
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 31 August 2017
Sec. Molecular and Cellular Oncology
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00180
This article is part of the Research Topic Inter-Organelle Calcium Communication in Cancer View all 14 articles
 Gaia Pedriali1†
Gaia Pedriali1† Alessandro Rimessi1†
Alessandro Rimessi1† Luigi Sbano1
Luigi Sbano1 Carlotta Giorgi1
Carlotta Giorgi1 Mariusz R. Wieckowski2
Mariusz R. Wieckowski2 Maurizio Previati3†
Maurizio Previati3† Paolo Pinton1*†
Paolo Pinton1*†
Inter-organelle membrane contact sites are emerging as major sites for the regulation of intracellular Ca2+ concentration and distribution. Here, extracellular stimuli operate on a wide array of channels, pumps, and ion exchangers to redistribute intracellular Ca2+ among several compartments. The resulting highly defined spatial and temporal patterns of Ca2+ movement can be used to elicit specific cellular responses, including cell proliferation, migration, or death. Plasma membrane (PM) also can directly contact mitochondria and endoplasmic reticulum (ER) through caveolae, small invaginations of the PM that ensure inter-organelle contacts, and can contribute to the regulation of numerous cellular functions through scaffolding proteins such as caveolins. PM and ER organize specialized junctions. Here, many components of the receptor-dependent Ca2+ signals are clustered, including the ORAI1-stromal interaction molecule 1 complex. This complex constitutes a primary mechanism for Ca2+ entry into non-excitable cells, modulated by intracellular Ca2+. Several contact sites between the ER and mitochondria, termed mitochondria-associated membranes, show a very complex and specialized structure and host a wide number of proteins that regulate Ca2+ transfer. In this review, we summarize current knowledge of the particular action of several oncogenes and tumor suppressors at these specialized check points and analyze anti-cancer therapies that specifically target Ca2+ flow at the inter-organelle contacts to alter the metabolism and fate of the cancer cell.
From the 1940s, when a link between Calcium (Ca2+) and cancer was observed for the first time (1), until today, its centrality of Ca2+ action as second messenger in carcinogenesis and tumor progression has been confirmed (2).
Under resting conditions, the cytosolic Ca2+ amount is maintained at a concentration of approximately 100 nM. Some organelles act as intracellular Ca2+ stores, like the Golgi apparatus and endoplasmic reticulum (ER), and the concentration of this cation rises to between 300 and 1,000 µM in such places (3). Because of this high concentration gradient, modulation of intracellular Ca2+ homeostasis at the ER level is fundamental to cellular life and destiny. Rapid release of Ca2+ from the ER determines transient waves in the cytoplasm and mitochondria with pro-survival effects. On the contrary, stimuli that massively increase the mitochondrial Ca2+ concentration for a prolonged time induce apoptotic or necrotic cell death triggered by the opening of the mitochondrial permeability transition pore (mPTP) (3–5).
The mitochondria can accumulate a significant amount of Ca2+ within their matrix, 10-fold higher than that measured in the cytosol (6). Ca2+ is transferred from the ER via specialized regions, called mitochondria-associated membranes (MAMs) where the two organelles organize dynamic contacts (7). ER Ca2+ depletion, initiated by the opening of reticular inositol 1,4,5 trisphosphate (IP3) receptors (IP3Rs), is recovered by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps that transport Ca2+ into the lumen of the ER (8).
Increases in cytosolic Ca2+ concentration occur fundamentally through the entry of Ca2+ from the extracellular space. This event is mediated by ligand-gated channels, such as the P2X purinergic-ionotropic receptor families (9), and transient receptor potential (TRP) channels. As a whole, they constitute a superfamily organized into seven subfamilies, where one is comprised of the “canonical” TRPs (TRPC subfamily) (10). Moreover, some TRP channels can be influenced by the residual amount of Ca2+ in the ER after its release in the cytosol. Their action, which ultimately consists in the refilling the depleted stores, is termed store-operated Ca2+ entry (SOCE) and is regulated by the Ca2+ release-activated calcium channel protein 1 (ORAI1) and the ER Ca2+ sensors stromal interaction molecule 1 (STIM1) and STIM2 (11). STIM1 has been shown to redistribute into clusters or puncta at ER-plasma membrane (PM) junctional sites (12), while the caveolar lipid rafts form flask-like invaginations 50–100 nm deep in the cell. These structures, reducing the gap between the two membranes, facilitate SOCE channel interaction with ER-associated STIM1 puncta (13), and constitute a proper tether between the ER and the PM (13, 14).
Therefore, the control mechanisms of intracellular Ca2+ homeostasis appear hierarchical, and their modulation and alteration as a cause or consequence of cancer induction and progression can change the sensitivity of cells to anti-tumor drugs.
Ca2+ exerts a complex regulatory role on the numerous cell functions, including cell death (15). In particular, the overload of cellular Ca2+ is mediated in its pro-apoptotic signaling role, which also relies on the presence of a wide array of intracellular transducers and the high spatiotemporal complexity of the increase in [Ca2+] evoked by different apoptotic stimuli. Such complexity is controlled primarily by the presence of ion channels located in the PM and by structured inter-organelle interactions, such as between the ER and mitochondria (16). The importance of caveolae in Ca2+ signaling was confirmed by the strategic localization of Ca2+ effectors, such as PM Ca2+ ATPase pumps and IP3Rs, providing a platform for the assembly of diverse Ca2+ signaling complexes (17–20). In particular, the tumor-suppressor caveolin-1, a fundamental member of caveolae, plays a key role in the control of the Ca2+-dependent apoptotic pathway and regulates fundamental mitochondrial functions during tumor growth (21). When the caveolin-1/Ca2+ axis is compromised, failure of both mitochondrial metabolism, and apoptotic route can occur.
At ER-PM junctional sites, STIM-ORAI can sense and respond to intracellular Ca2+ microenvironmental changes; this complex mediates Ca2+ influx, while STIM acts as an ER Ca2+ sensor, ORAI serves as a selective Ca2+-entry channel. The over-activation of ORAI channels and TRPC result in Ca2+ toxicity caused by excessive Ca2+ influx (22). ORAI channels have a dominant role in Ca2+ toxicity because ORAI1 is essential for Ca2+ influx and regulates the activity of TRPC channels (23–25). The most potent and immediate regulator of ORAI1 is Ca2+ itself, with a pivotal contribution of STIM1 (26).
The luminal Ca2+ level controls IP3R-mediated Ca2+ release, dampening or augmenting ER-mitochondrial Ca2+ transfer, and consequentially shifting the balancing between cell death and survival. In particular, several anti-apoptotic proteins and oncogenes, such as bax inhibitor-1 (BI-1), B-cell CLL/lymphoma 2 (Bcl-2), AKT, and RAS reduce [Ca2+] in the ER lumen as a survival mechanism (21, 27–29). The activity of BI-1 as an ER Ca2+-leak channel and/or the sensitization of IP3Rs channels can driven the reduction of [Ca2+] at ER level, as demonstrated by the redox-related proteins ERO1α and GPX8 (30–32). In particular, the IP3R isoform 3 and voltage-dependent anion channel (VDAC)1 are proposed to have a powerful role in this pro-apoptotic Ca2+ signaling (33, 34).
Excessive Ca2+-release from the ER triggers mitochondrial pathways which can lead to cell death. Pro-apoptotic proteins [such as Bax and fragile histidine triad protein (Fhit)] exert the opposite effect, potentiating the mitochondrial Ca2+ signals, albeit by molecularly distinct routes: Bax antagonizes the effect of Bcl-2 on ER Ca2+ reload (35) while Fhit increases the number of the initial sites of Ca2+-uptake in mitochondria. Ca2+ overload in mitochondria has long been known to be a critical event in the metabolic impairment associated with both necrosis and intrinsic pathways of apoptosis (2, 21, 36–39). Ca2+ triggers the release of caspase cofactors such as cytochrome c and SMAC/direct IAP binding protein with low Pi, thus allowing the assembly of the apoptosome and driving the cell toward the opening of the mPTP, organelle fragmentation, swelling, and ultimately to death. Another important trigger for PTP opening is oxidative stress (40). Changes in the redox state impact ER and mitochondrial physiology and Ca2+ signaling. In particular, (i) the ER redox-related proteins, ERO1α, and GPX8, are enriched in MAMs and regulate ER Ca2+ storage and flux to mitochondria (30, 32) and (ii) the mitochondrial calcium uniporter (MCU) is a mitochondrial luminal redox sensor whose oxidation promotes persistent channel activity and Ca2+-overload-induced cell death (41–43). MCU provides the rate-limiting step for mitochondrial Ca2+ accumulation and may be pivotal to apoptosis (44). However, a large number of studies showed that MCU downregulation or inhibition increases resistance to apoptosis, in colon cancer cells via the upregulation of miR-25, a microRNA targeting the MCU itself (6).
Endoplasmic reticulum Ca2+ depletion plays a pivotal role in preventing mitochondrial Ca2+ overload and programmed cell death and, for this reason, it is the main target of the action of several oncogenes or oncosuppressors (Figure 1). In particular, STIM1 levels are upregulated in colorectal cancer and are positively correlated with tumor invasion and metastasis in this malignancy (45). STIM1 knockdown inhibited cell migration and invasion in both gastric cancer and glioblastoma (46, 47). ORAI1 and STIM1 regulate focal adhesion kinase at the front or at the rear edge of migrating cells, respectively (48). A front-to-rear Ca2+ gradient exists in these cells and is associated with STIM/ORAI-dependent Ca2+ pulses at the leading edge of the cell. This promotes myosin II-mediated directional movements and formation of new focal adhesions (49). Consistently, the use of the pharmacological SOCE inhibitor SKF-96365 attenuated tumor metastasis from MDA-MB-231 (45) or cervical cancer cell (50) xenografts. Also, the ORAI1/STIM1-dependent calcium entry in melanoma may drive the cell from a proliferative to a migratory and invasive state (51).
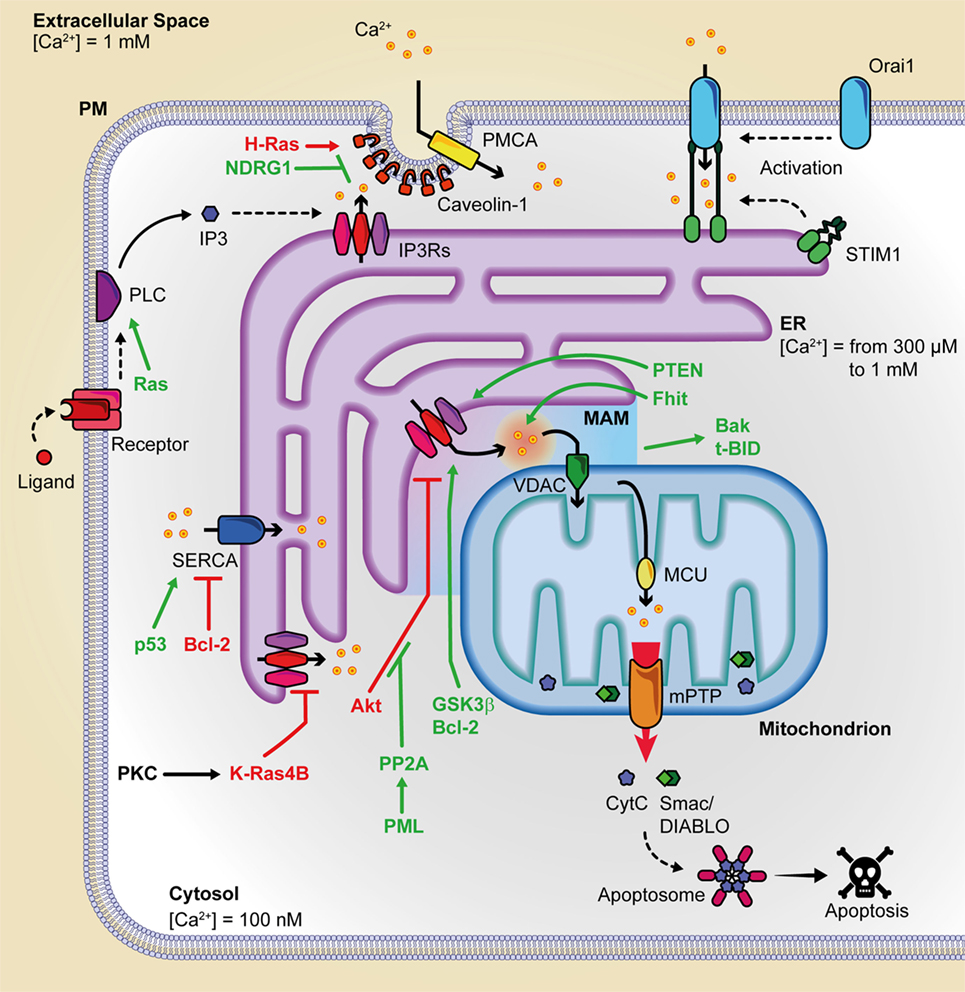
Figure 1. Summary of the principal oncogenes and oncosuppressors involved in inter-organelle Ca2+ transfer. This figure is a representation of the principal protein complexes involved in the intracellular Ca2+ transfer and of some of the various oncosuppressors and oncogenes that during cancer onset and progression can alter Ca2+ metabolism. Pro-apoptotic proteins are indicated in green, and anti-apoptotic proteins are depicted in purple. Bak, Bcl-2 antagonist/killer; Bcl-2, B-cell CLL/lymphoma 2; c; BID, BH3 interacting-domain death agonist; cyt. c, cytochrome c; ER, endoplasmic reticulum; Fhit, fragile histidine triad protein; GSK3, glycogen synthase kinase-3; IP3, inositol 1,4,5 trisphosphate; IP3Rs, inositol 1,4,5 trisphosphate receptors; MCU, mitochondrial calcium uniporter; mPTP, mitochondrial permeability transition pore; NDRG1, N-myc downregulated gene 1; PMCA, plasma membrane Ca2+ ATPase; PML, promyelocytic leukemia protein; PP2a, protein phosphatase 2PTEN, phosphatase, and tensin homolog deleted on chromosome 10; SERCA, sarco/endoplasmatic reticulum Ca2+ ATPase; SMAC/DIABLO, direct IAP binding protein with low Pi; VDAC, voltage-dependent anion channel.
The PM protein Cav1 regulates tumor-associated cellular processes. Many studies have shown that Cav1 is a growth inhibitory protein, and its gene locus is often deleted in many cancers (52). Alternatively, its tumor-promoting activity and its augmented expression have been confirmed in a variety of cancers (53). Induction of oncogenic H-Ras leads to Cav1-mediated variations in intracellular Ca2+, associated with the alteration of mitochondrial physiology (21).
N-myc downstream-regulated gene 1 is a cytoplasmic protein deregulated in prostatic and colorectal cancers (54) that appears to behave as a metastatic suppressor. It interacts with and promotes the ubiquitylation of Cav1, therefore reducing its expression, depressing epithelial-to-mesenchymal transition, and weakening the metastatic capacity of colorectal cells in vivo (55). The pro-metastatic capacity of Cav1 is mediated by the S100 calcium-binding protein P (54) and is stimulated by hypoxic conditions, which increase Cav1 expression in hepatocellular carcinoma.
Reticular IP3Rs represent a main target of the action of oncogenes or oncosuppressors. In particular, IP3R3 is enriched in MAMs regions and appears to be the major player in the pro-apoptotic transfer of Ca2+ from the ER to mitochondria (33). The fact that IP3R3 is not inhibited by high concentrations of Ca2+ (56) implicates IP3R3, among its family members, as the principal effector of supramaximal pro-apoptotic mitochondrial calcium loading. Also, this suggests that other mechanisms must be involved in the control of IP3R3 receptor opening.
The oncogene Ras is a small GTPase mutated in a high percentage of human cancers, including pancreatic, colorectal, and lung cancers (57). Mutated and constitutively active Ras hyper-activates PLCε, with a consequent increase in IP3 and the downstream pathway. In addition to targeting IP3-producing enzymes, K-Ras has also been reported to remodel the expression of IP3R isoforms and SERCA2b at the ER level. Again, protein kinase C (PKC)-mediated phosphorylation of K-Ras4B induces its translocation to the ER where it can reduce cell survival by targeting IP3R3. Strikingly, mutated K-Ras suppressed mitochondrial Ca2+ dynamics in a Bcl-XL-dependent manner (58, 59). As such, mutated K-Ras may exert its pro-oncogenic role by dampening ER-originated Ca2+ release, so promoting malignant cell survival (60). Besides K-Ras, PKC isoforms contribute to the modulation of mitochondrial Ca2+ entry in response to a plethora to cell stimuli (61, 62).
Glycogen synthase kinase-3β (GSK3β) has been identified as a novel component of the MAMs, where it phosphorylates the IP3Rs and directly regulates them (63). In cardiomyocytes, ischemia reperfusion increased GSK3β activity, enhanced GSK3β-mediated IP3R phosphorylation and, in turn, mitochondrial-dependent cell death. Concerning VDACs, the exposure to apoptotic stimuli increases mitochondrial Ca2+ uptake through only the VDAC1 isoform (34), while VDAC2 acts on apoptosis in a Ca2+ independent manner triggering the mitochondrial recruitment of Bcl-2 antagonist/killer in tBH3 interacting-domain death agonist-induced apoptosis (64).
The proto-oncogene Bcl-2, discovered in the chromosomal translocation breakpoint t(14; 18) in B-cell follicular lymphomas (65), is upregulated in several cancers through chromosomal translocation, gene hypomethylation, and miRNA dysregulation mechanisms. Bcl-2 can interact with the IP3Rs through its N-terminal BH4 domain (66), regulating its channel properties (67), and sensitizing IP3Rs toward their agonist IP3. Also, Bcl-2 has been shown to join with SERCA1 and SERCA2b isoforms directly, consequently lowering their ER Ca2+-uptake activity (68, 69). As a whole, Bcl-2 contributes to the regulation of intracellular calcium stores by increasing the efflux of Ca2+ from the ER and thereby lowering the steady-state Ca2+-storage content at ER (70–72). The effect of Bcl-2 on ER Ca2+-level appears to be mediated by phosphorylation. PKA and JunN-terminal protein kinase (73) are among the kinases involved. Also, Bcl-2 targets the sixth transmembrane domain of the IP3R, contributing to inducing pro-survival Ca2+ oscillations.
Several reports have described the interaction of Akt with IP3Rs at the ER, resulting in the inhibition of Ca2+ release from IP3Rs after its phosphorylation by Akt, without affecting histamine-induced Ca2+ release or Ca2+ content (28, 74). Akt activity is balanced by PTEN, a tumor suppressor localized to the ER and MAMs that restores Ca2+ transfer from the ER to mitochondria. Its action does not depend upon lipid dephosphorylation, but upon the protein, dephosphorylation activity exerted directly on IP3Rs (75). It has been recently shown that PTEN can also counteract the binding of the F-box protein FBXL2 to IP3R3, where FBXL2 can target IP3R3 to proteasomal degradation so limiting the Ca2+ flux to mitochondria (76). Another potent tumor-suppresser gene that can exert part of its action by regulating Ca2+ flux to mitochondria is BRCA1-associated protein 1, which localizes to ER and binds, deubiquitylates, and stabilizes IP3R3 (77).
Also, promyelocytic leukemia protein (PML) participated to complexes with the protein kinase Akt and protein phosphatase 2a (PP2a), and its binding was critical for Akt- and PP2a-dependent downregulation of IP3Rs phosphorylation. In fact, the amounts of phosphorylated IP3R3 were higher in PML−/− than in PML+/+ MEFs cells, and higher levels of active phosphorylated Akt together with reduced amounts of protein phosphatase PP2a were found to be associated with IP3R3 inhibition (78). Consistently, the overexpression of PML made the cells sensitive to ER stress-induced apoptosis but not to calcium-independent cell death (78).
Recent studies demonstrated for cytosolic p53 at ER level a role in the regulation of protein interactions (79, 80). After chemotherapy, p53 accumulated at the ER and MAMs and reinforced H2O2-induced apoptosis; overall, it increased calcium accumulation in the ER and, consequently, in the mitochondria. p53 acted through a non-transcriptional mechanism, interacting through the C-terminal regulatory domain with the SERCA pump at the ER. Interestingly, wild-type p53 boosted Ca2+ accumulation and promoted apoptosis increasing SERCA activity, but oncogenic p53 mutants failed to stimulate it. p53 was reported to tether PML (81, 82). More specifically, Missiroli et al. (83), using PML−/− and p53−/− animal models, showed that p53 is indispensable to the recruitment of PML at MAMs.
Given the number of different Ca2+-transport mechanisms, therapeutic strategies have many potential targets to bring calcium homeostasis to normality in cancer cells and re-sensitize them to cell death and chemotherapeutic drugs (Figure 2). Initially believed to support the refilling of intracellular Ca2+ stores solely, SOCE has now been shown to sustain multiple calcium-dependent cancer pathways including uncontrolled cell proliferation; consequently, its pharmacological control is a primary target for cancer therapy. SKF-96365 is an ORAI1 inhibitor that can inhibit breast cancer cell migration in vitro and reduce tumor growth and metastasis in vivo (45); it also inhibited ORAI1-mediated SOCE and intracellular Ca2+ oscillations in esophageal cancer cells (84). On the other hand, it has been reported that SKF-96365 can activate autophagy, so delaying apoptosis in colorectal cancer cells via inhibition of the calcium/CaMKIIγ/AKT-mediated pathway (85). An inhibitor of SOCE is ML-9, through its interference with STIM1 (86). Although its target and mechanism of action are unclear, it was proved to disperse STIM1 puncta and to effectively induce prostate cancer cell death. Moreover, a combination of ML-9 and anti-cancer drugs, such as docetaxel, significantly promoted cancer cell death (87).
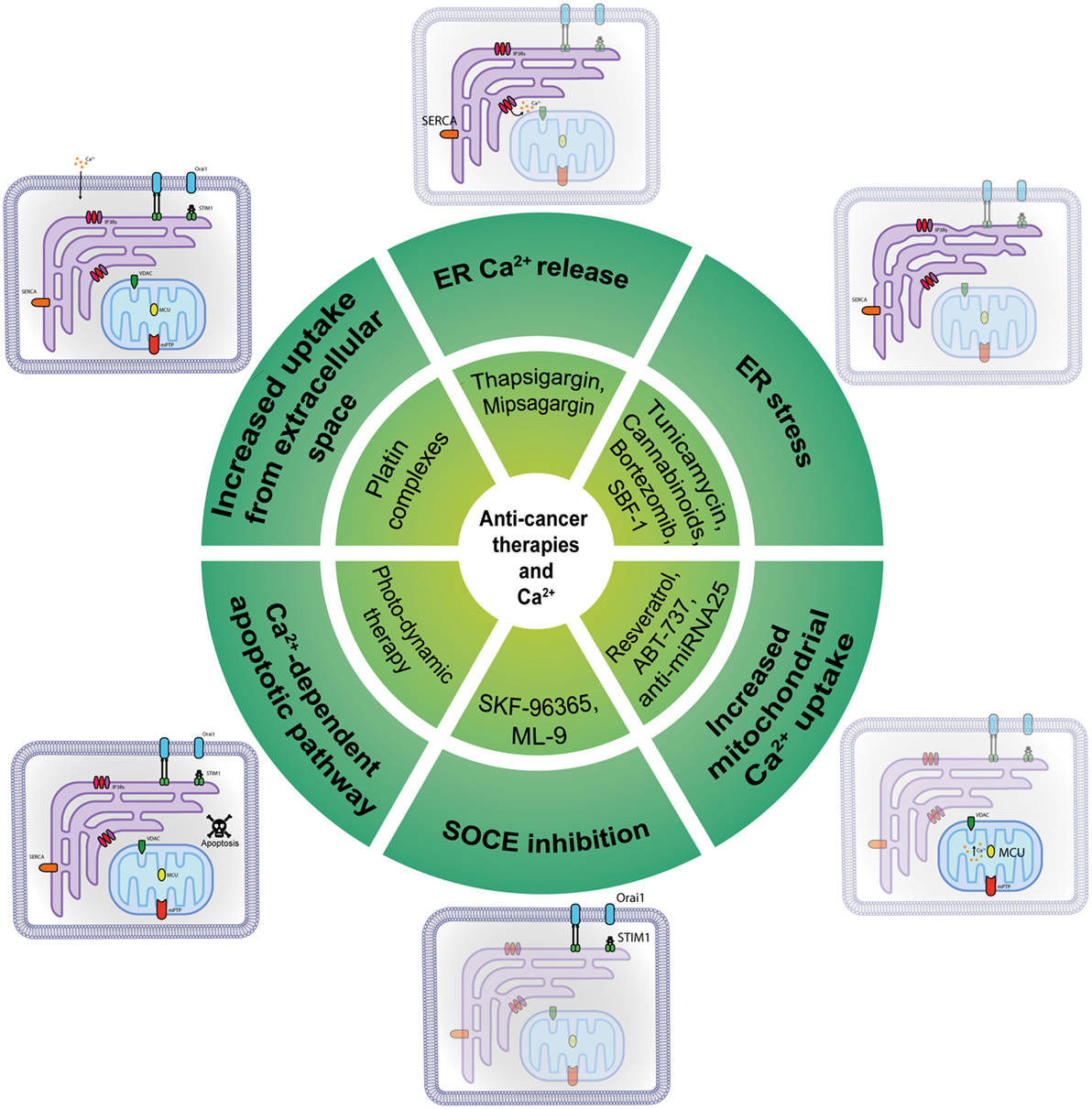
Figure 2. Anti-cancer molecules targeting intracellular Ca2+ transfer. Several classes of molecules can exert part of their anti-cancer action modulating at different levels the intracellular Ca2+ transfer.
Drugs containing metal compounds can modify Ca2+ signaling and are commonly used to treat different types of tumors: platin complexes, such as cisplatin, carboplatin, and oxaliplatin, are used clinically to treat various types of cancers, including sarcomas, carcinomas, lymphomas, and germ cell tumors (88). For example, cisplatin causes increased uptake from the extracellular space, opening a membrane-associated calcium pore; this process involves membrane-associated IP3Rs. Hence, all of the compounds that can regulate Ca2+ could be considered a new class of chemotherapeutics, but their effectiveness could be insufficient when ER-mitochondria signal transmission is constitutively worsen, as in the case of Akt hyper-activation, or PML and PTEN inactivation. To overcome this obstacle, it might be useful to stimulate artificial ER Ca2+ release using the SERCA inhibitor thapsigargin. More specifically, conjugating thapsigargin to peptide substrates for prostate-specific antigen or prostate-specific membrane antigen (PSMA), it was possible to develope mipsagargin (G-202). G-202 is an inactive non-toxic prodrug that is activated only in PSMA-expressing epithelial cells, and in tumor vasculature, giving a high precision in tumor killing, specific to hit prostate, and other cancer cells (89). G-202 is in at the moment in the clinical phase of testing in several cancers including hepatocellular carcinoma (NCT01777594), prostate cancer (NCT02381236, NCT01734681), glioblastoma (NCT02067156, NCT02876003), and others. The only published results refer to a multicentre, open-label phase I study advanced, refractory, or metastatic solid tumors (NCT01056029), that reported an acceptable safety profile but no clinical response (90). The synthetic steroidal glycoside called SBF-1 causes severe ER stress by binding to and inhibiting SERCA2 activity, thereby causing cervical cancer cell death (91). In fact, ER stress might be used to obtain an anti-cancer effect: tunicamycin potentiates cisplatin anti-cancer efficacy, inducing accumulation of unfolded proteins in the ER (92), while cannabinoids activate the ER stress-related genes ATF-4 and TRB3, inducing pancreatic tumor cell death (93). Bortezomib (Velcade), a proteasome inhibitor recently approved for multiple myeloma, provokes ER stress in addition to requiring MCU as a critical regulatory factor in its activity (94). Velcade has been involved in more than 100 clinical testing with results. Together with its efficacy, several adverse events emerged, including thrombosis and embolism events, neuropathies, and other primary malignancies. The downside of these approaches is that tumor cells can use sustained ER stress to become more tumorigenic, metastatic, and drug-resistant and to escape to immune cells (95).
Another anti-cancer molecule is resveratrol, which selectively increases mitochondrial Ca2+ uptake in cancer cells after suppression of SERCA activity at the MAMs, while healthy cells remained unaffected (96, 97). Recently, a peptide based on the BH4 domain, which is the IP3R binding site of Bcl-2, can disrupt the interaction between these proteins and enhance Ca2+ release and consequent apoptosis (98). A modified peptide called BIRD-2 has recently been synthetized: it was found to provokes apoptosis in chronic lymphocytic leukemia cells (99) and diffuse large B-cell lymphoma cells (100). Multiple myeloma, follicular lymphoma, and small cell lung cancer cells also appear to be sensitive to BIRD-2 treatment (101, 102).
Another example of a BH3 mimetic involved in calcium remodeling is ABT-737, a non-selective Bcl-2/Bcl-XL inhibitor (103, 104); it can enhance ER-mitochondrial contact sites leading to Ca2+ overload at mitochondria and improving cisplatin’s toxic effect in human ovarian cancer cells (105). Recently, it has been tested in a trial concerning samples from ovarian cancers (NCT01440504), with no published results. A recent discovery recognizes miR-25 as a cancer-related MCU-targeting microRNA family that can be targeted with anti-miRNA 25 oligonucleotides; it could be used as a potential agent against cancer, as an alternative approach to hit tumor cells (6, 106).
A growing number of findings indicate that several tumor suppressors and oncogenes can affect several levels of mechanisms regulating Ca2+ flow inside the cell, in addition to their well-known action on signal transduction pathways or on nuclear activities. The hierarchy of the Ca2+ transfer process involves several junctions in the highly compartmentalized cell interior, which warrant communication among different membrane systems. These junctions offer asylum to proteins that fill the role of oncogenes or tumor suppressors during cancer transformation, regulating the Ca2+ concentration inside the mitochondria and reticulum and consequently, altering cell metabolism, preventing apoptosis, and inducing cell migration. Specific therapies can target these junction complexes and revert Ca2+ flux to pro-apoptotic levels to sustain chemotherapy and other cancer therapies. These findings highlight the fundamental role of these inter-organelle junctions as hotspot domains, having pivotal, though not fully understood, roles in the regulation of cancer onset, and progression. Improving our knowledge of the regulation of Ca2+ transfer among these organelles will impact the search for new and more precise treatments for cancer.
Conception: GP, AR, MP, and PP. Design: GP, AR, LS, CG, MW, MP, and PP. Analysis and interpretation: GP, AR, LS, and CG. Drafting the manuscript for important intellectual content: GP, AR, LS, CG, MW, MP, and PP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
PP is grateful to Camilla degli Scrovegni for continuous support. PP is supported by the Italian Ministry of Education, University and Research, the Italian Ministry of Health, Telethon (GGP15219/B), the Italian Association for Cancer Research (IG-18624), and by local funds from the University of Ferrara. CG is supported by local funds from the University of Ferrara, the Italian Association for Cancer Research, the Italian Ministry of Health, and by Cariplo grant. AR is supported by local funds from the University of Ferrara, the Italian Ministry of Health (GR-2011-02346964), Italian Cystic Fibrosis Foundation (FFC # 20/2015). MW is supported by the National Science Center, Poland (grant 2014/15/B/NZ1/00490).
1. Carruthers C, Suntzeff V. The role of calcium in carcinogenesis summary. Science (1944) 99(2569):245–7. doi:10.1126/science.99.2569.245-a
2. Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Biophys Acta (2017) 1858(8):615–27. doi:10.1016/j.bbabio.2017.01.003
3. Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, et al. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc (2013) 8(11):2105–18. doi:10.1038/nprot.2013.127
4. Bonora M, Morganti C, Morciano G, Pedriali G, Lebiedzinska-Arciszewska M, Aquila G, et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep (2017) 18(7):1077–89. doi:10.15252/embr.201643602
5. Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, et al. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol (2015) 78:142–53. doi:10.1016/j.yjmcc.2014.08.015
6. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol (2012) 23(1):58–63. doi:10.1016/j.cub.2012.11.026
7. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal (2015) 22:995–1019. doi:10.1089/ars.2014.6223
8. Skryma R, Mariot P, Bourhis XL, Coppenolle FV, Shuba Y, Vanden Abeele F, et al. Store depletion and store-operated Ca2+ current in human prostate cancer LNCaP cells: involvement in apoptosis. J Physiol (2000) 527(Pt 1):71–83. doi:10.1111/j.1469-7793.2000.00071.x
9. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal (2013) 9(4):491–540. doi:10.1007/s11302-013-9372-5
10. Montell C. The TRP superfamily of cation channels. Sci STKE (2005) 2005(272):re3. doi:10.1126/stke.2722005re3
11. Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi-Seabrook GM, Muik M, et al. STIM1/ORAI1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun (2013) 4:2963. doi:10.1038/ncomms3963
12. Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol (2005) 15(13):1235–41. doi:10.1016/j.cub.2005.05.055
13. Lynes EM, Simmen T. Urban planning of the endoplasmic reticulum (ER): how diverse mechanisms segregate the many functions of the ER. Biochim Biophys Acta (2011) 1813(10):1893–905. doi:10.1016/j.bbamcr.2011.06.011
14. Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol (2012) 13(10):607–25. doi:10.1038/nrm3440
15. Rimessi A, Giorgi C, Pinton P, Rizzuto R. The versatility of mitochondrial calcium signals: from stimulation of cell metabolism to induction of cell death. Biochim Biophys Acta (2008) 1777(7–8):808–16. doi:10.1016/j.bbabio.2008.05.449
16. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280(5370):1763–6. doi:10.1126/science.280.5370.1763
17. Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J Cell Biol (1992) 119(6):1507–13. doi:10.1083/jcb.119.6.1507
18. Fujimoto T. Calcium pump of the plasma membrane is localized in caveolae. J Cell Biol (1993) 120(5):1147–57. doi:10.1083/jcb.120.5.1147
19. Isshiki M, Anderson RG. Function of caveolae in Ca2+ entry and Ca2+-dependent signal transduction. Traffic (2003) 4(11):717–23. doi:10.1034/j.1600-0854.2003.00130.x
20. Pulli I, Blom T, Lof C, Magnusson M, Rimessi A, Pinton P, et al. A novel chimeric aequorin fused with caveolin-1 reveals a sphingosine kinase 1-regulated Ca(2)(+) microdomain in the caveolar compartment. Biochim Biophys Acta (2015) 1853(9):2173–82. doi:10.1016/j.bbamcr.2015.04.005
21. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene (2013) 33(18):2329–40. doi:10.1038/onc.2013.192
22. Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, et al. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology (2011) 140(7):2107–15, 2115.e1–4. doi:10.1053/j.gastro.2011.02.052
23. Kim MS, Zeng W, Yuan JP, Shin DM, Worley PF, Muallem S. Native store-operated Ca2+ influx requires the channel function of ORAI1 and TRPC1. J Biol Chem (2009) 284(15):9733–41. doi:10.1074/jbc.M808097200
24. Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca(2)+ entry via ORAI1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol (2011) 9(3):e1001025. doi:10.1371/journal.pbio.1001025
25. Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, et al. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A (2013) 110(32):13186–91. doi:10.1073/pnas.1300910110
26. Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev (2005) 85(2):757–810. doi:10.1152/physrev.00057.2003
27. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J (2001) 20(11):2690–701. doi:10.1093/emboj/20.11.2690
28. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun (2008) 375(4):501–5. doi:10.1016/j.bbrc.2008.07.153
29. Reimers K, Choi CY, Bucan V, Vogt PM. The bax inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med (2008) 8(2):148–56. doi:10.2174/156652408783769562
30. Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, et al. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid Redox Signal (2011) 16(10):1077–87. doi:10.1089/ars.2011.4004
31. Bultynck G, Kiviluoto S, Henke N, Ivanova H, Schneider L, Rybalchenko V, et al. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J Biol Chem (2011) 287(4):2544–57. doi:10.1074/jbc.M111.275354
32. Yoboue ED, Rimessi A, Anelli T, Pinton P, Sitia R. Regulation of calcium fluxes by GPX8, a type-II transmembrane peroxidase enriched at the mitochondria-associated endoplasmic reticulum membrane. Antioxid Redox Signal (2017) 27(9):583–95. doi:10.1089/ars.2016.6866
33. Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem (2005) 280(49):40892–900. doi:10.1074/jbc.M506623200
34. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2011) 19(2):267–73. doi:10.1038/cdd.2011.92
35. Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, et al. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem (2004) 279(52):54581–9. doi:10.1074/jbc.M409663200
36. Vanlangenakker N, Vanden Berghe T, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med (2008) 8(3):207–20. doi:10.2174/156652408784221306
37. Rimessi A, Marchi S, Fotino C, Romagnoli A, Huebner K, Croce CM, et al. Intramitochondrial calcium regulation by the Fhit gene product sensitizes to apoptosis. Proc Natl Acad Sci U S A (2009) 106(31):12753–8. doi:10.1073/pnas.0906484106
38. Bhosale G, Sharpe JA, Sundier SY, Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann N Y Acad Sci (2015) 1350:107–16. doi:10.1111/nyas.12885
39. Missiroli S, Danese A, Iannitti T, Patergnani S, Perrone M, Previati M, et al. Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of the tumor cell fate. Biochim Biophys Acta (2017) 1864(6):858–64. doi:10.1016/j.bbamcr.2016.12.024
40. Zoratti M, Szabo I. Electrophysiology of the inner mitochondrial membrane. J Bioenerg Biomembr (1994) 26(5):543–53. doi:10.1007/BF00762739
41. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476(7360):341–5. doi:10.1038/nature10234
42. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476(7360):336–40. doi:10.1038/nature10230
43. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell (2017) 65(6):1014–28.e7. doi:10.1016/j.molcel.2017.01.032
44. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol (2013) 15(12):1464–72. doi:10.1038/ncb2868
45. Yang S, Zhang JJ, Huang XY. ORAI1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell (2009) 15(2):124–34. doi:10.1016/j.ccr.2008.12.019
46. Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, et al. STIM1 and ORAI1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch (2013) 465(9):1249–60. doi:10.1007/s00424-013-1254-8
47. Xu JM, Zhou Y, Gao L, Zhou SX, Liu WH, Li XA. Stromal interaction molecule 1 plays an important role in gastric cancer progression. Oncol Rep (2016) 35(6):3496–504. doi:10.3892/or.2016.4704
48. Schafer C, Rymarczyk G, Ding L, Kirber MT, Bolotina VM. Role of molecular determinants of store-operated Ca(2+) entry (ORAI1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. J Biol Chem (2012) 287(48):40745–57. doi:10.1074/jbc.M112.407155
49. Tsai FC, Seki A, Yang HW, Hayer A, Carrasco S, Malmersjo S, et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat Cell Biol (2014) 16(2):133–44. doi:10.1038/ncb2906
50. Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A (2011) 108(37):15225–30. doi:10.1073/pnas.1103315108
51. Stanisz H, Saul S, Muller CS, Kappl R, Niemeyer BA, Vogt T, et al. Inverse regulation of melanoma growth and migration by ORAI1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res (2014) 27(3):442–53. doi:10.1111/pcmr.12222
52. Engelman JA, Zhang XL, Galbiati F, Lisanti MP. Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3). Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett (1998) 429(3):330–6. doi:10.1016/S0014-5793(98)00619-X
53. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet (2000) 24(3):227–35. doi:10.1038/73432
54. Mao X, Wong SY, Tse EY, Ko FC, Tey SK, Yeung YS, et al. Mechanisms through which hypoxia-induced caveolin-1 drives tumorigenesis and metastasis in hepatocellular carcinoma. Cancer Res (2016) 76(24):7242–53. doi:10.1158/0008-5472.CAN-16-1031
55. Mi L, Zhu F, Yang X, Lu J, Zheng Y, Zhao Q, et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin-1 ubiquitylation in human colorectal cancer cells. Oncogene (2017) 36(30):4323–35. doi:10.1038/onc.2017.74
56. Missiaen L, DeSmedt H, Bultynck G, Vanlingen S, Desmet P, Callewaert G, et al. Calmodulin increases the sensitivity of type 3 inositol-1,4, 5-trisphosphate receptors to Ca(2+) inhibition in human bronchial mucosal cells. Mol Pharmacol (2000) 57(3):564–7. doi:10.1124/mol.57.3.564
57. Schmukler E, Kloog Y, Pinkas-Kramarski R. Ras and autophagy in cancer development and therapy. Oncotarget (2014) 5(3):577–86. doi:10.18632/oncotarget.1775
58. Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell (2006) 21(4):481–93. doi:10.1016/j.molcel.2006.01.012
59. Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, et al. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A (2013) 110(51):20593–8. doi:10.1073/pnas.1306431110
60. Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci (2014) 127(Pt 7):1607–19. doi:10.1242/jcs.141408
61. Pinton P, Leo S, Wieckowski MR, Di Benedetto G, Rizzuto R. Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J Cell Biol (2004) 165(2):223–32. doi:10.1083/jcb.200311061
62. Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, et al. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid Redox Signal (2010) 13(7):1051–85. doi:10.1089/ars.2009.2825
63. Gomez L, Thiebaut PA, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3beta during reperfusion injury. Cell Death Differ (2015) 22(11):1890. doi:10.1038/cdd.2015.118
64. Naghdi S, Varnai P, Hajnoczky G. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc Natl Acad Sci U S A (2015) 112(41):E5590–9. doi:10.1073/pnas.1510574112
65. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science (1985) 228(4706):1440–3. doi:10.1126/science.3874430
66. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A (2009) 106(34):14397–402. doi:10.1073/pnas.0907555106
67. Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol (2004) 166(2):193–203. doi:10.1083/jcb.200309146
68. Dremina ES, Sharov VS, Kumar K, Zaidi A, Michaelis EK, Schoneich C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem J (2004) 383(Pt 2):361–70. doi:10.1042/BJ20040187BJ20040187
69. Dremina ES, Sharov VS, Schoneich C. Heat-shock proteins attenuate SERCA inactivation by the anti-apoptotic protein Bcl-2: possible implications for the ER Ca2+-mediated apoptosis. Biochem J (2012) 444(1):127–39. doi:10.1042/BJ20111114
70. Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci U S A (2000) 97(11):5723–8. doi:10.1073/pnas.97.11.5723
71. Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, et al. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol (2000) 148(5):857–62. doi:10.1083/jcb.148.5.857
72. Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A (2004) 101(50):17404–9. doi:10.1073/pnas.0408030101
73. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy (2008) 4(7):949–51. doi:10.4161/auto.6788
74. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A (2008) 105(7):2427–32. doi:10.1073/pnas.0711324105
75. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ (2013) 20(12):1631–43. doi:10.1038/cdd.2013.77
76. Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumor growth. Nature (2017) 546(7659):554–8. doi:10.1038/nature22965
77. Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature (2017) 546(7659):549–53. doi:10.1038/nature22798
78. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330(6008):1247–51. doi:10.1126/science.1189157
79. Giorgi C, Bonora M, Pinton P. Inside the tumor: p53 modulates calcium homeostasis. Cell Cycle (2015) 14(7):933–4. doi:10.1080/15384101.2015.1010973
80. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112(6):1779–84. doi:10.1073/pnas.1410723112
81. Pearson M, Pelicci PG. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene (2001) 20(49):7250–6. doi:10.1038/sj.onc.1204856
82. Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med (2015) 20(2):167–74. doi:10.1038/nm.3441
83. Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R, et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep (2016) 16(9):2415–27. doi:10.1016/j.celrep.2016.07.082
84. Zhu H, Zhang H, Jin F, Fang M, Huang M, Yang CS, et al. Elevated ORAI1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget (2014) 5(11):3455–71. doi:10.18632/oncotarget.1903
85. Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou Y, et al. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIgamma/AKT-mediated pathway. Cancer Lett (2016) 372:226–38. doi:10.1016/j.canlet.2016.01.006
86. Smyth JT, Dehaven WI, Bird GS, Putney JW Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci (2008) 121(Pt 6):762–72. doi:10.1242/jcs.023903
87. Kondratskyi A, Yassine M, Slomianny C, Kondratska K, Gordienko D, Dewailly E, et al. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis (2014) 5:e1193. doi:10.1038/cddis.2014.156
88. Desoize B. Cancer and metals and metal compounds: part II—cancer treatment. Crit Rev Oncol Hematol (2002) 42(3):213–5. doi:10.1016/S1040-8428(02)00039-2
89. Doan NT, Paulsen ES, Sehgal P, Moller JV, Nissen P, Denmeade SR, et al. Targeting thapsigargin towards tumors. Steroids (2014) 97:2–7. doi:10.1016/j.steroids.2014.07.009
90. Mahalingam D, Wilding G, Denmeade S, Sarantopoulas J, Cosgrove D, Cetnar J, et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br J Cancer (2016) 114:986–94. doi:10.1038/bjc.2016.72
91. Li W, Ouyang Z, Zhang Q, Wang L, Shen Y, Wu X, et al. SBF-1 exerts strong anticervical cancer effect through inducing endoplasmic reticulum stress-associated cell death via targeting sarco/endoplasmic reticulum Ca(2+)-ATPase 2. Cell Death Dis (2014) 5:e1581. doi:10.1038/cddis.2014.538
92. Hou H, Sun H, Lu P, Ge C, Zhang L, Li H, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther (2013) 12(12):2874–84. doi:10.1158/1535-7163.MCT-13-0201
93. Carracedo A, Gironella M, Lorente M, Garcia S, Guzman M, Velasco G, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res (2006) 66(13):6748–55. doi:10.1158/0008-5472.CAN-06-0169
94. Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res (2005) 65(9):3828–36. doi:10.1158/0008-5472.CAN-04-3684
95. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell (2017) 168(4):692–706. doi:10.1016/j.cell.2016.12.004
96. Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, Eroglu E, Klec C, Waldeck-Weiermair M, et al. Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell Physiol Biochem (2016) 39(4):1404–20. doi:10.1159/000447844
97. Luyten T, Welkenhuyzen K, Roest G, Kania E, Wang L, Bittremieux M, et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2. Biochim Biophys Acta (2017) 1864(6):947–56. doi:10.1016/j.bbamcr.2017.02.013
98. Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell (2008) 31(2):255–65. doi:10.1016/j.molcel.2008.06.014
99. Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood (2011) 117(10):2924–34. doi:10.1182/blood-2010-09-307405
100. Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis (2013) 4:e632. doi:10.1038/cddis.2013.140
101. Lavik AR, Zhong F, Chang MJ, Greenberg E, Choudhary Y, Smith MR, et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget (2015) 6(29):27388–402. doi:10.18632/oncotarget.4489
102. Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis (2016) 6:e2034. doi:10.1038/cddis.2015.355
103. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature (2005) 435(7042):677–81. doi:10.1038/nature03579
104. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell (2006) 10(5):389–99. doi:10.1016/j.ccr.2006.08.027
105. Xie Q, Su J, Jiao B, Shen L, Ma L, Qu X, et al. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int J Oncol (2016) 49(6):2507–19. doi:10.3892/ijo.2016.3733
Keywords: mitochondria-associated membranes, calcium, oncogenes, tumor suppressors, cell death, ROS, endoplasmic reticulum
Citation: Pedriali G, Rimessi A, Sbano L, Giorgi C, Wieckowski MR, Previati M and Pinton P (2017) Regulation of Endoplasmic Reticulum–Mitochondria Ca2+ Transfer and Its Importance for Anti-Cancer Therapies. Front. Oncol. 7:180. doi: 10.3389/fonc.2017.00180
Received: 09 June 2017; Accepted: 07 August 2017;
Published: 31 August 2017
Edited by:
Giovanni Li Volti, University of Catania, ItalyReviewed by:
Roberto Bei, Università degli Studi di Roma Tor Vergata, ItalyCopyright: © 2017 Pedriali, Rimessi, Sbano, Giorgi, Wieckowski, Previati and Pinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Pinton, cGFvbG8ucGludG9uQHVuaWZlLml0
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.