 Eric Eidelman
Eric Eidelman Jeffrey Twum-Ampofo1
Jeffrey Twum-Ampofo1 Jamal Ansari
Jamal Ansari Mohummad Minhaj Siddiqui
Mohummad Minhaj Siddiqui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 June 2017
Sec. Cancer Molecular Targets and Therapeutics
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00131
This article is part of the Research Topic Cancer Metabolism: Molecular Targeting and Implications for Therapy View all 11 articles
Prostate cancer is the most common non-cutaneous cancer in men in the United States. Cancer metabolism has emerged as a contemporary topic of great interest for improved mechanistic understanding of tumorigenesis. Prostate cancer is a disease model of great interest from a metabolic perspective. Prostatic tissue exhibits unique metabolic activity under baseline conditions. Benign prostate cells accumulate zinc, and this excess zinc inhibits citrate oxidation and metabolism within the citric acid cycle, effectively resulting in citrate production. Malignant cells, however, actively oxidize citrate and resume more typical citric acid cycle function. Of further interest, prostate cancer does not exhibit the Warburg effect, an increase in glucose uptake, seen in many other cancers. These cellular metabolic differences and others are of clinical interest as they present a variety of potential therapeutic targets. Furthermore, understanding of the metabolic profile differences between benign prostate versus low- and high-grade prostate cancers also represents an avenue to better understand cancer progression and potentially develop new diagnostic testing. In this paper, we review the current state of knowledge on the metabolic phenotypes of prostate cancer.
Prostate cancer is one of the most commonly diagnosed cancers among men, particularly in the developed world. It represents not only a major factor in male morbidity and mortality but also an economic burden on the population (1, 2). Prostate cancer exhibits heterogenic properties among patients; however, there remain generalizable phenotypes associated with the majority of prostate cancers, including certain metabolic alterations (3, 4). With the rise of the field of study dedicated to cancer metabolomics, an effort has been made to evaluate prostate cancer through analysis of cellular metabolism in an effort to better understand the tumor pathogenesis as well as to create potential metabolically targeted treatments.
In addition to targeted treatments, the study of metabolism of cancer represents a new avenue for diagnostics (5). The tissue-specific activity of prostate epithelial cells has long been utilized in urologic oncology. A widely used prostate cancer marker is the prostate-specific antigen (PSA), which has been shown to be elevated in many men with prostatic disease (6). PSA testing is still used as an early screening method to determine the likelihood and progressive phenotype of prostate cancer. Recently, this method has undergone some scrutiny as it may lead to aggressive overtreatment in patients who may have been treated sufficiently with active surveillance (7). Improved tools for better risk stratification of prostate cancer patients are needed. This need emphasizes the importance of better understanding the metabolic mechanisms of prostate cancer (8). By better understanding the metabolism of prostate cancer, it may be possible to elucidate a metabolic biomarker whose levels may help to diagnose prostate cancer. This paper will discuss the current state of knowledge regarding prostate cancer metabolism and how it might relate to future treatment, screening, and diagnostic criteria.
In order to understand the metabolic alterations of prostate cancer, it is important to understand a key phenotype in benign prostate cells. Healthy prostate epithelial cells exhibit a highly specialized behavior regarding their metabolic pathways. Typically, cells rely on citrate oxidation as a key step in the Krebs cycle for the progression of aerobic respiration (9). However, prostate cells, notably the epithelial cells in the peripheral zone of the prostate are programmed to produce and not oxidize citrate (10). The citrate is subsequently secreted as a component of semen. The specialization of peripheral zone epithelium is of clinical interest as it is within this zone that prostate cancer begins (11). This unique process of citrate production is accomplished by another feature of prostate epithelial cells, the ability to accumulate large concentrations of zinc. Research on this pathway has indicated that high concentrations of zinc have an inhibitory effect on m-aconitase, the enzyme that catalyzes the oxidation of citrate within the Krebs cycle (Figure 1) (3). This zinc accumulation in benign prostatic epithelium is accomplished by increased amount of zinc transporter ZIP1 in this tissue (12). By accumulating citrate, normal prostate epithelial cells appear to halt the Krebs cycle and therefore act very different to the majority of cells in the body in the production of ATP.
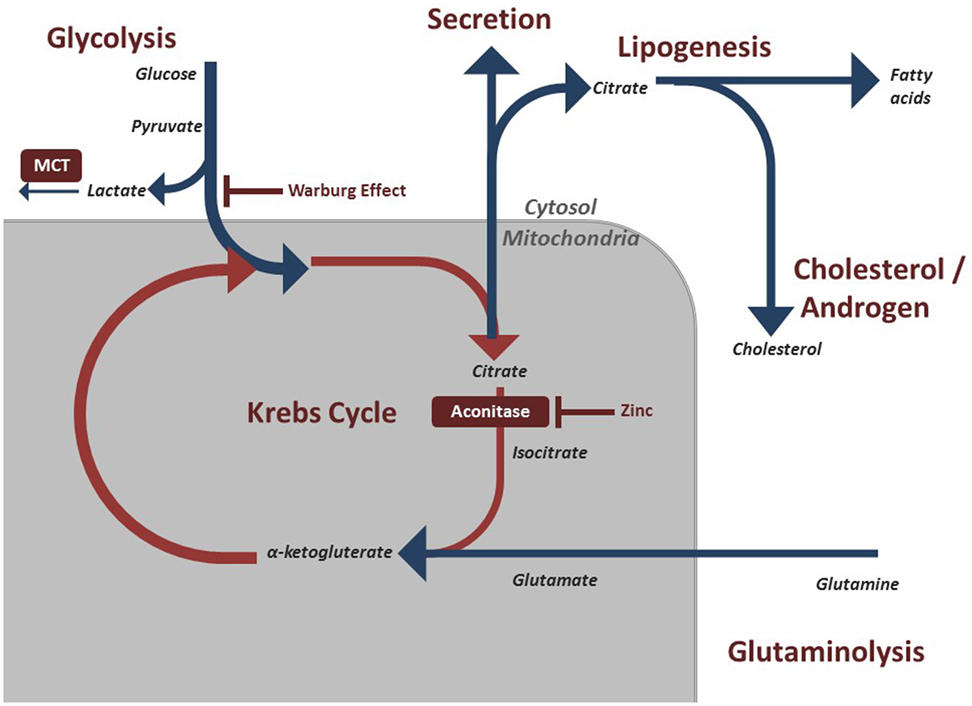
Figure 1. Regulated metabolic pathways involved in energy production and maintenance of prostatic cells. These pathways are often modified in malignant prostate cells to preserve cell division and growth.
Metabolic modifications allow cancer cells to accommodate the massive energy requirement for rapid proliferation. Most solid tumors experience the Warburg effect, described in the early to mid 1900s by Otto Warburg, whereby malignant cells shift their dominant ATP producing pathway away from oxidative phosphorylation to aerobic glycolysis (13). This results in lactic acid fermentation (Figure 1) (14). This switch means that the cancerous cells display a significant increase in the rate of glucose uptake in order to meet the energetic need (13). Clinically, F-deoxyglucose positron emission tomography (FDG PET) scans take advantage of this trait as infused radiolabeled glucose is absorbed by tumor cells at a much higher rate, which can then be detected on imaging. The exact explanation for this effect has been debated, but one possibility is that avoiding oxidative phosphorylation evades potential pathways and breakdown products that may result in mitochondrial stress that could halt cell division (13). Furthermore, incomplete breakdown of products such as glucose allows for rapid generation of cellular building blocks that are needed for proliferation. Complete oxidative phosphorylation, by contrast, results in breakdown of the energy source into carbon dioxide, which does not contribute to the biomass of the tumor. By relying on aerobic glycolysis, these cells can continue to divide rapidly and increase in biomass (15).
While a large number of solid tumor cells adhere to this Warburg effect, prostate cancer has a markedly different phenotype. As noted above, benign prostate cells evade oxidative phosphorylation at baseline. It has been shown that early prostate cancers rely on lipids and other energetic molecules for energy production and not on aerobic respiration (16, 17). Therefore, the Warburg effect does not hold consistent in the pathogenesis of prostate cancer, as these cells do not have the increased glucose uptake (18). Clinically this is relevant, as these cancers will not appear on FDG PET scans. It is only in the late stage with numerous mutation events that prostate cancer will begin exhibiting the Warburg effect and have a high glucose uptake. The exact nature of bioenergetics in early prostate cancer cells is still being worked out, and work is being done on specific pathways as discussed below.
As noted above, the hallmark of the healthy prostate epithelial cell is the zinc accumulating, citrate synthesizing phenotype. However, a well-noted shift occurs within malignant prostate cells. Prostate cancer cells reverse this phenotype and adopt a zinc wasting, citrate oxidizing phenotype, thereby representing a major shift in energy metabolism (19). This shift allows these cells to utilize the Krebs cycle and subsequent oxidative phosphorylation (Figure 1). It has long been identified that prostate cancer does not conform to the standard Warburg effect seen in most cancers. Unlike most cancer cells that resort to aerobic glycolysis, prostate cancer cells exhibit a higher level of citric acid cycle activity compared to benign cells (10).
Accumulation of zinc may also lead to a mitochondrial apoptotic phenotype within prostate cells. Therefore, malignant cells preferentially decrease the amount of stored zinc in order to avoid cell death (20). This reduction in zinc in malignant cells may be due to alterations in a zinc transporter ZIP1 (19). Altering these transporters does not allow the concentration of zinc to reach levels sufficient to inhibit m-aconitase. Thus, it has been argued that the altered zinc and citrate phenotype in prostate cancer has a dual role. Not only does the return to citrate oxidization increase the energy available for cell growth but also by avoiding an increased concentration of zinc these cells avoid apoptotic regulation (3). Further research is being done to better understand how dietary zinc can influence prostate malignancy as well as how the zinc/citrate metabolic path can be targeted therapeutically.
As noted above, later stage prostate cancer cells do begin utilizing aerobic glycolysis leading to an increased intracellular concentration of lactic acid. This build up of lactic acid is toxic so to compensate, cancer cells express monocarboxylate transporters (MCTs) to reduce intracellular lactate levels (Figure 1). Prostate cancer cells have shown to be heterogenic in the level of MCT expression between patients. A more avidly MCT-producing phenotype has been correlated with a more aggressive cancer and worse prognosis (21). By blocking MCT activity, cells may accumulate toxic metabolic products at a faster rate (21). Thus, the expression of lactate shuttles within prostate cancer represents a potential future target as well as a potential diagnostic and prognostic indicator (22).
As the building blocks of proteins and key intermediates, amino acids and their metabolism have been of an increasing interest in the field of cancer. Differential utilization of particular amino acids has been observed for promotion of cancer cell growth. Furthermore, the specific utilization of amino acids by tumor cells may be useful in determining the aggressiveness of the disease (23). Research into the amino acids glutamine and arginine has become an active area in the field of prostate cancer.
Glutamine is involved in numerous energetic pathways within cells. A key pathway within benign cells is the transformation into glutamate by glutaminase. This glutamate can transform into alpha-ketoglutarate that is then shuttled to the Krebs cycle (Figure 1).
Glutamine uptake and use has been shown to be elevated in multiple cancers including prostate cancer primarily for de novo lipid synthesis, which can contribute to such processes as building cell walls. Under hypoxic conditions commonly seen in tumors, reductive glutamine metabolism by the protein isocitrate dehydrogenase 1 has been shown to promote lipid formation. However, this mechanism is not fully understood (24). Lipids are a key fuel source of prostate cancer cells, particularly early cancer cells.
Glutaminolysis is a mechanism that cancer cells utilize to produce ATP. In prostate cancer cells, glutaminolysis is predominantly performed by the enzyme glutaminase-1. Upregulation of glutaminase-1 has been shown within prostate cancer (25, 26). Therefore, inhibiting the action of glutaminase-1 may theoretically dysregulate the glutamine-based energy production of prostate cancer cells. Indeed, an in vitro study has shown that inhibiting glutamine uptake limits the proliferation and invasiveness of prostate cancer (27). As such, interest has risen in the possibility of blocking glutamine uptake clinically.
Arginine is a non-essential amino acid that has been shown to have a variety of roles in the growth of normal cells. As an amino acid, it has an important role in the synthesis of proteins (28). Arginine can be converted to both proline and glutamine. It also has a unique function in the generation of nitric oxide (NO), a compound with a wide array of cellular effects. NO is believed to have an important role in various processes within cancerous cells; however, the exact role of NO in cancer is not well understood at this time (28). A variety of studies have shown that arginine plays an important role in the maintenance of a malignant phenotype in a variety of cancers, including prostate cancer.
While the exact impact that arginine expression has on cancer cells is not well understood, studies have shown that highly available supply of arginine is needed to continue the growth of prostate cancer (29). As such, it has become a potential therapeutic target as deprivation has been shown to lead to death of cancerous cells in multiple studies (28). Arginine can be synthesized from ornithine, a key component of the urea cycle. Ornithine and carbamoyl phosphate are catalyzed by ornithine carbamoyl transferase into citrulline. Citrulline is subsequently transformed into arginine by arginosuccinate synthase. In vitro, common prostate cancer cell lines have shown to produce a lower amount of ornithine carbamoyl transferase (28). They have subsequently been shown to be sensitive to treatment by recombinant human arginase (30). Arginine deiminase has also become a popular approach for treatment for prostate cancer, and in vitro studies have shown that arginine deiminase can kill susceptible cancer cells by starving the cells of arginine (31). A combination of arginine deprivation with arginine deiminase is being studied as combo therapy in cancer patients in Phase II clinical trials (32).
Many cancers consume lipids in order to produce energy and avoid the citric acid cycle. Prostate cancer cells often utilize lipids derived from androgens through the expression of an androgen receptor (33). However, these cells can also utilize de novo lipid synthesis to produce fatty acids in order to obtain energy. This shift to a lipid-producing phenotype is a key turning point in the progression of prostate cancer. The de novo lipid producers have ability to produce the key energetic molecules for growth without the regulation of androgens (Figure 1). Clinically, this is problematic as it represents a disease that is unresponsive to androgen deprivation therapy, known as castration-resistant prostate cancer (34).
Research has shown that certain prostate cancer cells overexpress certain markers that are key in the ability to produce de novo lipids (35). These include fatty acid synthase (FASN), sterol regulatory element binding protein 1 (SREBP1), and steroyl CoA desaturase among others. Steroyl CoA desaturase is a key enzyme in the formation of monounsaturated fatty acids from larger saturated fatty acids. In some animal models, steroyl CoA desaturase regulation has shown potential as a therapeutic target to inhibit the progression of prostate cancer (35).
The enzyme FASN functions to help synthesize long-chain fatty acids. It has long been accepted that a common phenotype within prostate cancer is the upregulation of FASN activity and is of clinical utility as a pathologic biomarker that is occasionally examined in a special stain. Furthermore, there appears to be a dysregulation of FASN where it no longer needs the external stimulation to activate. SREBP1 is stimulated by androgen signaling and epidermal growth factor and has been found to be overexpressed in prostate cancer, leading to the upregulation of FASN (36).
It is believed by some that unregulated FASN activity within prostate tissue is itself the beginning of malignant phenotype and has been argued to be necessary for prostate cancer growth maintenance (37). The use of lipid by the prostate cancer cells illustrates the anabolic pathways that these cells utilize in order to maintain energy and growth and bypass potential degenerative pathways.
Another element in metabolism of prostate cancer is PTEN is a tumor suppressor whose activation is of importance as it inhibits the activity of protein kinase B (PKB). The PKB signaling pathway promotes cell survival and proliferation as well as migration (38). It is of importance in signaling cells to progress toward apoptosis as well as arresting the cell cycle. PTEN has been shown to promote an anti-Warburg effect by promoting oxidative phosphorylation and downregulating glycolysis. Downregulation of PTEN has been correlated to a more severe progression and metastasis of prostate cancer (39).
The exact role of glucose metabolism and prostate cancer has not been well defined; however, it does appear to have involvement in the progression of the disease and cellular division (40). Further proof of this involvement is a correlation between individuals with diabetes mellitus and increased severity of cancer phenotype (41). While glucose uptake does not appear to be increased early in prostate cancer cells as mentioned above, there appears to be an important relationship between these factors, particularly in late stage disease (42).
It has been long understood that androgen receptor signaling leads to an increase in the growth of prostate cancer cells. Androgen signals are needed in order to promote de novo lipid synthesis, which is required for the growth and survival of these tumor cells (43). Androgen suppression therapy has been used clinically to prevent progression of disease. With prolonged treatment, cells may develop resistance to this treatment. Recently, prostate cancer cells have been shown to have the ability to self-produce steroids, a proposed path by which cells resist androgen deprivation treatment. Prostate cancers that are resistant to androgen deprivation therapy progress to have a poor prognosis, and further treatment options are limited (44).
5′ AMPK is a major site of convergence for many metabolic pathways within the cell and acts as a key energy sensor. Modulation of AMPK therefore leads to numerous alterations within cellular metabolic profiles. Within prostate cancer cells, multiple downstream effects from androgen signaling have been shown to act through AMPK activity (45). As the impact of androgen to prostate cancer has been well established, this has become an area of therapeutic interest. Recently, in vitro studies have shown that altering the metabolic activity of AMPK has shown to reduce the prostate cancer growth (44, 46).
Metabolomics of prostate cancer is an emerging and active area of research. Metabolomic analyses can theoretically be used to further refine the current understanding of prostate cancer cell metabolic activity by determining the concentration of the excreted products. As previously noted, the long used standard of PSA testing has come under increasing scrutiny recently. Better understanding of metabolomics profile can therefore have an immense benefit in potentially determining new screening and risk stratification methods both for initial diagnostic and follow-up care (47). Advancements in metabolomics technologies have further spurred interest in this field as the use of gas chromatography and mass spectroscopy and NMR spectroscopy has now been utilized in profiling prostate cancer products. Research has been conducted in using a urine sample to assess metabolic profiles (8). If efficacious, this would represent a less interventional testing modality for patients. However, the results from this method have been mixed and variability in urine collection represents a barrier to performance. Overall, metabolomics represents an exciting avenue for discovery as the presence of certain metabolites may underscore the importance of highly utilized pathways, and help determine future care (5, 8, 47–50).
As the leading form of cancer among men, prostate cancer represents a major burden of disease within the United States. An active area of research is in the metabolism of prostate cancer. There are a variety of metabolic alterations that occur when a benign cell becomes malignant. There is still a great deal of research to be done, as many of the mechanisms of cellular cancer metabolisms are not well understood. With better knowledge comes the possibility of creating better treatment options and diagnostic testing.
The authors were involved with the oversight and revision of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Part of this work was funded by support from the Department of Defense and the American Cancer Society (MS funding).
1. Roehrborn CG, Black LK. The economic burden of prostate cancer. BJU Int (2011) 108:806–13. doi:10.1111/j.1464-410X.2011.10365.x
2. Capper CP, Rae JM, Auchus RJ. The metabolism, analysis, and targeting of steroid hormones in breast and prostate cancer. Horm Cancer (2016) 7:149–64. doi:10.1007/s12672-016-0259-0
3. Costello LC, Franklin RB, Feng P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion (2005) 5:143–53. doi:10.1016/j.mito.2005.02.001
4. Ross RK, Pike MC, Coetzee GA, Reichardt JK, Yu MC, Feigelson H, et al. Androgen metabolism and prostate cancer: establishing a model of genetic susceptibility. Cancer Res (1998) 58:4497–504.
5. Giskeødegård GF, Hansen AF, Bertilsson H, Gonzalez SV, Kristiansen KA, Bruheim P, et al. Metabolic markers in blood can separate prostate cancer from benign prostatic hyperplasia. Br J Cancer (2015) 113:1712–9. doi:10.1038/bjc.2015.411
6. Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer (2008) 8:268–78. doi:10.1038/nrc2351
7. Carlsson SV, Kattan MW. Prostate cancer: personalized risk stratified screening or abandoning it altogether? Nat Rev Clin Oncol (2016) 13:140–2. doi:10.1038/nrclinonc.2016.11
8. Kumar D, Gupta A, Nath K. NMR-based metabolomics of prostate cancer: a protagonist in clinical diagnostics. Expert Rev Mol Diagn (2016) 16:651–61. doi:10.1586/14737159.2016.1164037
9. Dakubo GD, Parr RL, Costello LC, Franklin RB, Thayer RE. Altered metabolism and mitochondrial genome in prostate cancer. J Clin Pathol (2006) 59:10–6. doi:10.1136/jcp.2005.027664
10. Costello LC, Feng P, Milon B, Tan M, Franklin RB. Role of zinc in the pathogenesis and treatment of prostate cancer: critical issues to resolve. Prostate Cancer Prostatic Dis (2004) 7:111–7. doi:10.1038/sj.pcan.4500712
11. Costello LC, Franklin RB. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch Biochem Biophys (2016) 611:100–12. doi:10.1016/j.abb.2016.04.014
12. Costello LC, Franklin RB, Zou J, Feng P, Bok R, Swanson MG, et al. Human prostate cancer ZIP1/zinc/citrate genetic/metabolic relationship in the TRAMP prostate cancer animal model. Cancer Biol Ther (2011) 12:1078–84. doi:10.4161/cbt.12.12.18367
13. Asgari Y, Zabihinpour Z, Salehzadeh-Yazdi A, Schreiber F, Masoudi-Nejad A. Alterations in cancer cell metabolism: the Warburg effect and metabolic adaptation. Genomics (2015) 105:275–81. doi:10.1016/j.ygeno.2015.03.001
14. Warburn O, Dickens F. The metabolism of tumors. Am J Med Sci (1931) 182:123. doi:10.1097/00000441-193107000-00022
15. Ngo DC, Ververis K, Tortorella SM, Karagiannis TC. Introduction to the molecular basis of cancer metabolism and the Warburg effect. Mol Biol Rep (2015) 42:819–23. doi:10.1007/s11033-015-3857-y
16. Sadeghi RN, Karami-Tehrani F, Salami S. Targeting prostate cancer cell metabolism: impact of hexokinase and CPT-1 enzymes. Tumour Biol (2015) 36:2893–905. doi:10.1007/s13277-014-2919-4
17. Twum-Ampofo J, Fu D-X, Passaniti A, Hussain A, Siddiqui MM. Metabolic targets for potential prostate cancer therapeutics. Curr Opin Oncol (2016) 28:241–7. doi:10.1097/CCO.0000000000000276
18. Dueregger A, Schöpf B, Eder T, Höfer J, Gnaiger E, Aufinger A, et al. Differential utilization of dietary fatty acids in benign and malignant cells of the prostate. PLoS One (2015) 10:e0135704. doi:10.1371/journal.pone.0135704
19. Franz M-C, Anderle P, Bürzle M, Suzuki Y, Freeman MR, Hediger MA, et al. Zinc transporters in prostate cancer. Mol Aspects Med (2013) 34:735–41. doi:10.1016/j.mam.2012.11.007
20. Feng P, Li T-L, Guan Z-X, Franklin RB, Costello LC. Direct effect of zinc on mitochondrial apoptogenesis in prostate cells. Prostate (2002) 52:311–8. doi:10.1002/pros.10128
21. Sanità P, Capulli M, Teti A, Galatioto GP, Vicentini C, Chiarugi P, et al. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer (2014) 14:154. doi:10.1186/1471-2407-14-154
22. Pertega-Gomes N, Felisbino S, Massie CE, Vizcaino JR, Coelho R, Sandi C, et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: a role for monocarboxylate transporters as metabolic targets for therapy. J Pathol (2015) 236:517–30. doi:10.1002/path.4547
23. Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst (2013) 105:1463–73. doi:10.1093/jnci/djt241
24. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2012) 481:380–4. doi:10.1038/nature10602
25. Pan T, Gao L, Wu G, Shen G, Xie S, Wen H, et al. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun (2015) 456:452–8. doi:10.1016/j.bbrc.2014.11.105
26. Moncada S, Higgs EA, Colombo SL. Fulfilling the metabolic requirements for cell proliferation. Biochem J (2012) 446:1–7. doi:10.1042/BJ20120427
27. Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol (2015) 236:278–89. doi:10.1002/path.4518
28. Qiu F, Huang J, Sui M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett (2015) 364:1–7. doi:10.1016/j.canlet.2015.04.020
29. Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des (2008) 14:1049–57. doi:10.2174/138161208784246199
30. Hsueh EC, Knebel SM, Lo WH, Leung YC, Cheng PN, Hsueh CT. Deprivation of arginine by recombinant human arginase in prostate cancer cells. J Hematol Oncol (2012) 5:17. doi:10.1186/1756-8722-5-17
31. Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res (2009) 69:700–8. doi:10.1158/0008-5472.CAN-08-3157
32. Tomlinson BK, Thomson JA, Bomalaski JS, Diaz M, Akande T, Mahaffey N, et al. Phase I trial of arginine deprivation therapy with ADI-PEG 20 plus docetaxel in patients with advanced malignant solid tumors. Clin Cancer Res (2015) 21:2480–6. doi:10.1158/1078-0432.CCR-14-2610
33. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine Reviews (2011) 25(2):276–308. doi:10.1210/er.2002-0032
34. Griffin JE. Androgen resistance – the clinical and molecular spectrum. N Engl J Med (1992) 326:611–8. doi:10.1056/NEJM199202273260906
35. Deep G, Schlaepfer IR. Aberrant lipid metabolism promotes prostate cancer: role in cell survival under hypoxia and extracellular vesicles biogenesis. Int J Mol Sci (2016) 17(7):1061. doi:10.3390/ijms17071061
36. Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res (2009) 69:8133–40. doi:10.1158/0008-5472.CAN-09-0775
37. Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, et al. Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PLoS One (2013) 8:e64570. doi:10.1371/journal.pone.0064570
38. Wang H, Zhang L, Fu Y, Fang F, Jiang Y, Dong Y, et al. CSL regulates AKT to mediate androgen independence in prostate cancer progression. Prostate (2016) 76:140–50. doi:10.1002/pros.23104
39. Mithal P, Allott E, Gerber L, Reid J, Welbourn W, Tikishvili E, et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int J Urol (2014) 21:1209–14. doi:10.1111/iju.12571
40. Singh G, Lakkis CL, Laucirica R, Epner DE. Regulation of prostate cancer cell division by glucose. J Cell Physiol (1999) 180:431–8. doi:10.1002/(SICI)1097-4652(199909)180:3<431::AID-JCP14>3.0.CO;2-O
41. Murtola TJ, Wahlfors T, Haring A, Taari K, Stenman UH, Tammela TL, et al. Polymorphisms of genes involved in glucose and energy metabolic pathways and prostate cancer: interplay with metformin. Eur Urol (2015) 68:1089–97. doi:10.1016/j.eururo.2015.03.026
42. Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta (2013) 1831:1518–32. doi:10.1016/j.bbalip.2013.03.010
43. Hara N, Nishiyama T. Androgen metabolic pathway involved in current and emerging treatment for men with castration resistant prostate cancer: intraprostatic androgens as therapeutic targets and endocrinological biomarkers. Curr Drug Targets (2014) 15:1215–24. doi:10.2174/1389450115666141024114736
44. Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med (2014) 6:519–38. doi:10.1002/emmm.201302734
45. Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene (2014) 33:5251–61. doi:10.1038/onc.2013.463
46. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther (2014) 15:156–69. doi:10.4161/cbt.26726
47. McDunn JE, Li Z, Adam KP, Neri BP, Wolfert RL, Milburn MV, et al. Metabolomic signatures of aggressive prostate cancer. Prostate (2013) 73:1547–60. doi:10.1002/pros.22704
48. Truong M, Yang B, Jarrard DF. Toward the detection of prostate cancer in urine: a critical analysis. J Urol (2013) 189:422–9. doi:10.1016/j.juro.2012.04.143
49. Kumar D, Gupta A, Mandhani A, Sankhwar SN. Metabolomics-derived prostate cancer biomarkers: fact or fiction? J Proteome Res (2015) 14:1455–64. doi:10.1021/pr5011108
Keywords: prostatic neoplasms, metabolomics, metabolism, metabolic networks and pathways, precision medicine
Citation: Eidelman E, Twum-Ampofo J, Ansari J and Siddiqui MM (2017) The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 7:131. doi: 10.3389/fonc.2017.00131
Received: 22 March 2017; Accepted: 06 June 2017;
Published: 19 June 2017
Edited by:
Shanmugasundaram Ganapathy-Kanniappan, Johns Hopkins School of Medicine, United StatesReviewed by:
Lambros Stamatakis, MedStar Washington Hospital Center, United StatesCopyright: © 2017 Eidelman, Twum-Ampofo, Ansari and Siddiqui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohummad Minhaj Siddiqui, bXNpZGRpcXVpQHNvbS51bWFyeWxhbmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.