 Nicole Scholz
Nicole Scholz- Rudolf Schönheimer Institute of Biochemistry, Division of General Biochemistry, Faculty of Medicine, University Leipzig, Leipzig, Germany
In mammals, numerous organ systems are equipped with adhesion G protein-coupled receptors (aGPCRs) to shape cellular processes including migration, adhesion, polarity and guidance. All of these cell biological aspects are closely associated with tumor cell biology. Consistently, aberrant expression or malfunction of aGPCRs has been associated with dysplasia and tumorigenesis. Mounting evidence indicates that cancer cells comprise viscoelastic properties that are different from that of their non-tumorigenic counterparts, a feature that is believed to contribute to the increased motility and invasiveness of metastatic cancer cells. This is particularly interesting in light of the recent identification of the mechanosensitive facility of aGPCRs. aGPCRs are signified by large extracellular domains (ECDs) with adhesive properties, which promote the engagement with insoluble ligands. This configuration may enable reliable force transmission to the ECDs and may constitute a molecular switch, vital for mechano-dependent aGPCR signaling. The investigation of aGPCR function in mechanosensation is still in its infancy and has been largely restricted to physiological contexts. It remains to be elucidated if and how aGPCR function affects the mechanoregulation of tumor cells, how this may shape the mechanical signature and ultimately determines the pathological features of a cancer cell. This article aims to view known aGPCR functions from a biomechanical perspective and to delineate how this might impinge on the mechanobiology of cancer cells.
Introduction
Cancer is an ongoing threat to human health. In 2012, worldwide 14 Mio people were diagnosed with a form of malignant cancer (1) and the International Agency for Research on Cancer predicts an increase in cancer cases up to terrifying 20 Mio by 2030 (2).
Each cell in our body has the capacity to morph into a tumor cell if its genome accumulates a critical load of lesions to disturb the delicate balance between proliferation and apoptosis. Cancer scientists have long been aware of genetic and epigenetic changes as well as alterations in the perception and integration of biochemical signals as the source of tumorigenesis and metastasis. However, more recently it has been recognized that cell growth, invasion and metastasis are intricately linked to the constituent cells’ facility to detect, integrate and respond to intrinsic and extrinsic mechanical cues (3). Since 2009 the National Cancer Institute has been supporting 12 leading institutions in the US to establish Physical Science—Oncology Centers and to build a collaborative network to interrogate how the micromechanical interior and exterior of a tumor cell shapes its biochemical properties and vice versa. Thus, the realization that every cancer cell is subject to similar physical constraints may bring forth an exciting era of cancer research and has the potential to revolutionize the development of therapeutics.
Adhesion G protein-coupled receptors (aGPCR) comprise the second largest group within the superfamily of G protein-coupled receptors (GPCRs) and are expressed in most organ systems. aGPCRs are known to modulate pivotal cellular functions including migration, adhesion, polarity and guidance (4). Consistently, members from all aGPCR subfamilies have been reported in the context of dysplasia and tumorigenesis in one way or another (5–8). While for some receptors it is mere evidence of faulty expression or changes in their activity (e.g., VLGR1/ADGRV, LPHN/ADGRL), for others more detailed knowledge about the molecular mechanisms that make cells or tissues go awry have emerged [e.g., CD97/ADGRE5 (9–11), GPR116/ADGRF5 (12, 13), GPR133/ADGRD1 (14, 15), GPR56/ADGRG1 (16–19), and BAI1/ADRGB1 (20–22)].
A more recent development in aGPCR biology is the finding that these receptors possess mechanoceptive features (23–27), which contrasts the common view of GPCRs as chemosensors. Although most of the data that underpin this finding have been collected in physiological settings, it is conceivable that many of the seemingly unrelated pathophysiological phenotypes caused by aGPCR malfunction may be the result of defective mechanosensation. Thus, aGPCRs may constitute a connecting element between neoplastic, malignant, or metastatic properties and the mechanical phenotype of a given cell (Figure 1). This article aims to connect the current knowledge of aGPCR biology and cancer physics and will discuss potential intersections.
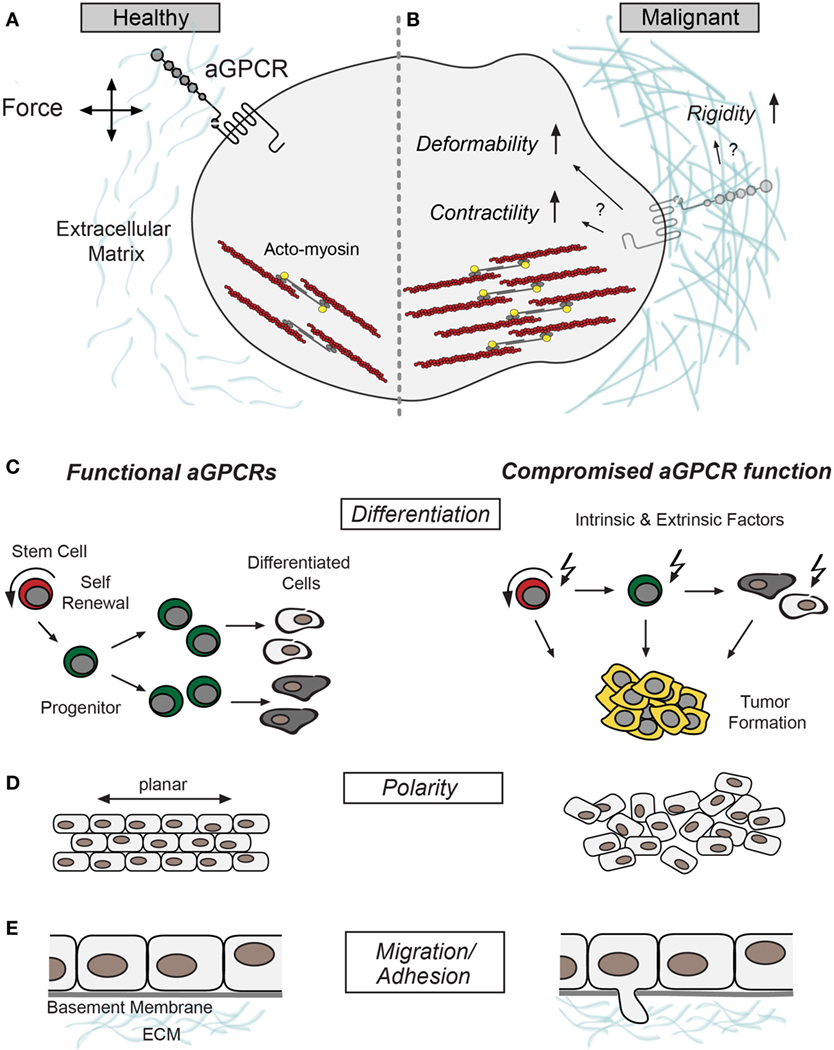
Figure 1. Putative roles of adhesion G protein-coupled receptors (aGPCRs) in cancer. (A) Schematic illustration of a healthy cell without structural abnormalities and proper aGPCR function aligned with (B) a malignant cell signified by increased contractility, compliance, and extracellular matrix rigidity (28). These changes could intersect with aGPCR dysfunction and/or altered expression. Several aGPCRs have been shown to signal through RhoA (18, 29), which indirectly stimulates myosin-light chain phosphorylation (yellow) to promote acto-myosin contractility (F-actin and myosin in red and dark gray, respectively) (30). (C–E) Show different cellular properties previously associated with aGPCR function and malignancies; left panel: intact aGPCR function. Right panel: cellular deficits related to compromised aGPCR function/expression. (C) Schemes of differentiation hierarchy. Adult stem cells (indicated in red) renew themselves and produce progenitor cells (indicated in green), which go through several rounds of division before they differentiate into specialized cells (31). Compromised aGPCRs may promote a shift from differentiation to proliferation, which may cause progenitor cell expansion and benign or malignant tumorigenesis (right). (D) Cell and tissue polarity is fundamental for oriented cell division and tissue formation, processes that have been associated with aGPCR function (32–34). (E) aGPCR malfunction often results in phenotypes linked to defective cell migration (35–38). Defective cell-cell and cell-matrix adhesion of tumor cells may be causally associated with aGPCR dysfunction and catalyzes tumor cell migration and invasion.
Mechanical Signature and Malignancy
En route from healthy over malignant to a metastatic state a cell undergoes an arduous ‘force journey’ signified by a mechanical reciprocity between cell contractility and extracellular matrix (ECM) rigidity. In other words, the biophysical signature of a cell and its ECM generates a lively biomechanical ‘conversation’ in which cellular contractile forces that impinge on the ECM counterbalance the elastic resistance of the ECM toward this mechanical stimulus (39).
To detect, integrate and react to mechanical inputs nature has invented an interconnected hierarchy of mechanical systems build from force sensors, adhesive molecules, focal adhesions as well as cytoskeletal and filamentous elements. Together these components comprise the molecular backbone of this delicate force balance between cells and their ECM, crucial for numerous cellular properties as well as the development and maintenance of tissues.
Dysplasia often leads to the generation of aberrant force values, which throw the system off balance leading to changes in the cytoskeletal and filamentous cellular architecture (40). An intriguing example of this phenomenon is the exchange from keratin-based to vimentin-based cytoskeleton in mammary tissue, which constitutes an indicator for epithelial-to-mesenchymal transition (EMT) (41–43). As malignancies progress cell and ECM feature specific changes common in many cancer types (Figures 1A,B): (i) The ECM exhibits increased stiffness (28). In breast malignancies, for example, this seems to be due to an integrin-dependent increase in collagen cross-linking and tissue fibrosis (44, 45). (ii) The ECM is remodeled due to the release of proteolytic enzymes (e.g. matrix metalloproteases) from protrusive cell processes (46, 47). (iii) Tumor cells show reduced adhesiveness to adjacent tumor and non-tumor cells, caused by loss in cell–cell contact integrity (48–51). (iv) Tumor cells often display increased compliance/deformability and contractility (52–55). This way metastatic cells are structurally fine-tuned to break through basal laminae, squeeze through tissues, and form secondary tumors at distant sites. Finally, elevation in ECM rigidity and cell contractility is attended by an increase in malignancy and metastatic potential (28, 56). Importantly, these cellular and extracellular anomalies manifest in many cancer types and appear as a prerequisite for cells to succumb to mechanical insults associated with dysplasia and the metastatic cascade.
Molecular Components and Mechanical Phenotype
According to the National Cancer Institute more than 30 tumor markers are currently used to diagnose and help manage different cancer types (https://www.cancer.gov/about-cancer/diagnosis-staging/diagnosis/tumor-markers-fact-sheet, accessed December 10, 2017). In light of the progress in genomic profiling technologies and tailor-made molecular targeting tumor markers will take an increasingly important role. However, to date we understand surprisingly little about the biological rationale behind their misexpression let alone the cellular signaling routes they employ and interactions they engage in to control tumor growth, invasion and metastasis. To this end it will be particularly interesting to decipher molecular players responsible for the mechanical metamorphosis of tumors. Several molecules have been documented in this context including constituents of focal adhesion complexes (FA), cytoskeletal and filamentous components as well as cell adhesion molecules (CAMs). Here, I will focus on FA protein complexes as mechanosensing entities and particularly integrin receptors, as they constitute bona fide mechanotransducers with intricate ties to the evolution of the malignant phenotype.
FAs comprise integrin-based macromolecular assemblies that physically link the cell’s exterior substrate and interior cytoskeleton. Integrins nucleate a plethora of molecules at the intracellular interface to build highly dynamic biochemical signaling hubs key to several cellular aspects including cell shape, proliferation, survival, differentiation, and motility (57–59). The composition and maintenance of FAs relies on the local force profile generated by myosin-driven contraction of the cytoskeleton (‘inside-out’) or by perturbations of the ECM (‘outside-in’) (60–62). The core of cellular FA mechanotransduction are integrins (63, 64). They act as direct and indirect mechanotransducers by reacting to mechanical force either with conformational changes or with aggregation to build a platform for force-transfer to the cytoskeleton, respectively. Application of external forces or changes in ECM stiffness stimulate integrins to activate RhoGTPase and Rho-associated kinase, which modulates myosin-light-chain phosphorylation (30). This way cell contractility increases further escalating ECM stiffness through aberrant integrin-signaling, collagen deposition and cross-linking (45). This deadly, self-sustaining positive feedback loop is intricately intertwined with Erk-dependent mitogenic signaling, which controls cell proliferation and the development of mammary cancer (44, 45). Moreover, due to their ability to regulate expression and activity of metalloproteases integrins play their part in invasion and metastasis (65, 66).
Among the canonical signaling routes triggered by integrins, the activation of the focal adhesion kinase (FAK) was shown to play a major role in several cancer types. FAK shapes the structure and function of FAs, e.g., through α-actinin-dependent changes in F-actin crosslinking and/or RhoGTPase-driven modulation of actomyosin contractility (67–69). Interestingly, a recent computational study advocates a novel mechanoenzymatic mode in which FAK localizes to PIP2 enriched FA-membranes to directly sense membrane perturbations that in turn induce specific conformational changes nourishing Ras-mediated delivery to the nucleus (70, 71).
Integrins have been in the limelight of mechanotransduction in cancer. However, nature has equipped cells with different membrane receptors to comply with cell-specific demands and to ensure reliable mechanotransduction. Hence, it is more likely than not that cancer cells utilize additional sophisticated bona fide force sensors.
Adhesion-GPCRs: Mechanotransduction and Malignancy
The aGPCR family counts 33 mammalian homologs serving functions in a multitude of developmental and physiological phenomena (4, 72). aGPCRs possess a modular architecture composed of an extracellular domain (ECD), a seven-transmembrane-spanning (7TM) domain and a intracellular domain (72). Their ECDs are exceptional in size and complexity generally imparted by the presence of various adhesive structural folds, which promote interactions with ECM constituents and receptors of adjacent cells (72). The ECDs of most aGPCRs contain a GPCR autoproteolysis-inducing (GAIN) domain (73), which usually includes a canonical GPCR proteolysis site (GPS; H/R−2–L/M/I−1 ↓ T/S/C+1) that catalyzes self-cleavage of the receptor (74–77). Once the resulting N- and C-terminal fragments (NTF and CTF) are delivered to the cell surface they reunite through a non-covalent bond to build heterodimeric receptor molecules (73). A seminal discovery with respect to aGPCR signaling was the identification of a short amino acid sequence C-terminal of the GPS motif as a cryptic tethered ligand (Stachel sequence) with agonistic capacity (78, 79). However, how the Stachel is freed from the envelop of the GAIN domain pocket it resides in, and whether receptor cleavage is involved is matter of ongoing debate (79, 80). Thus, while the functional relevance of GAIN/GPS-mediated cleavage remains in large part controversial, the GAIN domain in juxtaposition to the 7TM has become the defining aGPCR signature (72).
Another unifying property of aGPCRs may be their mechanosensitivity (23–27, 81). For example, GPR56/ADGRG1 was suggested to control muscle fiber size through its stimulation in response to mechanical preload (23). In Drosophila, LPHN/CIRL/ADGRL tunes the mechanosensitive profile of sensory neurons through cAMP-dependent modulation of ionotropic receptors (25, 80). EMR2/ADGRE2 was the first aGPCR, for which a mechano-dependent malfunction was directly linked to disease. Vibratory uticaria causes hives and systemic manifestations in patients when challenged with cutaneous vibration. The underlying cause is a single point mutation in the GAIN domain of EMR2, which might destabilize the NTF-CTF heterodimer to enhance receptor activity and induce histamine release from mast cells (27).
Physical anchorage constitutes a logical prerequisite for mechanosensing as it enables measurement of movements relative to the sensor-bearing structure to convey force information. Thus, attachment and size-dependent tensility of NTFs may be critical for mechanosensing through aGPCRs. NTF-size and domain layout vary across the receptor family. Moreover, untypical for canonical GPCRs, aGPCRs are vividly spliced producing a palette of differentially sized NTFs (74, 82, 83), which adds an additional level of structural flexibility and functional complexity. While the underlying reasons for this variability are unclear it is tempting to speculate that aGPCR-NTFs are built to sense chemical or mechanical signals from the ECM, bridge intercellular gaps or even read out the dimension of these gaps. The NTF size of each receptor may thus be tuned to meet the specific geometric demands of its expressing tissue enabling reliable and precise distance sensing and mechanotransduction. This notion is in line with the set-point of neuronal mechanosensitivity conferred by Drosophila CIRL, which inversely scales with the length of its NTF as shown by artificial elongation experiments (80). aGPCR function in diverse biological processes such as the establishment of cell and tissue polarity (32–34), synaptogenesis (84–86), and myelination of peripheral nerves (24) favors a palette of alternative aGPCR variants generated through transcriptional modifications.
The newfound mechanoceptive facility of aGPCRs in combination with cellular functions and implications in cancer is particularly daunting in light of the perilous force journey cells go on as they transform, turn malignant and potentially become metastatic (Figures 1C–E).
GPR56 presents one of the most well-studied aGPCRs with respect to cancer progression. In melanoma cells, GPR56 curtails cell growth and metastasis indicative of its suppressive function in tumorigenesis (16). While this phenotype was associated with PKCα/VEGF signaling-based defects in angiogenesis (17), other cellular routes, e.g., Gα12/13/RhoA seem to supplement the GPR56-dependent signaling profile in melanoma cells (18). Rho-dependent signaling plays roles in several steps of cancer progression (30), most of which can be traced back to its ability to control contractility, stress fiber formation and FA reinforcement (67); properties that confer cell migration through a heterogeneous stromal environment. This is in agreement with GPR56’s location at the leading edge of membrane filopodia and its colocalization with α-actinin in human glioma cells (87).
Recent work revealed that mesenchymal differentiation and radioresistance, defining features of glioblastoma, are repressed by GPR56 (19). The authors propose a model in which GPR56 obstructs nuclear factor kappa alpha (NF-κα) to inhibit mesenchymal differentiation. During differentiation, however, NF-κα levels are potentiated due to tumor (TNFα)-mediated activation and relief of GPR56 suppression. Atomic force microscopy of human colon cancer cells (HCT116) uncovered that TNFα-induced EMT is indeed associated with cytoskeletal rearrangements and changes in cell elasticity (88). Contrasting the situation in melanoma and glioblastoma, a recent in vivo study uncovered GPR56 as a novel marker for leukemic subpopulations signified by high repopulation capabilities typical for acute myloid leukemia (89). Interaction of leukemic stem cells and bone marrow niches is the leading cause of repopulation capacity and acute myloid leukemia-relapse. One hypothesis suggests that adhesive molecules [e.g., integrins (90), aGPCR (89)] retain leukemic stem cells in the niche, sheltering them from chemotherapy.
Brain angiogenesis inhibitor-1 (BAI1/ADGRB1), another cancer-associated aGPCR, is enriched in the brain and has been associated with tumor angiogenesis (8). In this process tumors induce vessel formation essential for oxygen and nutrient supply (91). Separation of BAI1’s NTF at the GPS results in the release of a fragment, vasculostatin-120, which interferes with angiogenesis through the inhibition of endothelial cell proliferation (92) and microvascular endothelial cell migration (21). Soluble BAI1 was also shown to ablate angiogenesis and glioma growth in vivo (20, 93–95). The BAI1-NTF is composed of one Arg-Gly-Asp integrin-binding motif (RGD) and five thrombospondin type 1 repeats. Interestingly, another functionally active fragment, vasculostatin-40, breaks away from the BAI1-NTF to mediate antiangiogenic effects in vitro and in vivo (94, 96). Thus, BAI1 N-termini negatively regulate vessel formation through a cell non-autonomous signaling mode. In contrast, ELTD1/ADGRL4 possesses proangiogenic capacities and is believed to modulate vascular sprouting (97). In glioblastoma, for example, ELTD1 levels are elevated and have been shown to modulate glioma growth (97–99). Interestingly, loss of Eltd1 in mice augments cardiac hypertrophy in response to pressure preload (100), which might indicate mechano-dependence of ELTD1 function in angiogenesis.
The Ingber lab established an intriguing model describing the mechanical control of angiogenesis (101). This model is based on the notion that local thinning of the ECM increases its compliance and causes local cell distortion generated by tractional forces of surrounding cells. Subsequently, integrin-dependent force transfer across the cell membrane alters the biochemical machinery to drive coordinated cell growth, motility and ultimately capillary patterning (102–106). This mechanical force concept applies to tumor angiogenesis as well, only here the ECM stiffens to shift the force balance. Strikingly, the anti-angiogenic potential of BAI1 seems to depend on the interaction of its soluble NTF with surface receptors (integrins and CD36) (92, 107). This begs the question whether BAI1 interacts with integrins to shape angiogenesis mechano-dependently.
BAI1 is downregulated in many forms of cancer (8), which may be due at least in part to gene silencing and somatic mutations (5, 95, 108). However, is it possible that cancer formation induces aberrant BAI1 expression after protein processing and trafficking? Could ECM stiffness break off the NTF on tumor cells to induce constitutive signaling and/or receptor internalization? BAI1 is known to direct cytoskeletal changes through direct and indirect signaling paths with PDZ-binding molecules as well as Rho and Rac molecules, respectively (109, 110). Hence, force-dependent misregulation of BAI1 could intersect with alterations in the cellular mechanical signature.
Here GPR56, BAI1, and ELTD1 serve to illustrate the putative role of aGPCRs in cancer cell mechanics. However, various aGPCRs have been associated with cancer development and progression including GPR133/ADGRD1, which is required for glioblastoma growth (14); GPR116/ADGRF5, which furthers breast cancer metastasis (12); and CD97/ADGRE5 seems to enhance tumor cell invasion in several human malignancies (111–114).
Integrins and aGPCRs
The force-sensing capabilities of tumor cells can be in large part traced back to integrins. Interestingly, several aGPCRs have been shown to interact with integrins; often by NTF shedding and cell non-autonomous signaling at ectopic sites. For example, soluble CD97/ADGRE5 was shown to interact with endothelial α5β1 and αvβ3 integrins to promote angiogenesis in vivo (9). The NTF of GPR124/ADGRA2 is released from cultured endothelial cells during angiogenesis and engages with ECM glycosaminoglycans. Subsequently, the NTF is truncated in a second cleavage event exposing a RGD motif to promote endothelial cell survival by interconnecting integrin αvβ3 and glycosaminoglycans (115). However, GPR124 was shown to modulate Wnt signaling to shape developmental CNS angiogenesis in vivo RGD motif-independently (35, 116, 117). In contrast, neuronal GPR56 and integrin α3β1 interact to regulate cell migration and cortical development in a cell autonomous manner (118). Collectively, the available data suggests a model in which aGPCRs engage in a cis or transcellular conversation with integrins. Moreover, aGPCRs may use RGD motifs as an anchor to target soluble aGPCR-NTFs to distant sites (9, 115, 119).
As aGPCRs function as sensors and probably transducers of mechanical information it seems plausible that they perform next to or in conjunction with integrins to shape the biophysical signature of tumor cells. Several properties of aGPCR are in line with this notion: (1) aGPCR interact with ECM molecules and are involved in the coordination of cytoskeletal architecture indirectly through G-protein, Rho and Rac dependent signaling cascades and directly, e.g., via PDZ proteins (120). Thus, similar to integrins aGPCRs may connect extracellular ECM and intracellular cytoskeleton. (2) aGPCR promote bidirectional signaling (119). (3) aGPCRs have the capacity to dimerize or oligomerize (121, 122). Thus, it is enticing to speculate that aGPCRs constitute novel mechanotransducers that shape the biomechanical profile of cells in health and disease.
Conclusion
Adhesion G protein-coupled receptors shape a number of cellular aspects intricately tied to tumorigenesis (Figures 1C–E), which is why they might hold tremendous potential as molecular targets for the development of novel therapeutic strategies to control cancer cell plasticity and malignancy. However, aGPCR research is still in its infancy and we are far from understanding their general roles in cancer and mechanobiology. Thus, it will be interesting to monitor putative aGPCR-dependent changes in the cytoskeleton during transformation and beyond using super-resolution and live imaging methodologies. Moreover, a complementary approach employing atomic force microscopy and subcellular laser ablation will help to analyze viscoelastic changes of malignant and metastatic cells with respect to loss or dysfunction of aGPCRs. Finally, this knowledge might contribute to a more comprehensive understanding of aGPCRs’ roles as mechanical force sensors in physiological and pathophysiological settings.
Author Contributions
NS wrote and revised/edited the manuscript; prepared the figure and approved the article for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank T. Langenhan, M. Bigl, D. Ljaschenko, and B. Blanco-Redondo for insightful discussions.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (FOR2149/P01, SCHO 1791/1-2) and the Medical Faculty, University of Leipzig (Junior Research grant).
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi:10.1002/ijc.29210
2. Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol (2012) 13:790–801. doi:10.1016/S1470-2045(12)70211-5
3. Fritsch A, Höckel M, Kiessling T, Nnetu KD, Wetzel F, Zink M, et al. Are biomechanical changes necessary for tumour progression? Nat Phys (2010) 6:730E–732. doi:10.1038/nphys1800
4. Hamann J, Aust G, Araç D, Engel FB, Formstone C, Fredriksson R, et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev (2015) 67:338–67. doi:10.1124/pr.114.009647
5. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature (2010) 466:869–73. doi:10.1038/nature09208
6. O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer (2013) 13:412–24. doi:10.1038/nrc3521
7. Lin H-H. Adhesion family of G protein-coupled receptors and cancer. Chang Gung Med J (2012) 35:15–27. doi:10.4103/2319-4170.106170
8. Aust G, Zhu D, Van Meir EG, Xu L. Adhesion GPCRs in tumorigenesis. In: Langenhan T, Schöneberg T, editors. Adhesion G Protein-Coupled Receptors: Molecular, Physiological and Pharmacological Principles in Health and Disease Handb Exp Pharmacol. (Vol. 234), Cham: Springer International Publishing (2016). p. 369–96.
9. Wang T, Ward Y, Tian L, Lake R, Guedez L, Stetler-Stevenson WG, et al. CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counterreceptors on endothelial cells. Blood (2005) 105:2836–44. doi:10.1182/blood-2004-07-2878
10. Hsiao C-C, Wang WC, Kuo WL, Chen HY, Chen T-C, Hamann J, et al. CD97 inhibits cell migration in human fibrosarcoma cells by modulating TIMP-2/MT1- MMP/MMP-2 activity – role of GPS autoproteolysis and functional cooperation between the N- and C-terminal fragments. FEBS J (2014) 281:4878–91. doi:10.1111/febs.13027
11. Li C, Liu D-R, Li G-G, Wang H-H, Li X-W, Zhang W, et al. CD97 promotes gastric cancer cell proliferation and invasion through exosome-mediated MAPK signaling pathway. World J Gastroenterol (2015) 21:6215–28. doi:10.3748/wjg.v21.i20.6215
12. Tang X, Jin R, Qu G, Wang X, Li Z, Yuan Z, et al. GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Gαq-p63RhoGEF-Rho GTPase pathway. Cancer Res (2013) 73:6206–18. doi:10.1158/0008-5472.CAN-13-1049
13. Yang L, Lin X-L, Liang W, Fu S-W, Lin W-F, Tian X-Q, et al. High expression of GPR116 indicates poor survival outcome and promotes tumor progression in colorectal carcinoma. Oncotarget (2017) 8:47943–56. doi:10.18632/oncotarget.18203
14. Bayin NS, Frenster JD, Kane JR, Rubenstein J, Modrek AS, Baitalmal R, et al. GPR133 (ADGRD1), an adhesion G-protein-coupled receptor, is necessary for glioblastoma growth. Oncogenesis (2016) 5:e263. doi:10.1038/oncsis.2016.63
15. Frenster JD, Inocencio JF, Xu Z, Dhaliwal J, Alghamdi A, Zagzag D, et al. GPR133 promotes glioblastoma growth in hypoxia. Neurosurgery (2017) 64:177–81. doi:10.1093/neuros/nyx227
16. Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci U S A (2006) 103:9023–8. doi:10.1073/pnas.0602681103
17. Yang L, Chen G, Mohanty S, Scott G, Fazal F, Rahman A, et al. GPR56 regulates VEGF production and angiogenesis during melanoma progression. Cancer Res (2011) 71:5558–68. doi:10.1158/0008-5472.CAN-10-4543
18. Chiang N-Y, Peng Y-M, Juang H-H, Chen T-C, Pan H-L, Chang G-W, et al. GPR56/ADGRG1 activation promotes melanoma cell migration via NTF dissociation and CTF-mediated Gα12/13/RhoA signaling. J Invest Dermatol (2017) 137:727–36. doi:10.1016/j.jid.2016.10.031
19. Moreno M, Pedrosa L, Paré L, Pineda E, Bejarano L, Martínez J, et al. GPR56/ADGRG1 inhibits mesenchymal differentiation and radioresistance in glioblastoma. Cell Rep (2017) 21:2183–97. doi:10.1016/j.celrep.2017.10.083
20. Kaur B, Brat DJ, Devi NS, Van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene (2005) 24:3632–42. doi:10.1038/sj.onc.1208317
21. Kaur B, Cork SM, Sandberg EM, Devi NS, Zhang Z, Klenotic PA, et al. Vasculostatin inhibits intracranial glioma growth and negatively regulates in vivo angiogenesis through a CD36-dependent mechanism. Cancer Res (2009) 69:1212–20. doi:10.1158/0008-5472.CAN-08-1166
22. Zhu D, Van Meir EG. BAI1: from cancer to neurological disease. Oncotarget (2016) 7:17288–9. doi:10.18632/oncotarget.8193
23. White JP, Wrann CD, Rao RR, Nair SK, Jedrychowski MP, You JS, et al. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc Natl Acad Sci U S A (2014) 111(44):15756–61. doi:10.1073/pnas.1417898111
24. Petersen SC, Luo R, Liebscher I, Giera S, Jeong S-J, Mogha A, et al. The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron (2015) 85:755–69. doi:10.1016/j.neuron.2014.12.057
25. Scholz N, Gehring J, Guan C, Ljaschenko D, Fischer R, Lakshmanan V, et al. The adhesion GPCR latrophilin/CIRL shapes mechanosensation. Cell Rep (2015) 11(6):866–74. doi:10.1016/j.celrep.2015.04.008
26. Wilde C, Fischer L, Lede V, Kirchberger J, Rothemund S, Schöneberg T, et al. The constitutive activity of the adhesion GPCR GPR114/ADGRG5 is mediated by its tethered agonist. FASEB J (2016) 30(2):666–73. doi:10.1096/fj.15-276220
27. Boyden SE, Desai A, Cruse G, Young ML, Bolan HC, Scott LM, et al. Vibratory urticaria associated with a missense variant in ADGRE2. N Engl J Med (2016) 374:656–63. doi:10.1056/NEJMoa1500611
28. Katira P, Bonnecaze RT, Zaman MH. Modeling the mechanics of cancer: effect of changes in cellular and extra-cellular mechanical properties. Front Oncol (2013) 3:145. doi:10.3389/fonc.2013.00145
29. Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK, et al. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res (2011) 71:7301–11. doi:10.1158/0008-5472.CAN-11-2381
30. Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett (2008) 582:2093–101. doi:10.1016/j.febslet.2008.04.039
31. White AC, Lowry WE. Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol (2015) 25:11–20. doi:10.1016/j.tcb.2014.08.008
32. Usui T, Shima Y, Shimada Y, Hirano S, Burgess RW, Schwarz TL, et al. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell (1999) 98:585–95. doi:10.1016/S0092-8674(00)80046-X
33. Langenhan T, Prömel S, Mestek L, Esmaeili B, Waller-Evans H, Hennig C, et al. Latrophilin signaling links anterior-posterior tissue polarity and oriented cell divisions in the C. elegans embryo. Dev Cell (2009) 17:494–504. doi:10.1016/j.devcel.2009.08.008
34. Curtin JA, Quint E, Tsipouri V, Arkell RM, Cattanach B, Copp AJ, et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol (2003) 13:1129–33. doi:10.1016/S0960-9822(03)00374-9
35. Kuhnert F, Mancuso MR, Shamloo A, Wang H-T, Choksi V, Florek M, et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science (2010) 330:985–9. doi:10.1126/science.1196554
36. Koirala S, Jin Z, Piao X, Corfas G. GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J Neurosci (2009) 29:7439–49. doi:10.1523/JNEUROSCI.1182-09.2009
37. Steimel A, Wong L, Najarro EH, Ackley BD, Garriga G, Hutter H. The Flamingo ortholog FMI-1 controls pioneer-dependent navigation of follower axons in C. elegans. Development (2010) 137:3663–73. doi:10.1242/dev.054320
38. Najarro EH, Wong L, Zhen M, Carpio EP, Goncharov A, Garriga G, et al. Caenorhabditis elegans Flamingo Cadherin fmi-1 regulates GABAergic neuronal development. J Neurosci (2012) 32:4196–211. doi:10.1523/JNEUROSCI.3094-11.2012
39. Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev (2009) 28:113–27. doi:10.1007/s10555-008-9173-4
40. Pawlak G, Helfman DM. Cytoskeletal changes in cell transformation and tumorigenesis. Curr Opin Genet Dev (2001) 11:41–7. doi:10.1016/s0959-437x(00)00154-4
41. Kokkinos MI, Wafai R, Wong MK, Newgreen DF, Thompson EW, Waltham M. Vimentin and epithelial-mesenchymal transition in human breast cancer-observations in vitro and in vivo. Cells Tissues Organs (2007) 185:191–203. doi:10.1159/000101320
42. Pagan R, Martín I, Alonso A, Llobera M, Vilaró S. Vimentin filaments follow the preexisting cytokeratin network during epithelial-mesenchymal transition of cultured neonatal rat hepatocytes. Exp Cell Res (1996) 222:333–44. doi:10.1006/excr.1996.0043
43. Willipinski-Stapelfeldt B, Riethdorf S, Assmann V, Woelfle U, Rau T, Sauter G, et al. Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin Cancer Res (2005) 11:8006–14. doi:10.1158/1078-0432.CCR-05-0632
44. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell (2005) 8:241–54. doi:10.1016/j.ccr.2005.08.010
45. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139:891–906. doi:10.1016/j.cell.2009.10.027
46. Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol (2007) 9:893–904. doi:10.1038/ncb1616
47. Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res (2008) 68:7247–9. doi:10.1158/0008-5472.CAN-08-0784
48. Coman DR. Decreased mutual adhesiveness, a property of cells from squamous cell carcinomas*. Cancer Res (1944) 4:625–9.
49. Brugmans M, Cassiman JJ, van den Berghe H. Selective adhesion and impaired adhesive properties of transformed cells. J Cell Sci (1978) 33:121–32.
50. Paredes J, Albergaria A, Oliveira JT, Jerónimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res (2005) 11:5869–77. doi:10.1158/1078-0432.CCR-05-0059
51. Ribeiro AS, Carreto LC, Albergaria A, Sousa B, Ricardo S, Milanezi F, et al. Co-expression of E- and P-cadherin in breast cancer: role as an invasion suppressor or as an invasion promoter? BMC Proc (2010) 4:47. doi:10.1186/1753-6561-4-S2-P47
52. Cross SE, Jin Y-S, Rao J, Gimzewski JK. Nanomechanical analysis of cells from cancer patients. Nat Nanotechnol (2007) 2:780–3. doi:10.1038/nnano.2007.388
53. Jonas O, Mierke CT, Käs JA. Invasive cancer cell lines exhibit biomechanical properties that are distinct from their noninvasive counterparts. Soft Matter (2011) 7:11488–95. doi:10.1039/C1SM05532A
54. Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PLoS One (2012) 7:e32572. doi:10.1371/journal.pone.0032572
55. Physical Sciences – Oncology Centers Network, Agus DB, Alexander JF, Arap W, Ashili S, Aslan JE, et al. A physical sciences network characterization of non-tumorigenic and metastatic cells. Sci Rep (2013) 3:1449. doi:10.1038/srep01449
56. Swaminathan V, Mythreye K, O’Brien ET, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res (2011) 71:5075–80. doi:10.1158/0008-5472.CAN-11-0247
57. Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci (2001) 114:3583–90.
58. Bershadsky AD, Balaban NQ, Geiger B. Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol (2003) 19:677–95. doi:10.1146/annurev.cellbio.19.111301.153011
59. Bershadsky A, Kozlov M, Geiger B. Adhesion-mediated mechanosensitivity: a time to experiment, and a time to theorize. Curr Opin Cell Biol (2006) 18:472–81. doi:10.1016/j.ceb.2006.08.012
60. Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev (2009) 23:397–418. doi:10.1101/gad.1758709
61. Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol (2001) 13:584–92. doi:10.1016/S0955-0674(00)00255-6
62. Anthis NJ, Campbell ID. The tail of integrin activation. Trends Biochem Sci (2011) 36:191–8. doi:10.1016/j.tibs.2010.11.002
63. Wang N, Butler J, Ingber D. Mechanotransduction across the cell surface and through the cytoskeleton. Science (1993) 260:1124–7. doi:10.1126/science.7684161
64. Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin–cytoskeleton linkages. Cell (1997) 88:39–48. doi:10.1016/S0092-8674(00)81856-5
65. Brooks PC, Strömblad S, Sanders LC, Schalscha von TL, Aimes RT, Stetler-Stevenson WG, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell (1996) 85:683–93. doi:10.1016/S0092-8674(00)81235-0
66. Yue J, Zhang K, Chen J. Role of integrins in regulating proteases to mediate extracellular matrix remodeling. Cancer Microenviron (2012) 5:275–83. doi:10.1007/s12307-012-0101-3
67. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol (2004) 265:23–32. doi:10.1016/j.ydbio.2003.06.003
68. Tilghman RW, Parsons JT. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin Cancer Biol (2008) 18:45–52. doi:10.1016/j.semcancer.2007.08.002
69. Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther (2015) 146:132–49. doi:10.1016/j.pharmthera.2014.10.001
70. Zhou J, Aponte-Santamaría C, Sturm S, Bullerjahn JT, Bronowska A, Gräter F. Mechanism of focal adhesion kinase mechanosensing. PLoS Comput Biol (2015) 11:e1004593. doi:10.1371/journal.pcbi.1004593
71. Herzog FA, Braun L, Schoen I, Vogel V. Structural insights how PIP2 imposes preferred binding orientations of FAK at lipid membranes. J Phys Chem B (2017) 121:3523–35. doi:10.1021/acs.jpcb.6b09349
72. Langenhan T, Aust G, Hamann J. Sticky signaling-adhesion class G protein-coupled receptors take the stage. Sci Signal (2013) 6(276):re3. doi:10.1126/scisignal.2003825
73. Araç D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC, et al. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J (2012) 31:1364–78. doi:10.1038/emboj.2012.26
74. Gray JX, Haino M, Roth MJ, Maguire JE, Jensen PN, Yarme A, et al. CD97 is a processed, seven-transmembrane, heterodimeric receptor associated with inflammation. J Immunol (1996) 157:5438–47.
75. Krasnoperov VG, Bittner MA, Beavis R, Kuang YN, Salnikow KV, Chepurny OG, et al. alpha-latrotoxin stimulates exocytosis by the interaction with a neuronal G-protein-coupled receptor. Neuron (1997) 18:925–37. doi:10.1016/S0896-6273(00)80332-3
76. Krasnoperov V, Bittner MA, Holz RW, Chepurny O, Petrenko AG. Structural requirements for -latrotoxin binding and -latrotoxin-stimulated secretion: a study with calcium-independent receptor of -latrotoxin (CIRL) deletion mutants. J Biol Chem (1999) 274:3590–6. doi:10.1074/jbc.274.6.3590
77. Krasnoperov V, Lu Y, Buryanovsky L, Neubert TA, Ichtchenko K, Petrenko AG. Post-translational proteolytic processing of the calcium-independent receptor of alpha -Latrotoxin (CIRL), a natural chimera of the cell adhesion protein and the g protein-coupled receptor. Role of the G protein-coupled receptor proteolysis site (GPS) motif. J Biol Chem (2002) 277:46518–26. doi:10.1074/jbc.M206415200
78. Liebscher I, Schön J, Petersen SC, Fischer L, Auerbach N, Demberg LM, et al. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep (2014) 9:2018–26. doi:10.1016/j.celrep.2014.11.036
79. Stoveken HM, Hajduczok AG, Xu L, Tall GG. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc Natl Acad Sci U S A (2015) 112(19):6194–99. doi:10.1073/pnas.1421785112
80. Scholz N, Guan C, Nieberler M, Grotemeyer A, Maiellaro I, Gao S, et al. Mechano-dependent signaling by Latrophilin/CIRL quenches cAMP in proprioceptive neurons. Elife (2017) 6:e28360. doi:10.7554/eLife.28360
81. Scholz N, Monk KR, Kittel RJ, Langenhan T. Adhesion GPCRs as a putative class of metabotropic mechanosensors. In: Langenhan T, Schöneberg T, editors. Adhesion G Protein-Coupled Receptors: Molecular, Physiological and Pharmacological Principles in Health and Disease Handb Exp Pharmacol. (Vol. 234), Cham: Springer International Publishing (2016). p. 221–47.
82. Bae B-I, Tietjen I, Atabay KD, Evrony GD, Johnson MB, Asare E, et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science (2014) 343:764–8. doi:10.1126/science.1244392
83. Mills JD, Kavanagh T, Kim WS, Chen BJ, Kawahara Y, Halliday GM, et al. Unique transcriptome patterns of the white and grey matter corroborate structural and functional heterogeneity in the human frontal lobe. PLoS One (2013) 8:e78480–78480. doi:10.1371/journal.pone.0078480
84. Silva J-P, Lelianova VG, Ermolyuk YS, Vysokov N, Hitchen PG, Berninghausen O, et al. Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc Natl Acad Sci U S A (2011) 108:12113–8. doi:10.1073/pnas.1019434108
85. Duman JG, Tzeng CP, Tu Y-K, Munjal T, Schwechter B, Ho TS-Y, et al. The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J Neurosci (2013) 33:6964–78. doi:10.1523/JNEUROSCI.3978-12.2013
86. Anderson GR, Maxeiner S, Sando R, Tsetsenis T, Malenka RC, Südhof TC. Postsynaptic adhesion GPCR latrophilin-2 mediates target recognition in entorhinal-hippocampal synapse assembly. J Cell Biol (2017) 216:3831–46. doi:10.1083/jcb.201703042
87. Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J, et al. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene (2005) 24:1673–82. doi:10.1038/sj.onc.1208395
88. Liu H, Wang N, Zhang Z, Wang H, Du J, Tang J. Effects of tumor necrosis factor-α on morphology and mechanical properties of HCT116 human colon cancer cells investigated by atomic force microscopy. Scanning (2017) 2017:2027079. doi:10.1155/2017/2027079
89. Pabst C, Bergeron A, Lavallée V-P, Yeh J, Gendron P, Norddahl GL, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood (2016) 127:2018–27. doi:10.1182/blood-2015-11-683649
90. Redondo-Muñoz J, Ugarte-Berzal E, García-Marco JA, del Cerro MH, Van den Steen PE, Opdenakker G, et al. Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood (2008) 112:169–78. doi:10.1182/blood-2007-08-109249
92. Koh JT, Kook H, Kee HJ, Seo Y-W, Jeong BC, Lee JH, et al. Extracellular fragment of brain-specific angiogenesis inhibitor 1 suppresses endothelial cell proliferation by blocking αvβ5 integrin. Exp Cell Res (2004) 294:172–84. doi:10.1016/j.yexcr.2003.11.008
93. Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, et al. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene (1997) 15:2145–50. doi:10.1038/sj.onc.1201542
94. Cork SM, Van Meir EG. Emerging roles for the BAI1 protein family in the regulation of phagocytosis, synaptogenesis, neurovasculature, and tumor development. J Mol Med (2011) 89:743–52. doi:10.1007/s00109-011-0759-x
95. Zhu D, Hunter SB, Vertino PM, Van Meir EG. Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res (2011) 71:5859–70. doi:10.1158/0008-5472.CAN-11-1157
96. Cork SM, Kaur B, Devi NS, Cooper L, Saltz JH, Sandberg EM, et al. A proprotein convertase/MMP-14 proteolytic cascade releases a novel 40 kDa vasculostatin from tumor suppressor BAI1. Oncogene (2012) 31:5144–52. doi:10.1038/onc.2012.1
97. Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell (2013) 24:229–41. doi:10.1016/j.ccr.2013.06.004
98. Dieterich LC, Mellberg S, Langenkamp E, Zhang L, Zieba A, Salomäki H, et al. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFβ2 in vascular abnormalization. J Pathol (2012) 228:378–90. doi:10.1002/path.4072
99. Towner RA, Jensen RL, Colman H, Vaillant B, Smith N, Casteel R, et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery (2013) 72:77–90. doi:10.1227/NEU.0b013e318276b29d discussion 91,
100. Xiao J, Jiang H, Zhang R, Fan G, Zhang Y, Jiang D, et al. Augmented cardiac hypertrophy in response to pressure overload in mice lacking ELTD1. PLoS One (2012) 7:e35779. doi:10.1371/journal.pone.0035779
101. Ingber DE. Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ Res (2002) 91:877–87. doi:10.1161/01.RES.0000039537.73816.E5
102. Lucitti JL, Jones EAV, Huang C, Chen J, Fraser SE, Dickinson ME. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development (2007) 134:3317–26. doi:10.1242/dev.02883
103. Mammoto A, Mammoto T, Ingber DE. Rho signaling and mechanical control of vascular development. Curr Opin Hematol (2008) 15:228–34. doi:10.1097/MOH.0b013e3282fa7445
104. Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature (2009) 457:1103–8. doi:10.1038/nature07765
105. Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development (2010) 137:1407–20. doi:10.1242/dev.024166
106. Mammoto T, Mammoto A, Ingber DE. Mechanobiology and developmental control. Annu Rev Cell Dev Biol (2013) 29:27–61. doi:10.1146/annurev-cellbio-101512-122340
107. Asch AS, Liu I, Briccetti FM, Barnwell JW, Kwakye-Berko F, Dokun A, et al. Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science (1993) 262:1436–40. doi:10.1126/science.7504322
108. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature (2010) 463:191–6. doi:10.1038/nature08658
109. Shiratsuchi T, Futamura M, Oda K, Nishimori H, Nakamura Y, Tokino T. Cloning and characterization of BAI-associated protein 1: a PDZ domain-containing protein that interacts with BAI1. Biochem Biophys Res Commun (1998) 247:597–604. doi:10.1006/bbrc.1998.8603
110. Stephenson JR, Paavola KJ, Schaefer SA, Kaur B, Van Meir EG, Hall RA. Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J Biol Chem (2013) 288:22248–56. doi:10.1074/jbc.M113.489757
111. Aust G, Steinert M, Schütz A, Boltze C, Wahlbuhl M, Hamann J, et al. CD97, but not its closely related EGF-TM7 family member EMR2, is expressed on gastric, pancreatic, and esophageal carcinomas. Am J Clin Pathol (2002) 118:699–707. doi:10.1309/A6AB-VF3F-7M88-C0EJ
112. Safaee M, Clark AJ, Oh MC, Ivan ME, Bloch O, Kaur G, et al. Overexpression of CD97 confers an invasive phenotype in glioblastoma cells and is associated with decreased survival of glioblastoma patients. PLoS One (2013) 8:e62765. doi:10.1371/journal.pone.0062765
113. Ward Y, Lake R, Martin PL, Killian K, Salerno P, Wang T, et al. CD97 amplifies LPA receptor signaling and promotes thyroid cancer progression in a mouse model. Oncogene (2013) 32:2726–38. doi:10.1038/onc.2012.301
114. Safaee M, Fakurnejad S, Bloch O, Clark AJ, Ivan ME, Sun MZ, et al. Proportional upregulation of CD97 isoforms in glioblastoma and glioblastoma-derived brain tumor initiating cells. PLoS One (2015) 10:e0111532. doi:10.1371/journal.pone.0111532
115. Vallon M, Essler M. Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin alpha(v)beta3 to glycosaminoglycans. J Biol Chem (2006) 281:34179–88. doi:10.1074/jbc.M605291200
116. Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev Cell (2014) 31:248–56. doi:10.1016/j.devcel.2014.08.018
117. Posokhova E, Shukla A, Seaman S, Suresh V, Hilton MB, Wu B, et al. GPR124 functions as a WNT7-specific coactivator of canonical β-catenin signaling. Cell Rep (2015) 10:123–30. doi:10.1016/j.celrep.2014.12.020
118. Jeong S-J, Luo R, Singer K, Giera S, Kreidberg J, Kiyozumi D, et al. GPR56 functions together with α3β1 integrin in regulating cerebral cortical development. PLoS One (2013) 8:e68781. doi:10.1371/journal.pone.0068781
119. Patra C, van Amerongen MJ, Ghosh S, Ricciardi F, Sajjad A, Novoyatleva T, et al. Organ-specific function of adhesion G protein-coupled receptor GPR126 is domain-dependent. Proc Natl Acad Sci U S A (2013) 110:16898–903. doi:10.1073/pnas.1304837110
120. Knapp B, Wolfrum U. Adhesion GPCR-related protein networks. In: Langenhan T, Schöneberg T, editors. Adhesion G Protein-Coupled Receptors: Molecular, Physiological and Pharmacological Principles in Health and Disease Handb Exp Pharmacol. (Vol. 234), Cham: Springer International Publishing (2016). p. 147–78.
121. Davies JQ, Chang G-W, Yona S, Gordon S, Stacey M, Lin H-H. The role of receptor oligomerization in modulating the expression and function of leukocyte adhesion-G protein-coupled receptors. J Biol Chem (2007) 282:27343–53. doi:10.1074/jbc.M704096200
Keywords: adhesion G protein-coupled receptors, mechanobiology, cancer, cytoskeleton, extracellular matrix
Citation: Scholz N (2018) Cancer Cell Mechanics: Adhesion G Protein-coupled Receptors in Action? Front. Oncol. 8:59. doi: 10.3389/fonc.2018.00059
Received: 22 December 2017; Accepted: 21 February 2018;
Published: 13 March 2018
Edited by:
Hsi-Hsien Lin, Chang Gung University, TaiwanReviewed by:
Mario Vallon, Stanford University, United StatesRandy Hall, Emory University School of Medicine, United States
Copyright: © 2018 Scholz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Scholz, bmljb2xlLnNjaG9sekBtZWRpemluLnVuaS1sZWlwemlnLmRl