 Anwen Howells
Anwen Howells Giulia Marelli
Giulia Marelli Nicholas R. Lemoine1,2
Nicholas R. Lemoine1,2 Yaohe Wang
Yaohe Wang- 1Centre for Molecular Oncology, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
- 2National Centre for International Research in Cell and Gene Therapy, Sino-British Research Centre for Molecular Oncology, Academy of Medical Sciences, Zhengzhou University, Zhengzhou, China
Oncolytic viruses (OVs) are an emerging treatment option for many cancer types and have recently been the focus of extensive research aiming to develop their therapeutic potential. The ultimate aim is to design a virus which can effectively replicate within the host, specifically target and lyse tumor cells and induce robust, long lasting tumor-specific immunity. There are a number of viruses which are either naturally tumor-selective or can be modified to specifically target and eliminate tumor cells. This means they are able to infect only tumor cells and healthy tissue remains unharmed. This specificity is imperative in order to reduce the side effects of oncolytic virotherapy. These viruses can also be modified by various methods including insertion and deletion of specific genes with the aim of improving their efficacy and safety profiles. In this review, we have provided an overview of the various virus species currently being investigated for their oncolytic potential and the positive and negative effects of a multitude of modifications used to increase their infectivity, anti-tumor immunity, and treatment safety, in particular focusing on the interaction of tumor cells and OVs.
Introduction
One of the most promising developments in cancer therapy to emerge over the past few decades is oncolytic virotherapy (OVT). Many of the more traditional treatment options routinely used to combat cancer in the clinic are not efficacious enough and have considerable side effects for patients. Although these treatments, such as chemotherapy and radiotherapy, are advancing and becoming more tolerable, we are yet to discover an alternative treatment option that has a high level of potency with minimal side effects that will dramatically change the overall survival of cancer patients.
Oncolytic viruses (OVs) have the potential to deliver this goal and much effort has been put into improvement of their efficacy and safety profiles in recent years. There are numerous viruses which either have naturally oncolytic properties or have been engineered to specifically lyse tumor cells. The great advantage of this therapy is that these viruses are able to specifically target tumor cells and therefore healthy tissue is not damaged during the course of the treatment. There are various ways to improve the specificity of OVs, for example, taking advantage of pathways which are upregulated in tumor cells and not healthy cells and engineering a virus which relies on such a pathway for successful infection thereby rendering the virus incapable of infecting healthy tissue (see Figure 1).
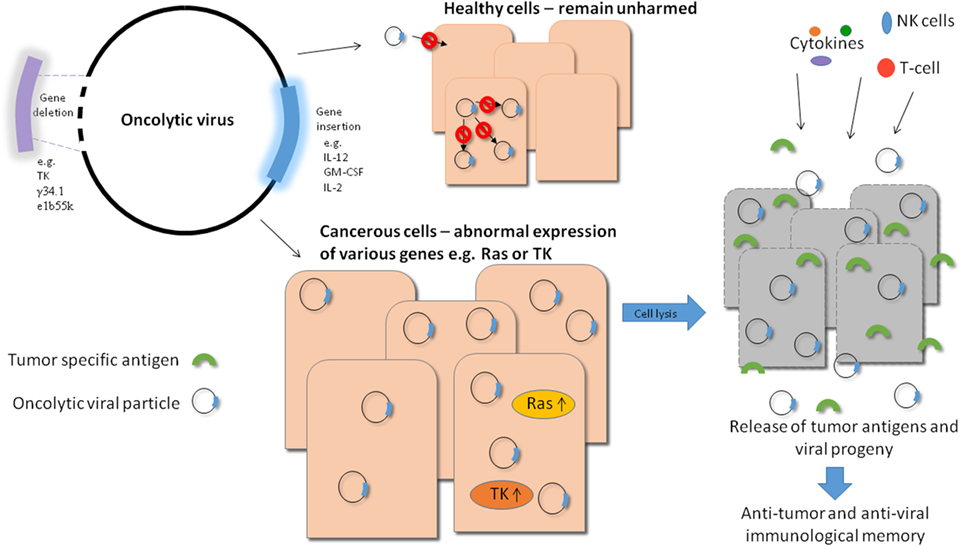
Figure 1. Overview of oncolytic viral therapy. Genetic engineering has made it possible to modify oncolytic viruses (OVs) to make them safer and more effective against tumor cells. Various genes can be deleted to produce a safer virus, e.g., thymidine kinase (TK), while others can be inserted into the viral genome to increase efficacy, e.g., immune stimulators like IL-12. By carrying out these modifications, OVs can be made safer as they require target cells to provide the essential deleted gene, e.g., TK which is upregulated in tumor cells and not in healthy cells. They can also generate long lasting immunity as a result of stimulation of a potent immune response in which tumor cell antigens (along with viral antigens) can be targeted by T-cells. Also, when the virally infected tumor cells die, they release progeny virions into the tumor microenvironment which can in turn infect neighboring cells, improving the efficiency of viral treatment. Systemically delivered OVs also have the potential to eliminate metastatic tumor cells.
Another factor of oncolytic viral therapy that makes it a promising candidate is that while viral infection can directly lyse tumor cells, the resultant immune response will be generated not only to viral antigens but also to tumor cell antigens. This unique feature of oncolytic viral therapy makes it a very exciting avenue of research as not only can the viruses act to eradicate existing tumors but they can also potentially generate lasting immunity in the form of memory T-cells which are primed against tumor cell antigens (see Figure 1).
In addition to causing direct lysis of cells, OVs can also be used as delivery vectors for therapeutic genes. In this case, the virus can be genetically modified to include the gene of interest which upon infection and viral replication will be produced at high levels in infected tumor cells where it can exert its function. There are a multitude of anti-cancer genes that can be incorporated into OVs in this way in order to maximize the efficacy of the virus and improve the anti-tumor response generated (see Figure 1).
Also, OVs have great potential as combination therapies used together with more traditional approaches. In this way, various treatment options can be used synergistically to combat cancer from more than one angle at a time which will likely give rise to a more positive response to treatment. This combination therapy approach can lead to improved tolerance of treatment in patients as the synergistic effect allows lower doses of each individual therapy to be used to gain similar effects compared with the use of one treatment alone.
As stated, much research effort has been put into improving this area of cancer therapy and the various viruses used and important advances made in their development are discussed here.
History of OV Therapy
The use of OVs was first conceived following the observation of the fact that during or after an infection, tumor regression is occasionally observed (1–3). Based on this observation, patients with Hodgkin’s lymphoma were treated with serum containing hepatitis virus (4).
In the following years, a lot of effort was put in to achieving better and safer results. For 30 years, from 1950 to 1980, many studies were performed without reaching good clinical outcomes or providing long-term results (5–7). This was mainly due to the fact that viral treatments were unsafe because there were no methods to control virulence nor to obtain tumor specificity. Finally, in the late 1980s, with the advent of genetic engineering, a renewed interest for OVT rose again and in recent years many advances have been made in this field.
Selectivity of OVs
There are various ways in which different OVs are able to infect cells. Some viruses, like vaccinia virus (VV) or Newcastle disease virus (NDV) lack specific receptors for attachment so enter cells via endocytosis. Other viruses have a specific receptor that they use to enter host cells; for example, adenoviruses (Ads) are able to bind coxsackie and adenovirus receptor (CAR), integrins, or cluster of differentiation 46 (CD46). Measles can also use CD46 for entry, whereas herpes simplex virus (HSV) uses nectin or herpesvirus entry mediator (8, 9). Despite the observed tendency of tumor cells to upregulate some of these receptors, they are also expressed on many normal cells.
There are a variety of ways in which OVs can be targeted to tumor cells in order to minimize damage to healthy cells. These include exploitation of various pathways which are aberrantly expressed in tumor cells to ensure engineered viruses are only capable of productive infection in cells which have abnormal levels of certain genes. Also, control of viral replication with microRNA differentially expressed in tumor cells compared with healthy cells can restrict viral replication specifically to tumor cells. Viral coat proteins can also be manipulated to ensure viral infection only occurs in cells with certain receptors, e.g., receptors found on tumor cells only. These strategies will be discussed in more detail here.
As it is essential that OVs only successfully infect tumor cells to avoid the spread of virus in healthy tissue, many different approaches have been investigated to increase specificity. One of these is to take advantage of the aberrant expression of various proteins in pathways which can have an effect on viral replication. Of these pathways, OVs commonly exploit aberrant expression of proteins involved in the Ras pathway. This pathway is generally silent in normal cells but activated in tumor cells and the downstream effects of this can be beneficial for OV infection (10, 11). There are a number of ways in which upregulation of the Ras pathway in tumor cells can influence the outcome of oncolytic viral infection. For example, it has been shown that the Ras/MEK pathway can downregulate specific interferon-inducible genes which may have an effect on anti-viral responses and apoptosis control (12). It has also been seen that apoptosis can be involved in the increased efficiency of OVs in tumor cells. In the case of Reovirus, this OV can cause an accumulation of Ras within the Golgi body which leads to triggering of apoptosis signaling pathways and subsequent release and spread of progeny virions (13).
Because of aberrant expression, genes involved in the Ras pathway (among others) can favor replication of viruses in tumor cells and many viruses have been engineered to exploit this to increase their selectivity for transformed cells. For example, engineering viruses which are only able to express certain critical viral proteins upon upregulation of transcription factors downstream of the Ras pathway renders the virus only able to replicate in cells with an upregulated Ras pathway (14).
Other strategies used to produce tumor-targeted replicating OVs include control of certain genes using microRNA. Hikicki et al. have shown that it is possible to place critical viral genes under the control of an miRNA which has low expression levels in tumor cells. This renders the virus unable to successfully infect healthy cells where normal levels of this miRNA are expressed, facilitating interference with production of the critical viral gene (15).
Also, modification of viral coat proteins can be used to specifically direct viral infection to tumor cells. There are various ways to achieve this, for example, covering the viral surface with polymer to “cloak” the existing receptor and addition of epidermal growth factor (EGF) to target the virus to tumor cells which tend to have upregulated EGF receptor (EGFR) expression (16). This strategy not only reduces the broad tropism conferred by the existing viral receptor but also replaces this with tumor specific receptors to direct oncolytic viral infection to target cells, leaving healthy tissue unharmed.
Another approach involves the use of antibodies to target OVs to tumor cells. As an example of this strategy, it was found by Watkins et al. that antibodies can be engineered which contain an Ad fiber protein targeting single-chain variable fragment (scFv), linked to EGF. This facilitated targeting of Ad to EGFR-upregulated tumor cells (17). This antibody focused approach was further developed to allow incorporation of scFv into the viral envelope. For example, HSV type-1 (HSV-1) relies on various glycoproteins for entry into cells and one of these glycoproteins (gD) is responsible for interaction with the viral entry receptors. If an scFv targeting EGFR is fused to this glycoprotein, the virus is then able to use EGFR as an entry receptor which improves tumor targeting (18).
In parallel to the attempt to create safer viruses, a new strategy is developing with the aim of incorporating transgenes within the virus to target the tumor microenvironment or to activate the immune system.
Modifications of OVs
This new strategy is achieved by modification of viral genomes by insertion or deletion of selected genes which aid or hinder oncolytic potential and some of the strategies being explored will be discussed in more detail here.
The ability to modify the genome to our advantage is one of the most promising aspects of OVs. These modifications have a variety of functions including improvement of tumor tropism and increased recruitment of the immune system to aid anti-tumor responses. Also, improved safety and efficacy of OVs are of utmost importance and are another aspect that can be controlled by genetic engineering. Some examples of gene editing include deleting replication-related genes to reduce replication efficiency (as attenuation improves safety) and/or addition of genes that induce pathways to promote tumor cell death, for example, the apoptosis pathway (19).
As previously mentioned, pathways which are alternatively regulated in tumor cells compared with healthy cells can be exploited to produce selective viruses. For example, genes can be deleted resulting in a virus that can only successfully infect certain tumor types which over-express MEK (20). This strategy can also be used with HSV whereby genes can be deleted to produce a virus which preferentially replicates in tumor cells which unlike healthy cells tend to have a constitutively activated Ras pathway (21). As this virus initiates apoptosis in infected and bystander cells and preferentially infects tumor cells, it can be used as oncolytic agent with this deletion (21, 22).
Gene Deletion Strategies
Many OVs have been modified with specific gene deletions to target the virus to tumor cells and inhibit infectivity in healthy cells. An example of this strategy is thymidine kinase (TK) deletion from VV. As the wild-type virus usually encodes this kinase it is able to replicate in healthy cells, however, when the gene is deleted the virus can no longer replicate efficiently in healthy cells. As tumor cells produce higher levels of TK, even though the gene is deleted the virus is still able to replicate in these cells (23, 24).
Another strategy used to improve tumor specificity is to delete apoptosis-inhibiting genes (usually used with Ad). In wild-type infections, Ad encodes genes which block apoptosis which is an advantage as infected cells then become viral factories given that they will not enter apoptosis in response to infection. As tumor cells often block apoptotic pathways as a survival mechanism, Ads with deleted apoptosis-inhibiting genes can undergo prolonged infection while infection of healthy cells will lead to lysis of the cell and clearance of virus (25).
Gene deletion strategies can also be used to improve the efficiency of virus delivery systems which are designed to deliver OVs to target cells without interference from the host immune system. In these systems, viruses are delivered within host cells (e.g., mesenchymal stem cells) that provide shelter from immune attack and subvert the problem of clearance of virus (by neutralizing antibodies) before they reach their target cells. This method has been improved in Ad by modification of the virus in order to make it more infective in MSC and more efficient at killing tumor cells. Oncolytic Ad can also be engineered through deletion of an anti-apoptotic gene, improving virus release from MSC, and allowing more potent anti-tumoral activity (26).
Although gene deletion often improves efficacy of OVs, the chosen candidates need to be selected very carefully. Various gene deletions have the potential to alter viral infectivity in ways which can either improve or diminish oncolytic potential. For example, deletion of E3-6.7K/gp19K leads to more rapid viral clearance which on one hand improves safety but on the other hand only allows a short time-frame for inserted therapeutic genes to be delivered to target cells and produced in a significant amount. Therefore, this particular deletion can only successfully be used for delivery of genes which can act quickly to have the desired effect (27). Another study has shown that deletion of a combination of viral genes will enhance tumor selectivity but reduce viral potency, highlighting the problems faced in engineering the perfect oncolytic viral therapy (28). There are also genes encoded by OVs that can act to inhibit oncolytic potential, for example, E4orf1 encoded by Ad leads to increased levels of survival in infected cells thereby reducing the ability of the virus to directly lyse infected tumor cells (29). This effect can lead to diminished efficacy and therefore needs to be addressed in order to maximize the potential of this virus to treat cancer.
Gene Insertion Strategies
Another advantage of gene deletion is the opportunity to insert therapeutic genes in their place without disrupting the reading frame (30). There are a multitude of therapeutic genes and immune stimulators which can be delivered within OVs to combat cancer (for example, the interleukin family of genes used to stimulate the immune system thereby improving anti-tumor immune responses). However, this approach is not perfect and the combination of deleting a gene and inserting a new one can result in problems of its own. For example, deletion of the E1B55K gene leads to improved virus spread (as it facilitates apoptosis), however, this may result in low levels of production of the inserted gene as the cell undergoes apoptosis before high quantities of the gene are expressed. The combinations of deletion and insertion need to be specifically studied in order to ascertain which ones complement each other and which have negative effects on each other (31, 32). Addition of cytokines or other genes may also give rise to toxicity, for example, the IL-12 cytokine has been seen to result in side effects that cause a poor safety profile. This problem has been combated in a range of ways with varying results; these include using a single-chain version of the cytokine and anchoring IL-12 to the membrane of cells through fusion with the CD4 trans-membrane region. These methods did not produce the desired reduction in toxicity without reducing anti-tumor efficacy, however, using a helper-dependent Ad vector with an inducible expression system was successful in allowing production of IL-12 without high levels of toxicity (33). Another strategy to reduce potency of the IL-12 cytokine is to deliver it within conditionally replicative Ad rather than replication competent strains. For example, on delivery within an adenoviral vector which can only replicate in hypoxic conditions typical of tumor masses, IL-12 was still effective but resulted in less toxicity and more specific delivery to target cells compared with replication competent viral delivery (34).
Other Strategies to Improve OVs
As well as addition and deletion, control of gene promoters can be used to modify viral behavior. For example, promoters that are activated more highly in tumor cells can be used to control an essential viral gene rendering that virus incapable of replicating in healthy cells (32). They can, however, replicate successfully in tumor cells as these cells have a higher activation of that promoter (e.g., use of Cyclin E promoter to target Ad infection to tumor cells) (35). It is also possible to use promoter control of virus genes in order to attenuate virus and make the therapy safer. If genes essential to virus replication are expressed under the control of a promoter downregulated in tumor cells, then delivery of such a tumor-selective virus into those cells will only allow a low level of infection which results in improved safety with retention of oncolytic activity (although at a lower level than wild-type infection) (15).
A constantly evolving area of research is the combination of OVs with other treatments for synergistic effect. For example, combining oncolytic Ad with a cytotoxic drug currently used in the clinic enhanced the anti-tumor efficacy of this treatment (36). These combinations can improve the activity of OVs in various ways, including promoting better replication or compensating for certain deletions without compromising tumor selectivity. For example, if a deleted gene has more than one function, it can be eliminated to improve selectivity in combination with delivery of a compound that can be administered to improve replication efficiency which may have been lost through deletion; an example is the use of 2-aminopurine to enhance the oncolytic activity of an E1B-deleted Ad (37). Combining treatments can also facilitate administration of each agent at lower and safer doses given their synergistic effect (38). Also, the combination of oncolytic Ad with CAR upregulation can improve the efficacy of this treatment due to the use of these receptor molecules for Ad infection (39). Various tumor cell types express different levels of CAR and this will have an impact on the ability of the virus to effectively treat different tumors. Therefore, administration of an agent which can upregulate CAR expression before oncolytic Ad therapy could increase the efficacy of adenoviral treatment.
There are numerous viruses which have been found to have natural or engineered oncolytic activity and some of the most promising candidates will be explored in more detail in the following sections.
Vaccinia Virus
Vaccinia virus is a naturally oncolytic virus which was found to have a natural tropism for tumor cells due to its sensitivity to type I interferon (40). It is a double-stranded DNA virus of the Poxviridae family. There are many different strains of the virus and of these; Lister, Wyeth, and Western Reserve strains are the most used in research. VV is a very promising anti-cancer agent (41) for many reasons including its very short life cycle (around 8 h) and its ability to replicate in hypoxic conditions (42). Moreover, it does not have a specific receptor and viral fusion with the plasma membrane facilitates entry (43) which makes it a potential candidate for treatment of all tumor types. Furthermore, VV does not depend on the host cell for mRNA transcription and its entire life cycle takes place in the cytoplasm, eliminating the risk of genomic integration (44). The virus, which can infect both human and mouse cells, is infectious at four different stages of its life cycle: intracellular mature virion released by cell disruption, intracellular enveloped virion, cell-associated enveloped virion, or extracellular enveloped virion released by endocytosis from the membrane.
To make oncolytic VV safer, two deletions have been made; one in the TK region (45) (as for HSV) and one in the vaccinia growth factor gene region increasing its specificity for tumor cells (23). As well as this, many efforts have been made to generate a strain that results in viral attenuation, rendering the virus harmless in normal tissue. For this purpose, mutations in the F14.5L and A56R genes have been engineered. The F14.5L gene encodes a secretory signal peptide, while the A56R gene generates hemagglutinin.
In addition to these safety measures, many different genes have been integrated into the VV genome in order to increase its anti-tumor efficacy such as cytokines and antibodies, as reviewed by Badrinath et al. (9). As an example, JX-594 is a VV with TK gene deletion and GM-CSF (a cytokine able to stimulate the immune system to kill tumor cells) gene insertion and is currently undergoing phase III trials. Phase I trials have shown promising results and acceptable safety profiles (46) and phase II studies were designed to investigate the optimal dose of intravenously delivered JX-594 (47). The results of a randomized, dose-finding phase II clinical trial reported by Heo et al. showed that high dose JX-594 resulted in higher overall survival duration than low dose administration in patients with advanced hepatocellular carcinoma. This study also demonstrated both anti-viral and anti-tumor immunity generated in clinical patients in response to OV administration (48).
There are many other modifications which can improve the anti-tumor efficacy of VV. For example, the addition of IL-10 was found to improve the oncolytic activity of VV through dampening of anti-viral immunity (prolonging viral infection) without reducing anti-tumor immunity (49).
The major concern in using VV as oncolytic therapy is the fact that it is easily recognized by the immune system. Indeed, the strains of VV exploited in clinical OVT are derived from the vaccine formulations used for smallpox eradication and so there is an activation of the immune system in those patients that were administered with the vaccination. Despite this, Breitbach et al. demonstrate in a clinical study that the intravenous administration of the modified VV JX-594 was safe and that the virus was able to replicate and to express the transgene only in tumor cells (50).
Adenovirus
Adenovirus is one of the most commonly studied viruses in oncolytic therapy and was the first to be given regulatory approval, granted by the State Food and Drug Administration in China in 2005 (51). It is a non-enveloped, double-stranded DNA virus of the Adenoviridae family. There are several strains of Ad and of these, Ad5 is the most commonly used in oncolytic therapy. As this virus is widely studied, there are a multitude of various modifications which have been shown to improve its efficacy and safety which will be further discussed below.
It has been seen that addition of various genes to oncolytic Ads can improve their anti-tumor efficacy. For example, the combination of p53 addition to suppress tumor growth with GM-CSF addition to induce the apoptotic pathway elicits a synergistic effect which is effective in combating hepatocellular cancer stem cells (52). This is especially exciting as it potentially provides a mechanism to combat cancer stem cells which are considered to be integral to cancer recurrence after current treatment options. GM-CSF addition has been tested in phase I clinical trials and was shown to be non-toxic and tolerable at the doses used, however, the efficacy showed room for improvement in terms of anti-tumor efficacy and long-term immunity (53).
It is also possible to improve oncolytic Ad by incorporation of a short-hairpin RNA which functions to downregulate Dicer (an endoribonuclease which has a role in processing virus-associated RNA). Downregulation of this protein inhibits the destruction of viral RNA and allows Ad to replicate efficiently and therefore improves the efficacy of this OV (54).
Gene silencing techniques can also be used in order to downregulate certain oncogenes in order to suppress tumor growth. For example, downregulation of EphA3 by insertion of siRNA targeting this gene into the genome of an Ad whose replication is made conditional under the control of TERTp (which increases specificity for tumor cells) results in increased levels of autophagy through inhibition of the AKT/mTOR pathway. This allows the virus to both inhibit tumor cell proliferation and kill infected cells (55).
In terms of important features of OVs on which to focus research efforts, it has been shown that the T-cell immune response to oncolytic adenoviral infection is more efficacious in combating tumors than direct lysis of tumor cells by viral infection (56).
It has been previously shown that treatment of tumors with immune checkpoint inhibitors (e.g., PD-L1 blockade) allows immune-boosting viral treatments to have a longer lasting effect. It has recently been found that encoding a PD-L1 blocking antibody within Ad vectors leads to production of this antibody in the local vicinity of tumors by host cells and this approach gives rise to better anti-tumor effects than with infusion of the antibody into tumors. When combined with chimeric antigen receptor-modified T-cell treatment targeting HER2 positive tumor cells, this approach to PD-L1 antibody delivery increased treatment efficacy (57).
In addition to investigating methods of improving current Ad strains used in oncolytic therapy, it is also important to find potentially new strains which may prove to be more appropriate for therapy. For example, Ad11 was found to have higher levels of receptor availability and lower levels of neutralizing antibody than Ad5 (which is the most commonly utilized strain in OV engineering). When this strain is modified to replace its E1A enhancer-promoter region with that of Ad5 (leading to higher levels of E1A mRNA) it can become a more potent OV. With additional modifications to increase tumor specificity, the Ad11/Ad5 strain could prove to be a more successful OV than the more commonly studied Ad5 strain (58). Also, another group B oncolytic Ad is enadenotucirev, this virus is able to infect both at apical and basolateral surfaces of polarized cells and results in progeny being released via the apical surface which directs them into the tumor mass rather than out into the blood stream (59). This proves to be an improvement on the traditional Ad5 strains used in the design of oncolytic therapy as type 5 Ads seem to infect preferentially via the apical surface which could pose a problem when delivered systemically.
Even once a promising candidate has been identified for oncolytic therapy, one of the major obstacles to effective use of OVs is clearance by the host immune response (namely anti-viral T-cells) but it would be extremely useful to use this response to our advantage. In a recent study, it has been found that it is possible to engage and therefore redirect these T-cells to react to specific antigens (in this case EGFR which is often overexpressed on tumor cells) in order to redirect the anti-viral response into an anti-tumor response (60). Also, the generation of T-cell responses both to virus and tumor was found to be more important in viral efficacy than direct oncolysis (56). Therefore, a balance between increasing T-cell production to improve anti-tumor immunity and controlling T-cell response to reduce viral clearance is necessary.
In the constant effort to find new and improved oncolytic therapies, it was found that certain cancer cell types produce a peptide which inhibits the ability of the Ad to escape endosomes and be released (61). This inhibition presents a barrier to effective viral spread and successful oncolytic therapy. However, type 3 Ad has evolved to manufacture a decoy capsid which sequesters HD5 and renders the virus able to escape endosomes (62). This finding suggests that the mechanism used by this strain of Ad can be mimicked in order to improve efficacy of existing or potential oncolytic Ad therapies.
Another obstacle facing oncolytic therapy is the tumor microenvironment and its immuno-suppressive properties. For example, expression of TGF-β in the tumor microenvironment results in diminished ability of virally delivered IL-12 to boost anti-tumor immune responses. One method to overcome this problem is to co-express decorin which leads to attenuated TGF-β expression in tumors (63). This is just one example of therapeutic gene addition which improves the action of oncolytic Ads armed with cytokines aimed at boosting anti-tumor immune responses.
Combination therapy, whereby OVs are used in combination with conventional therapy is a growing area of research with many promising leads. For example, it has been shown recently that Ad encoding pro-inflammatory IL-18 cytokine has a synergistic effect when delivered in combination with dacarbazine, which is conventionally used to treat melanoma by alkylation of DNA strands. When used in combination, it was seen that these treatments together result in inhibition of tumor cell growth and increase in apoptosis (64). A similar strategy was employed in another study whereby IL-12 was used as an immune stimulator and a VEGF-silencing ribonucleic acid was co-expressed in order to overcome the immune suppressive action of VEGF produced by tumor cells (65). Many studies have been undertaken to assess which combinations work best and promising results have been reported, for example, oncolytic Ad combined with Temozolomide treatment (alkylating agent) leads to increased levels of viral replication and tumor cell death via various mechanisms thought to include upregulation of autophagy and apoptosis pathways (66). Another combinatorial approach to cancer treatment is to combine OV with anti-tumor antibody treatment. It was found that the gene for an anti-HER2 antibody used in the clinic (Trastuzumab) to combat HER2 positive breast cancers could be inserted into an oncolytic vector and successfully translated into antibody within tumor cells. This was then released upon lysis of infected cells (aiding treatment specificity) and provided an answer to some of the difficulties in delivery of antibodies systemically, specifically the infiltration of tumor masses as virus can infect, and spread throughout the tumor delivering the antibody with it (67).
Another approach to combination therapy is to use two antigenically distinct OVs sequentially (whereby the second to be delivered is not cleared by immunological memory to the first dose). This strategy was reported to be successful when oncolytic VV was used after first administering adenoviral therapy. In this case, it was seen that the increased efficacy was dependent on T-cell activity (68). Subsequent to this finding, various other regimes have also been found to improve the efficacy of OVs. For example, sequential delivery of oncolytic Ad and NDV which are both engineered to express an immuno-stimulatory cytokine leads to significant anti-tumor responses even though when administered alone, each virus showed limited efficacy against tumors (69).
Another exciting avenue being explored to improve the oncolytic potential of Ads is to combine oncolytic therapy with induction of the autophagy pathway. It was found that this pathway is involved in viral antigen presentation and therefore its upregulation could increase presentation of virally delivered tumor-associated antigens (TAAs) at the cell surface in order to induce a more potent anti-tumor immune response than with antigen delivery alone (70).
Existing neutralizing antibodies pose yet another problem in the design of OVs. Certain viral strains may have been previously encountered/vaccinated against and therefore neutralizing antibodies can be rapidly produced by the host upon treatment (especially when virus is delivered intravenously) and clear the virus before it can have an effect on tumor cells. To combat this problem, it was found that coating virus in albumin provided a protective barrier from neutralizing antibodies in the host and therefore facilitated systemic delivery without the threat of viral clearance due to previously generated immunological memory (71).
One of the main barriers to viral spread in tumor masses is the interstitial matrix (including extracellular DNA). OVs have therefore been modified to encode proteins which will degrade the interstitial matrix along with expression of DNaseI to degrade the extracellular DNA, therefore allowing more efficient spread of OV throughout tumor masses (72).
Oncolytic viruses can also be exploited as carriers for anti-cancer drugs using more than one method. In a recent study, it was found that electrostatic attraction between viral capsid and the drug molecules themselves was an efficient way to deliver anti-cancer drugs which would then act synergistically with the oncolytic adenoviral therapy (73).
Herpes Simplex Virus
Herpes simplex virus type-1 is a double-stranded DNA virus belonging to the Herpesviridae family. HSV-1 was the first virus in which TK gene mutation was engineered. In 1991, Martuza et al. demonstrated that human glioblastoma cells can be destroyed by HSV-1 carrying a mutation in the TK region and this was observed in cell culture as well as in nude mice (74). A lot of effort has since been put into making HSV more active against tumor cells and safer for normal cells culminating in the approval of Talimogene laherparepvec (T-Vec), an engineered HSV-1 for the treatment of melanoma in 2015 (75). Phase III trials in patients with non-resectable melanoma showed that T-Vec had higher efficacy in patients with stage IIIB–IV melanoma than GM-CSF treatment alone (76). It was also found that T-Vec in combination with a CTLA-4 inhibitor (Ipilimumab) showed encouraging preliminary results in a phase Ib trial (77), though there is much more work to be done to fully evaluate the effects and outcomes of this treatment combination.
Talimogene laherparepvec has two viral gene deletions (one in the γ34.5 gene and one in the α47 gene) and it has the human GM-CSF gene inserted in place of the deleted γ34.5 gene. The function of γ34.5 is to prevent infected cells from switching off protein synthesis upon viral infection. Considering that tumor cells have a defect in this mechanism, a γ34.5-deleted virus is still able to replicate in these cells. This modified virus is therefore safer as it is only able to replicate in tumor cells. The α47 gene is associated with the downregulation of antigen presentation. A deletion in this region has a double function: first, it is able to increase the anti-tumoral immune response, and second, it is associated with the expression of another gene (US11) which results in boosted viral replication in tumor cells. Finally, the insertion of GM-CSF results in increased anti-tumoral immunity as demonstrated in a phase II study. The study reported an increase in tumor specific CD8+ lymphocytes along with a decrease in CD4+FoxP3+ regulatory T-cells and CD8+FoxP3+ T-suppressor cells (78, 79).
Another oncolytic HSV is G47Δ, a third generation HSV-1 with three different mutations. It was created by Todo et al. by the deletion of the ICP6 gene from the genome of G207 virus, which already has two mutations (γ34.5 and α47); the Escherichia coli LacZ gene was added in place of ICP6 (80). ICP6 encodes the large subunit of ribonucleotide reductase (RR), an enzyme essential for viral DNA synthesis. If this enzyme is missing, the virus fails to replicate. However, tumor cells synthesize a huge amount of RR which can compensate for its deletion from the viral genome. In this way, mutated virus is able to replicate only in tumor cells, becoming safer in normal tissue.
Another example of HSV used as OV is the NV1020 virus which is based on the R7020 construct developed by Meignier et al. (81). NV1020 virus has deletions in the ICP0 and ICP4 gene regions and has only one copy of the γ34.5 gene. Moreover, the α4 promoter which controls TK expression has been inserted, making the virus sensitive to common drugs (such as acyclovir) and improving its safety. This virus has been reported to stabilize metastasis in phase I/II clinical trials involving patients with advanced metastatic colorectal cancer and showed minimal levels of side effects (82).
It has also been found that modification of HSV to include an scFv fragment against HER2 increased viral tropism to HER2 positive tumor cells (83). HSV encodes various glycoproteins to facilitate viral entry and these are: gD, gH/gL, and gB. Usually, receptor recognition leads to modifications in gD and gH/gL which in turn activates gB. Interestingly, this redirected tropism to HER2 positive cells was conferred even when the scFv fragment was engineered into gB (a glycoprotein with a role in virus-cell fusion rather than receptor recognition), thereby bypassing the requirement for receptor mediated activation of gD and gH/gL. This provides a method of improving selectivity and therefore efficacy and safety of oncolytic HSV.
These data indicate a promising route for the clinical use of HSV-1. Many clinical trials have been carried out to treat different types of solid cancer with encouraging results (82, 84, 85). However, some limitations still have to be overcome. A major obstacle for HSV-1 is the way it infects host cells. This virus is able to spread from one cell to another without causing viremia which makes it suitable for intra-tumoral but not intra-venous injection. This could cause some problems in the treatment of tumor lesions which are very difficult to reach directly, like those of pancreatic cancer.
Alongside HSV-1, HSV-2 has also shown promise as an OV. For example, HSV-2 with a deletion of ICP10 to improve selectivity was engineered and found to be even more effective than HSV-1 for treatment of metastatic ovarian cancer in mouse models (86). Building on this initial success, many more studies have been conducted to improve HSV-2 as an OV. These include co-administration of cyclophosphamide, a drug which has chemotherapeutic effects as well as causing a dampening of the innate immune response. This combination works synergistically as reduced innate immunity can facilitate more potent viral infection. Li et al. showed that this combination of therapies leads to enhanced anti-tumor effects when used to treat lung carcinoma in mice and could prove to be a good combination in clinical trials (87). It has also more recently been found that HSV-2 can act in synergy with adoptive T-cell treatment. Administration of oncolytic HSV-2 directly into tumor sites was found to improve the homing of adoptively transferred T-cells (engineered to target tumor cells) to the tumor mass. This was achieved, at least in part, by elevated levels of various chemokines such as CXCL9 and CXCL10 (88, 89). It was also found that in addition to increasing attraction of these T-cells to the tumor, the various chemokines were also able to maintain persistence of these T-cells at the tumor site.
Newcastle Disease Virus
A more recently developed, naturally oncolytic virus is NDV. It has been found that NDV can effectively kill a variety of tumor cell types and that this activity occurs by induction of immunogenic cell death which in turn leads to adaptive anti-tumor immunity (90).
The initial suggestion of a mechanism for NDV tumor selectivity was that there is a lack of robust/normal anti-viral response in tumor cells. However, it seems that some tumor cells with intact anti-viral pathways are still killed by NDV. It was then found that the lack of apoptosis in tumor cells is what makes NDV tumor-selective (91). Interestingly, it was found that certain strains of NDV can induce apoptosis in tumor cells (92) and that the apoptosis pathway stimulated in infected tumor cells is p53-independent and perhaps triggered by endoplasmic stress (93). It was also found that apoptosis of infected cells occurs predominantly via the intrinsic mitochondrial pathway and is caspase dependent (94). This induction of apoptosis results in tumor cell death.
Although NDV is naturally tumor-selective, there are inherent problems with the virus which need to be overcome in order to improve its ability to infect and spread within solid tumors as viral spread is limited by factors such as the extra-cellular matrix (95). Again, there are many studies looking at ways to modify this virus to make it safer and more efficacious; for example, insertion of the IL-2 gene into the NP/P site has proved effective (96). Inclusion of IL-2 and/or TRAIL has been shown to increase apoptosis levels in infected cells resulting in the tumor-selective parental virus becoming an even more potent anti-cancer agent (97–101). Further improvement of oncolytic NDV is found with IL-2 addition in combination with expression of TAA. This combination, delivered by oncolytic NDV, improves tumor-specific T-cell responses leading to higher efficacy than delivery of TAA alone (102). Another strategy to boost the efficacy of oncolytic NDV is to insert the ICOS gene (usually upregulated by viral infection) in order to induce higher levels of T-cell infiltration into the local tumor and distant tumor sites (103).
As well as arming viruses with immune stimulators, other therapeutic genes are also able to increase anti-tumor effects of viral therapies. For example, NDV engineered to encode TNF receptor Fas shows greater oncolytic effect as Fas is responsible for increased apoptosis of infected cells via both the intrinsic and extrinsic apoptosis pathways, thereby increasing cell death and in turn anti-tumor efficacy (104). NDV’s naturally oncolytic properties could also be augmented by arming the virus with GM-CSF (105).
In addition to arming the OV with various therapeutic genes and immune stimulators, it is also beneficial to work toward increased virulence (within safety limits). Using a more infectious strain of NDV can produce better cytotoxic effects in tumor cells than arming less virulent strains with immune-modulating genes (106). However, a potential problem encountered with NDV is its ability to infect avian species. This introduces an added difficulty in that any oncolytic therapy involving NDV needs to ensure attenuation of the virus in avian hosts without reduced potency in mammalian cells (107).
It is also important to consider that modification of OVs does not always produce the desired result, for example, viral attenuation to improve safety needs to be carefully tested as production of an attenuated virus which may establish persistent infection could lead to tumors which are not effectively killed by OV and thereby become resistant to treatment. It has previously been reported that NDV has the potential to cause persistent infection in certain cell types (108). This highlights the fact that it is important to carefully engineer OVs in order to maximize efficiency and broaden tropism for a range of cancer types.
Frequently, viral therapies are able to target tumor cells but are not efficacious enough when administered alone or they are potent but toxic at high doses. These problems could potentially be overcome by combining various viral therapies which act synergistically to combat tumor growth. For example, two antigenically distinct viruses which both encode immune stimulators can be sequentially administered and allow two cycles of transgene expression without interference from neutralizing antibodies. This approach allows lower doses to be used (making the treatments safer) and allows a multi-faceted approach which is likely to be more effective than single treatments (69). Oncolytic NDV can also be used in combination with traditional therapies to gain synergistic effects which enhance its action and the overall effect of treatment (109). Also, localized NDV therapy was shown to sensitize distant tumors to treatment with immune checkpoint inhibitors through induction of inflammatory immune infiltrates in these distant sites (110). This study provides a strong basis for developing this combination treatment with potential for entry into clinical trials.
Retrovirus
In order to overcome the problem of viral infection causing lysis of infected cells and potentially harmful inflammatory responses, a method has been developed whereby retroviral particles which retain their replicative ability can be delivered and will selectively replicate only in cells which are undergoing proliferation (tumor cells) and are compromised in their ability to trigger innate immune responses (again, tumor cells are often unable to trigger innate immunity due to disruption in the signaling pathway). The ability of these particles to integrate into the host genome and replicate without causing lysis of the cell makes them efficient and long lasting producers of the therapeutic gene they are delivering without the consequences of productive viral infection (111).
As is the case for traditional oncolytic viral therapy, this method can be designed using a variety of retroviruses and with the addition of various therapeutic genes. For example, suicide genes which trigger cell death can be delivered to tumor cells via particles from various leukemia viruses with varying levels of success (112).
As well as delivering therapeutic genes, replicating retroviral vectors can also be used in order to enhance the response to anti-cancer drug therapy. For example, delivery of an activator of a therapeutic drug by replication competent retroviral vector resulted in significant anti-tumor effect and prolonged survival time in a murine model of malignant mesothelioma (113).
These vectors can also be delivered within a “gutted” Ad genome and the outcome of this combination is improved transfer efficiency of the retroviral genome into the tumor tissue and therefore increased production of the therapeutic gene and better treatment efficacy (114).
Measles Virus (MV)
Measles virus is a single-stranded, negative sense enveloped RNA virus of the Paramyxoviridae family. There are a number of receptors that can be utilized by MV to successfully infect cells including CD150, CD46, and nectin-4. Of these, CD46 has been attributed to increased specificity of MV to tumor cells that express increased levels of this receptor compared with healthy cells. This increased expression leads to increased levels of cell lysis upon infection of tumors compared with healthy tissue (115). Selectivity can also be increased by engineering a MV which is blinded to its usual receptors and redirected to recognize specific tumor cell markers as target antigens (116). Also, as tumor cells often have a defective interferon system they tend to be more susceptible to viral infection leading to increased lysis of these cells by MV in comparison with healthy cells (117). Another method of increasing oncolytic MV selectivity is to engineer miRNA sensitive viruses which can only successfully infect cells in which certain miRNAs are downregulated (i.e., cancer cells). For example, a virus has been developed which shows sensitivity to three host miRNAs through insertion of specific miRNA target sites into the viral genome, rendering the virus incapable of infecting healthy cells which express one or more of these miRNAs but still able to infect specific cancer cells which have downregulated levels of these miRNAs (118).
As well as showing selectivity to tumor cells, oncolytic MV therapy has been shown to recruit certain aspects of the host immune response including neutrophils (119) and dendritic cells (120) to augment tumor cell lysis by also stimulating anti-tumor immune responses to clear the tumor mass. This discovery has prompted development of oncolytic MVs which are engineered to stimulate the immune system at the tumor site in order to exploit the role it plays in anti-tumor immunity. For example, molecules known to stimulate potent immune responses, such as neutrophil-activating protein (NAP) derived from Helicobacter pylori, can be engineered into the MV genome to enhance anti-tumor effects generated by oncolytic MV (121). Immune responses can also be manipulated by encoding immune checkpoint blocking antibodies in oncolytic MV genomes. These viruses will result in soluble antibodies against inhibitory immune checkpoints being produced in infected cells which will act to dampen the ability of the immune response to limit itself. Blocking of inhibitory immune checkpoints allows OVs to exploit the immune system to a greater extent than would occur naturally as the immune cells are deprived of negative signals which usually regulate the immune response. This approach has been explored by Engeland et al. with promising results warranting further investigation (122).
One potential problem encountered with oncolytic MV is the widespread immunity in the population, gained from measles vaccinations. This immunity could dampen the effect of oncolytic MV therapy by rapidly clearing the virus before it can take effect on tumor cells. There are a number of potential ways to overcome this, for example, administering immuno-suppressants along with oncolytic MV, however, this approach has drawbacks as immuno-suppression must be carefully managed to ensure patients do not become susceptible to infection by otherwise harmless agents. The most promising solution so far is to “hide” the virus within mesenchymal stem cells, allowing delivery of the virus to the target site without recognition by the immune system and thereby bypassing the effect of neutralizing antibodies (123). It is also possible to exchange measles coat proteins (which are recognized by existing neutralizing antibodies) for those of a virus to which patients are not immune. For example, Miest et al. successfully replaced measles envelope glycoproteins with those from a related virus (canine distemper virus) which allowed the MV genome to be transported without detection by existing neutralizing antibodies against MV envelope proteins (124).
Much progress has been made with MV over the years and this has resulted in many clinical trials being started to ascertain the suitability of oncolytic MV as a clinical treatment. Various cancer types have been targeted for clinical trials of oncolytic MV including myeloma (125) and ovarian cancer (126). Encouraging results have been obtained, especially with regard to safety profiles, and this warrants further studies into optimal oncolytic MV treatments (127).
Other OVs
Reovirus is a double-stranded, non-enveloped RNA virus of the Reoviridae family and is considered a naturally occurring OV. Reoviruses are thought to selectively infect tumor cells because their oncolytic functions depend on the activation of the Ras pathway (128) which tends to be upregulated in transformed cells. Reolysin is a type 3 Reovirus and is so far the only wild-type Reovirus undergoing studies for use as a therapeutic agent. Many clinical trials have been performed or are ongoing and are being conducted on various tumor types as discussed by Gong et al. (129). In 2015, the FDA and EMA granted Reolysin an orphan drug designation for various cancers including gastric, pancreatic, and ovarian cancer (130).
Other viruses are now being tested as therapeutic agents for cancer treatment. As melanoma is easily targeted, many current studies are focused on this neoplasm (131). In recent years, two promising viruses have come out as possible OVs: coxsackievirus and echovirus. The former is responsible for the common cold and enters target cells via ICAM-1, while the latter is a positive sense single-stranded RNA virus from the Picornaviridae family responsible for many human disorders. As these two strains are the cause of very common diseases in humans, it is very important to ensure their safety.
Tumor Cell Biology and Viral Therapy
In the search for the most effective viral therapy to treat cancer, alongside the focus on how to improve the virus we can also attempt to influence tumor cell biology to our advantage. This strategy has been adopted by various groups and research has so far shown that there are many tumor cell genes which can be manipulated to increase the efficacy of OVs.
For example, tumor cell genes can play a role in the targeting of OVs to tumor cells. It was found by Cuddington and Mossman that a certain OV (Bovine herpesvirus-1) is better able to infect cells which have increased levels of KRAS expression (e.g., tumor cells) (132). This represents a method of tumor targeting which relies on tumor cell factors to ensure oncolytic therapy is delivered to tumor cells, leaving healthy cells unharmed. This knowledge could potentially be applied to other oncolytic viral therapies by engineering entry mechanisms specific to tumor cells.
The aberrant expression of components of the Raf/MEK/ERK pathway in tumor cells can also have an effect on the regulation of Ad receptor and therefore levels of viral infectivity. As this pathway tends to be upregulated in tumor cells compared with healthy cells, it can have a significant effect on oncolytic viral therapy. It was found that this pathway plays a role in downregulation of CAR (Ad receptor) and therefore oncolytic Ad will be less able to infect target tumor cells. To overcome this, it is possible to inhibit MEK either directly or indirectly in order to inhibit the Raf/MEK/ERK pathway and re-establish expression of CAR on the cell surface (133). However, it has subsequently been shown that phosphorylation of ERK during the later stage of adenoviral infection can actually play a role in facilitating the sustained levels of viral protein within the cell required to produce enhanced levels of progeny virions (134). Taken together, this evidence highlights the need to balance initial inhibition of this pathway to increase CAR expression with later enhancement of the same pathway to facilitate sustained progeny production.
As well as mutations, regulation of certain genes using microRNA can also be used to enhance viral specificity for tumor cells. For example, using an miRNA which is downregulated in tumor cells (such as let-7a) to control expression of an essential viral gene in VV (such as B5R which increases both pathogenicity and oncolytic activity) results in a virus which can only express sufficient amounts of B5R in cells which have low levels of let-7a expression, i.e., tumor cells (15).
Another gene found in tumor cells that can influence OV therapy is VEGF. Our group has demonstrated that VEGF-A increases VV internalization and in turn replication levels (135). Therefore, oncolytic VV can take advantage of the increased expression of VEGF by tumor cells to increase delivery of therapeutic genes which in turn increases the efficacy and potency of the treatment. In addition to this, Arulanandam et al. found that the increase in VEGF expression upon infection with VV leads to upregulation of PRD1-BF1 (a transcription repressor) which increases sensitivity of tumor vascular endothelial cells to infection with vaccinia via repression of type-1 interferon anti-viral signaling. This increase in viral tropism in turn allows the OV to spread through the tumor more efficiently and therefore increases the efficacy of this oncolytic therapy (136). This natural repression of interferon signaling highlights the potential of using interferon inhibitors to increase the efficacy of oncolytic viral therapy, as seen by Stewart et al. (137).
It has also been found that a properly functioning host interferon response pathway is a critical factor in measles infection of malignant pleural mesothelioma. It was seen that in cell lines, there is a correlation between sensitivity of cells to measles infection and an inability of the cell to elicit a full interferon response in the presence of MV (138). This warrants further investigation as it suggests that inhibition of the interferon pathway could prove to be critical in ensuring the efficiency of oncolytic therapy. Previous to this study, it was also found that VV infection is greatly increased through downregulation of c-Jun NH2-terminal kinase (JNK). Inhibition of the JNK signaling cascade leads to lower levels of double-stranded RNA dependent protein kinase which in turn allows increased replication of VV genomes (139). This knowledge provides an avenue of exploration for improvement of OV efficacy.
Also, it has been reported that another gene found in tumor cells (CEACAM6) has an effect on oncolytic viral therapy. This tumor-associated gene has various functions including a role in promotion of tumor adhesion and invasion among other factors (140). It was shown by Wang et al. that over-expression of CEACAM6 did not have an effect on Ad receptor expression or at attachment and internalization steps of infection but did interfere with cytoplasmic virus trafficking to the nucleus via reduced expression of cytoskeletal proteins (141). As this cell adhesion molecule is able to inhibit adenoviral infection of tumor cells, systemic pre-treatment with siRNA targeting this protein could significantly enhance the anti-tumor response generated by adenoviral vectors.
Another host system which can have a significant effect on oncolytic viral efficacy is the stress response pathway. In the case of oncolytic rhabdovirus, inhibition of certain ER stress response factors can significantly increase efficacy of subsequently delivered oncolytic rhabdovirus (142). This method of pre-conditioning tumor cells to improve subsequent viral infection warrants further study as a potential method of increasing efficacy of oncolytic therapy in patients.
In addition to this, induction of the unfolded protein response in tumor cells has been found to increase the efficacy of oncolytic Ad by improving viral spread and tumor cell killing (143).
Recently, it has been reported that host microRNAs are able to regulate infection of cells with various viruses and in various ways. One of these is the control of Ad replication by miR-27a/b which downregulates SNAP25 and TXN2. This leads to a reduction in Ad entry into cells and cell cycle arrest, respectively (which in turn reduce replication of Ad within the cell) (144). Controlling expression of host miRNA by inhibition of miRNA processing factors could therefore be a promising way to ensure maximal efficacy of oncolytic Ad therapies.
These examples open new insight in the field of oncolytic viral therapy revealing the possibility to manipulate virus–host interaction. We are still far from the optimum and new studies should be focused on the long-term effects of this interaction and the possible side effects.
Conclusion and the Future of the Field
Oncolytic viral therapy is a promising treatment for cancer. New knowledge regarding both viral biology and tumor cell biology has made it possible to improve several aspects of OV therapy including safety, potency, and delivery methods. Also, the possibility of relying not only on direct lysis of tumor cells by viral infection but also mounting a multi-faceted approach involving viral lysis, immune stimulation, and gene therapy has become an important aspect of research in this field.
As the current treatment options for cancer tend to rely on only one method of attack, formulating a therapy which has the ability to act in multiple ways against cancer is an extremely exciting prospect. In order to effectively kill tumor cells and also reduce the chance of recurrence, it is necessary to utilize both the ability of virus infection to lyse the cells which are infected along with the ability of virus infection to stimulate a potent immune response, generating long lasting immunity against the antigens present on tumor cells.
If we can also bring into play gene therapy which can act in multiple ways, ultimately leading to reduction in tumor volume, we can make oncolytic viral therapy an even more formidable weapon in the fight against cancer.
The next step in this area should be to improve efficacy through arming with immuno-modulatory genes. These genes can influence the host response to viral infection, stimulating long-term immunity in the form of memory T-cells and attracting immune cells to the tumor mass through stimulation of cytokine production. In addition, combined therapy with conventional agents should be another relevant field of research, in order to increase viral action. This area of research involves a multitude of possibilities including ways to increase the host response to virus (for example, blocking immune checkpoints) and also the use of oncolytic therapy to augment the action of conventional therapy like chemotherapy and radiotherapy.
We also need to focus on exploiting the range of genes found to be differentially expressed in tumor cells which have recently been seen to play a role in potency of OVs. If we are able to gain a better understanding of the interaction between virus and tumor cells, we can overcome some of the obstacles in OVT by modulating the expression of tumor cell genes to enhance viral infectivity and efficacy.
Ultimately, the long-term goal is to formulate a treatment which can target solid tumors and circulating tumor cells in a number of ways simultaneously (145), with the aim of mounting a response that is effective even if the tumor cells become resistant to one approach. In order to do this, we need to take into consideration the interactions between tumor cells, virus, and the host. The most effective treatments will be designed to improve viral infection of tumor cells, boost immune responses to tumor antigens and manipulate tumor cell gene expression and pathways to favor successful viral infection. This can improve on current therapies which are ineffective once the tumor cells gain resistance, an issue which occurs often given the heterogeneous and rapidly mutating nature of many cancers.
Author Contributions
YW conceived the study, interpreted the data, and finalized the manuscript. AH and GM designed the study, searched references, collected data, and wrote the manuscript. NL participated in interpretation of some data and critically reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the MRC DPFS grant (MR/M015696/1 and MR/N027655/1).
References
1. Dock G. The influence of complicating diseases on leukemia. Am J Med Sci (1904) 127:563–92. doi:10.1097/00000441-190412740-00001
2. Moore AE. The destructive effect of the virus of Russian far east encephalitis on the transplantable mouse sarcoma 180. Cancer (1949) 2:525–34. doi:10.1002/1097-0142(194905)2:3<525:AID-CNCR2820020317>3.0.CO;2-O
3. Moore AE. Viruses with oncolytic properties and their adaptation to tumors. Ann N Y Acad Sci (1952) 54:945–52. doi:10.1111/j.1749-6632.1952.tb39969.x
4. Hoster HA, Zanes RP, Von Haam E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. Cancer Res (1949) 9(8):473–80.
5. Taylor AW. Effects of glandular fever infection in acute leukaemia. Br Med J (1953) 1(4810):589–93. doi:10.1136/bmj.1.4810.589
6. Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents, with particular reference to Egypt 101 virus. Cancer (1952) 5:1025–34. doi:10.1002/1097-0142(195209)5:5<1025:AID-CNCR2820050518>3.0.CO;2-Q
7. Webb HE, Wetherley-Mein G, Smith CE, McMahon D. Leukaemia and neoplastic processes treated with Langat and Kyasanur forest disease viruses: a clinical and laboratory study of 28 patients. Br Med J (1966) 1(5482):258–66. doi:10.1136/bmj.1.5482.258
8. Dörig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell (1993) 75(2):295–305. doi:10.1016/0092-8674(93)80071-L
9. Badrinath N, Heo J, Yoo SY. Viruses as nanomedicine for cancer. Int J Nanomedicine (2016) 11:4835–47. doi:10.2147/IJN.S116447
10. Balachandran S, Porosnicu M, Barber GN. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol (2001) 75(7):3474–9. doi:10.1128/JVI.75.7.3474-3479.2001
11. Farassati F, Yang AD, Lee PW. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol (2001) 3(8):745–50. doi:10.1038/35087061
12. Christian SL, Zu D, Licursi M, Komatsu Y, Pongnopparat T, Codner DA, et al. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One (2012) 7(9):e44267. doi:10.1371/journal.pone.0044267
13. Garant KA, Shmulevitz M, Pan L, Daigle RM, Ahn DG, Gujar SA, et al. Oncolytic reovirus induces intracellular redistribution of Ras to promote apoptosis and progeny virus release. Oncogene (2016) 35(6):771–82. doi:10.1038/onc.2015.136
14. Pan W, Bodempudi V, Esfandyari T, Farassati F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One (2009) 4(8):e6514. doi:10.1371/journal.pone.0006514
15. Hikichi M, Kidokoro M, Haraguchi T, Iba H, Shida H, Tahara H, et al. MicroRNA regulation of glycoprotein B5R in oncolytic vaccinia virus reduces viral pathogenicity without impairing its antitumor efficacy. Mol Ther (2011) 19(6):1107–15. doi:10.1038/mt.2011.36
16. Morrison J, Briggs SS, Green N, Fisher K, Subr V, Ulbrich K, et al. Virotherapy of ovarian cancer with polymer-cloaked adenovirus retargeted to the epidermal growth factor receptor. Mol Ther (2008) 16(2):244–51. doi:10.1038/sj.mt.6300363
17. Watkins SJ, Mesyanzhinov VV, Kurochkina LP, Hawkins RE. The ‘adenobody’ approach to viral targeting: specific and enhanced adenoviral gene delivery. Gene Ther (1997) 4(10):1004–12. doi:10.1038/sj.gt.3300511
18. Conner J, Braidwood L, Brown SM. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther (2008) 15(24):1579–92. doi:10.1038/gt.2008.121
19. Xu HN, Huang WD, Cai Y, Ding M, Gu JF, Wei N, et al. HCCS1-armed, quadruple-regulated oncolytic adenovirus specific for liver cancer as a cancer targeting gene-viro-therapy strategy. Mol Cancer (2011) 10:133. doi:10.1186/1476-4598-10-133
20. Veerapong J, Bickenbach KA, Shao MY, Smith KD, Posner MC, Roizman B, et al. Systemic delivery of (gamma1)34.5-deleted herpes simplex virus-1 selectively targets and treats distant human xenograft tumors that express high MEK activity. Cancer Res (2007) 67(17):8301–6. doi:10.1158/0008-5472.CAN-07-1499
21. Fu X, Tao L, Cai R, Prigge J, Zhang X. A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol Ther (2006) 13(5):882–90. doi:10.1016/j.ymthe.2006.02.007
22. Fu X, Tao L, Zhang X. An HSV-2-based oncolytic virus deleted in the PK domain of the ICP10 gene is a potent inducer of apoptotic death in tumor cells. Gene Ther (2007) 14(16):1218–25. doi:10.1038/sj.gt.3302971
23. McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res (2001) 61(24):8751–7.
24. Hughes J, Wang P, Alusi G, Shi H, Chu Y, Wang J, et al. Lister strain vaccinia virus with thymidine kinase gene deletion is a tractable platform for development of a new generation of oncolytic virus. Gene Ther (2015) 22(6):476–84. doi:10.1038/gt.2015.13
25. Liu TC, Hallden G, Wang Y, Brooks G, Francis J, Lemoine N, et al. An E1B-19 kDa gene deletion mutant adenovirus demonstrates tumor necrosis factor-enhanced cancer selectivity and enhanced oncolytic potency. Mol Ther (2004) 9(6):786–803. doi:10.1016/j.ymthe.2004.03.017
26. Hammer K, Kazcorowski A, Liu L, Behr M, Schemmer P, Herr I, et al. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer (2015) 137(4):978–90. doi:10.1002/ijc.29442
27. Bortolanza S, Bunuales M, Alzuguren P, Lamas O, Aldabe R, Prieto J, et al. Deletion of the E3-6.7K/gp19K region reduces the persistence of wild-type adenovirus in a permissive tumor model in Syrian hamsters. Cancer Gene Ther (2009) 16(9):703–12. doi:10.1038/cgt.2009.12
28. Yang S, Guo ZS, O’Malley ME, Yin X, Zeh HJ, Bartlett DL. A new recombinant vaccinia with targeted deletion of three viral genes: its safety and efficacy as an oncolytic virus. Gene Ther (2007) 14(8):638–47. doi:10.1038/sj.gt.3302914
29. Thomas MA, Broughton RS, Goodrum FD, Ornelles DA. E4orf1 limits the oncolytic potential of the E1B-55K deletion mutant adenovirus. J Virol (2009) 83(6):2406–16. doi:10.1128/JVI.01972-08
30. Thomas MA, Nyanhete T, Tuero I, Venzon D, Robert-Guroff M. Beyond oncolytics: E1B55K-deleted adenovirus as a vaccine delivery vector. PLoS One (2016) 11(7):e0158505. doi:10.1371/journal.pone.0158505
31. Rohmer S, Quirin C, Hesse A, Sandmann S, Bayer W, Herold-Mende C, et al. Transgene expression by oncolytic adenoviruses is modulated by E1B19K deletion in a cell type-dependent manner. Virology (2009) 395(2):243–54. doi:10.1016/j.virol.2009.09.030
32. Zhang KJ, Zhang J, Wu YM, Qian J, Liu XJ, Yan LC, et al. Complete eradication of hepatomas using an oncolytic adenovirus containing AFP promoter controlling E1A and an E1B deletion to drive IL-24 expression. Cancer Gene Ther (2012) 19(9):619–29. doi:10.1038/cgt.2012.40
33. Poutou J, Bunuales M, Gonzalez-Aparicio M, Garcia-Aragoncillo E, Quetglas JI, Casado R, et al. Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther (2015) 22(9):696–706. doi:10.1038/gt.2015.45
34. Bortolanza S, Bunuales M, Otano I, Gonzalez-Aseguinolaza G, Ortiz-de-Solorzano C, Perez D, et al. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol Ther (2009) 17(4):614–22. doi:10.1038/mt.2009.9
35. Cheng PH, Rao XM, Duan X, Li XF, Egger ME, McMasters KM, et al. Virotherapy targeting cyclin E overexpression in tumors with adenovirus-enhanced cancer-selective promoter. J Mol Med (Berl) (2015) 93(2):211–23. doi:10.1007/s00109-014-1214-6
36. Oberg D, Yanover E, Adam V, Sweeney K, Costas C, Lemoine NR, et al. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res (2010) 16(2):541–53. doi:10.1158/1078-0432.CCR-09-1960
37. Sharon D, Schümann M, MacLeod S, McPherson R, Chaurasiya S, Shaw A, et al. 2-Aminopurine enhances the oncolytic activity of an E1b-deleted adenovirus in hepatocellular carcinoma cells. PLoS One (2013) 8(6):e65222. doi:10.1371/journal.pone.0065222
38. Leitner S, Sweeney K, Oberg D, Davies D, Miranda E, Lemoine NR, et al. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin Cancer Res (2009) 15(5):1730–40. doi:10.1158/1078-0432.CCR-08-2008
39. Wang B, Chen G, Zhou J, Wu P, Luo D, Huang X, et al. Deletion of the intracellular domain of coxsackie and adenovirus receptor (CAR) enhances the expression of itself and boosts the efficiency of current adenovirus-mediated gene therapy in ovarian cancer cell lines in vitro. Cancer Lett (2007) 248(2):299–307. doi:10.1016/j.canlet.2006.08.002
40. Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol (2004) 5(12):1266–74. doi:10.1038/ni1132
41. Al Yaghchi C, Zhang Z, Alusi G, Lemoine NR, Wang Y. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy (2015) 7(12):1249–58. doi:10.2217/imt.15.90
42. Hiley CT, Yuan M, Lemoine NR, Wang Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther (2010) 17(2):281–7. doi:10.1038/gt.2009.132
43. Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol (1998) 72(2):1577–85.
44. Broyles SS. Vaccinia virus transcription. J Gen Virol (2003) 84(Pt 9):2293–303. doi:10.1099/vir.0.18942-0
45. Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther (2000) 7(1):66–73. doi:10.1038/sj.cgt.7700075
46. Park SH, Breitbach CJ, Lee J, Park JO, Lim HY, Kang WK, et al. Phase 1b trial of biweekly intravenous Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther (2015) 23(9):1532–40. doi:10.1038/mt.2015.109
47. Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A phase 2, open-label, randomized study of Pexa-Vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol (2015) 1317:343–57. doi:10.1007/978-1-4939-2727-2_19
48. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19(3):329–36. doi:10.1038/nm.3089
49. Chard LS, Maniati E, Wang P, Zhang Z, Gao D, Wang J, et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res (2015) 21(2):405–16. doi:10.1158/1078-0432.CCR-14-0464
50. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature (2011) 477(7362):99–102. doi:10.1038/nature10358
51. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst (2006) 98(5):298–300. doi:10.1093/jnci/djj111
52. Lv SQ, Ye ZL, Liu PY, Huang Y, Li LF, Liu H, et al. 11R-P53 and GM-CSF expressing oncolytic adenovirus target cancer stem cells with enhanced synergistic activity. J Cancer (2017) 8(2):199–206. doi:10.7150/jca.16406
53. Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T, et al. Phase I study with ONCOS-102 for the treatment of solid tumors—an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer (2016) 4:17. doi:10.1186/s40425-016-0121-5
54. Machitani M, Sakurai F, Wakabayashi K, Tachibana M, Fujiwara T, Mizuguchi H. Enhanced oncolytic activities of the telomerase-specific replication-competent adenovirus expressing short-hairpin RNA against Dicer. Mol Cancer Ther (2017) 16(1):251–9. doi:10.1158/1535-7163.MCT-16-0383
55. Zhao Y, Li H, Wu R, Li S, Wang P, Wang H, et al. Antitumor effects of oncolytic adenovirus-carrying siRNA targeting potential oncogene EphA3. PLoS One (2015) 10(5):e0126726. doi:10.1371/journal.pone.0126726
56. Li X, Wang P, Li H, Du X, Liu M, Huang Q, et al. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian hamster model. Clin Cancer Res (2017) 23(1):239–49. doi:10.1158/1078-0432.CCR-16-0477
57. Tanoue K, Rosewell Shaw A, Watanabe N, Porter CE, Rana B, Gottschalk S, et al. Armed oncolytic adenovirus expressing PD-L1 mini-body enhances anti-tumor effects of chimeric antigen receptor T-cells in solid tumors. Cancer Res (2017) 77:2040–51. doi:10.1158/0008-5472.CAN-16-1577
58. Wong HH, Jiang G, Gangeswaran R, Wang P, Wang J, Yuan M, et al. Modification of the early gene enhancer-promoter improves the oncolytic potency of adenovirus 11. Mol Ther (2012) 20(2):306–16. doi:10.1038/mt.2011.242
59. Chia SL, Lei J, Ferguson DJ, Dyer A, Fisher KD, Seymour LW. Group B adenovirus enadenotucirev infects polarised colorectal cancer cells efficiently from the basolateral surface expected to be encountered during intravenous delivery to treat disseminated cancer. Virology (2017) 505:162–71. doi:10.1016/j.virol.2017.02.011
60. Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, et al. Oncolytic adenoviral delivery of an EGFR-targeting T cell engager improves antitumor efficacy. Cancer Res (2017) 77:2052–63. doi:10.1158/0008-5472.CAN-16-1708
61. Smith JG, Nemerow GR. Mechanism of adenovirus neutralization by human alpha-defensins. Cell Host Microbe (2008) 3(1):11–9. doi:10.1016/j.chom.2007.12.001
62. Vragniau C, Hübner JM, Beidler P, Gil S, Saydaminova K, Lu ZZ, et al. Studies on the interaction of tumor-derived HD5 alpha defensins with adenoviruses and implications for oncolytic adenovirus therapy. J Virol (2017) 91(6):e02030–16. doi:10.1128/JVI.02030-16
63. Oh E, Choi IK, Hong J, Yun CO. Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget (2017) 8(3):4730–46. doi:10.18632/oncotarget.13972
64. Yang C, Cao H, Liu N, Xu K, Ding M, Mao LJ. Oncolytic adenovirus expressing interleukin-18 improves antitumor activity of dacarbazine for malignant melanoma. Drug Des Devel Ther (2016) 10:3755–61. doi:10.2147/DDDT.S115121
65. Ahn HM, Hong J, Yun CO. Oncolytic adenovirus coexpressing interleukin-12 and shVEGF restores antitumor immune function and enhances antitumor efficacy. Oncotarget (2016) 7(51):84965–80. doi:10.18632/oncotarget.13087
66. Gomez-Gutierrez JG, Nitz J, Sharma R, Wechman SL, Riedinger E, Martinez-Jaramillo E, et al. Combined therapy of oncolytic adenovirus and temozolomide enhances lung cancer virotherapy in vitro and in vivo. Virology (2016) 487:249–59. doi:10.1016/j.virol.2015.10.019
67. Liikanen I, Tähtinen S, Guse K, Gutmann T, Savola P, Oksanen M, et al. Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol Cancer Ther (2016) 15(9):2259–69. doi:10.1158/1535-7163.MCT-15-0819
68. Tysome JR, Li X, Wang S, Wang P, Gao D, Du P, et al. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin Cancer Res (2012) 18(24):6679–89. doi:10.1158/1078-0432.CCR-12-0979
69. Nistal-Villan E, Bunuales M, Poutou J, Gonzalez-Aparicio M, Bravo-Perez C, Quetglas JI, et al. Enhanced therapeutic effect using sequential administration of antigenically distinct oncolytic viruses expressing oncostatin M in a Syrian hamster orthotopic pancreatic cancer model. Mol Cancer (2015) 14:210. doi:10.1186/s12943-015-0479-x
70. Klein SR, Jiang H, Hossain MB, Fan X, Gumin J, Dong A, et al. Critical role of autophagy in the processing of adenovirus capsid-incorporated cancer-specific antigens. PLoS One (2016) 11(4):e0153814. doi:10.1371/journal.pone.0153814
71. Rojas LA, Condezo GN, Moreno R, Fajardo CA, Arias-Badia M, San Martín C, et al. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J Control Release (2016) 237:78–88. doi:10.1016/j.jconrel.2016.07.004
72. Tedcastle A, Illingworth S, Brown A, Seymour LW, Fisher KD. Actin-resistant DNAse I expression from oncolytic adenovirus enadenotucirev enhances its intratumoral spread and reduces tumor growth. Mol Ther (2016) 24(4):796–804. doi:10.1038/mt.2015.233
73. Garofalo M, Iovine B, Kuryk L, Capasso C, Hirvinen M, Vitale A, et al. Oncolytic adenovirus loaded with L-carnosine as novel strategy to enhance the antitumor activity. Mol Cancer Ther (2016) 15(4):651–60. doi:10.1158/1535-7163.MCT-15-0559
74. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (1991) 252(5007):854–6. doi:10.1126/science.1851332
75. Coffin R. Interview with Robert Coffin, inventor of T-Vec: the first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy (2016) 8(2):103–6. doi:10.2217/imt.15.116
76. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33(25):2780–8. doi:10.1200/JCO.2014.58.3377
77. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen O, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol (2016) 34(22):2619–26. doi:10.1200/JCO.2016.67.1529
78. Barve M, Bender J, Senzer N, Cunningham C, Greco FA, McCune D, et al. Induction of immune responses and clinical efficacy in a phase II trial of IDM-2101, a 10-epitope cytotoxic T-lymphocyte vaccine, in metastatic non-small-cell lung cancer. J Clin Oncol (2008) 26(27):4418–25. doi:10.1200/JCO.2008.16.6462
79. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol (2010) 17(3):718–30. doi:10.1245/s10434-009-0809-6
80. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A (2001) 98(11):6396–401. doi:10.1073/pnas.101136398
81. Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: construction and evaluation in rodents. J Infect Dis (1988) 158(3):602–14. doi:10.1093/infdis/158.3.602
82. Geevarghese SK, Geller DA, de Haan HA, Horer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther (2010) 21(9):1119–28. doi:10.1089/hum.2010.020
83. Petrovic B, Gianni T, Gatta V, Campadelli-Fiume G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog (2017) 13(4):e1006352. doi:10.1371/journal.ppat.1006352
84. Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res (2006) 12(22):6737–47. doi:10.1158/1078-0432.CCR-06-0759
85. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther (2000) 7(10):867–74. doi:10.1038/sj.gt.3301205
86. Fu X, Tao L, Zhang X. An oncolytic virus derived from type 2 herpes simplex virus has potent therapeutic effect against metastatic ovarian cancer. Cancer Gene Ther (2007) 14(5):480–7. doi:10.1038/sj.cgt.7701033
87. Li H, Zeng Z, Fu X, Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res (2007) 67(16):7850–5. doi:10.1158/0008-5472.CAN-07-1087
88. Fu X, Rivera A, Tao L, Zhang X. An HSV-2 based oncolytic virus can function as an attractant to guide migration of adoptively transferred T cells to tumor sites. Oncotarget (2015) 6(2):902–14. doi:10.18632/oncotarget.2817
89. Fu X, Hu J, Han HY, Hua YJ, Zhou L, Shuai WD, et al. High expression of XPA confers poor prognosis for nasopharyngeal carcinoma patients treated with platinum-based chemoradiotherapy. Oncotarget (2015) 6(29):28478–90. doi:10.18632/oncotarget.4424
90. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer (2015) 136(5):E313–25. doi:10.1002/ijc.29202
91. Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol (2011) 85(12):6015–23. doi:10.1128/JVI.01537-10
92. Lazar I, Yaacov B, Shiloach T, Eliahoo E, Kadouri L, Lotem M, et al. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein livin. J Virol (2010) 84(1):639–46. doi:10.1128/JVI.00401-09
93. Fábián Z, Csatary CM, Szeberényi J, Csatary LK. p53-independent endoplasmic reticulum stress-mediated cytotoxicity of a Newcastle disease virus strain in tumor cell lines. J Virol (2007) 81(6):2817–30. doi:10.1128/JVI.02490-06
94. Elankumaran S, Rockemann D, Samal SK. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J Virol (2006) 80(15):7522–34. doi:10.1128/JVI.00241-06
95. Yaacov B, Lazar I, Tayeb S, Frank S, Izhar U, Lotem M, et al. Extracellular matrix constituents interfere with Newcastle disease virus spread in solid tissue and diminish its potential oncolytic activity. J Gen Virol (2012) 93(Pt 8):1664–72. doi:10.1099/vir.0.043281-0
96. Pan Z, He J, Rasoul LM, Liu Y, Che R, Ding Y, et al. Identification of optimal insertion site in recombinant Newcastle disease virus (rNDV) vector expressing foreign gene to enhance its anti-tumor effect. PLoS One (2016) 11(10):e0164723. doi:10.1371/journal.pone.0164723
97. Bian H, Fournier P, Moormann R, Peeters B, Schirrmacher V. Selective gene transfer to tumor cells by recombinant Newcastle disease virus via a bispecific fusion protein. Int J Oncol (2005) 26(2):431–9. doi:10.3892/ijo.26.2.431
98. Zhao H, Janke M, Fournier P, Schirrmacher V. Recombinant Newcastle disease virus expressing human interleukin-2 serves as a potential candidate for tumor therapy. Virus Res (2008) 136(1–2):75–80. doi:10.1016/j.virusres.2008.04.020
99. Zamarin D, Vigil A, Kelly K, García-Sastre A, Fong Y. Genetically engineered Newcastle disease virus for malignant melanoma therapy. Gene Ther (2009) 16(6):796–804. doi:10.1038/gt.2009.14
100. Bai F, Niu Z, Tian H, Li S, Lv Z, Zhang T, et al. Genetically engineered Newcastle disease virus expressing interleukin 2 is a potential drug candidate for cancer immunotherapy. Immunol Lett (2014) 159(1–2):36–46. doi:10.1016/j.imlet.2014.02.009
101. Bai FL, Yu YH, Tian H, Ren GP, Wang H, Zhou B, et al. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol Ther (2014) 15(9):1226–38. doi:10.4161/cbt.29686
102. Vigil A, Martinez O, Chua MA, García-Sastre A. Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol Ther (2008) 16(11):1883–90. doi:10.1038/mt.2008.181
103. Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun (2017) 8:14340. doi:10.1038/ncomms14340
104. Cuadrado-Castano S, Ayllon J, Mansour M, de la Iglesia-Vicente J, Jordan S, Tripathi S, et al. Enhancement of the proapoptotic properties of Newcastle disease virus promotes tumor remission in syngeneic murine cancer models. Mol Cancer Ther (2015) 14(5):1247–58. doi:10.1158/1535-7163.MCT-14-0913
105. Janke M, Peeters B, de Leeuw O, Moorman R, Arnold A, Fournier P, et al. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther (2007) 14(23):1639–49. doi:10.1038/sj.gt.3303026
106. Buijs P, van Nieuwkoop S, Vaes V, Fouchier R, van Eijck C, van den Hoogen B. Recombinant immunomodulating lentogenic or mesogenic oncolytic Newcastle disease virus for treatment of pancreatic adenocarcinoma. Viruses (2015) 7(6):2980–98. doi:10.3390/v7062756
107. Cheng X, Wang W, Xu Q, Harper J, Carroll D, Galinski MS, et al. Genetic modification of oncolytic Newcastle disease virus for cancer therapy. J Virol (2016) 90(11):5343–52. doi:10.1128/JVI.00136-16
108. Chia SL, Yusoff K, Shafee N. Viral persistence in colorectal cancer cells infected by Newcastle disease virus. Virol J (2014) 11:91. doi:10.1186/1743-422X-11-91
109. Al-Shammari AM, Rameez H, Al-Taee MF. Newcastle disease virus, rituximab, and doxorubicin combination as anti-hematological malignancy therapy. Oncolytic Virother (2016) 5:27–34. doi:10.2147/OV.S95250
110. Zamarin D, Holmgaard RB, Subudhi KS, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med (2014) 6(226):226ra32. doi:10.1126/scitranslmed.3008095
111. Logg CR, Robbins JM, Jolly DJ, Gruber HE, Kasahara N. Retroviral replicating vectors in cancer. Methods Enzymol (2012) 507:199–228. doi:10.1016/B978-0-12-386509-0.00011-9
112. Lu YC, Chen YJ, Yu YR, Lai YH, Cheng JC, Li YF, et al. Replicating retroviral vectors for oncolytic virotherapy of experimental hepatocellular carcinoma. Oncol Rep (2012) 28(1):21–6. doi:10.3892/or.2012.1789
113. Kawasaki Y, Tamamoto A, Takagi-Kimura M, Maeyama Y, Yamaoka N, Terada N, et al. Replication-competent retrovirus vector-mediated prodrug activator gene therapy in experimental models of human malignant mesothelioma. Cancer Gene Ther (2011) 18(8):571–8. doi:10.1038/cgt.2011.25
114. Kubo S, Haga K, Tamamoto A, Palmer DJ, Ng P, Okamura H, et al. Adenovirus-retrovirus hybrid vectors achieve highly enhanced tumor transduction and antitumor efficacy in vivo. Mol Ther (2011) 19(1):76–82. doi:10.1038/mt.2010.182
115. Anderson BD, Nakamura T, Russell SJ, Peng SW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res (2004) 64(14):4919–26. doi:10.1158/0008-5472.CAN-04-0884
116. Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol (2005) 23(2):209–14. doi:10.1038/nbt1060
117. Berchtold S, Lampe J, Weiland T, Smirnow I, Schleicher S, Handgretinger R, et al. Innate immune defense defines susceptibility of sarcoma cells to measles vaccine virus-based oncolysis. J Virol (2013) 87:3484–501. doi:10.1128/JVI.02106-12
118. Baertsch MA, Leber MF, Bossow S, Singh M, Engeland CE, Albert J, et al. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther (2014) 21(9):373–80. doi:10.1038/cgt.2014.40
119. Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res (2003) 63(19):6463–8.
120. Guillerme JB, Boisgerault N, Roulois D, Ménager J, Combredet C, Tangy F, et al. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res (2013) 19(5):1147–58. doi:10.1158/1078-0432.CCR-12-2733
121. Iankov ID, Allen C, Federspiel MJ, Myers RM, Peng KW, Ingle JN, et al. Expression of immunomodulatory neutrophil-activating protein of Helicobacter pylori enhances the antitumor activity of oncolytic measles virus. Mol Ther (2014) 20(6):1139–47. doi:10.1038/mt.2012.4
122. Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther (2014) 22(11):1949–59. doi:10.1038/mt.2014.160
123. Mader EK, Maeyama Y, Lin Y, Butler GW, Russell HM, Galanis E, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res (2009) 15(23):7246–55. doi:10.1158/1078-0432.CCR-09-1292
124. Miest TS, Yaiw KC, Frenzke M, Lampe J, Hudacek AW, Springfield C, et al. Envelope-chimeric entry-targeted measles virus escapes neutralization and achieves oncolysis. Mol Ther (2011) 19(10):1813–20. doi:10.1038/mt.2011.92
125. Dispenzieri A, Tong C, LaPlant B, Lacy MQ, Laumann K, Dingli D, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia (2017). doi:10.1038/leu.2017.120
126. Galanis E, Hartmann LC, Cliby WA, Long HA, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res (2010) 70(3):875–82. doi:10.1158/0008-5472.CAN-09-2762
127. Galanis E, Atherton PJ, Maurer MJ, Kuntson KL, Dowdy SC, Cliby WA, et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res (2015) 75(1):22–30. doi:10.1158/0008-5472.CAN-14-2533
128. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J (1998) 17(12):3351–62. doi:10.1093/emboj/17.12.3351
129. Gong J, Sachdev E, Mita AC, Mita MM. Clinical development of reovirus for cancer therapy: an oncolytic virus with immune-mediated antitumor activity. World J Methodol (2016) 6(1):25–42. doi:10.5662/wjm.v6.i1.25
130. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci (2016) 107(10):1373–9. doi:10.1111/cas.13027
131. Babiker HM, Riaz IB, Husnain M, Borad MJ. Oncolytic virotherapy including Rigvir and standard therapies in malignant melanoma. Oncolytic Virother (2017) 6:11–8. doi:10.2147/OV.S100072
132. Cuddington BP, Mossman KL. Permissiveness of human cancer cells to oncolytic bovine herpesvirus 1 is mediated in part by KRAS activity. J Virol (2014) 88(12):6885–95. doi:10.1128/JVI.00849-14
133. Anders M, Christian C, McMahon M, McCormick F, Korn WM. Inhibition of the Raf/MEK/ERK pathway up-regulates expression of the coxsackievirus and adenovirus receptor in cancer cells. Cancer Res (2003) 63(9):2088–95.
134. Schumann M, Dobbelstein M. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Res (2006) 66(3):1282–8. doi:10.1158/0008-5472.CAN-05-1484
135. Hiley CT, Chard LS, Gangeswaran R, Tysome JR, Briat A, Lemoine NR, et al. Vascular endothelial growth factor A promotes vaccinia virus entry into host cells via activation of the Akt pathway. J Virol (2013) 87(5):2781–90. doi:10.1128/JVI.00854-12
136. Arulanandam R, Batenchuk C, Angarita FA, Ottolino-Perry K, Cousineau S, Mottashed A, et al. VEGF-mediated induction of PRD1-BF1/Blimp1 expression sensitizes tumor vasculature to oncolytic virus infection. Cancer Cell (2015) 28(2):210–24. doi:10.1016/j.ccell.2015.06.009
137. Stewart CE, Randall RE, Adamson CS. Inhibitors of the interferon response enhance virus replication in vitro. PLoS One (2014) 9(11):e112014. doi:10.1371/journal.pone.0112014
138. Achard C, Boisgerault N, Delaunay T, Roulois D, Nedellec S, Royer PJ, et al. Sensitivity of human pleural mesothelioma to oncolytic measles virus depends on defects of the type I interferon response. Oncotarget (2015) 6(42):44892–904. doi:10.18632/oncotarget.6285
139. Hu W, Hofstetter W, Guo W, Li H, Pataer A, Peng HH, et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther (2008) 15(9):616–24. doi:10.1038/cgt.2008.32
140. Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. Overexpression of CEACAM6 promotes insulin-like growth factor I-induced pancreatic adenocarcinoma cellular invasiveness. Oncogene (2004) 23(34):5834–42. doi:10.1038/sj.onc.1207775
141. Wang Y, Gangeswaran R, Zhao X, Wang P, Tysome J, Bhakta V, et al. CEACAM6 attenuates adenovirus infection by antagonizing viral trafficking in cancer cells. J Clin Invest (2009) 119(6):1604–15. doi:10.1172/JCI37905
142. Mahoney DJ, Lefebvre C, Allan K, Brun J, Sanaei CA, Baird S, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell (2011) 20(4):443–56. doi:10.1016/j.ccr.2011.09.005
143. Prasad V, Suomalainen M, Pennauer M, Yakimovich A, Andriasyan V, Hemmi S, et al. Chemical induction of unfolded protein response enhances cancer cell killing through lytic virus infection. J Virol (2014) 88(22):13086–98. doi:10.1128/JVI.02156-14
144. Machitani M, Sakurai F, Wakabayashi K, Nakatani K, Tachibana M, Mizuguchi H. MicroRNA miR-27 inhibits adenovirus infection by suppressing the expression of SNAP25 and TXN2. J Virol (2017) 91:e159–17. doi:10.1128/JVI.00159-17
Keywords: oncolytic virus, tumor cells, selectivity, cancer treatment, host factors
Citation: Howells A, Marelli G, Lemoine NR and Wang Y (2017) Oncolytic Viruses—Interaction of Virus and Tumor Cells in the Battle to Eliminate Cancer. Front. Oncol. 7:195. doi: 10.3389/fonc.2017.00195
Received: 06 June 2017; Accepted: 15 August 2017;
Published: 08 September 2017
Edited by:
Joshua Pesach Rosenzweig, Cell-El Ltd., IsraelReviewed by:
Zong Sheng Guo, Harvard University, United StatesJean-Baptiste Ange, Philippe Guillerme, Université Nantes Angers Le Mans, France
Copyright: © 2017 Howells, Marelli, Lemoine and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaohe Wang, eWFvaGUud2FuZ0BxbXVsLmFjLnVr
†These authors have contributed equally to this work.