JCO.2015.61.6474
49. Hayashi T, Kobayashi Y, Kohsaka S, Sano K. The mutation in the ATP-binding region of JAK1, identified in human uterine leiomyosarcomas, results in defective interferon-gamma inducibility of TAP1 and LMP2. Oncogene (2006) 25:4016–26. doi: 10.1038/sj.onc.1209434
50. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature (2012) 481:157–63. doi: 10.1038/nature10725
51. Colaizzo D, Amitrano L, Tiscia GL, Grandone E, Guardascione MA, Margaglione M. A new JAK2 gene mutation in patients with polycythemia vera and splanchnic vein thrombosis. Blood (2007) 110:2768–9. doi: 10.1182/blood-2007-05-092502
52. Mercher T, Wernig G, Moore SA, Levine RL, Gu TL, Frohling S, et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood (2006) 108:2770–9. doi: 10.1182/blood-2006-04-014712
53. Wu QY, Ma MM, Fu L, Zhu YY, Liu Y, Cao J, et al. Roles of germline JAK2 activation mutation JAK2 V625F in the pathology of myeloproliferative neoplasms. Int J Biol Macromol. (2018) 116:1064–73. doi: 10.1016/j.ijbiomac.2018.05.120
54. Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood (2007) 109:2202–4. doi: 10.1182/blood-2006-09-045963
55. Walters DK, Mercher T, Gu TL, O'Hare T, Tyner JW, Loriaux M, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell (2006) 10:65–75. doi: 10.1016/j.ccr.2006.06.002
56. Bellanger D, Jacquemin V, Chopin M, Pierron G, Bernard OA, Ghysdael J, et al. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia (2014) 28:417–9. doi: 10.1038/leu.2013.271
57. Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science (2001) 293:300–3. doi: 10.1126/science.1061154
58. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. (2012) 366:1905–13. doi: 10.1056/NEJMoa1114885
59. Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stutz AM, Tal N, et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica (2014) 99:e188–92. doi: 10.3324/haematol.2014.104992
60. Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S, et al. Activating STAT6 mutations in follicular lymphoma. Blood (2015) 125:668–79. doi: 10.1182/blood-2014-06-582650
61. Oufkir T, Vaillancourt C. Phosphorylation of JAK2 by serotonin 5-HT (2A) receptor activates both STAT3 and ERK1/2 pathways and increases growth of JEG-3 human placental choriocarcinoma cell. Placenta (2011) 32:1033–40. doi: 10.1016/j.placenta.2011.09.005
62. Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de Ana A, Jones DR, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. (2001) 20:2497–507. doi: 10.1093/emboj/20.10.2497
63. Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, Moreno-Ortiz MC, Martinez AC, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. (1999) 13:1699–710.
64. Lukashova V, Asselin C, Krolewski JJ, Rola-Pleszczynski M, Stankova J. G-protein-independent activation of Tyk2 by the platelet-activating factor receptor. J Biol Chem. (2001) 276:24113–21. doi: 10.1074/jbc.M100720200
65. Lee JJ, Simborio HL, Reyes AW, Hop HT, Arayan LT, Lee HJ, et al. Influence of platelet-activating factor receptor (PAFR) on Brucella abortus infection: implications for manipulating the phagocytic strategy of B. abortus. BMC Microbiol. (2016) 16:70. doi: 10.1186/s12866-016-0685-8
66. Madamanchi NR, Li S, Patterson C, Runge MS. Thrombin regulates vascular smooth muscle cell growth and heat shock proteins via the JAK-STAT pathway. J Biol Chem. (2001) 276:18915–24. doi: 10.1074/jbc.M008802200
67. Ju H, Venema VJ, Liang H, Harris MB, Zou R, Venema RC. Bradykinin activates the Janus-activated kinase/signal transducers and activators of transcription (JAK/STAT) pathway in vascular endothelial cells: localization of JAK/STAT signalling proteins in plasmalemmal caveolae. Biochem J. (2000) 351(Pt 1):257–64. doi: 10.1042/bj3510257
68. Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature (1995) 375:247–50. doi: 10.1038/375247a0
69. Ali MS, Sayeski PP, Bernstein KE. Jak2 acts as both a STAT1 kinase and as a molecular bridge linking STAT1 to the angiotensin II AT1 receptor. J Biol Chem. (2000) 275:15586–93. doi: 10.1074/jbc.M908931199
70. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38:13–25. doi: 10.1016/j.immuni.2013.01.004
71. Barber DL, D'Andrea AD. Erythropoietin and interleukin-2 activate distinct JAK kinase family members. Mol Cell Biol. (1994) 14:6506–14.
72. Callus BA, Mathey-Prevot B. Interleukin-3-induced activation of the JAK/STAT pathway is prolonged by proteasome inhibitors. Blood (1998) 91:3182–92.
73. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. (2007) 178:2623–9. doi: 10.4049/jimmunol.178.5.2623
74. Pazdrak K, Stafford S, Alam R. The activation of the Jak-STAT 1 signaling pathway by IL-5 in eosinophils. J Immunol. (1995) 155:397–402.
75. Ogata N, Kouro T, Yamada A, Koike M, Hanai N, Ishikawa T, et al. JAK2 and JAK1 constitutively associate with an interleukin-5 (IL-5) receptor alpha and betac subunit, respectively, and are activated upon IL-5 stimulation. Blood (1998) 91:2264–71.
76. Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. (2003) 48:2092–6. doi: 10.1002/art.11095
77. Al-Rawi MA, Mansel RE, Jiang WG. Interleukin-7 (IL-7) and IL-7 receptor (IL-7R) signalling complex in human solid tumours. Histol Histopathol. (2003) 18:911–23. doi: 10.14670/HH-18.911
78. Yammani RR, Long D, Loeser RF. Interleukin-7 stimulates secretion of S100A4 by activating the JAK/STAT signaling pathway in human articular chondrocytes. Arthritis Rheum. (2009) 60:792–800. doi: 10.1002/art.24295
79. Demoulin JB, Grasso L, Atkins JM, Stevens M, Louahed J, Levitt RC, et al. Role of insulin receptor substrate-2 in interleukin-9-dependent proliferation. FEBS Lett. (2000) 482:200–4. doi: 10.1016/S0014-5793(00)02059-7
80. Fontaine RH, Cases O, Lelievre V, Mesples B, Renauld JC, Loron G, et al. IL-9/IL-9 receptor signaling selectively protects cortical neurons against developmental apoptosis. Cell Death Differ. (2008) 15:1542–52. doi: 10.1038/cdd.2008.79
81. Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. (1999) 274:16513–21.
82. Campbell CL, Jiang Z, Savarese DM, Savarese TM. Increased expression of the interleukin-11 receptor and evidence of STAT3 activation in prostate carcinoma. Am J Pathol. (2001) 158:25–32. doi: 10.1016/S0002-9440(10)63940-5
83. Lankford CS, Frucht DM. A unique role for IL-23 in promoting cellular immunity. J Leukoc Biol. (2003) 73:49–56. doi: 10.1189/jlb.0602326
84. Roy B, Bhattacharjee A, Xu B, Ford D, Maizel AL, Cathcart MK. IL-13 signal transduction in human monocytes: phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of Stats. J Leukoc Biol. (2002) 72:580–9. doi: 10.1189/jlb.72.3.580
85. Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell (2001) 104:9–19. doi: 10.1016/S0092-8674(01)00187-8
86. de Totero D Meazza, R, Zupo S, Cutrona G, Matis S, Colombo M, et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates proapoptotic signals in chronic lymphocytic leukemia B cells. Blood (2006) 107:3708–15. doi: 10.1182/blood-2005-09-3535
87. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
88. Ni B, Chen S, Xie H, Ma H. Functional polymorphisms in interleukin-23 receptor and susceptibility to esophageal squamous cell carcinoma in Chinese population. PLoS ONE (2014) 9:e89111. doi: 10.1371/journal.pone.0089111
89. Andoh A, Shioya M, Nishida A, Bamba S, Tsujikawa T, Kim-Mitsuyama S, et al. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J Immunol. (2009) 183:687–95. doi: 10.4049/jimmunol.0804169
90. Abdalla AE, Li Q, Xie L, Xie J. Biology of IL-27 and its role in the host immunity against Mycobacterium tuberculosis. Int J Biol Sci. (2015) 11:168–75. doi: 10.7150/ijbs.10464
91. Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. (2008) 19:347–56. doi: 10.1016/j.cytogfr.2008.08.003
92. Kotenko SV, Pestka S. Jak-Stat signal transduction pathway through the eyes of cytokine class II receptor complexes. Oncogene (2000) 19:2557–65. doi: 10.1038/sj.onc.1203524
93. Uze G, Monneron D. IL-28 and IL-29: newcomers to the interferon family. Biochimie (2007) 89:729–34. doi: 10.1016/j.biochi.2007.01.008
94. Guthridge MA, Stomski FC, Thomas D, Woodcock JM, Bagley CJ, Berndt MC, et al. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells (1998) 16:301–13. doi: 10.1002/stem.160301
95. Vosshenrich CA, Di Santo JP. Interleukin signaling. Curr Biol. (2002) 12:R760–3. doi: 10.1016/S0960-9822(02)01286-1
96. Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J Cell Biochem. (2008) 105:956–64. doi: 10.1002/jcb.21911
97. Andl CD, Mizushima T, Oyama K, Bowser M, Nakagawa H, Rustgi AK. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am J Physiol Gastrointest Liver Physiol. (2004) 287:G1227–37. doi: 10.1152/ajpgi.00253.2004
98. Velloso LA, Carvalho CR, Rojas FA, Folli F, Saad MJ. Insulin signalling in heart involves insulin receptor substrates-1 and−2, activation of phosphatidylinositol 3-kinase and the JAK 2-growth related pathway. Cardiovasc Res. (1998) 40:96–102.
99. Dudka AA, Sweet SM, Heath JK. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. (2010) 70:3391–401. doi: 10.1158/0008-5472.CAN-09-3033
100. Raju R, Palapetta SM, Sandhya VK, Sahu A, Alipoor A, Balakrishnan L, et al. A network map of FGF-1/FGFR signaling system. J Signal Transduct. (2014) 2014:962962. doi: 10.1155/2014/962962
101. Masamune A, Satoh M, Kikuta K, Suzuki N, Shimosegawa T. Activation of JAK-STAT pathway is required for platelet-derived growth factor-induced proliferation of pancreatic stellate cells. World J Gastroenterol. (2005) 11:3385–91. doi: 10.3748/wjg.v11.i22.3385
102. Sachsenmaier C, Sadowski HB, Cooper JA. STAT activation by the PDGF receptor requires juxtamembrane phosphorylation sites but not Src tyrosine kinase activation. Oncogene (1999) 18:3583–92. doi: 10.1038/sj.onc.1202694
103. Zhu X, Zhou W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front Endocrinol. (2015) 6:159. doi: 10.3389/fendo.2015.00159
104. Kim MS, Lee WS, Jeong J, Kim SJ, Jin W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget (2015) 6:40158–71. doi: 10.18632/oncotarget.5522
105. Korpelainen EI, Karkkainen M, Gunji Y, Vikkula M, Alitalo K. Endothelial receptor tyrosine kinases activate the STAT signaling pathway: mutant Tie-2 causing venous malformations signals a distinct STAT activation response. Oncogene (1999) 18:1–8. doi: 10.1038/sj.onc.1202288
106. Radhakrishnan A, Raju R, Tuladhar N, Subbannayya T, Thomas JK, Goel R, et al. A pathway map of prolactin signaling. J Cell Commun Signal. (2012) 6:169–73. doi: 10.1007/s12079-012-0168-0
107. Brooks AJ, Dai W, O'Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science (2014) 344:1249783. doi: 10.1126/science.1249783
108. Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. (2005) 280:27251–61. doi: 10.1074/jbc.M501376200
109. Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell (1998) 93:385–95.
110. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell (1998) 93:397–409.
111. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell (1998) 93:373–83.
112. Friedmann MC, Migone TS, Russell SM, Leonard WJ. Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proc Natl Acad Sci USA. (1996) 93:2077–82.
113. Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Interleukin-2 and its receptor complex (alpha, beta and gamma chains) in in situ and infiltrative human breast cancer: an immunohistochemical comparative study. Breast Cancer Res. (2004) 6:R1–7. doi: 10.1186/bcr730
114. D'Andrea AD, Fasman GD, Lodish HF. Erythropoietin receptor and interleukin-2 receptor beta chain: a new receptor family. Cell (1989) 58:1023–4.
115. O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell (2002) 109(Suppl.):S121–31. doi: 10.1016/S0092-8674(02)00701-8
116. Chen Z, Lund R, Aittokallio T, Kosonen M, Nevalainen O, Lahesmaa R. Identification of novel IL-4/Stat6-regulated genes in T lymphocytes. J Immunol. (2003) 171:3627–35. doi: 10.4049/jimmunol.171.7.3627
117. Kawakami K, Kawakami M, Puri RK. Specifically targeted killing of interleukin-13 (IL-13) receptor-expressing breast cancer by IL-13 fusion cytotoxin in animal model of human disease. Mol Cancer Ther. (2004) 3:137–47.
118. Amini-Vaughan ZJ, Martinez-Moczygemba M, Huston DP. Therapeutic strategies for harnessing human eosinophils in allergic inflammation, hypereosinophilic disorders, and cancer. Curr Allergy Asthma Rep. (2012) 12:402–12. doi: 10.1007/s11882-012-0290-3
119. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. (2003) 4:551–6. doi: 10.1038/ni938
120. Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. (2003) 4:540–5. doi: 10.1038/ni931
121. Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. (2005) 102:8686–91. doi: 10.1073/pnas.0500419102
122. Hanavadi S, Martin TA, Watkins G, Mansel RE, Jiang WG. Expression of interleukin 11 and its receptor and their prognostic value in human breast cancer. Ann Surg Oncol. (2006) 13:802–8. doi: 10.1245/ASO.2006.05.028
123. Bao L, Alexander JB, Shi VY, Mohan GC, Chan LS. Interleukin-4 up-regulation of epidermal interleukin-19 expression in keratinocytes involves the binding of signal transducer and activator of transcription 6 (Stat6) to the imperfect Stat6 sites. Immunology (2014) 143:601–8. doi: 10.1111/imm.12339
124. Wu S, Li Y, Yao L, Li Y, Jiang S, Gu W, et al. Interleukin-35 inhibits angiogenesis through STAT1 signalling in rheumatoid synoviocytes. Clin Exp Rheumatol. (2018) 36:223–227.
125. Briscoe J, Rogers NC, Witthuhn BA, Watling D, Harpur AG, Wilks AF, et al. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J. (1996) 15:799–809.
126. Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. (2008) 19:383–94. doi: 10.1016/j.cytogfr.2008.08.004
127. Arai KI, Lee F, Miyajima A, Miyatake S, Arai N, Yokota T. Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem. (1990) 59:783–836. doi: 10.1146/annurev.bi.59.070190.004031
128. Mullen M, and Gonzalez-Perez RR Leptin-induced JAK/STAT signaling and cancer growth. Vaccines (2016) 4:E26. doi: 10.3390/vaccines4030026
129. Wagner MA, Siddiqui MA. The JAK-STAT pathway in hypertrophic stress signaling and genomic stress response. JAKSTAT (2012) 1:131–41. doi: 10.4161/jkst.20702
130. Sonier B, Arseneault M, Lavigne C, Ouellette RJ, Vaillancourt C. The 5-HT2A serotoninergic receptor is expressed in the MCF-7 human breast cancer cell line and reveals a mitogenic effect of serotonin. Biochem Biophys Res Commun. (2006) 343:1053–9. doi: 10.1016/j.bbrc.2006.03.080
131. Mukherjee D, Zhao J. The role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res. (2013) 3:46–57.
132. Ueno H, Sasaki K, Miyagawa K, Honda H, Mitani K, Yazaki Y, et al. Antisense repression of proto-oncogene c-Cbl enhances activation of the JAK-STAT pathway but not the ras pathway in epidermal growth factor receptor signaling. J Biol Chem. (1997) 272:8739–43. doi: 10.1074/jbc.272.13.8739
133. Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. (2011) 18:971–6. doi: 10.1038/nsmb.2099
134. Wu J, Patmore DM, Jousma E, Eaves DW, Breving K, Patel AV, et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene (2014) 33:173–80. doi: 10.1038/onc.2012.579
135. Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. (2016) 49:1360–8. doi: 10.3892/ijo.2016.3632
136. Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene (2015) 34:3107–19. doi: 10.1038/onc.2014.257
137. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14:736–46. doi: 10.1038/nrc3818
138. Eyking A, Ey B, Runzi M, Roig AI, Reis H, Schmid KW, et al. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology (2011) 141:2154–65. doi: 10.1053/j.gastro.2011.08.043
139. Liu C, Gao F, Li B, Mitchel RE, Liu X, Lin J, et al. TLR4 knockout protects mice from radiation-induced thymic lymphoma by downregulation of IL6 and miR-21. Leukemia (2011) 25:1516–9. doi: 10.1038/leu.2011.113
140. Wang C, Cao S, Yan Y, Ying Q, Jiang T, Xu K, et al. TLR9 expression in glioma tissues correlated to glioma progression and the prognosis of GBM patients. BMC Cancer (2010) 10:415. doi: 10.1186/1471-2407-10-415
141. Herrmann A, Cherryholmes G, Schroeder A, Phallen J, Alizadeh D, Xin H, et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. (2014) 74:5218–28. doi: 10.1158/0008-5472.CAN-14-1151
142. Pezet A, Buteau H, Kelly PA, Edery M. The last proline of Box 1 is essential for association with JAK2 and functional activation of the prolactin receptor. Mol Cell Endocrinol. (1997) 129:199–208.
143. Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation of Janus kinase (JAK) activation. Biochem J. (2014) 462:1–13. doi: 10.1042/BJ20140712
144. Behrmann I, Smyczek T, Heinrich PC, Schmitz-Van de Leur H, Komyod, W, Giese, B, et al. Janus kinase (Jak) subcellular localization revisited: the exclusive membrane localization of endogenous Janus kinase 1 by cytokine receptor interaction uncovers the Jak.receptor complex to be equivalent to a receptor tyrosine kinase. J Biol Chem. (2004) 279:35486–93. doi: 10.1074/jbc.M404202200
145. LaFave LM, Levine RL. JAK2 the future: therapeutic strategies for JAK-dependent malignancies. Trends Pharmacol Sci. (2012) 33:574–82. doi: 10.1016/j.tips.2012.08.005
146. Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat Struct Mol Biol. (2014) 21:443–8. doi: 10.1038/nsmb.2807
147. Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. (2000) 20:3387–95. doi: 10.1128/MCB.20.10.3387-3395.2000
148. Li WX Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. (2008) 18:545–51. doi: 10.1016/j.tcb.2008.08.008
149. Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene (2000) 19:2628–37. doi: 10.1038/sj.onc.1203481
150. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi: 10.1038/nrc2734
151. Trevino JG, Gray MJ, Nawrocki ST, Summy JM, Lesslie DP, Evans DB, et al. Src activation of Stat3 is an independent requirement from NF-kappaB activation for constitutive IL-8 expression in human pancreatic adenocarcinoma cells. Angiogenesis (2006) 9:101–10. doi: 10.1007/s10456-006-9038-9
152. Liongue C, Ward AC. Evolution of the JAK-STAT pathway. JAKSTAT (2013) 2:e22756. doi: 10.4161/jkst.22756
153. Subramaniam PS, Torres BA, Johnson HM. So many ligands, so few transcription factors: a new paradigm for signaling through the STAT transcription factors. Cytokine (2001) 15:175–87. doi: 10.1006/cyto.2001.0905
154. Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. (2006) 20:3372–81. doi: 10.1101/gad.1485406
155. Hulsmann BB, Labokha AA, Gorlich D. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell (2012) 150:738–51. doi: 10.1016/j.cell.2012.07.019
157. Jans DA, Xiao CY, Lam MH. Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays (2000) 22:532–44. doi: 10.1002/(SICI)1521-1878(200006)22:6<532::AID-BIES6>3.0.CO;2-O
158. Ma J, Cao X. Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell Signal. (2006) 18:1117–26. doi: 10.1016/j.cellsig.2005.06.016
159. Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA. (2005) 102:8150–5. doi: 10.1073/pnas.0501643102
160. Ushijima R, Sakaguchi N, Kano A, Maruyama A, Miyamoto Y, Sekimoto T, et al. Extracellular signal-dependent nuclear import of STAT3 is mediated by various importin alphas. Biochem Biophys Res Commun. (2005) 330:880–6. doi: 10.1016/j.bbrc.2005.03.063
161. Meyer T, Vinkemeier U. STAT nuclear translocation: potential for pharmacological intervention. Expert Opin Ther Targets (2007) 11:1355–65. doi: 10.1517/14728222.11.10.1355
163. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
164. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. (2009) 6:587–95. doi: 10.1038/nrclinonc.2009.129
165. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21.
166. Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol. (1996) 16:1759–69.
167. Banninger G, Reich NC. STAT2 nuclear trafficking. J Biol Chem. (2004) 279:39199–206. doi: 10.1074/jbc.M400815200
168. Mowen K, David M. Role of the STAT1-SH2 domain and STAT2 in the activation and nuclear translocation of STAT1. J Biol Chem. (1998) 273:30073–6.
169. Sironi JJ, Ouchi T. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem. (2004) 279:4066–74. doi: 10.1074/jbc.M307774200
170. Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. (2002) 2:381–92. doi: 10.2174/1566524023362456
171. Bernabei P, Coccia EM, Rigamonti L, Bosticardo M, Forni G, Pestka S, et al. Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J Leukoc Biol. (2001) 70:950–60. doi: 10.1189/jlb.70.6.950
172. Sun Y, Qiao L, Xia HH, Lin MC, Zou B, Yuan Y, et al. Regulation of XAF1 expression in human colon cancer cell by interferon beta: activation by the transcription regulator STAT1. Cancer Lett. (2008) 260:62–71. doi: 10.1016/j.canlet.2007.10.014
173. Egwuagu CE, Li W, Yu CR, Che Mei Lin M, Chan, CC, Nakamura, T, et al. Interferon-gamma induces regression of epithelial cell carcinoma: critical roles of IRF-1 and ICSBP transcription factors. Oncogene (2006) 25:3670–9. doi: 10.1038/sj.onc.1209402
174. Burke F, Smith PD, Crompton MR, Upton C, Balkwill FR. Cytotoxic response of ovarian cancer cell lines to IFN-gamma is associated with sustained induction of IRF-1 and p21 mRNA. Br J Cancer (1999) 80:1236–44. doi: 10.1038/sj.bjc.6690491
175. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. (2007) 282:20059–63. doi: 10.1074/jbc.R700016200
176. Chan SR, Rickert CG, Vermi W, Sheehan KC, Arthur C, Allen JA, et al. Dysregulated STAT1-SOCS1 control of JAK2 promotes mammary luminal progenitor cell survival and drives ERalpha(+) tumorigenesis. Cell Death Differ. (2014) 21:234–46. doi: 10.1038/cdd.2013.116
177. Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon-gamma. Oncogene (2007) 26:594–603. doi: 10.1038/sj.onc.1209807
178. Li S, Xia X, Mellieon FM, Liu J, Steele S. Candidate genes associated with tumor regression mediated by intratumoral IL-12 electroporation gene therapy. Mol Ther. (2004) 9:347–54. doi: 10.1016/j.ymthe.2003.11.022
179. Cao ZH, Zheng QY, Li GQ, Hu XB, Feng SL, Xu GL, et al. STAT1-mediated down-regulation of Bcl-2 expression is involved in IFN-gamma/TNF-alpha-induced apoptosis in NIT-1 cells. PLoS ONE (2015) 10:e0120921. doi: 10.1371/journal.pone.0120921
180. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. (1998) 95:7556–61.
181. Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, et al. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J. (2000) 19:263–72. doi: 10.1093/emboj/19.2.263
182. Kominsky SL, Hobeika AC, Lake FA, Torres BA, Johnson HM. Down-regulation of neu/HER-2 by interferon-gamma in prostate cancer cells. Cancer Res. (2000) 60:3904–8.
183. Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. (2006) 66:3649–57. doi: 10.1158/0008-5472.CAN-05-3612
184. Huang S, Bucana CD, Van Arsdall M Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene (2002) 21:2504–12. doi: 10.1038/sj.onc.1205341
185. Liu H, Ma Y, Cole SM, Zander C, Chen KH, Karras J, et al. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood (2003) 102:344–52. doi: 10.1182/blood-2002-11-3396
186. Gao LF, Xu DQ, Wen LJ, Zhang XY, Shao YT, Zhao XJ. Inhibition of STAT3 expression by siRNA suppresses growth and induces apoptosis in laryngeal cancer cells. Acta Pharmacol Sin. (2005) 26:377–83. doi: 10.1111/j.1745-7254.2005.00053.x
187. Verma NK, Davies AM, Long A, Kelleher D, Volkov Y. STAT3 knockdown by siRNA induces apoptosis in human cutaneous T-cell lymphoma line Hut78 via downregulation of Bcl-xL. Cell Mol Biol Lett. (2010) 15:342–55. doi: 10.2478/s11658-010-0008-2
188. Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene (2006) 25:6817–30. doi: 10.1038/sj.onc.1209942
189. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
190. Huang JS, Chuang LY, Guh JY, Huang YJ, Hsu MS. Antioxidants attenuate high glucose-induced hypertrophic growth in renal tubular epithelial cells. Am J Physiol Renal Physiol. (2007) 293:F1072–82. doi: 10.1152/ajprenal.00020.2007
191. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. (2008) 118:3367–77. doi: 10.1172/JCI35213
192. Xie TX, Huang FJ, Aldape KD, Kang SH, Liu M, Gershenwald JE, et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. (2006) 66:3188–96. doi: 10.1158/0008-5472.CAN-05-2674
193. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. (2009) 206:1457–64. doi: 10.1084/jem.20090207
194. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell (2009) 15:114–23. doi: 10.1016/j.ccr.2008.12.018
195. Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells (2009) 27:857–65. doi: 10.1002/stem.23
196. Dalwadi H, Krysan K, Heuze-Vourc'h N, Dohadwala M, Elashoff D, Sharma S, et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin Cancer Res. (2005) 11:7674–82. doi: 10.1158/1078-0432.CCR-05-1205
197. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. (2006) 203:1651–6. doi: 10.1084/jem.20051848
198. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity (1999) 10:39–49.
199. Caprioli F, Sarra M, Caruso R, Stolfi C, Fina D, Sica G, et al. Autocrine regulation of IL-21 production in human T lymphocytes. J Immunol. (2008) 180:1800–7. doi: 10.4049/jimmunol.180.3.1800
200. Li H, Lee J, He C, Zou MH, Xie Z. Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids. Am J Physiol Endocrinol Metab. (2014) 306:E197–209. doi: 10.1152/ajpendo.00202.2013
201. Teng TS, Lin B, Manser E, Ng DC, Cao X. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator betaPIX. J Cell Sci. (2009) 122(Pt 22):4150–9. doi: 10.1242/jcs.057109
202. Kotenko SV, Langer JA. Full house: 12 receptors for 27 cytokines. Int Immunopharmacol. (2004) 4:593–608. doi: 10.1016/j.intimp.2004.01.003
203. Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. (2005) 25:7432–40. doi: 10.1128/MCB.25.17.7432-7440.2005
204. Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, et al. Distinct roles of different NF-kappa B subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol. (2007) 178:6777–88. doi: 10.4049/jimmunol.178.11.6777
205. Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell (2004) 118:453–64. doi: 10.1016/j.cell.2004.08.007
206. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. (2004) 10:48–54. doi: 10.1038/nm976
207. Kunigal S, Lakka SS, Sodadasu PK, Estes N, Rao JS. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int J Oncol. (2009) 34:1209–20. doi: 10.3892/ijo_00000249
208. Liang ZW, Guo BF, Li Y, Li XJ, Li X, Zhao LJ, et al. Plasmid-based Stat3 siRNA delivered by hydroxyapatite nanoparticles suppresses mouse prostate tumour growth in vivo. Asian J Androl. (2011) 13:481–6. doi: 10.1038/aja.2010.167
209. Chang HC, Han L, Goswami R, Nguyen ET, Pelloso D, Robertson MJ, et al. Impaired development of human Th1 cells in patients with deficient expression of STAT4. Blood (2009) 113:5887–90. doi: 10.1182/blood-2008-09-179820
210. de Groot RP, Raaijmakers, JA, Lammers, JW, Koenderman L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. (2000) 3:299–305. doi: 10.1006/mcbr.2000.0231
211. Creamer BA, Sakamoto K, Schmidt JW, Triplett AA, Moriggl R, Wagner KU. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol Cell Biol. (2010) 30:2957–70. doi: 10.1128/MCB.00851-09
212. Gatzka M, Piekorz R, Moriggl R, Rawlings J, Ihle JN. A role for STAT5A/B in protection of peripheral T-lymphocytes from postactivation apoptosis: insights from gene expression profiling. Cytokine (2006) 34:143–54. doi: 10.1016/j.cyto.2006.04.003
213. Nosaka T, Kawashima T, Misawa K, Ikuta K, Mui AL, Kitamura T. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. (1999) 18:4754–65. doi: 10.1093/emboj/18.17.4754
214. Xu M, Nie L, Kim SH, Sun XH. STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J. (2003) 22:893–904. doi: 10.1093/emboj/cdg094
215. Calabrese V, Mallette FA, Deschenes-Simard X, Ramanathan S, Gagnon J, Moores A, et al. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell (2009) 36:754–67. doi: 10.1016/j.molcel.2009.09.044
216. Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. (2005) 6:715–25. doi: 10.1038/nrm1714
217. Longmore GD A unique role for Stat5 in recovery from acute anemia. J Clin Invest. (2006) 116:626–8. doi: 10.1172/JCI27988
218. Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. (2007) 21:43–8. doi: 10.1101/gad.1487307
219. Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. (2004) 201:139–55. doi: 10.1111/j.0105-2896.2004.00192.x
220. Wurster AL, Rodgers VL, White MF, Rothstein TL, Grusby MJ. Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J Biol Chem. (2002) 277:27169–75. doi: 10.1074/jbc.M201207200
221. Maier E, Duschl A, and Horejs-Hoeck J STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol. (2012) 42:2827–33. doi: 10.1002/eji.201242433
222. Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem. (1997) 272:20070–6.
223. Van Gool SW, Vandenberghe, P, de, Boer M, Ceuppens JL. CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol Rev. (1996) 153:47–83.
224. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity (2000) 13:795–804. doi: 10.1016/S1074-7613(00)00077-7
225. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
226. Yu H, Jove R. The STATs of cancer–new molecular targets come of age. Nat Rev Cancer (2004) 4:97–105. doi: 10.1038/nrc1275
227. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. (1997) 94:3801–4.
228. Ivanov VN, Bhoumik A, Krasilnikov M, Raz R, Owen-Schaub LB, Levy D, et al. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol Cell (2001) 7:517–28.
229. Yokogami K, Yamashita S, Takeshima H. Hypoxia-induced decreases in SOCS3 increase STAT3 activation and upregulate VEGF gene expression. Brain Tumor Pathol. (2013) 30:135–43. doi: 10.1007/s10014-012-0122-0
230. Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. (2012) 13:290–9. doi: 10.1038/ni.2227
231. Thierfelder WE, van Deursen JM, Yamamoto, K, Tripp, RA, Sarawar, SR, Carson, RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature (1996) 382:171–4. doi: 10.1038/382171a0
232. Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. (2004) 24:8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004
233. Zeng R, Aoki Y, Yoshida M, Arai K, Watanabe S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. J Immunol. (2002) 168:4567–75. doi: 10.4049/jimmunol.168.9.4567
234. Ling MT, Wang X, Zhang X, Wong YC. The multiple roles of Id-1 in cancer progression. Differentiation (2006) 74:481–7. doi: 10.1111/j.1432-0436.2006.00083.x
235. Bruns HA, Kaplan MH. The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol. (2006) 57:245–53. doi: 10.1016/j.critrevonc.2005.08.005
236. Shimoda K, van Deursen J, Sangster, MY, Sarawar, SR, Carson, RT, Tripp, RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature (1996) 380:630–3. doi: 10.1038/380630a0
237. Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front Immunol. (2017) 8:29. doi: 10.3389/fimmu.2017.00029
238. Kim VN MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. (2005) 6:376–85. doi: 10.1038/nrm1644
239. Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell (2010) 39:493–506. doi: 10.1016/j.molcel.2010.07.023
240. Yang CH, Yue J, Fan M, Pfeffer LM. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res. (2010) 70:8108–16. doi: 10.1158/0008-5472.CAN-10-2579
241. Kohanbash G, Okada H. MicroRNAs and STAT interplay. Semin Cancer Biol. (2012) 22:70–5. doi: 10.1016/j.semcancer.2011.12.010
242. Passamonti F, Maffioli M. The role of JAK2 inhibitors in MPNs 7 years after approval. Blood (2018) 131:2426–2435. doi: 10.1182/blood-2018-01-791491
243. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. (2014) 370:2377–86. doi: 10.1056/NEJMoa1310476
244. Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol. (2016) 74:841–50. doi: 10.1016/j.jaad.2016.01.013
245. Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. (2015) 1:643–51. doi: 10.1001/jamaoncol.2015.1590
246. Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. (2015) 67:334–43. doi: 10.1002/art.38949
247. Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. (2016) 75:1057–64. doi: 10.1136/annrheumdis-2015-208279
248. Narla RK, Liu XP, Myers DE, Uckun FM. 4-(3'-Bromo-4'hydroxylphenyl)-amino-6,7-dimethoxyquinazoline: a novel quinazoline derivative with potent cytotoxic activity against human glioblastoma cells. Clin Cancer Res. (1998) 4:1405–14.
249. Seavey MM, Lu LD, Stump KL, Wallace NH, Hockeimer W, O'Kane TM, et al. Therapeutic efficacy of CEP-33779, a novel selective JAK2 inhibitor, in a mouse model of colitis-induced colorectal cancer. Mol Cancer Ther. (2012) 11:984–93. doi: 10.1158/1535-7163.MCT-11-0951
250. Park JS, Lee J, Lim MA, Kim EK, Kim SM, Ryu JG, et al. JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J Immunol. (2014) 192:4417–24. doi: 10.4049/jimmunol.1300514
251. Chai HT, Yip HK, Sun CK, Hsu SY, Leu S. AG490 suppresses EPO-mediated activation of JAK2-STAT but enhances blood flow recovery in rats with critical limb ischemia. J Inflamm. (2016) 13:18. doi: 10.1186/s12950-016-0126-3
252. Gupta V, Mesa RA, Deininger MW, Rivera CE, Sirhan S, Brachmann CB, et al. A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis. Haematologica (2017) 102:94–102. doi: 10.3324/haematol.2016.148924
253. Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko OV, Stanislavchuk M, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. (2017) 76:1009–1019. doi: 10.1136/annrheumdis-2016-210105
254. Vermeire S, Schreiber S, Petryka R, Kuehbacher T, Hebuterne X, Roblin X, et al. Clinical remission in patients with moderate-to-severe Crohn's disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet (2017) 389:266–275. doi: 10.1016/S0140-6736(16)32537-5
255. Kubo S, Nakayamada S, Tanaka Y. Baricitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Immunol. (2016) 12:911–9. doi: 10.1080/1744666X.2016.1214576
256. Sun Y, Moretti L, Giacalone NJ, Schleicher S, Speirs CK, Carbone DP, et al. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J Thorac Oncol. (2011) 6:699–706. doi: 10.1097/JTO.0b013e31820d9d11
257. Verstovsek S, Tam CS, Wadleigh M, Sokol L, Smith CC, Bui LA, et al. Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leuk Res. (2014) 38:316–22. doi: 10.1016/j.leukres.2013.12.006
258. Hay AE, Murugesan A, DiPasquale AM, Kouroukis T, Sandhu I, Kukreti V, et al. A phase II study of AT9283, an aurora kinase inhibitor, in patients with relapsed or refractory multiple myeloma: NCIC clinical trials group IND.191. Leuk Lymphoma (2016) 57:1463–6. doi: 10.3109/10428194.2015.1091927
259. Yang J, Ikezoe T, Nishioka C, Furihata M, Yokoyama A. AZ960, a novel Jak2 inhibitor, induces growth arrest and apoptosis in adult T-cell leukemia cells. Mol Cancer Ther. (2010) 9:3386–95. doi: 10.1158/1535-7163.MCT-10-0416
260. Plimack ER, Lorusso PM, McCoon P, Tang W, Krebs AD, Curt G, et al. AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist (2013) 18:819–20. doi: 10.1634/theoncologist.2013-0198
261. Baffert F, Regnier CH, De Pover A Pissot-Soldermann C, Tavares GA, Blasco F, et al. Potent and selective inhibition of polycythemia by the quinoxaline JAK2 inhibitor NVP-BSK805. Mol Cancer Ther. (2010) 9:1945–55. doi: 10.1158/1535-7163.MCT-10-0053
262. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. (2010) 363:1117–27. doi: 10.1056/NEJMoa1002028
263. Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood (2010) 115:1131–6. doi: 10.1182/blood-2009-10-246363
264. Goede V, Busch R, Bahlo J, Chataline V, Kremers S, Muller L, et al. Low-dose fludarabine with or without darbepoetin alfa in patients with chronic lymphocytic leukemia and comorbidity: primary results of the CLL9 trial of the German CLL Study Group. Leuk Lymphoma (2016) 57:596–603. doi: 10.3109/10428194.2015.1079314
265. Siddiquee, K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. (2007) 104:7391–6. doi: 10.1073/pnas.0609757104
266. Miyoshi K, Takaishi M, Nakajima K, Ikeda M, Kanda T, Tarutani M, et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest Dermatol. (2011) 131:108–17. doi: 10.1038/jid.2010.255
267. Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. (2015) 26:998–1005. doi: 10.1093/annonc/mdv026
268. Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, et al. Phase I Study of OPB-31121, an Oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res Treat. (2015) 47:607–15. doi: 10.4143/crt.2014.249
269. Rath KS, Naidu SK, Lata P, Bid HK, Rivera BK, McCann GA, et al. HO-3867, a safe STAT3 inhibitor, is selectively cytotoxic to ovarian cancer. Cancer Res. (2014) 74:2316–27. doi: 10.1158/0008-5472.CAN-13-2433
270. Yue P, Lopez-Tapia F, Paladino D, Li Y, Chen CH, Namanja AT, et al. Hydroxamic Acid and Benzoic acid-based STAT3 inhibitors suppress human glioma and breast cancer phenotypes in vitro and in vivo. Cancer Res. (2016) 76:652–63. doi: 10.1158/0008-5472.CAN-14-3558
271. Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. (2014) 349:8–14. doi: 10.1016/j.canlet.2014.04.003
272. Shen L, Zhang G, Lou Z, Xu G, Zhang G. Cryptotanshinone enhances the effect of Arsenic trioxide in treating liver cancer cell by inducing apoptosis through downregulating phosphorylated- STAT3 in vitro and in vivo. BMC Complement Altern Med. (2017) 17:106. doi: 10.1186/s12906-016-1548-4
273. Zhang Q, Zhang C, He J, Guo Q, Hu D, Yang X, et al. STAT3 inhibitor stattic enhances radiosensitivity in esophageal squamous cell carcinoma. Tumour Biol. (2015) 36:2135–42. doi: 10.1007/s13277-014-2823-y
274. Frank DA, Mahajan S, Ritz J. Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med. (1999) 5:444–7. doi: 10.1038/7445
275. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. (2017) 4:e225–36. doi: 10.1016/S2352-3026(17)30027-3
276. Akahane K, Li Z, Etchin J, Berezovskaya A, Gjini E, Masse CE, et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br J Haematol. (2017) 177:271–82. doi: 10.1111/bjh.14563
277. Musumeci F, Greco C, Giacchello I, Fallacara AL, Ibrahim MM, Grossi G, et al. An Update on JAK Inhibitors. Curr Med Chem. (2018). doi: 10.2174/0929867325666180327093502
278. Sen M, Grandis JR. Nucleic acid-based approaches to STAT inhibition. JAKSTAT (2012) 1:285–91. doi: 10.4161/jkst.22312
279. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. (2015) 7:314ra185. doi: 10.1126/scitranslmed.aac5272
280. Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. (2012) 2:694–705. doi: 10.1158/2159-8290.CD-12-0191
281. Quick L, Young R, Henrich IC, Wang X, Asmann YW, Oliveira AM, et al. Jak1-STAT3 signals are essential effectors of the USP6/TRE17 oncogene in tumorigenesis. Cancer Res. (2016) 76:5337–47. doi: 10.1158/0008-5472.CAN-15-2391
282. Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer (2007) 7:673–83. doi: 10.1038/nrc2210
283. Fernandez-Perez L, Guerra B, Diaz-Chico JC, Flores-Morales A. Estrogens regulate the hepatic effects of growth hormone, a hormonal interplay with multiple fates. Front Endocrinol. (2013) 4:66. doi: 10.3389/fendo.2013.00066
284. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. (2014) 13:379–95. doi: 10.1038/nrd4296
285. Vansaun MN Molecular pathways: adiponectin and leptin signaling in cancer. Clin Cancer Res. (2013) 19:1926–32. doi: 10.1158/1078-0432.CCR-12-0930
286. Platanias LC Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
287. Swiatek-Machado K, Mieczkowski J, Ellert-Miklaszewska A, Swierk P, Fokt I, Szymanski S, et al. Novel small molecular inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and exhibit a potent antitumor activity in glioma cells. Cancer Biol Ther. (2012) 13:657–70. doi: 10.4161/cbt.20083
288. Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. (2014) 16:1229–43. doi: 10.1093/neuonc/nou046
289. Zhu T, Goh EL, Lobie PE. Growth hormone stimulates the tyrosine phosphorylation and association of p125 focal adhesion kinase (FAK) with JAK2. Fak is not required for stat-mediated transcription. J Biol Chem. (1998) 273:10682–9.
290. Bjorge JD, Pang AS, Funnell M, Chen KY, Diaz R, Magliocco AM, et al. Simultaneous siRNA targeting of Src and downstream signaling molecules inhibit tumor formation and metastasis of a human model breast cancer cell line. PLoS ONE (2011) 6:e19309. doi: 10.1371/journal.pone.0019309
Keywords: janus tyrosine kinases (JAKs), signal transducers and activators of transcription (STATs), cancer, signaling pathways, proliferation, survival
Citation: Bousoik E and Montazeri Aliabadi H (2018) “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 8:287. doi: 10.3389/fonc.2018.00287
Received: 09 June 2018; Accepted: 09 July 2018;
Published: 31 July 2018.
Edited by:
Anna Rita Migliaccio, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Alessandro Malara, University of Pavia, ItalyFrancesca Palandri, Università degli Studi di Bologna, Italy
Copyright © 2018 Bousoik and Montazeri Aliabadi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hamidreza Montazeri Aliabadi, bW9udGF6ZXJAY2hhcG1hbi5lZHU=