 Lukas F. Mager1
Lukas F. Mager1 Marie-Hélène Wasmer
Marie-Hélène Wasmer Philippe Krebs
Philippe Krebs
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 19 April 2016
Sec. Gastrointestinal Cancers
Volume 6 - 2016 | https://doi.org/10.3389/fonc.2016.00096
The emergence of novel immunomodulatory cancer therapies over the last decade, above all immune checkpoint blockade, has significantly advanced tumor treatment. For colorectal cancer (CRC), a novel scoring system based on the immune cell infiltration in tumors has greatly improved disease prognostic evaluation and guidance to more specific therapy. These findings underline the relevance of tumor immunology in the future handling and therapeutic approach of malignant disease. Inflammation can either promote or suppress CRC pathogenesis and inflammatory mediators, mainly cytokines, critically determine the pro- or anti-tumorigenic signals within the tumor environment. Here, we review the current knowledge on the cytokines known to be critically involved in CRC development and illustrate their mechanisms of action. We also highlight similarities and differences between CRC patients and murine models of CRC and point out cytokines with an ambivalent role for intestinal cancer. We also identify some of the future challenges in the field that should be addressed for the development of more effective immunomodulatory therapies.
Colorectal cancer is the second and third most common malignant disease in women and men, respectively (1). Estimations predict that there will be over 750,000 new cases and more than 350,000 deaths from colorectal cancer (CRC) in developed countries in 2015 alone [reviewed in Ref. (2)]. A large number of genetic aberrations have been identified that underlie CRC (3, 4). Somatically altered genes or polymorphisms that are repeatedly found in CRC often affect the KRAS-, MYC-, Wnt-, mitogen-activated protein kinase (MAPK)-, or TGF-β/bone morphogenetic protein (BMP)-signaling pathways, lamina structural proteins or components of the DNA repair machinery [reviewed in Ref. (2, 5)]. Recently, several factors have received increased attention that are distinct from tumor cells and that substantially contribute to cancer progression. These include non-cancerous cells in the vicinity of the tumor, which are commonly referred to as tumor microenvironment or stroma, and the microbiota. Non-malignant cells in the tumor mass comprise (myo)fibroblasts, endothelial cells, and immune cells (6). Although fibroblasts and endothelial cells can promote CRC (7–9), the immune infiltrates in the tumor microenvironment appear to differently modulate CRC development, depending on their nature. Initial studies reporting a correlation between the inflammatory cell pattern in CRC tumors and the prognosis (10, 11) have been further elaborated by Galon and colleagues into the “Immunoscore,” a classification that has improved the prognostication of CRC development (12–14). In essence, this work established that analysis of the composition of the immune infiltrates enhances the accuracy of prognostic information and predictability of response to therapy. Therefore, it provides complementary information to the traditional American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC)-TNM classification applied for more than 80 years. The Immunoscore analyzes the regional density of all (CD3+) T cells, including CD8+ cytotoxic T cells, in human CRC. This allows in a next step to associate T cell differentiation with CRC progression; e.g., a T helper 1 (Th1) signature correlates with better disease-free survival, whereas a Th17 signature is predictive of the opposite (15).
A central feature of activated immune cells is the production and release of growth factors and cytokines that modulate the inflammatory milieu in tumor tissues. Systemic and local changes in the cytokine profile have been shown in CRC (16–18). Recent work indicates that multiple pro-tumorigenic and also anti-tumorigenic cytokines are differently expressed in distinct CRC tissues (19). Therefore, it is critical to study the specific contribution of individual cytokines to CRC development, progression, and patient survival.
Here, we review the current knowledge on cytokines known to modulate intestinal tumor development. Each listed cytokine is systemically presented according to the following scheme: (1) role as a biomarker in samples of CRC patients, (2) phenotype of knockout or transgenic mice in experimental models of CRC or intestinal inflammation, (3) tissue of origin or type of the secreting cells, (4) nature of the receptor-expressing cells in the tumor stroma, (5) biological effect on human/murine primary cells or cell lines, and (6) molecular mechanisms and pathways activated upon receptor engagement.
As a synthesis, we highlight two major, functionally different inflammatory networks: a network of inflammatory mediators driving (antigen)-specific anti-tumor immunity to inhibit tumor development (19, 20) and a nexus of cytokines supporting chronic, unspecific, pro-tumorigenic inflammation in the CRC microenvironment (Figure 1), which is associated with a disruption of the intestinal barrier and invasion by microbial products (21). It is likely that the balance between these two opposite inflammatory networks within the tumor stroma determines the course of CRC development. We also propose that these different groups of cytokines may be used as biomarkers to strengthen the diagnostic based on the Immunoscore, and that they represent targets for the design of therapeutic approaches.
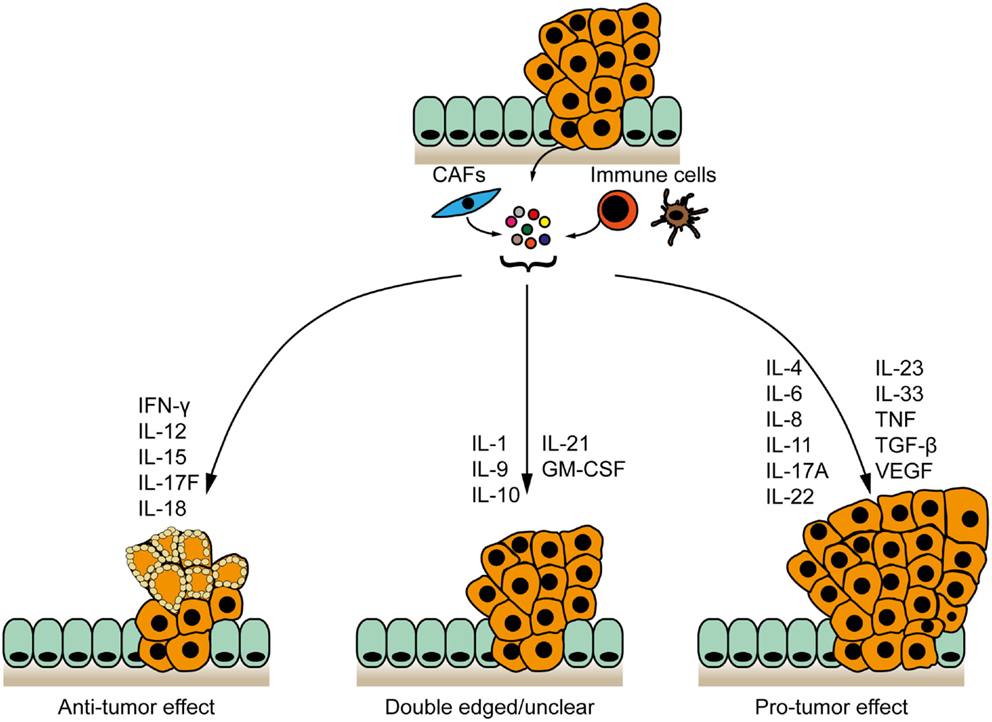
Figure 1. Cytokine networks in the pathogenesis of colorectal cancer. Cytokines expressed by tumor and/or stromal cells cluster to form networks with anti-tumor, pro-tumor, or bivalent properties. IFN-γ, interleukin-12 (IL-12), IL-15, IL-17F, and IL-18 inhibit CRC development. IL-4, IL-6, IL-8, IL-11, IL-17A, IL-22, IL-23, IL-33, TNF, TGF-β, and VEGF are pro-tumorigenic. The contribution of IL-1, IL-9 IL-10, IL21 and GM-CSF to intestinal cancer remains unclear.
The role of IL-1 in CRC is controversial. IL1B transcripts are increased in tumor biopsies of patients with metastatic CRC (22) and polymorphisms in IL-1 receptor antagonist (IL-1RA) (encoded by IL1RN) may be associated with CRC (23). IL-1β is activated by caspase-mediated cleavage subsequent to the activation of the leucine-rich repeat (NLR) inflammasome as a response to cell stress or infection. The NLR inflammasome plays an important role in the development of colitis-associated cancer (CAC). In vivo blockade of IL-1β using recombinant IL-1RA significantly decreased tumor development in the AOM/DSS mouse model of CAC (24), indicating a pro-tumorigenic role of IL-1β in this setting. IL-1β is expressed by tumor-associated macrophages (TAMs) (25) and neutrophils (24). IL-1β can act on intestinal epithelial cells (IECs) (24) and directly on tumor cells (25) to induce their proliferation. IL-1β also promotes the recruitment of myeloid-derived suppressor cells (MDSCs) to tumors, which supports cancer progression (26–28). Furthermore, colon cancer cell-derived IL-1α may upregulate angiogenesis by modulating stromal cells within the tumor microenvironment (29).
Interleukin-1 binds to either IL-1Rα (encoded by IL1R1) that has a long cytoplasmic domain to relay signaling or to IL-1Rβ (encoded by IL1R2) that may act as a decoy receptor. Upon IL-1 binding, IL-1Rα forms a complex with IL-1 receptor accessory protein (encoded by IL1RAP). This induces the recruitment and activation of the IRAK and TRAF6 adapter molecules and the activation of nuclear factor κB (NF-κB), JNK, AP-1, and p38 MAPK pathways [reviewed in Ref. (30)]. Furthermore, IL-1β induces the activation of the Wnt signaling pathway by phosphorylation of GSK3β (25). Importantly, all these signaling pathways are key for intestinal tumorigenesis (31–33), thereby further supporting the central role of IL-1 for CRC pathogenesis.
Interleukin-4 is overexpressed in early events of CRC development, including hyperplastic polyp, adenoma, and serrated adenomas, whereas in adenocarcinomas, IL-4 levels are not elevated compared with normal mucosa (34). In addition, higher serum levels of IL-4 were found in CRC patients with distant metastases (M1) compared with patients without metastases (M0) (16). However, presence of a Th2 gene signature – comprising IL4, IL5, and IL13 – in human CRC does not appear to have a prognostic value (15).
In experimental animal models of CRC, Il4-deficient mice treated with AOM developed fewer tumors compared with wild-type (WT) mice (35). In the AOM/DSS model of tumorigenesis, signaling through IL-4 receptor α (IL-4Rα) promoted intestinal tumor growth (36). However, both IL-4 and IL-13 engage IL-4Rα, which prevents a clear estimation of IL-4 function in CRC in this study. To bypass this issue, Ingram et al. evaluated intestinal tumor formation in AOM-treated Il13−/− mice, thus only allowing signaling of IL-4 trough IL-4Rα. They found that intestinal tumor development was markedly increased in Il13−/− compared with WT mice (37), which further indicates a pro-tumorigenic effect of IL-4 in CRC.
Th2 and double-positive CD4+ CD8αβ+ αβ T cells as well as cancer-initiating cells are important sources of secreted IL-4 in CRC (38, 39). IL-4 signaling occurs through either the type I or type II IL-4 receptor. The type I IL-4 receptor complex is composed of IL-4Rα and the common gamma chain, whereas the type II IL-4 receptor consists of the IL-4Rα and IL-13Rα1 subunits (40). Type I IL-4 receptor is predominately expressed on hematopoietic cells, whereas type II IL-4 receptor expression is high on transformed IECs (36). This expression pattern suggests that IL-4 may have a direct and an indirect effect on CRC development. Indeed, the proliferation of several CRC cell lines was increased after IL-4 stimulation (36). Furthermore, in vitro coculture of IL-4-secreting CRC-derived tumor-initiating cells with peripheral blood mononuclear cells (PBMCs) was found to inhibit the proliferation of these PBMCs, which could be restored upon addition of IL-4 blocking antibodies. This may serve as a mechanism for tumor-initiating cells to escape immune surveillance and in turn promote CRC progression (39).
Mechanistically, IL-4/IL-4 receptor engagement leads to signal transducer and activator of transcription (STAT)-6 phosphorylation in hematopoietic (41) and epithelial cells (42). Increased STAT6 phosphorylation in CRC tumors negatively correlates with survival in patients (43). Of note, IL-4 has also been shown to inhibit tumor growth and progression in other tissues, such as renal cancer (44) and glioblastoma (45). This was dependent on tumor-specific CD8+ T cells or associated with a marked eosinophil infiltrate, respectively. In addition, Th2 immune responses have been shown to induce IL-4- and eosinophil-dependent anti-tumor activity (46). Thus, IL-4 may have distinct functions, depending on the tumor environment. However, in CRC IL-4 rather appears to drive tumor development.
Interleukin-6 is a prototypic inflammatory cytokine clearly involved in the development of sporadic CRC and CAC (47). IL-6 is overexpressed in CRC tissues (48, 49) and elevated levels of serum IL-6 correlate with larger tumor size, occurrence of liver metastases, and reduced survival (50). Moreover, increased blood concentration of IL-6 is an independent adverse prognostic marker of survival in CRC patients (51). Patients suffering from inflammatory bowel disease (IBD) – who have an increased risk for developing CAC – show elevated levels of IL-6 in the serum and lamina propria (52, 53).
In the AOM/DSS model of CAC, genetic ablation of Il6 or treatment with anti-IL-6 receptor ameliorated tumor development. This is explained by the fact that IECs critically depend on IL-6 trans-signaling for their survival (54, 55). Lamina propria T cells, macrophages, and cancer-associated fibroblasts (CAFs) can all secrete IL-6 in the CRC stroma (53, 55–57). IL-6 was also described to promote angiogenesis (56) and DNA mismatch repair defects (58). In addition, IL-6 directly promotes the accumulation of MDSCs in tumors, thereby facilitating tumor progression (26). Furthermore, MDSC-produced IL-6 limits the development of CD4+ Th1 cells (59).
Intestinal epithelial cells do not express IL-6R themselves but rely on IL-6 trans-signaling. IL-6 trans-signaling requires soluble IL-6R generated through alternative mRNA splicing of IL6R or ADAM17-dependent limited proteolysis of IL-6R on other cells. Soluble IL-6R, then, associates with the ubiquitously expressed glycoprotein 130 (IL-6 ST/GP130) (60). Engagement of IL-6 with its receptor leads to activation of STAT3, which promotes the proliferation of cancer cells and tumor progression (55, 61). Thus, it is not surprising that the level of STAT3 phosphorylation in CRC patients negatively correlates with survival and is therefore indicative of a poor prognosis (62, 63).
In CRC patients, IL-8 expression is upregulated in tumor tissue compared with adjacent healthy colonic tissue (64). Moreover, germline polymorphisms of IL-8 and vascular endothelial growth factor (VEGF), two genes involved in tumor angiogenesis, are associated with increased risk of recurrence in stage III CRC patients treated with adjuvant chemotherapy (65).
Upon AOM/DSS treatment, transgenic mice expressing human IL8 were found to show increased CRC tumor numbers and load compared with WT counterparts. IL-8 resulted in higher IEC proliferation in these transgenic mice (64).
In humans, the epithelial–mesenchymal transition (EMT) activator protein SNAIL regulates Il8 expression in CRC stem-like cells (66).
Interleukin-8 mainly acts on myeloid cells to enhance their mobilization (64). These myeloid cells have immune suppressive functions and promote tumor progression (64, 67).
Mechanistically, IL-8 promotes tumor growth, metastasis, chemoresistance, and angiogenesis, as assayed in CRC cell line models (68, 69). IL-8 signals through its receptor CXCR1/2 to activate the Akt and MAPK pathways and promote the expression of genes responsible for cell proliferation, invasion, and angiogenesis (70). Therefore, blockade of IL-8 signaling pathway may represent a promising therapeutic strategy to restrain CRC development.
Interleukin-9 seems to have a dual role for cancer development. Although IL-9 possibly promotes lymphomagenesis (71), it can also inhibit the growth of melanoma either directly or by promoting anti-tumor immunity (72, 73). Compared with healthy individuals, CRC patients showed decreased levels of IL-9 in the serum and intestinal tissues, and IL-9 expression in these samples negatively correlated with tumor progression (74). By contrast, another study found no significant changes in IL-9 expression in serum of CRC patients, with possibly a trend toward higher levels of IL-9 in patients with high-grade tumors (16). Interestingly, in a heterotopic tumor model using the colon carcinoma cell line CT26, Il9−/− animals were protected from CRC development and showed better survival compared with challenged WT animals, a phenotype that was T cell-dependent (75).
Various immune cells in the colon have been shown to produce IL-9, such as T cells, dendritic cells, and natural killer (NK) T cells (76–78). Moreover, stromal cells, including CAFs, also produce IL-9 during CRC (79). IL-9 receptor is expressed both on hematopoietic cells and IECs. Therefore, IL-9 may have a direct or indirect mode of action for CRC development (80). Of note, stimulation of human CRC cell lines with IL-9 has also yielded conflicting data. Caco-2 cells showed reduced proliferation and wound closure in the presence of recombinant IL-9 (80), whereas the proliferation of KM12C and KM12SM cell was enhanced upon stimulation with IL-9 (79).
Engagement of the IL-9 receptor leads to STAT5 phosphorylation (80), the expression of which is predictive of poor prognosis and shorter survival in CRC patients (81).
In conclusion, IL-9 may either enhance or inhibit CRC development. Therefore, further studies are required to clearly dissect the role of IL-9 during intestinal tumorigenesis.
In patient samples, IL-10 serum levels increase over time during CRC progression (82, 83), and high preoperative serum levels of IL-10 correlate with poor survival of CRC patients (84). This suggests a tumor-promoting role of IL-10 in CRC patients. In contrast, IL-10 appears to play a protective role in animal models of CRC. IL-10 was required in regulatory T cells (Treg) to reduce tumor burden in ApcMin/+ mice (85). However, during CRC development, Treg cells may switch their cytokine production from IL-10 to IL-17, which promotes tumor development (86). Furthermore, oral administration of IL-10 microparticles decreased polyposis in the ApcMin/+ model by suppressing the development of IL-17-producing Treg and inducing conventional, IL-17-negative Treg (87). In the ApcΔ468 model, T cell-restricted ablation of IL-10 increased the number of intestinal polyps by promoting the accumulation of microbes and eosinophils in intestinal tumors (88). In CAC, Il10-deficient mice were shown to be more susceptible to spontaneous intestinal tumor development compared with WT animals (89).
In the intestine, a number of cells, including T cells, monocytes, macrophages, and epithelial cells, have been shown to produce IL-10 (90–92). The IL-10 receptor is a heterotetramer complex consisting of two IL-10Rα and two IL-10Rβ molecules. Although IL-10Rα is specific for IL-10, IL-10Rβ is also used for IL-22 and IL-26 signaling. IL-10Rα is constitutively expressed on most hematopoietic cells and colonic IECs (93); yet, it can also be induced on a number of other cells [reviewed in Ref. (94)]. Binding of IL-10 to its receptor activates STAT1, STAT3, and STAT5 (95), and it is not clear which of these pathways is preferentially activated by IL-10 in CRC. This may explain the divergent findings on IL-10 in CRC, and elucidation of the precise role of IL-10 for CRC remains one of the future challenges in the field.
Interleukin-11, a family member of the IL-6 family of cytokines, has recently been implicated in CRC pathogenesis. IL-11 and its receptor have both been shown to be overexpressed in sporadic CRC specimens (96, 97). Moreover, multiple cell types upregulate IL11 transcript levels during CRC development, including hematopoietic cells, CAFs, and also tumor cells (7, 97). The pro-tumorigenic effect of IL-11 on tumorigenesis was found to be stronger than the one of IL-6, both in models of CAC and sporadic CRC (97). IL-11 has thus emerged as a novel cytokine driving STAT3-dependent intestinal tumorigenesis similarly to IL-6, and further investigation will help to evaluate its role as a diagnostic or therapeutic target for CRC.
Active IL-12 is constituted of two subunits, IL-12p35 and IL-12p40 that are encoded by IL12A and IL12B, respectively. These subunits may form either an agonistic IL-12p70 heterodimer or an antagonistic IL-12p80 homodimer (98). The IL-12p35 subunit is shared to generate IL-35 (99, 100), whereas IL-12p40 is shared to form IL-23 (101). Therefore, it is difficult to study the effect of IL-12 without interfering at the same time with IL-23 or IL-35 signaling. Nevertheless, in CRC patients high preoperative IL-12p40 serum levels predicted a better survival (102), whereas a low production of IL-12p70 in dendritic cells was associated with a poor prognosis (103).
In mice, the anti-tumor effect of IL-12 has been described in a variety of murine tumor models, including melanoma, sarcoma, renal cell carcinomas, and lymphomas (104–107). Moreover, in a heterotopic tumor model of CRC metastasis, recombinant IL-12 inhibited metastatic events to the lung (105). In the intestine, dendritic cells, macrophages, and B cells have been reported to produce IL-12p35 and IL-12p40. Of note, stimulus by microbial products, such as lipopolysaccharide (LPS) and CpG oligodeoxynucleotide, and also IL-10 are necessary for IL-12 production (108, 109).
Natural killer cells and γδ T cells are the main type of cells expressing IL-12 receptor (110), thus suggesting an indirect action of IL-12 on tumor cells. IL-12 plays a central role both for the induction and the expansion of Th1 responses as well as the activation of cytotoxic immune effectors, such as NK and CD8+ T cells (111). IL-12 activates and induces IFN-γ production in these cells, which limits tumor growth and metastasis (112–115).
Taken together, these data indicate a protective role of IL-12 in CRC. Although administration of IL-12 as an adjuvant leads to specific immunity against tumor antigens in some patients (116), the net clinical benefit of IL-12 treatment was found to be rather moderate (117, 118). The immunosuppressive effect of the tumor environment and tumor escape mechanisms appear to be the reasons for the failure of the IL-12-based immunotherapy applied so far in human cancers [reviewed in Ref. (119)]. Whether improved administration of IL-12 or combined immunomodulatory approaches that may induce more potent anti-tumor activity remains to be addressed.
Colorectal cancer patients with genomic deletion of IL15 have a significantly higher risk of tumor recurrence and show reduced survival compared with patients with intact IL15 (19). Moreover, IL-15 is expressed in human CRC cells in situ (120).
In the AOM/DSS model of CAC, Il15−/− but not Il15ra−/− mice showed higher tumor incidence and tumor size than WT counterparts. Therefore, residual low-affinity IL-15 signaling via the shared IL-2Rβ/γc subunits of the IL-15 receptor appears to be able to decrease CAC pathogenesis in Il15ra−/− mice. Furthermore, in the same study, loss of IL-15 was found to be associated with an upregulation of inflammatory mediators involved in CRC progression (120). Transgenic mice overexpressing Il15 better controlled transferred MC-38 colon carcinoma cells and showed no pulmonary metastasis compared with WT mice. Accordingly, in the same model, Il15−/− mice showed more rapid tumor growth and died from lung metastases (121). In another study, IL-15 was found to potentiate the effect of immune checkpoint blockade using combined anti-PD-L1 and anti-CTLA-4, thereby leading to prolonged survival in a metastatic murine CT26 CRC model (122). Similarly, IL-15 lead to a synergistic enhancement of agonistic anti-CD40 therapeutic treatment in mouse models of lung metastasis using CT26 or MC38 CRC cells (123). Consequently, IL-15 may have a promising immunotherapeutic potential in the treatment of CRC.
Interleukin-15 is produced by a broad range of cells, including stromal and epithelial cells, and also myeloid cells, such as monocytes, macrophages, and dendritic cells. Dendritic cells, effector CD8+ T cells, and NK cells all express the IL-15 receptor that is composed of three distinct receptor chains (124–126). IL-15 can either bind directly to the IL-2Rβ/γc heterodimer with low-affinity or it can be trans-presented to this same heterodimer after binding the high-affinity IL-15Rα subunit (125). Cytotoxic T and NK cells represent the most important immune effectors to integrate the anti-tumorigenic function of IL-15 by activating the APO-1/FAS- or granule-mediated cytotoxic pathway (124, 126). Thus, IL-15 regulates anti-tumor cytotoxicity and modulates the inflammatory tumor microenvironment.
Of the six members of the IL-17 family identified so far, namely IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F (127), only IL-17A and IL-17F have been studied in greater detail for their contribution to CRC development. Expression of IL17A transcripts is enhanced stepwise along the adenoma-to-carcinoma sequence in the stroma and adenomatous/cancerous intestinal epithelium of CRC patients (128). Furthermore, IL-17A serum levels were elevated in CRC patients compared with healthy individuals, positively correlated with tumor size (129) or circulating tumor cells (130), and predicted poor survival (130). Patients with high expression of genes associated with a Th17 signature in CRC tissues have a poor prognosis (15, 131).
In mice, Il17a deficiency partially protected from CRC in the ApcMin/+ as well as AOM/DSS models (132, 133). Furthermore, there is probably a link between IL-17A and IL-6 expression, as Il17a−/− mice showed markedly reduced IL-6 levels in the AOM/DSS model of CAC (133). Thus, IL-17A may indirectly activate STAT3 through IL-6.
CD4+ Th17 immune cells are the main source of IL-17A in CRC tumors and adjacent tissues (134, 135); yet, CD8+ T cell subsets, γδ T cells, and innate lymphoid cells (ILCs) are also important producers of IL-17A (136–139). IL-17A binds to a heterodimeric receptor comprising an IL-17RA and an IL-17RC chain. The IL-17 receptor is expressed on a variety of cells, such as hematopoietic, fibroblastic, and epithelial cells (140, 141). Although this indicates that engagement of IL-17 receptor signaling may act directly and indirectly on tumor cells, a recent study reports that direct signaling on transformed colonic epithelial cells is sufficient to promote CRC development (140).
Contrarily to IL-17A, IL-17F, another IL-17 family member, appears to have anti-tumorigenic properties. In a cohort of 102 Tunisian CRC patients, individuals with WT IL17F had a longer overall survival compared with patients with polymorphisms in IL17F (142). Similarly, the IL17F TT genotype was associated with a lower risk of CRC compared with the TC genotype and C allele in CRC patients from Iran (129). Furthermore, Il17f-deficient mice developed more and larger intestinal tumors compared with WT animals in the AOM/DSS model of CAC (143). In a heterotopic tumor model where HCT 116 CRC cells were engineered to overexpress IL-17F, tumor growth was inhibited likely through inhibition of angiogenesis (143). Even though IL-17F is strongly expressed in the colon by activated T cells and colonic epithelial cells (143, 144), IL-17F is downregulated in colorectal tumors (143). As for IL-17A, IL-17F binds to a receptor complex comprising IL-17RA and IL-17RC. Thus, IL-17F may also affect CRC development directly and indirectly.
It is so far still elusive how IL-17A and IL-17F exert such opposing functions through a same receptor. The differential regulation of IL-17A and IL-17F expression during CRC may provide a partial explanation to this paradox. Furthermore, IL-17F has a reduced ability to activate the downstream signaling cascade compared with IL-17A. Finally, IL-17RA and IL-17RC have different binding affinities for IL-17A and IL-17F, which may also account for the different roles of these cytokines [reviewed in Ref. (145)].
Mechanistically, IL-17A activates the NF-κB pathway in CRC cells (146), thereby driving tumor survival and growth [see also tumor necrosis factor (TNF)]. Of note, IL-17F does not lead to the activation of NF-κB in CRC cells, further highlighting the difference between IL-17A and IL-17F signaling (146).
Overall, these studies highlight the differential effect of IL-17A and IL-17F on CRC development. Il17ra−/− animals, which are insensitive to IL-17A and IL-17F, show reduced intestinal tumor number, size, and load during sporadic CRC (21). Therefore, the pro-tumorigenic effect of IL-17A is probably dominant over IL-17F in this model. Given different binding affinities of IL-17RA and IL-17RC for IL-17A and IL-17F, it may be relevant to assess CRC development also in Il17rc−/−animals.
Interleukin-18 is an anti-tumorigenic cytokine. Humans heterozygous for the IL-18 A607C polymorphism exhibit increased risk for CRC development (147). Compared with normal mucosa, IL-18 production is decreased in colon adenocarcinomas. In half of the cases, this reduction in IL-18 expression correlated with lack of IFNG and FASLG expression in the CRC tissue and the presence of distant metastases (148). Of note, mutations in the IL-18 receptor accessory protein (IL18RAP) gene are associated with Crohn’s disease and IBD (149).
Damage of IECs promotes the formation of the NLRP3 inflammasome, which in turn leads to the caspase-1-dependent processing and secretion of active IL-1β and IL-18 (150, 151). Mice deficient in Il18 or Il18r1 developed more tumors upon AOM/DSS treatment, compared with WT controls (152). In this model of CAC, IL-18 seems to be particularly important during the early inflammatory phase of the treatment. Indeed, Il18−/− mice showed a more severe clinical and histopathological manifestation of colitis than WT animals upon challenge with DSS, a phenotype recapitulated in Nlrp3−/− or Casp1−/− mice (153).
Murine IECs produce IL-18 under steady state. Secreted IL-18 stimulates IL-18 receptor (IL18-R1) that is expressed by CD4+ cells in the lamina propria. IL-18 signaling restricts inflammation in the intestine by limiting the differentiation of Th17 cells and promoting the expression of effector molecules in Treg (154). In addition, IL-18 is involved in the repair of the intestinal epithelium (155, 156), possibly via IL-22 (157), and it downregulates intestinal Il22bp expression (157). IL-18 also promotes protective host immunity mediated by cytotoxic cells, including CD8+ T lymphocytes, and NK cells (158).
Binding of IL-18 to its receptor IL-18Rα leads to the recruitment of IL-18Rβ to form a high affinity complex that recruits intracellular signaling effectors, including MyD88, IRAK, and TRAF6. As for IL-1 signaling, this cascade eventually results in the activation of NF-κB, JNK, and p38 MAPK (30). Although IL-1 and IL-18 pathways use the same signaling molecules downstream of their respective receptor complexes, they exert opposite function in CRC. This apparent contradiction may be explained by the distinct types of cells each of these cytokines activates within the tumor stroma.
Interleukin-21 appears to have an opposite role for CRC development in humans versus mice. IL-21 is upregulated in patients with ulcerative colitis-associated colon cancer and in the murine model of CAC based on AOM/DSS treatment (159). A systems biology approach aiming at quantifying different immune cell subpopulations in situ in human tumor found a positive correlation between IL-21 expression and disease-free survival (131). A preclinical study using IL-21 combined with cetuximab (an anti-EGFR monoclonal antibody) indicated an activation of immune response biomarkers on NK and T cells in stage IV CRC patients, yet it did not evaluate treatment efficacy (160).
Compared with WT controls, reduced tumor size and numbers were found in Il21−/− mice treated with AOM/DSS. Il21-deficient tumors show higher cell apoptosis and reduced cell proliferation, with high IFNγ and low IL-17 (161). In another study, resistance to CAC in Il21−/− mice was associated with reduced CD4+ T cell infiltration and decreased production of IL-6 and IL-17A in the intestinal mucosa (159). This suggests a pro-tumorigenic role of IL-21 in the setting of AOM/DSS-induced CAC.
CD4+ T helper cells, including Th1 and Th17 cells, activated NKT cells and T follicular helper cells all can secrete IL-21. IL-21 can act on a broad range of cells, such as B cells, NK cells, activated T cells, dendritic cells, macrophages, fibroblasts, and IECs (159, 162, 163). IL-21 enhances anti-tumor NK and CD8+ T cell responses (164, 165). In tumors of AOM/DSS-treated WT mice, lamina propria mononuclear cells and tumor-infiltrating immune cells, mostly T and myeloid cells, express the IL-21 receptor (159).
Interleukin-21 signals via heterodimers of the IL-21 receptor (IL-21R) and the common cytokine receptor γ-chain (IL-2Rγc). Upon receptor engagement, IL-21 induces the activation of JAK1, JAK3, and mainly STAT3 [reviewed in Ref. (163)].
Taken together, IL-21 has a possible impact on the polarization of the T helper cell response in CRC. Further investigation on the role of IL-21 for CRC may reconcile the current discrepancies from human and mouse studies, e.g., by using animal models of CRC in which inflammation is less central than for the AOM/DSS model.
Interleukin-22 has recently emerged as a novel player in CRC development. In patients, the accumulation of Th22 cells is associated with CRC development (166). Furthermore, high levels of IL-22 in the serum or CRC tissue are predictive of a poor survival of patients, and IL-22 promotes resistance to chemotherapy (167). Moreover, the rs1179251 polymorphism in IL22 gene is associated with an increased risk for CRC (168).
In animal models, IL-22 has been shown to ameliorate experimentally induced colitis and enhance wound healing (169–171). In Helicobacter hepaticus/AOM-induced intestinal tumors, IL-22 released by ILCs supported the growth of intestinal tumors in T and B cell-deficient Rag2−/− mice (138). Il22−/−; ApcMin/+ mice develop smaller tumors than controls. However, deficiency in Il22 leads to delayed wound healing and increased inflammation and therefore promotes intestinal tumor development in the AOM/DSS model of CAC (157). In addition, animal deficient in Il22bp, an antagonist of IL-22 signaling, developed strongly enhanced tumorigenesis. This likely relies on the IL-22BP-dependent regulation of IL-22 activity during intestinal tissue damage and tumorigenesis (157). Subcutaneous injection of primary CRC cells together with anti-IL-22 antibody treatment strongly inhibited tumor development and growth (172).
CD3+CD4+IL-22+ ILCs represent the major source of IL-22 in the intestine (138). IL-22 signaling is transmitted through a receptor complex comprised of the IL-10Rβ and IL-22Rα1 chains. Of note, IL-10Rβ and IL-22Rα1 were recently shown to be upregulated in primary CRC tissue samples (173). IL-22 signaling directly activates STAT3 in epithelial cells and increases stemness and tumorigenic potential in tumor cells through the methyltransferase DOT1L (172). In addition, IL-22 protects CRC cells from chemotherapy via STAT3-dependent autocrine secretion of IL-8 (167).
In sum, IL-22 seems to promote CRC development via induction of stemness in tumor cells. The mechanisms how IL-22 confers stemness in tumor cells compared with other pro-tumorigenic cytokines also signaling through STAT3, such as IL-6 and IL-11, warrant further investigation. This may be addressed by stratified analysis of the expression of IL-6R or IL-11R compared with IL-22R on intestinal stem cells. Alternatively, the IL-22 signaling is not negatively regulated by suppressor of cytokine signaling 3 (SOCS3), which is the case for IL-6 and IL-11 signaling. Consequently, IL-22 signaling may be more sustained and more pro-tumorigenic compared with other STAT3 activators [reviewed in Ref. (174)].
Bioactive IL-23 is a heterodimeric complex consisting of IL-23p19 (encoded by IL23A) and IL-12p40 (encoded by IL12B), which are specific for IL-23 or shared with IL-12, respectively. In the serum of CRC patients, IL-23 levels are increased and positively correlate with VEGF (175). In primary CRC tissue, IL23A and IL12B transcripts are overexpressed, whereas IL12A mRNA is not upregulated (176). Moreover, high IL-23 levels together with low SOCS3 expression in primary tumor tissue were predictive of a higher rate of CRC metastasis (177).
Il23a deficiency in mice resulted in fewer and smaller tumors compared with WT controls in a model relying on heterozygous loss of Apc (21). Furthermore, IL-23 enhanced the metastatic capabilities of SW620 CRC cells after injection into nude mice. Of note, the pro-metastatic effect of IL-23 was dependent on SOCS3, as concomitant overexpression of IL-23 and SOCS3 in SW620 cells reduced the number of metastases (177).
Dendritic cells, macrophages, and neutrophils have been shown to produce IL-23 during intestinal inflammation (178–180). A variety of hematopoietic cells in the intestine can react to IL-23, among them ILCs, Treg, and Th17 cells. The net biological effect of IL-23 signaling may vary in different cell populations. For instance, IL-23 signaling promotes IL-22 secretion by ILCs and IL-17 production by Th17 cells, while it abrogates Treg cell activation (181–183).
Overall, this suggests that IL-23 may indirectly promote tumor cell survival. Indeed, IL-23 has been reported to drive intestinal inflammation by inducing other pro-inflammatory cytokines, such as IL-6, IL-17, and IL-22 (184–186). These cytokines may in turn activate tumor cell proliferation through STAT3 and NF-κB.
Interleukin-33 is a member of the IL-1 family of cytokines, which has recently received greater attention for its contribution to intestinal inflammation and CRC. IL-33 is thought to function as an “alarmin” released upon cellular stress or damage to promote or amplify inflammation (187, 188). Unlike other IL-1 family members, such as IL1-β and IL-18, IL-33 becomes inactivated through caspase-1 cleavage. Cathepsin G and neutrophil elastase have been shown to cleave IL-33 and enhance it bioactivity (189, 190). The role of IL-33 for CRC development and its function in intestinal inflammation are still ambiguous. In IBD patients, expression of IL-33 and its receptor, ST2, positively correlate with the extent of inflammation (191, 192). In animal models of colitis, activation of the IL-33/ST2 pathway either promotes or restrains immunopathology and different phases of intestinal disease (188, 192, 193). Furthermore, IL-33 promotes intestinal Treg function in the inflamed intestine (183). Therefore, the IL-33/ST2 pathway may either promote or inhibit CRC development and was therefore the focus of several recent studies.
In CRC patients, we found overexpression of both IL-33 and ST2 in intestinal adenomas and adenocarcinomas. Although several high-grade adenocarcinomas showed a strong expression of IL-33 and ST2, both proteins were predominately overexpressed in low-grade adenocarcinomas (194). Similar results were reported in a smaller CRC cohort where increased microvessel density of IL-33-positive and ST2-positive tumors was additionally observed (195). Higher amounts of serum IL-33 were measured in CRC patients compared with healthy individuals, thereby further suggesting a tumor-promoting effect of IL-33 (196).
In AOM/DSS-treated mice we could show that intestinal tumor number, size, and grade were markedly reduced in St2−/− compared with WT mice (194). Similarly, ApcMin/+ mice on an Il33-deficient background developed fewer and smaller intestinal tumors. In the same study, CRC development was also abrogated by treatment with ST2-blocking antibody (196). This indicates that the nuclear function of IL-33 as a regulator of gene transcription (197) and its role as a soluble cytokine upon secretion (198) may promote CRC pathogenesis in this model. Other studies using heterotopic models suggest that IL-33 signaling may also play a protective role (199) or promote metastasis (200) in CRC.
Epithelial cells and myofibroblasts are the main IL-33-expressing cells in the CRC microenvironment (196, 201, 202). Extracellular IL-33 binds to the IL-33 receptor consisting of ST2 and IL-1 receptor accessory protein (IL-1RAP). ST2, the IL-33-specific subunit of the IL-33 receptor, is expressed on epithelial cells, myofibroblasts, and immune cells (183, 194, 196). Our data suggest that IL-33 does not directly affect the proliferation of tumor cells, but rather decreases the barrier function of the intestine. This in turn allows for increased translocation of bacterial products to normally sterile tissues and induces the production of pro-tumorigenic cytokines, such as IL-6, by immune cells (194). Cytokines, such as IL-6, may then activate STAT3 to promote tumor growth. IL-6 is likely not the only downstream target of IL-33 in CRC. IL-33 stimulation of subepithelial myofibroblasts induced the expression of extracellular matrix components and growth factors associated with intestinal tumor progression (196). Furthermore, IL-33 may foster angiogenesis in CRC (195). As IL-33 and angiogenesis have been linked in previous studies (203, 204), it is conceivable that IL-33 triggers the production of or synergize with pro-angiogenic factors, such as VEGF, which may promote CRC progression and metastasis.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) has an ambivalent role for CRC in humans versus mice. GM-CSF expression is elevated in primary colon tumors compared with healthy controls. Moreover, overexpression of GM-CSF and its receptor in intestinal tissue correlates with improved overall survival of CRC patients (205). Interestingly, patient stratification revealed that increased levels of GM-CSF benefit mismatch repair-proficient, yet not repair-deficient patients. This favorable prognostic effect of GM-CSF production by CRC cells was independent from CD16+ myeloid and CD8+ T cell infiltrations (206).
GM-CSF promotes CAC in AOM/DSS-challenged mice as treatment with a neutralizing anti-GM-CSF antibody decreased tumor development and colitis score in this model (207).
GM-CSF is produced by IECs and even more by neoplastic colonic epithelial cells (206, 207). Commensal microbiota-derived LPS triggers GM-CSF expression in IECs (207). Stromal fibroblasts and lymphocytes adjacent to the CRC tumor have also been found to be positive for GM-CSF (208). More recently, ILC3-derived GM-CSF has been shown to play an important role for intestinal inflammation (209).
Cancerous epithelial cells, monocytes, and antigen-presenting cells all express the GM-CSF receptor in the CRC microenvironment (205–207). GM-CSF induces autocrine or paracrine VEGF release by IECs (207), thereby promoting angiogenesis; yet, it does not have a direct proliferative effect on these cells (208). GM-CSF can act on dendritic cells to promote an anti-tumor response (210) and on monocytes/macrophages to inhibit CRC cell proliferation (206). GM-CSF also recruits and activates eosinophils in the intestine to induce colitis in mice (211), yet high eosinophil counts in human CRC tumor infiltrate are associated with favorable patient outcome (212).
The GM-CSF receptor is a heterodimer consisting of a major binding subunit (GMRα, encoded by CSF2RA) and a signaling subunit (βc, encoded by CSF2RB and shared with IL-3 and IL-5). GM-CSF binding to its receptor activates the JAK-STAT, the MAPK, and the PI3K pathways, which results in cell survival and proliferation [reviewed in Ref. (213)].
IFN-γ expression is reduced in PBMCs of CRC patients (214). There is an association between high serum IFN-γ and absence of nodal metastases in CRC patients (16) and CRC patients with high IFN-γ levels in the supernatant of stimulated PBMC cultures indicate a trend toward better survival in donor CRC patients (215). These data suggest that IFN-γ induces a protective, anti-tumor response in CRC patients. Animal models of CRC support these clinical findings as Ifng−/− mice show more and larger intestinal tumors compared with WT controls (35). Moreover, in the sporadic ApcMin/+ tumor model, heterozygous loss of Ifng promoted adenoma progression and induced adenocarcinoma development (216).
Lymphocytes and activated dendritic cells represent the main sources of IFN-γ in the colon (217–219). The IFN-γ receptor (IFNGR) is expressed on almost all nucleated cells, including mature T cells, B cells, macrophages, endothelial, and epithelial tissues (220). IFN-γ signaling is a key determinant for a Th1 polarization of immune responses. It enhances MHC class I antigen representation and promotes CD8+ T-, NK cell-, and macrophage-mediated cytotoxicity. Hence, IFN-γ robustly stimulates anti-tumor immunity [reviewed in Ref. (221)]. Of note, exogenous IFN-γ inhibited whereas knockdown of Ifngr1 promoted the growth of HT-29 CRC cells (216).
Mechanistically, IFN-γ acts on CRC cells by inducing STAT1 phosphorylation and inhibiting the EGFR/Erk1/2 and Wnt/β-catenin signaling pathways, thereby restraining cell proliferation (216). Since increased nuclear STAT1 is associated with a better survival in CRC patients (222) and Th1 polarization – also induced through IFN-γ – correlates with prolonged survival of CRC patients (15), immunomodulatory approaches selectively inducing IFN-γ production may be considered for CRC therapy. However, IFN-γ can also compromise the barrier function of IEC monolayers in vitro (223–225) and possibly alter the colonic epithelial barrier in vivo (226). As increased intestinal permeability can drive intestinal inflammation to foster CRC formation (21), approaches promoting IFN-γ production likely represent a balancing act and need to be tightly controlled.
TGF-β has a dual function during intestinal tumorigenesis. In early tumors, TGF-β is a potent tumor suppressor that induces induced cell cycle arrest (227). The relevance of TGF-β for cancer is corroborated by the frequent occurrence of inactivating mutations found in molecular components of TGF-β signaling (228, 229).
In contrast, high TGF-β levels in the primary tumor or serum correlate with poor survival of CRC patients (7, 230–232). In animal models, a dual function of TGF-β for CRC has also been observed. Loss of TGF-β signaling induced more tumors in AOM-treated mice compared with controls (228). Moreover, heterozygous loss of Smad4, a downstream effector molecule of TGF-β receptor, caused larger and more invasive intestinal carcinomas in ApcΔ716/+ mice (233). This indicates a tumor-suppressive activity of TGF-β in these models. On the other hand, TGF-β was recently shown to promote EMT and metastasis of CRC cell lines in vivo, through mechanisms involving SOX4 and miR-1269a (234). Furthermore, TGF-β acting on CAFs promoted the growth of HT-29 cells in a xenograft model of CRC (9). Therefore, TGF-β also has a pro-tumorigenic function. TGF-β and its receptors TGFBR1 and TGFBR2 are commonly expressed on epithelial cells [reviewed in Ref. (235, 236)]. However, other cells in the tumor microenvironment, including CAFs, respond to or secrete TGF-β (237).
Although the exact mechanisms underlying the dual role of TGF-β for CRC have yet to be delineated, a recent study by Calon et al. sheds new light on this paradox. While activation of TGF-β signaling in epithelial cells rapidly induces the expression of cell-cycle checkpoint genes leading to growth arrest [reviewed in Ref. (238)], Calon et al. showed that activation of TGF-β signaling in fibroblasts promotes the metastatic capabilities of intestinal tumor cells (9). Therefore, TGF-β may indirectly exert a pro-tumorigenic effect on CRC cells, via the stroma. Indeed, TGF-β may for instance promote IL-11 secretion by CAFs (7), which in turn activates STAT3 and drives the proliferation of tumor cells (97).
As indicated by its name, TNF was first identified as an anticancer agent. Yet, it was thereafter recognized as a key cytokine linking inflammation and cancer (239). TNF expression is increased in CRC tissues and TNF serum levels positively correlate with CRC progression and reduced patient survival (240, 241). Accordingly, TNF blockers may possibly reduce the frequency of CAC in treated IBD patients (242–244), yet additional IBD cases are needed to strengthen this initial observation. Another open question remains whether patients with sporadic CRC may also benefit from a therapy based on TNF blockade. In addition to the evaluation of clinical parameters, the efficacy of such a blockade may be also directly assessed in vivo, via confocal laser endomicroscopy, as already performed in IBD patients (245).
Hematopoietic cell-produced TNF is critical for intestinal polyp formation in ApcΔ468 mice, a model of sporadic CRC development (246). Furthermore, TNF blockade strongly diminished tumor development in AOM/DSS-treated mice (247). Similarly, TNF neutralization in obese mice reduced the growth of tumors in a xenograft model using the human CRC line HT-29 (248).
Activated macrophages are the main producers of TNF in CRC (249, 250). TNF signals through TNF receptor 1, which is expressed on most cells, and TNF receptor 2 (TNFR2), which is mainly expressed on hematopoietic cells (251). However, in colitic mice and in IBD patients, TNFR2 expression becomes upregulated on IECs (252). Accordingly, binding of TNF to TNFR2 triggers the proliferation of IECs and CRC cell lines in a STAT3-dependent manner (252, 253) – similar to the IL-6-dependent activation cascade discussed above. In addition, TNF signaling drives the accumulation of MDSCs by promoting their survival (254). Activation of the signaling cascade downstream of the TNF receptors results either in the nuclear translocation of NF-κB and AP-1, which promotes cell survival and proliferation, or induces cell death through caspase activation [reviewed in Ref. (255)].
Most studies in the field of CRC suggest that TNF rather promotes cell survival and thus promotes CRC development. Nevertheless, few reports indicate that TNF may also have an anti-tumor effect in CAC, possibly by providing early antibacterial protection in Il10−/− mice (256). In addition, TNF may also serve as a marker of tumor-specific T cells (20). Therefore, the net contribution of TNF to CRC may be determined by the timing of its secretion during tumorigenesis or the type of the immune cells secreting it.
Vascular endothelial growth factor is a potent angiogenic factor that is frequently upregulated in cancer where it promotes tumor angiogenesis. In CRC, VEGF expression is elevated in tumor tissue and positively correlates with advanced tumor stage as well as positive lymph node and liver metastasis (257). Furthermore, CRC patients with VEGF-positive tumors show reduced life expectancy (257, 258). VEGF plasma levels are elevated in CRC patients compared with healthy individuals, and high preoperative VEGF plasma levels predict reduced survival (259).
In AOM/DSS-treated mice, both VEGF and its receptor VEGFR2 are strongly upregulated and anti-VEGF treatment reduced tumor growth (260). Antibody-mediated or genetic blockade of VEGF limited tumor growth and increased the survival of ApcMin/+ mice (261). Inhibition of VEGF receptor signaling similarly reduced intestinal tumor burden in ApcMin/+ animals (262).
Various cells, such as TAMs, CAFs, tumor cells, platelets, and mast cells, produce VEGF (6). There are several different molecules comprising the VEGF family, namely VEGF-A, VEGF-B, VEGF-C, and VEGF-D, as well as different isoforms of VEGF-A and VEGF-B with potentially different function. Their mode of action has already been reviewed in detail elsewhere (263). Of note, VEGFA, VEGFB, VEGFC, and VEGFD expression is modulated during the adenoma–carcinoma sequence in CRC. For instance, VEGFA is upregulated in adenomas and carcinomas, whereas VEGFD is more abundant in normal tissues (264).
Secreted VEGF binds to either VEGFR1 and VEGFR2 or VGFR3, which are expressed on endothelial cells. In CRC, VEGFR2 is thought to mainly drive angiogenesis to foster tumor development, whereas the function of VEGFR1 is still unclear. However, more recent data reveal that VEGFR1 is expressed on CRC cells and that VEGFR1 signaling actives the β-catenin/Wnt signaling pathway to promote tumor growth (265). In addition, β-catenin/Wnt signaling regulates VEGF expression in CRC (266). Furthermore, engagement of VEGF/VEGFR2 signaling directly in CRC cells leads to STAT3 phosphorylation and promotes tumor development (260).
Taken together, VEGF signaling can support CRC tumor development indirectly, by acting on endothelial cells, to stimulate angiogenesis (267). In addition, VEGF may also directly promote tumor growth through STAT3 and Wnt signaling and represents, therefore, an interesting target for CRC therapy.
Tumors are comprised of tumor cells, immune cells, endothelial cells, and (myo)fibroblasts. These cells form together a microenvironment that determines tumorigenesis and they interact, among others, through the intermediate of cytokines. These cytokines in the tumor stroma critically influence CRC development and progression either by directly stimulating neoplastic epithelial cells or by altering the function or activity of non-tumor cells in the CRC microenvironment. Particular cytokines inhibit, whereas other promote CRC progression (Figure 1). Therefore, it is critical to identify cytokine networks to be either repressed or enhanced using combinatorial therapeutic approaches, as neutralization of individual cytokines may not suffice for therapeutic efficacy (146). Moreover, the net effect of the inflammatory response on the tumor cells may be better predicted by analyzing the molecular signatures downstream of cytokine receptors, rather than solely quantifying individual cytokines. Eventually, further investigations aiming at elucidating the precise reasons for the apparent ambivalent function of certain cytokines are critical, as these studies may also reveal novel mechanisms to be leveraged for therapy.
We discuss below five aspects that we consider to be of particular relevance for the development of future therapeutic strategies for CRC.
Genetic aberrations are necessary for malignancy to be established and to determine the general behavior of the different subtypes of CRC identified so far (268). Of note, differences in the quantity of lesion-infiltrating immune cells are already distinguishable at a very early stage of CRC tumorigenesis, in dependence on subtypes of CRC (269). There is increasing evidence that distinct genetic signatures may be associated with specific cytokine networks. Enterocyte-restricted loss of Trp53, which encodes murine p53, is associated with increased intestinal permeability that results in an NF-κB-dependent inflammatory tumor environment (270). Moreover, p53 can negatively regulate IL-6 signaling (271), whereas gain-of-function mutant p53 prolongs TNF-induced NF-κB activation to increase susceptibly to CAC (272). Therefore, inhibition of the IL-6 and TNF pathways may represent an adjuvant therapy specific for CRC with p53 mutations, i.e., CRC subtype with “canonical” molecular signature (CMS2) (268).
As presented earlier, DNA mismatch repair-proficient CRC patients may benefit from a treatment activating the GM-CSF pathway (206), whereas IL-6 blockade may be more suitable in cases of mismatch repair-deficiency (58), i.e., for the subtype of CRC characterized by microsatellite instability (CMS1) (268).
Importantly, several of the studies performed so far using human samples or data from The Cancer Genome Atlas may have overlooked the presence of certain cytokines specifically produced by hematopoietic cells, since immune infiltrates are proportionally low in CRC tissues compared to cancerous and non-malignant stromal cells (268). Further research may unveil additional connections between molecular signatures that segregate CRC subtypes (268) and distinct cytokine networks, which may be used for immunomodulatory regimens more specific to tumor subtypes.
The pro- versus anti-tumorigenic effect of inflammation is not only determined by the mere presence of define cytokines at a given time in CRC tumors but also by the duration of the induced inflammatory stimulus. Indeed, prolonged or constant exposure to cytokines promotes tumorigenesis, as illustrated by a higher risk for CRC in IBD patients who suffer from chronic intestinal inflammation (273). Chronic inflammation contributes to the generation of MDSCs (274), which are also found in increased frequency in the blood of IBD patients (275). As a matter of fact, CRC tumor-elicited inflammation and MDSC-meditated immunosuppression are closely associated (139). Inflammation-induced accumulation of MDSCs in tumors leads a downregulation of immune surveillance and anti-tumor immunity, thereby facilitating tumor growth (276). Myeloid cells, in particular the so-called TAMs, are mainly responsible for the activation and maintenance of the chronic inflammatory process in tumors. The TAM-induced chronic inflammatory microenvironment may additionally promote genetic instability within the developing tumor epithelial cells [reviewed in Ref. (277–279)].
In contrast to this pro-tumorigenic chronic inflammation, short cytokine exposure or acute inflammatory signals may rather stimulate an anti-tumor response, e.g., by promoting T cell function (280, 281). For instance, treatment with endotoxin was shown to activate lymphocytes and induce an anti-tumor effect in some CRC patients (282). Therefore, therapeutic strategies aiming at changing the pro-tumorigenic chronic inflammation to an acute, antitumor inflammatory state may be beneficial.
Immunomodulation via inhibition of CTLA-4 or PD-1/PD-L1 signaling has recently emerged as a potent means for the treatment of malignancies (283). A beneficial effect of CTLA-4/PD-1 blockade has been shown in melanoma, lung, renal, and kidney prostate (283–285). CRC tumors with RAS mutation have a low expression of inhibitory molecules and fewer infiltrating immune cells (286). On the other hand, defects in mismatch repair are associated not only with a Th1 microenvironment in CRC tumors but also with upregulated expression of multiple immune checkpoints (287). This implies that particular CRC subtypes may show different susceptibility for immune checkpoint therapy. Nevertheless, immune checkpoint blockade for CRC therapy has not yielded promising results so far (288, 289).
Approaches targeting selected cytokine pathways or networks may directly restrain CRC tumorigenesis or improve the response rate of CRC tumors to chemotherapies or checkpoint inhibitors. As a matter of fact, clinical trials are completed or ongoing to evaluate the effect of blocking cytokines, such as IL-1α, IL-1β, IL-6, IL-10, IL-21, TNF, or VEGF. In spite of the above-presented multiple function of VEGF signaling in CRC, therapeutic antibody-mediated blockade of VEGF-A (using bevacizumab) did not prolong the survival of stages II and III CRC patients when given in combination with chemotherapy (290). Nevertheless, treatment with the same VEGF-A blocking antibody (bevacizumab) in combination with a different chemotherapy improved the outcome in patients with metastatic CRC (291). Furthermore, a phase I/II trial and a phase III trial have been completed in patients with solid malignancies to evaluate a blockade of IL-6 or TNF, respectively. Unfortunately, both trials showed little to no effect in disease control (292, 293). Indeed, inhibition of single cytokines will probably not yield strong results as cytokine signals are often overlapping. For instance, IL-6 exerts its pro-tumorigenic function through STAT3, a signaling pathway also switched on by multiple other cytokines, such as IL-11, IL-21, or IL-22. Therefore, combinatorial approaches targeting multiple cytokines or their downstream signaling molecules, also conjointly with chemotherapy or immune checkpoint blockade, may prove to be more effective. Such approaches may possibly also permit a lower treatment dose of checkpoint inhibitors, thereby reducing the risk for immune-related adverse events (294, 295). Such type of therapeutic strategy has actually already shown increased anti-tumor activity in a murine model of metastatic CRC (122).
Manipulation of the microbiota may represent an additional future therapeutic strategy for the treatment of CRC. Overall changes in microbial communities have been found in the adenoma-to-carcinoma sequence of CRC, compared with healthy controls (296). Different bacterial communities are either under- or overrepresented in cancerous versus adjacent non-cancerous intestinal tissues (297). Importantly, certain types of commensal bacteria can influence the host immune system by promoting the accumulation of Th17 or Treg in the intestinal mucosa (298, 299). In addition, commensal and pathogenic bacteria species induce different cytokine responses in IECs (300, 301). Alterations in the microbiota have been shown to drive IL-17C production from IECs to promote tumorigenesis (302). Intestinal tumors are frequently covered by microbial biofilms, which correlates with enhanced epithelial cell IL-6 and STAT3 activation (303). Moreover, bacteria-derived butyrate induces IL-18 in the colonic epithelium (304). Finally, the intestinal microbiota can influence the outcome of tumor immunotherapy, possibly by augmenting dendritic cell function and subsequent priming of anti-tumor T cells (305, 306). Therefore, it is conceivable that modulation of the microbiome may permit to influence the composition of the immune effectors and the cytokine networks within the CRC stroma, which may be used as adjuvant for therapy. A future objective in the field will, thus, be to advance our understanding on how microbes cross talk with the host to either promote or inhibit CRC formation. Improved knowledge of such communication pathways will likely ameliorate the treatment of CRC.
PK: conception and writing of the manuscript; LM: writing of the manuscript; MHW: writing of the manuscript; and TR: critical review of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Christoph Müller, Aurel Perren, Alessandro Lugli, and Inti Zlobec for continuous support and valuable discussions.
This work was supported by grants from the Swiss National Science Foundation (310030_138188 and 314730_163086), the Bern University Research Foundation, and the San Salvatore Foundation (to PK); the Foundation Johanna Duermueller-Bol (to PK and LM); and by a stipend from the Gertrud-Hagmann Foundation for Malignoma Research (to LM).
AOM, azoxymethane; CAC, colitis-associated (colorectal) cancer; CAF, cancer-associated fibroblasts; CRC, colorectal cancer; DSS, dextran sulfate sodium; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; IL, interleukin; ILC(s), innate lymphoid cell(s); LPS, lipopolysaccharide; MDSCs, myeloid-derived suppressor cells; Th, CD4+ T helper cell; TNF, tumor necrosis factor; Treg, regulatory T cell(s); WT, wild-type.
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer (2010) 127:2893–917. doi:10.1002/ijc.25516
2. Peters U, Bien S, Zubair N. Genetic architecture of colorectal cancer. Gut (2015) 64:1623–36. doi:10.1136/gutjnl-2013-306705
3. de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer (2004) 4:769–80. doi:10.1038/nrc1453
4. Vasovcak P, Pavlikova K, Sedlacek Z, Skapa P, Kouda M, Hoch J, et al. Molecular genetic analysis of 103 sporadic colorectal tumours in Czech patients. PLoS One (2011) 6:e24114. doi:10.1371/journal.pone.0024114
5. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci (2013) 14:16365–85. doi:10.3390/ijms140816365
6. Peddareddigari VG, Wang D, Dubois RN. The tumor microenvironment in colorectal carcinogenesis. Cancer Microenviron (2010) 3:149–66. doi:10.1007/s12307-010-0038-3
7. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell (2012) 22:571–84. doi:10.1016/j.ccr.2012.08.013
8. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell (2013) 23:171–85. doi:10.1016/j.ccr.2012.12.021
9. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet (2015) 47:320–9. doi:10.1038/ng.3225
10. Jass JR, Atkin WS, Cuzick J, Bussey HJ, Morson BC, Northover JM, et al. The grading of rectal cancer: historical perspectives and a multivariate analysis of 447 cases. Histopathology (1986) 10:437–59. doi:10.1111/j.1365-2559.1986.tb02497.x
11. Klintrup K, Makinen JM, Kauppila S, Vare PO, Melkko J, Tuominen H, et al. Inflammation and prognosis in colorectal cancer. Eur J Cancer (2005) 41:2645–54. doi:10.1016/j.ejca.2005.07.017
12. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
13. Galon J, Pages F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med (2012) 10:205. doi:10.1186/1479-5876-10-205
14. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol (2014) 232:199–209. doi:10.1002/path.4287
15. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res (2011) 71:1263–71. doi:10.1158/0008-5472.CAN-10-2907
16. Kantola T, Klintrup K, Vayrynen JP, Vornanen J, Bloigu R, Karhu T, et al. Stage-dependent alterations of the serum cytokine pattern in colorectal carcinoma. Br J Cancer (2012) 107:1729–36. doi:10.1038/bjc.2012.456
17. De Simone V, Pallone F, Monteleone G, Stolfi C. Role of T17 cytokines in the control of colorectal cancer. Oncoimmunology (2013) 2:e26617. doi:10.4161/onci.26617
18. Krzystek-Korpacka M, Diakowska D, Kapturkiewicz B, Bebenek M, Gamian A. Profiles of circulating inflammatory cytokines in colorectal cancer (CRC), high cancer risk conditions, and health are distinct. Possible implications for CRC screening and surveillance. Cancer Lett (2013) 337:107–14. doi:10.1016/j.canlet.2013.05.033
19. Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med (2014) 6:228ra237. doi:10.1126/scitranslmed.3007240
20. Reissfelder C, Stamova S, Gossmann C, Braun M, Bonertz A, Walliczek U, et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J Clin Invest (2015) 125:739–51. doi:10.1172/JCI74894
21. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature (2012) 491:254–8. doi:10.1038/nature11465
22. Elaraj DM, Weinreich DM, Varghese S, Puhlmann M, Hewitt SM, Carroll NM, et al. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin Cancer Res (2006) 12:1088–96. doi:10.1158/1078-0432.CCR-05-1603
23. Viet HT, Wagsater D, Hugander A, Dimberg J. Interleukin-1 receptor antagonist gene polymorphism in human colorectal cancer. Oncol Rep (2005) 14:915–8. doi:10.3892/or.14.4.915
24. Wang Y, Wang K, Han GC, Wang RX, Xiao H, Hou CM, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol (2014) 7:1106–15. doi:10.1038/mi.2013.126
25. Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene (2009) 28:3892–902. doi:10.1038/onc.2009.247
26. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res (2007) 67:10019–26. doi:10.1158/0008-5472.CAN-07-2354
27. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell (2008) 14:408–19. doi:10.1016/j.ccr.2008.10.011
28. Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol (2006) 176:284–90. doi:10.4049/jimmunol.176.1.284
29. Matsuo Y, Sawai H, Ma J, Xu D, Ochi N, Yasuda A, et al. IL-1alpha secreted by colon cancer cells enhances angiogenesis: the relationship between IL-1alpha release and tumor cells’ potential for liver metastasis. J Surg Oncol (2009) 99:361–7. doi:10.1002/jso.21245
30. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi:10.1111/j.1600-065X.2008.00624.x
31. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer (2009) 9:537–49. doi:10.1038/nrc2694
32. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev (2010) 21:11–9. doi:10.1016/j.cytogfr.2009.11.005
33. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell (2012) 149:1192–205. doi:10.1016/j.cell.2012.05.012
34. Marszalek A, Szylberg L, Wisniewska E, Janiczek M. Impact of COX-2, IL-1beta, TNF-alpha, IL-4 and IL-10 on the process of carcinogenesis in the large bowel. Pol J Pathol (2012) 63:221–7. doi:10.5114/pjp.2012.32768
35. Osawa E, Nakajima A, Fujisawa T, Kawamura YI, Toyama-Sorimachi N, Nakagama H, et al. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int J Cancer (2006) 118:2232–6. doi:10.1002/ijc.21639
36. Koller FL, Hwang DG, Dozier EA, Fingleton B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis (2010) 31:1010–7. doi:10.1093/carcin/bgq044
37. Ingram N, Northwood EL, Perry SL, Marston G, Snowden H, Taylor JC, et al. Reduced type II interleukin-4 receptor signalling drives initiation, but not progression, of colorectal carcinogenesis: evidence from transgenic mouse models and human case-control epidemiological observations. Carcinogenesis (2013) 34:2341–9. doi:10.1093/carcin/bgt222
38. Sarrabayrouse G, Corvaisier M, Ouisse LH, Bossard C, Le Mevel B, Potiron L, et al. Tumor-reactive CD4+ CD8alphabeta+ CD103+ alphabetaT cells: a prevalent tumor-reactive T-cell subset in metastatic colorectal cancers. Int J Cancer (2011) 128:2923–32. doi:10.1002/ijc.25640
39. Volonte A, Di Tomaso T, Spinelli M, Todaro M, Sanvito F, Albarello L, et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J Immunol (2014) 192:523–32. doi:10.4049/jimmunol.1301342
40. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol (1999) 17:701–38. doi:10.1146/annurev.immunol.17.1.701
41. Wick KR, Berton MT. IL-4 induces serine phosphorylation of the STAT6 transactivation domain in B lymphocytes. Mol Immunol (2000) 37:641–52. doi:10.1016/S0161-5890(00)00088-2
42. Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ, et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development (2007) 134:2739–50. doi:10.1242/dev.003194
43. Wang CG, Ye YJ, Yuan J, Liu FF, Zhang H, Wang S. EZH2 and STAT6 expression profiles are correlated with colorectal cancer stage and prognosis. World J Gastroenterol (2010) 16:2421–7. doi:10.3748/wjg.v16.i19.2421
44. Golumbek PT, Lazenby AJ, Levitsky HI, Jaffee LM, Karasuyama H, Baker M, et al. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science (1991) 254:713–6. doi:10.1126/science.1948050
45. Yu JS, Wei MX, Chiocca EA, Martuza RL, Tepper RI. Treatment of glioma by engineered interleukin 4-secreting cells. Cancer Res (1993) 53:3125–8.
46. Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science (1992) 257:548–51. doi:10.1126/science.1636093
47. Waldner MJ, Foersch S, Neurath MF. Interleukin-6 – a key regulator of colorectal cancer development. Int J Biol Sci (2012) 8:1248–53. doi:10.7150/ijbs.4614
48. Komoda H, Tanaka Y, Honda M, Matsuo Y, Hazama K, Takao T. Interleukin-6 levels in colorectal cancer tissues. World J Surg (1998) 22:895–8. doi:10.1007/s002689900489
49. Maihofner C, Charalambous MP, Bhambra U, Lightfoot T, Geisslinger G, Gooderham NJ, et al. Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis (2003) 24:665–71. doi:10.1093/carcin/bgg006
50. Chung YC, Chang YF. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J Surg Oncol (2003) 83:222–6. doi:10.1002/jso.10269
51. Belluco C, Nitti D, Frantz M, Toppan P, Basso D, Plebani M, et al. Interleukin-6 blood level is associated with circulating carcinoembryonic antigen and prognosis in patients with colorectal cancer. Ann Surg Oncol (2000) 7:133–8. doi:10.1007/s10434-000-0133-7
52. Heits F, Stahl M, Ludwig D, Stange EF, Jelkmann W. Elevated serum thrombopoietin and interleukin-6 concentrations in thrombocytosis associated with inflammatory bowel disease. J Interferon Cytokine Res (1999) 19:757–60. doi:10.1089/107999099313604
53. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med (2000) 6:583–8. doi:10.1038/75068
54. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity (2004) 21:491–501. doi:10.1016/j.immuni.2004.07.020
55. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell (2009) 15:103–13. doi:10.1016/j.ccr.2009.01.001
56. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer (2014) 110:469–78. doi:10.1038/bjc.2013.748
57. Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng Y, et al. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer (2014) 14:330. doi:10.1186/1471-2407-14-330
58. Tseng-Rogenski SS, Hamaya Y, Choi DY, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology (2015) 148:579–89. doi:10.1053/j.gastro.2014.11.027
59. Tsukamoto H, Nishikata R, Senju S, Nishimura Y. Myeloid-derived suppressor cells attenuate TH1 development through IL-6 production to promote tumor progression. Cancer Immunol Res (2013) 1:64–76. doi:10.1158/2326-6066.CIR-13-0030
60. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci (2012) 8:1237–47. doi:10.7150/ijbs.4989
61. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia (2005) 7:545–55. doi:10.1593/neo.04571
62. Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Inoue K, et al. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol Rep (2006) 15:1445–51. doi:10.3892/or.15.6.1445
63. Morikawa T, Baba Y, Yamauchi M, Kuchiba A, Nosho K, Shima K, et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin Cancer Res (2011) 17:1452–62. doi:10.1158/1078-0432.CCR-10-2694
64. Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology (2013) 144:155–66. doi:10.1053/j.gastro.2012.09.057
65. Lurje G, Zhang W, Schultheis AM, Yang D, Groshen S, Hendifar AE, et al. Polymorphisms in VEGF and IL-8 predict tumor recurrence in stage III colon cancer. Ann Oncol (2008) 19:1734–41. doi:10.1093/annonc/mdn368
66. Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology (2011) 141:e271–5. doi:10.1053/j.gastro.2011.04.008
67. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
68. Li A, Varney ML, Singh RK. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin Cancer Res (2001) 7:3298–304.
69. Ning Y, Manegold PC, Hong YK, Zhang W, Pohl A, Lurje G, et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int J Cancer (2011) 128:2038–49. doi:10.1002/ijc.25562
70. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res (2008) 14:6735–41. doi:10.1158/1078-0432.CCR-07-4843
71. Lv X, Wang X. The role of interleukin-9 in lymphoma. Leuk Lymphoma (2013) 54:1367–72. doi:10.3109/10428194.2012.745072
72. Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med (2012) 18:1248–53. doi:10.1038/nm.2856
73. Fang Y, Chen X, Bai Q, Qin C, Mohamud AO, Zhu Z, et al. IL-9 inhibits HTB-72 melanoma cell growth through upregulation of p21 and TRAIL. J Surg Oncol (2015) 111:969–74. doi:10.1002/jso.23930
74. Huang Y, Cao Y, Zhang S, Gao F. Association between low expression levels of interleukin-9 and colon cancer progression. Exp Ther Med (2015) 10:942–6. doi:10.3892/etm.2015.2588
75. Hoelzinger DB, Dominguez AL, Cohen PA, Gendler SJ. Inhibition of adaptive immunity by IL9 can be disrupted to achieve rapid T-cell sensitization and rejection of progressive tumor challenges. Cancer Res (2014) 74:6845–55. doi:10.1158/0008-5472.CAN-14-0836
76. Leech MD, Grencis RK. Induction of enhanced immunity to intestinal nematodes using IL-9-producing dendritic cells. J Immunol (2006) 176:2505–11. doi:10.4049/jimmunol.176.4.2505
77. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol (2008) 9:1341–6. doi:10.1038/ni.1659
78. Kim HS, Chung DH. IL-9-producing invariant NKT cells protect against DSS-induced colitis in an IL-4-dependent manner. Mucosal Immunol (2013) 6:347–57. doi:10.1038/mi.2012.77
79. Torres S, Bartolome RA, Mendes M, Barderas R, Fernandez-Acenero MJ, Pelaez-Garcia A, et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res (2013) 19:6006–19. doi:10.1158/1078-0432.CCR-13-1130
80. Nalleweg N, Chiriac MT, Podstawa E, Lehmann C, Rau TT, Atreya R, et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut (2015) 64:743–55. doi:10.1136/gutjnl-2013-305947
81. Mao Y, Li Z, Lou C, Zhang Y. Expression of phosphorylated Stat5 predicts expression of cyclin D1 and correlates with poor prognosis of colonic adenocarcinoma. Int J Colorectal Dis (2011) 26:29–35. doi:10.1007/s00384-010-1090-7
82. O’Hara RJ, Greenman J, MacDonald AW, Gaskell KM, Topping KP, Duthie GS, et al. Advanced colorectal cancer is associated with impaired interleukin 12 and enhanced interleukin 10 production. Clin Cancer Res (1998) 4:1943–8.
83. Stanilov N, Miteva L, Deliysky T, Jovchev J, Stanilova S. Advanced colorectal cancer is associated with enhanced IL-23 and IL-10 serum levels. Lab Med (2010) 41:159–63. doi:10.1309/Lm7t43aqziupiowz
84. Miteva LD, Stanilov NS, Deliysky TS, Stanilova SA. Significance of -1082A/G polymorphism of IL10 gene for progression of colorectal cancer and IL-10 expression. Tumour Biol (2014) 35:12655–64. doi:10.1007/s13277-014-2589-2
85. Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res (2005) 65:3998–4004. doi:10.1158/0008-5472.CAN-04-3104
86. Gounaris E, Blatner NR, Dennis K, Magnusson F, Gurish MF, Strom TB, et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res (2009) 69:5490–7. doi:10.1158/0008-5472.CAN-09-0304
87. Chung AY, Li Q, Blair SJ, De Jesus M, Dennis KL, LeVea C, et al. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res (2014) 74:5377–85. doi:10.1158/0008-5472.CAN-14-0918
88. Dennis KL, Wang Y, Blatner NR, Wang S, Saadalla A, Trudeau E, et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res (2013) 73:5905–13. doi:10.1158/0008-5472.CAN-13-1511
89. Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest (1996) 98:1010–20. doi:10.1172/JCI118861
90. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389:737–42. doi:10.1038/39614
91. Van Montfrans C, Rodriguez Pena MS, Pronk I, Ten Kate FJ, Te Velde AA, Van Deventer SJ. Prevention of colitis by interleukin 10-transduced T lymphocytes in the SCID mice transfer model. Gastroenterology (2002) 123:1865–76. doi:10.1053/gast.2002.37067
92. Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z, et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat Commun (2015) 6:7055. doi:10.1038/ncomms8055
93. Denning TL, Campbell NA, Song F, Garofalo RP, Klimpel GR, Reyes VE, et al. Expression of IL-10 receptors on epithelial cells from the murine small and large intestine. Int Immunol (2000) 12:133–9. doi:10.1093/intimm/12.2.133
94. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol (2014) 122:177–210. doi:10.1016/B978-0-12-800267-4.00005-5
95. Wehinger J, Gouilleux F, Groner B, Finke J, Mertelsmann R, Weber-Nordt RM. IL-10 induces DNA binding activity of three STAT proteins (Stat1, Stat3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS Lett (1996) 394:365–70. doi:10.1016/0014-5793(96)00990-8
96. Yoshizaki A, Nakayama T, Yamazumi K, Yakata Y, Taba M, Sekine I. Expression of interleukin (IL)-11 and IL-11 receptor in human colorectal adenocarcinoma: IL-11 up-regulation of the invasive and proliferative activity of human colorectal carcinoma cells. Int J Oncol (2006) 29:869–76. doi:10.3892/ijo.29.4.869
97. Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell (2013) 24:257–71. doi:10.1016/j.ccr.2013.06.017
98. Gillessen S, Carvajal D, Ling P, Podlaski FJ, Stremlo DL, Familletti PC, et al. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol (1995) 25:200–6. doi:10.1002/eji.1830250133
99. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450:566–9. doi:10.1038/nature06306
100. Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol (2007) 37:3021–9. doi:10.1002/eji.200737810
101. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity (2000) 13:715–25. doi:10.1016/S1074-7613(00)00070-4
102. Stanilov N, Miteva L, Jovchev J, Cirovski G, Stanilova S. The prognostic value of preoperative serum levels of IL-12p40 and IL-23 for survival of patients with colorectal cancer. APMIS (2014) 122:1223–9. doi:10.1111/apm.12288
103. Michielsen AJ, Ryan EJ, O’Sullivan JN. Dendritic cell inhibition correlates with survival of colorectal cancer patients on bevacizumab treatment. Oncoimmunology (2012) 1:1445–7. doi:10.4161/onci.21318
104. Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med (1993) 178:1223–30. doi:10.1084/jem.178.4.1223
105. Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol (1994) 153:1697–706.
106. Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol (2000) 165:2665–70. doi:10.4049/jimmunol.165.5.2665
107. Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol (2010) 11:1030–8. doi:10.1038/ni.1947
108. Rigby RJ, Knight SC, Kamm MA, Stagg AJ. Production of interleukin (IL)-10 and IL-12 by murine colonic dendritic cells in response to microbial stimuli. Clin Exp Immunol (2005) 139:245–56. doi:10.1111/j.1365-2249.2004.02674.x
109. Sugimoto K, Ogawa A, Shimomura Y, Nagahama K, Mizoguchi A, Bhan AK. Inducible IL-12-producing B cells regulate Th2-mediated intestinal inflammation. Gastroenterology (2007) 133:124–36. doi:10.1053/j.gastro.2007.03.112
110. Chognard G, Bellemare L, Pelletier AN, Dominguez-Punaro MC, Beauchamp C, Guyon MJ, et al. The dichotomous pattern of IL-12r and IL-23R expression elucidates the role of IL-12 and IL-23 in inflammation. PLoS One (2014) 9:e89092. doi:10.1371/journal.pone.0089092
111. Engel MA, Neurath MF. Anticancer properties of the IL-12 family – focus on colorectal cancer. Curr Med Chem (2010) 17:3303–8. doi:10.2174/092986710793176366
112. Gately MK, Wolitzky AG, Quinn PM, Chizzonite R. Regulation of human cytolytic lymphocyte responses by interleukin-12. Cell Immunol (1992) 143:127–42. doi:10.1016/0008-8749(92)90011-D
113. Trinchieri G. Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood (1994) 84:4008–27.
114. Hayes MP, Wang J, Norcross MA. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-gamma of lipopolysaccharide-inducible p35 and p40 genes. Blood (1995) 86:646–50.
115. Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol (1995) 13:251–76. doi:10.1146/annurev.iy.13.040195.001343
116. Gajewski TF, Fallarino F, Ashikari A, Sherman M. Immunization of HLA-A2+ melanoma patients with MAGE-3 or MelanA peptide-pulsed autologous peripheral blood mononuclear cells plus recombinant human interleukin 12. Clin Cancer Res (2001) 7:895s–901s.
117. Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol (2004) 22:1389–97. doi:10.1200/JCO.2004.04.059
118. Bekaii-Saab TS, Roda JM, Guenterberg KD, Ramaswamy B, Young DC, Ferketich AK, et al. A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol Cancer Ther (2009) 8:2983–91. doi:10.1158/1535-7163.MCT-09-0820
119. Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother (2014) 63:419–35. doi:10.1007/s00262-014-1523-1
120. Bahri R, Pateras IS, D’Orlando O, Goyeneche-Patino DA, Campbell M, Polansky JK, et al. IL-15 suppresses colitis-associated colon carcinogenesis by inducing antitumor immunity. Oncoimmunology (2015) 4:e1002721. doi:10.1080/2162402X.2014.1002721
121. Kobayashi H, Dubois S, Sato N, Sabzevari H, Sakai Y, Waldmann TA, et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood (2005) 105:721–7. doi:10.1182/blood-2003-12-4187
122. Yu P, Steel JC, Zhang M, Morris JC, Waldmann TA. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin Cancer Res (2010) 16:6019–28. doi:10.1158/1078-0432.CCR-10-1966
123. Zhang M, Yao Z, Dubois S, Ju W, Muller JR, Waldmann TA. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc Natl Acad Sci U S A (2009) 106:7513–8. doi:10.1073/pnas.0902637106
124. Pagliari D, Frosali S, Landolfi R, Cianci R. The role of IL-15 in human cancer: friend or foe? Int Trends Immun (2013) 1:35–42.
125. Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res (2014) 20:2044–50. doi:10.1158/1078-0432.CCR-12-3603
126. Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol (2015) 15:771–83. doi:10.1038/nri3919
127. Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity (2004) 21:467–76. doi:10.1016/j.immuni.2004.08.018
128. Cui G, Yuan A, Goll R, Florholmen J. IL-17A in the tumor microenvironment of the human colorectal adenoma-carcinoma sequence. Scand J Gastroenterol (2012) 47:1304–12. doi:10.3109/00365521.2012.725089
129. Nemati K, Golmoghaddam H, Hosseini SV, Ghaderi A, Doroudchi M. Interleukin-17FT7488 allele is associated with a decreased risk of colorectal cancer and tumor progression. Gene (2015) 561:88–94. doi:10.1016/j.gene.2015.02.014
130. Tseng JY, Yang CY, Liang SC, Liu RS, Yang SH, Lin JK, et al. Interleukin-17A modulates circulating tumor cells in tumor draining vein of colorectal cancers and affects metastases. Clin Cancer Res (2014) 20:2885–97. doi:10.1158/1078-0432.CCR-13-2162
131. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013) 39:782–95. doi:10.1016/j.immuni.2013.10.003
132. Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A (2010) 107:5540–4. doi:10.1073/pnas.0912675107
133. Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis (2012) 33:931–6. doi:10.1093/carcin/bgs106
134. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
135. Cui G, Yang H, Zhao J, Yuan A, Florholmen J. Elevated proinflammatory cytokine IL-17A in the adjacent tissues along the adenoma-carcinoma sequence. Pathol Oncol Res (2015) 21:139–46. doi:10.1007/s12253-014-9799-1
136. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15:1016–22. doi:10.1038/nm.2015
137. Zhuang Y, Peng LS, Zhao YL, Shi Y, Mao XH, Chen W, et al. CD8(+) T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology (2012) 143:951e–62e. doi:10.1053/j.gastro.2012.06.010
138. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210:917–31. doi:10.1084/jem.20122308
139. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity (2014) 40:785–800. doi:10.1016/j.immuni.2014.03.013
140. Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity (2014) 41:1052–63. doi:10.1016/j.immuni.2014.11.009
141. Al-Samadi A, Moossavi S, Salem A, Sotoudeh M, Tuovinen SM, Konttinen YT, et al. Distinctive expression pattern of interleukin-17 cytokine family members in colorectal cancer. Tumour Biol (2015) 1–7. doi:10.1007/s13277-015-3941-x
142. Omrane I, Medimegh I, Baroudi O, Ayari H, Bedhiafi W, Stambouli N, et al. Involvement of IL17A, IL17F and IL23R polymorphisms in colorectal cancer therapy. PLoS One (2015) 10:e0128911. doi:10.1371/journal.pone.0128911
143. Tong Z, Yang XO, Yan H, Liu W, Niu X, Shi Y, et al. A protective role by interleukin-17F in colon tumorigenesis. PLoS One (2012) 7:e34959. doi:10.1371/journal.pone.0034959
144. Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol (2001) 167:4137–40. doi:10.4049/jimmunol.167.8.4137
145. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol (2009) 9:556–67. doi:10.1038/nri2586
146. De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene (2015) 34:3493–503. doi:10.1038/onc.2014.286
147. Nikiteas N, Yannopoulos A, Chatzitheofylaktou A, Tsigris C. Heterozygosity for interleukin-18 -607 A/C polymorphism is associated with risk for colorectal cancer. Anticancer Res (2007) 27:3849–53.
148. Pages F, Berger A, Henglein B, Piqueras B, Danel C, Zinzindohoue F, et al. Modulation of interleukin-18 expression in human colon carcinoma: consequences for tumor immune surveillance. Int J Cancer (1999) 84:326–30. doi:10.1002/(SICI)1097-0215(19990621)84:3<326::AID-IJC22>3.3.CO;2-B
149. Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, et al. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet (2008) 82:1202–10. doi:10.1016/j.ajhg.2008.03.016
150. Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol (2010) 42:792–5. doi:10.1016/j.biocel.2010.01.008
151. Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol (2011) 11:9–20. doi:10.1038/nri2891
152. Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med (2010) 207:1625–36. doi:10.1084/jem.20100199
153. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol (2010) 185:4912–20. doi:10.4049/jimmunol.1002046
154. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol (2015) 8:1226–36. doi:10.1038/mi.2015.13
155. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32:367–78. doi:10.1016/j.immuni.2010.02.012
156. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
157. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi:10.1038/nature11535
158. Pages F, Berger A, Lebel-Binay S, Zinzindohoue F, Danel C, Piqueras B, et al. Proinflammatory and antitumor properties of interleukin-18 in the gastrointestinal tract. Immunol Lett (2000) 75:9–14. doi:10.1016/S0165-2478(00)00285-6
159. Stolfi C, Rizzo A, Franze E, Rotondi A, Fantini MC, Sarra M, et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J Exp Med (2011) 208:2279–90. doi:10.1084/jem.20111106
160. Steele N, Anthony A, Saunders M, Esmarck B, Ehrnrooth E, Kristjansen PE, et al. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br J Cancer (2012) 106:793–8. doi:10.1038/bjc.2011.599
161. Jauch D, Martin M, Schiechl G, Kesselring R, Schlitt HJ, Geissler EK, et al. Interleukin 21 controls tumour growth and tumour immunosurveillance in colitis-associated tumorigenesis in mice. Gut (2011) 60:1678–86. doi:10.1136/gutjnl-2011-300612
162. Pallikkuth S, Parmigiani A, Pahwa S. Role of IL-21 and IL-21 receptor on B cells in HIV infection. Crit Rev Immunol (2012) 32:173–95. doi:10.1615/CritRevImmunol.v32.i2.50
163. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov (2014) 13:379–95. doi:10.1038/nrd4296
164. Moroz A, Eppolito C, Li Q, Tao J, Clegg CH, Shrikant PA. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol (2004) 173:900–9. doi:10.4049/jimmunol.173.2.900
165. Skak K, Kragh M, Hausman D, Smyth MJ, Sivakumar PV. Interleukin 21: combination strategies for cancer therapy. Nat Rev Drug Discov (2008) 7:231–40. doi:10.1038/nrd2482
166. Huang YH, Cao YF, Jiang ZY, Zhang S, Gao F. Th22 cell accumulation is associated with colorectal cancer development. World J Gastroenterol (2015) 21:4216–24. doi:10.3748/wjg.v21.i14.4216
167. Wu T, Wang Z, Liu Y, Mei Z, Wang G, Liang Z, et al. Interleukin 22 protects colorectal cancer cells from chemotherapy by activating the STAT3 pathway and inducing autocrine expression of interleukin 8. Clin Immunol (2014) 154:116–26. doi:10.1016/j.clim.2014.07.005
168. Thompson CL, Plummer SJ, Tucker TC, Casey G, Li L. Interleukin-22 genetic polymorphisms and risk of colon cancer. Cancer Causes Control (2010) 21:1165–70. doi:10.1007/s10552-010-9542-5
169. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest (2008) 118:534–44. doi:10.1172/JCI33194
170. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity (2008) 29:947–57. doi:10.1016/j.immuni.2008.11.003
171. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med (2009) 206:1465–72. doi:10.1084/jem.20082683
172. Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity (2014) 40:772–84. doi:10.1016/j.immuni.2014.03.010
173. Khare V, Paul G, Movadat O, Frick A, Jambrich M, Krnjic A, et al. IL10R2 overexpression promotes IL22/STAT3 signaling in colorectal carcinogenesis. Cancer Immunol Res (2015) 3:1227–35. doi:10.1158/2326-6066.CIR-15-0031
174. Koltsova EK, Grivennikov SI. IL-22 gets to the stem of colorectal cancer. Immunity (2014) 40:639–41. doi:10.1016/j.immuni.2014.04.014
175. Ljujic B, Radosavljevic G, Jovanovic I, Pavlovic S, Zdravkovic N, Milovanovic M, et al. Elevated serum level of IL-23 correlates with expression of VEGF in human colorectal carcinoma. Arch Med Res (2010) 41:182–9. doi:10.1016/j.arcmed.2010.02.009
176. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature (2006) 442:461–5. doi:10.1038/nature04808
177. Zhang L, Li J, Li L, Zhang J, Wang X, Yang C, et al. IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis (2014) 35:1330–40. doi:10.1093/carcin/bgu017
178. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118:2269–80. doi:10.1172/JCI34610
179. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity (2012) 36:276–87. doi:10.1016/j.immuni.2011.12.011
180. Kvedaraite E, Lourda M, Idestrom M, Chen P, Olsson-Akefeldt S, Forkel M, et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut (2015) 1–10. doi:10.1136/gutjnl-2014-309014
181. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity (2010) 33:279–88. doi:10.1016/j.immuni.2010.08.010
182. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208:1127–33. doi:10.1084/jem.20101712
183. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513:564–8. doi:10.1038/nature13577
184. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med (2006) 203:2473–83. doi:10.1084/jem.20061099
185. Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med (2006) 203:2485–94. doi:10.1084/jem.20061082
186. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest (2006) 116:1310–6. doi:10.1172/JCI21404
187. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol (2010) 10:103–10. doi:10.1038/nri2692
188. Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A (2010) 107:18581–6. doi:10.1073/pnas.1003059107
189. Lefrancais E, Cayrol C. Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur Cytokine Netw (2012) 23:120–7. doi:10.1684/ecn.2012.0320
190. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A (2012) 109:1673–8. doi:10.1073/pnas.1115884109
191. Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A (2010) 107:8017–22. doi:10.1073/pnas.0912678107
192. Sedhom MA, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut (2013) 62:1714–23. doi:10.1136/gutjnl-2011-301785
193. Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis (2012) 18:1900–9. doi:10.1002/ibd.22900
194. Mertz KD, Mager LF, Wasmer MH, Thiesler T, Koelzer VH, Ruzzante G, et al. The IL-33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology (2016) 5:e1062966. doi:10.1080/2162402X.2015.1062966
195. Cui G, Qi H, Gundersen MD, Yang H, Christiansen I, Sorbye SW, et al. Dynamics of the IL-33/ST2 network in the progression of human colorectal adenoma to sporadic colorectal cancer. Cancer Immunol Immunother (2015) 64:181–90. doi:10.1007/s00262-014-1624-x
196. Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci U S A (2015) 112:E2487–96. doi:10.1073/pnas.1422445112
197. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A (2007) 104:282–7. doi:10.1073/pnas.0606854104
198. Mager LF, Riether C, Schurch CM, Banz Y, Wasmer MH, Stuber R, et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J Clin Invest (2015) 125:2579–91. doi:10.1172/JCI77347
199. O’Donnell C, Mahmoud A, Keane J, Murphy C, White D, Carey S, et al. An antitumorigenic role for the IL-33 receptor, ST2L, in colon cancer. Br J Cancer (2016) 114:37–43. doi:10.1038/bjc.2015.433
200. Liu X, Zhu L, Lu X, Bian H, Wu X, Yang W, et al. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun (2014) 453:486–92. doi:10.1016/j.bbrc.2014.09.106
201. Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarstrom C, Loos T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol (2010) 177:2804–15. doi:10.2353/ajpath.2010.100378
202. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol (2012) 188:3488–95. doi:10.4049/jimmunol.1101977
203. Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood (2009) 114:3117–26. doi:10.1182/blood-2009-02-203372
204. Stojkovic S, Kaun C, Heinz M, Krychtiuk KA, Rauscher S, Lemberger CE, et al. Interleukin-33 induces urokinase in human endothelial cells – possible impact on angiogenesis. J Thromb Haemost (2014) 12:948–57. doi:10.1111/jth.12581
205. Urdinguio RG, Fernandez AF, Moncada-Pazos A, Huidobro C, Rodriguez RM, Ferrero C, et al. Immune-dependent and independent antitumor activity of GM-CSF aberrantly expressed by mouse and human colorectal tumors. Cancer Res (2013) 73:395–405. doi:10.1158/0008-5472.CAN-12-0806
206. Nebiker CA, Han J, Eppenberger-Castori S, Iezzi G, Hirt C, Amicarella F, et al. GM-CSF production by tumor cells is associated with improved survival in colorectal cancer. Clin Cancer Res (2014) 20:3094–106. doi:10.1158/1078-0432.CCR-13-2774
207. Wang Y, Han G, Wang K, Liu G, Wang R, Xiao H, et al. Tumor-derived GM-CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res (2014) 74:716–26. doi:10.1158/0008-5472.CAN-13-1459
208. Trutmann M, Terracciano L, Noppen C, Kloth J, Kaspar M, Peterli R, et al. GM-CSF gene expression and protein production in human colorectal cancer cell lines and clinical tumor specimens. Int J Cancer (1998) 77:378–85. doi:10.1002/(SICI)1097-0215(19980729)77:3<378::AID-IJC12>3.0.CO;2-4
209. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife (2016) 5:e10066. doi:10.7554/eLife.10066
210. Le DT, Pardoll DM, Jaffee EM. Cellular vaccine approaches. Cancer J (2010) 16:304–10. doi:10.1097/PPO.0b013e3181eb33d7
211. Griseri T, Arnold IC, Pearson C, Krausgruber T, Schiering C, Franchini F, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity (2015) 43:187–99. doi:10.1016/j.immuni.2015.07.008
212. Fernandez-Acenero MJ, Galindo-Gallego M, Sanz J, Aljama A. Prognostic influence of tumor-associated eosinophilic infiltrate in colorectal carcinoma. Cancer (2000) 88:1544–8. doi:10.1002/(SICI)1097-0142(20000401)88:7<1544::AID-CNCR7>3.3.CO;2-J
213. Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood (2009) 114:1289–98. doi:10.1182/blood-2008-12-164004
214. Ganapathi SK, Beggs AD, Hodgson SV, Kumar D. Expression and DNA methylation of TNF, IFNG and FOXP3 in colorectal cancer and their prognostic significance. Br J Cancer (2014) 111:1581–9. doi:10.1038/bjc.2014.477
215. Evans C, Morrison I, Heriot AG, Bartlett JB, Finlayson C, Dalgleish AG, et al. The correlation between colorectal cancer rates of proliferation and apoptosis and systemic cytokine levels; plus their influence upon survival. Br J Cancer (2006) 94:1412–9. doi:10.1038/sj.bjc.6603104
216. Wang L, Wang Y, Song Z, Chu J, Qu X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J Interferon Cytokine Res (2015) 35:273–80. doi:10.1089/jir.2014.0132
217. Wang HC, Zhou Q, Dragoo J, Klein JR. Most murine CD8+ intestinal intraepithelial lymphocytes are partially but not fully activated T cells. J Immunol (2002) 169:4717–22. doi:10.4049/jimmunol.169.9.4717
218. Moretto MM, Weiss LM, Combe CL, Khan IA. IFN-gamma-producing dendritic cells are important for priming of gut intraepithelial lymphocyte response against intracellular parasitic infection. J Immunol (2007) 179:2485–92. doi:10.4049/jimmunol.179.4.2485
219. Zufferey C, Erhart D, Saurer L, Mueller C. Production of interferon-gamma by activated T-cell receptor-alphabeta CD8alphabeta intestinal intraepithelial lymphocytes is required and sufficient for disruption of the intestinal barrier integrity. Immunology (2009) 128:351–9. doi:10.1111/j.1365-2567.2009.03110.x
220. Valente G, Ozmen L, Novelli F, Geuna M, Palestro G, Forni G, et al. Distribution of interferon-gamma receptor in human tissues. Eur J Immunol (1992) 22:2403–12. doi:10.1002/eji.1830220933
221. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6:836–48. doi:10.1038/nri1961
222. Gordziel C, Bratsch J, Moriggl R, Knosel T, Friedrich K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br J Cancer (2013) 109:138–46. doi:10.1038/bjc.2013.274
223. Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest (1989) 83:724–7. doi:10.1172/JCI113938
224. Youakim A, Ahdieh M. Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol (1999) 276:G1279–88.
225. Willemsen LE, Hoetjes JP, van Deventer SJ, van Tol EA. Abrogation of IFN-gamma mediated epithelial barrier disruption by serine protease inhibition. Clin Exp Immunol (2005) 142:275–84. doi:10.1111/j.1365-2249.2005.02906.x
226. Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, et al. Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology (2003) 125:795–804. doi:10.1016/S0016-5085(03)01057-6
227. Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res (1999) 59:3379–86.
228. Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res (2004) 64:4687–92. doi:10.1158/0008-5472.CAN-03-3255
229. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res (2013) 73:725–35. doi:10.1158/0008-5472.CAN-12-2706
230. Friedman E, Gold LI, Klimstra D, Zeng ZS, Winawer S, Cohen A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol Biomarkers Prev (1995) 4:549–54.
231. Robson H, Anderson E, James RD, Schofield PF. Transforming growth factor beta 1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. Br J Cancer (1996) 74:753–8. doi:10.1038/bjc.1996.432
232. Tsushima H, Ito N, Tamura S, Matsuda Y, Inada M, Yabuuchi I, et al. Circulating transforming growth factor beta 1 as a predictor of liver metastasis after resection in colorectal cancer. Clin Cancer Res (2001) 7:1258–62.
233. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell (1998) 92:645–56. doi:10.1016/S0092-8674(00)81132-0
234. Bu P, Wang L, Chen KY, Rakhilin N, Sun J, Closa A, et al. miR-1269 promotes metastasis and forms a positive feedback loop with TGF-beta. Nat Commun (2015) 6:6879. doi:10.1038/ncomms7879
235. Xu Y, Pasche B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet (2007) 16(Spec No 1):R14–20. doi:10.1093/hmg/ddl486
236. Feagins LA. Role of transforming growth factor-beta in inflammatory bowel disease and colitis-associated colon cancer. Inflamm Bowel Dis (2010) 16:1963–8. doi:10.1002/ibd.21281
237. Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, van der Ploeg K, et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene (2014) 33:97–107. doi:10.1038/onc.2012.536
238. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell (2000) 103:295–309. doi:10.1016/S0092-8674(00)00121-5
239. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer (2009) 9:361–71. doi:10.1038/nrc2628
240. Al Obeed OA, Alkhayal KA, Al Sheikh A, Zubaidi AM, Vaali-Mohammed MA, Boushey R, et al. Increased expression of tumor necrosis factor-alpha is associated with advanced colorectal cancer stages. World J Gastroenterol (2014) 20:18390–6. doi:10.3748/wjg.v20.i48.18390
241. Stanilov N, Miteva L, Dobreva Z, Stanilova S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol Biotechnol Equip (2014) 28:911–7. doi:10.1080/13102818.2014.965047
242. Biancone L, Orlando A, Kohn A, Colombo E, Sostegni R, Angelucci E, et al. Infliximab and newly diagnosed neoplasia in Crohn’s disease: a multicentre matched pair study. Gut (2006) 55:228–33. doi:10.1136/gut.2005.075937
243. Biancone L, Petruzziello C, Calabrese E, Zorzi F, Naccarato P, Onali S, et al. Long-term safety of Infliximab for the treatment of inflammatory bowel disease: does blocking TNFalpha reduce colitis-associated colorectal carcinogenesis? Gut (2009) 58:1703. doi:10.1136/gut.2008.176461
244. Fidder H, Schnitzler F, Ferrante M, Noman M, Katsanos K, Segaert S, et al. Long-term safety of infliximab for the treatment of inflammatory bowel disease: a single-centre cohort study. Gut (2009) 58:501–8. doi:10.1136/gut.2008.163642
245. Atreya R, Neumann H, Neufert C, Waldner MJ, Billmeier U, Zopf Y, et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat Med (2014) 20:313–8. doi:10.1038/nm.3462
246. Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med (2012) 4:164ra159. doi:10.1126/scitranslmed.3004566
247. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118:560–70. doi:10.1172/JCI32453
248. Flores MB, Rocha GZ, Damas-Souza DM, Osorio-Costa F, Dias MM, Ropelle ER, et al. Obesity-induced increase in tumor necrosis factor-alpha leads to development of colon cancer in mice. Gastroenterology (2012) 143(741–753):e741–4. doi:10.1053/j.gastro.2012.05.045
249. Cox GW, Melillo G, Chattopadhyay U, Mullet D, Fertel RH, Varesio L. Tumor necrosis factor-alpha-dependent production of reactive nitrogen intermediates mediates IFN-gamma plus IL-2-induced murine macrophage tumoricidal activity. J Immunol (1992) 149:3290–6.
250. Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res (2007) 67:1038–45. doi:10.1158/0008-5472.CAN-06-2295
251. Wang Y, Han G, Chen Y, Wang K, Liu G, Wang R, et al. Protective role of tumor necrosis factor (TNF) receptors in chronic intestinal inflammation: TNFR1 ablation boosts systemic inflammatory response. Lab Invest (2013) 93:1024–35. doi:10.1038/labinvest.2013.89
252. Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology (2002) 122:134–44. doi:10.1053/gast.2002.30347
253. Hamilton KE, Simmons JG, Ding S, Van Landeghem L, Lund PK. Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol Cancer Res (2011) 9:1718–31. doi:10.1158/1541-7786.MCR-10-0210
254. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest (2012) 122:4094–104. doi:10.1172/JCI64115
255. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15:362–74. doi:10.1038/nri3834
256. Hale LP, Greer PK. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLoS One (2012) 7:e41797. doi:10.1371/journal.pone.0041797
257. Cao D, Hou M, Guan YS, Jiang M, Yang Y, Gou HF. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer (2009) 9:432. doi:10.1186/1471-2407-9-432
258. Bendardaf R, Buhmeida A, Hilska M, Laato M, Syrjanen S, Syrjanen K, et al. VEGF-1 expression in colorectal cancer is associated with disease localization, stage, and long-term disease-specific survival. Anticancer Res (2008) 28:3865–70.
259. Werther K, Christensen IJ, Nielsen HJ, Danish RANX05 Colorectal Cancer Study Group. Prognostic impact of matched preoperative plasma and serum VEGF in patients with primary colorectal carcinoma. Br J Cancer (2002) 86:417–23. doi:10.1038/sj.bjc.6600075
260. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R, et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med (2010) 207:2855–68. doi:10.1084/jem.20100438
261. Korsisaari N, Kasman IM, Forrest WF, Pal N, Bai W, Fuh G, et al. Inhibition of VEGF-A prevents the angiogenic switch and results in increased survival of Apc+/min mice. Proc Natl Acad Sci U S A (2007) 104:10625–30. doi:10.1073/pnas.0704213104
262. Goodlad RA, Ryan AJ, Wedge SR, Pyrah IT, Alferez D, Poulsom R, et al. Inhibiting vascular endothelial growth factor receptor-2 signaling reduces tumor burden in the ApcMin/+ mouse model of early intestinal cancer. Carcinogenesis (2006) 27:2133–9. doi:10.1093/carcin/bgl113
263. Cao Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci Signal (2009) 2:re1. doi:10.1126/scisignal.259re1
264. Hanrahan V, Currie MJ, Gunningham SP, Morrin HR, Scott PA, Robinson BA, et al. The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J Pathol (2003) 200:183–94. doi:10.1002/path.1339
265. Naik S, Dothager RS, Marasa J, Lewis CL, Piwnica-Worms D. Vascular endothelial growth factor receptor-1 is synthetic lethal to aberrant {beta}-catenin activation in colon cancer. Clin Cancer Res (2009) 15:7529–37. doi:10.1158/1078-0432.CCR-09-0336
266. Easwaran V, Lee SH, Inge L, Guo L, Goldbeck C, Garrett E, et al. Beta-catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res (2003) 63:3145–53.
267. Rmali KA, Puntis MC, Jiang WG. Tumour-associated angiogenesis in human colorectal cancer. Colorectal Dis (2007) 9:3–14. doi:10.1111/j.1463-1318.2006.01089.x
268. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21:1350–6. doi:10.1038/nm.3967
269. Rau TT, Atreya R, Aust D, Baretton G, Eck M, Erlenbach-Wünsch K, et al. Inflammatory response in serrated precursor lesions of the colon classified according to WHO entities, clinical parameters and phenotype–genotype correlation. J Pathol Clin Res (2016). doi:10.1002/cjp2.41
270. Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell (2013) 23:93–106. doi:10.1016/j.ccr.2012.11.014
271. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest (2014) 124:1853–67. doi:10.1172/JCI73531
272. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell (2013) 23:634–46. doi:10.1016/j.ccr.2013.03.022
273. Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front Immunol (2012) 3:107. doi:10.3389/fimmu.2012.00107
274. Ostanin DV, Bhattacharya D. Myeloid-derived suppressor cells in the inflammatory bowel diseases. Inflamm Bowel Dis (2013) 19:2468–77. doi:10.1097/MIB.0b013e3182902b11
275. Haile LA, von Wasielewski R, Gamrekelashvili J, Kruger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology (2008) 135:e871–5. doi:10.1053/j.gastro.2008.06.032
276. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
277. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today (1992) 13:265–70. doi:10.1016/0167-5699(92)90008-U
278. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30:1073–81. doi:10.1093/carcin/bgp127
279. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
280. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008) 27:5904–12. doi:10.1038/onc.2008.271
281. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
282. Otto F, Schmid P, Mackensen A, Wehr U, Seiz A, Braun M, et al. Phase II trial of intravenous endotoxin in patients with colorectal and non-small cell lung cancer. Eur J Cancer (1996) 32A:1712–8. doi:10.1016/0959-8049(96)00186-4
283. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi:10.1016/j.ccell.2015.03.001
284. Fong L, Kwek SS, O’Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res (2009) 69:609–15. doi:10.1158/0008-5472.CAN-08-3529
285. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32:1020–30. doi:10.1200/JCO.2013.53.0105
286. Lal N, Beggs AD, Willcox BE, Middleton GW. An immunogenomic stratification of colorectal cancer: implications for development of targeted immunotherapy. Oncoimmunology (2015) 4:e976052. doi:10.4161/2162402X.2014.976052
287. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov (2015) 5:43–51. doi:10.1158/2159-8290.CD-14-0863
288. Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol (2010) 28:3485–90. doi:10.1200/JCO.2010.28.3994
289. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
290. Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol (2011) 29:11–6. doi:10.1200/JCO.2010.30.0855
291. Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med (2014) 371:1609–18. doi:10.1056/NEJMoa1403108
292. Jatoi A, Dakhil SR, Nguyen PL, Sloan JA, Kugler JW, Rowland KM Jr, et al. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: results from N00C1 from the North Central Cancer Treatment Group. Cancer (2007) 110:1396–403. doi:10.1002/cncr.22944
293. Angevin E, Tabernero J, Elez E, Cohen SJ, Bahleda R, van Laethem JL, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2014) 20:2192–204. doi:10.1158/1078-0432.CCR-13-2200
294. Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol (2010) 37:499–507. doi:10.1053/j.seminoncol.2010.09.007
295. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
296. Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun (2015) 6:6528. doi:10.1038/ncomms7528
297. Gao Z, Guo B, Gao R, Zhu Q, Qin H. Microbiota disbiosis is associated with colorectal cancer. Front Microbiol (2015) 6:20. doi:10.3389/fmicb.2015.00020
298. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi:10.1016/j.cell.2009.09.033
299. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi:10.1126/science.1198469
300. Haller D, Bode C, Hammes WP, Pfeifer AM, Schiffrin EJ, Blum S. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut (2000) 47:79–87. doi:10.1136/gut.47.1.79
301. Bahrami B, Macfarlane S, Macfarlane GT. Induction of cytokine formation by human intestinal bacteria in gut epithelial cell lines. J Appl Microbiol (2011) 110:353–63. doi:10.1111/j.1365-2672.2010.04889.x
302. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity (2014) 40:140–52. doi:10.1016/j.immuni.2013.11.018
303. Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A (2014) 111:18321–6. doi:10.1073/pnas.1406199111
304. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40:128–39. doi:10.1016/j.immuni.2013.12.007
305. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350:1084–9. doi:10.1126/science.aac4255
Keywords: colorectal cancer, inflammation, cytokine, tumor microenvironment, biomarker
Citation: Mager LF, Wasmer MH, Rau TT and Krebs P (2016) Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 6:96. doi: 10.3389/fonc.2016.00096
Received: 28 January 2016; Accepted: 02 April 2016;
Published: 19 April 2016
Edited by:
Giuseppe Valentino Masucci, Karolinska Institute, SwedenReviewed by:
Viktor Umansky, German Cancer Research Center (DKFZ), GermanyCopyright: © 2016 Mager, Wasmer, Rau and Krebs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Krebs, cGhpbGlwcGUua3JlYnNAcGF0aG9sb2d5LnVuaWJlLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.