 Stephen M. Madamba
Stephen M. Madamba Kevin N. Damri
Kevin N. Damri Laurent M. Dejean
Laurent M. Dejean Pablo M. Peixoto
Pablo M. Peixoto- 1Department of Natural Sciences, Baruch College, City University of New York, New York, NY, USA
- 2City University of New York Graduate Center, New York, NY, USA
- 3Department of Chemistry, College of Science and Mathematics, California State University Fresno, Fresno, CA, USA
- 4Department of Basic Sciences, New York University College of Dentistry, New York, NY, USA
Cancer transformation involves reprograming of mitochondrial function to avert cell death mechanisms, monopolize energy metabolism, accelerate mitotic proliferation, and promote metastasis. Mitochondrial ion channels have emerged as promising therapeutic targets because of their connection to metabolic and apoptotic functions. This mini review discusses how mitochondrial channels may be associated with cancer transformation and expands on the possible involvement of mitochondrial protein import complexes in pathophysiological process.
Introduction
The eukaryotic merger that gave rise to mitochondria was arguably the major contributor to the origin of multicellular organisms (1, 2), afforded by the increased efficiency in cellular energy conversion. However, maintaining multicellular life required more than power. Mitochondria had to specialize in eliminating malfunctioning cells and policing unrestrained growth of once-single cells. An insurgence against the newfound multicellular way of life would be punished with a death sentence.
Power, or a death sentence, is released from mitochondria through channels spanning the inner and the outer membranes. For example, mitochondria deliver ATP and other high-energy metabolites to the cell through a voltage-dependent anion channel [VDAC, reviewed in Ref. (3)], while death signals like cytochrome c are unleashed through the mitochondrial apoptosis-induced channel [MAC; (4)] (Figure 1). Comprehensive reviews on the structure–function relationships of ion channels in mitochondria are available elsewhere (5–11). This mini review will focus on the possible involvement of mitochondrial ion channels in cancer transformation.
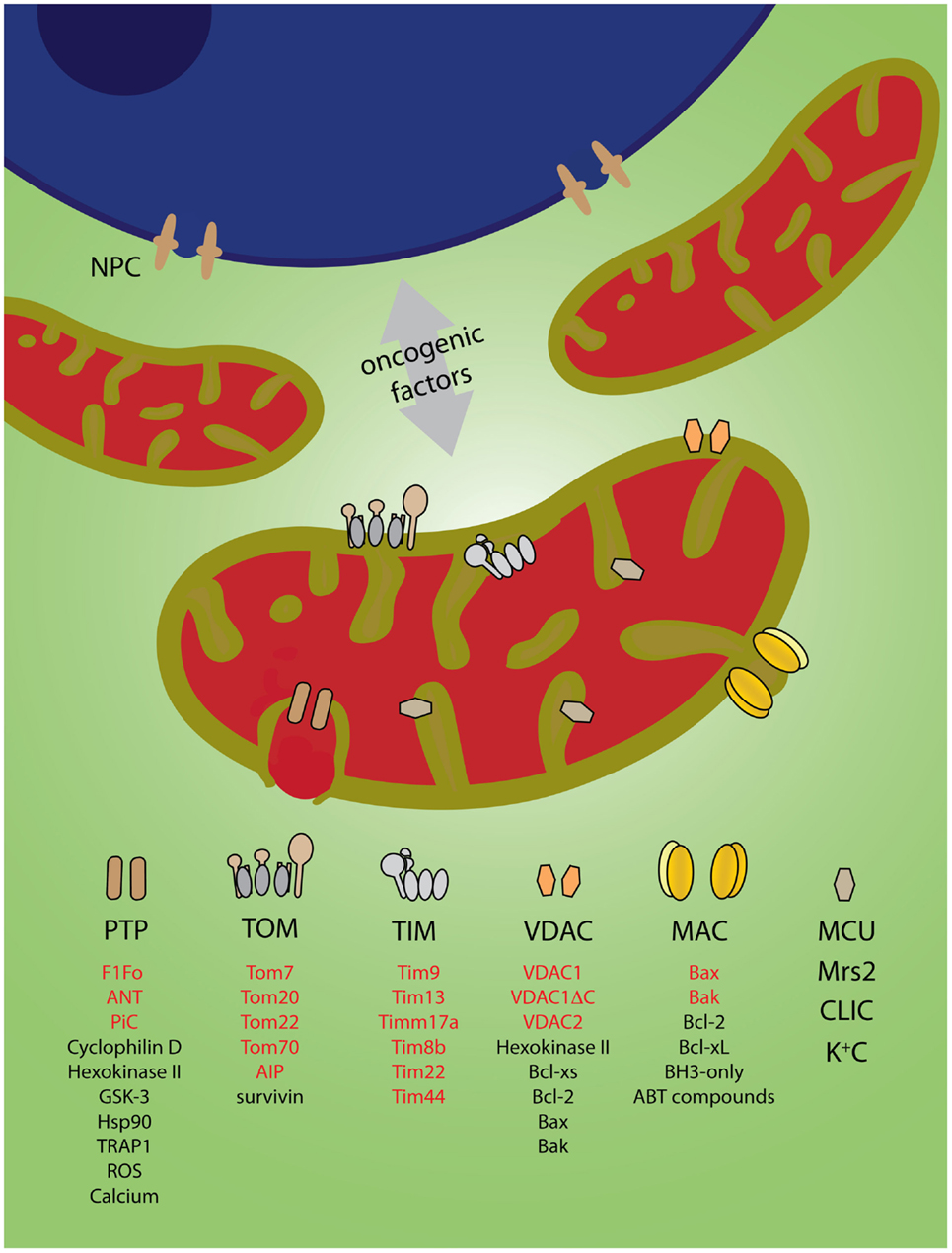
Figure 1. Mitochondrial ion channels might mediate exchange of oncogenic factors with the nucleus. Nuclear oncogenic factors either directly (via nuclear pore complexes, NPC) (12) or indirectly (via oncogenic expression) are transmitted to mitochondria to reprogram metabolism and cell death mechanisms. Channels in the outer and the inner membranes might provide the pathways for production and/or bi-directional transport of oncogenic factors. Alternatively, they might be direct targets (see text). The legend indicates channel components (red) or modulators (black) associated with cancer (see text for references). Components and regulators of TIM22 and TIM23 channels were listed in the same category. K+C represents the potassium channels BKCa and IKCa, ATP-dependent KATP, Kv1.3, two-pore TWIK-related acid-sensitive K+ channel-3 (TASK-3). Inspired by Odra Noel’s “Mitochondrial Dawn”.
If an insurgent cell is to survive, it must reprogram the power plant. Some cancer cells accomplish this task by decreasing the open probability of mitochondrial channels involved in the transport of energy metabolites (13–15) and relying on glycolytic metabolism to fuel competitive reproduction (3). One would think that the payoff for relinquishing energy conversion efficiency is that cancer cells suffer lower oxidative stress. However, oxidative stress seems to be one of the determinants of tumor cell transformation (16, 17). As discussed below and elsewhere (18), respiring mitochondria are the major sources of cytotoxic reactive oxygen species (ROS). Interestingly, exacerbated ROS emission causes the opening of pores in the inner membrane that breach the permeability barrier to solutes and the ensuing uncontrolled matrix swelling, membrane rupture, and spillage of mitochondrial proteins into the cytosol. Although the exact mechanism of permeability transition of the inner membrane is still being deciphered, cancer cells expertly avert them by equally mysterious ways.
Another feature of cancer mitochondria is that they are coated with sentinel proteins, which prevent the formation of MAC in the outer membrane, rendering the power plant oblivious to cancer transformation and unrestrained growth. Interestingly, much of our current understanding about the regulation of MAC arose from studying cancer cells, which over express the sentinel protein Bcl-2. It may also sound counterintuitive that the MAC components Bax and Bak are structurally related and belong to the Bcl-2 family of proteins. The truth is that this multifaceted family of proteins controls permeabilization of the outer membrane by either inducing or preventing it. In cancer cells, the second mode prevails and might even go beyond the outer membrane, as some anti-apoptotic members (Bcl-xL and Mcl-1) have been shown to interact with and regulate inner membrane proteins, including putative components of the permeability transition pore complex (19–21).
Induction and maintenance of tumor transformation might involve more than reprograming of metabolism and containment of apoptotic signaling. Recent studies suggest that mitochondrial dysfunction might be causally linked to the characteristic genomic instability of many human cancer cells (22–27). One could envision the involvement of additional mitochondrial channels responsible for, e.g., control of calcium, potassium and magnesium flux, organelle volume, and nucleic acid transport. The development of new tools has enabled the identification of elusive mitochondrial channels like the calcium uniporter (MCU) (28, 29), creating momentum for deeper understanding of the role of mitochondria in cancer cell transformation. Excellent reviews have been recently published on the therapeutic potential of mitochondrial ion channels in cancer (30–32). This mini review will focus on the putative role that these channels might exert on the reciprocal transmission of oncogenic factors between mitochondria and the nucleus (Figure 1).
Metabolic Channels
Voltage-Dependent Anion Channel
Sometimes coined the food channel, VDAC was discovered in mitochondria almost 40 years ago (33). There is currently no definitive evidence that indicates the steady state conformation of VDAC in intact cells, but a pioneering study suggests an open state (34). When fully open, VDAC allows the exchange of ATP and ADP as well as electron transport chain substrates pyruvate, malate, succinate, and NADH across the outer mitochondrial membrane (35–41). In this state, the channel is normally slightly anion-selective but becomes cation-selective in its half-open state, possibly favoring calcium over metabolites (42). Interestingly, this cationic conductance has also been observed when VDAC is fully open (42), suggestive of a possible role in transport of larger positively charged metabolites. Although more sporadic in reconstituted systems, this behavior was observed in preparations from yeast and mammalian VDAC and might explain the overall positive net conductance of mitochondria (42–44). In mammals, three isoforms of VDAC have been discovered: VDAC1, VDAC2, and VDAC3 (45). These isoforms display similarities in both structure and function, but each has been found to play a distinct role in mitochondria-mediated apoptosis and metabolism. Little is known of the function of VDAC3, which is abundant in sperm cells. Is any particular isoform up/down regulated in different tumors? The answer seems to be yes, at least for VDAC1. Recent studies found that the vdac1 gene is up regulated in breast, colon, liver, lung, pancreatic, and thyroid cancer tissue (46, 47). Knockdown of vdac1 caused slowed proliferation of HeLa cells and reduced tumor size in vivo, suggesting a role of VDAC1 in the loss of growth control commonly observed in cancer cells (48).
Another possible role for VDAC1 may exist in tumor microenvironments, which are often hypoxic. VDAC1 with a truncated C-terminus (VDAC1-ΔC) has been identified in both cancer cell lines and patient tumor tissue samples (41). VDAC1-ΔC was present in cancer cell lines that had high-cytoplasmic levels of adenosine triphosphate and in lung cancer biopsies that showed a strong resistance to chemotherapy-induced apoptosis (49).
Voltage-dependent anion channel has become an attractive target for anticancer therapy due to its essential role in apoptosis and metabolism, processes deregulated in cancer (50). Cancer cells exhibit high levels of aerobic glycolysis, a phenomenon first observed by Otto Warburg in 1956. The glycolytic enzyme hexokinase 2 (HK2), which is overexpressed in cancer cells, is a key regulator of the Warburg effect and binds to VDAC, forming HK–VDAC complexes (51). It remains to be determined if HK2 binds to all VDAC isoforms. However, an additional link to VDAC1 and VDAC2 involvement can be inferred from their regulation by free tubulin, suggesting that it might underlie the cytotoxic effect of chemotherapeutic drugs affecting microtubule formation (15). It has also been recently suggested that the low-ATP/ADP ratio resulting from free tubulin-mediated VDAC closure is a contributing factor to the Warburg effect (12–15). VDAC2 also regulates apoptosis through its interactions with Bak, namely, the localization of Bak to the mitochondria and inhibition of Bak-mediated apoptosis (49, 52). As demonstrated in melanoma cells, overexpressing Bcl-xS disrupts the VDAC2-Bak interaction by binding to VDAC2, releasing Bak, and allowing it to initiate apoptosis (53). Less is known about interaction between VDAC2 and the primarily cytosolic Bcl-2 protein Bax, but recent studies have shed some light on Bax regulation by VDAC2. Cells deficient in Bak and VDAC2, but not Bax, displayed resistance to apoptotic stimuli, suggesting a role for VDAC2 in Bax activation (54). More recently, it has been shown that VDAC2 facilitates recruitment of both Bak and Bax to the mitochondria and that VDAC2 and Bak are required for Bax-mediated apoptosis (52, 53, 55).
Cell Death Channels
The two mitochondria-permeabilizing structures MAC and PTP are considered to be the main death channels. Of note, these two functional entities allow the release in the cytosol of mitochondrial factors such as cytochrome c during apoptosis and/or necrosis (56).
Mitochondrial Apoptosis-Induced Channel
Broadly recognized as the main site through which death signals like cytochrome c are released into the cytosol, MAC is a dynamic channel formed in the outer membrane by at least two pro-apoptotic members of the Bcl-2 protein family, Bak and Bax (57, 58). However, the MAC pore may not be entirely of protein nature. Some studies showed that ceramides or intermediates of ceramide biosynthesis pathway had the potential to stimulate cytochrome c release in a Bak- and/or Bax-dependent fashion, indicating that these lipids are potentially an integral part of MAC (59, 60). In theory, MAC function antagonizes cancer cell transformation and is, in fact, kept at bay by oncogenic gene expression. Up regulation of the Bcl-2 or Bcl-xL proto-oncogenes is associated with tumor formation and more particularly with B-cell non-Hodgkin’s lymphoma (61–63). Pro-lymphocytic cell lines overexpressing Bcl-2 exhibit resistance to MAC formation and the ability to induce lymphoma in mice (44, 63–65). Paradoxically, it was shown that an over-abundance of Bcl-2 not only caused mitochondrial sequestration of BH3-only proteins, which are activators of MAC formation, but also that of the MAC core component Bax (66–68). This phenomenon seems to be a common feature of anti-apoptotic Bcl-2 family members as an over-abundance of Bcl-xL was also recently shown to induce both Bax accumulation and functional activation at the mitochondria (69, 70). This accumulation has been coined “priming for death,” and this concept is currently being used to develop new cancer therapies (66, 67). The first approach to pharmacologically trigger Bax-mediated apoptosis in primed cancer cells comes from the group of Stanley Korsmeyer. In one of these studies, a stabilized stapled BIM helix peptide was even shown to efficiently compete for binding to Bcl-2 with mitochondrial native BIM in a model of hematologic cancer (71). Another approach taken was to screen for small organic molecules, which can act as BH3 mimetics on anti-apoptotic multi-domain Bcl-2 family members. The first of these BH3 mimetic compounds to be developed was ABT-737, which binds Bcl-2, Bcl-xL, and Bcl-w (69). An orally bioavailable variant (ABT-263 or Navitoclax) was later developed (12) with promising clinical trial results but the caveat of causing thrombocytopenia (15).
ABT-199 is another recently developed drug that has shown antitumor activity in vitro and in vivo (31) and is perhaps the most promising BH3 mimetic. Unlike ABT-263 and ABT-737, ABT-199 is Bcl-2 selective and does not seem to cause thrombocytopenia (72). Its efficacy was first demonstrated in chronic lymphocytic leukemia (CLL), but its cytotoxic activity has since been reported in T-cell acute lymphoblastic leukemia (T-ALL) (73), acute myeloid leukemia (74), and chronic myeloid leukemia (CML) progenitor cells (15). In the CML study, treatment with ABT-199 in combination with the tyrosine kinase inhibitor imatinib enhanced inhibitory effects. It has also been used in combination with tamoxifen to induce apoptosis in estrogen receptor-positive breast cancer cells, in which Bcl-2 is overexpressed (70). ABT-199, alone or in combination with other anticancer agents, represents a promising strategy against growth of Bcl-2 dependent cancers.
Tumorigenesis and the accompanying resistance to apoptosis can also be initiated by down-regulation of pro-apoptotic Bax and Bak. In this case, the death program fails as MAC formation is prevented by a lack of building blocks (75). Chemotherapies in this latter case may involve the activation of Nur77 by the drug 3-Cl-AHPC; activation of Nur77 leading to the conversion of Bcl-2 into a core component of a new type of cytochrome c release channel (76).
PTP
The selective induction of permeability transition in the inner membrane has obvious onco-therapeutic potential. However, most cancer mitochondria seem to be desensitized to signals that trigger permeability transition in normal mitochondria (discussed below). Interestingly, a high incidence of cancer is found in transplant patients treated with cyclosporine A, an immunosuppressant known to inhibit permeability transition of the inner membrane (77). The mechanisms of PTP inhibition in cancer cells are expertly summarized in two recent reviews by the Pinton and Bernardi groups: cancer cells down regulate PTP inducers (i.e., ROS and calcium), alter expression of chaperones that regulate PTP opening, desensitize the channel through kinase signaling pathways (GSK-3 and hexokinase II), leading to reduced mitochondrial oxidative phosphorylation regimes characterized by high ATP/ADP ratios, low-inorganic phosphate, and high-ROS levels (74, 78). Intriguingly, some of the currently proposed PTP components seem to be up regulated in cancer cells. That is the case, for example, for the c subunit of the ATP synthase (79). Other studies showed that increased levels of the c subunit induce cell death via PTP (80, 81). One possible explanation to this seemingly discrepancy is that cancer cells initially benefit from PTP function (mitochondrial dysfunction and ROS emission) to generate genome instability. As the cancer phenotype strengthens, PTP is silenced, possibly by a combination of the four mechanisms listed above, a process sometimes potentiated by an increase in mitophagy (74). It should be noted, however, that the structural composition of PTP is still a matter of debate and it might involve interactions between cyclophilin D, the adenine nucleotide transporter (ANT), the phosphate carrier (PiC), and the F1Fo ATP synthase in the inner membrane (82, 83).
Protein Import Channels
Mitochondrial biogenesis relies on the import of over 99% of the organelle proteome from the cytoplasm. The import pathway is through water-filled channel complexes, namely, the translocase of the outer membrane (TOM), and the translocases of the inner membrane (TIM22, and TIM23) [for a comprehensive review, see Ref. (84)]. The TIM23 channel was the first electrophysiological demonstration of the link between protein import and water-filled channels in mitochondria (85, 86) followed by reports on the channel activity of TOM, the protein import complex of the outer membrane (87, 88). These two channels enable the coordinated passage of proteins across both membranes and into the matrix space.
Translocase of the outer membrane channels act as gatekeepers of protein import as they open in response to binding of precursor proteins carrying a signal peptide (89). This signal can be a cysteine motif (90), a presequence (91), or an internal targeting element (92). Most of the precursor proteins pass the outer membrane via the TOM complex. Precursors with cleavable N-terminal presequences are further sorted to the matrix or inner membrane by TIM23. Precursors of β-barrel proteins bind to small Tim chaperones in the inter membrane space and are then inserted into the outer membrane via the sorting and assembly machinery (SAM). If the precursor carries specific cysteine motifs, it will be transferred from TOM to the MIA complex for sorting into the intermembrane space. Finally, precursors of the metabolite carrier family are transferred via the Tim chaperones to the TIM22 translocase which then mediates their insertion into the inner membrane (93). A few α-helical outer membrane proteins seem to be imported independently from the TOM complex but may instead require the SAM complex (94).
A number of subunits of the protein import machineries have been found to be overexpressed in mitochondria of cancer cells, including Timm17a, Tim9, Tim13, Tim8b, Tim22, Tom20, Tom70, and Tom7 (79). While it might sound logical that enhanced biogenesis of mitochondrial proteins during cancer transformation requires up-regulation of the protein import machineries, it is surprising that, out of the list mentioned above, the only channel forming protein is Tim22. Another study found splice variants of Tim44 in oncotic thyroid carcinomas and suggests this TIM23 peripheral component might have a role in cancer transformation possibly by induction of ROS production or impairment of protein import (95). Furthermore, metabolic reprograming during tumorigenesis often requires redistribution of kinases or transcription factors to mitochondria in a process that depends on the mitochondrial import machinery (12–15).
Translocase of the outer membrane complex proteins have been shown to interact with both pro- and anti-apoptotic Bcl-2 family proteins, but their role in mitochondrial apoptosis is unclear. Tom22, essential for maintaining TOM structure, has been established as the mitochondrial receptor of Bax (96). In HeLa cells treated with the proteasome inhibitor celastrol, Tom22 was up regulated, yet Bax translocation still occurred in the absence of Tom22, Tom70, and Tom40 (97, 98). Interestingly, metaxins 1 and 2, components of the SAM complex, were found to be required for Bak activation in TNF-induced apoptosis in Bax deficient glioma cells (99).
Another TOM subunit, Tom20, might have a role in cancer transformation via interaction with the arylhydrocarbon receptor-interacting protein (AIP) (100). AIP is also associated with the inhibitor of apoptosis protein survivin, which localizes to the mitochondria and when released in response to apoptotic stimuli, prevents cell death by inhibiting caspase activation (101). Survivin is overexpressed in cancer cells and its import into the mitochondria is mediated by the AIP–Tom20 complex; this pathway may be a potential target for cancer cells (102), provided that the AIP–survivin interaction can be specifically targeted, as AIP has been shown to bind other mitochondrial pre-proteins (100).
Small Ion Channels
Voltage-dependent anion channel is thought to allow unrestrained ion flux across the outer membrane. However, ion flow is tightly regulated across the inner membrane as to allow the vital establishment of the hydrogen proton gradient. Inner membrane ion channels control metabolic pace, emission of ROS, organelle volume, and other functions that seem to be modified in cancer cells; they are therefore natural candidates for activity modulation through cancer transformation. A non-exhaustive list of potential targets includes mitochondrial potassium channels [calcium-dependent BKCa and IKCa, ATP-dependent KATP, Kv1.3, two-pore TWIK-related Acid-Sensitive K+ channel-3 (TASK-3)], calcium uniporter MCU, magnesium channels (Mrs2), anion channels (CLIC), and channel activities that await molecular identification (mCs, AAA, ACA, and IMAC) (30–32).
Concluding Remarks
Mitochondrial ion channels are emerging as promising onco-therapeutic targets. Compelling and mounting literature suggests that reprogramed metabolic and apoptotic signaling from mitochondria is present in cancer cells and might underlie their characteristic genome instability. The field should benefit from studies that aim at identifying oncogenic factors that are either released from mitochondria or that affect the function of these organelles. Also helpful would be the ability to manipulate specific mitochondrial functions in animal models of cancer cell transformation; and new electrophysiological and imagery techniques allowing live monitoring of mitochondrial channels in cancer cells. Cancer cell transformation, rather than being solely induced by the nucleus or the mitochondria, might involve crosstalk between these cellular compartments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Weissman School of Arts & Sciences – Baruch College startup funds to PP and by startup funds from CSU-Fresno College of Science and Mathematics to LD.
References
1. Lane N. Power, Sex, and Suicide: Mitochondria and the Origin of Life. Oxford: Oxford University Press (2005).
2. Lane N. Bioenergetic constraints on the evolution of complex life. Cold Spring Harb Perspect Biol (2014) 6:a015982. doi: 10.1101/cshperspect.a015982
3. Maldonado EN, Lemasters JJ. ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion (2014) 19(Pt A):78–84. doi:10.1016/j.mito.2014.09.002
4. Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ (2006) 13:1387–95. doi:10.1038/sj.cdd.4401949
5. Peixoto PM, Ryu SY, Kinnally KW. Mitochondrial ion channels as therapeutic targets. FEBS Lett (2010) 584:2142–52. doi:10.1016/j.febslet.2010.02.046
6. Ryu SY, Beutner G, Dirksen RT, Kinnally KW, Sheu SS. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett (2010) 584:1948–55. doi:10.1016/j.febslet.2010.01.032
7. Tomaskova Z, Ondrias K. Mitochondrial chloride channels – what are they for? FEBS Lett (2010) 584:2085–92. doi:10.1016/j.febslet.2010.01.035
8. Colombini M. Mitochondrial outer membrane channels. Chem Rev (2012) 112:6373–87. doi:10.1021/cr3002033
9. Szabo I, Leanza L, Gulbins E, Zoratti M. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch (2012) 463:231–46. doi:10.1007/s00424-011-1058-7
10. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol (2014) 592:829–39. doi:10.1113/jphysiol.2013.268235
11. Szabo I, Zoratti M. Mitochondrial channels: ion fluxes and more. Physiol Rev (2014) 94:519–608. doi:10.1152/physrev.00021.2013
12. Simon DN, Rout MP. Cancer and the nuclear pore complex. Adv Exp Med Biol (2014) 773:285–307. doi:10.1007/978-1-4899-8032-8_13
13. Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, et al. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene (2008) 27:4636–43. doi:10.1038/onc.2008.108
14. Rostovtseva TK, Bezrukov SM. VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta (2012) 1818:1526–35. doi:10.1016/j.bbamem.2011.11.004
15. Maldonado EN, Sheldon KL, Dehart DN, Patnaik J, Manevich Y, Townsend DM, et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem (2013) 288:11920–9. doi:10.1074/jbc.M112.433847
16. Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal (2012) 16:1295–322. doi:10.1089/ars.2011.4414
17. Yang Y, Karakhanova S, Werner J, Bazhin AV. Reactive oxygen species in cancer biology and anticancer therapy. Curr Med Chem (2013) 20:3677–92. doi:10.2174/0929867311320999165
18. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi:10.1152/physrev.00026.2013
19. Vento MT, Zazzu V, Loffreda A, Cross JR, Downward J, Stoppelli MP, et al. Praf2 is a novel Bcl-xL/Bcl-2 interacting protein with the ability to modulate survival of cancer cells. PLoS One (2010) 5:e15636. doi:10.1371/journal.pone.0015636
20. Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs (2011) 20:1397–411. doi:10.1517/13543784.2011.609167
21. Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem (2012) 287:23152–61. doi:10.1074/jbc.M112.345918
22. Delsite R, Kachhap S, Anbazhagan R, Gabrielson E, Singh KK. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol Cancer (2002) 1:6. doi:10.1186/1476-4598-1-6
23. Rasmussen AK, Chatterjee A, Rasmussen LJ, Singh KK. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res (2003) 31:3909–17. doi:10.1093/nar/gkg446
24. Miceli MV, Jazwinski SM. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci (2005) 46:1765–73. doi:10.1167/iovs.04-1327
25. Singh KK, Kulawiec M, Still I, Desouki MM, Geradts J, Matsui S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene (2005) 354:140–6. doi:10.1016/j.gene.2005.03.027
26. Kulawiec M, Safina A, Desouki MM, Still I, Matsui S, Bakin A, et al. Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol Ther (2008) 7:1732–43. doi:10.4161/cbt.7.11.6729
27. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8:754–66. doi:10.1016/j.celrep.2014.06.043
28. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
29. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
30. Peixoto PM, Dejean LM, Kinnally KW. The therapeutic potential of mitochondrial channels in cancer, ischemia-reperfusion injury, and neurodegeneration. Mitochondrion (2012) 12:14–23. doi:10.1016/j.mito.2011.03.003
31. Leanza L, Biasutto L, Manago A, Gulbins E, Zoratti M, Szabo I. Intracellular ion channels and cancer. Front Physiol (2013) 4:227. doi:10.3389/fphys.2013.00227
32. Leanza L, Zoratti M, Gulbins E, Szabo I. Mitochondrial ion channels as oncological targets. Oncogene (2014) 33:5569–81. doi:10.1038/onc.2013.578
33. Schein SJ, Colombini M, Finkelstein A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol (1976) 30:99–120. doi:10.1007/BF01869662
34. Jonas EA, Buchanan J, Kaczmarek LK. Prolonged activation of mitochondrial conductances during synaptic transmission. Science (1999) 286:1347–50. doi:10.1126/science.286.5443.1347
35. Holden MJ, Colombini M. The mitochondrial outer membrane channel, VDAC, is modulated by a soluble protein. FEBS Lett (1988) 241:105–9. doi:10.1016/0014-5793(88)81040-8
36. Liu MY, Colombini M. Regulation of mitochondrial respiration by controlling the permeability of the outer membrane through the mitochondrial channel, VDAC. Biochim Biophys Acta (1992) 1098:255–60. doi:10.1016/S0005-2728(05)80344-5
37. Blachly-Dyson E, Zambronicz EB, Yu WH, Adams V, McCabe ER, Adelman J, et al. Cloning and functional expression in yeast of two human isoforms of the outer mitochondrial membrane channel, the voltage-dependent anion channel. J Biol Chem (1993) 268:1835–41.
38. Zizi M, Forte M, Blachly-Dyson E, Colombini M. NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J Biol Chem (1994) 269:1614–6.
39. Ahmadzadeh M, Horng A, Colombini M. The control of mitochondrial respiration in yeast: a possible role of the outer mitochondrial membrane. Cell Biochem Funct (1996) 14:201–8. doi:10.1002/(SICI)1099-0844(199609)14:3<201::AID-CBF673>3.0.CO;2-Q
40. Rostovtseva T, Colombini M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J Biol Chem (1996) 271:28006–8. doi:10.1074/jbc.271.45.28006
41. Sampson MJ, Lovell RS, Craigen WJ. Isolation, characterization, and mapping of two mouse mitochondrial voltage-dependent anion channel isoforms. Genomics (1996) 33:283–8. doi:10.1006/geno.1996.0193
42. Pavlov E, Grigoriev SM, Dejean LM, Zweihorn CL, Mannella CA, Kinnally KW. The mitochondrial channel VDAC has a cation-selective open state. Biochim Biophys Acta (2005) 1710:96–102. doi:10.1016/j.bbabio.2005.09.006
43. Campo ML, Kinnally KW, Tedeschi H. The effect of antimycin A on mouse liver inner mitochondrial membrane channel activity. J Biol Chem (1992) 267:8123–7.
44. Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, et al. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol (2001) 155:725–31. doi:10.1083/jcb.200107057
45. Quast SA, Berger A, Buttstadt N, Friebel K, Schonherr R, Eberle J. General sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS One (2012) 7:e39290. doi:10.1371/journal.pone.0039290
46. Rusznak Z, Bakondi G, Kosztka L, Pocsai K, Dienes B, Fodor J, et al. Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch (2008) 452:415–26. doi:10.1007/s00428-007-0545-x
47. Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, et al. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med (2012) 4:577–93. doi:10.1002/emmm.201200235
48. Kosztka L, Rusznak Z, Nagy D, Nagy Z, Fodor J, Szucs G, et al. Inhibition of TASK-3 (KCNK9) channel biosynthesis changes cell morphology and decreases both DNA content and mitochondrial function of melanoma cells maintained in cell culture. Melanoma Res (2011) 21:308–22. doi:10.1097/CMR.0b013e3283462713
49. Brahimi-Horn MC, Mazure NM. Hypoxic VDAC1: a potential mitochondrial marker for cancer therapy. Adv Exp Med Biol (2014) 772:101–10. doi:10.1007/978-1-4614-5915-6_5
50. Leanza L, Venturini E, Kadow S, Carpinteiro A, Gulbins E, Becker KA. Targeting a mitochondrial potassium channel to fight cancer. Cell Calcium (2014). doi:10.1016/j.ceca.2014.09.006
51. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med (2011) 208:313–26. doi:10.1084/jem.20101470
52. Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S, Stroud DA, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ (2014) 21:1925–35. doi:10.1038/cdd.2014.119
53. Roy SS, Ehrlich AM, Craigen WJ, Hajnoczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep (2009) 10:1341–7. doi:10.1038/embor.2009.219
54. Yamagata H, Shimizu S, Nishida Y, Watanabe Y, Craigen WJ, Tsujimoto Y. Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax. Oncogene (2009) 28:3563–72. doi:10.1038/onc.2009.213
55. Leanza L, O’Reilly P, Doyle A, Venturini E, Zoratti M, Szegezdi E, et al. Correlation between potassium channel expression and sensitivity to drug-induced cell death in tumor cell lines. Curr Pharm Des (2014) 20:189–200. doi:10.2174/13816128113199990032
56. Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta (2011) 1813:616–22. doi:10.1016/j.bbamcr.2010.09.013
57. Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, et al. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell (2005) 16:2424–32. doi:10.1091/mbc.E04-12-1111
58. Martinez-Caballero S, Dejean LM, Kinnally MS, Oh KJ, Mannella CA, Kinnally KW. Assembly of the mitochondrial apoptosis-induced channel, MAC. J Biol Chem (2009) 284:12235–45. doi:10.1074/jbc.M806610200
59. Siskind LJ, Mullen TD, Romero Rosales K, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, et al. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Chem (2010) 285:11818–26. doi:10.1074/jbc.M109.078121
60. Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell (2012) 148:988–1000. doi:10.1016/j.cell.2012.01.038
61. Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood (2004) 103:2276–83. doi:10.1182/blood-2003-07-2469
62. Swanson PJ, Kuslak SL, Fang W, Tze L, Gaffney P, Selby S, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol (2004) 172:6684–91. doi:10.4049/jimmunol.172.11.6684
63. Meijerink JP, Van Lieshout EM, Beverloo HB, Van Drunen E, Mensink EJ, Macville M, et al. Novel murine B-cell lymphoma/leukemia model to study BCL2-driven oncogenesis. Int J Cancer (2005) 114:917–25. doi:10.1002/ijc.20822
64. Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, et al. Regulated targeting of BAX to mitochondria. J Cell Biol (1998) 143:207–15. doi:10.1083/jcb.143.1.207
65. Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J (1998) 17:3878–85. doi:10.1093/emboj/17.14.3878
66. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9:351–65. doi:10.1016/j.ccr.2006.03.027
67. Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer (2008) 8:121–32. doi:10.1038/nrc2297
68. Teijido O, Dejean L. Upregulation of Bcl2 inhibits apoptosis-driven BAX insertion but favors BAX relocalization in mitochondria. FEBS Lett (2010) 584:3305–10. doi:10.1016/j.febslet.2010.07.002
69. Gautier F, Guillemin Y, Cartron PF, Gallenne T, Cauquil N, Le Diguarher T, et al. Bax activation by engagement with, then release from, the BH3 binding site of Bcl-xL. Mol Cell Biol (2011) 31:832–44. doi:10.1128/MCB.00161-10
70. Renault TT, Teijido O, Antonsson B, Dejean LM, Manon S. Regulation of Bax mitochondrial localization by Bcl-2 and Bcl-x(L): keep your friends close but your enemies closer. Int J Biochem Cell Biol (2013) 45:64–7. doi:10.1016/j.biocel.2012.09.022
71. LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest (2012) 122:2018–31. doi:10.1172/JCI46231
72. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell (2007) 11:37–51. doi:10.1016/j.ccr.2006.10.020
73. Opalinska M, Meisinger C. Metabolic control via the mitochondrial protein import machinery. Curr Opin Cell Biol (2015) 33:42–8. doi:10.1016/j.ceb.2014.11.001
74. Bonora M, Pinton P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol (2014) 4:302. doi:10.3389/fonc.2014.00302
75. Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood (2008) 111:3322–30. doi:10.1182/blood-2007-09-078162
76. Han YH, Cao X, Lin B, Lin F, Kolluri SK, Stebbins J, et al. Regulation of Nur77 nuclear export by c-Jun N-terminal kinase and Akt. Oncogene (2006) 25:2974–86. doi:10.1038/sj.onc.1209358
77. Norman KG, Canter JA, Shi M, Milne GL, Morrow JD, Sligh JE. Cyclosporine A suppresses keratinocyte cell death through MPTP inhibition in a model for skin cancer in organ transplant recipients. Mitochondrion (2010) 10:94–101. doi:10.1016/j.mito.2009.10.001
78. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium (2015). doi:10.1016/j.ceca.2015.03.004
79. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, et al. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle (2012) 11:4390–401. doi:10.4161/cc.22777
80. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle (2013) 12:674–83. doi:10.4161/cc.23599
81. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A (2014) 111:10580–5. doi:10.1073/pnas.1401591111
82. Halestrap AP. The C ring of the F1Fo ATP synthase forms the mitochondrial permeability transition pore: a critical appraisal. Front Oncol (2014) 4:234. doi:10.3389/fonc.2014.00234
83. Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol (2015) 78:129–41. doi:10.1016/j.yjmcc.2014.08.018
84. Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol (2010) 11:655–67. doi:10.1038/nrm2959
85. Lohret TA, Jensen RE, Kinnally KW. Tim23, a protein import component of the mitochondrial inner membrane, is required for normal activity of the multiple conductance channel, MCC. J Cell Biol (1997) 137:377–86. doi:10.1083/jcb.137.2.377
86. Truscott KN, Kovermann P, Geissler A, Merlin A, Meijer M, Driessen AJ, et al. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat Struct Biol (2001) 8:1074–82. doi:10.1038/nsb726
87. Hill K, Model K, Ryan MT, Dietmeier K, Martin F, Wagner R, et al. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins [see comment]. Nature (1998) 395:516–21. doi:10.1038/26780
88. Kunkele KP, Heins S, Dembowski M, Nargang FE, Benz R, Thieffry M, et al. The preprotein translocation channel of the outer membrane of mitochondria. Cell (1998) 93:1009–19. doi:10.1016/S0092-8674(00)81206-4
89. Andreoli C, Prokisch H, Hortnagel K, Mueller JC, Munsterkotter M, Scharfe C, et al. MitoP2, an integrated database on mitochondrial proteins in yeast and man. Nucleic Acids Res (2004) 32:D459–62. doi:10.1093/nar/gkh137
90. Herrmann JM, Hell K. Chopped, trapped or tacked – protein translocation into the IMS of mitochondria. Trends Biochem Sci (2005) 30:205–11. doi:10.1016/j.tibs.2005.02.005
92. Brix J, Rudiger S, Bukau B, Schneider-Mergener J, Pfanner N. Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non- cleavable preprotein. J Biol Chem (1999) 274:16522–30. doi:10.1074/jbc.274.23.16522
93. Peixoto PM, Grana F, Roy TJ, Dunn CD, Flores M, Jensen RE, et al. Awaking TIM22, a dynamic ligand-gated channel for protein insertion in the mitochondrial inner membrane. J Biol Chem (2007) 282:18694–701. doi:10.1074/jbc.M700775200
94. Stojanovski D, Guiard B, Kozjak-Pavlovic V, Pfanner N, Meisinger C. Alternative function for the mitochondrial SAM complex in biogenesis of alpha-helical TOM proteins. J Cell Biol (2007) 179:881–93. doi:10.1083/jcb.20070604320071212c
95. Handa N, Kishishita S, Morita S, Akasaka R, Jin Z, Chrzas J, et al. Structure of the human Tim44 C-terminal domain in complex with pentaethylene glycol: ligand-bound form. Acta Crystallogr D Biol Crystallogr (2007) 63:1225–34. doi:10.1107/S0907444907051463
96. Bellot G, Cartron PF, Er E, Oliver L, Juin P, Armstrong LC, et al. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ (2007) 14:785–94. doi:10.1038/sj.cdd.4402055
97. Ross K, Rudel T, Kozjak-Pavlovic V. TOM-independent complex formation of Bax and Bak in mammalian mitochondria during TNF[alpha]- induced apoptosis. Cell Death Differ (2009) 16:697–707. doi:10.1038/cdd.2008.194
98. Feng L, Zhang D, Fan C, Ma C, Yang W, Meng Y, et al. ER stress-mediated apoptosis induced by celastrol in cancer cells and important role of glycogen synthase kinase-3[beta] in the signal network. Cell Death Dis (2013) 4:e715. doi:10.1038/cddis.2013.222
99. Cartron P-F, Petit E, Bellot G, Oliver L, Vallette FM. Metaxins 1 and 2, two proteins of the mitochondrial protein sorting and assembly machinery, are essential for Bak activation during TNF alpha triggered apoptosis. Cell Signal (2014) 26:1928–34. doi:10.1016/j.cellsig.2014.04.021
100. Yano M, Terada K, Mori M. AIP is a mitochondrial import mediator that binds to both import receptor Tom20 and preproteins. J Cell Biol (2003) 163:45–56. doi:10.1083/jcb.200305051
101. Dohi T, Beltrami E, Wall NR, Plescia J, Altieri DC. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest (2004) 114:1117–27. doi:10.1172/JCI22222
Keywords: cancer cell transformation, VDAC, MAC, PTP, TIM, TOM
Citation: Madamba SM, Damri KN, Dejean LM and Peixoto PM (2015) Mitochondrial ion channels in cancer transformation. Front. Oncol. 5:120. doi: 10.3389/fonc.2015.00120
Received: 07 March 2015; Accepted: 15 May 2015;
Published: 04 June 2015
Edited by:
Gavin P. McStay, New York Institute of Technology, USACopyright: © 2015 Madamba, Damri, Dejean and Peixoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo M. Peixoto, Department of Natural Sciences, Baruch College, CUNY, 17 Lexington Avenue, New York, NY 10010, USA,cGFibG8ucGVpeG90b0BiYXJ1Y2guY3VueS5lZHU=