 Veronika Mladenova
Veronika Mladenova Emil Mladenov
Emil Mladenov George Iliakis
George Iliakis- Institute of Medical Radiation Biology, University of Duisburg-Essen Medical School, Essen, Germany
The adverse biological effects of ionizing radiation (IR) are commonly attributed to the generation of DNA double-strand breaks (DSBs). IR-induced DSBs are generated by clusters of ionizations, bear damaged terminal nucleotides, and frequently comprise base damages and single-strand breaks in the vicinity generating a unique DNA damage-clustering effect that increases DSB “complexity.” The number of ionizations in clusters of different radiation modalities increases with increasing linear energy transfer (LET), and is thought to determine the long-known LET-dependence of the relative biological effectiveness (RBE). Multiple ionizations may also lead to the formation of DSB clusters, comprising two or more DSBs that destabilize chromatin further and compromise overall processing. DSB complexity and DSB-cluster formation are increasingly considered in the development of mathematical models of radiation action, which are then “tested” by fitting available experimental data. Despite a plethora of such mathematical models the ultimate goal, i.e., the “a priori” prediction of the radiation effect, has not yet been achieved. The difficulty partly arises from unsurmountable difficulties in testing the fundamental assumptions of such mathematical models in defined biological model systems capable of providing conclusive answers. Recently, revolutionary advances in methods allowing the generation of enzymatic DSBs at random or in well-defined locations in the genome, generate unique testing opportunities for several key assumptions frequently fed into mathematical modeling – including the role of DSB clusters in the overall effect. Here, we review the problematic of DSB-cluster formation in radiation action and present novel biological technologies that promise to revolutionize the way we address the biological consequences of such lesions. We describe new ways of exploiting the I-SceI endonuclease to generate DSB-clusters at random locations in the genome and describe the possible utility of Zn-finger nucleases and of TALENs in generating DSBs at defined genomic locations. Finally, we describe ways to harness the revolution of CRISPR/Cas9 technology to advance our understanding of the biological effects of DSBs. Collectively, these approaches promise to improve the focus of mathematical modeling of radiation action by providing testing opportunities for key assumptions on the underlying biology. They are also likely to further strengthen interactions between experimental radiation biologists and mathematical modelers.
Introduction
All living organisms are continuously exposed to background ionizing radiation (IR) deriving from space, solar activity, or emitted by certain minerals and soils. Although it is generally accepted that IR “per se” can be harmful, IR is nevertheless extensively used for diagnostic purposes and in cancer therapy. Therefore, it is of a great importance to investigate and rationalize the mechanisms of IR action on living organisms, as this will directly help to maximize human radiation protection and to optimize approaches to cancer treatment.
Ionizing radiation generates a broad spectrum of DNA damages, encompassing single-strand breaks (SSB), a variety of oxidative base lesions, DNA–DNA crosslinks as well as DNA–Protein crosslinks, and double-strand breaks (DSBs) (1, 2). However, from the variety of lesions induced by IR, the DSB elicits most of the documented detrimental effects (3, 4), including genomic rearrangements, chromosome aberrations, cell death, genetic mutations, and cancer (5–8).
It has long been known that different IR modalities generate markedly different biological responses although they generate in principle the same basic lesions described above. Thus, α-particles, neutrons, or high-charge and energy particles (HZE ions) are significantly more effective in killing cells than high-energy electrons or protons, γ-rays, or X-rays (9, 10). This increased efficacy, typically described by the higher relative biological effectiveness (RBE), depends on the linear energy transfer (LET) of the radiation modality – which for charged particles is the energy absorbed per unit particle track-length, expressed as kiloelectron-volts/micrometer. Typically, RBE increases with increasing LET of radiation up to a maximum and declines subsequently (11–14).
In the recent years, charged particles such as carbon ions are increasingly considered as main modality for cancer radiotherapy and inflammation treatment in an effort to harness in a targeted manner their higher LET (15–18).
Complexity of IR Induced DSBs: The Role of LET
Bacteria harness the severity of DSB as a lesion to protect themselves from foreign DNA. A family of enzymes, known as restriction endonucleases (RE), recognize and cut specific DNA sequences generating DSBs with blunt or staggered ends. During this process, nucleotides are not altered and the phosphodiester bond retains the 5′-phosphate and 3′-OH groups at each DNA strand. As a result, processing and removal of the DSB by simple ligation is in principle possible. RE-generated DSBs have been used to model IR-induced DSBs (see below) (19, 20) and found to have reparability that depends on the type of ends generated (21–23).
The approach to model DSBs using nucleases gained ground with the introduction of I-SceI homing endonuclease, whose 18-bp long recognition sequence (5′-TAGGGATAA/CAGGGTAAT-3′) is not present in the mammalian genome. The I-SceI recognition sequence can nevertheless be inserted into a mammalian genome according to a pre-conceived design using molecular biology approaches (24–27). I-SceI recognition sequences artificially introduced in a genome can be cut to generate DSBs by expressing constitutive or inducible forms of the endonuclease (28, 29). The biological consequences of these DSBs can then be analyzed using molecular biology approaches. The strength of the method lies in the fact that DSBs are generated at defined locations in the genome, and that combination with appropriately constructed reporters allows analysis of the underlying processing mechanisms.
When DSBs are induced by IR via oxidation reactions – either direct loss of an electron or attack from an ⋅OH produced from the radiolysis of water – they frequently comprise ends with a 3′-damaged sugar in the form of phosphoglycolate and a 5′-OH groups (30–33). Such ends prevent direct DNA ligation and necessitate end-processing during repair (34). Moreover, the adverse biological effects of X-rays or γ-rays are thought to derive from DSBs generated within ionization clusters (35, 36), and not by the coincidence of independently generated ionizations on opposite DNA strands. Indeed, track-structure calculations using computational approaches (37–39) show that secondary electrons, at the end of their tracks generate clusters of ionizations, i.e., multiple ionizations confined in a small volume.
Despite the generation of ionization clusters at the ends of low-energy electron tracks, X-rays and γ-rays still deposit 50–70% of their energy in well-separated ionization events that generate a relatively even ionization pattern within the cell (35, 36). Consequently, X-rays and γ-rays are considered low-LET forms of IR. On the other hand, charged particles (e.g., α-particles or carbon ions) are considered as high-LET forms of radiation because they ionize along their tracks at a higher rate than the electrons generated by X-rays (40).
This increased clustering of ionizations generates DNA damage that is more complex than that induced by low-LET radiations, in the sense that it comprises more DNA lesions within one or two turns of the DNA helix (33). It constitutes what is sometimes called clustered damage sites (CDS) or multiply damaged sites (MDS) (41, 42). While MDS are generated by low-LET radiation such as X-rays, they occur more frequently after exposure to high-LET radiations and are implicated in their enhanced biological effects.
Indeed, about 30% of DSBs contain additional lesions following exposure to low-energy electrons; notably, this fraction increases up to 70% at the same dose of α-particles. In addition, the ratio of the number of SSBs to DSBs decreases from 22.8 for 60Co γ-rays to 3.4 for 50 MeV 12C-ions (39, 43). Since these shifts in the spectrum of lesions do not increase the yields of DSBs in a manner corresponding to the increased killing after exposure to high- versus low-LET radiation, it can be inferred that increased clustering of DNA damage is an important determinant of the biological effect (see also below) (44).
Complexity at a DSB may compromise repair through the simultaneous recruitment of multiple repair-pathway-factors (e.g., from one of the DSB repair pathways together with factors of BER) to close-by lesions in the DNA. Moreover, it may even generate a DSB indirectly when in a complexly damaged DNA, individual lesions in the two strands are processed independently (6, 43, 45, 46). There is evidence that this form of clustered DNA damage outnumbers direct DSBs after exposure to low-LET radiation by nearly 4:1. Similarly, delayed formation of DSBs can occur from the chemical evolution in cells of thermally unstable lesions, which initially do not break the DNA, but which do so minutes after irradiation as they become chemically modified in the cellular milieu (47–52). DSB repair models considering DSB complexity have been also developed to describe radiation effects and DSB repair kinetics throughout the cell cycle (53).
It is thus evident that IR-induced DSBs are the products of ionization clusters that generate clustered DNA damage, which can present in different forms of complexity including modified ends, presence of other lesions in the vicinity of the break, as well lesions that generate DSBs only after enzymatic or chemical processing. Since the size of ionization clusters that generate complex DSBs increases with increasing LET, it is plausible to consider this form of DNA damage complexity as a relevant determinant of the increased effectiveness of high-LET radiation.
Higher Order of DNA Damage Complexity: DSB Clusters
An additional level of DSB complexity is generated by clusters of DSBs (33, 54). This form of DNA damage severely undermines local chromatin stability and thus overall processing in a chromatin-location and composition-dependent manner. DSB clustering as a cause of irreversible radiation effects has been considered by several investigators [see Ref. (39) for a review]. Thus, Bryant and his group developed a non-ionic neutral filter elution assay to generate histone-depleted nuclear structures retaining higher order nuclear matrix organization, and used it to measure DNA fragment loss from two or more DSBs within a single-looped chromatin domain (55–57). They proposed that the spatial distribution of DSBs in higher order chromatin loops affects their reparability, and that misrepair involves DNA fragment-loss at such DSB clusters.
Holley and Chatterjee also considered DSB clusters as a particularly consequential form of radiation damage particularly for high-LET radiations (58). In their calculations, they found fragmentation peaks at 85 bp and then again at multiples of 1000 bp, which they interpreted as reflecting aspects of chromatin structure. Notably, such fragments could indeed be detected by pulsed-field gel electrophoresis in irradiated cells (59, 60) and have also been postulated using alternative modeling approaches (14, 39, 61, 62).
Atomic force microscopy imaging shows clustered DSBs and formation of short DNA fragments – even when irradiating “naked” DNA (63). Small (<30 bp) DNA fragments generated from clustered DSBs have also been propose to compromise Ku function (64). Further work shows that DNA–PK, a complex between the Ku and DNA–PKcs, is also inhibited by short (14–20 bp) DNA fragments (63).
The contribution of DSB clusters to the adverse effects of IR has been the focus of extensive mathematical modeling (39). Ostashevsky developed a model according to which DSB clusters generate small DNA fragments, which can be lost from the chromatin context, thus compromising repair of the constituent DSBs (65, 66). A more specialized induction of DSB clusters within chromatin loops, similar to that considered by Bryant, has been used to develop alternative mathematical models (39, 61, 62, 67, 68). In addition, Scholz and his group (69–71) use an extension of the Giant LOop Binary LEsion (GLOBLE) model (72) and classify DNA lesions with respect to their distribution in giant chromatin loops as single DSBs or DSB clusters (~2 Mbp in size) (73–75). These assumptions generally allow successful fitting of cell survival data (76, 77), including fluctuations of radiosensitivity throughout the cell cycle (72).
Mathematical models to analyze DSB repair kinetics based on DSB complexity have been also recently developed (78). In the synapsis formation (SF) model, the rejoining of complex DSBs is not simulated as a first order event (break filling/joining). Rather the rejoining of complex DSBs is assumed to be realized through SF, similar to a second-order reaction between DNA ends. This approach allows DNA ends to be clearly defined before the SF, which is essential for predicting higher number of chromosomal aberrations after high- as compared to low-LET radiation.
Notably, the generation of DSB clusters represents a form of chromothripsis, defined as chromosome shattering and subsequent incorrect rejoining that underpins carcinogenesis (79–82).
The satisfactory fitting of experimental data achieved under these assumptions points to the biological relevance of DSB clusters as a level of DNA damage complexity that likely explains the increased biological efficacy of high LET radiation.
Mathematical Modeling of Radiation Action Will Benefit from Molecular Biology Approaches Directly Testing Their Basic Assumptions
Collectively, the above outline shows how the physical clustering of ionizations generates DSBs of different complexity, as well as DSB-clusters, and places these forms of DNA damage to the center of responses elicited by radiation modalities of different LET. The recognition that discontinuities in the genome may be caused by DSBs of widely different complexity, immediately implies different biological consequences.
Information on the molecular underpinnings of the responses elicited by genomic breaks of different complexity is scarce despite the central contribution widely attributed to this parameter in the overall radiation effect. As a result, DNA damage complexity is typically only mathematically “modeled” in radiation response formalisms, without direct knowledge of the biological effects of each complexity level. As a consequence, quality of fitting is the only way for testing the validity of the basic assumptions on which these models rest. Yet, this approach is not satisfactory due to the large spectrum of DNA damages induced by IR and their dependence on LET that increases the number of parameters required for complete mathematical modeling.
Furthermore, IR-dependent DSB induction by nature precludes mechanistic molecular biology experiments on the molecular processing of individual lesions, as irradiated cells sustain DSBs in a stochastic manner at different numbers and severity that are randomly distributed throughout the genome. As a result, analysis of effects is only possible by theoretical modeling (39).
The above difficulties and shortcomings suggest that the field will benefit from molecular biology approaches allowing induction and processing analysis for specific forms of DSBs generated at specific locations in the genome. With such model-DSBs, the probability associated with each form to be processed correctly or incorrectly by each of the available repair pathways can be estimated. This information may subsequently be fed as a defined constant in mathematical models, reducing thus the number of free parameters and increasing the predictive power of the model. In the following sections, we describe such biological approaches and explain strengths and limitations.
Endonuclease-Induced DSBs: The Simplest Form
Almost a decade ago, a fundamentally new approach for analyzing the effects of DSBs in living cells was introduced using rare cutting homing endonucleases. The most widely used member of this family of enzymes is the Saccharomyces cerevisiae I-SceI endonuclease. As already mentioned, I-SceI recognizes a unique 18-bp long DNA sequence (Figure 1A), which is absent from the human and mouse genomes. Thus, in order for I-SceI to generate a DSB in these genomes, its recognition sequence must first be inserted using molecular biology approaches. Subsequent expression of I-SceI will generate a DSB, specifically at the site of integration of the recognition sequence, which can be preselected or random (83, 84). Expression of I-SceI must be transient and can be mediated by transient transfection of constitutively expressing vectors, or by the proper activation of an inducible enzyme.
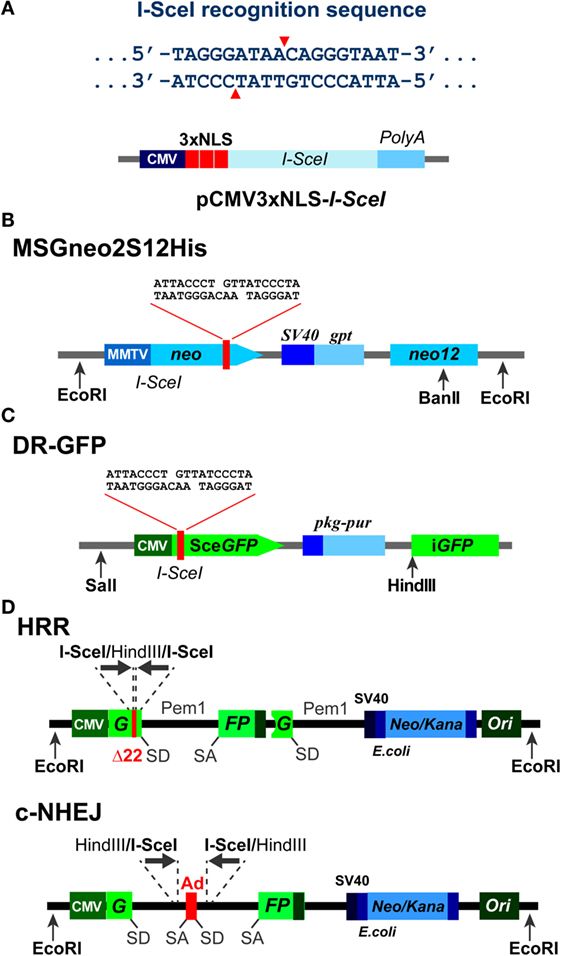
Figure 1. Background information on I-SceI based constructs. (A) Top: recognition sequence of I-SceI endonuclease. Note the generation of 3′ 4-bp overhangs. Bottom: domain structure of an expression vector, pCMV3xnls-I-SceI, used frequently in transient transfection experiments to express I-SceI (85). Note the three NLS sites that ensure the nuclear localization of the expressed enzyme. (B) Schematic representation of the MSGneo2S12His neomycin reporter vector developed to specifically analyze repair of the I-SceI DSB by HRR (86). (C) Schematic representation of the DR-GFP vector developed to specifically analyze repair of the I-SceI DSB by HRR (87). The GFP signal allows analysis by flow cytometry 1–3 days after transfection. (D) Schematic representation of reporter constructs utilizing the Pem1 intron and the Ad2 exon elements, and specifically developed to analyze repair of the I-SceI-DSB by HRR and c-NHEJ, respectively (88).
In the most typical application of this model system, the I-SceI recognition sequence is combined in a construct including a reporter gene [e.g., neomycin or in recent reporter assays, green fluorescence protein (GFP)] and located to interrupt its expression. Restoration of reporter expression serves as readout for the operation of a particular repair pathway in the processing of the DSB. These reporter constructs are in their majority integrated in the genome and are appropriately designed to evaluate repair efficiency through homologous recombination repair (HRR), classical non-homologous end joining (c-NHEJ), alternative end joining (alt-EJ), or single-strand annealing (SSA). Thus, analysis of DSB processing by a specific DSB repair pathway requires the construction of the appropriate vector and its integration into the genome.
In initial studies, I-SceI was utilized to induce a DSB between two inactive neomycin (neo) direct repeat genes integrated into the genome of CHO cells, processing of which by homologous recombination generated a functional neo gene (86, 89) (Figure 1B). In these constructs, the fist neo allele is inactivated by the I-SceI recognition sequence. The second neo allele is promoterless or truncated and may carry silent single-base substitutions that create restriction sites useful in product characterization through restriction fragment length polymorphism analysis. In the native state of this construct, neo is not expressed, and cells are sensitive to neomycin. However, after I-SceI-mediated DSB induction, gene conversion may generate a functional neo gene and thus also neomycin-resistant clones. Such events are considered to reflect successful processing of the DSB by HRR.
More recently, DR-GFP reporter systems based on two directly repeated copies of the gene encoding GFP have been developed in the laboratory of Dr. Jasin (87) (Figure 1C) and find in different forms wide application in the field. In this system, gene conversion events result in expression of GFP, which can be quantitated by flow cytometry. The two mutated GFP genes are oriented as direct repeats and are separated by a puromycin N-acetyltransferase gene, which allows selection for cells carrying the construct. Distinct advantage of this version of the assay is that results are typically available 1–3 days after transfection of the I-SceI expression plasmid. Analysis of neomycin resistance, on the other hand, requires 1–2 weeks.
The DR-GFP reporter system has been successfully adapted to human cells, resulting in generation of U2OS–DR-GFP cells (90) (Figure 2A). Similar to the previously described system, one of the GFP genes in DR-GFP, SceGFP, is mutated by the insertion of the I-SceI recognition site, while the second, internal GFP fragment, iGFP, located 821 bp downstream from SceGFP, has lost its active promotor element.
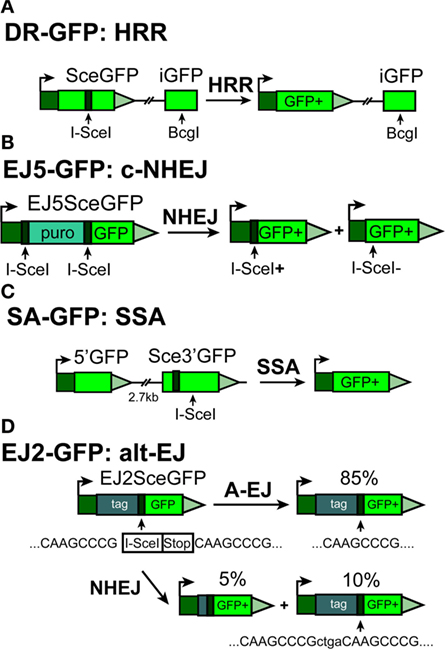
Figure 2. Outline of reporter constructs developed in the laboratory of Dr. J. Stark (90) to analyze efficiency of I-SceI-DSB-processing by different DSB repair pathways in human cells. (A) DR-GFP construct for analyzing HRR. (B) EJ5-GFP for analyzing c-NHEJ (C) SA-GFP construct for analyzing single-strand annealing (SSA) and (D) EJ2-GFP construct for analyzing alt-EJ.
All reporter systems described above are designed to determine the activity of HRR in the processing of a single DSB induced by I-SceI endonuclease. In addition to the above systems, a set of GFP-based fluorescent reporter constructs has been generated by Gorbunova et al. (88, 91) (Figure 1D), allowing analysis of NHEJ and HRR. These constructs are also based on artificially engineered GFP genes containing I-SceI recognition sites for the induction of a DSB. In their native state, the integrated constructs do not express GFP as a result of an N-terminal truncation and mutations in the duplicated gene (in the case of the HRR construct), or by the integration within the GFP gene of an exon (Ad) flanked by the Pem1 intron elements (in case of the NHEJ construct). Here again, successful repair of the I-SceI-induced DSB by NHEJ or HRR will restore the GFP gene, an event that is quantitated by flow cytometry.
Green fluorescence protein-based reporter substrates are now also available to specifically assess c-NHEJ, SSA, or alt-EJ (29, 90, 92, 93). Most of these constructs rely on the principles described above, but include elements allowing analysis of a specific DSB repair pathway (Figures 2A–D).
As already mentioned, use of I-SceI as DSB inducer requires integration of its recognition sequence in the genome of human, mouse, or hamster cells. During the last few years, alternative approaches have been developed using endonucleases for which recognition sequences are present in the genome. One of these systems utilizes the I-PpoI endonuclease, a member of a His–Cys box family of homing endonucleases isolated from the myxomycetous Physarum polycephalum (94), to induce multiple DSBs in the human genome (95–97).
I-PpoI is a relatively small enzyme (18–20 kDa), operating as a homodimer, which in its natural host functions to cleave the highly conserved 15-bp ribosomal DNA homing sites (Figure 3A) to generate target intron transposition or “homing” (98). Expression of I-PpoI in human cells causes cleavage of approximately 10% of the identified I-PpoI genomic target sites (200–300 per genome) (95), generating 20–30 DSBs per cell, equivalent to the number of DSBs introduced by 1 Gy of X-rays. For increased versatility, a system has been developed using an I-PpoI fused to a mutant estrogen receptor hormone-binding domain. The fusion protein stays constitutively in the cytoplasm unable to generate DSBs. Translocation into the nucleus can be mediated by incubation with 4-hydroxytamoxifen (4-OHT) (99–101), allowing thus the regulated induction of DSBs.
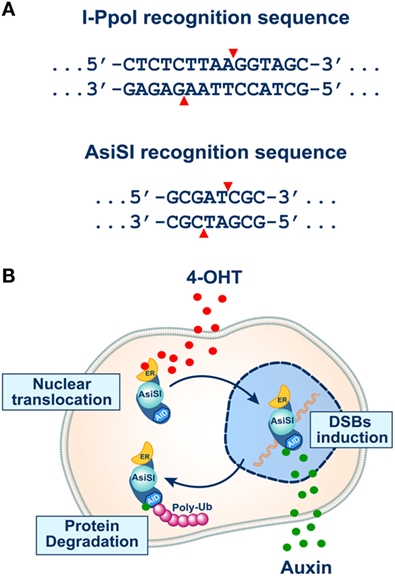
Figure 3. (A) Recognition sequences of I-PpoI (top) and AsiSI (bottom) endonucleases (see text for details). Note that both endonucleases generate 3′ overhangs. (B) The inducible system of AsiSI endonuclease developed in the laboratory of Dr. Legube (102). The DIvA cell line expresses a form of the AsiSI endonuclease fused to estrogen receptor (ER) and the auxin-inducible degron (AID). The enzyme sequesters under normal conditions in the cytoplasm unable to reach the nucleus and thus to induce DSBs. Administration of tamoxifen (4-OHT) causes efficient translocation of the enzyme to the cell nucleus and the induction of DSBs (top part of the schematic). In this system, the endonuclease activity of AsiSI can be rapidly turned off by removing 4-OHT and administering auxin that activates the degron element and causes ubiquitin-mediated degradation of the enzyme (bottom part of the schematic).
This system allows characterization of several features of the DSB response in human cells and has certain advantages over I-SceI-based systems. First, I-PpoI sites are present at well-known locations in the genome, which obviates their artificial introduction. Second, evolutionary conservation of the endogenous I-PpoI sites permits DSBs to be introduced and assays to be performed in virtually any eukaryotic cell line. Third, as I-PpoI induces multiple DSBs in the genome, full activation of the DNA damage response ensues, which allows analyses that go beyond repair pathway utilization.
An elegant assay along similar lines has also been proposed by Aymard et al. (103). These investigators developed a cellular system harboring a stable integration of a gene expressing the rare-cutting AsiSI restriction nuclease, which targets an 8-bp double-stranded DNA sequence (Figure 3A) and cleaves between the T and C to generate a 2-bases, 3′ overhanging ends (25, 104, 105). The genome-integrated AsiSI endonuclease in this model system is also fused to a modified estrogen-receptor ligand-binding domain. Thus, treatment of cells with 4-OHT triggers nuclear localization of the AsiSI enzyme and the rapid induction of approximately 150 sequence-specific DSBs dispersed across the genome (25, 104). This system provides a unique opportunity to simultaneously study, at a molecular level, repair events that transpire at many different DSBs located within various known chromatin locations (103).
Furthermore, as the AsiSI cleavage-sites are known, it is possible to use chromatin immunoprecipitation (ChIP) to directly monitor recruitment of repair factors onto damaged chromatin (103). Using this approach, it could be demonstrated that DSBs induced across the genome are not repaired by the same DSB repair pathway, and that transcriptionally active, H3K36me3 enriched, chromatin is preferentially repaired by homologous recombination, thereby pointing out a critical role of pre-existing chromatin state as determinant of DSB repair pathway selection (103).
As with the I-SceI and I-PpoI systems, the AsiSI system does not allow analysis of DSB repair kinetics because the enzyme remains present in the nucleus for prolonged periods of time and can cut repeatedly. To reduce this limitation, the same group added an auxin-inducible degron (AID) to AsiSI-ER fusion nuclease, thus allowing fast and efficient degradation upon auxin addition (102, 106) (Figure 3B). A similar improvement has also been successfully introduced in the I-SceI system and was coupled with an extension allowing parallel analysis of HRR and alt-EJ (29).
Zn-F Nucleases and TALENs: Tools for Site-Specific DSB Generation
Before the discovery of the CRISPR/Cas9 system that rapidly overtakes all previous systems (see below), significant effort and investment in resources was placed in two families of site-specific nucleases: the zinc-finger nucleases (ZFNs) and the transcription activator-like effector nucleases (TALENs). Both families of engineered proteins have a chimeric design with a common nuclease domain and a DNA-binding domain (Figure 4). In both families, DSB formation is mainly catalyzed by FOK1 endonuclease (107, 108) that generates, depending on the design, cohesive, or blunt DNA ends without sequence specificity (Figure 4). Yet, the concepts underlying the DNA-binding characteristics of ZFNs and TALENs are distinct and responsible for their inherent strengths and limitations. Their versatility arises from the ability to customize through molecular biology approaches their DNA-binding domains in ways that allow the recognition of virtually any DNA sequence.
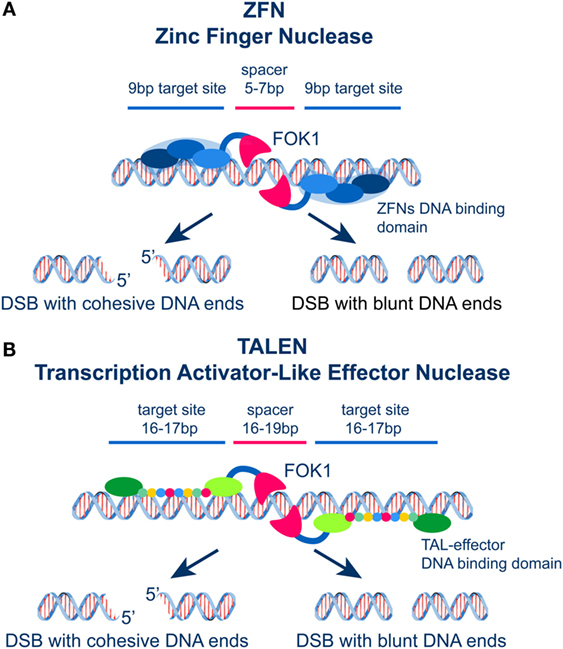
Figure 4. (A) Schematic representation of ZFNs showing the DNA binding and the FOK1 nuclease domains. Two zinc fingers bind left and right the site of DSB generation and localize the activity of FOK1 in a DNA molecule. The DNA binding domains are frequently designed to recognize a 9 bp target sequence. The FOK1 nuclease cuts the DNA strand 5–7 bp 3′ of the target site. Depending on the design of the target sites, expression of a ZNF nuclease will result in the formation of cohesive or blunt DNA ends. (B) Schematic representation of TALENs showing the DNA binding and the FOK1 nuclease domains. Targeting sites and spacer regions are indicated. Here again, cohesive or blunt ends are generated at the DSB depending on the selection of the recognition sites left and right the DSB.
The Cys2–His2 zinc-finger domain is among the most common types of DNA-binding motifs in eukaryotes operating in a widely different array of DNA sequences. It actually represents the second most frequently encoded protein domain in the human genome. The ZFN technology harnesses this biological evolution. An individual zinc-finger consists of approximately 30 amino acids in a conserved ββa (beta-sheet, beta-sheet, and alpha-helix) motif configuration. Each zinc-finger domain contacts 3 or 4 bp in the major groove of the DNA (108). In this technology, different zinc-finger motifs are combined to generate ZFNs that recognize the desired sequence 9 bp left and right from the target region (Figure 4A). The two components of the ZFN cut the corresponding DNA strands using FOK1, which is attached to the C-terminus of the ZFN modules (Figure 4A). Moreover, the cleavage domain requires the 5′ edge of each binding site to be separated by a 5–7 bp spacer region (Figure 4A).
Despite distinct strengths, the construction of ZFNs is complex requiring extensive know-how; it is very time consuming and shows limited flexibility in terms of engineering proteins recognizing any DNA sequence. As a result, their utilization, even before the advent of the CRIPSR/Cas9 system, had given way to the much more flexible TALENS.
TALENs contain TALE repeats of about 33–35 amino acids that recognize a single base pair via two hypervariable residues (repeat-variable di-residues, RVDs) (108). Combined TALE repeats can recognize a specific DNA target site of about 17 bp in length (Figure 4B). As a result of this unique property, TALENs can be easily and flexibly engineered to recognize DNA sequences of arbitrarily chosen lengths and compositions. For the application of TALENs discussed here, the number and location of the induced DSBs will depend on the frequency and the location in the genome of the selected sequence used to design the nuclease (109, 110). Thus, sequences can be selected and TALENs designed inducing in the genome a single DSB or multiple DSBs in variable configurations, depending on the specific question addressed.
The TALEN technology is powerful and flexible, and engineering of a site specific TALEN can be accelerated by “off-the-shelf” components that are combined to generate a functional protein within 1–2 weeks. Although the second generation of TALEN technology further improves on the distinct advantages of the approach, the work required to generate and test a single site specific TALEN nuclease is still considerable (111). As a result, this technology is also rapidly losing ground to CRIPR/Cas9 technology.
The CRISPR/Cas9 System: Great Versatility, Short Learning Curve
The CRISPR/Cas9 system with all its applications and potential is arguably the most rapidly expanding and evolving field in modern biology (Figure 5). From the initial discovery of the system as part of bacteria immunity to its modification for sequence specific genome editing, the technology has gone through a series of revolutionary developments that have been extensively reviewed (112–114). Relevant for the present outline is the potential of the system to generate a DSB anywhere in the genome by targeting at the specific location the Cas9 nuclease. Cas9 cuts the DNA as shown in Figure 5 to generate blunt DNA ends, guided by a partially complementary RNA molecule (gRNA). In its current stage of development, gRNA carries in addition to the sequence required for the proper targeting of Cas9 to the DNA molecule also a sequence for its activation.
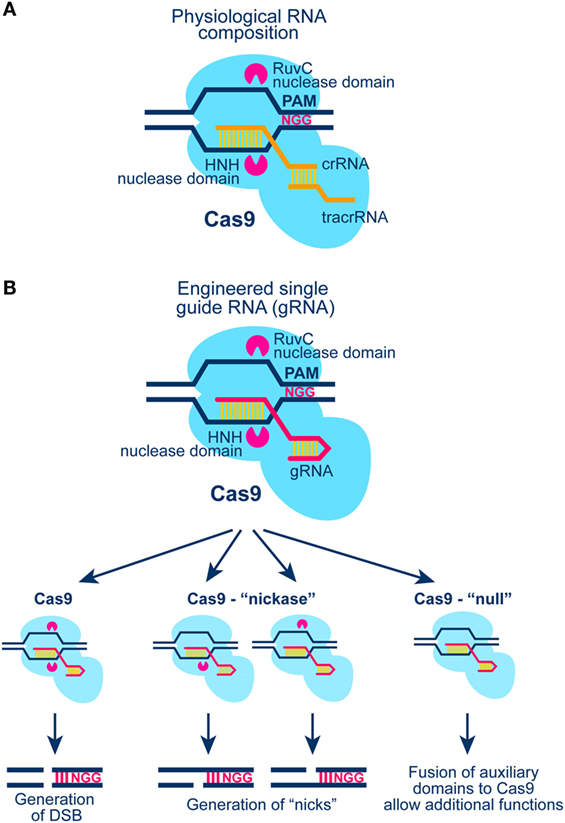
Figure 5. (A) Schematic representation of the CRISPR/Cas9 system in S. Pyogenes (see text for details). Cas9 generates blunt ends by cutting 3 bp downstream the PAM region of the target DNA molecule. (B) Schematic representation of Cas9 activation with a chimeric gRNA combining crRNA and tracrRNA (see A; see also text for more details). Cas9 nuclease harbors two nuclease domains: HNH and RuvC-like; mutation of one or both of these nuclease domains results in Cas9 enzymes with “nickase” properties, or with null nuclease activity. The latter form of Cas9 can be tethered to other proteins for gain-of-function DNA sequence-specific operations.
Originally, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) were found in the genome of Escherichia coli, but their function remained unknown until recently, when it was shown that these genetic elements are essential for the development of resistance against bacteriophages (115). Moreover, the CRISPR-associated protein 9 (Cas9) was described as a RNA-guided DNA endonuclease associated with the CRISPR-adaptive “immune” system in Streptococcus pyogenes (115, 116).
Three types of CRISPR systems have been identified thus far; from these forms type II is the most widely studied and the most relevant to the present outline. In type II CRISPR system, invading DNA is nucleolytically processed into small fragments (approximately 20 bp) that are incorporated into the CRISPR locus. This locus is transcribed, and transcripts are then processed to generate small CRISPR-RNAs (crRNA), which together with a trans-activating-CRISPR-RNA (tracrRNA) guide Cas9 to digest invading DNA upon repeat encounter (Figure 5A).
An important element of Cas9 activation is the Protospacer Adjacent Motif (PAM) at the target DNA sequence, which is essential for interactions between Cas9 and DNA (Figure 5A). Early work revealed that all three components (Cas9 protein, mature crRNA and tracrRNA) are required for efficient recruitment to and digestion of the target DNA sequence. A major development in the field was the recognition that crRNA and tracrRNA can be combined to generate a single guide RNA (gRNA) that enables all operations required for the targeted function of Cas9 (Figure 5B). This development greatly simplified the evolution of a large array of applications and forms the basis of the applications described here.
The number of applications utilizing CRISPR/Cas9-related genome editing/manipulation approaches is increasing exponentially with time. The technology is also very powerful for studies on the effects of single DSBs and DSB clusters in the mammalian genome (117, 118). Thus, existing CRISPR/Cas9 systems (116, 119) can be combined with appropriately designed gRNAs with the aim of inducing single DSBs or DSB clusters of different complexity within exons or introns of selected genes and study consequences in cell survival, genome integrity, DSB-response, or gene function. Figures 6A,B show as an example possible site selections for DSB induction within the HPRT gene, since it has been extensively used in the past to study IR-induced mutation induction (120, 121). Here, single DSBs and DSB clusters are induced at selected locations throughout the gene and at various constellations by combining different gRNAs (Figure 6).
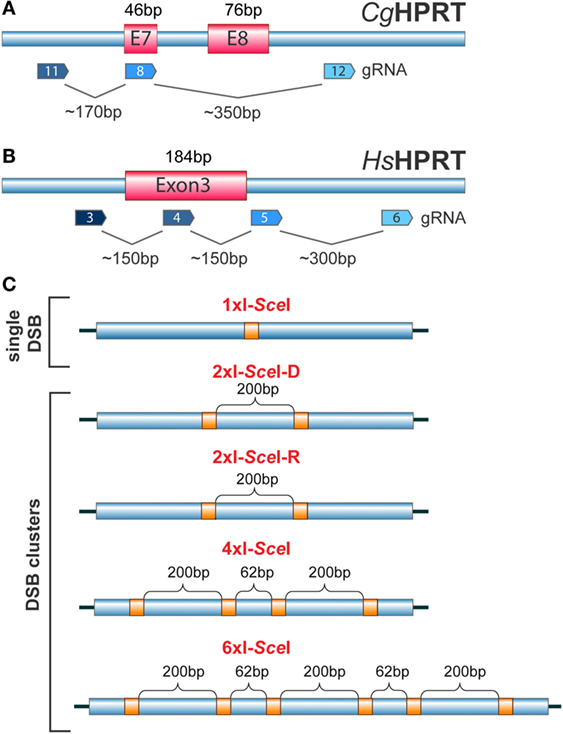
Figure 6. (A) Organization of the HPRT gene in the Chinese hamster C. griseous (Cg) near exons 7 and 8. Possible recognition sites of gRNAs allowing the generation of DSBs at different locations within exons and introns are indicated. (B) Exon 3 of the HPRT gene in H. sapiens (Hs). Possible recognition sites for gRNAs allowing the generation of single DSBs or DSB clusters within exons and introns are indicated. (C) Constructs carrying different combinations of I-SceI sites engineered at different distances to model DSB clusters of increasing complexity. The schematic shows I-SceI constructs that would generate upon integration in the genome of a cell, single DSBs, DSB pairs, DSB quadruplets or a cluster of six DSBs. The distances shown are arbitrary and chosen only for illustration purposes.
The CRISPR/Cas9 technology is extremely powerful and promises to revolutionize the field by virtue of its ability to generate DSBs with great ease at any location of a known genome. In addition, mutated forms of Cas9 can be introduced in which one or both endonuclease domains are inactivated, thus generating enzymes with “nickase” or “null” activity (Figure 5B). Combination of Cas9 “nickase” with other systems of DSB generation, including the I-SceI system described below, will allow testing of the biological consequences of a single-strand break in the vicinity of a DSB (complex DSB). Finally, fusions between a non-functional Cas9 enzyme and protein domains generating additional forms of DNA damage will further expand the spectrum of experiments investigating DSB complexity as a parameter in biological responses (Figure 5B). As with the I-SceI, I-PpoI, and the AsiSI systems, Cas9 will also generate repeated DSBs in the genome and its prolonged presence in the cell nucleus precludes analysis of DSB repair kinetics.
The fact that the generation of each DSB using the CRISPR/Cas9 technology requires individual gRNAs restricts somewhat its application for generating multiple DSBs, which at times may be a desirable outcome (see next session). This is because the generation of multiple single DSBs or DSB clusters at different genomic locations will require a number of gRNAs. This problem may be partly overcome by designing gRNAs, which recognize sequences in the genome that are repeated several times – excluding of course highly repeated DNA sequences. For example, many proteins contain common functional domains, encoded by similar if not identical DNA sequences, which could be targeted at once using a single gRNA molecule. An alternative solution for this limitation is offered by the model system described in the following section.
I-SceI-Based Models of DSB Clustering
We conclude this overview by outlining a recently introduced I-SceI-based model system, complementary to the system outlined in the previous section using the CRISPR/Cas9 technology that allows direct analysis of assumptions regarding the biological effects of multiple single DSBs and DSB clusters (54). The model system utilizes transposon technology (122) to generate clonal cell lines with multiple genomic integrations of constructs carrying I-SceI restriction sites at arrangements selected depending on the specific question addressed (Figure 6C). Cleavage of these sites by transient or conditional expression of I-SceI to generate single DSBs or DSB clusters at different numbers (typically 1–15) and constellations allow analysis of the biological consequences at different endpoints. First results obtained using this model system (54) indicate that DSB clusters compromise c-NHEJ and possibly HRR, leaving alt-EJ as last resort in DSB processing.
Application of the same technology to mutant cell lines with defects in different aspects of DSB repair will allow extensive analysis of the DSB repair pathways handling this form of damage (4, 123, 124).
Concluding Remarks
It is evident from the above outline that numerous novel technologies are available that promise to revive and revolutionize the ways we address fundamental questions of radiation damage and the associated radiation responses. These technologies allow the testing in well-defined systems of key hypotheses of mathematical models of radiation action, and the generation of data that may help to reduce their free variables. Such systems will be particularly useful in the analysis and characterization of the role of single DSBs and DSB cluster formation in the biological responses of high LET radiation.
The system utilizing the I-SceI meganuclease to generate single DSBs and DSB clusters at random locations in the genome will extend the successful application this enzyme saw during the past 15 years to new questions relevant to DNA damage response. Zn-finger nucleases and TALENs will perhaps remain useful in addressing related questions at specific settings. Certainly, the most promising technology is the one utilizing the CRISPR/Cas9 system to introduce DSBs at pre-selected locations in the unmodified genome, with a pre-defined constellation.
One aspect with all systems of enzymatic DSB generation that needs to be considered in comparisons with the effects of IR concerns the specifics of DSB induction. Thus, IR by virtue of its well defined and typically short exposure times induces DSBs through non-recurring, distinct energy deposition events; DSBs induced in this way are subsequently processed and terminally removed from the genome. Nucleases, on the other hand, through their prolonged presence after expression or activation in the cell nucleus, will generate cycles in which initial DSB induction and subsequent processing will be followed by additional cutting and processing cycles, which will in principle continue until repeat processing mutates the nuclease recognition sequence, or until enzyme expression or activity subside. Since both “solutions” require a relatively long window of time, which is also likely to be different for each individual DSB, they generate a condition of chronic assault to the DNA generating “chronic” DSBs. Such chronic DSBs may induce responses with facets not present to those generated by the single events of IR, and which may engage distinct processing mechanisms that change the ultimate outcome. Additional problems may arise from off-target effects, variable on-target cutting frequencies, and the induction of a single form of DSB these systems allow.
Despite these inherent limitations, the approaches described above promise to enrich our knowledge of the biological responses to DSBs, to improve the focus on this form of DNA damage, and to enhance the power and utility of mathematical modeling by generating first principles that can be used as starting points. Last but not least, they are likely to strengthen interactions between experimental radiation biologists and mathematical modelers.
Author Contributions
VM and EM wrote the manuscript and participated in preparation of the figures. GI generated the main outline of the article and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Work supported by grants from BMBF (02NUK043B –COLLAR) and the DFG (GRK1739).
References
1. Goodhead DT, Thacker J, Cox R. Effects of radiations of different qualities on cells: molecular mechanisms of damage and repair. Int J Radiat Biol (1993) 63:543–56. doi:10.1080/09553009314450721
2. Prise KM, Ahnström G, Belli M, Carlsson J, Frankenberg D, Kiefer J, et al. A review of dsb induction data for varying quality radiations. Int J Radiat Biol (1998) 74:173–84. doi:10.1080/095530098141564
3. Kinner A, Wu W, Staudt C, Iliakis G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res (2008) 36:5678–94. doi:10.1093/nar/gkn550
4. Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res (2011) 711:61–72. doi:10.1016/j.mrfmmm.2011.02.005
5. Bryant PE, Riches AC, Terry SYA. Mechanisms of the formation of radiation-induced chromosomal aberrations. Mutat Res (2010) 701:23–6. doi:10.1016/j.mrgentox.2010.03.016
6. Sage E, Harrison L. Clustered DNA lesion repair in eukaryotes: relevance to mutagenesis and cell survival. Mutat Res (2011) 711:123–33. doi:10.1016/j.mrfmmm.2010.12.010
7. Byrne M, Wray J, Reinert B, Wu Y, Nickoloff J, Lee S-H, et al. Mechanisms of oncogenic chromosomal translocations. Ann N Y Acad Sci (2014) 1310:89–97. doi:10.1111/nyas.12370
8. Soni A, Siemann M, Pantelias GE, Iliakis G. Marked contribution of alternative end-joining to chromosome-translocation-formation by stochastically induced DNA double-strand-breaks in G(2)-phase human cells. Mutat Res (2015) 793:2–8. doi:10.1016/j.mrgentox.2015.07.002
9. Barendsen GW. Responses of cultured cells, tumours, and normal tissues to radiations of different linear energy transfer. Curr Top Radiat Res (1968) 4:293–356.
10. Bertsche U, Iliakis G, Kraft G. Inactivation of Ehrlich ascites tumor cells by heavy ions. Radiat Res (1983) 95:57–67. doi:10.2307/3576071
11. Bedford JS, Goodhead DT. Breakage of human interphase chromosomes by alpha particles and X-rays. Int J Radiat Biol (1989) 55(2):211–6. doi:10.1080/09553008914550261
12. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. Philadelphia, Baltimore, New York, London, Buenos Aires, Hong Kong, Sydney, Tokyo: Lippincott Williams & Wilkins (2006).
13. Franken N, Ten Cate R, Krawczyk P, Stap J, Haveman J, Aten J, et al. Comparison of RBE values of high- LET α-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiat Oncol (2011) 6:64. doi:10.1186/1748-717X-6-64
14. Li Y, Qian H, Wang Y, Cucinotta FA. A stochastic model of DNA fragments rejoining. PLoS One (2012) 7:e44293. doi:10.1371/journal.pone.0044293
15. Brenner DJ, Doll R, Goodhead DT, Hall EJ, Land CE, Little JB, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci USA (2003) 100:13761–6. doi:10.1073/pnas.2235592100
16. Durante M, Cucinotta FA. Heavy ion carcinogenesis and human space exploration. Nat Rev Cancer (2008) 8:465–72. doi:10.1038/nrc2391
17. Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol (2010) 7:37–43. doi:10.1038/nrclinonc.2009.183
18. Loeffler JS, Durante M. Charged particle therapy – optimization, challenges and future directions. Nat Rev Clin Oncol (2013) 10:411–24. doi:10.1038/nrclinonc.2013.79
19. Obe G, Winkel E-U. The chromosome-breaking activity of the restriction endonuclease Alu I in CHO cells is independent of the S-phase of the cell cycle. Mutat Res (1985) 152:25–9. doi:10.1016/0027-5107(85)90042-9
20. Bryant PE, Johnston PJ. Restriction-endonuclease-induced DNA double-strand breaks and chromosomal aberrations in mammalian cells. Mutat Res (1993) 299:289–96. doi:10.1016/0165-1218(93)90105-M
21. Pfeiffer P, Feldmann E, Odersky A, Kuhfittig-Kulle S, Goedecke W. Analysis of double-strand break repair by non-homologous DNA end joining in cell-free extracts from mammalian cells. Methods Mol Biol (2005) 291:351–71. doi:10.1007/978-1-62703-739-6_39
22. Van Gent DC, Van Der Burg M. Non-homologous end-joining, a sticky affair. Oncogene (2007) 26:7731–40. doi:10.1038/sj.onc.1210871
23. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:1.1–1.31. doi:10.1146/annurev.biochem.052308.093131
24. Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet (1996) 12:224–8. doi:10.1016/0168-9525(96)10019-6
25. Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, et al. High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J (2010) 29:1446–57. doi:10.1038/emboj.2010.38
26. Fenina M, Simon-Chazottes D, Vandormael-Pournin S, Soueid J, Langa F, Cohen-Tannoudji M, et al. I-SceI-mediated double-strand break does not increase the frequency of homologous recombination at the Dct locus in mouse embryonic stem cells. PLoS One (2012) 7:e39895. doi:10.1371/journal.pone.0039895
27. Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand-breaks. Methods Mol Biol (2012) 920:379–91. doi:10.1007/978-1-61779-998-3_27
28. Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science (2008) 320:1507–10. doi:10.1126/science.1159051
29. Bindra RS, Goglia AG, Jasin M, Powell SN. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res (2013) 41:e115. doi:10.1093/nar/gkt255
30. Henner WD, Grunberg SM, Haseltine WA. Sites and structure of γ radiation-induced DNA strand breaks. J Biol Chem (1982) 257:11750–4.
31. Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. Gamma-ray Induced deoxyribonucleic acid strand breaks. J Biol Chem (1983) 258:711–3.
32. Von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. Berlin, Heidelberg: Springer (2006).
33. Schipler A, Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res (2013) 41:7589–605. doi:10.1093/nar/gkt556
34. Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JNM. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci (2011) 36:262–71. doi:10.1016/j.tibs.2011.01.006
35. Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucl Acid Res (1988) 35:95–125. doi:10.1016/S0079-6603(08)60611-X
36. Goodhead DT, Nikjoo H. Track structure analysis of ultrasoft X-rays compared to high- and low-LET radiations. Int J Radiat Biol (1989) 55:513–29. doi:10.1080/09553008914550571
37. Paretzke HG. Radiation track structure theory. In: Freeman GR, editor. Kinetics of Nonhomogeneous Processes. New York City, NY: John Wiley & Sons, Inc (1987). p. 89–170.
38. Friedland W, Jacob P, Kundrá TP. Stochastic simulation of DNA double-strand break repair by non-homologous end joining based on track structure calculations. Radiat Res (2010) 173:677–88. doi:10.1667/RR1965.1
39. Friedland W, Dingfelder M, Kundrát P, Jacob P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat Res (2011) 711:28–40. doi:10.1016/j.mrfmmm.2011.01.003
40. Allen C, Borak TB, Tsujii H, Nickoloff JA. Heavy charged particle radiobiology: using enhanced biological effectiveness and improved beam focusing to advance cancer therapy. Mutat Res (2011) 711:150–7. doi:10.1016/j.mrfmmm.2011.02.012
42. Ward JF, Evans JW, Limoli CL, Calabro-Jones PM. Radiation and hydrogen peroxide induced free radical damage to DNA. Br J Cancer (1987) 55:105–12.
43. Georgakilas AG, O’neill P, Stewart RD. Induction and repair of clustered DNA lesions: what do we know so far? Radiat Res (2013) 180:100–9. doi:10.1667/RR3041.1
44. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol (1994) 65:7–17. doi:10.1080/09553009414550021
45. Eccles LJ, O’neill P, Lomax ME. Delayed repair of radiation induced clustered DNA damage: friend or foe? Mutat Res (2011) 711:134–41. doi:10.1016/j.mrfmmm.2010.11.003
46. Wilson DM 3rd, Kim D, Berquist BR, Sigurdson AJ. Variation in base excision repair capacity. Mutat Res (2011) 711:100–12. doi:10.1016/j.mrfmmm.2010.12.004
47. Singh SK, Wu W, Wu W, Wang M, Iliakis G. Extensive repair of DNA double-strand breaks in cells deficient in the DNA-PK dependent pathway of NHEJ after exclusion of heat-labile sites. Radiat Res (2009) 172:152–64. doi:10.1667/RR1745.1
48. Singh SK, Wang M, Staudt C, Iliakis G. Post-irradiation chemical processing of DNA damage generates double-strand breaks in cells already engaged in repair. Nucleic Acids Res (2011) 39:8416–29. doi:10.1093/nar/gkr463
49. Singh SK, Wu W, Stuschke M, Bockisch A, Iliakis G. Reduced contribution of thermally-labile sugar lesions to DNA double-strand break formation after exposure to neutrons. Radiat Res (2012) 178:581–90. doi:10.1667/RR2996.1
50. Singh SK, Bencsik-Theilen A, Mladenov E, Jakob B, Taucher-Scholz G, Iliakis G. Reduced contribution of thermally labile sugar lesions to DNA double strand break formation after exposure to heavy ions. Radiat Oncol (2013) 8:77. doi:10.1186/1748-717X-8-77
51. Cheng Y, Li F, Mladenov E, Iliakis G. The yield of DNA double strand breaks determined after exclusion of those forming from heat-labile lesions predicts tumor cell radiosensitivity to killing. Radiother Oncol (2015) 116:366–73. doi:10.1016/j.radonc.2015.08.004
52. Li F, Cheng Y, Iliakis G. The contribution of thermally labile sugar lesions to DNA double-strand break formation in cells grown in the presence of BrdU. Int J Radiat Biol (2015) 91:312–20. doi:10.3109/09553002.2014.996260
53. Taleei R, Nikjoo H. Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat Res (2013) 756:206–12. doi:10.1016/j.mrgentox.2013.06.004
54. Schipler A, Mladenova V, Soni A, Nikolov V, Saha J, Mladenov E, et al. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res (2016). doi:10.1093/nar/gkw487
55. Johnston PJ, Bryant PE. A component of DNA double-strand break repair is dependent on the spatial orientation of the lesions within the higher-order structures of chromatin. Int J Radiat Biol (1994) 66:531–6. doi:10.1080/09553009414551571
56. Johnston PJ, Olive PL, Bryant PE. Higher-order chromatin structure-dependent repair of DNA double-strand breaks: modeling the elution of DNA from nucleoids. Radiat Res (1997) 148:561–7. doi:10.2307/3579731
57. Johnston PJ, Macphail SH, Banáth JP, Olive PL. Higher-order chromatin structure-dependent repair of DNA double-strand breaks: factors affecting elution of DNA from nucleoids. Radiat Res (1998) 149:533–42. doi:10.2307/3579785
58. Holley WR, Chatterjee A. Clusters of DNA damage induced by ionizing radiation: formation of short DNA fragments. 1. Theoretical modeling. Radiat Res (1996) 145:188–99. doi:10.2307/3579174
59. Löbrich M, Cooper PK, Rydberg B. Non-random distrubution of DNA double-strand breaks induced by particle irradiation. Int J Radiat Biol (1996) 70:493–503. doi:10.1080/095530096144680
60. Rydberg B. Clusters of DNA damage induced by ionizing radiation: formation of short DNA fragments. 11. Experimental detection. Radiat Res (1996) 145:200–9. doi:10.2307/3579175
61. Friedland W, Jacob P, Paretzke HG, Stork T. Monte carlo simulation of the production of short DNA fragments by low-linear energy transfer radiation using higher-order DNA models. Radiat Res (1998) 150:170–82. doi:10.2307/3579852
62. Friedland W, Dingfelder M, Jacob P, Paretzke HG. Calculated DNA double-strand break and fragmentation yields after irradiation with He ions. Radiat Phys Chem (2005) 72:279–86. doi:10.1016/j.radphyschem.2004.05.053
63. Pang D, Winters TA, Jung M, Purkayastha S, Cavalli LR, Chasovkikh S, et al. Radiation-generated short DNA fragments may perturb non-homologous end-joining and induce genomic instability. J Radiat Res (2011) 52:309–19. doi:10.1269/jrr.10147
64. Wang H, Zhang X, Wang P, Yu X, Essers J, Chen D, et al. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res (2010) 38:3245–51. doi:10.1093/nar/gkq069
65. Ostashevsky JY. A model relating cell survival to DNA fragment loss and unrepaired double-strand breaks. Radiat Res (1989) 118:437–66. doi:10.2307/3577405
66. Ostashevsky JY. Higher-order structure of interphase chromosomes and radiation-induced chromosomal exchange aberrations. Int J Radiat Biol (2000) 76:1179–87. doi:10.1080/09553000050134410
67. Friedland W, Jacob P, Bernhardt P, Paretzke HG, Dingfelder M. Simulation of DNA damage after proton irradiation. Radiat Res (2003) 159:401–10. doi:10.1667/0033-7587(2003)159[0401:SODDAP]2.0.CO;2
68. Ponomarev AL, Cucinotta FA. Chromatin loops are responsible for higher counts of small DNA fragments induced by high-LET radiation, while chromosomal domains do not affect the fragment sizes. Int J Radiat Biol (2006) 82:293–305. doi:10.1080/09553000600637716
69. Elsässer T, Brons S, Psonka K, Scholz M, Gudowska-Nowak E, Taucher-Scholz G. Biophysical modeling of fragment length distributions of DNA plasmids after X and heavy-ion irradiation analyzed by atomic force microscopy. Radiat Res (2008) 169:649–59. doi:10.1667/RR1028.1
70. Friedrich T, Durante M, Scholz M. Modeling cell survival after photon irradiation based on double-strand break clustering in megabase pair chromatin loops. Radiat Res (2012) 178:385–94. doi:10.1667/RR2964.1
71. Friedrich T, Scholz U, Elsässer T, Durante M, Scholz M. Calculation of the biological effects of ion beams based on the microscopic spatial damage distribution pattern. Int J Radiat Biol (2012) 88:103–7. doi:10.3109/09553002.2011.611213
72. Hufnagl A, Herr L, Friedrich T, Durante M, Taucher-Scholz G, Scholz M. The link between cell-cycle dependent radiosensitivity and repair pathways: a model based on the local, sister-chromatid conformation dependent switch between NHEJ and HR. DNA Repair (2015) 27:28–39. doi:10.1016/j.dnarep.2015.01.002
73. Sachs RK, Van Den Engh G, Trask B, Yokota H, Hearst JE. A random-walk/giant-loop model for interphase chromosomes. Proc Natl Acad Sci USA (1995) 92:2710–4. doi:10.1073/pnas.92.7.2710
74. Ostashevsky J. A polymer model for the structural organization of chromatin loops and minibands in interphase chromosomes. Mol Biol Cell (1998) 9:3031–40. doi:10.1091/mbc.9.11.3031
75. Gauter B, Zlobinskaya O, Weber K-J. Rejoining of radiation-induced DNA double-strand breaks: pulsed-field electrophoresis analysis of fragment size distributions after incubation for repair. Radiat Res (2002) 157:721–33. doi:10.1667/0033-7587(2002)157[0721:RORIDD]2.0.CO;2
76. Menegakis A, Yaromina A, Eicheler W, Dörfler A, Beuthien-Baumann B, Thames HD, et al. Prediction of clonogenic cell survival curves based on the number of residual DNA double strand breaks measured by γH2AX staining. Int J Radiat Biol (2009) 85:1032–41. doi:10.3109/09553000903242149
77. Koch U, Höhne K, Von Neubeck C, Thames HD, Yaromina A, Dahm-Daphi J, et al. Residual γH2AX foci predict local tumour control after radiotherapy. Radiother Oncol (2013) 108:434–9. doi:10.1016/j.radonc.2013.06.022
78. Li Y, Reynolds P, O’neill P, Cucinotta FA. Modeling damage complexity-dependent non-homologous end-joining repair pathway. PLoS One (2014) 9:e85816. doi:10.1371/journal.pone.0085816
79. Stephens PJ, Greenman CD, Fu B, Yang F, Rignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell (2011) 144:27–40. doi:10.1016/j.cell.2010.11.055
80. Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer (2012) 12:663–70. doi:10.1038/nrc3352
81. Kloosterman WP, Tavakoli-Yaraki M, Van Roosmalen MJ, Van Binsbergen E, Renkens I, Duran K, et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep (2012) 1:648–55. doi:10.1016/j.celrep.2012.05.009
82. Molenaar JJ, Koster J, Zwijnenburg DA, Van Sluis P, Valentijn LJ, Van Der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature (2012) 483:589–93. doi:10.1038/nature10910
83. Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA (1994) 91:6064–8. doi:10.1073/pnas.91.13.6064
84. Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature (2000) 405:697–700. doi:10.1038/35015097
85. Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci USA (1998) 95:5172–7. doi:10.1073/pnas.95.9.5172
86. Taghian DG, Nickoloff JA. Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol (1997) 17:6386–93. doi:10.1128/MCB.17.11.6386
87. Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev (1999) 13:2633–8. doi:10.1101/gad.13.20.2633
88. Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (2008) 7:1765–71. doi:10.1016/j.dnarep.2008.06.018
89. Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature (1999) 401:397–9. doi:10.1038/43932
90. Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet (2008) 4:e1000110. doi:10.1371/journal.pgen.1000110
91. Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci USA (2007) 104:13068–73. doi:10.1073/pnas.0702410104
92. Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res (2010) 38:6065–77. doi:10.1093/nar/gkq387
93. Gomez-Cabello D, Jimeno S, Fernández-Ávila MJ, Huertas P. New tools to study DNA double-strand break repair pathway choice. PLoS One (2013) 8:e77206. doi:10.1371/journal.pone.0077206
94. Muscarella DE, Ellison EL, Ruoff BM, Vogt VM. Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol Cell Biol (1990) 10:3386–96. doi:10.1128/MCB.10.7.3386
95. Monnat RJ Jr, Hackmann AFM, Cantrell MA. Generation of highly site-specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochem Biophys Res Commun (1999) 255:88–93. doi:10.1006/bbrc.1999.0152
96. Berkovich E, Monnat RJ Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol (2007) 9:683–90. doi:10.1038/ncb1599
97. Berkovich E, Monnat RJ Jr, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc (2008) 3:915–22. doi:10.1038/nprot.2008.54
98. Belfort M, Roberts RJ. Homing endonucleases: keeping the house in order. Nucleic Acids Res (1997) 25:3379–88. doi:10.1093/nar/25.17.3379
99. Vigo E, Müller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol (1999) 19:6379–95. doi:10.1128/MCB.19.9.6379
100. Berkovich E, Lamed Y, Ginsberg D. E2F and Ras synergize in transcriptionally activating p14ARF expression. Cell Cycle (2003) 2:127–34. doi:10.4161/cc.2.2.293
101. Goldstein M, Derheimer FA, Tait-Mulder J, Kastan MB. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc Natl Acad Sci USA (2013) 110:16874–9. doi:10.1073/pnas.1306160110
102. Caron P, Choudjaye J, Clouaire T, Bugler B, Daburon V, Aguirrebengoa M, et al. Non-redundant functions of ATM and DNA-PKcs in response to DNA double-strand breaks. Cell Rep (2015) 13:1598–609. doi:10.1016/j.celrep.2015.10.024
103. Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol (2014) 21:366–74. doi:10.1038/nsmb.2796
104. Massip L, Caron P, Iacovoni JS, Trouche D, Legube G. Deciphering the chromatin landscape induced around DNA double strand breaks. Cell Cycle (2010) 9:2963–72. doi:10.4161/cc.9.15.12412
105. Caron P, Aymard F, Iacovoni JS, Briois S, Canitrot Y, Bugler B, et al. Cohesin protects genes against γH2AX induced by DNA double-strand breaks. PLoS Genet (2012) 8:e1002460. doi:10.1371/journal.pgen.1002460
106. Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods (2009) 6:917–22. doi:10.1038/nmeth.1401
107. Wood AJ, Lo T-W, Zeitler B, Pickle CS, Ralston EJ, Lee AH, et al. Targeted genome editing across species using ZFNs and TALENs. Science (2011) 333:307. doi:10.1126/science.1207773
108. Gaj T, Gersbach CA, Barbas Iii CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol (2013) 31:397–405. doi:10.1016/j.tibtech.2013.04.004
109. Ghezraoui H, Piganeau M, Renouf B, Renaud J-B, Sallmyr A, Ruis B, et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell (2014) 55:829–42. doi:10.1016/j.molcel.2014.08.002
110. Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun (2014) 5:5560. doi:10.1038/ncomms6560
111. Sun N, Bao Z, Xiong X, Zhao H. SunnyTALEN: a second-generation TALEN system for human genome editing. Biotechnol Bioeng (2014) 111(4):683–91. doi:10.1002/bit.25154
112. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science (2014) 346:1077. doi:10.1126/science.1258096
113. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer (2015) 15:387–95. doi:10.1038/nrc3950
114. Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet (2015) 16:299–311. doi:10.1038/nrg3899
115. Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature (2015) 526:55–61. doi:10.1038/nature15386
116. Mali P, Yang L, Esvelt KM, Aach J, Guell M, Dicarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Science (2013) 339:823–6. doi:10.1126/science.1232033
117. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han Y-C, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature (2014) 516:423–7. doi:10.1038/nature13902
118. Li Y, Park AI, Mou H, Colpan C, Bizhanova A, Akama-Garren E, et al. A versatile reporter system for CRISPR-mediated chromosomal rearrangements. Genome Biol (2015) 16:111. doi:10.1186/s13059-015-0680-7
119. Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods (2013) 10:957–63. doi:10.1038/nmeth.2649
120. Iliakis G. The influence of conditions affecting repair and expression of potentially lethal damage on the induction of 6-thioguanine resistance after exposure of mammalian cells to x-rays. Mutat Res (1984) 126:215–25. doi:10.1016/0027-5107(84)90063-0
121. Thacker J. The molecular nature of mutations in cultured mammalian cells: a review. Mutat Res (1985) 159:431–42. doi:10.1016/0027-5107(85)90140-X
122. Hyland KA, Olson ER, Clark KJ, Aronovich EL, Hackett PB, Blazar BR, et al. Sleeping Beauty-mediated correction of Fanconi anemia type C. J Gene Med (2011) 13:462–9. doi:10.1002/jgm.1589
123. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol (2013) 3:113. doi:10.3389/fonc.2013.00113
124. Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: implications for the formation of chromosome translocations. Mutat Res (2015) 793:166–75. doi:10.1016/j.mrgentox.2015.07.001
Keywords: radiation effects, high-LET, RBE, DSB clusters, DSB repair, I-SceI, CRISPR/Cas9
Citation: Mladenova V, Mladenov E and Iliakis G (2016) Novel Biological Approaches for Testing the Contributions of Single DSBs and DSB Clusters to the Biological Effects of High LET Radiation. Front. Oncol. 6:163. doi: 10.3389/fonc.2016.00163
Received: 22 March 2016; Accepted: 15 June 2016;
Published: 28 June 2016
Edited by:
Francis A. Cucinotta, University of Nevada Las Vegas, USAReviewed by:
Janice Marie Pluth, Lawrence Berkeley National Laboratory, USAAsaithamby Aroumougame, UT Southwestern Medical Center, USA
Copyright: © 2016 Mladenova, Mladenov and Iliakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Iliakis, georg.iliakis@uk-essen.de