 Cristina Mammucari
Cristina Mammucari Gaia Gherardi
Gaia Gherardi Rosario Rizzuto
Rosario Rizzuto
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 10 July 2017
Sec. Molecular and Cellular Oncology
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00139
This article is part of the Research Topic Inter-Organelle Calcium Communication in Cancer View all 14 articles
Mitochondrial Ca2+ uptake plays a pivotal role both in cell energy balance and in cell fate determination. Studies on the role of mitochondrial Ca2+ signaling in pathophysiology have been favored by the identification of the genes encoding the mitochondrial calcium uniporter (MCU) and its regulatory subunits. Thus, research carried on in the last years on one hand has determined the structure of the MCU complex and its regulation, on the other has uncovered the consequences of dysregulated mitochondrial Ca2+ signaling in cell and tissue homeostasis. Whether mitochondrial Ca2+ uptake can be exploited as a weapon to counteract cancer progression is debated. In this review, we summarize recent research on the molecular structure of the MCU, the regulatory mechanisms that control its activity and its relevance in pathophysiology, focusing in particular on its role in cancer progression.
The role of mitochondria in cancer was long debated. In the 20s of the last century, Otto Warburg made the pivotal observation that cancer cells mostly rely on glycolysis for ATP production even in the presence of optimal pO2. Warburg proposed that mitochondrial dysfunction was the primary cause of preferential aerobic glycolysis. In addition, only cells able to increase glycolytic ATP production during periods of respiratory failure would progress toward cancer. Thus, cancer would be a consequence of progressive and irreversible defect in oxidative metabolism. However, experimental evidence revealed the weaknesses of this model. First, in some tumors, cells have normal respiratory capacity and/or are able to reactivate oxidative phosphorylation in case of impaired glycolysis. Second, in alternative to glucose metabolism, certain cancers utilize glutamine as an energy source (1). In addition, Warburg’s theory did not explain the existence of cancer-related gene mutations and, finally, it was unclear how impaired respiration would lead to uncontrolled cell growth. As a result, for many years, tumor metabolism was overlooked, in favor of a more accepted view of cancer as genetic disease. Recently, the importance of metabolism in cancer progression has been reconsidered. Mutations in genes encoding mitochondrial proteins and mitochondrial dysfunctions are linked to cancer progression and respiration defects contribute to genome instability. In addition, damage in respiration underlies genome instability which, in turn, sustains respiratory impairment and cancer progression (2). Mitochondrial Ca2+ signaling plays different roles in pathophysiology. On one hand, by buffering cytosolic [Ca2+], it regulates cell processes controlled by intracellular [Ca2+], like those regulated by Ca2+-binding proteins. On the other side, within mitochondria, Ca2+ regulates three TCA cycle enzymes, thus exerting a positive role on oxidative metabolism. However, excessive mitochondrial Ca2+ entry consequent to stress stimuli causes opening of the mitochondrial permeability transition pore (mPTP) and release of proapoptotic factors which eventually lead to cell death (3). On this basis, for a long time, the study of mitochondrial Ca2+ signaling in cancer biology has been primarily focused on the Ca2+-dependent activation of the proapoptotic axis as a putative therapeutic strategy. However, once stimulated with an appropriate agonist, cancer cells display large mitochondrial Ca2+ peaks, raising the question whether effective mitochondrial Ca2+ accumulation might play a permissive role in cancer progression. Thus, the scenario is quite complex, and recent literature suggests that mitochondrial Ca2+ homeostasis plays different roles in cancer, depending on the type and stage of the tumor and on whether primary tumors or metastasis are being analyzed.
Once the electrophysiological properties of the mitochondrial calcium uniporter (MCU), the highly selective channel responsible for mitochondrial Ca2+ uptake, were described (4), the genes encoding both the pore and the regulatory subunits have been eventually identified (3). Therefore, studies aimed at further delving into the role of mitochondrial Ca2+ homeostasis in pathophysiology, including cancer progression, were conducted based on the molecular modulation of the MCU complex components.
Genetic modulation of MCU expression confirmed the previously recognized role of mitochondrial Ca2+ overload in triggering cell death. Thus, the old paradigm of excessive Ca2+ uptake being permissive for apoptotic cell death still holds true. However, other studies highlighted novel and still incompletely defined aspects of MCU in cancer, indicating that mitochondrial Ca2+ uptake plays multiple and complex roles in cancer biology. This review will summarize the molecular structure of MCU, the regulatory mechanisms underlying its activity, and will uncover its role in pathophysiology, with particular attention to cancer biology.
Voltage-dependent anion channels (VDACs) are the most abundant proteins of the outer mitochondrial membrane. VDAC1, VDAC2, and VDAC3 are permeable to <5 kDa metabolites and are characterized by a weak anion selectivity in the open channel conformation, while they show strong cation preference in the closed states (5). The three VDAC isoforms share comparable channel properties in Ca2+ transport from the cytosol to the intermembrane space (6); however, they differ in specific functions. In particular, VDAC1, by interacting with the InsP3R located at the ER, specifically senses low amplitude proapoptotic Ca2+ signals (7). In addition, isoform-specific interactions of VDAC with apoptotic regulators contribute to the selective control of cell fate (3). VDACs are involved in many other functions, including metabolism regulation and energy production. For further discussion on VDACs, the reader is referred to specialized publications (8–11).
A 480 kDa inner mitochondrial membrane (IMM) multimeric complex, i.e., the MCU, comprising channel pore-forming subunits and regulatory elements, ensures Ca2+ entry into mitochondria (Figure 1). Long before the electrophysiological properties and the molecular structure of MCU were disclosed, cell Ca2+ signaling was already extensively characterized. The driving force for Ca2+ entry into mitochondria is represented by the electrochemical gradient across the IMM, which is negative in the mitochondrial matrix (about −180 mV). However, despite this huge driving force, resting mitochondrial [Ca2+] is in the nanomolar range, indicating that the affinity of the channel for Ca2+ is low. In addition, the mitochondrial Na+/Ca2+ exchanger ensures efficient extrusion of Ca2+ from mitochondria (12). Upon stimulation with InsP3-generating agonists, cytosolic Ca2+ transients reach the micromolar range and are paralleled by sharp mitochondrial [Ca2+] increases. This apparent conundrum is explained by the fact that ER and mitochondria share sites of near proximity, where the distance between the two organelles is extremely close (13, 14). These sites are named mitochondrial associated membranes (MAMs). At MAMs, Ca2+ released from the ER generates microdomains of high [Ca2+], which are sensed by mitochondria and which are responsible for the significant increase in mitochondrial [Ca2+]. The amount of Ca2+ uptake by mitochondria follows a non-linear relationship related to the amount of Ca2+ present at microdomains. Ca2+ uptake is inefficient not only at low [Ca2+], for the reasons explained above, and for the newly described role of MCU modulators (see below), but also in the presence of too high [Ca2+] at which the inhibitory effect of Ca2+ on InsP3R prevails. Thus, efficient mitochondrial Ca2+ uptake is warranted by the prompt removal of Ca2+ from microdomains exerted by mitochondria.
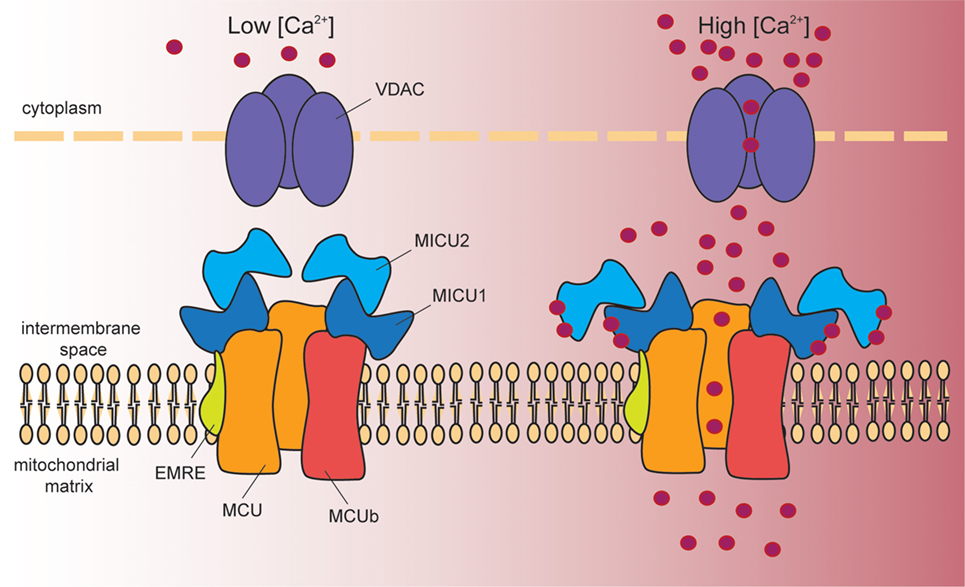
Figure 1. Schematic representation of the proteins involved in mitochondrial Ca2+ entry. Cytosolic Ca2+ enters into the mitochondrial matrix according to the mitochondrial membrane potential (−180 mV, negative inside). At the outer mitochondrial membrane, voltage-dependent anion channels (VDAC1–3) ensure permeability to solutes smaller than 5 kDa, including Ca2+. At the inner mitochondrial membrane, a multiprotein complex controls mitochondrial Ca2+ uptake. This complex is composed of the pore-forming subunits MCU and MCUb, and by the regulatory proteins MICU1, MICU2, and EMRE. In resting conditions (left hand side), MICU1/MICU2 heterodimers act as MCU gatekeeper, due to the inhibitory effect of MICU2. Once Ca2+ signaling is activated (right hand side), the increase in cytosolic [Ca2+] induces a conformational change in the dimer that releases MICU2-dependent inhibition. At the same time, MICU1 acts as a cooperative activator of the channel, and thus stimulates channel activity. EMRE stabilizes MCU–MICU1 complex contributing to fine-tuned Ca2+ entry into mitochondria.
In 2004, Clapham and colleagues, by patch-clamping mitoplasts preparations, unequivocally identified a Ca2+-selective mitochondrial channel whose current had the properties of that mediated by MCU (4). Subsequent characterization demonstrated that different tissues display different MCU activity, according to the specific physiological properties of Ca2+ signaling in each tissue, suggesting that tissue-specific regulation of the channel occurs (15).
More recently, the Mootha and Rizzuto groups, by different methods, in parallel achieved the identification of ccdc109a as the gene encoding the pore-forming subunit of MCU, sufficient per se to ensure channel activity (16, 17). MCU was co-expressed with the previously described regulatory subunit MICU1 (18), it was required for mitochondrial Ca2+ uptake and, most importantly, the purified protein reconstituted in planar lipid bilayers recapitulated the known electrophysiological properties of MCU. Structurally, MCU protein contains two coil-coiled domains and two transmembrane (TM) domains separated by a short hydrophilic loop enriched in acidic amino acids (16, 17). Electron microscopy of mitochondria overexpressing tagged MCU demonstrated that both the N- and the C-terminus of MCU face the matrix (19). By combining structural bioinformatics techniques and molecular dynamic simulations, a quaternary structure was proposed, and tridimensional localization of critical residues of the pore was modeled (20). Each pore-forming subunit consists of two helical membrane spanning domains connected by a short loop containing a “DIME” motif. This region contains negatively charged residues which have been previously identified to play an essential role in MCU-mediated Ca2+ uptake (16, 17). These residues are part of a narrow selectivity filter which accommodates a single Ca2+ ion. One additional feature is represented by the C-terminal tails, which define a pore of 1 Å of diameter, suggesting that this MCU model stably adopts a close conformation, as it commonly occurs for Ca2+ channels (20). Subsequently, a crystal structure at a 1.80 Å resolution of the MCU N-terminal domain (NTD) was released (21). This analysis identified one α-helix and six β-strands that form the central core, two highly conserved loops and one leucine-rich short α-helix, which stabilizes the hydrophobic interior of MCU NTD. One additional α-helix and a C-terminal tail are also present. MCU NTD is essential for regulation of Ca2+ uptake, either through interaction with regulators, or by post-transcriptional modifications. Finally, the structure of the pore domain of MCU from Caenorhabditis elegans was determined by nuclear magnetic resonance (NMR) and electron microscopy. Differently from the quaternary complex proposed for mammalian MCU (20), a pentameric organization was proposed (22), in which the TM domain consists of 10 helices arranged in two concentric layers. Specific aspartate and glutamate residues were identified as forming the selectivity filter. The proposed structure is completely different from other Ca2+ channels and leaves many open questions concerning Ca2+ binding and transport, and regulation of MCU conformation by its interactors. Further details were recently added by an NMR-based analysis aimed at better characterizing the properties of the selectivity filter. This analysis conclusively demonstrated that the double carboxylate rings formed by the “DXXE” motif is the ion selectivity filter and that cooperativity of the two ion binding sites confers high ion affinity (23).
Genetic manipulation of MCU in animal models gave interesting results. In lower organisms, i.e., Trypanosoma brucei and Danio rerio, MCU knockdown or knockout caused developmental defects. In the parasite, MCU silencing or conditional knockout impaired mitochondrial Ca2+ uptake as expected, reduced energy production and increased AMP/ATP ratio and autophagosome formation. As a result, growth was markedly inhibited as well as infectivity in mice (24). In zebrafish, MCU silencing caused major developmental defects, impinging on RhoA signaling and F-actin dynamics (25). In Drosophila melanogaster, overexpression of a dominant-negative isoform of MCU (DN-MCU) in mushroom body neurons during pupation caused memory impairment associated with a decrease in synaptic vesicles and an increased length in the axons (26), again highlighting the importance of efficient mitochondrial Ca2+ uptake for development. On the contrary, C. elegans MCU null mutants are viable and fertile. Studies performed in the worm help elucidating the involvement of mitochondrial Ca2+ uptake and ROS production in the wound repair mechanism, demonstrating that wounding is paralleled by an increase in mitochondrial [Ca2+] and ROS production. ROS are required for proper repair, and MCU deletion causes impaired wound healing (27). Deletion of MCU in the mouse has opposite developmental effects depending on the strain. MCU−/− embryos in the C57BL/6 background die around E 11.5–E 13.5, for causes that are not completely clear (28, 29). Viable mice were instead obtained in the outbred CD1 background, although not at Mendelian ratio (30). In the latter case, defects in MCU−/− mice were mostly apparent in the diminished performance of skeletal muscle. MCU−/− hearts were as susceptible as wild-type hearts to ischemia/reperfusion (I/R) injury, although a difference was apparent in the effects of the mPTP inhibitor cyclosporin A (CsA). In contrast to wild-type hearts, MCU−/− hearts were not protected from I/R injury by CsA treatment. These data indicate that, besides mPTP opening by mitochondrial Ca2+ overload, alternative damaging pathways are activate by I/R which are not counteracted by MCU deletion (30). One of the more obvious and more explored interpretation for the unexpected lack of marked phenotype is that MCU−/− animals could have developed compensatory mechanisms that allow them to cope with developmental challenges and with stresses encountered during postnatal life.
Comparison of MCU activity in patch-clamped mitoplasts from different tissues demonstrated great variability (15). In particular, heart is characterized by very low MCU current density, which is 30-fold higher in skeletal muscle. However, the biophysical properties of heart and skeletal muscle MCU currents are identical, indicating that different tissues share the same Ca2+ channel and that tissue-specific regulatory mechanisms are responsible for the differences in channel activity. These differences are explained by the fact that MCU is part of a large protein complex, comprising both other channel-forming subunits and regulatory components, as detailed hereafter.
A closely related protein with inhibitory activity on channel function is encoded by the ccdc109b gene and is named MCUb (20). MCUb shares 50% similarity with MCU and, like MCU, it contains two TM domains. However, once inserted in lipid bilayers, it is not able to conduct any current and, co-expressed with MCU, completely abolishes MCU currents, thus exerting a dominant-negative effect on the channel activity. MCU forms both homo-oligomers and hetero-oligomers with MCUb. However, MCUb is expressed at different levels compared to MCU in different tissues, indicating that it plays a tissue-dependent modulatory role.
By quantitative mass spectrometry of the uniporter complex, a 10 kDa protein containing a single TM domain was purified (31). This protein was named EMRE (essential MCU regulator), since its abrogation depleted uniporter channel activity and interaction of MCU with MICU1 and MICU2 (which are described below). However, whether EMRE is really essential for uniporter activity has been questioned, since organisms like plants and fungi, in which MCU and MICU1 are highly conserved, do not express EMRE. In addition, mice deleted for both EMRE alleles had normal body weight and no signs of ataxia or skeletal muscle defects (32). To clarify this point, the yeast Saccharomyces cerevisiae, which lacks MCU, was used as in vivo system to reconstitute uniporter activity. While expression of Dictyostelium discoideum MCU alone was sufficient to reconstitute uniporter activity, the human homolog required co-expression of EMRE (33). This observation clarifies the difference in uniporter component expression among different species and sheds light on the evolution of the MCU. According to recent data, EMRE not only keeps in place MCU and MICUs, but actively plays a regulatory role on channel function. Specifically, one report suggested that EMRE, in complex with MCU and MICUs, spans the inner membrane and acts as a sensor of [Ca2+] at both sides. By sensing matrix [Ca2+] it inhibits MCU activity. EMRE mutants depleted of acidic residues facing the matrix loose this function, resulting in MCU activation and rising of matrix [Ca2+]. Thus, in this configuration, EMRE contributes to mitochondrial Ca2+ uptake avoiding mitochondrial Ca2+ overload (34). Studies performed with mouse uniporter proteins reconstituted in yeast proposed an alternative model. In contrast to the previously proposed topology with the C-terminus facing the mitochondrial matrix (34), in yeast mitochondria, mouse EMRE spans the inner membrane with the NTD facing the matrix, and specific residues located both at the C-terminal and NTDs are required for interaction with MCU. From genetic studies, the authors suggested that the role of EMRE is to keep MCU in an open conformation, thus ensuring Ca2+ entry (35). Further details were added by a study performed in HEK 293 cells, in which the topology of EMRE with the N-terminal facing the matrix was confirmed. Mutants in which either the N- or the C-terminal domains were deleted still supported uniporter activity, while substitutions of critical amino acids of the TM helix, which appeared to be essential for interaction with MCU, caused complete loss of channel activity (36). However, in this case, EMRE mutant deleted at the NTD still contained the critical amino acid previously reported as essential for interaction with MCU (35). EMRE was required to ensure that Ca2+ entries mitochondria only above a certain cytosolic [Ca2+], and this was achieved by binding of the C-terminal tail to MICU1. Thus, EMRE was proposed to act as stabilizer or MCU–MICU1 interaction (36) and assembly of the MCU complex, comprising MCU, EMRE, and regulators of the MICU family, ensures fine-tuned mitochondrial Ca2+ entry. Physiological EMRE turnover, ensured by mitochondrial AAA (m-AAA) protease, is essential for correct assembly of the gatekeeping subunits (37, 38). In detail, m-AAA protease degrades the surplus fraction of EMRE that would interact with MCU alone and that would increase the amount of constitutively open channel with consequent Ca2+ leakage into mitochondria.
Mitochondrial calcium uniporter channel activity is characterized by a sigmoidal response respective to changes in cytosolic [Ca2+]. Little Ca2+ enters through the channel at low cytosolic [Ca2+]. Upon cell stimulation with an appropriate stimulus, cytosolic [Ca2+] increases, followed by rapid and efficient mitochondrial Ca2+ entry (3). These properties are determined by the presence of MCU regulators, which exert either a gatekeeping or a stimulatory function on channel activity.
In 2010, the Mootha group applied an integrated approach based on data from comparative physiology, evolutionary genomics, and organelle proteomics, to identified human genes involved in mitochondrial Ca2+ entry. Targeted RNAi screen leads to the identification of MICU1, to which an essential role in regulating agonist-induced mitochondrial Ca2+ uptake was recognized (18). Subsequent work revealed the complexity of MCU regulation and, in this context, the role of MICU1 was reassessed. MICU1 silencing was reported to increase basal mitochondrial [Ca2+], causing mitochondrial Ca2+ overload and increasing sensitivity to apoptotic stimuli (39). Accordingly, it was proposed that, by interacting with MCU, MICU1 sets the Ca2+ threshold of MCU activation, without affecting the kinetic properties of MCU-mediate Ca2+ uptake. Thus, the concept of “gatekeeper” for MICU1 function was introduced. Analysis of MICU1/MCU interaction established that MICU1 localizes in the inner membrane space and forms homo-oligomers. MICU1 domains responsible for interaction with MCU and for gatekeeper function were also identified (40). Further work confirmed the gatekeeping function that MICU1 exerts at low cytosolic [Ca2+] but also demonstrated that MICU1 contributes to cooperative activation of mitochondrial Ca2+ uptake at high cytosolic [Ca2+]. MICU1 plays this dual function by sensing cytosolic [Ca2+] at the outer surface of the IMM (41). Crystal structure studies of Ca2+-free and Ca2+-bound human MICU1 demonstrated that Ca2+-free MICU1 forms a hexamer capable of binding to MCU. Upon Ca2+ binding, large conformational changes occur within one of the two canonical Ca2+ binding EF-hand domains present in the MICU1 sequence. Consequently, disassembly of the hexamer and formation of multiple oligomers of MICU1 dimer occurs after Ca2+ binding. The affinity of MICU1 for Ca2+ was calculated in the range of 15–20 µM. Thus, at resting cytosolic [Ca2+], which is approximately 0.1 µM, MICU1 is free of Ca2+ and keeps the MCU channel closed. Upon cell stimulation, cytosolic [Ca2+] rises and Ca2+ binds to MICU1, inducing MICU1 conformational changes and eventually MCU activation (42). Concerning the pathophysiological function of MICU1, loss-of-function mutations cause a disease phenotype comprising muscle weakness associated with myofiber regeneration, learning difficulties and extra-pyramidal movement disorder. This is due to the fact that loss of MICU1 activity causes an increase in basal mitochondrial [Ca2+] and a reduction in cytosolic [Ca2+], as demonstrated by studies carried on in patient fibroblasts. The mitochondrial network is fragmented despite maintenance of normal ΔΨ (43). In the available MICU1−/− mouse models, MICU1 deletion causes either complete (44) or significant (32) perinatal mortality. Importantly, the phenotype of surviving MICU1−/− animals mirrors the muscle weakness and neurological defects observed in patients. Similarly to patient fibroblasts, MICU1−/− mice display increased resting mitochondrial Ca2+ levels and altered mitochondria morphology (32). Interestingly, in aging animals, resting mitochondrial [Ca2+] is no longer significantly different between MICU1−/− and wild-type mice, and phenotypic parameters are also ameliorated, suggesting that MICU1−/− mitochondria undergo a functional remodeling. At the same time, MICU1 deletion causes a marked decrease in EMRE protein levels in mitochondria of young animals, which is even more evident in older mice. Genetic evidence indicates that EMRE heterozygosity rescues the partial lethality and the muscle and brain defects caused by MICU1 deletion. However, the scenario is quite complex, as combined homozygous MICU1 and EMRE deletions causes complete mice lethality (32). Finally, liver-specific downregulation of MICU1 causes an impairment in liver regeneration after injury, due to mPTP opening by mitochondrial Ca2+ overload. Indeed, similarly to previously reported cell models (39, 41, 45, 46), MICU1 knockdown in hepatocytes lowers the threshold and decreases the cooperativity of mitochondrial Ca2+ accumulation (44). Recently, further insights of the importance of MICU1:MCU ratio in determining tissue-specific mitochondrial Ca2+ responses were provided. Comparison between liver and heart tissues lead to the conclusion that the tissue specificity of MICU1:MCU ratio directly reflects the amount of MICU1-free channel. This is turn determines tissue-specific decoding of cytosolic [Ca2+]. In particular, while in hepatocytes MICU1 is highly expressed, ensuring high threshold and cooperativity, heart mitochondria display low levels of MICU1, which, in turn, determine low threshold and cooperativity necessary for proper heart function. Interestingly, forced expression of MICU1 induced heart mitochondria to acquire a “liver-like” mitochondrial Ca2+ pattern, which caused contractile dysfunction (47).
Studies on flies added further insights on the pathophysiological role of MICU1. In D. melanogaster, the CG4495 gene was identified as putative MICU1 homolog based on the high similarity of essential domains. Silencing of MICU1 in dopaminergic neurons caused shortening of life span and impaired climbing ability, the latter worsened with age (48).
The MICU1 paralog MICU2, identified by bioinformatics in the human genome together with MICU3 (which is mainly expressed in brain, and, to a lesser extent, in skeletal muscle) (49), participates to MCU activity regulation. Similarly to MICU1 silencing (18), silencing of MICU2 impairs mitochondrial Ca2+ uptake, and depletion of both MICU1 and MICU2 has an additive effect (49). MICU1 and MICU2 expression levels are dependent one to each other. In detail, MICU2 protein expression shows a direct correlation with MICU1 expression levels in HeLa and HEK293T cell lines, while the effect of MICU2 silencing on MICU1 expression are cell-type dependent (49). However, MICU2 protein levels were unaffected in MICU1−/− MEFs and in primary mouse hepatocytes of MICU1 knockdown mice (44). The physical and functional interaction between MICU1 and MICU2 and their coordinate effects on MCU activity were further examined (46). Patron et al. demonstrated that MICU1 and MICU2 form a heterodimer and play opposing effects on mitochondrial Ca2+ entry. In particular, MICU1 has a stimulatory role, while MICU2 inhibits Ca2+ uptake. Both are regulated by Ca2+ binding at their EF-hand domains. At low cytosolic [Ca2+], the inhibitory role of MICU2 prevails, while at high cytosolic [Ca2+] conformational modifications ensure prompt MICU1-dependent activation of mitochondrial Ca2+ uptake (46). This work also confirmed that MICU1 is essential for MICU2 stability in HeLa cells, and thus, in light of this observation, it reassessed previous data based on MICU1 downregulation. In particular, the increase in mitochondrial Ca2+ uptake triggered by MICU1 silencing was due to concomitant destabilization of MICU2 protein. Similarly, Kamer and Mootha demonstrated that MICU1 and MICU2 act together in setting the threshold of the Ca2+ response, that MICU2 requires MICU1 for binding to the pore and that the Ca2+-binding subunits of both proteins are essential for mitochondrial Ca2+ uptake (45). In addition, disulfide bonds catalyzed by the oxidoreductase Mia40/CHCHD4 contribute to MICU1/MICU2 dimer formation and to mitochondrial Ca2+ uptake (50). However, contrary to MICU1:MCU ratio, which, as described above, determines tissue-specific mitochondrial Ca2+ uptake profiles, MICU1:MICU2 and MICU2:MCU relative abundance does not dictate the mitochondrial Ca2+ uptake properties of different tissues (47).
Specifically in skeletal muscle, MICU1 gene encodes an alternative splice variant, MICU1.1, which differs from MICU1 for a tiny four amino acid sequence encoded by an alternative exon. This short sequence is sufficient to confer MICU1.1 unique properties. Overexpression of MICU1.1 in HeLa cells causes high increases in mitochondrial Ca2+ uptake, which exceed those triggered by MICU1. While MICU2 overexpression reduces MICU1-induced mitochondrial Ca2+ uptake (46), it does not exert similar effects on MICU1.1. A detailed analysis demonstrated that MICU1.1–MICU2 heterodimer acts as gatekeeper at resting cytosolic [Ca2+], but sets the Ca2+ threshold for channel activation at a lower [Ca2+] compared to that of the MICU1–MICU2 dimer. This effect is due to the one magnitude higher affinity for Ca2+ of MICU1.1 compared to MICU1. MICU1.1 ensures prompt mitochondrial Ca2+ uptake in skeletal muscle, which is translated in more efficient ATP synthesis required for muscle contraction (51).
Finally, MCUR1 is an IMM protein which possesses two TM domains. In mammalian cells, MCUR1 acts as a positive modulator of MCU. Downregulation of MCUR1 decreases mitochondrial Ca2+ uptake, oxidative phosphorylation, and ATP production, while AMP kinase-dependent pro-survival autophagy is activated (52, 53). However, the negative effect of MCUR1 downregulation on mitochondrial Ca2+ uptake and oxidative phosphorylation has been questioned, and it has been attributed to its dramatic effect on the mitochondrial membrane potential. Rather, a role for MCUR1 as cytochrome c oxidase assembly factor was recognized, which would justify the collapse in membrane potential and mitochondrial Ca2+ uptake observed upon its suppression (54). The demonstration that MCUR1 actually plays a role in regulating MCU activity and that the effect on membrane potential is negligible, was provided by recording MCU-mediated Ca2+ currents by patch clamp electrophysiology of mitoplasts derived from cells in which MCUR1 was silenced. In experiments in which membrane potential was constantly monitored, stable knockdown of MCUR1 diminished MCU Ca2+ currents by 65%, while membrane potential was unaffected (55). In addition, both cardiac and endothelial mitochondria of tissue-specific MCUR1−/− mice showed minimal mitochondrial Ca2+ uptake upon extramitochondrial Ca2+ pulses despite constant Δψ (53). Ectopic MCUR1 co-immunoprecipitates with MCU (52) and EMRE and functions as scaffold factor required for proper complex assembly (53). However, the 480 kDa complex obtained upon digitonin permeabilization and native gel electrophoresis of MCU-Flag overexpressing HEK293 cells, contained all known MCU regulators with the exception of MCUR1 (56). Finally, MCUR1 plays species-specific functions, since in D. melanogaster it controls the Ca2+ threshold for permeability transition, independently of its effects on Ca2+ uptake rates (57). The intricated results of these studies are likely due to cell type-specific processes, and/or to methodological differences and surely deserve further investigation.
Besides the role of the abovementioned modulatory proteins, additional mechanisms control the channel activity, both transcriptionally and post-transcriptionally. Transcriptional regulation of MCU complex components is far from being completely elucidated; however, some important insights are already available. MCU transcription is controlled by the transcription factor CREB. Specifically, CREB binds to Mcu promoter, and this binding is regulated by cytosolic Ca2+. Accordingly, dysregulation of Ca2+ homeostasis, triggered by lack of expression of either InsP3R or Orai1 or STIM1, determines lower MCU expression and consequently altered mitochondria metabolism (58). In one study, in which the contribution of MCU to excitotoxicity was assessed, it was also shown that Mcu is subject to transcriptional repression by neuroprotective Ca2+ signals. The regulation mechanism requires the induction of immediate-early gene Npas4, which is dependent on nuclear Ca2+ and CaM kinase. MCU overexpression exacerbates excitotoxic cell death, while MCU silencing is protective. These findings suggest that MCU activity in neurons is regulated to prevent mitochondrial Ca2+-overload and subsequent cell death (59).
Post-transcriptionally, Mcu was reported to be a target of miR-25. In colon cancer, expression of miR-25 and Mcu are inversely correlated. Overexpression of miR-25, by downregulating Mcu, reduces mitochondrial Ca2+ uptake and protects cells from apoptotic stimuli. Vice versa, anti-miR-25 expression increases mitochondrial Ca2+ uptake and sensitizes cells to apopotic death (60). Similarly, miR-25 targets MCU in cardiomyocytes, presumably as a protective effect against oxidative damage (61). Together with miR-25, a second miRNA, i.e., miR-138, was reported to target MCU in pulmonary artery smooth muscle cells (PASMCs). MiR–MCU axis would play a regulatory role in pulmonary arterial hypertension (PAH) by controlling the rate of PASMC proliferation, migration, and sensitivity to apoptosis (62).
Post-translational modifications have also been described. MCU has been proposed to be a target of Ca2+/calmodulin-dependent protein kinase II (CamKII). CamKII would positively regulate MCU activity, and CamKII inhibition protects cardiomyocytes from I/R injury (63, 64). However, the role of CamKII in regulating MCU currents has been questioned (65). A second post-translational modification of MCU was observed to be mediated by proline-rich tyrosine kinase 2 (Pyk2) which, in cardiac cells, translocates from the cytosol to the mitochondrial matrix by activation of α1-adrenoceptor. Within the mitochondria, Pyk2 phosphorylates MCU, thus stimulating MCU oligomerization and activity (66).
Mitochondrial calcium uniporter is also regulated by mitochondria ROS production. In particular, Cys-97 in the MCU sequence has been identified as a target of mROS and undergoes S-glutathionylation. This modification promotes high-order MCU oligomerization and persistent MCU activity, which in turn triggers mitochondrial Ca2+ overload and eventually cell death (67).
Finally, MICU1 appears to be target of protein arginine methyl transferase 1. Accordingly, methylation of MICU1 would represent an additional mechanism of mitochondrial Ca2+ uptake regulation (68).
The idea that mitochondrial Ca2+ signaling regulates crucial hallmarks of cancer finds its roots in seminal studies investigating cell functions regulated by mitochondrial Ca2+ uptake. In particular, one of the first observations was that mitochondrial Ca2+ overload sensitizes cells to apoptotic stimuli, and this has been always viewed as a promising strategy to eliminate aberrant cells, including cancer cells, which would have otherwise escaped apoptotic death (3). In recent years, the role of mitochondrial Ca2+ uptake has evolved in parallel to novel hallmark of cancer (69), including the renewed interest for the deregulation of cellular energetics. On this topic, one of the conundrums that continue to puzzle researchers in the field is related to the so-called Warburg effect, i.e., the fact that cancer cells mainly rely on glycolysis-produced ATP despite normal pO2 and despite being provided with functional mitochondria, in principle capable of efficient oxidative phosphorylation. The matter is even more intricate if one considers the fact that cancer cell lines (e.g., HeLa, MDA-MB-231, and others) promptly respond to InsP3-generating stimuli with high mitochondrial Ca2+ peaks. Clearly, efficient mitochondrial Ca2+ uptake in these cells is not primarily utilized to stimulate TCA cycle and oxidative phosphorylation and should have other functions, if any. Partial answers derive from studies of mitochondria metabolism that have unraveled important roles for mROS production, regulation of NADH/NADPH, and utilization of metabolic intermediates in cancer.
To verify whether correlations between MCU complex components expression, tumor progression, and prognosis exist, different approaches have been used. In a study by Hall et al., the Breastmark algorithm, which integrates many DNA microarray experiments for which clinical data are available, was interrogated to determine the relationship between expression of MCU and MICU1 and survival (70). The results of this study indicate that combined overexpression of MCU and underexpression of MICU1 is a feature of patients with poor prognosis, while overexpression of MICU1 and of MCUb, alone or in combination, correlates with better prognosis. A similar analysis was performed by interrogating the Oncomine microarray database bank. In this case, MCU expression was analyzed in different breast cancer subtypes in comparison to normal tissue. The analysis revealed that MCU is overexpressed in breast cancer and also that invasive ductal breast cancer expresses more MCU compared to ductal carcinoma in situ. Finally, MCU expression correlates with lymph node invasion (71). When different breast cancer subtypes where compared, a higher expression of MCU was highlighted in estrogen receptor-negative patient samples, especially in basal-like breast cancers compared to luminal A and B subtypes (72). A fourth study correlated mRNA levels of MCU and related proteins (MCUb, MICU1–3, and EMRE) from the TCGA breast cancer dataset with clinical stages (73) demonstrating that MCU expression increases with tumor size and lymph node infiltration, while MCUb expression decreases. Expression of related proteins, i.e., MICU1, MICU2, MICU3, and EMRE did not show significative correlation with tumor stages. In head and neck squamous cell carcinoma, MICU1 expression correlates with the expression of EZH2, which is a negative prognostic factor. EZH2 as well as MICU1 inhibition triggers cell cycle arrest and apoptosis. The EZH2 chemical inhibitor DZNep reduced MICU1 protein levels, suggesting that MICU1 could be involved in EZH2-regulated cell-death pathway (74). Finally, in pancreatic cancer MCU and MICU1 genes undergo loss of heterozygosity (75), although whether MCU and MICU1 are oncogenes or tumor suppressor genes still needs to be clarified. Further studies are required to clarify the expression profile of MCU components in a wider array of cancers.
The metabolic and biological effects of MCU complex modulation have been investigated in different cancers, although the heterogeneity intrinsic of tumor development suggests that much still needs to be explored. In addition, analyses of the expression profiles of the uniporter components in cancer specimens have unravel additional information on the role of MCU on cancer progression.
One of the first reports investigating the role of MCU in cancer progression identified miR-25 as a cancer-related MCU-targeting miRNA family. In detail, in colon adenocarcinoma, miR-25 is overexpressed and, accordingly, MCU expression is inhibited. In agreement with the role of mitochondrial Ca2+ uptake in controlling cell survival and death, both in prostate and in colon carcinoma cells, miR-25 overexpression reduces mitochondrial Ca2+ uptake and sensitization to apoptotic stimuli, while expression of an “antagomiR” against miR-25 had a deleterious effect on cells that where stressed with an apoptotic challenge (60).
Celastrol, a triterpene extracted from the plant Tripterygium wilfordii, was reported to induce mitochondrial Ca2+ uptake and ER stress in cancer cells. Celastrol treatment induced paraptosis of these cells, a form of programmed cell-death morphologically and biochemically distinct from apoptosis (76). Intriguingly, MCU silencing partially decreased celastrol-induced cancer cell death, supporting the view of a deleterious effect of Ca2+ uptake in stress conditions.
Besides resistance to apoptosis, a fundamental step toward tumor initiation and progression is escape from senescence, a stable proliferation arrest status that exerts a protective function against many pathologies, including cancer. MCU was identified, together with ITPR2, as a novel regulator of both replicative and oncogene-induced senescence (OIS) (77). During OIS, ITPR2 activity triggers Ca2+ release from the ER, which is followed by mitochondrial Ca2+ accumulation, loss of membrane potential, increased ROS production, and senescence. Vice versa, loss of MCU enables escape from OIS.
In a particular cancer model, i.e., triple negative breast cancer (TNBC), the role of MCU has been extensively investigated both in terms of cell viability and on migration/invasion properties (Table 1). Based on the expression data reported above, suggesting increased MCU expression in basal-like breast cancer, Curry et al. hypothesized a survival advantage triggered by MCU (72). Accordingly, MCU was silenced in MDA-MB-231 cell line and cell viability and resistance to stress stimuli was measured. MCU silencing per se did not affect at all cell proliferation and viability. Also, when cells were treated with the BCL-2 inhibitor ABT-263, caspase-dependent cell death was unaffected. However, caspase-independent death triggered by treatment with the Ca2+ ionophore ionomycin was potentiated by MCU knockdown, suggesting that MCU inhibition could in principle be viewed as a therapeutical strategy against TNBC.
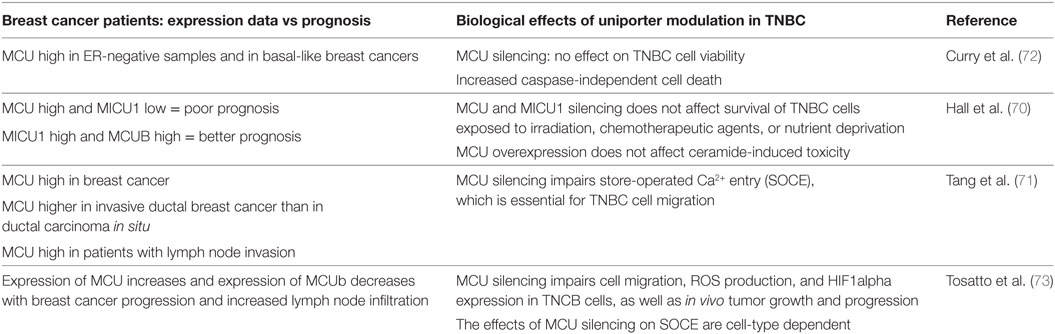
Table 1. Summary table on the role of MCU in triple negative breast cancer (TNBC).
A similar hypothesis was tested by Hall et al. (70). As reported above, their expression study convincingly correlated combined overexpression of MCU and underexpression of MICU1 with poor patient survival. Thus, MCU and MICU1 were silenced in MDA-MB-231 cells, and the effects on survival of cells exposed to irradiation, chemotherapeutic agents, or nutrient deprivation were measured. However, while cervical cancer HeLa cells and non-cancerous breast epithelial HMEC cells showed reduced survival, MDA-MB-231 cells were insensitive to changes in MCU activity. In addition, also overexpression of MCU was ineffective in altering cell response to ceramide-induced toxicity. Thus, the role of mitochondrial Ca2+ uptake in cancer cell survival depends on cancer type and on stress stimuli, not to mention the role that cancer niche and extracellular environment exert. In this context, the role of MCU is still obscure.
A different issue is the role that MCU plays in modulating motility and invasion potential of cancer cells. Two reports agreed in indicating MCU as an essential modulator of MDA-MB-231 cell migration (71, 73); however, different mechanisms were proposed. According to Tang et al., MCU silencing inhibited serum- or thapsigargin-induced store-operated Ca2+ entry (SOCE) which, in turn, was required for proper cell migration. Tosatto et al. confirmed the data that MCU is required for MDA-MB-231 migration and extended this observation to other triple-negative breast cancer cell lines. However, despite the consistent role on cell motility, the effect of MCU silencing on SOCE was cell line dependent, indicating that different mechanisms are involved, likely directly dependent on the strong reduction in mitochondrial Ca2+ uptake, rather than on a secondary effect on global Ca2+ homeostasis. MCU inhibition caused a strong reduction in mROS production and HIF-1α expression. In addition, restoring HIF-1α levels was sufficient to overcome the decrease in motility. The essential role of MCU in TNBC progression was demonstrated by in vivo experiments in which MDA-MB-231 cells were injected into the fat pad of immune-deficient SCID mice. Deletion of MCU markedly retarded primary tumor formation. In addition, at equal primary tumor size, absence of MCU impaired lung and lymph node colonization (73).
These studies indicate that the MCU complex plays different roles in different cancer types and stages. Research is still needed to extend the analysis to in vivo models and to additional cancers and to clarify whether mitochondrial Ca2+ uptake impinges on the tumor microenvironment.
An important amount of work has been done to disclose the pathophysiological role of MCU in different tissues (Figure 2).
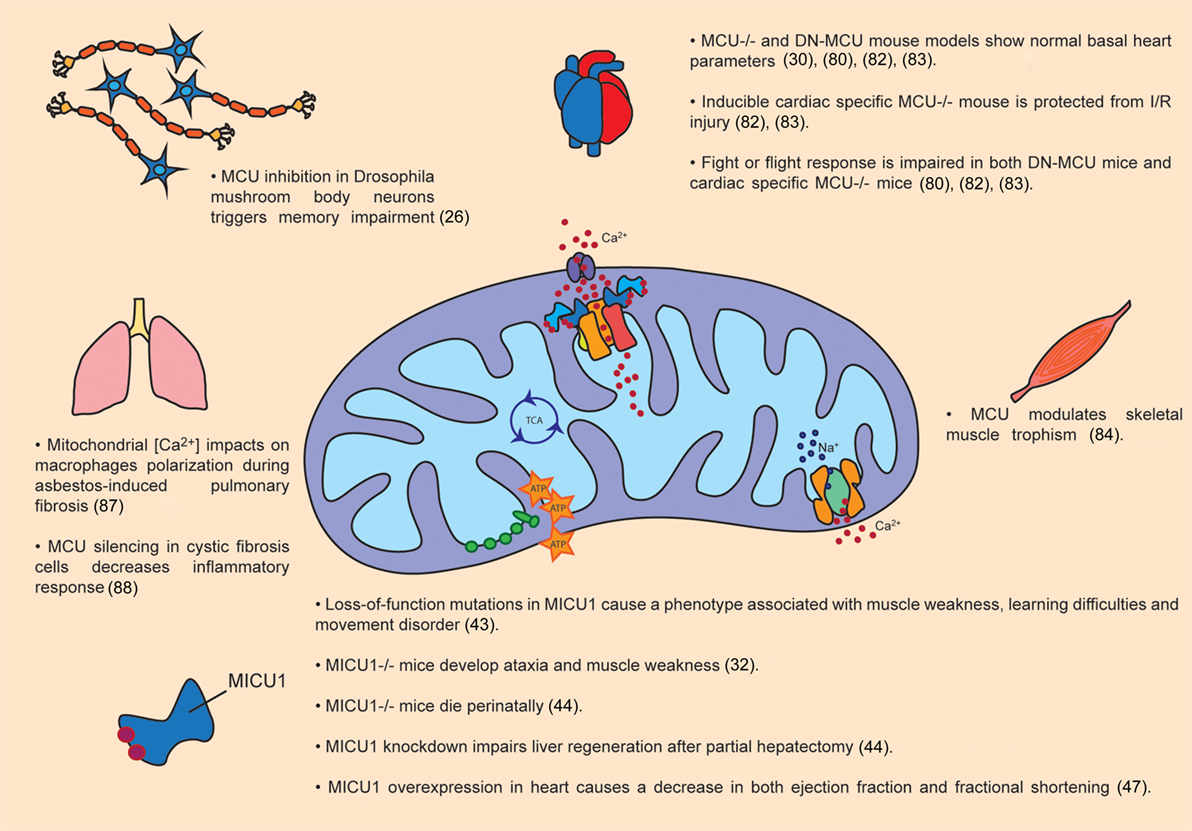
Figure 2. Pathophysiological roles of MCU complex. The figure summarizes the main effects of MCU activity modulation in organs and tissues, both in animal models as well as in patients.
On this issue, one of the first studies after the identification of MCU was carried out in neonatal cardiomyocytes ex vivo, demonstrating that MCU expression regulates the buffering of cytoplasmic Ca2+ during systole. In detail, cytoplasmic Ca2+ peaks are increased or reduced by MCU silencing or overexpression, respectively (78). However, the first in vivo analysis of MCU−/− hearts gave unexpected results that are still debated. MCU−/− hearts show no sign of protection against I/R injury (30). Interestingly, while wild-type hearts were protected from I/R injury when treated with CsA, which inhibits Ca2+-dependent cell death mediated by the opening of mPTP, MCU−/− hearts lacked CsA protection. This result suggests that a Ca2+-independent death pathway occurs in the absence of MCU. Experiments on isolated heart mitochondria showed impaired mitochondrial Ca2+ accumulation and reduced matrix Ca2+ levels in MCU−/− strain (79), consistent with the data previously reported on skeletal muscle isolated mitochondria (30). However, basal ATP levels were similar between MCU−/− and wild-type hearts, suggestive of preserved basal metabolic function. In addition, an accurate study of heart morphology by echocardiographic analysis of 12- and 20-month-old mice revealed similar stroke volume, ventricular volumes, and wall thickness between MCU−/− and wild-type controls, indicating that MCU−/− mice maintain basal cardiac function over at least 20 months of life (79). Upon physiological stress induced by isoproterenol stimulation, left ventricular cardiac output and heart rate were undistinguishable from those of wild-type controls even though MCU−/− cardiomyocytes did not accumulate Ca2+. Also upon chronic stress, induced by transverse aortic constriction (TAC), MCU−/− and wild-type genotypes display similar decline in ejection fraction, fractional shortening, hypertrophy and fibrosis.
Additional studies were carried out in a transgenic mouse model, expressing a DN-MCU (80, 81). As expected, the heart mitochondria from DN-MCU mice showed loss of mitochondrial Ca2+ accumulation (80), although DN-MCU does not completely abolish mitochondrial Ca2+ uptake when expressed in HeLa cells (16). DN-MCU mice showed hearts with normal chamber size and heart beat. Increased heart rate triggered by isoproterenol treatment requires enhanced ATP production necessary to support SERCA activity and appropriate Ca2+ load of the sarcoplasmic reticulum in the SAN (sinus atrial node) cells. MCU inhibition impaired fight or flight heart rate by decreasing ATP levels, and dialysis of ATP was sufficient to rescue the phenotype in cardiac pacemaking cells (80). Enhanced oxygen consumption was a peculiar feature of DN-MCU perfused hearts, while it was not observed in isolated mitochondria or permeabilized cardiomyocytes of the same animals, suggesting that the increased energetic demand is related to extramitochondrial Ca2+ homeostasis. In addition, DN-MCU ventricular cardiomyocytes exhibited increased cytosolic [Ca2+], in line with the Ca2+ buffering effect of mitochondria, which was partially overturned by ATP dialysis (81). Consistent with results obtained in MCU−/− mouse (30), DN-MCU expression was not protective against myocardial death induced by I/R injury, despite maintenance of ΔΨm and decreased ROS production (81).
In both mouse models discussed above, embryonic adaptations to MCU modulation could not be excluded. To overcome this issue, in 2015 a new mouse model was generated, with two LoxP sites flanking exons 5 and 6 of MCU gene. These animals were crossed with mice expressing a tamoxifen-inducible Cre recombinase driven by a cardiomyocyte-specific promoter (α-myosin heavy chain promoter). MCU gene deletion was induced in adult mice, and cardiac function was evaluated (82, 83). Similarly to DN-MCU and MCU total knockout mice, basal cardiac function and heart morphology were normal. On the other hand, studies of inducible cardiac-specific MCU−/− mice confirmed the impairment in the fight or flight response. Cardiomyocytes derived from these mice had normal respiration rate in basal conditions, while decreased oxygen consumption rate after β-adrenergic stimulation suggesting a MCU pivotal role during increase heart workload. However, no differences were observed in fractional shortening, cardiomyocytes area and heart weight upon TAC. Finally, as opposed to constitutive MCU−/− mice and DN-MCU animals, inducible MCU ablation strongly protected hearts from I/R-induced cell death.
Besides heart, another tissue strictly dependent on oxidative metabolism is skeletal muscle. Skeletal muscle is the most affected tissue in constitutive MCU−/− mice although at very mild levels. Skeletal muscle mitochondrial matrix [Ca2+] was reduced compared to wild-type controls, although not completely abolished. A decrease maximal oxygen consumption rate was detected. In addition, PDH activity was impaired in agreement with the pivotal role of mitochondrial Ca2+ accumulation in the control of ATP production. Finally, MCU−/− animals show defective maximal power output generation. Subsequently, to discriminate the compensatory effects occurring during embryonic development, MCU modulation was achieved after birth by means of adeno-associated viral vectors for both overexpression and silencing (84). Modulation of mitochondrial Ca2+ accumulation was shown to contribute to skeletal muscle trophism. Indeed, MCU overexpression and downregulation trigger muscle hypertrophy and atrophy, respectively, both during postnatal development and in adulthood. Most importantly, MCU overexpression protects from denervation-induced muscle atrophy caused by sciatic nerve excision suggesting a possible therapeutic role of mitochondrial Ca2+ uptake in muscle atrophy. Surprisingly, these effects are independent from the control of aerobic metabolism as demonstrated by different evidence. First, PDH activity, although defective in MCU silenced muscles, was unaffected in MCU overexpressing muscles. Second, hypertrophy was comparable in both oxidative and glycolytic muscles and, finally, analyses of aerobic metabolism revealed no major alterations. Conversely, the control of skeletal muscle mass by mitochondrial Ca2+ modulation is due to the role of two major hypertrophic pathways of skeletal muscle, PGC-1α4 and IGF1-AKT/PKB. Taken together, these results demonstrate the existence of a Ca2+-dependent mitochondria-to-nucleus signaling route that clearly links organelle physiology to the control of muscle mass. As mentioned above, loss-of function mutations in MICU1 genes cause a disease which includes neuromuscular defects and learning difficulties (43), and MICU1 deletion in mice is either lethal (44) or causing similar phenotype to the human disease (32). Finally, a recent work carried out by Zampieri and coworkers reported increased skeletal muscle MCU protein levels in human seniors subjected to muscle training, suggesting that MCU represents a potential pharmacological target to counteract sarcopenia (85).
Beside striated muscles, the role of MCU has been studied also in other organs in which mitochondria play a crucial role for tissue-specific functions. One case is represented by endocrine pancreas, specifically β-cells. Soon after MCU discovery, experiments carried on in pancreatic β-cells demonstrated that MCU-mediated mitochondrial Ca2+ uptake is essential for cell depolarization-induced increase in ATP/ADP ratio (86). In addition, culture conditions mimicking diabetic milieu delayed mitochondrial Ca2+ uptake. Thus, MCU is an essential component of glucose sensing by pancreatic β-cells.
In a totally different context, a recent article reported evidence that mitochondrial [Ca2+] could have an impact on macrophage polarization (87). Gu and coworkers demonstrated that MCU+/- mice are protected from asbestos-induced pulmonary fibrosis. In detail, macrophages of MCU+/- mice showed attenuated profibrotic polarization after asbestos exposure together with a decrease in ATP production. In addition, histological analysis revealed normal lung architecture without collagen deposition in MCU+/- mice. These results suggest that the modulation of mitochondrial Ca2+ accumulation could be a promising therapeutic target for human fibrosis.
Additional demonstration of the pivotal role of mitochondrial Ca2+ uptake in pathophysiology is described by Rimessi and coworkers (88). This study analyzes the Pseudomonas aeruginosa-induced inflammatory process that takes place in cystic fibrosis cells. When inflammation is triggered in a cystic fibrosis cell line model, MCU silencing causes a decrease in inflammasome activation, indicating that mitochondrial Ca2+ accumulation is a potential therapeutic target.
Finally, MCU is a target of miR-25, and miRNA-dependent MCU modulation has consequences on different physiopathological settings, as detailed in the Sections “MCU Activity Modulation” and “MCU and Cancer” (60, 61). Also miR-138 has been reported to play a regulatory role on MCU in the context of PAH (62).
The seminal discoveries of cbara1 as the gene encoding MICU1, and of ccdc109a as the one encoding MCU, followed by the identification of the other components of the uniporter, determined the possibility of conducting genetic and molecular studies on mitochondrial Ca2+ uptake mechanism and significance. In a simplified view, so far research has progressed in different directions to define: (a) the identity and role of uniporter channel components and interacting subunits, and their mutual regulation; (b) the transcriptional and posttranscriptional mechanisms that contribute to channel activity control; and (c) the pathophysiological consequences of dysregulated mitochondrial Ca2+ signaling. For any of these topics, impressive progresses have been achieved in a relatively short time. However, many questions are still unanswered. First, work is still needed to better clarify how the different channel subunits and regulatory components functionally interact with each other and to determine cell-type specificity of these mechanisms. Second, the regulation of uniporter components expression and activity by transcriptional and posttranscriptional events is a still largely unexplored field. Third, the pathophysiological role of MCU in different tissues is still unclear. Specifically on tumor development and progression, the role and significance of mitochondrial Ca2+ signaling has been studied only in few cancer types, and nothing is known on the effects of MCU activity in the tumor niche. In addition, although patient-derived samples have been analyzed for the expression levels of MCU components and correlation data between expression and prognosis have been obtained, in vivo tumorigenesis experiments aimed to determine the significance of aberrant mitochondrial Ca2+ uptake at different cancer stages have been rarely performed. Research in the next years will uncover these and others aspects of mitochondrial Ca2+ signaling.
CM, GG, and RR wrote the manuscript. GG prepared figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research in Rizzuto laboratory is supported by fundings from the Italian Telethon Foundation, the Italian Association for Cancer Research (AIRC), the Italian Ministry of Education, University, and Research, the French Muscular Dystrophy Association (AFM-Téléthon), the National Institutes of Health (NIH).
1. Singleterry J, Sreedhar A, Zhao Y. Components of cancer metabolism and therapeutic interventions. Mitochondrion (2014) 17:50–5. doi:10.1016/j.mito.2014.05.010
2. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) (2010) 7:7. doi:10.1186/1743-7075-7-7
3. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13:566–78. doi:10.1038/nrm3412
4. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature (2004) 427:360–4. doi:10.1038/nature02246
5. Mertins B, Psakis G, Essen L-O. Voltage-dependent anion channels: the wizard of the mitochondrial outer membrane. Biol Chem (2014) 395:1435–42. doi:10.1515/hsz-2014-0203
6. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi:10.1083/jcb.200205091
7. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2012) 19:267–73. doi:10.1038/cdd.2011.92
8. Shoshan-Barmatz V, De S, Meir A. The mitochondrial voltage-dependent anion channel 1, Ca2+ transport, apoptosis, and their regulation. Front Oncol (2017) 7:60. doi:10.3389/fonc.2017.00060
9. Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta (2016) 1863:2498–502. doi:10.1016/j.bbamcr.2016.01.019
10. Naghdi S, Hajnóczky G. VDAC2-specific cellular functions and the underlying structure. Biochim Biophys Acta (2016) 1863:2503–14. doi:10.1016/j.bbamcr.2016.04.020
11. Reina S, Guarino F, Magrì A, De Pinto V. VDAC3 as a potential marker of mitochondrial status is involved in cancer and pathology. Front Oncol (2016) 6:264. doi:10.3389/fonc.2016.00264
12. Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A (2010) 107:436–41. doi:10.1073/pnas.0908099107
13. Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, et al. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell (2010) 38:280–90. doi:10.1016/j.molcel.2010.04.003
14. Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell (2010) 39:121–32. doi:10.1016/j.molcel.2010.06.029
15. Fieni F, Bae Lee S, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun (2012) 3:1317. doi:10.1038/ncomms2325
16. De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
17. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
18. Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature (2010) 467:291–6. doi:10.1038/nature09358
19. Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol (2012) 30:1143–8. doi:10.1038/nbt.2375
20. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J (2013) 32:2362–76. doi:10.1038/emboj.2013.157
21. Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep (2015) 16:1318–33. doi:10.15252/embr.201540436
22. Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, et al. Architecture of the mitochondrial calcium uniporter. Nature (2016) 533(7602):269–73. doi:10.1038/nature17656
23. Cao C, Wang S, Cui T, Su X-C, Chou JJ. Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A (2017) 114(14):E2846–51. doi:10.1073/pnas.1620316114
24. Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun (2013) 4:2865. doi:10.1038/ncomms3865
25. Prudent J, Popgeorgiev N, Bonneau B, Thibaut J, Gadet R, Lopez J, et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat Commun (2013) 4:2330. doi:10.1038/ncomms3330
26. Drago I, Davis RL. Inhibiting the mitochondrial calcium uniporter during development impairs memory in adult Drosophila. Cell Rep (2016) 16:2763–76. doi:10.1016/j.celrep.2016.08.017
27. Xu S, Chisholm AD. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell (2014) 31:48–60. doi:10.1016/j.devcel.2014.08.002
28. Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun (2014) 449:384–5. doi:10.1016/j.bbrc.2014.04.144
29. Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. High-throughput discovery of novel developmental phenotypes. Nature (2016) 537:508–14. doi:10.1038/nature19356
30. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol (2013) 15:1464–72. doi:10.1038/ncb2868
31. Sancak Y, Markhard ALL, Kitami T, Kovacs-Bogdan E, Kamer KJJ, Udeshi NDD, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 342:1379–82. doi:10.1126/science.1242993
32. Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep (2016) 16:1561–73. doi:10.1016/j.celrep.2016.07.011
33. Kovacs-Bogdan E, Sancak Y, Kamer KJJ, Plovanich M, Jambhekar A, Huber RJJ, et al. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A (2014) 111:8985–90. doi:10.1073/pnas.1400514111
34. Vais H, Mallilankaraman K, Mak D-OD, Hoff H, Payne R, Tanis JE, et al. EMRE is a matrix Ca(2+) sensor that governs gatekeeping of the mitochondrial Ca(2+) uniporter. Cell Rep (2016) 14:403–10. doi:10.1016/j.celrep.2015.12.054
35. Yamamoto T, Yamagoshi R, Harada K, Kawano M, Minami N, Ido Y, et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim Biophys Acta (2016) 1857:831–9. doi:10.1016/j.bbabio.2016.03.019
36. Tsai M-F, Phillips CB, Ranaghan M, Tsai C-W, Wu Y, Williams C, et al. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife (2016) 5:e15545. doi:10.7554/eLife.15545
37. König T, Tröder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, et al. The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol Cell (2016) 64:148–62. doi:10.1016/j.molcel.2016.08.020
38. Tsai C-W, Wu Y, Pao P-C, Phillips CB, Williams C, Miller C, et al. Proteolytic control of the mitochondrial calcium uniporter complex. Proc Natl Acad Sci U S A (2017) 114(17):4388–93. doi:10.1073/pnas.1702938114
39. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell (2012) 151:630–44. doi:10.1016/j.cell.2012.10.011
40. Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep (2013) 5:1576–88. doi:10.1016/j.celrep.2013.11.026
41. Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab (2013) 17:976–87. doi:10.1016/j.cmet.2013.04.020
42. Wang L, Yang X, Li S, Wang Z, Liu Y, Feng J, et al. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J (2014) 33:2014. doi:10.1002/embj.201386523
43. Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs A-M, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet (2013) 46:188–93. doi:10.1038/ng.2851
44. Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat Commun (2016) 7:10955. doi:10.1038/ncomms10955
45. Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep (2014) 15:299–307. doi:10.1002/embr.201337946
46. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell (2014) 53:726–37. doi:10.1016/j.molcel.2014.01.013
47. Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep (2017) 18:2291–300. doi:10.1016/j.celrep.2017.02.032
48. M’Angale PG, Staveley BE. Inhibition of mitochondrial calcium uptake 1 in Drosophila neurons. Genet Mol Res (2017) 16(1). doi:10.4238/gmr16019436
49. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One (2013) 8:e55785. doi:10.1371/journal.pone.0055785
50. Petrungaro C, Zimmermann KM, Küttner V, Fischer M, Dengjel J, Bogeski I, et al. The Ca(2+)-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab (2015) 22:721–33. doi:10.1016/j.cmet.2015.08.019
51. Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol Cell (2016) 64:760–73. doi:10.1016/j.molcel.2016.10.001
52. Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol (2012) 14:1336–43. doi:10.1038/ncb2622
53. Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep (2016) 15:1673–85. doi:10.1016/j.celrep.2016.04.050
54. Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab (2015) 21:109–16. doi:10.1016/j.cmet.2014.12.004
55. Vais H, Tanis JE, Müller M, Payne R, Mallilankaraman K, Foskett JK. MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab (2015) 22:533–5. doi:10.1016/j.cmet.2015.09.015
56. Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 30:1143–8. doi:10.1126/science.1242993
57. Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A (2016) 113:E1872–80. doi:10.1073/pnas.1602264113
58. Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal (2015) 8:1–15. doi:10.1126/scisignal.2005673
59. Qiu J, Tan Y-W, Hagenston AM, Martel M-A, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun (2013) 4:2034. doi:10.1038/ncomms3034
60. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol (2013) 23:58–63. doi:10.1016/j.cub.2012.11.026
61. Pan L, Huang B-J, Ma X-E, Wang S-Y, Feng J, Lv F, et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int J Mol Sci (2015) 16:5420–33. doi:10.3390/ijms16035420
62. Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, et al. MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med (2017) 195:515–29. doi:10.1164/rccm.201604-0814OC
63. Joiner MA, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. CaMKII determines mitochondrial stress responses in heart. Nature (2012) 491:269–73. doi:10.1038/nature11444
64. Joiner MA, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. Joiner et al. reply. Nature (2014) 513:E3–3. doi:10.1038/nature13627
65. Fieni F, Johnson DE, Hudmon A, Kirichok Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature (2014) 513:E1–2. doi:10.1038/nature13626
66. O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal (2014) 21:863–79. doi:10.1089/ars.2013.5394
67. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell (2017) 65:1014–1028.e7. doi:10.1016/j.molcel.2017.01.032
68. Madreiter-Sokolowski CT, Klec C, Parichatikanond W, Stryeck S, Gottschalk B, Pulido S, et al. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca2+ uptake in immortalized cells. Nat Commun (2016) 7:12897. doi:10.1038/ncomms12897
69. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
70. Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One (2014) 9:e96866. doi:10.1371/journal.pone.0096866
71. Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, et al. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun (2015) 458:186–93. doi:10.1016/j.bbrc.2015.01.092
72. Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun (2013) 434:695–700. doi:10.1016/j.bbrc.2013.04.015
73. Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med (2016) 8:569–85. doi:10.15252/emmm.201606255
74. Zhou X, Ren Y, Kong L, Cai G, Sun S, Song W, et al. Targeting EZH2 regulates tumor growth and apoptosis through modulating mitochondria dependent cell-death pathway in HNSCC. Oncotarget (2015) 6:33720–32. doi:10.18632/oncotarget.5606
75. Long J, Zhang Z-B, Liu Z, Xu Y-H, Ge C-L. Loss of heterozygosity at the calcium regulation gene locus on chromosome 10q in human pancreatic cancer. Asian Pac J Cancer Prev (2015) 16:2489–93. doi:10.7314/APJCP.2015.16.6.2489
76. Yoon MJ, Lee AR, Jeong SA, Kim Y-S, Kim JY, Kwon Y-J, et al. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget (2014) 5:6816–31. doi:10.18632/oncotarget.2256
77. Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calvé B, Augert A, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun (2014) 5:3792. doi:10.1038/ncomms4792
78. Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A (2012) 109:12986–91. doi:10.1073/pnas.1210718109
79. Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol (2015) 85:178–82. doi:10.1016/j.yjmcc.2015.05.022
80. Wu Y, Rasmussen TP, Koval OM, Joiner M-LA, Hall DD, Chen B, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun (2015) 6:6081. doi:10.1038/ncomms7081
81. Rasmussen TP, Wu Y, Joiner MA, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A (2015) 112:9129–34. doi:10.1073/pnas.1504705112
82. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep (2015) 12:15–22. doi:10.1016/j.celrep.2015.06.002
83. Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep (2015) 12:23–34. doi:10.1016/j.celrep.2015.06.017
84. Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep (2015) 10:1269–79. doi:10.1016/j.celrep.2015.01.056
85. Zampieri S, Mammucari C, Romanello V, Barberi L, Pietrangelo L, Fusella A, et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol Rep (2016) 4:e13005. doi:10.14814/phy2.13005
86. Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, et al. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One (2012) 7:e39722. doi:10.1371/journal.pone.0039722
87. Gu L, Larson-Casey JL, Carter AB. Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J (2017). doi:10.1096/fj.201601371R
Keywords: mitochondrial calcium uniporter, mitochondrial calcium uniporter pathophysiology, mouse models, breast cancer, mitochondrial calcium uniporter regulators
Citation: Mammucari C, Gherardi G and Rizzuto R (2017) Structure, Activity Regulation, and Role of the Mitochondrial Calcium Uniporter in Health and Disease. Front. Oncol. 7:139. doi: 10.3389/fonc.2017.00139
Received: 12 May 2017; Accepted: 19 June 2017;
Published: 10 July 2017
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Varda Shoshan-Barmatz, Ben-Gurion University of the Negev, IsraelCopyright: © 2017 Mammucari, Gherardi and Rizzuto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Mammucari, Y3Jpc3RpbmEubWFtbXVjYXJpQHVuaXBkLml0;
Rosario Rizzuto, cm9zYXJpby5yaXp6dXRvQHVuaXBkLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.