 Tianyuan Hu
Tianyuan Hu Rebecca Murdaugh
Rebecca Murdaugh Daisuke Nakada
Daisuke Nakada- 1Department of Molecular & Human Genetics, Baylor College of Medicine, Houston, TX, United States
- 2Program in Developmental Biology, Baylor College of Medicine, Houston, TX, United States
Leukemia is characterized by the uncontrolled production of leukemic cells and impaired normal hematopoiesis. Although the combination of chemotherapies and hematopoietic stem cell transplantation has significantly improved the outcome of leukemia patients, a proportion of patients still suffer from relapse after treatment. Upon relapse, a phenomenon termed “lineage switch” is observed in a subset of leukemia patients, in which conversion of lymphoblastic leukemia to myeloid leukemia or vice versa is observed. A rare entity of leukemia called mixed-phenotype acute leukemia exhibits co-expression of markers representing two or three lineages. These two phenotypes regarding the lineage ambiguity suggest that the fate of some leukemia retain or acquire a certain degree of plasticity. Studies using animal models provide insight into how lineage specifying transcription factors can enforce or convert a fate in hematopoietic cells. Modeling lineage conversion in normal hematopoietic progenitor cells may improve our current understanding of how lineage switch occurs in leukemia. In this review, we will summarize the role of transcription factors and microenvironmental signals that confer fate plasticity to normal hematopoietic progenitor cells, and their potential to regulate lineage switching in leukemias. Future efforts to uncover the mechanisms contributing to lineage conversion in both normal hematopoiesis and leukemia may pave the way to improve current therapeutic strategies.
Introduction
Hematopoietic stem cells (HSCs) establish and maintain the hematopoietic system through differentiation into the multi-lineage progenitors and committed progenitors from which all the mature lineage cell types arise. In the classical model of hematopoiesis, long-term HSCs, short-term HSCs, and multipotent progenitors (MPPs) reside at the apex of the hierarchy (1–5). MPPs are able to differentiate into lineage-committed progenitors, including common lymphoid progenitors (CLPs) (6) and common myeloid progenitors, which further differentiate into granulocyte-monocyte progenitors (GMPs) and megakaryocyte-erythroid progenitors (MEPs) (7). A characteristic feature of this model is that, as progenitors differentiate through this pathway, their developmental potential narrows. For example, MEPs lack the granulocyte-monocyte (GM) potential of GMPs and instead have differentiation potential that is restricted to the megakaryocyte and erythroid (Meg/E) lineages. Importantly, although many studies have provided evidence supporting this classical hierarchy, studies have shown that the committed state can be canceled or reprogramed by the action of lineage specifying cytokines and transcription factors. This raises the question to what extent the committed states are fixed, and whether or not oncogenic mutations exploit the lineage promiscuous state of normal progenitor cells to change their phenotypes upon therapies.
Lineage Commitment and Switch in Normal Hematopoiesis
The increasingly narrowed lineage potential results from a precise combination of gene expression signatures and epigenetic modification. Several transcription factors have been found to be involved in the fate decision of hematopoietic progenitors (8, 9). Among these are the lineage-specific master regulator transcription factors, such as Pu.1 (also known as Spi-1; spleen focus forming virus proviral integration oncogene 1), C/ebp-α, Gata1, Pax5, and Ikaros (Figure 1). Pu.1 and C/ebp-α are master regulators of the myeloid cell fate, and not only do these transcription factors promote myeloid differentiation of progenitor cells (10) but also ectopic expression of these transcription factors confer a myeloid cell fate to cells of other lineages, such as T-cells, B-cells, or fibroblasts (11–14). Gata1 is a master regulator of erythroid cell fate that is required and sufficient to confer the erythroid fate. Deletion of Gata1 in mice causes defective erythropoiesis (15–21), whereas ectopic expression of Gata1 confers Meg/E fate to cells of other lineages, such as monocytic cells (22, 23). Loss of B-cell master regulators Pax5 and Ikaros disrupts the B-cell transcriptome and reprograms B-cells into myeloid (24, 25) or epithelial-like cells (26), respectively. Lineage conversion by ectopic expression or loss of these master regulator transcription factors is often associated with a change in a network of transcription factors that governs cell fate. Pu.1 promotes multipotent hematopoietic progenitors to differentiate into myeloid cells by activating multiple myeloid-lineage-related genes, including C/ebp-β and suppressing erythroid factors such as Gata1 (10), while C/ebp-α and C/ebp-β converts differentiated B-cells into macrophages by inhibiting Pax5 (12). The extent to which these transcription factors over-ride the lineage-committed state of differentiated cells illustrates the extensive potential these master regulators possess.
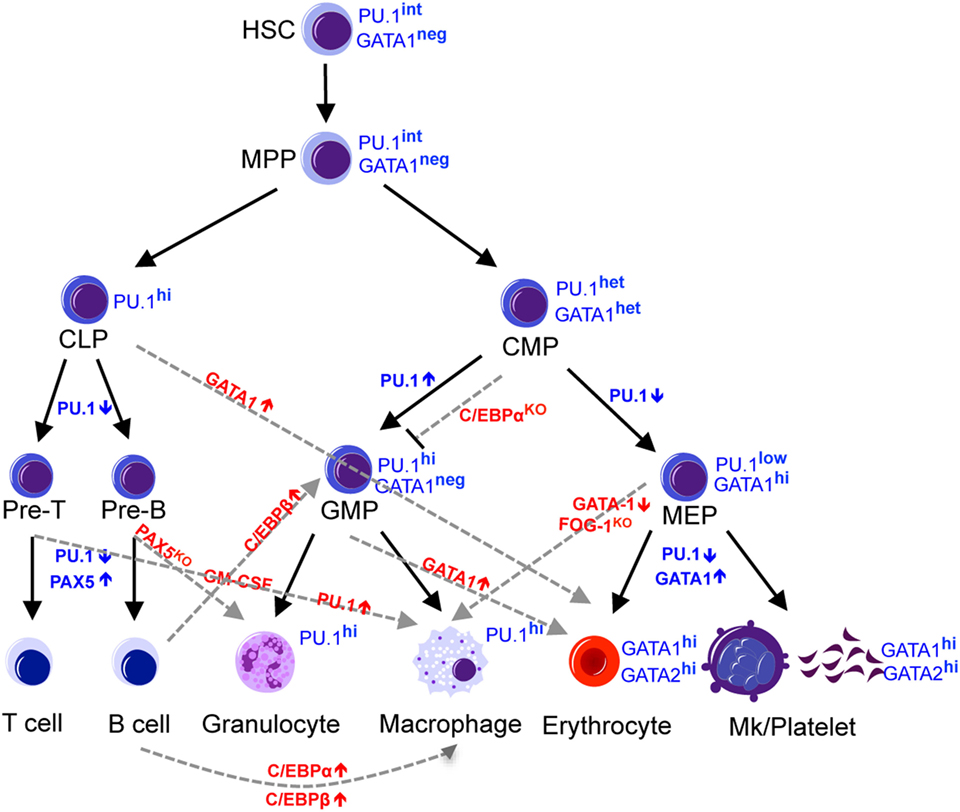
Figure 1. Regulators of lineage commitment in normal hematopoiesis. The hematopoietic system is maintained by HSCs, which gradually lose developmental potential through differentiation into downstream progenitors and mature cells. This narrowed lineage potential is controlled by a precise combination of transcription factors and can be reprogrammed by manipulating the expression level of certain transcription factors. Black solid lines indicate the normal commitment steps, while the gray dashed lines indicate altered lineage potential by manipulating the levels of transcription factors. HSC, hematopoietic stem cells; MPP, multipotent progenitors; CLP, common lymphoid progenitors; CMP, common myeloid progenitors, MEP, megakaryocyte-erythroid progenitors; GMP, granulocyte-monocyte progenitors.
Lineage Ambiguity in Malignant Hematopoiesis
Leukemias develop as a consequence of mutations that cooperatively confer aberrant self-renewal capacity to leukemic cells and allow them to proliferate indefinitely without differentiation. Recent advances in high-throughput sequencing of leukemia genomes have revealed numerous mutations in cytokine signaling, epigenetic regulators, and transcription factors (27, 28). Genetic studies using murine models have established that mutations in epigenetic and transcriptional regulators upregulate self-renewal and block differentiation of hematopoietic stem/progenitor cells (HSPCs) (29). The ability of transcription factor levels and external cytokine milieu to influence hematopoietic progenitor plasticity raises the question of whether lineage conversion plays any role in malignant hematopoiesis that often carry mutations in these regulators. In fact, a phenomenon called lineage switch has been reported, in which patients with acute leukemia that meet the French–American–British classification for being lymphoid or myeloid leukemias relapse with acute leukemia of the other lineage. Most cases of lineage switches are from ALL to AML (30–34), but AML to ALL switches have also been reported (33, 35–37). Additionally, some leukemias show no clear evidence of differentiation into a single lineage. These leukemias, termed mixed-phenotype acute leukemia (MPAL) (38) exhibit cells of at least two lineages; MPALs involving B-cell and myeloid lineages are the most frequent but some rare cases involve B- and T-cells, or B/T/myeloid cells. Patients with lineage switch leukemia or MPAL have poor prognosis, due to the difficulty in diagnosis and the lack of set protocols to guide treatments (38–41). Understanding the molecular mechanism behind lineage switch and ambiguity should pave a way for better treatment. We will discuss several hypotheses that have been proposed to explain the lineage switch and ambiguity in leukemias. These mechanisms are not mutually exclusive and likely occur in parallel. For example, dysregulation of lineage-specific transcription factors may generate an aberrant bi-potential leukemic clone, and therapies may facilitate the selection of bi-potential clones that are better equipped to survive the therapy by changing their phenotype.
Multipotency of Leukemic Clones
One potential mechanism to explain how some leukemias switch their lineages is that these leukemias were derived from bi-potential clones. When leukemia cells from a patient who exhibited T-cell acute lymphoblastic leukemia (T-ALL) to AML switch upon chemotherapy were transplanted into SCID mice, engrafted AML cells exhibited myeloid cell markers (such as CD33) as expected, but the cells exhibited T-cell markers (such as CD2, CD4, and CD7) similar to the T-ALL at diagnosis if the recipient mice were treated with cytokines GM-CSF or interleukin 3 (IL-3) (42). Interestingly, although a common NRAS mutation was identified at every time point during the study (T-ALL at diagnosis, AML upon lineage switch, and T-ALL or AML in SCID mice) suggesting that both T-ALL and AML were derived from a common founding clone, the TCR rearrangement observed in T-ALL at diagnosis was not observed in patient’s AML cells upon switch nor the cells in SCID mice. This results indicate that the AML emerged in the patient upon lineage switch were not derived from T-ALL cells with TCR rearrangement, and suggests that a common NRAS mutated bi-potent leukemia clone with T-ALL and AML potential existed (42).
On the other hand, other studies have shown the presence of TCR rearrangements in myeloid leukemia cells upon lineage switch from lymphoid leukemias (43, 44). In these cases, it remains unclear whether the lymphoid leukemia clones with TCR rearrangements had bi-potential at diagnosis, or whether the lymphoid clones gained myeloid potential through potential mechanisms discussed below. Similarly, reports on B-cell precursor acute lymphoblastic leukemia (BCP-ALL) to AML switch have suggested that a bi-potential B-myeloid progenitor, which has been detected in fetal and adult mice (45, 46), may have become transformed, but evidence that such bi-potential progenitor cells are the origin of the disease and existed at diagnosis is lacking. It is equally possible that a B-ALL clone changed the phenotype upon treatment due to selective pressure.
Clonal Selection and Therapies
Lineage switch is often associated with therapy relapse, suggesting that clones with altered phenotypes emerge as a consequence of the selective pressure imposed by the therapy. Therapies can eradicate the dominant clone(s) but select for a latent clone that survived the therapy, or the dominant clone may acquire additional mutations to evolve. While this has been elegantly demonstrated in relapsed AML (47), therapy-related selection has also been reported in lineage switched leukemia. For example, Podgornik et al. reported a B-ALL patient who relapsed with AML from a separate clone that survived the B-ALL therapy (48), while Mantadakis et al. reported on a pediatric patient with T-ALL who relapsed with AML after chemotherapy (49). Recent findings with B-ALL immunotherapy further lend insight into this mechanism (Figure 2). Chromosomal rearrangements at 11q23 are found in both AML and ALL and result in the fusion of the MLL1 gene with approximately 80 partner genes, among which AF4 is the most common partner (50). MLL–AF4 fusion is mostly associated with pro-B-ALL expressing some myeloid cell markers such as CD15 (51). Recent studies reported cases of MLL–AF4 rearranged pro-B-ALL patients treated with blinatumomab, a bispecific antibody that targets CD19 on B-cells, who relapsed with AML (52–54). Transplantation of the relapsed AML into NSG mice caused CD19+ B-ALL that was genetically related to the relapsed AML, suggesting that the anti-CD19 treatment selected for clones that downregulated CD19 and acquired phenotypic changes toward the myeloid lineage (52). Moreover, CD19 CAR-T therapy caused relapse accompanied by a phenotypic change from pre-B-ALL to a myeloid phenotype (55, 56). The authors also used a mouse E2a–PBX1 transgenic B-ALL model and demonstrated that mouse CD19 CAR-T treatment relapses by either causing an alternative splicing of CD19 in B-ALL cells to escape CAR-T cells, or by causing a switch to myeloid leukemias with low Pax5 and Ebf1 expression and increased expression of CD11b and Gr-1 (56). Relapsed myeloid leukemia cells were not detected in E2a–Pax5 pre-B-ALL cells using single cell approaches, suggesting that CAR-T unlikely selected for rare myeloid leukemia cells but instead reprogrammed the B-ALL cells into a myeloid fate. These findings reinforce the idea that some leukemias retain the plasticity to drastically change their phenotype in the face of a strong selection imposed by therapies, and that lineage switch might represent a novel mechanism of resistance against immune therapies.
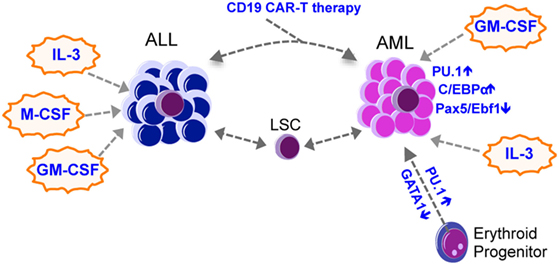
Figure 2. Lineage switch in leukemia. Most lineage switch in leukemia is from ALL to AML, with occasional AML to ALL switches being observed. Lineage switch could be mediated by therapy-mediated (such as CAR-T immunotherapies) selection of heterogeneous leukemic clones that survive the therapy and expand to produce an altered lineage output. Transcription factors such as Pu.1, C/ebp-α, Pax5, and Ebf1, together with cytokines such as IL-3, M-CSF, and GM-CSF, may mediate the lineage switch. AML, acute myeloid leukemia; ALL, acute lymphoid leukemia; CML, chronic myeloid leukemia; LSC, leukemia stem cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; IL-3, interleukin 3.
Cell Reprogramming by Transcription Factors
Similar to how lineage-specific transcription factors reprogram the fate of normal hematopoietic progenitors, these transcription factors have a large impact upon the fate of leukemia cells. Leukemogenic mutations appear to tip the balance created by the network of transcription factors to block and/or bias the differentiation program toward a particular lineage. Dysregulated expression of Pu.1, Gata1, and C/ebp-α all contribute to leukemogenesis (57), and some of these factors are used by leukemic fusion genes to promote aberrant self-renewal of leukemia. For example, AML caused by MLL-fusion genes or MOZ-TIF2 depends upon Pu.1 for their maintenance (58, 59). Recent studies also indicate that these lineage-specific transcription factors regulate the fate choice of leukemia, and may contribute to the lineage switch of leukemias observed upon therapies.
In zebrafish models, AML1–ETO upregulates Pu.1 and downregulates Gata1 to convert the fate of erythroid cells into granulocytic cells, causing a phenotypic change similar to human AML (60). Overexpression of Pu.1 was reported in a rare case of adult Philadelphia chromosome-positive bilineage leukemia (myeloid and T-cell), in which TCR rearrangement was detected in both the myeloid and T-cell compartments of the disease, suggesting that the AML population emerged from T-ALL cells, potentially due to Pu.1 expression (61). Similar to Pu.1, C/ebp-α is also found to be involved in leukemia lineage switch. Slamova et al. reported cases of BCP-ALL that underwent lineage switch upon therapy to monocytic leukemias, which carried the same Ig/TCR rearrangements as the original BCP-ALL (43). The monocytic leukemias with Ig/TCR rearrangements had C/ebp-α promoter hypomethylation accompanied by increased C/ebp-α expression compared to the original BCP-ALL (43). These results suggest that myeloid transcription factors, such as Pu.1 and C/ebp-α, are involved in promoting lymphoid leukemias to switch their fate to a myeloid fate. Additional evidence suggests that the lineage switch can be promoted by the loss of lymphoid transcription factor expression. In the reported case of CD19 CAR-T-induced lineage switch of B-ALL to AML (56), expression levels of lymphoid transcription factors Pax5 and Ebf1 were reduced. Mouse model of CAR-T-induced lineage switch also revealed epigenetic changes leading to loss of Pax5/Ebf1 and increase of C/ebp-α expression and demonstrated that deletion of Pax5 or Ebf1 promoted the lineage switch from B to myeloid fate without CAR-T therapy (56). However, it is still unclear to what extent myeloid transcription factors contribute to the switch toward the myeloid fate, and whether the lineage-specific transcription factors can be exploited to block switching or target the switched leukemias.
Microenvironment
The local microenvironment of HSPCs regulates the maintenance of HSPCs and is often altered in hematological malignancies (62–64). The microenvironment influences disease initiation (65), progression (63), and the efficacy of the therapies (66) by modulating the cytokine milieu and the metabolic parameters. Moreover, similar to how lineage-instructing cytokines can affect the fate of normal HSPCs, leukemia cells with certain plasticity exhibit different lineage output depending on the cytokine milieu.
MLL-translocated leukemias appear to retain lineage plasticity that can be tapped to direct the differentiation toward either B-cell or myeloid lineages using different cytokines. Expression of MLL-fusion oncogenes in cord blood HSPCs induces B-ALL upon xenotransplantation (67). The types of leukemias these oncogenes caused was affected by the culture conditions, as MLL–ENL expressing cells that are prone to cause B-ALL initiated AML with rearranged IgH when cultured in myeloid-promoting conditions. The ability of MLL–AF9 oncogene to produce B-ALL or AML is also affected by the culture condition, as well as the humanized cytokines expressed in the recipient immunocompromised mice (68). The fusion product of human MLL and murine Af4 (MLL–Af4) initiates pro-B-ALL that recapitulates the human pathology but causes AML when the cells were culture in myeloid-promoting conditions (52). Interestingly, MLL–Af4 transformed myeloid cells cultured in a myelopoietic condition had increased expression of lymphoid regulators, such as Ebf1, compared to AML cells transformed by MLL–AF9, suggesting that the myelopoietic condition cannot fully rewire the lymphoid program imposed by MLL–Af4.
A recent study demonstrated that BCR–ABL1 rearranged B-ALL can be reprogrammed to a myeloid fate by myeloid-instructing and proinflammatory cytokines (69–73). Purified B-ALL blasts exhibited myeloid cell marker expression and phagocytotic phenotypes upon stimulation by IL-3, M-CSF, and GM-CSF. The reprogrammed macrophage-like cells (termed MLCs) had increased expression of myeloid master regulators C/ebp-α and Pu.1, and corresponding overexpression of C/ebp-α and Pu.1 significantly induced myeloid reprogramming, suggesting that the myeloid cytokines and myeloid transcription factors cooperate to confer a myeloid cell fate to B-ALL, consistent with the ability of these two transcription factors to confer a myeloid fate to lymphocytes (11, 12, 14). Interestingly, although the original B-ALL cells that failed to reprogram into MLCs had the ability to cause B-ALL in recipient mice upon xenotransplantation, the reprogrammed MLCs had negligible ability to engraft. Since lineage switch is often associated with relapse and worse clinical outcome, it is unclear whether promoting myeloid reprogramming can be used as a therapeutic strategy. Nonetheless, depending on how deeply leukemias can be directed to differentiate, instructing cells to differentiate into other lineages may provide a novel therapeutic option.
Conclusion
Similar to normal hematopoietic cells, leukemia cells also exhibit lineage plasticity and reversibility aided by master transcriptional regulators that control lineage determination of normal hematopoietic progenitor cells, by the instructive cytokine milieu and also by the strong selective pressure imposed by therapies. Although some improvements in treatment outcome have been reported by the use of intensified ALL therapy followed by AML therapy upon lineage switch, treatment of these leukemia remains challenging (74, 75). The ability of B-ALL cells to change their lineage-specific cell surface marker expression in response to immunotherapies underscores the clinical challenges posed by the plasticity of leukemia. Molecular characterization of the fundamental requirements of leukemias may reveal new strategies to target the disease regardless of their lineages.
Author Contributions
TH, RM, and DN reviewed the literature and wrote the manuscript. TH designed the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health (CA193235 and DK107413), the Cancer Prevention Research Institute of Texas (RP160035), and the Edward P. Evans Foundation.
References
1. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414:105–11. doi:10.1038/35102167
2. Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell (2007) 1:635–45. doi:10.1016/j.stem.2007.10.001
3. Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity (1994) 1:661–73. doi:10.1016/1074-7613(94)90037-X
4. Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science (1996) 273:242–5. doi:10.1126/science.273.5272.242
5. Randall TD, Lund FE, Howard MC, Weissman IL. Expression of murine CD38 defines a population of long-term reconstituting hematopoietic stem cells. Blood (1996) 87:4057–67.
6. Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell (1997) 91:661–72. doi:10.1016/S0092-8674(00)80453-5
7. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature (2000) 404:193–7. doi:10.1038/35004599
8. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol (2007) 7:105–17. doi:10.1038/nri2024
9. Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev (2006) 20:3010–21. doi:10.1101/gad.1493506
10. Nerlov C, Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev (1998) 12:2403–12. doi:10.1101/gad.12.15.2403
11. Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBP alpha and PU.1 transcription factors. Immunity (2006) 25:731–44. doi:10.1016/j.immuni.2006.09.011
12. Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell (2004) 117:663–76. doi:10.1016/S0092-8674(04)00419-2
13. Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, et al. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci U S A (2008) 105:6057–62. doi:10.1073/pnas.0711961105
14. Cirovic B, Schönheit J, Kowenz-Leutz E, Ivanovska J, Klement C, Pronina N, et al. C/EBP-induced transdifferentiation reveals granulocyte-macrophage precursor-like plasticity of B cells. Stem Cell Reports (2017) 8:346–59. doi:10.1016/j.stemcr.2016.12.015
15. Orkin SH, Shivdasani RA, Fujiwara Y, McDevitt MA. Transcription factor GATA-1 in megakaryocyte development. Stem Cells (1998) 16(Suppl 2):79–83. doi:10.1002/stem.5530160710
16. Wang X, Crispino JD, Letting DL, Nakazawa M, Poncz M, Blobel GA. Control of megakaryocyte-specific gene expression by GATA-1 and FOG-1: role of Ets transcription factors. EMBO J (2002) 21:5225–34. doi:10.1093/emboj/cdf527
17. Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood (1999) 93:2867–75.
18. Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, et al. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood (2004) 104:3136–47. doi:10.1182/blood-2004-04-1603
19. Rhodes J, Hagen A, Hsu K, Deng M, Liu TX, Look AT, et al. Interplay of PU.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev Cell (2005) 8:97–108. doi:10.1016/j.devcel.2004.11.014
20. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci U S A (1996) 93:12355–8. doi:10.1073/pnas.93.22.12355
21. Gutiérrez L, Tsukamoto S, Suzuki M, Yamamoto-Mukai H, Yamamoto M, Philipsen S, et al. Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood (2008) 111:4375–85. doi:10.1182/blood-2007-09-115121
22. Kulessa H, Frampton J, Graf T. GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev (1995) 9:1250–62. doi:10.1101/gad.9.10.1250
23. Visvader JE, Elefanty AG, Strasser A, Adams JM. GATA-1 but not SCL induces megakaryocytic differentiation in an early myeloid line. EMBO J (1992) 11:4557–64.
24. Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature (1999) 401:556–62. doi:10.1038/44076
25. Lindeman GJ, Adams JM, Cory S, Harris AW. B-lymphoid to granulocytic switch during hematopoiesis in a transgenic mouse strain. Immunity (1994) 1:517–27. doi:10.1016/1074-7613(94)90094-9
26. Hu Y, Zhang Z, Kashiwagi M, Yoshida T, Joshi I, Jena N, et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev (2016) 30:1971–90. doi:10.1101/gad.283762.116
27. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374:2209–21. doi:10.1056/NEJMoa1516192
28. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368:2059–74. doi:10.1056/NEJMoa1301689
29. Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, et al. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell (2012) 11:302–17. doi:10.1016/j.stem.2012.08.006
30. Grammatico S, Vitale A, La Starza R, Gorello P, Angelosanto N, Negulici AD, et al. Lineage switch from pro-B acute lymphoid leukemia to acute myeloid leukemia in a case with t(12;17)(p13;q11)/TAF15-ZNF384 rearrangement. Leuk Lymphoma (2013) 54:1802–5. doi:10.3109/10428194.2012.753450
31. Imataki O, Ohnishi H, Yamaoka G, Arai T, Kitanaka A, Kubota Y, et al. Lineage switch from precursor B cell acute lymphoblastic leukemia to acute monocytic leukemia at relapse. Int J Clin Oncol (2010) 15:112–5. doi:10.1007/s10147-009-0007-3
32. Shivarov V, Stoimenov A, Galabova I, Balatzenko G, Guenova M. Very early onset of an acute myeloid leukemia in an adult patient with B-cell lymphoblastic leukemia. Int J Lab Hematol (2009) 31:106–13. doi:10.1111/j.1751-553X.2007.00968.x
33. van den Ancker W, Terwijn M, Regelink J, Westers TM, Ossenkoppele GJ, van de Loosdrecht AA, et al. Uncommon lineage switch warrants immunophenotyping even in relapsing leukemia. Leuk Res (2009) 33:e77–80. doi:10.1016/j.leukres.2008.11.029
34. Della Starza I, Ceglie G, Nunes V, Gianfelici V, Marinelli M, Fuligni F, et al. A case of lineage switch from B-cell acute lymphoblastic leukaemia to acute myeloid leukaemia. Role of subclonal/clonal gene mutations. Br J Haematol (2016) 174:648–51. doi:10.1111/bjh.13800
35. Dorantes-Acosta E, Arreguin-Gonzalez F, Rodriguez-Osorio CA, Sadowinski S, Pelayo R, Medina-Sanson A. Acute myelogenous leukemia switch lineage upon relapse to acute lymphoblastic leukemia: a case report. Cases J (2009) 2:154. doi:10.1186/1757-1626-2-154
36. Krawczuk-Rybak M, Zak J, Jaworowska B. A lineage switch from AML to ALL with persistent translocation t(4;11) in congenital leukemia. Med Pediatr Oncol (2003) 41:95–6. doi:10.1002/mpo.10276
37. Lounici A, Cony-Makhoul P, Dubus P, Lacombe F, Merlio JP, Reiffers J. Lineage switch from acute myeloid leukemia to acute lymphoblastic leukemia: report of an adult case and review of the literature. Am J Hematol (2000) 65:319–21. doi:10.1002/1096-8652(200012)65:4<319::AID-AJH13>3.0.CO;2-1
38. Weinberg OK, Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia (2010) 24:1844–51. doi:10.1038/leu.2010.202
39. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood (2015) 125:2477–85. doi:10.1182/blood-2014-10-551465
40. Matutes E, Pickl WF, Van’t Veer M, Morilla R, Swansbury J, Strobl H, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood (2011) 117:3163–71. doi:10.1182/blood-2010-10-314682
41. Mejstrikova E, Volejnikova J, Fronkova E, Zdrahalova K, Kalina T, Sterba J, et al. Prognosis of children with mixed phenotype acute leukemia treated on the basis of consistent immunophenotypic criteria. Haematologica (2010) 95:928–35. doi:10.3324/haematol.2009.014506
42. Fujisaki H, Hara J, Takai K, Nakanishi K, Matsuda Y, Ohta H, et al. Lineage switch in childhood leukemia with monosomy 7 and reverse of lineage switch in severe combined immunodeficient mice. Exp Hematol (1999) 27:826–33. doi:10.1016/S0301-472X(99)00008-9
43. Slamova L, Starkova J, Fronkova E, Zaliova M, Reznickova L, van Delft FW, et al. CD2-positive B-cell precursor acute lymphoblastic leukemia with an early switch to the monocytic lineage. Leukemia (2014) 28:609–20. doi:10.1038/leu.2013.354
44. Rossi JG, Bernasconi AR, Alonso CN, Rubio PL, Gallego MS, Carrara CA, et al. Lineage switch in childhood acute leukemia: an unusual event with poor outcome. Am J Hematol (2012) 87:890–7. doi:10.1002/ajh.23266
45. Cumano A, Paige CJ, Iscove NN, Brady G. Bipotential precursors of B cells and macrophages in murine fetal liver. Nature (1992) 356:612–5. doi:10.1038/356612a0
46. Montecino-Rodriguez E, Leathers H, Dorshkind K. Bipotential B-macrophage progenitors are present in adult bone marrow. Nat Immunol (2001) 2:83–8. doi:10.1038/83210
47. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481:506–10. doi:10.1038/nature10738
48. Podgornik H, Debeljak M, Zontar D, Cernelc P, Prestor VV, Jazbec J. RUNX1 amplification in lineage conversion of childhood B-cell acute lymphoblastic leukemia to acute myelogenous leukemia. Cancer Genet Cytogenet (2007) 178:77–81. doi:10.1016/j.cancergencyto.2007.06.015
49. Mantadakis E, Danilatou V, Stiakaki E, Paterakis G, Papadhimitriou S, Kalmanti M. T-cell acute lymphoblastic leukemia relapsing as acute myelogenous leukemia. Pediatr Blood Cancer (2007) 48:354–7. doi:10.1002/pbc.20543
50. Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol (2012) 7:283–301. doi:10.1146/annurev-pathol-011811-132434
51. Sanjuan-Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, et al. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood (2015) 126:2676–85. doi:10.1182/blood-2015-09-667378
52. Lin S, Luo RT, Ptasinska A, Kerry J, Assi SA, Wunderlich M, et al. Instructive role of MLL-fusion proteins revealed by a model of t(4;11) pro-B acute lymphoblastic leukemia. Cancer Cell (2016) 30:737–49. doi:10.1016/j.ccell.2016.10.008
53. Balducci E, Nivaggioni V, Boudjarane J, Bouriche L, Rahal I, Bernot D, et al. Lineage switch from B acute lymphoblastic leukemia to acute monocytic leukemia with persistent t(4;11)(q21;q23) and cytogenetic evolution under CD19-targeted therapy. Ann Hematol (2017) 96(9):1579–81. doi:10.1007/s00277-017-3050-6
54. Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer (2016) 63:1113–5. doi:10.1002/pbc.25953
55. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood (2016) 127:2406–10. doi:10.1182/blood-2015-08-665547
56. Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun (2016) 7:12320. doi:10.1038/ncomms12320
57. Burda P, Laslo P, Stopka T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia (2010) 24:1249–57. doi:10.1038/leu.2010.104
58. Aikawa Y, Yamagata K, Katsumoto T, Shima Y, Shino M, Stanley ER, et al. Essential role of PU.1 in maintenance of mixed lineage leukemia-associated leukemic stem cells. Cancer Sci (2015) 106:227–36. doi:10.1111/cas.12593
59. Aikawa Y, Katsumoto T, Zhang P, Shima H, Shino M, Terui K, et al. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat Med (2010) 16:580–585, 581p following 585. doi:10.1038/nm.2122
60. Yeh JR, Munson KM, Chao YL, Peterson QP, Macrae CA, Peterson RT. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development (2008) 135:401–10. doi:10.1242/dev.008904
61. Monma F, Nishii K, Ezuki S, Miyazaki T, Yamamori S, Usui E, et al. Molecular and phenotypic analysis of Philadelphia chromosome-positive bilineage leukemia: possibility of a lineage switch from T-lymphoid leukemic progenitor to myeloid cells. Cancer Genet Cytogenet (2006) 164:118–21. doi:10.1016/j.cancergencyto.2005.06.021
62. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505:327–34. doi:10.1038/nature12984
63. Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell (2015) 16:254–67. doi:10.1016/j.stem.2015.02.014
64. Jiang Y, Nakada D. Cell intrinsic and extrinsic regulation of leukemia cell metabolism. Int J Hematol (2016) 103:607–16. doi:10.1007/s12185-016-1958-6
65. Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature (2016) 539:304–8. doi:10.1038/nature20131
66. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol (2012) 14:276–86. doi:10.1038/ncb2432
67. Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science (2007) 316:600–4. doi:10.1126/science.1139851
68. Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell (2008) 13:483–95. doi:10.1016/j.ccr.2008.04.020
69. McClellan JS, Dove C, Gentles AJ, Ryan CE, Majeti R. Reprogramming of primary human Philadelphia chromosome-positive B cell acute lymphoblastic leukemia cells into nonleukemic macrophages. Proc Natl Acad Sci U S A (2015) 112:4074–9. doi:10.1073/pnas.1413383112
70. Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, et al. IL-6 blocks a discrete early step in lymphopoiesis. Blood (2005) 106:879–85. doi:10.1182/blood-2005-02-0456
71. Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell (2011) 20:661–73. doi:10.1016/j.ccr.2011.10.012
72. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell (2014) 14:460–72. doi:10.1016/j.stem.2014.01.002
73. de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood (2012) 119:1543–54. doi:10.1182/blood-2011-07-367706
74. Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D. Lineage switch in acute leukemia. Blood (1984) 64:701–6.
Keywords: lineage switch leukemia, mixed-phenotype acute leukemia, hematopoietic stem cells, acute myeloid leukemia, acute lymphoid leukemia, CAR-T cells
Citation: Hu T, Murdaugh R and Nakada D (2017) Transcriptional and Microenvironmental Regulation of Lineage Ambiguity in Leukemia. Front. Oncol. 7:268. doi: 10.3389/fonc.2017.00268
Received: 31 August 2017; Accepted: 23 October 2017;
Published: 06 November 2017
Edited by:
Keisuke Ito, Albert Einstein College of Medicine, United StatesReviewed by:
Takaomi Sanda, National University of Singapore, SingaporeFrancesco Grignani, University of Perugia, Italy
Copyright: © 2017 Hu, Murdaugh and Nakada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisuke Nakada, bmFrYWRhQGJjbS5lZHU=