 Dawid Walerych
Dawid Walerych Kamil Lisek
Kamil Lisek Giannino Del Sal
Giannino Del Sal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 21 December 2015
Sec. Molecular and Cellular Oncology
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00289
This article is part of the Research Topic Human tumor-derived p53 mutants: a growing family of oncoproteins View all 13 articles
Encoded by the mutated variants of the TP53 tumor suppressor gene, mutant p53 proteins are getting an increased experimental support as active oncoproteins promoting tumor growth and metastasis. p53 missense mutant proteins are losing their wild-type tumor suppressor activity and acquire oncogenic potential, possessing diverse transforming abilities in cell and mouse models. Whether various mutant p53s differ in their oncogenic potential has been a matter of debate. Recent discoveries are starting to uncover the existence of mutant p53 downstream programs that are common to different mutant p53 variants. In this review, we discuss a number of studies on mutant p53, underlining the advantages and disadvantages of alternative experimental approaches that have been used to describe the numerous mutant p53 gain-of-function activities. Therapeutic possibilities are also discussed, taking into account targeting either individual or multiple mutant p53 proteins in human cancer.
Mutations in the TP53 gene occur in almost every type of cancer, with frequencies that vary between 10% (hematopoietic malignancies) and 96% (high grade ovarian serous carcinoma) (1). Cancer genome sequencing studies confirm that TP53 is the most commonly mutated tumor suppressor gene in human cancers (2). The majority of studies indicate that the presence of mutated TP53 is associated with bad prognosis in various cancer types (3). TP53 mutations are known first and foremost to inactivate the oncosuppressive properties of the wild-type p53 protein as a transcription factor (loss-of-function – LOF). However, since p53 acts as a tetramer, expressed TP53 mutant variants can also exert a dominant negative (DN) effect over their wild-type counterpart, and additionally they can arm cancer cells with novel oncogenic gain-of-function (GOF) activities (4–6).
In over 70% of cases, the TP53 mutations are missense, most frequently within the region encoding the core domain of the p53 protein, which is responsible for binding DNA (7). Although the spectrum of the TP53 missense mutations is vast – counting about 1,800 different amino-acid changes (8) – several hotspot p53 mutants, in particular, affecting residues R273, R248, R175, and G245 of the p53 protein, are present with a higher frequency both in sporadic tumors (together over 21% of total missense mutations) and in individuals with the Li–Fraumeni syndrome (LFS), a genetic disorder caused by inherited TP53 mutations that predispose carriers to an early-onset development of various cancers (9).
The hotspot changes in p53 are traditionally classified as “conformational” or “DNA contact” mutations. This notion comes from the biophysical observation that the former group disturbs the proper folding of the core domain of p53, thus depriving it of the ability to bind the DNA and transactivate its target genes, while the latter group is composed of mutations in residues that are responsible for directly binding DNA, with a near-native core domain structure (10, 11). In the LFS, a wild-type TP53 allele is usually present, whereas in LFS tumors, it is often (in the 40–60% of cases) subjected to inactivation (loss of heterozygosity – LOH) – a process that is observed both in mouse LFS models (12) and in humans (13), involving various mechanisms of wild-type TP53 inactivation (14). Interestingly, it has been recently noted that in the embryonic stem cells from LFS mice the lost allele is often the mutant one, suggesting that a bi-directional TP53 LOH process may function as a cell-fate checkpoint and that there exists a selective pressure against the heterozygous TP53 state (15).
p53 mutant proteins are stabilized and protected from degradation in a tumor microenvironment by various oncogenic signaling pathways (16, 17), and several studies in mutant p53 knock-in (KI) mice showed that the presence of p53 mutants promotes tumor growth with higher metastasis rate and different tissue spectrum than the absence of wild-type p53 (12, 18). These in vivo proofs of mutant p53 GOF came as confirmation of the initial observations in cell models that mutant p53 missense variants may actively support cell transformation (19, 20).
Even though the oncogenic activity related to GOF p53 mutants has been described many times in the last 25 years of research on p53, there are still doubts concerning its significance. Current approaches are only starting to resolve whether missense p53 mutants can be regarded as essentially one oncoprotein endowed with a conserved tumorigenic activity, or they represent a population of different oncoproteins, each exerting its unique oncogenic potential. Mutant p53 is still not used in standard clinical practice as a target of anti-cancer therapies. We discuss these issues in the following sections of this review.
The rising importance of the GOF of p53 mutants in cancer has led to numerous studies describing their mechanisms of action and a brought forward question how much the obtained results can be generalized across different mutant p53 variants and cellular or cancer backgrounds.
A minority of these studies is based on mutant p53 KI mouse models and led to a number of discoveries in the field, including (i) the inhibitory role of mutant p53 on MRE11 protein and the induction of genomic instability (human TP53 KI “HupKI” mouse model) (21), (ii) the transcription-based activation of PDGFRβ signaling in pancreatic cancer model (22), (ii) the transcriptional activation of oncogenic Pla2g16 phospholipase (23), and (iv) the confirmation of prior cell-based reports on a mutant p53-mediated inhibition of the p63/p73 oncosuppressive activity (12, 18). The LFS mouse-model-based studies underlined differences between GOF properties of different p53 mutants and among the consequences of TP53 mutations in human and in mouse. Comparative studies of the R270H and R172H variants in KI mice showed different tumor spectra confirming the notion that the GOF of p53 mutants may differ (12). These spectra, however, turned out to be different also from the spectra caused by human counterparts of these mutant p53 variants – R273H and R175H – in patients with LFS (e.g., lack of mammary carcinomas in mice – frequent in humans) (9). On the other hand, the investigation in KI mice of the R246S p53 mutant, corresponding to the human R249S p53 hotspot mutant, showed no clear indication of GOF (24), whereas in human cell-based experiments, this variant was demonstrated to induce growth, chemoresistance, and a specific mutant p53 transcriptional program in several studies (25–27). Altogether these results indicate that mouse models – albeit very informative – may have their limitations and require careful confirmation of their significance in human systems.
Most of the human cell line-based studies on mutant p53 are based on initial phenotype-related experiments or large scale analysis, such as gene expression microarray or ChIP sequencing, leading to discovery of mechanisms/targets associated with a particular mutant variant in its endogenous background. In most cases, validation in other mutant p53 variants/backgrounds is also reported. Such studies have led to describing important roles of mutant p53 in direct inhibition of the p63/p73-mediated tumor suppression (28, 29), activation of the cell cycle drivers, such as Cyclins (30, 31), the vitamin D3 receptor signaling (32), steroid synthesis (mevalonate pathway) (33), the ID4-mediated angiogenesis (34), or nucleotide homeostasis (26), to name a few. A comprehensive list of these studies published since 2005 – with the indication of the initially tested mutant p53 variant(s) and p53 mutants used for validation – is shown in Table 1.
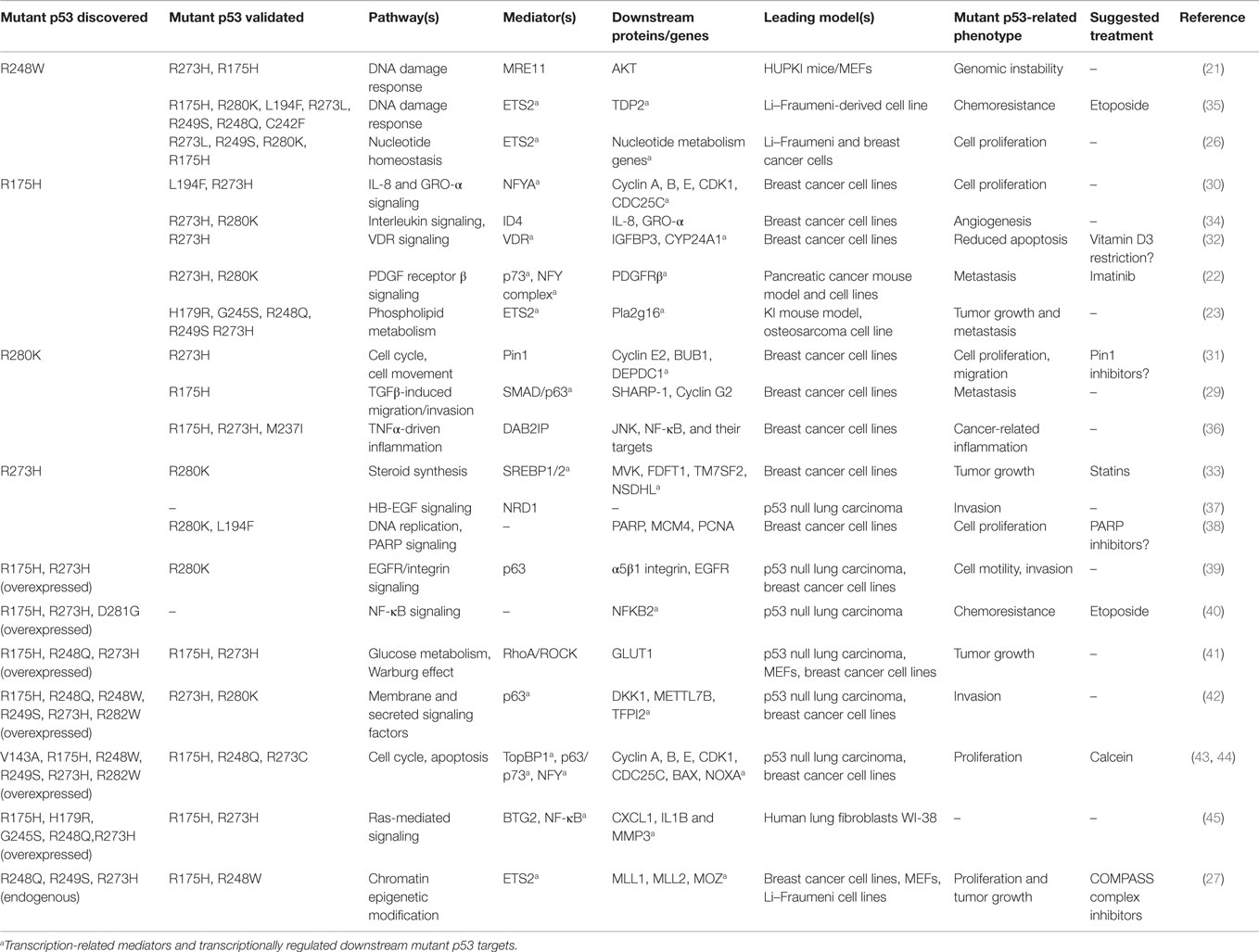
Table 1. Selected mutant p53 gain-of-function effects, mediators, and related therapeutic opportunities, published since 2005.
Mutant p53 activities have been described both in the cell’s cytoplasm and in the nucleus. The reported cytoplasm-specific activities include the DAB2IP protein regulation affecting TNFα-dependent signaling (36) and the regulation of PARP localization and activity (38). Nuclear activities are related to more general influence on the chromatin function (the example of the above-mentioned MRE11 regulation) but, in most cases, are related to a specific transcriptional regulation. The available mutant p53 ChIP-sequencing data and other DNA-interaction data have not defined a mutant p53 target site analogous to that of wild-type p53, and currently the main hypothesis is that mutant p53 transactivation takes place through interaction with several transcription factors – among them NFY complex, SREBP 1 and 2, or ETS2 (Table 1). In most cases, mutant p53 proteins boost basic properties of these transcription factors, leading to the aberrant activation of their downstream programs and to the intersection with other key oncogenic pathways, as shown for the mutant p53-SREBP or NFY causing activation of the YAP/TAZ pathway (46, 47).
Even though experimental approaches using single initial in vivo and in vitro models led to the discovery of numerous pathways controlled by mutant p53, it is unclear whether these pathways have the same central role in diverse cellular contexts.
In an attempt to fill this gap, studies have been conducted involving the overexpression of multiple mutant p53 variants in a p53-null or wild-type background (Table 1). Investigation in the p53-null background of non-small lung carcinoma H1299 cells led to discovering the role of mutant p53 in integrin recycling (39), in the NF-κB signaling (40), and in the Warburg effect (41) as well as a role of TopBP1 in the upstream regulation of mutant p53 (43). These studies largely confirmed that the mutant p53 GOF is exerted indirectly at the level of transcription by cooperation with transcription factors. Neilsen et al. showed that genes activated by mutant p53 largely overlap between mutant variants overexpressed in H1299 cells, but interestingly also frequently share promoter sequences with p63 and wild-type p53 (42). This indicates that the mutant p53-mediated promoter activation may be an aberrant representation of the interaction of wild-type p53 with transcription factors in normal cells. Other mutant p53 overexpression studies led to uncovering regulation of the epithelial-to-mesenchymal transition (EMT) phenotype by mutant p53 upon wild-type TP53 silencing in MCF10A mammary epithelium cells (48) as well as the cooperation of mutant p53 with the Ras oncogenic program in WI-38 human embryonic lung fibroblasts (45). Much of this evidence, however, still awaits confirmation in experimental settings in which mutant p53 variants are endogenously expressed. During the course of transformation, cell lines carrying endogenous TP53 mutations become addicted to the mutant p53 GOF – as often their growth or migration/invasion abilities are compromised upon mutant p53 knock-down (27, 31, 36, 49). Conversely, p53-null and wild-type p53 cell lines survive and proliferate without mutant p53, suggesting that very likely the GOF program observed under such conditions only partially resembles the cancer-related one. Therefore, the lack of the cellular context in which p53 mutants are naturally embedded and background-associated effects represent relevant weaknesses of the studies in a p53-null or p53 wild-type background.
A solution to these limitations may be represented by studies that include an initial analysis using different p53 mutants in their endogenous backgrounds. Analyzing downstream programs – both at the phenotypic and the molecular level – may help to understand to what extent p53 mutants possess a “core” oncogenic program, and whether some mutants display specific features. A recent study by Zhu et al. focuses on the common DNA interaction pattern of three distinct p53 mutants, in their endogenous context of breast cancer cell lines, using as term of comparison the pattern obtained from two cell lines bearing wild-type p53 (27). As a highlight of this multi-mutant p53–DNA interaction pattern, the group identified the chromatin regulatory genes that are activated by the transcription factor ETS2, a previously known mutant p53 interactor (23, 26, 35). The relevance of a mutant p53/ETS2 cooperation has been confirmed as a general feature in several mutant p53 expressing cell lines and thanks to the transcriptional program perturbed, as a critical modulator of the chromatin modification (27).
Even with these many studies published this is apparently only the beginning of a deeper understanding of both specificity and general picture of mutant p53 GOF in cancer. Multiple cellular/cancer models have to be studied simultaneously in unbiased, large-scale manner, by comparing more mutant p53 variants, including non-hotspot mutations. Another important issue is how these discoveries could be transferred into clinical applications.
The issue regarding how widely the GOF effects are shared between multiple mutant p53 variants extends to the experimental targeted therapies based on the presence of mutant p53. Since TP53 is one of the most frequently mutated genes in cancer, reactivation of the wild-type p53 oncosuppressive properties and eliminating the mutant p53 GOF are potentially instrumental in personalized treatment of hundreds of thousands cancer patients worldwide. In this context, the possibility to distinguish mutant p53-specific processes from those shared by at least hotspot mutant p53 variants seems of relevance in order to develop and test drugs targeting properties and/or downstream pathways that are common to as many mutant p53 variants as possible.
Research on widely acting molecules targeting mutant p53 began over two decades ago. Some of the first approaches included inhibitors of Hsp90, a molecular chaperone that participates in a multiprotein complex stabilizing GOF p53 mutants with distorted DNA-binding domain structure (50). Hsp90 inhibitor geldanamycin was shown to lower levels and nuclear translocation of mutant p53 (51, 52). The interest toward Hsp90 inhibitors remains high, as the recent study by Alexandrova et al. describes significantly increased survival of mutant p53 KI mice treated with the geldanamycin derivative 17-DMAG or with a new generation Hsp90 inhibitor – ganetespib (53). Other drugs – such as the histone deacetylase inhibitor SAHA (Vorinostat) (54) and sodium butyrate (NaB) (55) – have been also shown to downregulate the levels of mutant p53 variants.
Different suggested strategies involve blocking the mutant p53 activation by targeting proteins, such as Pin1 (31) or TopBP1 (43). Inhibitor of TopBP1 – Calcein (44) – and experimental inhibitors of Pin1 (56) are examples of molecules targeting specific upstream activators of mutant p53. Among compounds that have been shown to efficiently directly target mutant p53 are small peptides (57–59). None of them, however, is so advanced in experimentation as small molecules that directly modify mutant p53 promoting its transition into a wild-type like form, capable of activating the tumor suppressive wild-type p53 transcriptional targets. The first described micromolecule targeting mutant p53 was CP-31398 (60) that, despite turning out not to directly interact with mutant p53 but rather with its target DNA sequences (58), is still considered as a promising drug candidate (61, 62). Most studies were, however, performed on the PRIMA-1 molecule (63) and later on its more potent and less toxic derivative PRIMA-1MET/APR-246 (64). Experiments showed that this molecule is able to directly bind and modify thiol residues in mutant p53 transforming it into a wild-type-like protein (65), thus becoming able to activate wild-type p53 targets, such as GADD45B, NOXA, or CDKN1A (p21), and induce in vitro and in vivo cell cycle arrest or apoptosis (66, 67).
In the case of drugs targeting mutant p53, most studied molecules, as those mentioned above, target several mutant p53 missense variants, while drug candidates focusing on particular mutants are rare. NSC319726 is one such these compounds. Identified by screening studies, NSC319726 possesses specific activity toward the R175H mutant p53 and induces apoptosis in human cells (68). Two other studies led to discovering PhiKan083 (69) and PK7088 (70) as molecules that specifically target and reactivate mutant p53 hotspot variant Y220C, which is found at a relatively high frequency in breast cancer (5). The low number of such studies and the fact that other mutant p53 reactivating compounds target various mutant p53 variants may suggest that the classic distinction of contact and structural p53 mutants may not be decisive and these mutant types may, in fact, represent structural extremes of a spectrum of distortions in the DNA-binding domain, leading to similar GOF effects.
Another important strategy to mutant p53 targeting is based on the treatment with drugs that downregulate oncogenic pathways activated by the means of mutant p53 GOF (listed in Table 1). This activation in general leads to two types of therapeutically relevant outcome – chemoresistance and chemosensitization (Figure 1). In the first case, the sensitivity to either specific or broad activity anti-cancer compounds, including doxorubicin, cisplatin, or etoposide, is dampened in the presence of mutant p53 (35, 49). In the latter case, a number of pathway targeting drugs – such as statins that inhibit the mevalonate pathway (33), imatinib inhibiting PDGFRβ (22), or COMAPSS complex inhibitors (27) – can cause increased cell death in mutant p53 vs. wild-type p53-bearing cancer cells. The performance of these drugs is often promising, but their drawback is the limited number of mutant p53/cell backgrounds tested.
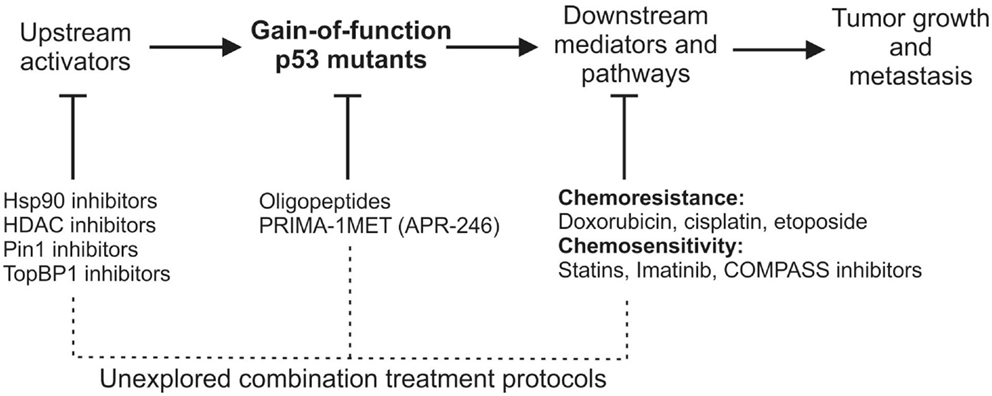
Figure 1. A schematic view of gain-of-function mutant p53 activation, mutant p53 downstream effectors/pathways, and therapeutic opportunities of targeting each of the processes. Below the largely unexplored possibilities of mutant p53-related combinational anti-cancer therapies are suggested.
A big issue of the mutant p53-oriented therapies is their slow progress toward the clinics, most of them being still at an early stage of development (71). The only drug directly targeting mutant p53 that has reached the clinical stage is PRIMA-1MET/APR-246. This compound successfully went through phase I/II clinical trial in hematological malignancies and prostate cancer that included mutant p53 patients (72). An approach targeting triple negative breast cancer (TNBC) cells with p53 deficiency or mutant status using Chk1 inhibitors showed promising results in in vitro and mouse tests (73, 74), while it failed to show significant improvement in human patients (75). At the same time, many of the drugs that could be beneficial for mutant p53 patients – Hsp90 inhibitors, HDAC inhibitors, or statins – are undergoing clinical trials in cancer in which the mutant p53 status is not considered or even known (76–78).
The combination of drugs directly targeting mutant p53 with drugs inhibiting mutant p53-related pathways is surprisingly avoided (Figure 1), although it might favor the decrease of compensatory responses and dosage toxicity, and thus an increase in the therapeutic efficacy. This notion is supported by a number of experiments showing that the combination of PRIMA-1 and PRIMA1-MET/APR-246 with cisplatin (CDDP) results in synergistic effects in cancer cells and xenografts (79–81). Taking into account that mutant p53 is known to increase chemoresistance to cisplatin (49), it is not surprising that targeting the cause of this chemoresistance opens the window to more effective treatments. This combinational approach may suggest that other compounds are worth being tested together with mutant p53 targeting drugs, such as PRIMA-1MET/APR-246 (Figure 1).
Even though TP53 is one of the most frequently mutated genes in human cancer and mutant p53 emerges as a major oncoprotein controlling an exceptionally vast network of tumor-promoting activities, it still possesses underused potential as a drug target and much effort is needed to bring it to a prominent position on the map of personalized therapeutic solutions for cancer patients.
DW, KL, and GS wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank A. Testa for discussing and reading the manuscript. This work was supported by the Italian Health Ministry and Italian Association for Cancer Research (AIRC) Special Program Molecular Clinical Oncology “5 per mille” (Grant no. 10016), to GS; DW was a recipient of the FEBS postdoctoral fellowship.
1. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer (2011) 2:466–74. doi: 10.1177/1947601911408889
2. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502:333–9. doi:10.1038/nature12634
3. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer (2009) 9:701–13. doi:10.1038/nrc2693
4. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev (2012) 26:1268–86. doi:10.1101/gad.190678.112
5. Walerych D, Napoli M, Collavin L, Del Sal G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis (2012) 33:2007–17. doi:10.1093/carcin/bgs232
6. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi:10.1016/j.ccr.2014.01.021
7. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat (2007) 28:622–9. doi:10.1002/humu.20495
8. Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Adv Cancer Res (2011) 110:107–39. doi:10.1016/B978-0-12-386469-7.00005-0
10. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (1994) 265:346–55. doi:10.1126/science.8023157
11. Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene (2000) 19:1245–56. doi:10.1038/sj.onc.1203434
12. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell (2004) 119:847–60. doi:10.1016/j.cell.2004.11.004
13. Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, et al. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene (1997) 14:865–71. doi:10.1038/sj.onc.1201041
14. Schlegelberger B, Kreipe H, Lehmann U, Steinemann D, Ripperger T, Gohring G, et al. A child with Li-Fraumeni syndrome: modes to inactivate the second allele of TP53 in three different malignancies. Pediatr Blood Cancer (2015) 62:1481–4. doi:10.1002/pbc.25486
15. Shetzer Y, Kagan S, Koifman G, Sarig R, Kogan-Sakin I, Charni M, et al. The onset of p53 loss of heterozygosity is differentially induced in various stem cell types and may involve the loss of either allele. Cell Death Differ (2014) 21:1419–31. doi:10.1038/cdd.2014.57
16. Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev (2008) 22:1337–44. doi:10.1101/gad.1662908
17. Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, et al. Multiple stress signals activate mutant p53 in vivo. Cancer Res (2011) 71:7168–75. doi:10.1158/0008-5472.CAN-11-0459
18. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell (2004) 119:861–72. doi:10.1016/j.cell.2004.11.006
19. Halevy O, Michalovitz D, Oren M. Different tumor-derived p53 mutants exhibit distinct biological activities. Science (1990) 250:113–6. doi:10.1126/science.2218501
20. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet (1993) 4:42–6. doi:10.1038/ng0593-42
21. Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol (2007) 9:573–80. doi:10.1038/ncb1571
22. Weissmueller S, Manchado E, Saborowski M, Morris JPT, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell (2014) 157:382–94. doi:10.1016/j.cell.2014.01.066
23. Xiong S, Tu H, Kollareddy M, Pant V, Li Q, Zhang Y, et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc Natl Acad Sci U S A (2014) 111:11145–50. doi:10.1073/pnas.1404139111
24. Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell (2012) 22:751–64. doi:10.1016/j.ccr.2012.10.022
25. Yan W, Chen X. Characterization of functional domains necessary for mutant p53 gain of function. J Biol Chem (2010) 285:14229–38. doi:10.1074/jbc.M109.097253
26. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun (2015) 6:7389. doi:10.1038/ncomms8389
27. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature (2015) 525:206–11. doi:10.1038/nature15251
28. Di Agostino S, Cortese G, Monti O, Dell’Orso S, Sacchi A, Eisenstein M, et al. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle (2008) 7:3440–7. doi:10.4161/cc.7.21.6995
29. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137:87–98. doi:10.1016/j.cell.2009.01.039
30. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell (2006) 10:191–202. doi:10.1016/j.ccr.2006.08.013
31. Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, et al. A pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell (2011) 20:79–91. doi:10.1016/j.ccr.2011.06.004
32. Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell (2010) 17:273–85. doi:10.1016/j.ccr.2009.11.025
33. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. doi:10.1016/j.cell.2011.12.017
34. Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol (2009) 16:1086–93. doi:10.1038/nsmb.1669
35. Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev (2012) 26:830–45. doi:10.1101/gad.181685.111
36. Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol Cell (2014) 56:617–29. doi:10.1016/j.molcel.2014.10.013
37. Coffill CR, Muller PA, Oh HK, Neo SP, Hogue KA, Cheok CF, et al. Mutant p53 interactome identifies nardilysin as a p53R273H-specific binding partner that promotes invasion. EMBO Rep (2012) 13:638–44. doi:10.1038/embor.2012.74
38. Polotskaia A, Xiao G, Reynoso K, Martin C, Qiu WG, Hendrickson RC, et al. Proteome-wide analysis of mutant p53 targets in breast cancer identifies new levels of gain-of-function that influence PARP, PCNA, and MCM4. Proc Natl Acad Sci U S A (2015) 112(11):E1220–9. doi:10.1073/pnas.1416318112
39. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell (2009) 139:1327–41. doi:10.1016/j.cell.2009.11.026
40. Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF, et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol (2005) 25:10097–110. doi:10.1128/MCB.25.22.10097-10110.2005
41. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun (2013) 4:2935. doi:10.1038/ncomms3935
42. Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget (2011) 2(12):1203–17. doi:10.18632/oncotarget.382
43. Liu K, Ling S, Lin WC. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol Cell Biol (2011) 31:4464–81. doi:10.1128/MCB.05574-11
44. Chowdhury P, Lin GE, Liu K, Song Y, Lin FT, Lin WC. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat Commun (2014) 5:5476. doi:10.1038/ncomms6476
45. Solomon H, Buganim Y, Kogan-Sakin I, Pomeraniec L, Assia Y, Madar S, et al. Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature. J Cell Sci (2012) 125:3144–52. doi:10.1242/jcs.099663
46. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol (2014) 16:357–66. doi:10.1038/ncb2936
47. Di Agostino S, Sorrentino G, Ingallina E, Valenti F, Ferraiuolo M, Bicciato S, et al. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep (2015).
48. Zhang Y, Yan W, Chen X. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem (2011) 286:16218–28. doi:10.1074/jbc.M110.214585
49. Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene (2006) 25:304–9. doi:10.1038/sj.onc.1209026
50. Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A (1996) 93:8379–83. doi:10.1073/pnas.93.16.8379
51. Blagosklonny MV, Toretsky J, Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene (1995) 11:933–9.
52. Dasgupta G, Momand J. Geldanamycin prevents nuclear translocation of mutant p53. Exp Cell Res (1997) 237:29–37. doi:10.1006/excr.1997.3766
53. Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature (2015) 523:352–6. doi:10.1038/nature14430
54. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ (2011) 18(12):1904–13. doi:10.1038/cdd.2011.71
55. Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene (2013) 32:599–609. doi:10.1038/onc.2012.81
56. Wei S, Kozono S, Kats L, Nechama M, Li W, Guarnerio J, et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med (2015) 21:457–66. doi:10.1038/nm.3839
57. Selivanova G, Iotsova V, Okan I, Fritsche M, Strom M, Groner B, et al. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med (1997) 3:632–8. doi:10.1038/nm0697-632
58. Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, et al. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci U S A (2002) 99:937–42. doi:10.1073/pnas.241629998
59. Guida E, Bisso A, Fenollar-Ferrer C, Napoli M, Anselmi C, Girardini JE, et al. Peptide aptamers targeting mutant p53 induce apoptosis in tumor cells. Cancer Res (2008) 68:6550–8. doi:10.1158/0008-5472.CAN-08-0137
60. Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science (1999) 286:2507–10. doi:10.1126/science.286.5449.2507
61. He X, Kong X, Yan J, Zhang Y, Wu Q, Chang Y, et al. CP-31398 prevents the growth of p53-mutated colorectal cancer cells in vitro and in vivo. Tumour Biol (2015) 36:1437–44. doi:10.1007/s13277-014-2389-8
62. Li P, Zhao M, Parris AB, Feng X, Yang X. p53 is required for metformin-induced growth inhibition, senescence and apoptosis in breast cancer cells. Biochem Biophys Res Commun (2015) 464:1267–74. doi:10.1016/j.bbrc.2015.07.117
63. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8:282–8. doi:10.1038/nm0302-282
64. Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett (2014) 588:2622–7. doi:10.1016/j.febslet.2014.04.017
65. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell (2009) 15:376–88. doi:10.1016/j.ccr.2009.03.003
66. Zache N, Lambert JM, Wiman KG, Bykov VJ. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol (2008) 30:411–8. doi:10.3233/CLO-2008-0440
67. Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene (2010) 29:1329–38. doi:10.1038/onc.2009.425
68. Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell (2012) 21:614–25. doi:10.1016/j.ccr.2012.03.042
69. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A (2008) 105:10360–5. doi:10.1073/pnas.0805326105
70. Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res (2013) 41:6034–44. doi:10.1093/nar/gkt305
71. Girardini JE, Marotta C, Del Sal G. Disarming mutant p53 oncogenic function. Pharmacol Res (2014) 79:75–87. doi:10.1016/j.phrs.2013.11.003
72. Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol (2012) 30:3633–9. doi:10.1200/JCO.2011.40.7783
73. Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest (2012) 122:1541–52. doi:10.1172/JCI58765
74. Origanti S, Cai SR, Munir AZ, White LS, Piwnica-Worms H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene (2013) 32:577–88. doi:10.1038/onc.2012.84
75. Ma CX, Ellis MJ, Petroni GR, Guo Z, Cai SR, Ryan CE, et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res Treat (2013) 137:483–92. doi:10.1007/s10549-012-2378-9
76. Gonyeau MJ. The spectrum of statin therapy in cancer patients: is there a need for further investigation? Curr Atheroscler Rep (2014) 16:383. doi:10.1007/s11883-013-0383-z
77. Sidera K, Patsavoudi E. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov (2014) 9:1–20. doi:10.2174/15748928113089990031
78. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest (2014) 124:30–9. doi:10.1172/JCI69738
79. Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene (2005) 24:3484–91. doi:10.1038/sj.onc.1208419
80. Messina RL, Sanfilippo M, Vella V, Pandini G, Vigneri P, Nicolosi ML, et al. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int J Cancer (2012) 130:2259–70. doi:10.1002/ijc.26228
Keywords: p53 mutation, gain-of-function, cancer, drug therapy, combination, oncogenes, tumor suppressor proteins
Citation: Walerych D, Lisek K and Del Sal G (2015) Mutant p53: One, No One, and One Hundred Thousand. Front. Oncol. 5:289. doi: 10.3389/fonc.2015.00289
Received: 19 October 2015; Accepted: 07 December 2015;
Published: 21 December 2015
Edited by:
Ygal Haupt, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Alessandro Rimessi, University of Ferrara, ItalyCopyright: © 2015 Walerych, Lisek and Del Sal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giannino Del Sal, ZGVsc2FsQGxuY2liLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.