 Assia L. Angelova
Assia L. Angelova Mathias Witzens-Harig2
Mathias Witzens-Harig2 Jean Rommelaere
Jean Rommelaere- 1Department of Tumor Virology, German Cancer Research Center, Heidelberg, Germany
- 2Department of Hematology, Oncology and Rheumatology, University Hospital Heidelberg, Heidelberg, Germany
- 3Department of Virology, The Stephan Angeloff Institute of Microbiology, Bulgarian Academy of Sciences, Sofia, Bulgaria
Non-Hodgkin lymphoma (NHL) and leukemia are among the most common cancers worldwide. While the treatment of NHL/leukemia of B-cell origin has much progressed with the introduction of targeted therapies, few treatment standards have been established for T-NHL/leukemia. As presentation in both B- and T-NHL/leukemia patients is often aggressive and as prognosis for relapsed disease is especially dismal, this cancer entity poses major challenges and requires innovative therapeutic approaches. In clinical trials, oncolytic viruses (OVs) have been used against refractory multiple myeloma (MM). In preclinical settings, a number of OVs have demonstrated a remarkable ability to suppress various types of hematological cancers. Most studies dealing with this approach have used MM or B- or myeloid-cell-derived malignancies as models. Only a few describe susceptibility of T-cell lymphoma/leukemia to OV infection and killing. The rat H-1 parvovirus (H-1PV) is an OV with considerable promise as a novel therapeutic agent against both solid tumors (pancreatic cancer and glioblastoma) and hematological malignancies. The present perspective article builds on previous reports of H-1PV-driven regression of Burkitt’s lymphoma xenografts and on unpublished observations demonstrating effective killing by H-1PV of cells from CHOP-resistant diffuse large B-cell lymphoma, cutaneous T-cell lymphoma, and T-cell acute lymphoblastic leukemia. On the basis of these studies, H-1PV is proposed for use as an adjuvant to (chemo)therapeutic regimens. Furthermore, in the light of a recently completed first parvovirus clinical trial in glioblastoma patients, the advantages of H-1PV for systemic application are discussed.
Introduction
Viruses and Human Health, a Two-Edged Sword: Chronology of Virus Rehabilitation
1898: viruses are discovered as “minute living things capable of reproducing themselves.” After the pioneering work of Adolf Eduard Mayer, Dmitri Ivanovsky, and Martinus Beijerinck, two German researchers, Friedrich Loeffler and Paul Frosch, were the first to contradict the “contagium vivum fluidum” (contagious living fluid) hypothesis to define a virus (the foot-and-mouth disease virus) as a tiny particle and to suggest that “the causative agents of numerous other infectious diseases of man and animals may also belong to this group of minute organisms” (1). Thus, at the beginning of the 20th century, the door opened to a new and exciting research area: virology.
Twentieth century: viruses as triggers of human infectious diseases. In the course of the 20th century and as predicted by Loeffler and Frosch, viruses were identified as the unquestionable causative agents of many human infectious diseases, from yellow fever (2), rabies (3), and poliomyelitis (4) to the acquired immunodeficiency syndrome (5). And this was not the end of the story: new disease-causing viruses, such as human bocaviruses, continue to emerge (6). It never rains but it pours…
Further bad news: viruses and human cancer. In addition to their vicious role as causative agents of numerous human infectious diseases, viruses are also involved in cancer development. This was first demonstrated at the beginning of the 20th century. Some 15–20% of all human cancers are attributed to viruses, notably Epstein–Barr virus, papilloma viruses, hepatitis B and C viruses, human herpesviruses, and human T-lymphotropic virus 1 (7). The molecular mechanisms underlying virus-induced carcinogenesis are diverse and complex. In addition to causing direct effects such as induction of genomic instability, DNA damage, and viral oncogene-triggered cell transformation (8, 9), oncogenic viruses can establish a chronic infection allowing them to escape from the host’s immune system while producing proteins that control cell death and proliferation. Chronic infection also leads to inflammatory reactions promoting cancer development (10). In nasopharyngeal cancer, certain lymphomas, cervical cancer, liver cancer, Kaposi’s sarcoma, and human adult T-cell leukemia/lymphoma, malignant transformation is likely to be initiated by host cell infection by an oncogenic virus. And yet…
Two sides to every coin: viruses have a bad side and an “oncolytic” side. Breakthrough observations at the start of the 20th century and findings peaking in the 1950s made it clear that “severe (virus) infections may on occasion favorably modify the course of far-advanced neoplastic disease…” (11). A significant drop in leukocyte counts associated with some clinical improvement was documented in children diagnosed with acute lymphoblast leukemia (ALL) having simultaneously acquired a varicella virus infection (11). At least five cases showing Hodgkin’s disease regression after measles virus infection were described (12–14). Similar observations were made in patients having developed viral hepatitis during Hodgkin’s lymphoma progression (15). In 1971, Bluming and Ziegler published a case report on Burkitt’s lymphoma (BL) regression associated with measles virus infection (16). Today, more than a century after the first report on virus infection-associated clinical remission in cancer patients, virotherapy with oncolytic viruses (OVs) is the focus of a rapidly growing research field. Studies in this field have brought convincing evidence that oncolytic virotherapy, alone or in combination with surgery, chemotherapy, or radiotherapy, may significantly impact cancer mortality and improve patients’ quality of life.
Oncolytic Viruses As Anticancer Tools: From Bench to Clinical Trials
Oncolytic viruses form a diverse biological group whose members belong to at least 10 different virus families, contain either an RNA or a DNA genome, and vary considerably as regards genome size, particle complexity, and natural host preferences (17). OVs naturally possess? or are engineered to acquire the capacity to selectively infect, replicate in, and destroy tumor cells (oncolysis) while sparing their normal counterparts (17, 18). Multiple factors explain this oncoselectivity: altered expression by tumor cells of virus entry receptors and/or intracellular permissiveness factors, rapid tumor cell division and high metabolic activity, deficient antiviral type I interferon responses in tumor cells, etc. (19). Furthermore, there is mounting evidence that OV infection of tumor cells induces an immunogenic process, with neo-antigen recognition and establishment of specific antitumor immune responses (20). The remarkable potential of OVs as cancer therapeutics has been well documented in a number of preclinical studies, and the resulting knowledge has been translated into an expanding wave of clinical trials (21, 22). In 2015, talimogene laherparepvec was the first OV to receive FDA approval as an anticancer drug (23) based on the fact that this herpes simplex virus type 1-based oncolytic immunotherapy has demonstrated therapeutic benefit against metastatic melanoma in a phase III clinical trial (24). In 2016, there were about 40 OV-based clinical trials recruiting cancer patients (19).
H-1 Parvovirus (H-1PV) Against Pancreatic Cancer and Glioma: The Brave Little Cancer Fighter
With a particle diameter of only 22 nm, the non-enveloped ssDNA-containing H-1 parvovirus is the smallest of the OVs. Its natural host is the rat. Humans are not naturally infected with H-1PV, no firm association between this virus and any human disease has been established, and no preexisting H-1PV immunity has been detected in the human population (25). Failure to observe any virus-related pathogenic effects in two early studies of H-1PV administration to human cancer patients (26, 27) prompted further therapy-oriented H-1PV research. Considerable preclinical evidence has accumulated over the last 30 years [reviewed in Ref. (28–30)] providing straightforward proof that H-1PV has broad oncosuppressive potential. In particular, pancreatic carcinoma and glioblastoma have attracted major attention as parvovirotherapy targets. In the respective preclinical models, efforts have been made to unravel the mechanisms and improve the efficacy of H-1PV treatment.
Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive tumor, often unresectable at the time of initial diagnosis. Median overall survival is only 6–9 months. As current therapies for PDAC patients fail to improve significantly their quality of life and to prolong survival (31), it is urgent to develop novel curative strategies. Extensive work by our team on H-1PV-based virotherapy for PDAC has yielded the following key findings: (i) H-1PV efficiently kills PDAC cells, including gemcitabine-resistant ones (32); (ii) H-1PV infection of pancreatic cancer cells results in active cathepsin B translocation to the cytosol (32) and in extracellular HMGB1 danger signaling (33); (iii) some predictive markers of PDAC permissiveness to H-1PV infection and lysis, e.g., SMAD4, have been identified (34); (iv) in an orthotopic PDAC model, H-1PV causes tumor regression and prolongs animal survival, without affecting bone marrow activity, liver function, or kidney function (32); (v) H-1PV-induced tumor suppression is potentiated under conditions of gemcitabine pretreatment (the current gold standard in pancreatic cancer therapy) (32); (vi) H-1PV oncosuppressive effects involve the participation of immune cells, which become activated either after an abortive infection with the virus (35) or through induction of immunogenic factors such as NK cytotoxicity receptor ligands (36) in H-1PV-infected PDAC cells; (vii) the vaccination potential of H-1PV, in combination with IFN-γ, extends to the treatment of peritoneal carcinomatosis, an untreatable condition traditionally managed with palliative measures only (37). Current preclinical achievements and prospects for pancreatic cancer parvovirotherapy are summarized in Ref (38, 39).
Glioma
Glioblastoma is the most aggressive human primary brain tumor. Life expectancy remains very poor, despite standard and alternative therapeutic attempts (40). Our team has shown that oncolytic H-1PV infection of human glioma cells results in efficient cell killing (41). High-grade glioma stem cell models are also permissive to lytic H-1PV infection (42). The cellular mechanism of virus-induced glioma cell killing has been elucidated and is based on active lysosomal cathepsin B translocation and accumulation in the cytosol of H-1PV-infected glioma cells but not normal cells (astrocytes) (43). Enhanced glioma cell killing has been observed when the virus was applied shortly after tumor cell irradiation, suggesting that this protocol might be translated to cases of non-resectable recurrent glioblastoma (44). In animal models, local, systemic, or intranasal administration of H-1PV has been found to cause regression of advanced tumors, virus replication being restricted to tumor tissues (45, 46). The favorable safety profile of local or systemic treatment with medical-grade GMP-produced H-1PV has been confirmed in a study using a permissive animal model (47, 48).
On the basis of the above preclinical evidence, the first phase I/IIa clinical trial (ParvOryx01) of an oncolytic parvovirus (H-1PV) in recurrent glioblastoma patients was launched in 2011 (49) and successfully completed in 2015. This trial, in addition to confirming the excellent safety and tolerability of H-1PV, yielded valuable observations, which strongly encourage further clinical development of this virus as an anticancer therapeutic. Particularly essential is the evidence suggesting that H-1PV (i) crosses the blood–brain barrier after systemic administration and (ii) may induce immunogenic conversion of the tumor microenvironment. In 2015, a second phase I/IIa trial was launched in inoperable metastatic PDAC patients. The outcome of this study is eagerly awaited.
Glioblastoma and pancreatic cancer are far from being the only tumor types sensitive to H-1PV-induced oncotoxicity, since it has also been demonstrated in preclinical models of breast, gastric, cervical (29), and colorectal (50, 51) cancer. H-1PV thus has the potential to treat not only brain and pancreatic but also a variety of other tumors.
Oncolytic (Parvo)Viruses Against Hematological Malignancies
Preclinical Experience
Lymphoma and leukemia are the two cancer types tightly associated with the dawn of the oncolytic virotherapy era. Later, however, they were superseded as oncolytic virotherapy targets by solid tumors, such as breast, ovarian, bladder, skin, colon, and lung carcinomas. Nevertheless, a substantial set of preclinical data shows that several OVs can selectively lyse hematopoietic stem cells or downstream blood cell lineages (Table 1). As shown in the table, the predominant preclinical model is multiple myeloma (MM), followed by leukemia/lymphoma of B-lymphoid, myeloid, or T-lymphoid origin. Myxoma virus, a poxvirus whose natural tropism is restricted to European rabbits and is non-pathogenic for other vertebrates, has been demonstrated to selectively induce apoptotic death in MM cells (52–54). MM has also been successfully targeted by a double-deleted vaccinia virus (55), adenovirus serotype 5 (56), coxsackievirus A21 (57), reovirus (58–60), vesicular stomatitis (VSV) virus (61), and measles virus (62–65). Furthermore, myxoma and VSV infections are oncotoxic to acute myeloid leukemia cells (66), while adeno- (67, 68), herpes- (69), reo- (70), VSV (71), and measles virus (72–76) are reported to induce killing/suppression of B- and T-lymphoma or leukemia-derived cells/xenografts.
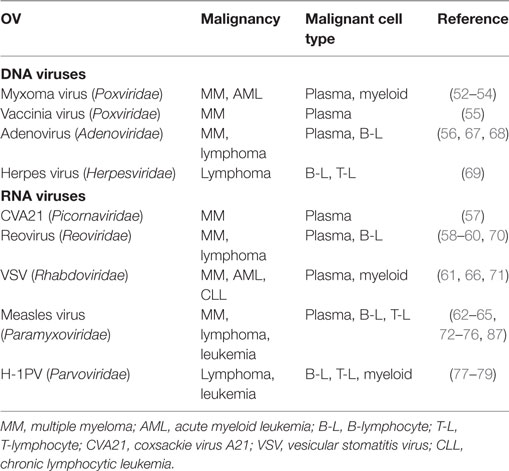
Table 1. Oncolytic viruses (OVs) targeting hematological malignancies: preclinical evidence.
First proofs of the capacity of H-1PV to infect and destroy human blood cancer-derived cells date back to the 1980s, when Faisst et al. screened for H-1PV permissiveness and killing a panel of BL, adult T cell leukemia-derived, and in vitro-transformed lymphoblastoid cell lines (77, 78). Further proof-of-concept was provided by Angelova et al., who showed that African and European BL cells, including those lacking CD20 and hence resistant to the CD20-targeting therapeutic rituximab, are highly susceptible to H-1PV-induced killing, in contrast to normal B lymphocytes from healthy donors. In a SCID mouse lymphoma model, a single intratumoral H-1PV injection was sufficient to cause full tumor suppression and disease-free survival for the whole period of observation (70 days). This striking oncosuppression was observed even when the virus was applied late after tumor initiation, so as to mimic an advanced disease stage (79).
Clinical State of the Art
The rapid development of gene therapy and immune modulation approaches in recent years has led to greatly improving the management of many hematological cancer types. Several clinical trials are currently examining the effects of RNA interference, suicide gene therapy, and immune modulation in myeloma, lymphoma, and leukemia patients (80). In the development of new therapies, the most progress has been made in the treatment of B-cell leukemia/lymphoma. These account for over 80% of all non-Hodgkin lymphomas (NHL). The current standard treatment is a combination of the anti-CD20 antibody rituximab and chemotherapy, e.g., the CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) (81, 82). In contrast, NHL/leukemia of T-cell origin remains a therapeutic challenge, and treatment advances lag behind those for B-NHL. For example, treatment outcome is worse in pediatric T-ALL patients than in pediatric B-ALL patients (83). Adult T-ALL poses even greater treatment difficulties and no current option prolongs survival satisfactorily (84). In both B- and T-NHL/leukemia patients, outcomes of relapsed disease are usually dismal. Late effects and systemic toxicities related to conventional strategies (chemo- and radiotherapy) must also be considered. This spells out a continuing need for innovative approaches, especially for patients with relapsed B-NHL or newly diagnosed/relapsed T-NHL. Targeted therapies (85), immunotherapy (86), and oncolytic virotherapy have triggered growing interest and are the focus of much attention. Two OVs, the wild-type reovirus and an engineered measles virus, have successfully reached the clinical testing phase (87).1 , 2 In particular, a non-randomized phase I study conducted in Switzerland and involving cutaneous T-cell lymphoma (CTCL) patients with accessible lesions allowing intratumoral measles virus application has already yielded promising results as regards both the safety and efficacy of this OV treatment (87). One should note, however, that OV trials currently recruiting hematological cancer patients are restricted to refractory MM and that they are strikingly fewer than, for instance, melanoma or glioma OV trials. Given the promising preclinical data that demonstrate the potential of several other OVs to induce oncolytic effects in myeloid, B- and T-cell lymphoma/leukemia models, further clinical development of this anticancer approach is to be expected, and also hoped for, in the case of hematological malignancies. A recent study by Kishore and Kishor, comparing mortality rates between parvovirus-B19-infected and uninfected pediatric ALL patients has raised the intriguing hypothesis that natural B19 infection may exert unexplored oncolytic effects (88).
Oncolytic H-1PV as a Candidate for Further Development in Oncohematology
After the first demonstration that H-1PV could induce efficient BL cell killing in vitro (77, 78) and BL regression in animal models (79), the question arose: might H-1PV be used against other types of hematological cancers? This question is of general interest, since BL is mostly seen in Uganda and Nigeria and is a rare condition outside Africa (89). It prompted us to conduct further studies to assess the capacity of this virus to target cells derived from other hematological malignancies. A panel of commercially available ATCC cell lines derived from aggressive or indolent lymphomas/leukemias of B- or T-cell origin was tested in vitro (A. Angelova, Z. Raykov, J. Rommelaere, unpublished data). First, encouraging results were obtained as shown in Table 2. Only one mixed type B-cell lymphoma and one Sézary syndrome CTCL were resistant to H-1PV-induced cell death. This resistance was associated with either the absence (Hut78 cells) or a low level (Farage cells) of progeny virion production and was not due to blockage of virus entry. In contrast, large B-cell-lymphoma-derived cells supported high levels of H-1PV progeny virion production and were almost totally eradicated by very low virus doses. Notably, DLBCL cell lines (e.g., Pfeiffer) with upregulated expression of aldehyde dehydrogenase 1A1 conferring CHOP resistance (90) were among the most sensitive H-1PV targets. These results suggest a potential use of H-1PV in chemoresistant DLBCL cases. Furthermore, H-1PV was able to replicate in T-ALL and some CTCL cells, with striking cytopathic effects. Although CTCL is a relatively rare condition, its incidence has increased about threefold over the last 2–3 decades in the United States (91) and in other regions of the world (92). Advanced disease stages with blood involvement require systemic therapies and, in general, the quality of life of CTCL patients is greatly affected. We are, therefore, now expanding the panel of in vitro models to test the antineoplastic potential of H-1PV in several, mostly T-cell-derived, types of hematological cancers, including CTCL. The failure of CHOP-based chemotherapies in CTCL patients has led to the development and subsequent FDA approval of two histone deacetylase inhibitors (HDACis), vorinostat and romidepsin (93–95). As patients often fail to reach or sustain a 50% partial response to these drugs, other agents have to be added in a combinatorial manner, in order to overcome resistance to HDACi (94). OVs, notably H-1PV, appear as potential candidates, as it was recently shown by Li et al. that another HDACi, valproic acid, when combined with oncolytic H-1PV, increases parvovirus-mediated cytotoxicity toward cervical and pancreatic cancer cells, thus resulting in synergistic killing (96). Further preclinical studies are worth conducting to determine whether these findings can be extended to CTCL and other clinically challenging T-cell malignancies such as T-ALL. Interestingly, it was recently reported that expression of the transcription factor TAL-1 (associated with poor prognosis in T-ALL) is markedly downregulated upon HDAC inhibition (97).
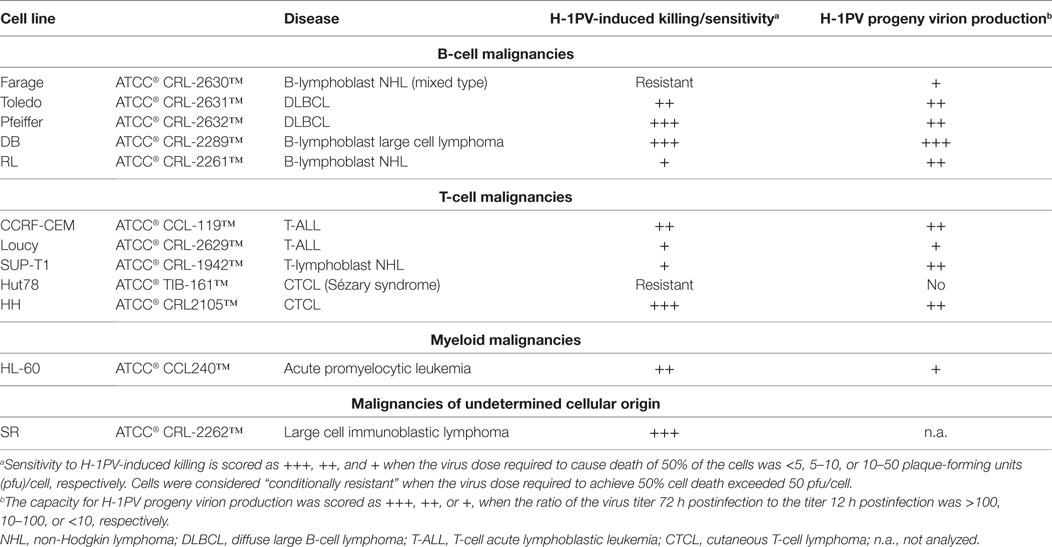
Table 2. Responsiveness of lymphoma- and leukemia-derived cell lines to oncolytic H-1PV infection.
Conclusion and Perspective
In conclusion, oncolytic H-1PV has shown outstanding oncosuppressive activity in preclinical models of various solid tumors. Data from the first H-1PV clinical trial in recurrent glioblastoma patients have confirmed the excellent safety and tolerability of this virus upon local or systemic application. Accumulating preclinical evidence shows, furthermore, that H-1PV can efficiently kill, via productive infection, cancer cells derived from different hematological malignancies. These include both rituximab- and chemotherapy-resistant B-cell lymphomas and T-cell leukemia/lymphoma, which currently pose a major therapeutic challenge. These first results strongly encourage further preclinical studies aimed at substantiating the oncolytic and adjuvant potential of H-1PV against hematological cancers, both as single agent and as a component of combination treatments. These studies should pave the way toward innovative improvements of current standard therapies, for the benefit of chemotherapy-resistant and relapsing patients.
Author Contributions
AA, MW-H, JR, and AG contributed to writing the present article. AA contributed to generating, analyzing, and presenting the unpublished data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Rita Hörlein for assistance with H-1PV plaque assay experiments and Kathleen Broman for critical reading of the manuscript.
Funding
The lymphoma/leukemia research was supported by a DKFZ guest researcher fellowship to AA on leave from the Stephan Angeloff Institute of Microbiology, Sofia, Bulgaria.
Abbreviations
ALL, acute lymphoblastic leukemia; BL, Burkitt’s lymphoma; OV(s), oncolytic virus(es); H-1PV, parvovirus H-1; PDAC, pancreatic ductal adenocarcinoma; MM, multiple myeloma; CV, coxsackievirus; VSV, vesicular stomatitis virus; NHL, non-Hodgkin lymphoma; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; DLBCL, diffuse large B-cell lymphoma; CTCL, cutaneous T-cell lymphoma; HDACi, histone deacetylase inhibitor.
Footnotes
- ^ Oncolytics Biotech Inc. – Clinical Trials. Available from: http://www.oncolyticsbiotech.com/reolysin/clinical-trials/
- ^ ClinicalTrials.gov: Vaccine Therapy With or Without Cyclophosphamide in Treating Patients With Recurrent or Refractory Multiple Myeloma. Available from: https://clinicaltrials.gov/ct2/show/NCT00450814
References
1. Rott R, Siddell S. One hundred years of animal virology. J Gen Virol (1998) 79:2871–4. doi: 10.1099/0022-1317-79-11-2871
2. Reed W, Carroll J, Agramonte A, Lazear JW. The etiology of yellow fever – a preliminary note. Public Health Pap Rep (1900) 26:27–53.
3. Remlinger P. Le passage du virus rabique à travers les filtres. Annales de L’Institut Pasteur (Paris) (1903) 101:765–74.
4. Landsteiner K, Popper E. Übertragung der Poliomyelitis acuta auf Affen. Zeitschrift für Immunitätsforschung und Experimentelle Therapie (1909) 2:377–90.
5. Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science (1983) 220:868–71. doi:10.1126/science.6189183
6. Woolhouse M, Scott F, Hudson Z, Howey R, Chase-Topping M. Human viruses: discovery and emergence. Philos Trans R Soc Lond B Biol Sci (2012) 367:2864–71. doi:10.1098/rstb.2011.0354
8. Hoppe-Seyler F, Butz K. Molecular mechanisms of virus-induced carcinogenesis: the interaction of viral factors with cellular tumor suppressor proteins. J Mol Med (Berl) (1995) 73:529–38. doi:10.1007/BF00195138
9. Chen Y, Williams V, Filippova M, Filippov V, Duerksen-Hughes P. Viral carcinogenesis: factors inducing DNA damage and virus integration. Cancers (2014) 6:2155–86. doi:10.3390/cancers6042155
10. McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta (2008) 1782:127–50. doi:10.1016/j.bbadis.2007.12.005
11. Bierman HR, Crile DM, Dod KS, Kelly KH, Petrakis NL, White LP, et al. Remissions in leukemia of childhood following acute infectious disease. Cancer (1953) 6:591–605. doi:10.1002/1097-0142(195305)6:3<591::AID-CNCR2820060317>3.0.CO;2-M
12. Hernandez SA. Observación de un caso de enfermedad de Hodgkin, con regresión de las sítomas e infartos ganglionares, post-sarampión. Arch Cubanos Cancer (1949) 8:26–31.
13. Zygiert Z. Hodgkin’s disease: remissions after measles. Lancet (1971) 1:593. doi:10.1016/S0140-6736(71)91186-X
14. Taqi AM, Abdurrahman MB, Yakubu AM, Fleming AF. Regression of Hodgkin’s disease after measles. Lancet (1981) 1:1112. doi:10.1016/S0140-6736(81)92286-8
15. Hoster HA, Zanes RP Jr, Von Haam E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. Cancer Res (1949) 9:473–80.
16. Bluming AZ, Ziegler JL. Regression of Burkitt’s lymphoma in association with measles infection. Lancet (1971) 2:105–6. doi:10.1016/S0140-6736(71)92086-1
17. Russel SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30:658–70. doi:10.1038/nbt.2287
18. Kelly E, Russel SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther (2007) 15:651–9. doi:10.1038/sj.mt.6300108
19. Lawler SE, Speranza M-C, Cho C-F, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol (2016). doi:10.1001/jamaoncol.2016.2064
20. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer (2014) 14:559–67. doi:10.1038/nrc3770
21. Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol (2014) 12:23–34. doi:10.1038/nrmicro3140
22. Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe (2014) 15:260–5. doi:10.1016/j.chom.2014.01.002
23. US Food and Drug Administration. FDA Approves First-of-Its-Kind Product for the Treatment of Melanoma. (2015). Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm469571.htm
24. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33:2780–8. doi:10.1200/JCO.2014.58.3377
25. Neuman SJ, McCallin PF, Sever JL. Attempts to isolate H-1 virus from spontaneous human abortions: a negative report. Teratology (1970) 3:279–82. doi:10.1002/tera.1420030311
26. Toolan HW, Saunders EL, Southam CM, Moore AE, Levin AG. H-1 virus viremia in the human. Proc Soc Exp Biol Med (1965) 119:711–5. doi:10.3181/00379727-119-30278
27. Le Cesne A, Dupressoir T, Janin N, Spielmann M, Le Chevalier T, Sancho-Garnier H, et al. Intra-lesional administration of a live virus, parvovirus H1 (PVH1) in cancer patients: a feasibility study. Proc Am Soc Clin Oncol (1993) 12:297.
28. Rommelaere J, Geletneky K, Angelova AL, Daeffler L, Kiprianova I, Schlehofer JR, et al. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev (2010) 21:185–95. doi:10.1016/j.cytogfr.2010.02.011
29. Angelova AL, Geletneky K, Nüesch JP, Rommelaere J. Tumor selectivity of oncolytic parvoviruses: from in vitro and animal models to cancer patients. Front Bioeng Biotechnol (2015) 3:55. doi:10.3389/fbioe.2015.00055
30. Geletneky K, Nüesch JP, Angelova A, Kiprianova I, Rommelaere J. Double-faceted mechanism of parvoviral oncosuppression. Curr Opin Virol (2015) 13:17–24. doi:10.1016/j.coviro.2015.03.008
31. Hartwig W, Hackert T, Hinz U, Gluth A, Bergmann F, Strobel O, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg (2011) 254:311–9. doi:10.1097/SLA.0b013e31821fd334
32. Angelova AL, Aprahamian M, Grekova SP, Hajri A, Leuchs B, Giese NA, et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res (2009) 15:511–9. doi:10.1158/1078-0432.CCR-08-1088
33. Angelova AL, Grekova SP, Heller A, Kuhlman O, Soyka E, Giese T, et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J Virol (2014) 88:5263–76. doi:10.1128/JVI.03688-13
34. Dempe S, Stroh-Dege AY, Schwarz E, Rommelaere J, Dinsart C. SMAD4: a predictive marker of PDAC cell permissiveness for oncolytic infection with parvovirus H-1PV. Int J Cancer (2010) 126:2914–27. doi:10.1002/ijc.24992
35. Grekova S, Aprahamian M, Giese N, Schmitt S, Giese T, Falk CS, et al. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol Ther (2010) 10:1280–9. doi:10.4161/cbt.10.12.13455
36. Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer (2011) 128:908–19. doi:10.1002/ijc.25415
37. Grekova SP, Aprahamian M, Daeffler L, Leuchs B, Angelova A, Giese T, et al. Interferon γ improves the vaccination potential of oncolytic parvovirus H-1PV for the treatment of peritoneal carcinomatosis in pancreatic cancer. Cancer Biol Ther (2011) 12:888–95. doi:10.4161/cbt.12.10.17678
38. Raykov Z, Grekova SP, Angelova AL, Rommelaere J. Parvoviruses: the friendly anticancer immunomodulator. In: Giese M, editor. Molecular Vaccines. Vienna: Springer-Verlag Wien (2013). p. 413–23.
39. Akladios C, Aprahamian M. Virotherapy of digestive tumors with rodent parvovirus: overview and perspectives. Expert Opin Biol Ther (2016) 16:645–53. doi:10.1517/14712598.2016.1151492
40. Clarke JL, Ennis MM, Yung WK, Chang SM, Wen PY, Cloughesy TF, et al. Is surgery at progression a prognostic marker for improved 6-month progression-free survival or overall survival for patients with recurrent glioblastoma? Neuro Oncol (2011) 13:1118–24. doi:10.1093/neuonc/nor110
41. Herrero Y, Calle M, Cornelis JJ, Herold-Mende C, Rommelaere J, Schlehofer JR, et al. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int J Cancer (2004) 109:76–84. doi:10.1002/ijc.11626
42. Josupeit R, Bender S, Kern S, Leuchs B, Hielscher T, Herold-Mende C, et al. Pediatric and adult high-grade glioma stem cell culture models are permissive to lytic infection with parvovirus H-1. Viruses (2016) 8. doi:10.3390/v8050138
43. Di Piazza M, Mader C, Geletneky K, Herrero Y, Calle M, Weber E, et al. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J Virol (2007) 81:4186–98. doi:10.1128/JVI.02601-06
44. Geletneky K, Hartkopf AD, Krempien R, Rommelaere J, Schlehofer JR. Improved killing of human high-grade glioma cells by combining ionizing radiation with oncolytic parvovirus H-1 infection. J Biomed Biotechnol (2010) 2010:350748. doi:10.1155/2010/350748
45. Geletneky K, Kiprianova I, Ayache A, Koch R, Herrero Y, Calle M, et al. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol (2010) 12:804–14. doi:10.1093/neuonc/noq023
46. Kiprianova I, Thomas N, Ayache A, Fischer M, Leuchs B, Klein M, et al. Regression of glioma in rat models by intranasal application of parvovirus H-1. Clin Cancer Res (2011) 17:5333–42. doi:10.1158/1078-0432.CCR-10-3124
47. Geletneky K, Leoni AL, Pohlmeyer-Esch G, Loebhard S, Leuchs B, Hoefer C, et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp Med (2015) 65:36–45.
48. Geletneky K, Leoni AL, Pohlmeyer-Esch G, Loebhard S, Baetz A, Leuchs B, et al. Pathology, organ distribution, and immune response after single and repeated intravenous injection of rats with clinical-grade parvovirus H1. Comp Med (2015) 65:23–35.
49. Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer (2012) 12:99. doi:10.1186/1471-2407-12-99
50. Malerba M, Daeffler L, Rommelaere J, Iggo RD. Replicating parvoviruses that target colon cancer cells. J Virol (2003) 77:6683–91. doi:10.1128/JVI.77.12.6683-6691.2003
51. Heinrich B, Goepfer K, Delic M, Galle PR, Moehler M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer. Onco Targets Ther (2013) 6:1119–27. doi:10.2147/OTT.S49371
52. Kim M, Madlambayan GJ, Rahman MM, Smallwood SE, Meacham AM, Hosaka K, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia (2009) 23:2313–7. doi:10.1038/leu.2009.219
53. Bartee MY, Dunlap KM, Bartee E. Myxoma virus induces ligand independent extrinsic apoptosis in human myeloma cells. Clin Lymphoma Myeloma Leuk (2016) 16:203–12. doi:10.1016/j.clml.2015.12.005
54. Bartee E, Bartee MY, Bogen B, Yu X-Z. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol Ther Oncolytics (2016) 3:16032. doi:10.1038/mto.2016.32
55. Deng H, Tang N, Stief AE, Mehta N, Baig E, Head R, et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia (2008) 22:2261–4. doi:10.1038/leu.2008.120
56. Senac JS, Doronin K, Russel SJ, Jelinek DF, Greipp RP, Barry MA. Infection and killing of multiple myeloma by adenoviruses. Hum Gene Ther (2010) 21:179–90. doi:10.1089/hum.2009.082
57. Au GG, Lincz LF, Enno A, Shafren DR. Oncolytic coxsackievirus A21 as a novel therapy for multiple myeloma. Br J Haematol (2007) 137:133–41. doi:10.1111/j.1365-2141.2007.06550.x
58. Thirukkumaran CM, Luider JM, Stewart DA, Cheng T, Lupichuk SM, Nodwell MJ, et al. Reovirus oncolysis as a novel purguing strategy for autologous stem cell transplantation. Blood (2003) 102:337–87. doi:10.1182/blood-2002-08-2508
59. Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res (2012) 18:4962–72. doi:10.1158/1078-0432.CCR-11-3085
60. Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy (2013) 9:413–4. doi:10.4161/auto.22867
61. Naik S, Nace R, Barber GN, Russel SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer Gene Ther (2012) 19:443–50. doi:10.1038/cgt.2012.14
62. Peng K-W, Ahmann GJ, Pham L, Greipp RP, Cattaneo R, Russel SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood (2001) 98:2002–7. doi:10.1182/blood.V98.7.2002
63. Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood (2004) 103:1641–6. doi:10.1182/blood-2003-07-2233
64. Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicity of intravenoues MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther (2007) 82:700–10. doi:10.1038/sj.clpt.6100409
65. Russel SJ, Federspiel MJ, Peng K-W, Tong C, Dingli D, Morice WG, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc (2014) 89:926–33. doi:10.1016/j.mayocp.2014.04.003
66. Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther (2004) 15:821–31. doi:10.1089/hum.2004.15.821
67. Medina DJ, Sheay W, Osman M, Goodell L, Martin J, Robson AB, et al. Adenovirus infection and cytotoxicity of primary mantle cell lymphoma cells. Exp Hematol (2005) 33:1337–47. doi:10.1016/j.exphem.2005.07.009
68. Qian W, Liu J, Tong Y, Yan S, Yang C, Yang M, et al. Enhanced antitumor activity by a selective conditionally replicating adenovirus combining with MDA-7/interleukin-24 for B-lymphoblastic leukemia via induction of apoptosis. Leukemia (2008) 22:361–9. doi:10.1038/sj.leu.2405034
69. Esfandyari T, Tefferi A, Szmidt A, Alain T, Zwolak P, Lasho T, et al. Transcription factors down-stream of Ras as molecular indicators for targeting malignancies with oncolytic herpes virus. Mol Oncol (2009) 3:464–8. doi:10.1016/j.molonc.2009.07.002
70. Allain T, Hirasawa KJ, Pon J, Nishikawa SG, Urbanski SJ, Auer Y, et al. Reovirus therapy of lymphoid malignancies. Blood (2002) 100:4146–53. doi:10.1182/blood-2002-02-0503
71. Tumilasci VF, Olière S, Nguyen TL-A, Shamy A, Bell J, Hiscott J. Targeting the apoptotic pathway with BCL-2 inhibitors sensitizes primary chronic lymphocytic leukemia cells to vesicular stomatitis virus-induced oncolysis. J Virol (2008) 82:8487–99. doi:10.1128/JVI.00851-08
72. Grote D, Russel SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood (2001) 97:3746–54. doi:10.1182/blood.V97.12.3746
73. Yaiw K-C, Miest TS, Frenzke M, Timm M, Johnston PB, Cattaneo R. CD20-targeted measles virus shows high oncolytic specificity in clinical samples from lymphoma patients independent of prior rituximab therapy. Gene Ther (2011) 18:313–7. doi:10.1038/gt.2010.150
74. Patel B, Dey A, Ghorani E, Kumar S, Malam Y, Rai L, et al. Differential cytopathology and kinetics of measles oncolysis in two primary B-cell malignancies provides mechanistic insights. Mol Ther (2011) 19:1034–40. doi:10.1038/mt.2011.44
75. Miest TS, Frenzke M, Cattaneo R. Measles virus entry through the signaling lymphocyte activation molecule governs efficacy of mantle cell lymphoma radiovirotherapy. Mol Ther (2013) 21:2019–31. doi:10.1038/mt.2013.171
76. Castleton A, Dey A, Beaton B, Patel B, Aucher A, Davis DM, et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood (2014) 123:1327–35. doi:10.1182/blood-2013-09-528851
77. Faisst S, Schlehofer JR, zur Hausen H. Transformation of human cells by oncogenic viruses supports permissiveness for parvovirus H-1 propagation. J Virol (1989) 63:2152–8.
78. Faisst S, Bartnitzke S, Schlehofer JR, zur Hausen H. Persistence of parvovirus H-1 DNA in human B- and T-lymphoma cells. Virus Res (1990) 16:211–23. doi:10.1016/0168-1702(90)90024-6
79. Angelova AL, Aprahamian M, Balboni G, Delecluse H-J, Feederle R, Kiprianova I, et al. Oncolytic rat rarvovirus H-1PV, a candidate for the treatment of human lymphoma: in vitro and in vivo studies. Mol Ther (2009) 17:1164–72. doi:10.1038/mt.2009.78
80. Domingo-Musibay E, Yamamoto M. Gene and virotherapy for hematological malignancies. Int J Hematol (2016) 104:29–41. doi:10.1007/s12185-016-2031-1
81. Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Niedhart JA, et al. IDEC-C2B8 (rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin lymphoma. Blood (1997) 90:2188–95.
82. Dotan E, Aggarwal C, Smith MR. Impact of rituximab (rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T (2010) 35:148–57.
83. Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol (2013) 50:185–96. doi:10.1053/seminhematol.2013.06.007
84. Litzow M, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood (2015) 126:833–41. doi:10.1182/blood-2014-10-551895
85. Hoelzer D. Targeted therapy with monoclonal antibodies in acute lymphoblastic leukemia. Curr Opin Oncol (2013) 25:701–6. doi:10.1097/CCO.0000000000000009
86. McLaughlin L, Russel Cruz C, Bollard CM. Adoptive T-cell therapies for refractory/relapsed leukemia and lymphoma: current strategies and recent advances. Ther Adv Hematol (2015) 6:295–307. doi:10.1177/2040620715594736
87. Heinzerling L, Künzi V, Oberholzer PA, Kündig T, Naim H, Dummer R, et al. Oncolytic measles virus in cutaneous T-cell lymphoma mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood (2005) 106:2287–94. doi:10.1182/blood-2004-11-4558
88. Kishore J, Kishor D. Can parvovirus B19 infection be naturally oncolytic: clinical findings raise such a possibility in leukaemic children. Indian J Med Res (2014) 139:952–53.
89. Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol (2012) 156:744–56. doi:10.1111/j.1365-2141.2011.09013.x
90. Jiang J, Liu Y, Tang Y, Li L, Zeng R, Zeng S, et al. ALDH1A1 induces resistance to CHOP in diffuse large B-cell lymphoma through activation of the JAK2/STAT3 pathway. Onco Targets Ther (2016) 9:5349–60. doi:10.2147/OTT.S107957
91. Wilson LD, Hinds GA, Yu JB. Age, race, sex, stage, and incidence of cutaneous lymphoma. Clin Lymphoma Myeloma Leuk (2012) 12:291–6. doi:10.1016/j.clcl.2012.06.010
92. Riou-Gotta MO, Fournier E, Mermet I, Pelletier F, Humbert P, Danzon A, et al. Primary cutaneous lymphomas: a population-based descriptive study of 71 consecutive cases diagnosed between 1980 and 2003. Leuk Lymphoma (2008) 49:1537–44. doi:10.1080/10428190802136368
93. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol (2009) 27:5410–7. doi:10.1200/JCO.2008.21.6150
94. Rangwala S, Zhang C, Duvic M. HDAC inhibitors for the treatment of cutaneous T-cell lymphoma. Future Med Chem (2012) 4:471–86. doi:10.4155/fmc.12.6
95. Duvic M. Histone deacetylase inhibitors for cutaneous T-cell lymphoma. Dermatol Clin (2015) 33:757–64. doi:10.1016/j.det.2015.05.010
96. Li J, Bonifati S, Hristov G, Marttila T, Valmary-Degano S, Stanzel S, et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol Med (2013) 5:1537–55. doi:10.1002/emmm.201302796
Keywords: oncolytic virotherapy, oncolytic H-1 parvovirus, glioblastoma, pancreatic ductal adenocarcinoma, oncolytic (parvo)virotherapy of hematological malignancies, diffuse large B-cell lymphoma, cutaneous T-cell lymphoma
Citation: Angelova AL, Witzens-Harig M, Galabov AS and Rommelaere J (2017) The Oncolytic Virotherapy Era in Cancer Management: Prospects of Applying H-1 Parvovirus to Treat Blood and Solid Cancers. Front. Oncol. 7:93. doi: 10.3389/fonc.2017.00093
Received: 22 March 2017; Accepted: 25 April 2017;
Published: 12 May 2017
Edited by:
Joshua Pesach Rosenzweig, Cell-El Ltd., IsraelReviewed by:
Emily R. Levy, National Institutes of Health, USADaniel Olive, Institut national de la santé et de la recherche médicale (INSERM), France
Copyright: © 2017 Angelova, Witzens-Harig, Galabov and Rommelaere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Assia L. Angelova, YS5hbmdlbG92YUBka2Z6LWhlaWRlbGJlcmcuZGU=