 Alejandro J. Cagnoni
Alejandro J. Cagnoni Juan M. Pérez Sáez
Juan M. Pérez Sáez Gabriel A. Rabinovich
Gabriel A. Rabinovich Karina V. Mariño
Karina V. Mariño
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 13 May 2016
Sec. Molecular and Cellular Oncology
Volume 6 - 2016 | https://doi.org/10.3389/fonc.2016.00109
This article is part of the Research Topic Glycosylation Changes in Cancer: An Innovative Frontier at the Interface of Cancer and Glycobiology View all 16 articles
Aberrant glycosylation, a common feature associated with malignancy, has been implicated in important events during cancer progression. Our understanding of the role of glycans in cancer has grown exponentially in the last few years, concurrent with important advances in glycomics and glycoproteomic technologies, paving the way for the validation of a number of glycan structures as potential glycobiomarkers. However, the molecular bases underlying cancer-associated glycan modifications are still far from understood. Glycans exhibit a natural heterogeneity, crucial for their diverse functional roles as specific carriers of biologically relevant information. This information is decoded by families of proteins named lectins, including sialic acid-binding immunoglobulin (Ig)-like lectins (siglecs), C-type lectin receptors (CLRs), and galectins. Siglecs are primarily expressed on the surface of immune cells and differentially control innate and adaptive immune responses. Among CLRs, selectins are a family of cell adhesion molecules that mediate interactions between cancer cells and platelets, leukocytes, and endothelial cells, thus facilitating tumor cell invasion and metastasis. Galectins, a family of soluble proteins that bind β-galactoside-containing glycans, have been implicated in diverse events associated with cancer biology such as apoptosis, homotypic cell aggregation, angiogenesis, cell migration, and tumor-immune escape. Consequently, individual members of these lectin families have become promising targets for the design of novel anticancer therapies. During the past decade, a number of inhibitors of lectin–glycan interactions have been developed including small-molecule inhibitors, multivalent saccharide ligands, and more recently peptides and peptidomimetics have offered alternatives for tackling tumor progression. In this article, we review the current status of the discovery and development of chemical lectin inhibitors and discuss novel strategies to limit cancer progression by targeting lectin–glycan interactions.
Cancer is a leading cause of death worldwide and represents one of the biggest challenges faced by medicine. Novel biological therapies such as tumor-antigen targeted vaccines (1, 2) and immune checkpoint blockade [i.e., monoclonal antibody (mAb)-based therapies targeting cytotoxic T-lymphocyte antigen 4 (CTLA-4) or programed cell death protein-1 (PD-1) (3–5)] have been designed to target specific determinants expressed by different tumor types and their associated stroma and immune compartments. However, due to the complexity of the tumor microenvironment (TME) and the intrinsic or acquired resistance mechanisms, only certain types of cancers can be effectively treated by these therapies (3–5). Furthermore, variable responses in patients with a similar malignancy reflect inherent heterogeneity among different tumor types (5). As technology advances, genomics studies have unveiled specific genetic signatures, which enabled tailored treatments and personalized cancer therapy to move a step closer to fruition. In time, other high-throughput technologies have emerged to expand personalized medicine beyond genomics, including proteomics and more recently, glycomics (6).
Glycosylation is the most abundant posttranslational modification: all cell surface and secreted glycoproteins must travel through the endoplasmic reticulum and the Golgi compartments, where addition of carbohydrates take place. The structure and nature of glycans strongly influence various functional aspects of glycoproteins such as cellular localization, turnover, protein quality control, and receptor–ligand interactions. The structural diversity of glycans, a key aspect that governs their role as information carriers, results from the concerted action of a number of glycosyltransferases and/or glycosylhydrolases that build and remodel their structure, generating a variety of glycoforms for a specific peptide sequence and allowing both cell-type and protein-specific glycan expression patterns (7, 8). Taking into consideration the ubiquitous presence of glycoconjugates on the cell surface, the fact that certain human diseases (including cancer) display altered glycan processing pathways is not surprising to glycobiologists (9, 10).
During the last decades, and as a result of advances in glycomics and glycoproteomics technologies, aberrant cell surface glycosylation has been considered an important hallmark of cellular oncogenesis and tumor progression. Simultaneous alterations of the overall glycome were identified in several types of cancer, where a differential glycan profile could be found not only in tumor cells themselves and the associated microenvironment (stromal fibroblasts, endothelial cells, and immune infiltrating cells) but also in serum glycoproteins (i.e., acute phase proteins), revealing potential glycobiomarkers of malignancy (11–13). It is now well established that aberrant glycosylation can promote tumor cell invasion and metastasis, as these processes involve cell detachment, intravasation, transport, attachment, extravasation, and angiogenesis (14). A long-standing and still unresolved question is whether aberrant glycosylation is a cause or a consequence of tumorigenesis.
Aberrant glycosylation in cancer is usually associated with poor prognosis, and may be present in different glycoconjugates, not only in N- and O-glycans on cell surface glycoproteins (15) but also in glycolipids and glycosaminoglycans (GAGs) (13, 16). These altered structures constitute the so-called tumor-associated cancer antigens (TACAs; Figure 1), and implicate not only the under- or overexpression of naturally occurring glycans but also the neo-expression of others. Glycan alterations vary depending on the type of cancer, but for N-glycans, they can include differential expression of blood group Lewis-related antigens such as Lewis X (LeX), Lewis Y (LeY), sialyl Lewis X (SLeX), and sialyl Lewis A (SLeA), increased synthesis of polylactosamine chains, increased β(1 → 6) branching of N-linked glycans, core α(1 → 6)-fucosylation, outer arm α(1 → 2)- and α(1 → 3)-fucosylation, and changes in sialylation, among others (11, 17, 18) (Figure 1A). For O-glycans and glycolipids, expression of TACA include mucin-related (O-linked) GalNAc (Tn), sialyl Tn (STn), Thomsen–Friedenreich antigen (Tf), polysialic acid (PSA), glycosphingolipid Globo-H, and gangliosides GM2 and GD2/GD3 (2, 9, 14, 19, 20) (Figures 1B,C).
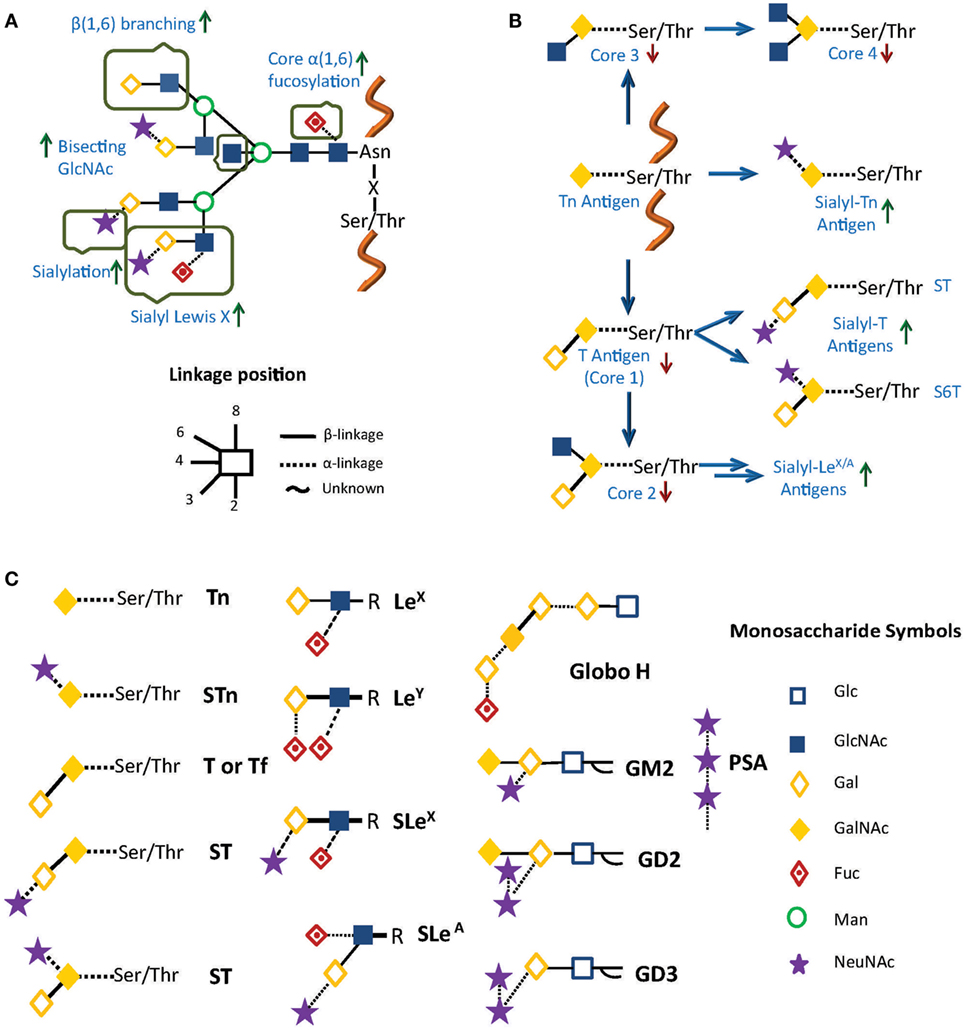
Figure 1. Common alterations observed in cancer in (A) N-glycosylation and (B) O-glycosylation. Green and red arrows represent increased and decreased expression of glycan structures, respectively. (C) Principal tumor-associated carbohydrate antigens (TACAs). In all cases, glycans are represented using a combination of the Oxford (21) and the Consortium for Functional Glycomics formats (http://www.functionalglycomics.org/).
It has been clearly demonstrated that these changes in glycosylation are dependent on biochemical factors such as availability of nucleotide sugar pools (activated donors for glycan biosynthesis), and differential expression of certain glycosyltransferases (17). As examples of important glycosylatransferases involved in O-glycan aberrant biosynthesis, ppGalNAc6 (GALNT6, one of the UDP-N-acetyl-d-galactosamine: polypeptide N-acetylgalactosaminyltransferases responsible for the initiation of mucin-type O-glycosylation) is upregulated in breast cancer (22) and has been also postulated as a potential marker associated with venous invasion in gastric carcinoma (23). On the other hand, GALNT9, another member of this family, has been described as a prognostic marker in neuroblastoma (24). In prostate cancer, overexpression of GCNT1 [β(1 → 6)-N-acetylglucosaminyltransferase, involved in core 2 O-glycan biosynthesis] was associated with higher levels of core 2 O-SLex in prostate specific antigen (PSA) (25). Regarding N-glycans, bisecting GlcNAc has been identified as a hallmark of epithelial ovarian cancer and mannosyl β(1 → 4)-glycoprotein β(1 → 4)-N-acetylglucosaminyltransferase (MGAT3), the glycosyltransferase involved in its biosynthesis, showed a clear upregulation in ovarian cancer (26). Finally, Wang et al. described an upregulation of Fut8, a fucosyltransferase that decorates the N-glycan core, in hepatocellular carcinoma (HCC), and core fucosylation was proposed as a prognostic marker as well as a therapeutic target for HCC (27). The role of N-glycans and O-glycans in cancer has been thoroughly reviewed in previous publications (17, 28, 29).
Even though there is abundant evidence on the important role of altered expression of glycosyltransferases in tumor cells, the regulation of glycan-related pathways is still far from clear. In the last few years, studies describing the influence of DNA methylation (26, 30) as well as cytokine levels (31) have shed light on these issues. Finally, a recent article describing the mutational landscape of aberrant glycosylation in colon cancer has shown specific mutations associated to glycosyltransferases in patients, particularly in B3GNT2, B4GALT2, and ST6GALNAc2. These significant findings indicate that functionally deleterious mutations in glycosyltransferase genes in part underlie aberrant glycosylation and contribute to the pathogenesis of molecular subsets of colon and other gastrointestinal malignancies (32).
These glycosylation changes, complex as they are, trigger different biological processes via interaction with an evolutionarily divergent family of glycan-binding proteins or lectins. Lessons learned from knockout and transgenic models in physiologic and pathologic settings revealed major roles for lectin–glycan interactions in immune cell homeostasis, controlling regulatory cell programs, and activating tolerogenic circuits that orchestrate tumor-immune escape mechanisms (33, 34). In this review, we focus on therapeutic strategies, based on chemical inhibition of three different lectin families, namely sialic acid-binding immunoglobulin (Ig)-like lectins (siglecs), C-type lectin receptors (CLRs), and galectins, which play relevant roles in cancer (Figure 2).
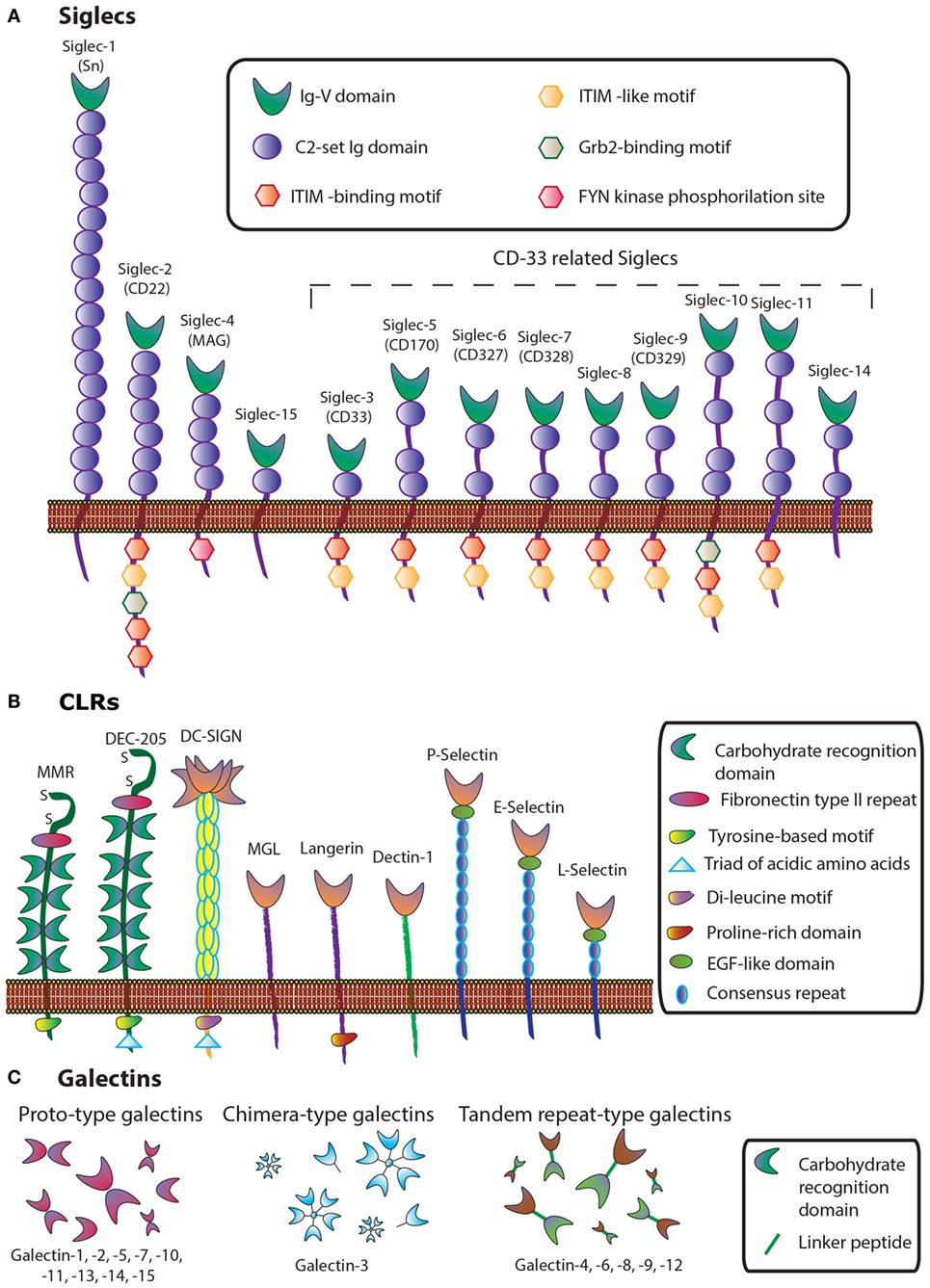
Figure 2. Schematic representation of three lectin families: (A) siglecs, (B) C-type lectins, and (C) galectins.
Siglecs, also known as the I-type lectin family, constitute a family of sialic acid binding Ig domain-containing lectins that are mainly found on cells of the immune and hematopoietic system (35) (Figure 2). From a structural viewpoint, siglecs are transmembrane type I receptors bearing 2–16 extracellular C2-set Ig domains, with an extracellular N-terminal V-set Ig (Ig-V) domain responsible for the binding of sialoside ligands (36), a single transmembrane domain, and varying lengths of cytosolic tails (37) (Figure 2A). Siglecs are typically classified into two functionally diverse subsets. The most distantly interrelated group (25–30% sequence identity) includes Siglec-1 (Sialoadhesin, Sn), -2 (CD22), -4 [myelin-associated glycoprotein (MAG)], and -15. The second group represents the rapidly evolving CD33-related Siglecs, which have high homology to CD33 in their extracellular domains (50–85% identity) and comprises Siglec-3 (CD33), -5, -6, -7, -8, -9, -10, -11, and -14 (35, 37, 38).
Siglecs are primarily expressed in B cells, macrophages, dendritic cells (DCs), and eosinophils and have been implicated in both innate and adaptive immunity. They play important roles in host–pathogen interactions, cell–cell communication, and regulation of immune tolerance (39), maintaining immune homeostasis and regulating inflammatory processes (37). With respect to innate immunity, Siglecs have been involved in pathogen internalization and immune evasion, attenuation of damage-associated molecular pattern (DAMP)-mediated inflammation, and inhibition of natural killer (NK) cell function. In adaptive immunity, they act as modulators of T-cell activation and polarization as well as regulators of B cells and plasmacytoid DCs (38).
Many siglecs have been studied as potential targets for the design of therapeutic agents for the treatment of inflammatory, autoimmune, allergic, and infectious diseases (35). Even though changes in sialylation may modulate tumor cell invasion or metastasis, the involvement of siglecs in tumor immunity is currently being explored. For example, Siglec-2 (CD22) has been implicated in B-cell activation in non-Hodgkin Lymphoma (40), and Siglec-7 has been shown to exert a pivotal role in tumor escape by inactivation of NK cells (41) (Figure 3A). Siglec-3 (CD33) is expressed on malignant blast cells in 85–90% of Acute Myeloid Leukemia cases, while is absent on normal hematopoietic pluripotent stem cells (42). Takamiya et al. reported that Siglec-15, which preferentially recognizes sialyl-Tn antigen (Figure 1), induced a M2-like immunosuppressive macrophage phenotype and upregulated TGF-β secretion in human monocytic leukemia cells and human lung carcinoma cells (43) (Figure 3B). Furthermore, interactions between Siglec-4a (MAG) and the mucin MUC1 enhanced adhesion of pancreatic cells and stimulated pancreatic cancer cell perineural invasion (44). Other siglecs have been correlated with tumor progression, such as Siglec-9, involved in tumor-immune evasion, and Siglec-12, which was found to be overexpressed on human prostate epithelial carcinomas (45).
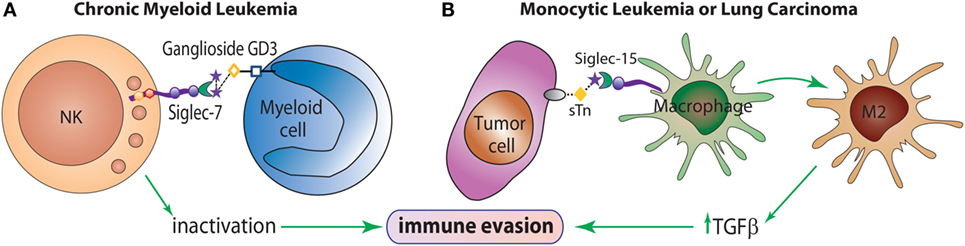
Figure 3. The role of siglecs in immune evasion mechanisms. (A) Siglec-7 is expressed predominantly on NK cells and inhibits NK cell cytotoxicity toward target cells overexpressing the α(2 → 8)-disialic acid-bearing ganglioside, GD3. (B) Siglec-15 recognizes the tumor sialyl-Tn (sTn) antigen and transduces an intracellular signal leading to enhance TGF-β secretion and polarization toward an M2-like macrophage profile, which contributes to tumor progression.
C-type lectins comprise a diverse family of calcium-dependent glycan-binding proteins that play essential immunological roles as adhesion and signaling receptors in inflammation, tumor progression, and viral infections (46). This lectin family is classified into 17 different subgroups depending on their C-type lectin domains and their structures. Some representative subsets are collectins, endocytic receptors [Mannose Receptor (MR), Dec-205], DC receptors [dendritic-cell specific intracellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)], macrophage galactose-binding lectin (MGL), Langerin, and selectins (L-Selectin, P-Selectin, and E-Selectin) (Figure 2B).
Given their biological and clinical relevance, we will focus here on Selectins, C-type transmembrane lectins that mediate leukocyte trafficking and specific adhesive interactions of leukocytes, platelets, and endothelial cells with tumor cells (47). These lectins are present on endothelial cells (E-Selectin), leukocytes (L-Selectin), and platelets (P-Selectin) (46), and preferentially bind glycans containing SLeX and SLeA glycoepitopes (Figure 1), which are abundantly expressed in several tumor types. In the TME, selectins are functionally relevant in the context of leukocyte recruitment, tumor-promoting inflammation, and acquisition of metastatic potential (36) (Figure 4).
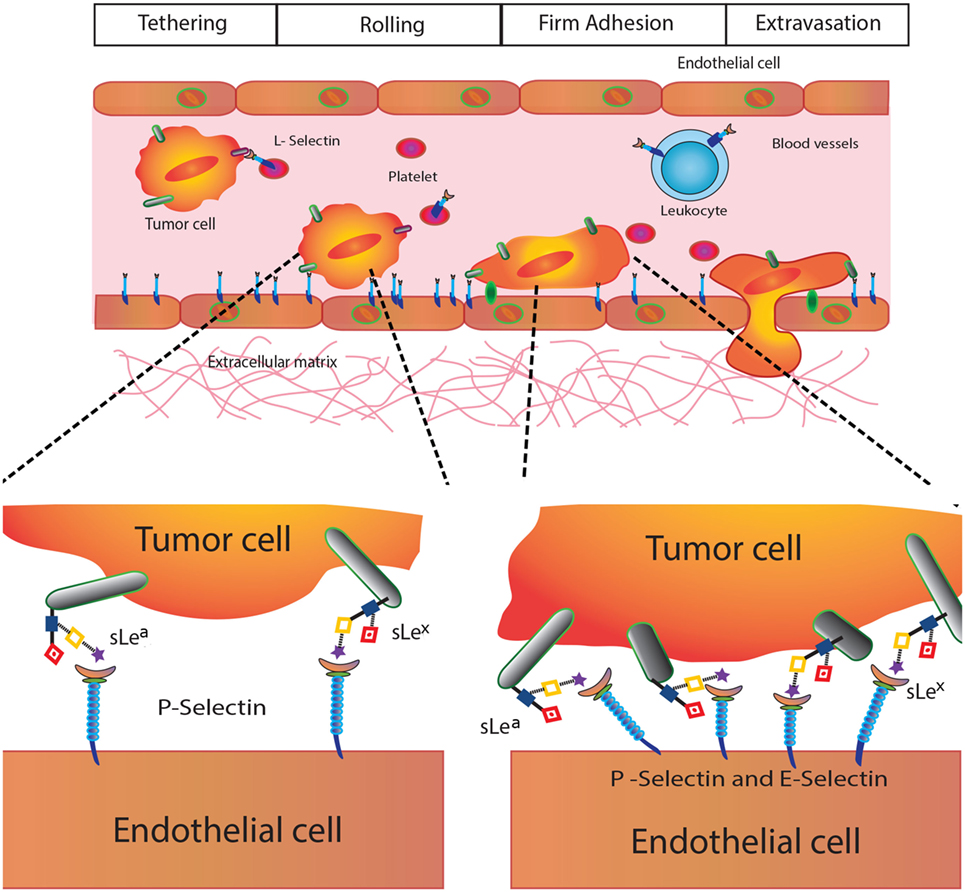
Figure 4. Selectins in cancer biology. Selectins play different roles in tumor biology including modulation of platelet–cancer cell interactions (P-selectin), promotion of tumor cell adhesiveness, extravasation and metastasis (E-selectin), and leukocyte trafficking and hematogenous metastasis (L-selectin).
P-Selectin (CD62P) is involved in tumor growth and metastasis, as it mediates interactions between activated platelets and cancer cells contributing to tumorigenesis (47). E-Selectin (CD62E) also play major roles in cancer cell adhesiveness at different events of the metastatic cascade, promoting tumor cell extravasation (48). Finally, L-Selectin (CD62L), constitutively expressed on leukocytes, regulates tumor–leukocyte interactions and promotes cell adhesion and hematogenous metastasis by favoring emboli formation (49).
Because of their critical involvement in cancer metastasis, several research groups have developed therapeutic strategies based on disruption of selectin–glycan interactions with the ultimate goal of controlling inflammation and metastasis (48).
Galectins, a family of highly conserved glycan-binding soluble lectins, are defined by a conserved carbohydrate recognition domain (CRD) and a common structural fold (50). Based on structural features, mammalian galectins have been classified into three types: prototype galectins (Gal-1, -2, -5, -7, -10, -11, -13, -14, and -15, containing one CRD and existing as monomers or dimerizing through non-covalent interactions), tandem repeat-type galectins (Gal-4, -6, -8, -9, and -12), which exist as bivalent galectins containing two different CRDs connected by a linker peptide, and finally, Gal-3, the only chimera-type member of the galectin family (Figure 2). Their distribution in mammalian tissues is diverse. While Gal-1 and -3 are detected ubiquitously, other galectins are more specifically located, such as Gal-2 and -4, which are preferentially found in the gastrointestinal tract (51, 52), Gal-7 is highly abundant in the skin (53), Gal-10 in eosinophils (54), and Gal-12 in adipose tissue (55, 56).
The ability of galectins to modulate different events in tumorigenesis and metastasis makes them attractive targets for cancer therapy (57, 58), controlling malignant transformation (59), apoptosis (60), cell-cycle progression (61), angiogenesis (62, 63), tumor metastasis (64, 65), and tumor immune escape (66). Galectins contribute to immune tolerance and escape through apoptosis of effector T cells (67), regulation of clonal expansion, function of regulatory T cells (Tregs) (64), and control of cytokine secretion (68). Expression levels for some galectins also change during malignant transformation, confirming their essential roles in cancer progression (69). Among the galectin family members, in this review, we will focus on Gal-1 and Gal-3, the two most extensively studied galectins, which have key roles during cancer progression.
Gal-1, abundantly secreted by almost all malignant tumor cells, has been characterized as a major promoter of an immunosuppressive protumorigenic microenvironment (67). This lectin induces selective apoptosis of TH1 and TH17 effector T cells, without affecting TH2 cells due to differential sialylation of cell surface glycoproteins (67, 70) (Figure 5). In recent years, the immunosuppressive activity of Gal-1 has also been extended to differentiation and expansion of CD4+CD25+Foxp3+ Tregs (64) and, similarly to other galectins, controls cell surface retention and signaling thresholds of a number of glycosylated receptors. However, its immunoregulatory activity is not limited to T cell populations: Gal-1 also promotes differentiation of tolerogenic dendritic cells (tDCs) (71) and controls tissue emigration of immunogenic, but not tDCs (72). The tolerogenic effects induced by this lectin have been substantiated by the work of Kuo and colleagues, who found that Gal-1-induced tDCs in lung cancer also favored the induction of Tregs (73). Furthermore, in addition to its immune inhibitory effects, Gal-1 can also favor tumor development and progression through promotion of tumor angiogenesis, favoring vascular endothelial growth factor (VEGF) signaling, and promoting endothelial cell proliferation, adhesion, migration, and resistance to apoptosis (34, 74). Finally, Gal-1 has been reported to contribute to heterotypic adhesion of tumor cells to extracellular matrix and endothelial cells, critical steps during the early stages of tumor invasion and metastasis (74, 75). Finally, within the intracellular compartment, Gal-1 also interacts with oncogenic H-Ras–guanosine triphosphate (H-Ras–GTP) through its farnesyl group, enhancing H-Ras-mediated cell transformation through ERK1/2 signaling (76) (Figure 5).
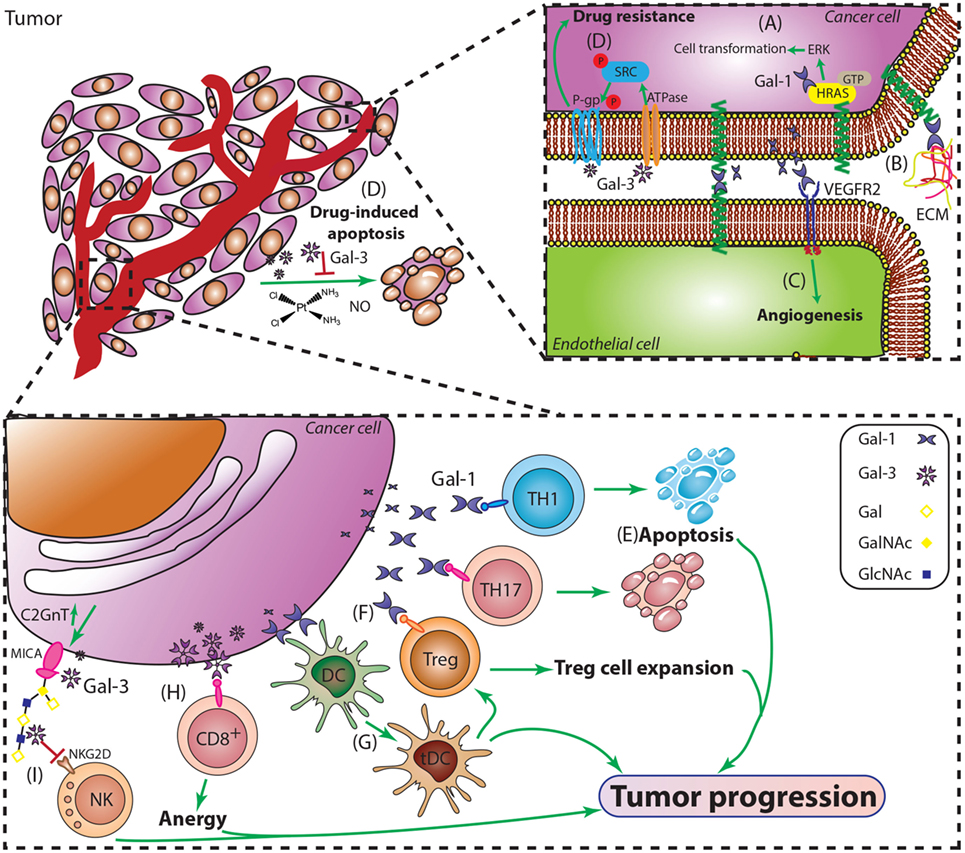
Figure 5. Major roles of Gal-1 and Gal-3 during cancer progression. (A) Intracellular Gal-1 triggers activation of the H-Ras/ERK cascade leading to malignant transformation. (B) Gal-1 promotes adhesion of tumor cells to extracellular matrix and endothelial cells, critical steps during early stages of tumor invasion and metastasis. (C) Gal-1 induces tumor angiogenesis by engaging key proangiogenic pathways including VEGF-like signaling. (D) Resistance to apoptosis is essential for cancer cell survival and plays a role in tumor progression. Gal-3 suppresses apoptosis induced by cisplatin, nitric oxide (NO), or radiation, through interactions with P-gp and Na+/K+-ATPase, thus promoting tumor cell survival. (E) Gal-1 selectively deletes TH1 and TH17 cells and (G) promotes the differentiation of tolerogenic DCs (tDCs). (F) Gal-1 promotes expansion of CD4+CD25+Foxp3+ regulatory T cells (Tregs) and amplifies their immunosuppressive activity. (H) Gal-3 induces anergy of CD8+ T cells by distancing the TCR from CD8 molecules. (I) Gal-3 impairs NK cell function by inhibiting the interactions between the heavily O-glycosylated tumor-derived MICA and the NK cell activating receptor NKG2D.
Gal-3, another member of the family, has shown prominent protumorigenic effects in a multiplicity of tumors. This multifunctional protein has demonstrated different effects depending on its subcellular localization (77) (Figure 5). A pioneer work by Raz et al. reported a correlation between higher Gal-3 expression and increase incidence of experimental lung metastases in mouse models (78). Since then, compelling evidence has implicated Gal-3 expression with different aspects of cancer biology including cell adhesion, migration, angiogenesis, and immune escape (79, 80).
Thomsen–Friedenreich glycoantigen (Tf; Figure 1) is expressed in up to 90% of human carcinomas (81) and is a major cancer cell surface carbohydrate ligand for Gal-3. Similarly to Gal-1, Gal-3 signaling contributes to tilt the balance toward immunosuppressive TMEs by interacting with specific glycans, and impairing antitumor responses. In this regard, Gal-3 has been shown to promote anergy of tumor infiltrating lymphocytes (TILs) (82). Furthermore, Tsuboi et al. described a new mechanism of tumor escape involving Gal-3 and NK cells in bladder cancer (83); the authors demonstrated that overexpression of core 2 β(1→6)-N-acetylglucosaminyl transferase 1 (C2GnT1), a glycosyltransferase responsible of generating branched core-2 O-glycans that can be elongated with poly-N-acetyllactosamine (LacNAc) sequences, negatively controls the activity of tumor-associated major histocompatibility complex class I-related chain A (MICA). Interactions between polyLacNAc and Gal-3 reduced the affinity of MICA to the NK cell receptor NKG2D, thereby impairing NK cell activation and their antitumor activity [(71) Figure 5]. However, Gal-3 not only assists tumor escape by inhibiting immune responses; it also promotes tumor cell survival by hampering drug-induced apoptosis by cisplatin, nitric oxide (NO) or radiation, through phosphorylation, translocation, and regulation of survival signaling pathways (84, 85) (Figure 5). The mechanisms underlying Gal-3 protection of drug-induced apoptosis has recently been reported by Harazono et al. (86, 87), who showed that interaction of this lectin with Na+/K+-ATPase activated SRC tyrosine kinase, subsequently inducing phosphorylation of P-glycoprotein (P-gp) and enhancing its ATPase activity. These effects contribute to decrease sensitivity to doxorubicin-mediated cell death (72, 73).
A variety of approaches have been designed to tackle selectin-mediated inflammation and metastasis through chemical inhibition. As a result, sulfated polysaccharides, modified glycans, and glycopeptides have been postulated as pharmacological selectin inhibitors (Table 1).
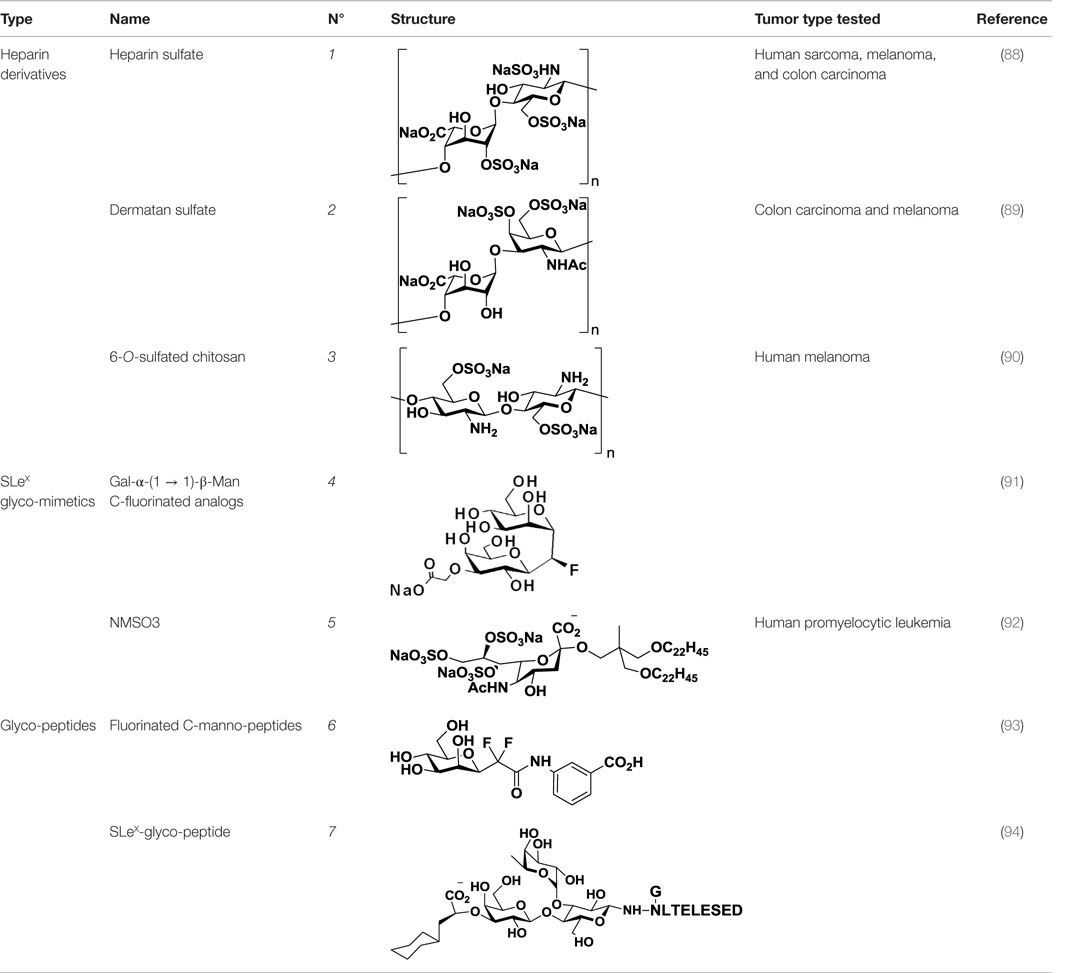
Table 1. Structure and clinical applications of different anti-selectin agents described in this review.
Heparin (1, Table 1), a highly sulfated polysaccharide from the GAG family, is composed of a repetitive disaccharide unit containing glucosamine and glucuronic/iduronic acid residues with a high degree of sulfation (95). Heparins and its derivatives, traditionally used as anticoagulants, have recently emerged as attractive compounds for selectin inhibition showing promising antimetastatic activity through disruption of P- and L-selectin-mediated adhesion of tumor cells (96). In an attempt to reduce the anticoagulant activity of heparin and enhance its affinity toward selectins, different chemical modifications such as N-acetylation, succinylation, O-desulfation, and reduction have been performed (97, 98). Heparins of different molecular weight have been successfully tested in tumor models such as melanoma, sarcoma, breast, and colon adenocarcinoma demonstrating inhibitory effects on tumor metastasis. However, they had limited effects on tumor growth, showing that heparin mediates interactions of tumor cells with host cells and the extracellular matrix during the metastatic process (99). Heparin and its derivatives have been tested in numerous clinical trials, but mostly as anticoagulant agents for prevention of cancer-related thrombosis (100).
Other negatively charged polysaccharides have also been evaluated as selectin inhibitors. Dermatan sulfate (2, Table 1), a sulfated polysaccharide bearing a [→4)-IdoA(2S)-β(1 → 3)-GalNAc-β-(1→] repetitive unit, has shown to be an effective P-selectin inhibitor in vitro for colon carcinoma and melanoma in experimental models (89). 6-O-Sulfated chitosan (3, Table 1), a modified linear β-(1 → 4)-glucosamine polysaccharide, has also demonstrated the ability to reduce interactions of P-selectin with melanoma cells in vitro (90).
Taken together, these findings suggest that inhibition of selectin–glycan interactions by charged polysaccharidic agents could become an alternative therapy for inhibition of tumor metastasis. However, further analyses are necessary not only to unravel the mechanisms underlying the pharmacological activity of these compounds but also to reduce the risk of heparin-induced anticoagulant-related side effects.
As selectins preferentially bind to sulfated or fucosylated oligosaccharides such as SLeX or SLeA (Figure 1), Lewis-type glycomimetic analogs emerged as interesting decoys for disruption of selectin-mediated processes. Non-reductive disaccharide Gal-α-(1 → 1)-β-Man became a promising building block for SLeX glycomimetics, and a number of research groups have explored fluorinated C-glycosyl analogs (4, Table 1) in an attempt to improve the pharmacokinetics of these ligands (91, 101). On a different approach, Shodai et al. designed NMSO3 (5, Table 1), a sulfated derivative of sialic acid, which demonstrated to be a good inhibitor of P-selectin-mediated tumor cell adhesion (92).
Besides carbohydrates, glycopeptides were also evaluated as selectin blockers. Flexible difluorinated C-mannopeptides (6, Table 1) were synthesized and tested as E- and P-selectin inhibitors, exhibiting moderate binding affinities (93). Starting from SLeX, Filser et al. designed high-affinity synthetic glycopeptides for E-selectin inhibition, bearing a peptide sequence from the natural ligand E-selectin ligand-1 (ESL-1) and replacing the sialic acid with a cyclohexyl moiety (7, Table 1). These compounds confirmed that the peptide moiety was essential for selectin binding and exhibited encouraging IC50 values in the low micromolar range (94). However, no experiments have yet corroborated their efficacy in vitro or in vivo.
Compared to the advances made for selectins or galectins, the role of siglecs during cancer progression has not been studied in such detail. Thus, there have been fewer reports on siglec inhibitors as anticancer agents. Promising approaches reported in literature include C4- and C9-modified sialic acid derivatives (Figure 6). Siglec-7, which belongs to the CD33-related siglec type, preferentially binds internally branched α(2 → 6) sialic acid and is primarily expressed on NK cells. This lectin has been reported to favorably interact with melanoma or neuroblastoma cancer cells that overexpress GD3, an α(2 → 8) disialic acid-bearing ganglioside, thus inhibiting NK cell cytotoxicity as an immune evasion mechanism (102). In an attempt to disrupt Siglec-7–GD3 interactions as a potential cancer therapeutic strategy, Attrill et al. described the design of sialic acid derivatives as inhibitors of Siglec-7 signaling (103). One of these ligands, oxamido-Neu5Ac [8, Table 2, methyl α-9-(amino-oxalyl-amino)-9-deoxy-Neu5Ac] exhibited a twofold decrease in the IC50 value (1.6 mM) for inhibition of Siglec-7 in vitro, compared to the canonical ligand methyl-α-Neu5Ac (>3.0 mM) (103).
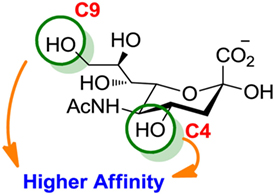
Figure 6. Modifications of neuraminic acid leading to high-affinity siglec blockers. Green circles denote the main positions modified in search of high-binding affinity and selective siglec inhibitors.
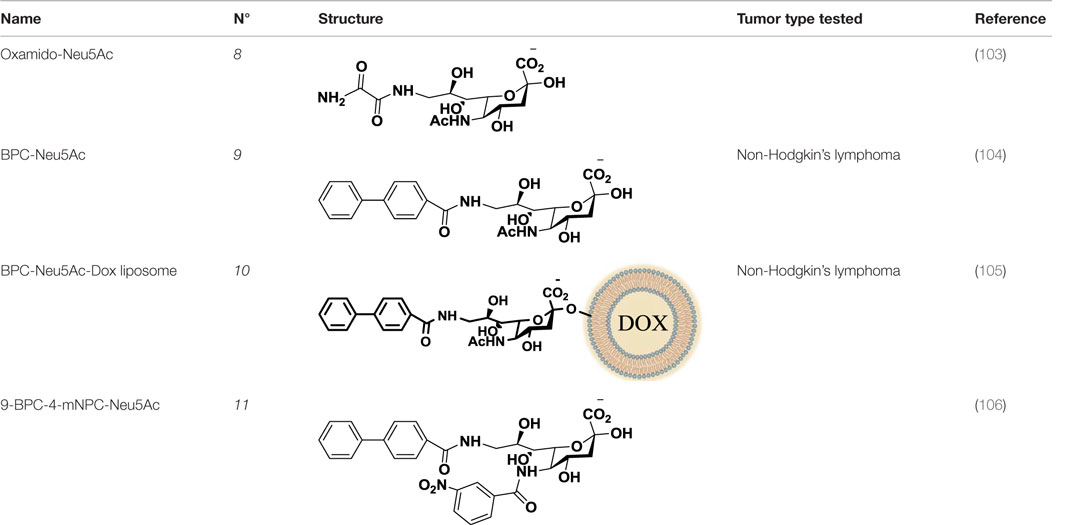
Table 2. Anti-siglec agents described in this review.
Another interesting approach was reported by Kelm et al. who described the synthesis of sialic acid derivatives as high-affinity inhibitors of Siglec-2 (106). CD22 or Siglec-2 is an antigen widely expressed on normal and malignant B cells and plays a primary role in B-cell activation. Thus, it has become an interesting target for the treatment of autoimmune diseases and B-cell derived non-Hodgkin’s Lymphoma (40). In 2002, Kelm et al. reported the synthesis of a C9-modified Neu5Ac, namely methyl-α-9-N-(biphenyl-4-carbonyl)-amino-9-deoxy-Neu5Ac (BPC-Neu5Ac, 9, Table 2), which exhibited a >200-fold relative inhibitory potency (rIP) for human CD22 than Me-α-Neu5Ac (104). More recently, Paulson and colleagues used this potent Siglec-2 inhibitor for the design of doxorubicin-loaded liposomal nanoparticles bearing BPC-Neu5Ac (9, Table 2) ligands to target B cell lymphoma (105). The CD22-targeted BPC-Neu5Ac-Dox liposomes (10, Table 2) provided a significant increase in survival rates when injected into a xenograft model of human B-cell lymphoma. Interestingly, this delivery strategy exhibited cytotoxic effects toward malignant B cells in patients with hairy cell leukemia, marginal zone lymphoma, and chronic lymphocytic leukemia. In 2013, Kelm et al. presented a further optimized novel glycan inhibitor with modifications on both Neu5Ac C4 and C9 positions (106). This compound, methyl 9-biphenylcarboxamido-4-m-nitrophenylcarboxamido-4,9-dideoxy Neu5Ac (9-BPC-4-mNPC-Neu5Ac, 11, Table 2) presented a 14-fold decrease in the IC50 value toward Siglec-2, thus emerging as a promising lead agent, although its activity has not yet been tested in vitro or in vivo.
Given the key protumorigenic, prometastatic, and immunosuppressive activities of galectins and their roles in tumor resistance to antiangiogenic therapies, important efforts have been made in the design of high-binding affinity specific inhibitors. Here, we describe a number of successful strategies to chemically disrupt galectin–ligand interactions and discuss advantages, limitations, and obstacles in their translation to clinical settings.
A First Step: β-Galactoside Ligands
Since galectins share the structurally conserved CRD that defines their natural ligands, the initial approach was the design of β-galactoside inhibitors targeting the carbohydrate-binding site (107). Specific modifications of the galactose-based structures have led to the design of good inhibitors with dissociation constants in the micromolar range (20–300 μM) (108, 109). However, the diverse chemical modifications tested on this monosaccharide did not achieve enough improvement on galectin affinity, and in consequence, galactose-based monosaccharide inhibitors have not been tested in cultured cells or in vivo.
The second approach for the design of galectin inhibitors was the use of chemically modified natural galectin ligands, such as the disaccharides lactose (Lac) or N-acetyllactosamine (LacNAc). Ingrassia et al. reported an unspecific lactosylated steroid (12, Table 3) that induced increased survival rates when administered in experimental models of mouse lymphoma and human glioblastoma (110). Furthermore, Iurisci et al. reported the use of allyl-lactoside (13, Table 3) as ligand of Gal-1 and Gal-3, inhibiting homotypic cell aggregation in human melanoma cells, and promoting apoptosis of small cell lung carcinoma cells (111). Although Lac and LacNAc are more promising scaffolds than galactose for the design of galectin inhibitors since both disaccharides present higher binding affinity than galactose (KD ≈ 90–500 μM), they are both sensitive to enzymatic hydrolysis by glycosidases.
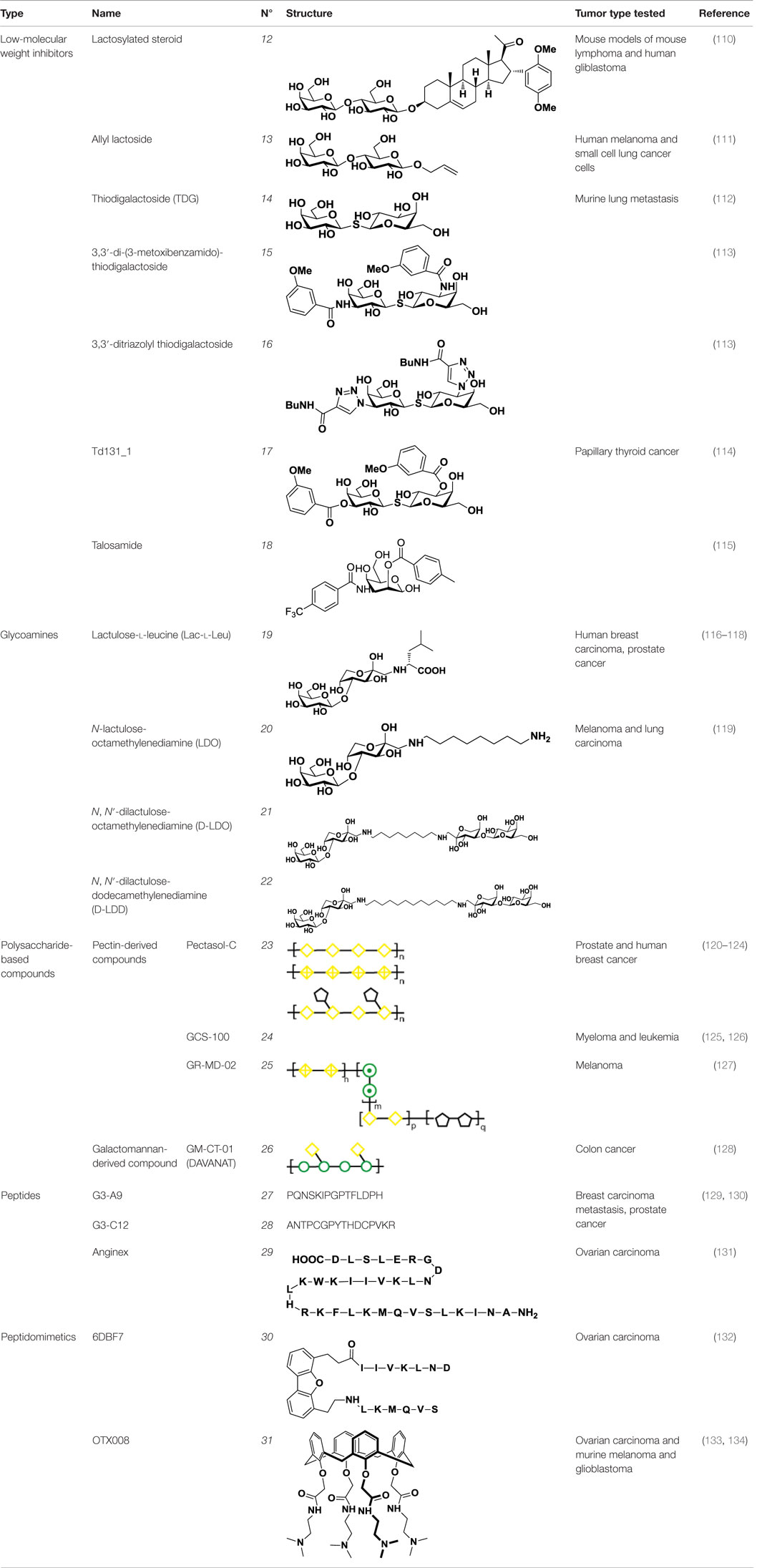
Table 3. Structure and applications of different anti-galectin agents described in this review.
Chemical modifications in O2 and O3 of the galactose residue in lactose (named O2′ and O3′; Figure 7) afforded novel structures with anticancer biological activities. The Nilsson group explored galactose O3′ modifications for Gal-3 binding, showing that lactose O3′ modification with aromatic groups led to active Gal-3 inhibitors with binding affinities as low as 300–600 nM, due to additional favorable cation–π interactions with a conserved arginine residue, Arg-144, in the carbohydrate-binding site (113). However, these Gal-3 blockers showed affinities in the micromolar/low millimolar range (100–1000 μM) when tested in vitro.
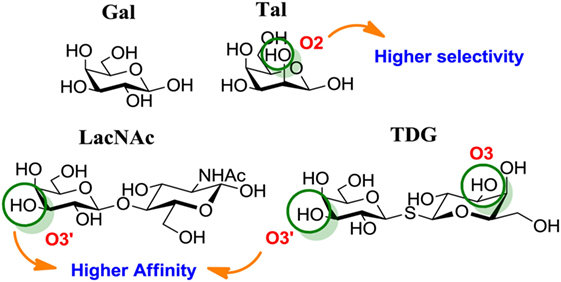
Figure 7. Mono and disaccharides as building blocks to design galectin inhibitors. Structures of galactose (Gal), talose (Tal), N-acetyllactosamine (LacNAc), and thiodigalactoside (TDG). Green circles denote critical modified positions in search for high-binding affinity and selective galectin inhibitors.
In order to overcome the poor bioavailability of lactose derivatives, thiosugars, and in particular, thiodigalactoside (TDG; 14, Table 3) have emerged as interesting building blocks for the design of potent galectin inhibitors, although with low selectivity (135, 136). Ito et al. demonstrated that administration of TDG significantly reduced tumor progression and metastasis via Gal-1 inhibition, using murine models of breast and colon adenocarcinoma (112). Moreover, intratumoral injection of TDG raised the level TILs and reduced tumor growth in models of B16 melanoma as well as 4T1 orthotopic breast cancer models (137). Symmetrical modifications of TDG at O3 and O3′ positions led to increased galectin-binding affinities, affording the most potent Gal-3 inhibitors in vitro reported to date (136), with KD values as low as 29 nM (3,3′-ditriazolyl thiodigalactoside, 15, Table 3) and 50 nM (3,3′-di-3-metoxibenzamido thiodigalactoside) (16, Table 3). In spite of their promising biochemical performance, in vivo experiments were not as encouraging. For instance Td131_1 (17, Table 3), an ester-modified TDG derivative, was evaluated as a Gal-3 inhibitor in papillary thyroid cancer. This compound showed concomitant increased apoptosis of cancer cells, although requiring concentrations up to 1000 times than the KD value (114).
Changing Stereochemistry for Enhanced Selectivity: Talose-Based Ligands
Carbohydrates are characterized for containing multiple stereocenters, and many stereoisomers are possible including enantiomers and diastereoisomers. Talose (a C-2 epimer of galactose; Figure 7) provides an axially disposed O2 and additional protein–ligand interactions, potentially making talose-based inhibitors more selective than galactose-based ones. This approach was taken by Nilsson and collaborators, who created a new family of synthetic talosides that showed affinity and selectivity toward Gal-4 (C-terminal CRD), Gal-8 (N-terminal CRD), and Gal-3 (138). In 2011, Öberg et al. reported the synthesis of a new family of talosamides with additional modifications at C2 and C3 with aromatic moieties. The best matches for these talosamide galectin inhibitors were found against Galectin-4C (C-terminal) (115): compound 18 (Table 3) presented a KD value of 94 μM and a good selectivity when compared to Gal-1 (1900 μM), Gal-2 (1700 μM), Gal-3 (570 μM), Gal-7 (>4000 μM), and Gal-9 (>4000 μM). These promising talose-based compounds still await detailed in vitro and in vivo evaluation.
Mimicking Glycans: Glycoamines on the Spot
A different approach to carbohydrate-based galectin inhibitors was proposed by Glinsky et al., who showed that a synthetic β-galactoside-containing disaccharide-amino acid conjugate, the glycoamine lactulose-l-leucine (Lac-l-Leu, 19, Table 3), binds and inhibits Gal-3 by mimicking cancer-associated Tf antigen (Figure 1). This glycomimetic decreased the incidence of lung metastasis in human breast carcinoma xenografts (118) and also inhibited homotypic and heterotypic aggregation of prostate cancer cells via Gal-3 inhibition (81). Furthermore, intraperitoneal administration of Lac-l-Leu reduced bone metastasis of human prostate carcinoma cells in a mouse model (116). However, it required high concentrations (daily injections of 200 μL of Lac-l-Leu 20 μM) to disrupt Gal-3–Tf interactions.
Further studies reported the design and synthesis of a new family of lactulose amines based on N-lactulose-octamethylenediamine (LDO, 20, Table 3) (119). In order to assess multivalency as potential modification to increase local concentration of these galactose-containing ligands, N, N′-dilactulose-octamethylenediamine (D-LDO, 21, Table 3) and N, N′-dilactulose-dodecamethylenediamine (D-LDD, 22; Table 3) were also synthesized. Compounds 10 and 11 (Table 3) exhibited interesting regulatory effects in vitro, such as prevention of Gal-1-mediated homotypic aggregation in melanoma cells and apoptosis of small cell lung carcinoma cells (119). In order to further develop these compounds and finally reach clinical status, additional structural studies clarifying the relevance of the aglycone moiety, and their selectivity even beyond galectins are required.
In addition to low-molecular weight carbohydrate-based synthetic inhibitors, natural polysaccharides have also emerged as high affinity galectin inhibitors with low toxicity for cancer treatment. Modified citrus pectin (MCP, Pectasol-C) and Davanat (GM-CT-01) are the best studied galectin blockers derived from natural sources.
Pectin-Based Compounds
Pectins compose a family of complex polysaccharides, which are found in high amounts in the plant primary wall. Three main pectic polysaccharides have been isolated from plant walls: homogalacturonan (HG), rhamnogalacturonan-I (RG-I), and substituted galacturonans (GS) (120). Pectins can be modified by pH and heat treatments, both of which expose galactoside residues by hydrolysis (139). The most studied modified pectin is MCP, which is obtained by partial degradation of citrus pectin polysaccharide chain and hydrolysis of the galacturonic acid esters from the HG regions. MCP through a multivalent display of galactoside residues has shown affinity for Gal-3 and has been tested as an anticancer agent in galectin-mediated tumorigenic processes (120).
The biological activity of MCP in cancer was extensively studied by Raz and coworkers (140–142). MCP has been shown to act as a ligand for Gal-3, while citrus pectin (CP) is unable to interact with this galectin (142). The authors showed that intravenous injection of MCP into mice bearing B16 melanoma resulted in significant decrease of lung colonization, while CP led to the opposite effect (142). In 2002, they expanded their findings to nude mice injected either with human breast carcinoma cells or with colon cancer cells, showing that the ability of MCP to inhibit primary tumor growth and metastasis in vivo is not restricted to prostate cancer (140). The antitumor effects of MCP were associated with an antiangiogenic activity, since MCP also inhibited capillary tube formation in vitro in human endothelial cells. Recently, Menachem et al. studied in vivo the combined inhibition of Ras and Gal-3 with FTS (S-trans farnesylthiosalicylic acid, Salirasib) and MCP, respectively, for the treatment of aggressive anaplastic thyroid carcinoma (143). Interestingly, FTS and MCP inhibited tumor growth in nude mice showing decreased levels of Gal-3, K-Ras-GTP, and p-ERK. However, the structure–function relationship of MCP and the molecular mechanisms underlying its effects are not yet completely understood, probably due to the lack of thorough structural characterization of pectin fractions. Nevertheless, MCP emerged as a very promising anticancer agent since it was the first reported inhibition of tumor growth by a soluble, orally ingested dietary carbohydrate fiber. In fact, two different commercial forms of MCP, PectaSol-C, and GCS-100, have been incorporated into clinical trials (112, 114, 115, 137, 138).
PectaSol (23, Table 3), and its most recent version Pectasol-C, commercial forms of MCP developed by EcoNugenics® (Santa Rosa, CA, USA), showed cytotoxic activity on different prostate cancer cell lines (122, 123). Additionally, Jiang et al. demonstrated that the combination of PectaSol-C with two polybotanical compounds for breast and prostate health, BreastDefend (BD) and ProstaCaid (PC), synergistically inhibited metastatic phenotype of human breast and prostate cancer cell lines, respectively (121). In 2003, Guess and colleagues developed a Phase II clinical trial to investigate the tolerability and effect of PectaSol in patients with prostate cancer, showing that prostate cancer patients exhibited a serial increase in PSA after localized treatment (124). The results from this pilot clinical trial suggested that PectaSol may slow down PSA increase, as it improves prostate-specific antigen doubling time (PSADT) in patients with recurrent prostate cancer. However, PSADT is an indirect measure of tumor progression and the detailed effect that PectaSol may have on prostate cancer progression is still poorly understood. Currently, there is an ongoing patient recruitment by EcoNugenics for a Phase III clinical trial pointing at the effects of orally administered PectaSol-C for improving PSA kinetics in men with biochemical relapsed prostate cancer and serial increasing PSA (NCT: NCT01681823) (120).
GCS-100 (24, Table 3), another commercial MCP derivative, is also in clinical development for the treatment of cancer. GCS-100 inhibits cell growth in various multiple myeloma cell lines (126). Demotte et al. showed that GCS-100 influences the reinvigoration of anergic TILs: treatment with GCS-100 successfully detached Gal-3 from CD8+ TILs and boosted cytotoxicity and secretion of proinflammatory cytokines (144). Furthermore, in tumor-bearing mice vaccinated with a tumor antigen, injections of GCS-100 led to increased tumor rejection compared to control mice, showing the potential application of pectin-derived agents in combination with therapeutic vaccination as a combined cancer treatment (145).
GCS-100 was evaluated as an anticancer agent in three clinical trials, but two of them were suspended because of lack of funding (NCT00776802 and NCT00609817). The third one (La Jolla Pharmaceutical Company, NCT00514696) was a Phase II study of the safety of GCS-100 in 24 subjects with chronic lymphocytic leukemia. GCS-100 was delivered intravenously, showing excellent overall tolerability, partial remission in 25% of the patients and >50% shrinkage of lymph node lesions in 16% of patients (125, 146). Another interesting approach was reported by Zomer et al., who developed GR-MD-02 (25, Table 3), a pectin-derived galactoarabino-rhamnogalacturonan polysaccharide (127). GR-MD-02 is currently undergoing two clinical trials in combination with immune checkpoints inhibitors: the anti-PD-1 mAb Pembrolizumab and the anti-CTLA-4 mAb Ipilimumab for melanoma treatment (NCT02575404 and NCT02117362, respectively).
Galactomannan-Derived Compounds
Besides pectin-derived agents, β-d-(1 → 4)-galactomannan-based compounds, such as GM-CT-01 (trade name DAVANAT, 26, Table 3), offer another alternative for galectin inhibition. GM-CT-01 is isolated from seeds of Cyamopsis tetragonoloba (Guar gum), and subjected to a controlled partial chemical degradation (147). With an average size of 51 kDa, this β-d-(1 → 4)-galactomannan is composed by galactose residues α(1 → 6)-linked to the mannose backbone at recurring intervals (148). GM-CT-01 has been mainly tested as an allosteric inhibitor of Gal-1 (KD = 10 mM), but has also affinity for Gal-3, -7, and -9 (128, 149). GM-CT-01 has been evaluated alone or in combination with the chemotherapeutic agent 5-fluorouracil (5-FU) in preclinical studies. Phase I and II clinical studies for colon cancer treatment showed 70% higher stability for patients administered with GM-CT-01 and a 46% increase in survival of patients with end stage colon cancer (NCT00110721) (128). This galectin inhibitor also influenced human TILs from patients with various cancers, boosting IFN-γ secretion upon ex vivo stimulation in ~80% of CD8+ TILs and ~50% of CD4+ TILs (145). The response observed suggested that administration of GM-CT-01 may contribute to correct impaired TIL functions in cancer patients.
Peptides exhibit several advantages when compared to carbohydrate-based ligands. While carbohydrate–lectin interactions occur in the mid-micromolar range, peptide–protein or protein–protein interactions occur in the nanomolar range. Therefore, and since peptide synthesis has advanced considerably in the past few years, several research groups have developed an alternative approach using peptides or peptidomimetics to target galectins at their CRD or at distant sites.
In 2005, a pioneer work by Zou and colleagues reported binding of small peptides to Gal-3, with a concomitant reduction in cancer cell adhesion in vitro (150). Considering that Tf glycoantigen (Figure 1) is exposed on up to 90% of human carcinomas (81) and is a major cancer cell surface carbohydrate ligand for Gal-3, the authors obtained synthetic peptides mimicking the Tf structure using combinatorial bacteriophage display technology. The most promising compounds, G3-A9 (27, Table 3) and G3-C12 (28, Table 3), blocked interactions between Tf and Gal-3 and presented high affinity (72 nM) and good selectivity toward Gal-3 when compared to Gal-1, Gal-4, and LacNAc-binding plant lectins (123). These peptides effectively inhibited heterotopic adhesion of human breast carcinoma cells to endothelial cells, as well as homotypic tumor cell aggregation in vitro. Some years later, these authors also reported the ability of peptide G3-C12 (28) to modulate tumor establishment and growth of human breast carcinoma cells in mice, demonstrating a significant reduction of lung metastatic tumors (72%) by in vivo bioluminiscent imaging (129). G3-C12 (28, Table 3) was also studied in metastatic prostate cancer by Deutscher et al., who proposed radiolabeled G3-C12 as a marker of Gal-3-expressing prostate tumors (130). 111In-labeled G3-C12 peptide showed good tumor uptake in severe combined immunodeficient (SCID) mice bearing human prostate tumor xenografts. A major limiting factor for its application in prostate tumor imaging was its non-specific uptake, as it was also found in kidneys. Yang et al. solved this issue by coupling G3-C12 to water-soluble N-(2-hydroxypropyl)methacrilamide (HPMA) copolymers as drug carriers (151). These copolymers were further modified either with 131I radioisotope for in vivo SPECT-imaging (151), or with 5-FU for enhanced anticancer activity (152). 131I-G3-C12–HPMA copolymer showed higher tumor accumulation when compared to controls, although the authors experienced some difficulties with the adjustment of the ligand modification degree and the molecular size of the copolymer. On the other hand, 5-FU modified G3-C12–HPMA copolymer displayed a superior intracellular internalization followed by enhanced cytotoxicity and apoptosis induction in the PC-3 tumor-bearing mouse model (152). Recently, Sun et al. reported that the conjugation of G3-C12–HPMA copolymer with doxorubicin (G3-C12–HPMA–Dox) improved internalization into Gal-3-overexpressing PC-3 cells, but stimulated the translocation of Gal-3 to the mitochondria to prevent apoptosis (153). However, as time progressed, G3-C12–HPMA–Dox conjugates delivered increasing amounts of Dox into the mitochondria and overcame the anti-apoptotic effects of Gal-3. In spite of the lack of structural studies characterizing Gal-3/G3-C12 peptide binding, this peptide seems to be very promising not only as Gal-3-targeted imaging agent but also as therapeutic agent for Gal-3-overexpressing cancers such as melanoma and breast, ovarian, and gastric carcinomas (154).
Another example of peptide-based galectin ligand for cancer treatment is Anginex (29, Table 3), a 33-mer synthetic peptide originally designed to reproduce the β-sheet structure of antiangiogenic proteins like platelet factor 4 (PF4), interleukin (IL)-8, and bactericidal/permeability-increasing protein (BPI) (155, 156). This synthetic peptide has antiangiogenic and anti-tumor effects in vitro and in vivo (131, 157) and has been shown to bind Gal-1 (158), although it may also recognize other galectins such as Gal-2, -7, -8 (N-terminal), and -9 (N-terminal) (159). Later on, 6DBF7 (30, Table 3), a partial peptidomimetic of Anginex, bearing a hydrophobic dibenzofuran scaffold required for the β-sheet peptide configuration exhibited a better antiangiogenic performance in a mouse xenograft model of ovarian carcinoma (132).
In order to overcome the intrinsic susceptibility of peptides to hydrolysis by proteases, Dings et al. designed a non-peptidic topomimetic of Anginex and 6DBF7, based on a calixarene scaffold (134). This compound, named calixarene 0118 or OTX008 (31, Table 3), demonstrated potent angiogenesis inhibition in two mouse models of human ovarian carcinoma and murine melanoma. In addition, OTX008 has shown synergistic effects with sunitinib (a tyrosine kinase inhibitor) in ovarian carcinoma and glioblastoma mouse xenografts (133). Interestingly, OTX008 downregulates cancer cell proliferation, invasion, and tumor angiogenesis in a variety of tumor cells (160) and is undergoing a Phase I clinical trial by OncoTx Inc. (NCT01724320) (161).
In the past few years, the field of cancer therapy has experienced an impressive breakthrough with the development of targeted therapies and immune checkpoint blockers (162). However, although significant improvements have been achieved with these therapeutic agents, some patients have not shown clinical benefits, presumably because of the development of adaptive resistance mechanisms and acquisition of compensatory pathways.
Aberrant glycosylation has emerged as a hallmark of cancer progression, and the presence of a tumor-associated glycome is deeply associated with malignant transformation and metastasis-associated processes including tumor cell migration, invasiveness, angiogenesis, and immune escape (14). However, the translational and clinical applications of altered glycan structures in cancer have not yet been completely accomplished, probably due to the complex regulation of the glycosylation machinery including glycosyltransferases and glycosidases. Heterogeneity is inherent to the language of glycans and crucial for their diverse biological roles as information carrier for lectins. On the other hand, lectins, glycan-binding receptors responsible for deciphering the glycome, arose as feasible targets for cancer therapy. As a result, considerable efforts have been made to disrupt glycan–lectin interactions by designing pharmacological anticancer agents that target different lectins including selectins, siglecs, and galectins. Further studies are still necessary to understand the precise underlying mechanisms of the antitumor effects displayed by lectin blockers, and to explore the potential complementation or synergy of turning-off lectin signaling with immune checkpoint blockade therapies, targeted therapies (e.g., small molecule inhibitors of receptor tyrosine kinases) and antiangiogenic therapies, as well as with chemotherapy, radiotherapy, and vaccination strategies. As of today, different approaches have generated chemical inhibitors for lectin–glycan interactions, resulting in diverse selectivity, affinity, and therapeutic efficacy in cancer models.
Small glycan derivatives and glycomimetics, which represent the majority of lectin blockers reported to date, target the carbohydrate-binding site and showed promising affinities and lectin selectivity in vitro. However, these carbohydrate-based ligands suffer certain disadvantages for their use in the clinics, i.e., low in vivo bioavailability, susceptibility to glycosidase hydrolysis, and fast clearance. For instance, for galectin inhibition, no galactose-based ligand has reached clinical trials up to date. Thus, alternative approaches have been made to circumvent these drawbacks including the design of hydrolytically stable N-, S-, and C-saccharides and the development of neoglycoproteins with higher bioavailability (163), but only a few of them have been successfully tested in vivo. Although not included in this review, metabolic inhibitors for sugar nucleotide biosynthesis were also proposed and efficiently tested: for example, per-acetylated 4-fluoro-glucosamine (4-F-GlcNAc) as a GlcNAc biosynthetic inhibitor was evaluated in cancer melanoma- or lymphoma-bearing mice, showing enhanced tumor lymphocytic infiltration and increased the frequency of cytotoxic T cells and IFN-γ production (164). However, these chemical inhibitors do not affect protein glycosylation in a specific way. Therefore, small carbohydrate inhibitors have to face diverse intrinsic drawbacks before reaching clinical settings.
Multivalent display by polysaccharide inhibitors has become an interesting approach for galectin and selectin inhibition. Heparin, dextran, and chitosan sulfated derivatives have been successfully tested as selectin inhibitors. Nevertheless, no detailed clinical trials have been yet designed to determine the antimetastatic effects of heparin derivatives in cancer. On the other hand, galactomannan-derived compounds, together with pectin-derived agents, are probably the most successful galectin chemical inhibitors that reached clinical evaluation in cancer patients. DAVANAT, GCS-100, and PectaSol currently being tested in clinical trials (among several other polysaccharides-based inhibitors) seem very promising anticancer agents, since they present high-binding affinities and can easily be obtained from natural sources. Moreover, these ligands stress the importance that dietary carbohydrate compounds may reach as therapeutic agents. Looking to the future, critical structure–function relationships and selectivity studies are still necessary, as well as elucidation of the precise mechanisms underlying interactions with these polysaccharide compounds. For example, the use of MCP as an anticancer agent is still limited because of lack of selectivity and structure variability (165). In fact, there are multiple MCPs on the market with different molecular weight range, diverse esterification degrees, and limited characterization at the molecular level. Although the affinity of MCPs for Gal-3 has been documented, there is still no study showing selectivity with regard to other lectins. In fact, MCP demonstrated the ability to detach cell surface binding of Gal-1 and Gal-3 to melanoma cells at comparable concentrations (144). In order to completely understand the biological modes of action of MCPs as well as other polysaccharide-based ligands in cancer-related processes, further studies are required to assess their affinity toward other lectins.
As alternative inhibitors, peptides and peptidomimetics have emerged as interesting agents capable of overcoming some of the intrinsic disadvantages of glycans. Among them, fluorinated C-manno-peptides represent promising therapeutic agents as selectin inhibitors, while G3-C12 (28) and the Anginex-derivative OTX008 (31) represent the most successful approaches for inhibition of Gal-3 and Gal-1, respectively; both are currently being tested in clinical trials.
With the advances in molecular biology and immunology, anti-sense nucleotides, mAbs, and lectin modifications have also been proposed as potential strategies for lectin inhibition. Small-interfering RNA (siRNA) has recently been offered as an alternative approach for galectin inhibition, but literature is still limited. In a recent study, Park et al. demonstrated that siRNA-mediated silencing of Gal-3 in a human osteosarcoma cell line resulted in decreased tumor cell migration and invasiveness (84). On the other hand, anti-E-selectin and anti-L-selectin mAbs have been successfully tested in the treatment of liver metastasis and ovarian cancer, respectively (49, 166, 167). In addition, modifications of natural selectin ligands such as P-selectin Glycoprotein Ligand-1 (PSGL-1) (i.e., by conjugation with IgG) afforded an auspicious selective inhibitor for interrupting leukocyte rolling and adhesion (168). In the case of galectin-inhibition, availability of recombinant proteins (i.e., truncated form of Gal-3) as well as mAbs (i.e., F8.G7 for Gal-1) opened new avenues for more specific and efficient blockers (34, 169–171). These therapies have been successfully tested in vivo and demonstrated a very good performance, suggesting their potential translation to clinical trials.
In summary, the data presented in this article stress the importance of understanding the biochemistry, biology, and glycobiology involved in tumor development and progression. The current major challenge faced by this field is designing selective inhibitors of lectin–glycan interactions with increased bioavailability. In turn, combined therapies of chemical inhibitors with biological therapeutics may tailor cancer treatments according to the unique features of individual tumors and patients.
AC and KM drafted the manuscript. AC and JS designed the figures and tables. All authors contributed with literature research, writing, discussion, and conclusions of the review. KM and GR performed a critical revision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the Argentinean Agency for Promotion of Science and Technology (PICT V 2014-367 and PICT 2012-2440) to GR, and PICT 2012-0555 to KM. All authors acknowledge support from Sales, Bunge and Born, and René Baron Foundations. KM and GR are researchers from the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina. AC is a post-doctoral fellow from CONICET.
1. Kissick HT, Sanda MG. The role of active vaccination in cancer immunotherapy: lessons from clinical trials. Curr Opin Immunol (2015) 35:15–22. doi:10.1016/j.coi.2015.05.004
2. Liu C-C, Ye X-S. Carbohydrate-based cancer vaccines: target cancer with sugar bullets. Glycoconj J (2012) 29(5–6):259–71. doi:10.1007/s10719-012-9399-9
3. Miller JFAP, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell (2015) 27(4):439–49. doi:10.1016/j.ccell.2015.03.007
4. Koster BD, de Gruijl TD, van den Eertwegh AJM. Recent developments and future challenges in immune checkpoint inhibitory cancer treatment. Curr Opin Oncol (2015) 27(6):482–8. doi:10.1097/cco.0000000000000221
5. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi:10.1038/nature13988
6. Hart GW, Copeland RJ. Glycomics hits the big time. Cell (2010) 143(5):672–6. doi:10.1016/j.cell.2010.11.008
7. Corfield AP, Berry M. Glycan variation and evolution in the eukaryotes. Trends Biochem Sci (2015) 40(7):351–9. doi:10.1016/j.tibs.2015.04.004
8. Stanley P, Schachter H, Taniguchi N. N-Glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2nd edn, Chapter 8. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1917/
9. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer (2015) 15(9):540–55. doi:10.1038/nrc3982
10. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell (2006) 126(5):855–67. doi:10.1016/j.cell.2006.08.019
11. Mariño K, Saldova R, Adamczyk B, Rudd PM. Changes in serum N-glycosylation profiles: functional significance and potential for diagnostics. In: Rauter AP, Lindhorst TK, editors. Carbohydrate Chemistry. (Vol. 37). Cambridge: The Royal Society of Chemistrys (2012). p. 57–93.
12. Reis CA, Osorio H, Silva L, Gomes C, David L. Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol (2010) 63(4):322–9. doi:10.1136/jcp.2009.071035
13. Peracaula R, Barrabás S, Sarrats A, Rudd PM, de Llorens R. Altered glycosylation in tumours focused to cancer diagnosis. Dis Markers (2008) 25(4–5):207–18. doi:10.1155/2008/797629
14. Häuselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol (2014) 4:28. doi:10.3389/fonc.2014.00028
15. Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics (2014) 14(4–5):525–46. doi:10.1002/pmic.201300387
16. Daniotti JL, Vilcaes AA, Torres Demichelis V, Ruggiero FM, Rodriguez-Walker M. Glycosylation of glycolipids in cancer: basis for development of novel therapeutic approaches. Front Oncol (2013) 3:306. doi:10.3389/fonc.2013.00306
17. Vasconcelos-dos-Santos A, Oliveira IA, Lucena MC, Mantuano NR, Whelan SA, Dias WB, et al. Biosynthetic machinery involved in aberrant glycosylation: promising targets for developing of drugs against cancer. Front Oncol (2015) 5:138. doi:10.3389/fonc.2015.00138
18. Brooks SA, Carter TM, Royle L, Harvey DJ, Fry SA, Kinch C, et al. Altered glycosylation of proteins in cancer: what is the potential for new anti-tumour strategies. Anticancer Agents Med Chem (2008) 8(1):2–21. doi:10.2174/187152008783330860
19. Boligan K, Mesa C, Fernandez L, von Gunten S. Cancer intelligence acquired (CIA): tumor glycosylation and sialylation codes dismantling antitumor defense. Cell Mol Life Sci (2015) 72(7):1231–48. doi:10.1007/s00018-014-1799-5
20. Mechref Y, Hu Y, Garcia A, Hussein A. Defining putative glycan cancer biomarkers by mass spectrometry. Bioanalysis (2012) 4(20):2457–69. doi:10.4155/bio.12.246
21. Harvey DJ, Merry AH, Royle L, Campbell MP, Dwek RA, Rudd PM. Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics (2009) 9(15):3796–801. doi:10.1002/pmic.200900096
22. Patani N, Jiang W, Mokbel K. Prognostic utility of glycosyltransferase expression in breast cancer. Cancer Genomics Proteomics (2008) 5(6):333–40.
23. Gomes J, Marcos NT, Berois N, Osinaga E, Magalhães A, Pinto-de-Sousa J, et al. Expression of UDP-N-acetyl-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-6 in gastric mucosa, intestinal metaplasia, and gastric carcinoma. J Histochem Cytochem (2009) 57(1):79–86. doi:10.1369/jhc.2008.952283
24. Berois N, Gattolliat C-H, Barrios E, Capandeguy L, Douc-Rasy S, Valteau-Couanet D, et al. GALNT9 gene expression is a prognostic marker in neuroblastoma patients. Clin Chem (2013) 59(1):225–33. doi:10.1373/clinchem.2012.192328
25. Chen Z, Gulzar ZG, St. Hill CA, Walcheck B, Brooks JD. Increased expression of GCNT1 is associated with altered O-glycosylation of PSA, PAP, and MUC1 in human prostate cancers. Prostate (2014) 74(10):1059–67. doi:10.1002/pros.22826
26. Anugraham M, Jacob F, Nixdorf S, Everest-Dass AV, Heinzelmann-Schwarz V, Packer NH. Specific glycosylation of membrane proteins in epithelial ovarian cancer cell lines: glycan structures reflect gene expression and DNA methylation status. Mol Cell Proteomics (2014) 13(9):2213–32. doi:10.1074/mcp.M113.037085
27. Wang Y, Fukuda T, Isaji T, Lu J, Im S, Hang Q, et al. Loss of α1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J (2015) 29(8):3217–27. doi:10.1096/fj.15-270710
28. Chia J, Goh G, Bard F. Short O-GalNAc glycans: regulation and role in tumor development and clinical perspectives. Biochim Biophys Acta (2016). doi:10.1016/j.bbagen.2016.03.008
29. Taniguchi N, Kizuka Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res (2015) 126:11–51. doi:10.1016/bs.acr.2014.11.001
30. Saldova R, Dempsey E, Pérez-Garay M, Mariño K, Watson JA, Blanco-Fernández A, et al. 5-AZA-2′-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics (2011) 6(11):1362–72. doi:10.4161/epi.6.11.17977
31. Bassagañas S, Allende H, Cobler L, Ortiz MR, Llop E, de Bolós C, et al. Inflammatory cytokines regulate the expression of glycosyltransferases involved in the biosynthesis of tumor-associated sialylated glycans in pancreatic cancer cell lines. Cytokine (2015) 75(1):197–206. doi:10.1016/j.cyto.2015.04.006
32. Venkitachalam S, Revoredo L, Varadan V, Fecteau RE, Ravi L, Lutterbaugh J, et al. Biochemical and functional characterization of glycosylation-associated mutational landscapes in colon cancer. Sci Rep (2016) 6:23642. doi:10.1038/srep23642
33. Dimitroff CJ. Galectin-binding O-glycosylations as regulators of malignancy. Cancer Res (2015) 75(16):3195–202. doi:10.1158/0008-5472.CAN-15-0834
34. Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell (2014) 156(4):744–58. doi:10.1016/j.cell.2014.01.043
35. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol (2007) 7(4):255–66. doi:10.1038/nri2056
36. Rachel H, Chang-Chun L. Recent advances toward the development of inhibitors to attenuate tumor metastasis via the interruption of lectin–ligand interactions. In: Derek H, editor. Advances in Carbohydrate Chemistry and Biochemistry. Oxford: Academic Press (2013). p. 125–207.
37. Magesh S, Ando H, Tsubata T, Ishida H, Kiso M. High-affinity ligands of siglec receptors and their therapeutic potentials. Curr Med Chem (2011) 18(23):3537–50. doi:10.2174/092986711796642580
38. Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annu Rev Immunol (2012) 30(1):357–92. doi:10.1146/annurev-immunol-020711-075018
39. Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol (2014) 14(10):653–66. doi:10.1038/nri3737
40. Merli M, Ferrario A, Maffioli M, Arcaini L, Passamonti F. Investigational therapies targeting lymphocyte antigens for the treatment of non-Hodgkin’s lymphoma. Expert Opin Investig Drugs (2015) 24(7):897–912. doi:10.1517/13543784.2015.1038342
41. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell–dependent tumor immunosurveillance. J Clin Invest (2014) 124(4):1810–20. doi:10.1172/JCI65899
42. Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia (2004) 19(2):176–82. doi:10.1038/sj.leu.2403598
43. Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12–Syk pathway. Glycobiology (2013) 23(2):178–87. doi:10.1093/glycob/cws139
44. Swanson BJ, McDermott KM, Singh PK, Eggers JP, Crocker PR, Hollingsworth MA. MUC1 is a counter-receptor for myelin-associated glycoprotein (Siglec-4a) and their interaction contributes to adhesion in pancreatic cancer perineural invasion. Cancer Res (2007) 67(21):10222–9. doi:10.1158/0008-5472.can-06-2483
45. Mitra N, Banda K, Altheide TK, Schaffer L, Johnson-Pais TL, Beuten J, et al. SIGLEC12, a human-specific segregating (Pseudo)gene, encodes a signaling molecule expressed in prostate carcinomas. J Biol Chem (2011) 286(26):23003–11. doi:10.1074/jbc.M111.244152
46. Cummings RD, McEver RP. C-type lectins. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. Chapter 31. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1943/
47. Chen M, Geng J-G. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz) (2006) 54(2):75–84. doi:10.1007/s00005-006-0010-6
48. Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets (2007) 11(11):1473–91. doi:10.1517/14728222.11.11.1473
49. Chen ZP, Jing YMS, Song BMS, Han YMS, Chu YP. Chemically modified heparin inhibits in vitro L-selectin-mediated human ovarian carcinoma cell adhesion. Int J Gynecol Cancer (2009) 19(4):540–6. doi:10.1111/IGC.0b013e3181a44bc8
50. Vasta GR. Galectins as pattern recognition receptors: structure, function, and evolution. Adv Exp Med Biol (2012) 946:21–36. doi:10.1007/978-1-4614-0106-3_2
51. Huflejt M, Leffler H. Galectin-4 in normal tissues and cancer. Glycoconj J (2003) 20(4):247–55. doi:10.1023/B:GLYC.0000025819.54723.a0
52. Oka T, Murakami S, Arata Y, Hirabayashi J, Kasai K-I, Wada Y, et al. Identification and cloning of rat galectin-2: expression is predominantly in epithelial cells of the stomach. Arch Biochem Biophys (1999) 361(2):195–201. doi:10.1006/abbi.1998.0968
53. Madsen P, Rasmussen HH, Flint T, Gromov P, Kruse TA, Honoré B, et al. Cloning, expression, and chromosome mapping of human galectin-7. J Biol Chem (1995) 270(11):5823–9. doi:10.1074/jbc.270.11.5823
54. Dyer KD, Rosenberg HF. Eosinophil charcot-leyden crystal protein binds to beta-galactoside sugars. Life Sci (1996) 58(23):2073–82. doi:10.1016/0024-3205(96)00201-9
55. Yang R-Y, Hsu DK, Yu L, Chen H-Y, Liu F-T. Galectin-12 is required for adipogenic signaling and adipocyte differentiation. J Biol Chem (2004) 279(28):29761–6. doi:10.1074/jbc.M401303200
56. Hotta K, Funahashi T, Matsukawa Y, Takahashi M, Nishizawa H, Kishida K, et al. Galectin-12, an adipose-expressed galectin-like molecule possessing apoptosis-inducing activity. J Biol Chem (2001) 276(36):34089–97. doi:10.1074/jbc.M105097200
57. Rabinovich Gabriel A, Croci Diego O. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity (2012) 36(3):322–35. doi:10.1016/j.immuni.2012.03.004
58. Salatino M, Rabinovich GA. Fine-tuning antitumor responses through the control of galectin–glycan interactions: an overview. In: Cuturi CM, Anegon I, editors. Suppression and Regulation of Immune Responses: Methods and Protocols. Totowa, NJ: Humana Press (2011). p. 355–74.
59. Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene (2001) 20:7486–93. doi:10.1038/sj.onc.1204950
60. Harazono Y, Kho DH, Balan V, Nakajima K, Zhang T, Hogan V, et al. Galectin-3 leads to attenuation of apoptosis through Bax heterodimerization in human thyroid carcinoma cells. Oncotarget (2014) 5(20):9992–10001. doi:10.18632/oncotarget.2486
61. Kopitz J, von Reitzenstein C, André S, Kaltner H, Uhl J, Ehemann V, et al. Negative regulation of neuroblastoma cell growth by carbohydrate-dependent surface binding of galectin-1 and functional divergence from galectin-3. J Biol Chem (2001) 276(38):35917–23. doi:10.1074/jbc.M105135200
62. Markowska AI, Liu F-T, Panjwani N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J Exp Med (2010) 207(9):1981–93. doi:10.1084/jem.20090121
63. Thijssen VL, Barkan B, Shoji H, Aries IM, Mathieu V, Deltour L, et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res (2010) 70(15):6216–24. doi:10.1158/0008-5472.can-09-4150
64. Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Méndez-Huergo SP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res (2013) 73(3):1107–17. doi:10.1158/0008-5472.CAN-12-2418
65. Hittelet A, Legendre H, Nagy N, Bronckart Y, Pector J-C, Salmon I, et al. Upregulation of galectins-1 and -3 in human colon cancer and their role in regulating cell migration. Int J Cancer (2003) 103(3):370–9. doi:10.1002/ijc.10843
66. Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: a potential mechanism of tumor-immune privilege. Cancer Cell (2004) 5(3):241–51. doi:10.1016/S1535-6108(04)00024-8
67. Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol (2007) 8(8):825–34. doi:10.1038/ni1482
68. Stowell SR, Qian Y, Karmakar S, Koyama NS, Dias-Baruffi M, Leffler H, et al. Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. J Immunol (2008) 180(5):3091–102. doi:10.4049/jimmunol.180.5.3091
69. Liu F-T, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer (2005) 5(1):29–41. doi:10.1038/nrc1527
70. Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature (1995) 378(6558):736–9. doi:10.1038/378736a0
71. Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol (2009) 10(9):981–91. doi:10.1038/ni.1772
72. Thiemann S, Man JH, Chang MH, Lee B, Baum LG. Galectin-1 regulates tissue exit of specific dendritic cell populations. J Biol Chem (2015) 290:22662–77. doi:10.1074/jbc.M115.644799
73. Kuo P-L, Hung J-Y, Huang S-K, Chou S-H, Cheng D-E, Jong Y-J, et al. Lung cancer-derived galectin-1 mediates dendritic cell anergy through inhibitor of DNA binding 3/IL-10 signaling pathway. J Immunol (2011) 186(3):1521–30. doi:10.4049/jimmunol.1002940
74. Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Neuzillet C, Albert S, et al. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat Rev (2014) 40(2):307–19. doi:10.1016/j.ctrv.2013.07.007
75. Ito K, Stannard K, Gabutero E, Clark A, Neo S-Y, Onturk S, et al. Galectin-1 as a potent target for cancer therapy: role in the tumor microenvironment. Cancer Metastasis Rev (2012) 31(3–4):763–78. doi:10.1007/s10555-012-9388-2
76. Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. Galectin-1 is a novel structural component and a major regulator of H-Ras nanoclusters. Mol Biol Cell (2008) 19(4):1404–14. doi:10.1091/mbc.E07-10-1053
77. Nakahara S, Oka N, Raz A. On the role of galectin-3 in cancer apoptosis. Apoptosis (2005) 10(2):267–75. doi:10.1007/s10495-005-0801-y
78. Raz A, Zhu D, Hogan V, Shah N, Raz T, Karkash R, et al. Evidence for the role of 34-kDa galactoside-binding lectin in transformation and metastasis. Int J Cancer (1990) 46(5):871–7. doi:10.1002/ijc.2910460520
79. Oliveira JTD, Ribeiro C, Gärtner F. Role of galectin-3 in cancer metastasis. Glycobiol Insights (2015) 5:1–13. doi:10.4137/GBI.S13916
80. Fukumori T, Kanayama H-O, Raz A. The role of galectin-3 in cancer drug resistance. Drug Resist Updat (2007) 10(3):101–8. doi:10.1016/j.drup.2007.04.001
81. Glinsky VV, Glinsky GV, Rittenhouse-Olson K, Huflejt ME, Glinskii OV, Deutscher SL, et al. The role of thomsen-friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res (2001) 61(12):4851–7.
82. Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity (2008) 28(3):414–24. doi:10.1016/j.immuni.2008.01.011
83. Tsuboi S, Sutoh M, Hatakeyama S, Hiraoka N, Habuchi T, Horikawa Y, et al. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J (2011) 30(15):3173–85. doi:10.1038/emboj.2011.215
84. Park GB, Kim D, Kim Y, Lee H, Kim CW, Hur DY. Silencing of galectin-3 represses osteosarcoma cell migration and invasion through inhibition of FAK/Src/Lyn activation and β-catenin expression and increases susceptibility to chemotherapeutic agents. Int J Oncol (2015) 46:185–94. doi:10.3892/ijo.2014.2721
85. Moon B-K, Lee YJ, Battle P, Jessup JM, Raz A, Kim H-RC. Galectin-3 protects human breast carcinoma cells against nitric oxide-induced apoptosis: implication of galectin-3 function during metastasis. Am J Pathol (2001) 159(3):1055–60. doi:10.1016/S0002-9440(10)61780-4
86. Harazono Y, Hyo Kho D, Balan V, Nakajima K, Hogan V, Raz A. Extracellular galectin-3 programs multidrug resistance through Na+/K+-ATPase and P-glycoprotein signaling. Oncotarget (2015) 6(23):19592–604. doi:10.18632/oncotarget.4285
87. Harazono Y, Nakajima K, Raz A. Why anti-Bcl-2 clinical trials fail: a solution. Cancer Metastasis Rev (2014) 33(1):285–94. doi:10.1007/s10555-013-9450-8
88. Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci U S A (2001) 98(6):3352–7. doi:10.1073/pnas.061615598
89. Kozlowski EO, Pavao MSG, Borsig L. Ascidian dermatan sulfates attenuate metastasis, inflammation and thrombosis by inhibition of P-selectin. J Thromb Haemost (2011) 9(9):1807–15. doi:10.1111/j.1538-7836.2011.04401.x
90. Wang R, Huang J, Wei M, Zeng X. The synergy of 6-O-sulfation and N- or 3-O-sulfation of chitosan is required for efficient inhibition of P-selectin-mediated human melanoma A375 cell adhesion. Biosci Biotechnol Biochem (2010) 74(8):1697–700. doi:10.1271/bbb.100140
91. Perez-Castells J, Hernandez-Gay JJ, Denton RW, Tony KA, Mootoo DR, Jimenez-Barbero J. The conformational behaviour and P-selectin inhibition of fluorine-containing sialyl LeX glycomimetics. Org Biomol Chem (2007) 5(7):1087–92. doi:10.1039/B615752A
92. Shodai T, Suzuki J, Kudo S, Itoh S, Terada M, Fujita S, et al. Inhibition of P-selectin-mediated cell adhesion by a sulfated derivative of sialic acid. Biochem Biophys Res Commun (2003) 312(3):787–93. doi:10.1016/j.bbrc.2003.10.188
93. Gouge-Ibert V, Pierry C, Poulain F, Serre A-L, Largeau C, Escriou V, et al. Synthesis of fluorinated C-mannopeptides as sialyl Lewisx mimics for E- and P-selectin inhibition. Bioorg Med Chem Lett (2010) 20(6):1957–60. doi:10.1016/j.bmcl.2010.01.141
94. Filser C, Kowalczyk D, Jones C, Wild MK, Ipe U, Vestweber D, et al. Synthetic glycopeptides from the E-selectin ligand 1 with varied sialyl Lewisx structure as cell-adhesion inhibitors of E-selectin. Angew Chem Int Ed (2007) 46(12):2108–11. doi:10.1002/anie.200604442
95. Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest (2001) 108(2):169–73. doi:10.1172/JCI200113530
96. Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol (2012) 2012:10. doi:10.1155/2012/676731
97. Hostettler N, Naggi A, Torri G, Ishai-Michaeli R, Casu B, Vlodavsky I, et al. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J (2007) 21(13):3562–72. doi:10.1096/fj.07-8450com
98. Kragh M, Binderup L, Vig Hjarnaa P, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep (2005) 14:99–104. doi:10.3892/or.14.1.99
99. Mousa SA, Petersen LJ. Anti-cancer properties of low-molecular-weight heparin: preclinical evidence. Thromb Haemost (2009) 102(8):258–67. doi:10.1160/TH08-12-0832
100. Belting M. Glycosaminoglycans in cancer treatment. Thromb Res (2014) 133(Suppl 2):S95–101. doi:10.1016/S0049-3848(14)50016-3
101. Denton RW, Cheng X, Tony KA, Dilhas A, Hernández JJ, Canales A, et al. C-disaccharides as probes for carbohydrate recognition – investigation of the conformational requirements for binding of disaccharide mimetics of sialyl Lewis X. European J Org Chem (2007) 2007(4):645–54. doi:10.1002/ejoc.200600825
102. Nicoll G, Avril T, Lock K, Furukawa K, Bovin N, Crocker PR. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur J Immunol (2003) 33(6):1642–8. doi:10.1002/eji.200323693
103. Attrill H, Takazawa H, Witt S, Kelm S, Isecke R, Brossmer R, et al. The structure of siglec-7 in complex with sialosides: leads for rational structure-based inhibitor design. Biochem J (2006) 397(2):271–8. doi:10.1042/bj20060103
104. Kelm S, Gerlach J, Brossmer R, Danzer C-P, Nitschke L. The ligand-binding domain of CD22 is needed for inhibition of the B cell receptor signal, as demonstrated by a novel human CD22-specific inhibitor compound. J Exp Med (2002) 195(9):1207–13. doi:10.1084/jem.20011783
105. Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood (2010) 115(23):4778–86. doi:10.1182/blood-2009-12-257386
106. Kelm S, Madge P, Islam T, Bennett R, Koliwer-Brandl H, Waespy M, et al. C-4 modified sialosides enhance binding to siglec-2 (CD22): towards potent Siglec inhibitors for immunoglycotherapy. Angew Chem Int Ed (2013) 52(13):3616–20. doi:10.1002/anie.201207267
107. Blanchard H, Yu X, Collins PM, Bum-Erdene K. Galectin-3 inhibitors: a patent review (2008–present). Expert Opin Ther Pat (2014) 24(10):1053–65. doi:10.1517/13543776.2014.947961
108. Blanchard H, Bum-Erdene K, Hugo MW. Inhibitors of galectins and implications for structure-based design of galectin-specific therapeutics. Aust J Chem (2014) 67(12):1763–79. doi:10.1071/CH14362
109. Tellez-Sanz R, Garcia-Fuentes L, Vargas-Berenguel A. Human galectin-3 selective and high affinity inhibitors. Present state and future perspectives. Curr Med Chem (2013) 20(24):2979–90. doi:10.2174/09298673113209990163
110. Ingrassia L, Nshimyumukiza P, Dewelle J, Lefranc F, Wlodarczak L, Thomas S, et al. A lactosylated steroid contributes in vivo therapeutic benefits in experimental models of mouse lymphoma and human glioblastoma. J Med Chem (2006) 49(5):1800–7. doi:10.1021/jm050971v
111. Iurisci IDA, Cumashi A, Sherman AA, Tsvetkov YE, Tinari N, Piccolo E, et al. Synthetic inhibitors of galectin-1 and -3 selectively modulate homotypic cell aggregation and tumor cell apoptosis. Anticancer Res (2009) 29(1):403–10.
112. Ito K, Ralph S. Inhibiting galectin-1 reduces murine lung metastasis with increased CD4+ and CD8+ T cells and reduced cancer cell adherence. Clin Exp Metastasis (2012) 29(6):561–72. doi:10.1007/s10585-012-9471-7
113. Leffler H, Nilsson UJ. Low-molecular weight inhibitors of galectins. In: Klyosov AA, Traber PG, editors. Galectins and Disease Implications for Targeted Therapeutics. Washington, DC: Oxford University Press (2012). p. 47–59.
114. Lin C-I, Whang EE, Donner DB, Jiang X, Price BD, Carothers AM, et al. Galectin-3 targeted therapy with a small molecule inhibitor activates apoptosis and enhances both chemosensitivity and radiosensitivity in papillary thyroid cancer. Mol Cancer Res (2009) 7(10):1655–62. doi:10.1158/1541-7786.MCR-09-0274
115. Öberg CT, Noresson A-L, Leffler H, Nilsson UJ. Synthesis of 3-amido-3-deoxy-β-d-talopyranosides: all-cis-substituted pyranosides as lectin inhibitors. Tetrahedron (2011) 67(47):9164–72. doi:10.1016/j.tet.2011.09.098
116. Glinskii OV, Sud S, Mossine VV, Mawhinney TP, Anthony DC, Glinsky GV, et al. Inhibition of prostate cancer bone metastasis by synthetic TF antigen mimic/galectin-3 inhibitor lactulose-l-leucine. Neoplasia (2012) 14(1):65–73. doi:10.1593/neo.111544
117. Glinsky VV, Kiriakova G, Glinskii OV, Mossine VV, Mawhinney TP, Turk JR, et al. Synthetic galectin-3 inhibitor increases metastatic cancer cell sensitivity to taxol-induced apoptosis in vitro and in vivo. Neoplasia (2009) 11(9):901–9. doi:10.1593/neo.09594
118. Glinsky GV, Price JE, Glinsky VV, Mossine VV, Kiriakova G, Metcalf JB. Inhibition of human breast cancer metastasis in nude mice by synthetic glycoamines. Cancer Res (1996) 56(23):5319–24.
119. Rabinovich GA, Cumashi A, Bianco GA, Ciavardelli D, Iurisci I, D’Egidio M, et al. Synthetic lactulose amines: novel class of anticancer agents that induce tumor-cell apoptosis and inhibit galectin-mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology (2006) 16(3):210–20. doi:10.1093/glycob/cwj056
120. Zhang W, Xu P, Zhang H. Pectin in cancer therapy: a review. Trends Food Sci Technol (2015) 44(2):258–71. doi:10.1016/j.tifs.2015.04.001
121. Jiang J, Eliaz I, Sliva D. Synergistic and additive effects of modified citrus pectin with two polybotanical compounds, in the suppression of invasive behavior of human breast and prostate cancer cells. Integr Cancer Ther (2013) 12(2):145–52. doi:10.1177/1534735412442369
122. Jun Y, Katz A. PectaSol-C modified citrus pectin induces apoptosis and inhibition of proliferation in human and mouse androgen-dependent and- independent prostate cancer cells. Integr Cancer Ther (2010) 9(2):197–203. doi:10.1177/1534735410369672
123. Jackson CL, Dreaden TM, Theobald LK, Tran NM, Beal TL, Eid M, et al. Pectin induces apoptosis in human prostate cancer cells: correlation of apoptotic function with pectin structure. Glycobiology (2007) 17(8):805–19. doi:10.1093/glycob/cwm054
124. Guess BW, Scholz MC, Strum SB, Lam RY, Johnson HJ, Jennrich RI. Modified citrus pectin (MCP) increases the prostate-specific antigen doubling time in men with prostate cancer: a phase II pilot study. Prostate Cancer Prostatic Dis (2003) 6(4):301–4. doi:10.1038/sj.pcan.4500679
125. Grous JJ, Redfern CH, Mahadevanm D, Schindler J. GCS-100, a galectin-3 antagonist, in refractory solid tumors: a phase I study. 2006 ASCO Annual Meeting Proceedings. June 2–6. Atlanta (2006). 13023.
126. Chauhan D, Li G, Podar K, Hideshima T, Neri P, He D, et al. A novel carbohydrate-based therapeutic GCS-100 overcomes bortezomib resistance and enhances dexamethasone-induced apoptosis in multiple myeloma cells. Cancer Res (2005) 65(18):8350–8. doi:10.1158/0008-5472.CAN-05-0163
127. Zomer E, Traber PG, Klyosov AA, Chekhova E. Google Patents. Composition of Novel Carbohydrate Drug for Treatment of Human Diseases. (2013).
128. Klyosov A, Zomer E, Platt D. DAVANAT (GM-CT-01) and colon cancer: preclinical and clinical (phase I and II) studies. In: Klyosov AA, editor. Glycobiology and Drug Design. Washington, DC: Oxford University Press (2012). p. 89–130.
129. Newton-Northup J, Dickerson M, Ma L, Besch-Williford C, Deutscher S. Inhibition of metastatic tumor formation in vivo by a bacteriophage display-derived galectin-3 targeting peptide. Clin Exp Metastasis (2013) 30(2):119–32. doi:10.1007/s10585-012-9516-y
130. Deutscher SL, Figueroa SD, Kumar SR. Tumor targeting and SPECT imaging properties of an 111In-labeled galectin-3 binding peptide in prostate carcinoma. Nucl Med Biol (2009) 36(2):137–46. doi:10.1016/j.nucmedbio.2008.10.015
131. Dings RPM, van der Schaft DWJ, Hargittai B, Haseman J, Griffioen AW, Mayo KH. Anti-tumor activity of the novel angiogenesis inhibitor anginex. Cancer Lett (2003) 194(1):55–66. doi:10.1016/S0304-3835(03)00015-6
132. Mayo KH, Dings RPM, Flader C, Nesmelova I, Hargittai B, van der Schaft DWJ, et al. Design of a partial peptide mimetic of anginex with antiangiogenic and anticancer activity. J Biol Chem (2003) 278(46):45746–52. doi:10.1074/jbc.M308608200
133. Zucchetti M, Bonezzi K, Frapolli R, Sala F, Borsotti P, Zangarini M, et al. Pharmacokinetics and antineoplastic activity of galectin-1-targeting OTX008 in combination with sunitinib. Cancer Chemother Pharmacol (2013) 72(4):879–87. doi:10.1007/s00280-013-2270-2
134. Dings RPM, Chen X, Hellebrekers DMEI, van Eijk LI, Zhang Y, Hoye TR, et al. Design of nonpeptidic topomimetics of antiangiogenic proteins with antitumor activities. J Natl Cancer Inst (2006) 98(13):932–6. doi:10.1093/jnci/djj247
135. Cagnoni AJ, Kovensky J, Uhrig ML. Design and synthesis of hydrolytically stable multivalent ligands bearing thiodigalactoside analogues for peanut lectin and human galectin-3 binding. J Org Chem (2014) 79(14):6456–67. doi:10.1021/jo500883v
136. Cumpstey I, Salomonsson E, Sundin A, Leffler H, Nilsson UJ. Double affinity amplification of galectin–ligand interactions through arginine–arene interactions: synthetic, thermodynamic, and computational studies with aromatic diamido thiodigalactosides. Chemistry (2008) 14(14):4233–45. doi:10.1002/chem.200701932
137. Ito K, Scott S, Cutler S, Dong L-F, Neuzil J, Blanchard H, et al. Thiodigalactoside inhibits murine cancers by concurrently blocking effects of galectin-1 on immune dysregulation, angiogenesis and protection against oxidative stress. Angiogenesis (2011) 14(3):293–307. doi:10.1007/s10456-011-9213-5
138. Öberg CT, Blanchard H, Leffler H, Nilsson UJ. Protein subtype-targeting through ligand epimerization: talose-selectivity of galectin-4 and galectin-8. Bioorg Med Chem Lett (2008) 18(13):3691–4. doi:10.1016/j.bmcl.2008.05.066
139. Leclere L, Van Cutsem P, Michiels C. Anti-cancer activities of pH- or heat-modified pectin. Front Pharmacol (2013) 4:128. doi:10.3389/fphar.2013.00128
140. Nangia-Makker P, Hogan V, Honjo Y, Baccarini S, Tait L, Bresalier R, et al. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J Natl Cancer Inst (2002) 94(24):1854–62. doi:10.1093/jnci/94.24.1854
141. Pienta KJ, Nailk H, Akhtar A, Yamazaki K, Replogle TS, Lehr J, et al. Inhibition of spontaneous metastasis in a rat prostate cancer model by oral administration of modified citrus pectin. J Natl Cancer Inst (1995) 87(5):348–53. doi:10.1093/jnci/87.5.348
142. Platt D, Raz A. Modulation of the lung colonization of B16-F1 melanoma cells by citrus pectin. J Natl Cancer Inst (1992) 84(6):438–42. doi:10.1093/jnci/84.6.438
143. Menachem A, Bodner O, Pastor J, Raz A, Kloog Y. Inhibition of malignant thyroid carcinoma cell proliferation by Ras and galectin-3 inhibitors. Cell Death Discov (2015) 1:15047. doi:10.1038/cddiscovery.2015.47
144. Demotte N, Wieërs G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K, et al. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res (2010) 70(19):7476–88. doi:10.1158/0008-5472.CAN-10-0761
145. Demotte N, Bigirimana R, Wieërs G, Stroobant V, Squifflet J-L, Carrasco J, et al. A short treatment with galactomannan GM-CT-01 corrects the functions of freshly isolated human tumor–infiltrating lymphocytes. Clin Cancer Res (2014) 20(7):1823–33. doi:10.1158/1078-0432.CCR-13-2459
146. Cotter F, Smith DA, Boyd TE, Richards DA, Alemany C, Loesch D, et al. Single-agent activity of GCS-100, a first-in-class galectin-3 antagonist, in elderly patients with relapsed chronic lymphocytic leukemia. 2009 ASCO Annual Meeting Proceedings. May 29–June 2. Orlando (2009). 7006.
147. Zomer E, Klyosov AA, Platt D. Development of a galactomannan polysaccharide as a vehicle to improve the efficacy of chemotherapeutics. In: Klyosov AA, editor. Glycobiology and Drug Design. Washington, DC: Oxford University Press (2012). p. 69–87.
148. Miller MC, Klyosov A, Platt D, Mayo KH. Using pulse field gradient NMR diffusion measurements to define molecular size distributions in glycan preparations. Carbohydr Res (2009) 344(10):1205–12. doi:10.1016/j.carres.2009.04.010
149. Miller MC, Klyosov A, Mayo KH. The α-galactomannan davanat binds galectin-1 at a site different from the conventional galectin carbohydrate binding domain. Glycobiology (2009) 19(9):1034–45. doi:10.1093/glycob/cwp084
150. Zou J, Glinsky VV, Landon LA, Matthews L, Deutscher SL. Peptides specific to the galectin-3 carbohydrate recognition domain inhibit metastasis-associated cancer cell adhesion. Carcinogenesis (2005) 26(2):309–18. doi:10.1093/carcin/bgh329
151. Yang Y, Li L, Zhou Z, Yang Q, Liu C, Huang Y. Targeting prostate carcinoma by G3-C12 peptide conjugated N-(2-Hydroxypropyl)methacrylamide copolymers. Mol Pharm (2014) 11(10):3251–60. doi:10.1021/mp500083u
152. Yang Y, Zhou Z, He S, Fan T, Jin Y, Zhu X, et al. Treatment of prostate carcinoma with (Galectin-3)-targeted HPMA copolymer-(G3-C12)-5-Fluorouracil conjugates. Biomaterials (2012) 33(7):2260–71. doi:10.1016/j.biomaterials.2011.12.007
153. Sun W, Li L, Yang Q, Shan W, Zhang Z, Huang Y. G3-C12 peptide reverses galectin-3 from foe to friend for active targeting cancer treatment. Mol Pharm (2015) 12(11):4124–36. doi:10.1021/acs.molpharmaceut.5b00568
154. Balan V, Nangia-Makker P, Raz A. Galectins as cancer biomarkers. Cancers (Basel) (2010) 2(2):592. doi:10.3390/cancers2020592
155. Wang JB, Wang MD, Li EX, Dong DF. Advances and prospects of anginex as a promising anti-angiogenesis and anti-tumor agent. Peptides (2012) 38(2):457–62. doi:10.1016/j.peptides.2012.09.007
156. Mayo KH. From carbohydrate to peptidomimetic inhibitors of galectins. In: Klyosov AA, Traber PG, editors. Galectins and Disease Implications for Targeted Therapeutics. Washington, DC: Oxford University Press (2012). p. 61–77.
157. Griffioen AW, van der Schaft DW, Barendsz-Janson AF, Cox A, Struijker Boudier HA, Hillen HF, et al. Anginex, a designed peptide that inhibits angiogenesis. Biochem J (2001) 354(Pt 2):233–42. doi:10.1042/bj3540233
158. Brandwijk RJMGE, Dings RPM, van der Linden E, Mayo KH, Thijssen VLJL, Griffioen AW. Anti-angiogenesis and anti-tumor activity of recombinant anginex. Biochem Biophys Res Commun (2006) 349(3):1073–8. doi:10.1016/j.bbrc.2006.08.154
159. Salomonsson E, Thijssen VL, Griffioen AW, Nilsson UJ, Leffler H. The anti-angiogenic peptide anginex greatly enhances galectin-1 binding affinity for glycoproteins. J Biol Chem (2011) 286(16):13801–4. doi:10.1074/jbc.C111.229096
160. Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Rabinovich GA, Bieche I, et al. OTX008, a selective small-molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur J Cancer (2014) 50(14):2463–77. doi:10.1016/j.ejca.2014.06.015
161. Rezai K, Durand S, Lachaux N, Raymond E, Herait P, Lokiec F. OTX008 pharmacokinetics (PK) during the first-in-man phase I study in patients with advanced solid tumors. Proceedings of the 104th Annual Meeting of the American Association for Cancer Research April 6–10, 2013. Washington, DC (2013).
162. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
163. Böcker S, Laaf D, Elling L. Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3. Biomolecules (2015) 5(3):1671. doi:10.3390/biom5031671
164. Cedeno-Laurent F, Opperman M, Barthel SR, Hays D, Schatton T, Zhan Q, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates anti-tumor immunity. J Invest Dermatol (2012) 132(2):410–20. doi:10.1038/jid.2011.335
165. Gao X, Zhi Y, Zhang T, Xue H, Wang X, Foday A, et al. Analysis of the neutral polysaccharide fraction of MCP and its inhibitory activity on galectin-3. Glycoconj J (2012) 29(4):159–65. doi:10.1007/s10719-012-9382-5
166. Martín-Satué M, Marrugat R, Cancelas JA, Blanco J. Enhanced expression of α(1,3)-fucosyltransferase genes correlates with E-selectin-mediated adhesion and metastatic potential of human lung adenocarcinoma cells. Cancer Res (1998) 58(7):1544–50.
167. Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer (1997) 71(4):612–9. doi:10.1002/(SICI)1097-0215(19970516)71:4<612:AID-IJC17>3.0.CO;2-D
168. Eppihimer MJ, Schaub RG. Soluble P-selectin antagonist mediates rolling velocity and adhesion of leukocytes in acutely inflamed venules. Microcirculation (2001) 8(1):15–24. doi:10.1111/j.1549-8719.2001.tb00154.x
169. Mirandola L, Yu Y, Cannon MJ, Jenkins MR, Rahman RL, Nguyen DD, et al. Galectin-3 inhibition suppresses drug resistance, motility, invasion and angiogenic potential in ovarian cancer. Gynecol Oncol (2014) 135(3):573–9. doi:10.1016/j.ygyno.2014.09.021
170. Croci DO, Salatino M, Rubinstein N, Cerliani JP, Cavallin LE, Leung HJ, et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J Exp Med (2012) 209(11):1985–2000. doi:10.1084/jem.20111665
Keywords: glycans, cancer, siglecs, C-type lectins, selectins, galectins
Citation: Cagnoni AJ, Pérez Sáez JM, Rabinovich GA and Mariño KV (2016) Turning-Off Signaling by Siglecs, Selectins, and Galectins: Chemical Inhibition of Glycan-Dependent Interactions in Cancer. Front. Oncol. 6:109. doi: 10.3389/fonc.2016.00109
Received: 09 March 2016; Accepted: 18 April 2016;
Published: 13 May 2016
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Fabio Grizzi, Humanitas Clinical and Research Center, ItalyCopyright: © 2016 Cagnoni, Pérez Sáez, Rabinovich and Mariño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel A. Rabinovich, Z2FieXJhYmlAZ21haWwuY29t;
Karina V. Mariño, a21hcmlub0BkbmEudWJhLmFy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.