 Xiao-Jian Sun
Xiao-Jian Sun Na Man1,3
Na Man1,3 Lan Wang
Lan Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 26 May 2015
Sec. Cancer Endocrinology
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00108
This article is part of the Research TopicTranscriptional Regulation in Cancers and Metabolic DiseasesView all 13 articles
Histone, and non-histone, protein acetylation plays an important role in a variety of cellular events, including the normal and abnormal development of blood cells, by changing the epigenetic status of chromatin and regulating non-histone protein function. Histone acetyltransferases (HATs), which are the enzymes responsible for histone and non-histone protein acetylation, contain p300/CBP, MYST, and GNAT family members. HATs are not only protein modifiers and epigenetic factors but also critical regulators of cell development and carcinogenesis. Here, we will review the function of HATs such as p300/CBP, Tip60, MOZ/MORF, and GCN5/PCAF in normal hematopoiesis and the pathogenesis of hematological malignancies. The inhibitors that have been developed to target HATs will also be reviewed here. Understanding the roles of HATs in normal/malignant hematopoiesis will provide the potential therapeutic targets for the hematological malignancies.
Histone acetyltransferases (HATs) acetylate histone proteins by transferring acetyl group from acetyl-CoA to specific lysine residues (1, 2). The acetylation of histones by HATs results in a dispersed structure of chromatin, which becomes accessible by transcriptional factors. Besides histones, a variety of non-histone substrates also have been shown to be acetylated by HATs, thus the HATs are now generally categorized as lysine acetyltransferases (3). The acetylome studies have led to discovery of many new substrates of HATs, and a lot of non-histone substrates of HATs, such as AML1, AML1-ETO (AE), p53, c-MYC, NF-κB, Cohesin and Tubulin, have been found to play important roles in different cellular processes (4–10). Based on the cellular localization, HATs are classified into type A and type B HATs. The type A HATs show nuclear localization and likely catalyze the processes related to transcription (11). The type A HATs are further divided into five families according to their homology and acetylation mechanisms. The GNAT family members include PCAF, Gnc5 and ELP3. CBP and p300 form the CBP/p300 family (12). Tip60, MOZ, MORF, HBO1 and HMOF belong to the MYST family (13). The transcriptional factor related HAT family includes TAF1 and TIFIIIC90. In addition, several steroid receptor co-activators, such as p600, SRC1, CLOCK and AIB1/ACTR/SCR3 etc., are also HATs. (14, 15). Type B HATs are localized in the cytoplasm and they are shown to acetylate the newly synthesized histones. For example, HAT1 is one of type B HAT members and functions in DNA repair and histone deposition (16).
Histone acetyltransferases play key roles in normal and malignant hematopoiesis. The acetylation of histones and non-histone proteins has been shown to regulate normal blood cell development (17–19) (Table 1). Analysis of chromatin factor interaction network in hematopoietic development shows multiple chromatin factor complexes, including NuA4/P300/CBP/HBO1, are required for normal hematopoiesis (20). Protein acetylation regulates hematopoietic stem cell (HSC) self-renewal, proliferation, and their differentiation into committed hematopoietic progenitors. In line with the critical functions of HATs in normal hematopoiesis, chromosomal translocations that involve HAT genes are frequently found in hematological malignancies. Recent cancer genome studies have identified HATs as common targets for mutations in these diseases. Meanwhile, the acetylation states of some oncoproteins and tumor suppressor proteins have been correlated with hematological malignancies manifestation (5, 19, 21–25) (Table 2). Notably, most leukemogenic fusion proteins physically interact with HATs, even though they are not directly fused with HATs, suggesting that the aberrant acetylation regulation by these fusion proteins are critically important in leukemogenesis (26). In this article, we will first briefly review a few examples of interesting findings that potentially lead to development of new therapeutic strategies for hematological malignancies, and then provide an overview of the functions of HATs in normal and malignant hematopoiesis (Figure 1).
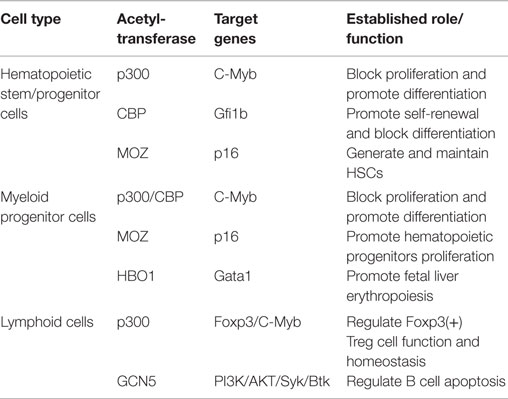
Table 1. The role of HATs in hematopoietic stem/progenitor cells.

Table 2. The role of HATs in hematological malignancies.
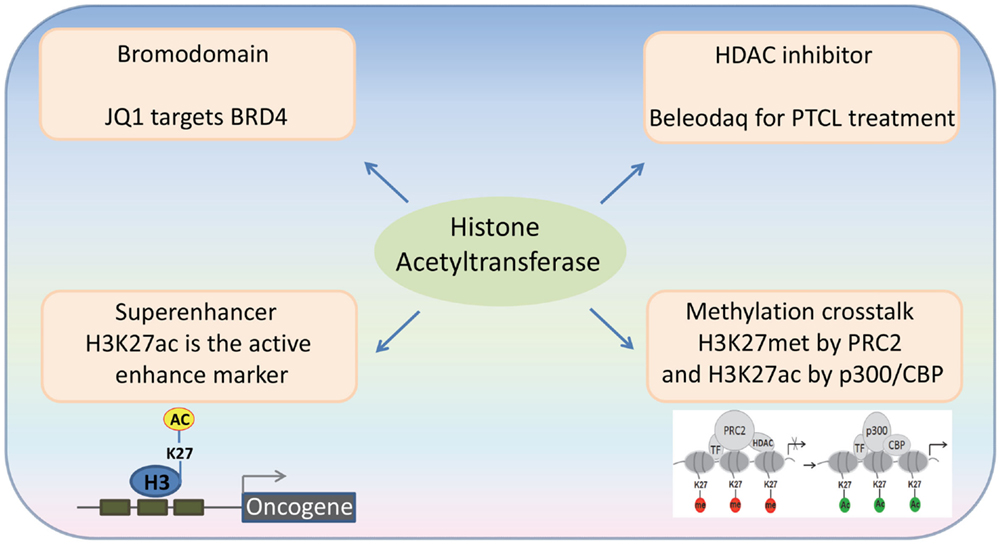
Figure 1. Recent advances in the emerging fields of histone acetylation in hematopoiesis: (1) Bromodomains are a promising target for the therapy of hematological malignancies; (2) HATs generate histone marks found in active enhancers; (3) Histone acetylation-methylation crosstalk in hematopoiesis; (4) Third generation HDAC inhibitors for the therapy of hematological malignancies.
Acetylated lysine residues generated by HATs can be specifically bound by some protein domains (“readers”). Bromodomains have been identified as an important type of the readers of acetyl lysine. The human genome encodes over 60 bromodomain proteins, including HATs, HAT-associated proteins (such as GCN5L2, PCAF, and BRD9), histone methyltransferases (such as ASH1L and MLL), transcriptional co-activators (such as TRIMs and TAFs), as well as the BET family proteins (27). BRD4, a member of the BET family proteins, is shown to locate at the enhancer and/or promoter regions of many active genes. The bromodomain inhibitor JQ1 is able to disassociate BRD4 from acetylated histones, leading to downregulation of gene transcription and decreased phosphorylation of RNA polymerase II (28) (Figure 2). JQ1 is also able to remove BRD4 from the super-enhancers and thus repress many super-enhancer regulated oncogenes. These findings have been verified in hematological malignancies, such as diffuse large B cell lymphoma (DLBCL), and provide a basis for using JQ1, as well as other BET bromodomain inhibitors, in the treatment of hematological malignancies (29–31).
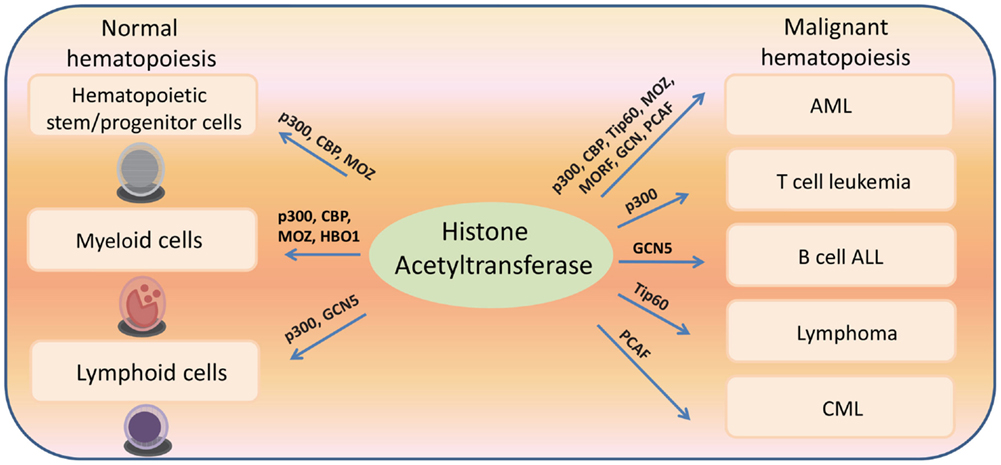
Figure 2. Histone acetyltransferases regulate both normal and malignant hematopoiesis.
Super-enhancers are clusters of active enhancers bound by master transcription factors and cofactors, including the mediator complex, and can promote the high-level expression of genes that control cell identity (32, 33). Cancer cells can acquire super-enhancers to active oncogenes, which suggest that super-enhancer-associated genes could be candidate oncogenic drivers (29, 34–36). Histone H3 lysine 27 acetylation (H3K27ac) by p300/CBP is used as an active enhancer mark, which biophysically facilitates opening of chromatin and recruits the co-activators that recognize ϵ-acetyl lysine through bromodomain (37). BRD4 is most commonly associated with enhancer regions, defined by the presence of H3K27ac by p300/CBP and the absence of H3K4me3. In a genome-wide study of DLBCL, a rank ordering of the enhancer regions by H3K27ac enrichment reveals that BRD4 binds to the majority of active enhancers and that the genome-wide correlation between BRD4 occupancy and H3K27ac is extremely strong. Importantly, BRD4 is highly loaded at the super-enhancers (29). These findings suggest that the HAT-mediated H3K27ac and the BRD4 recruitment may play an important role in the formation and function of the super-enhancers.
Polycomb group (PcG) proteins are transcriptional repressors, and their abnormal expression is frequently associated with hematological malignancies. The PcG proteins can induce transcription repression through disassociating HATs from their target genes (38). The Polycomb repressive complex 2 (PRC2) catalyzes trimethylation of H3K27, and this activity is important for transcriptional repression. Depletion of PRC2 leads to a global increase of H3K27 acetylation, which is catalyzed by p300 and CBP. In MLL-AF9-transduced HSCs, the transcriptional activation of PcG-targeted genes has been found to correlate with a methylation-to-acetylation change (Figure 1). These findings suggest that the acetylation–methylation crosstalk plays an important role in hematological malignancies.
The initial studies showed that CBP, instead of p300, is pivotal for the self-renewal of HSCs. However, it has been shown that p300, but not CBP, is essential for hematopoietic differentiation (39). However, it has been recently reported that CBP regulates both self-renewal and differentiation in adult HSCs and that loss of CBP leads to an increase in apoptosis, differentiation, and quiescence in HSCs through regulating Gfi1b (40). The numbers of colony-forming cells and erythroid cells are reduced in mouse embryos expressing the truncated CBP protein (1–1084 amino acids), which suggests that CBP mutations disrupt primitive hematopoiesis. Abnormal endothelial precursors have been found when the CBP mutant para-aortic splanchnopleural mesoderm was cultured with stromal cells, suggesting defects in the hematopoietic microenvironment (39). CBP+/− mice develop highly penetrant, multilineage defects in hematopoietic differentiation (41). These findings indicate that CBP has important functions in normal hematopoiesis. No such pathology was observed in p300+/− mice. Thus, p300 and CBP play essential but distinct roles in maintaining normal hematopoiesis (42).
MOZ contains two coactivation domains and a HAT catalytic domain. The fetal liver cells with moz mutation were able to help reconstitute the hematopoietic system in the lethally irradiated recipient mice. In the Moz-deficient mice, the number of hematopoietic progenitors in all lineages was reduced, and defects in HSCs were found (43). Loss of MOZ HAT activity causes abnormalities in hematopoietic stem/progenitor cell (HSPC) numbers in mice since HSPCs lacking MOZ HAT activity cannot expand. Loss of MOZ HAT activity also leads to the disruption of B cell development in mice. MOZ-mediated acetylation has been found to play an important role, controlling the balance between differentiation and proliferation in normal hematopoiesis (26, 44). MOZ controls the proliferation of HSCs at least in part by repressing the transcription of p16. The expression level of p16 is increased in HSPCs without MOZ HAT activity, which can induce the senescence of HSPCs. Loss of p16 rescues the proliferative abnormality in the hematopoietic progenitors lacking the MOZ HAT activity. These findings indicate an important role of MOZ HAT activity in the transcription of p16 and HSPC senescence (45). Together, MOZ is essential for a fundamental property of HSCs and the development of hematopoietic progenitors.
The KIX domains in p300 and CBP are responsible for interacting with other proteins, and they regulate c-Myb-mediated transcription activation and repression. Loss of the CH1 or KIX domain in p300 leads to profound abnormalities in hematopoiesis, while deletion of other portions of p300 only affects some specific lineages (46). Certain site specific point mutations in the KIX domain of p300 can disrupt the interaction between p300 and CREB/c-Myb, and mice homozygous for these mutations have many hematopoietic defects, such as anemia, thrombocytosis, megakaryocytosis, thymic hypoplasia, and B cell deficiency. However, no defects are detected in mice carrying the same point mutations in CBP. The interaction between the KIX domain of p300 and c-Myb is important for the function and development of megakaryocytes, and a synergistic genetic interaction has been found between the mutations in the KIX domain of p300 and mutations in c-Myb. CBP KIX domain mutations affect platelets, B cells, T cells, and red cells. Therefore, the KIX domains in p300 and CBP have their unique functions in normal hematopoiesis (47). Altogether, the KIX domains in p300 and CBP are essential for the normal hematopoiesis through regulating c-Myb-mediated transcription activation and repression (48).
HBO1 is responsible for the acetylation of histone H4K5, K8, and K12. The interaction between ING4 and histone H3K4me3 augments the ability of HBO1 to acetylate histone H3 (49, 50). HBO1 and BRD1 can form a HAT complex and control erythropoiesis. Loss of Brd1 leads to severe anemia in mouse embryos due to abnormal erythropoiesis in the fetal liver. HBO1 and BRD1 are found to mostly co-localize in the erythroblast genome, and regulate critical developmental genes. Loss of Brd1 or depletion of Hbo1 significantly decreases the levels of H3K14 acetylation in erythroblasts. Loss of Brd1 leads to reduced expression of Gata1, the key erythroid developmental regulator, and the forced expression of Gata1 can partially rescue the abnormal erythropoiesis induced by loss of Brd1. Taken together, the Brd1–Hbo1 HAT complex is an important H3K14 HAT, which is essential for the transcriptional activation of key erythroid regulators (17).
Forkhead box P3 (Foxp3) is acetylated by p300 and is essential for the development of a Treg suppressor phenotype. Hyperacetylation of Foxp3 prevents its ubiquitination and proteasome mediated degradation, which leads to a significant increase in the Foxp3 protein level. Foxp3 acetylation can rapidly control Foxp3 protein levels in T cells, which provides a new mechanism for regulating the number and function of Treg cells (51). In the presence of a p300 inhibitor, Garcinol, p300 becomes disassociated from the FOXP3 protein complex, and subsequently FOXP3 is degraded through the lysosome-dependent system. A subset of four lysine residues, which together control the total acetylation of FOXP3, could also be acetylated by p300 (52, 53). The conditional deletion or pharmacologic inhibition of p300, was able to increase apoptosis induced by the T cell receptor in Foxp3(+) Treg cells, and inhibit tumor growth in immunodeficient mice. Together, p300 is critical for the function and homeostasis of Foxp3(+) Treg cells, and thus p300 inhibitors are able to impair the function of Treg cells without affecting T effector cells suggesting a new approach for cancer immunotherapy (54).
GCN5 controls the PI3K/AKT pathway activation through regulating the transcription of Btk and Syk, which are involved in PI3K/AKT pathway activation in B cells under oxidative stress. GCN5 deficiency significantly induced apoptosis in chicken DT40 cells treated with hydrogen peroxide. GCN5 is localized at the proximal 5′-upstream regions of Btk and Syk, and the expression levels of Syk and Btk were significantly decreased in GCN5-deficient chicken DT40 cells exposed to exogenous hydrogen peroxide. Moreover, the phosphorylation level of AKT was also significantly decreased in hydrogen peroxide-treated GCN5-deficient chicken DT40 cells. Together, GCN5 regulates the transcription of Btk and Syk, and is crucial for the epigenetic regulation of the PI3K/AKT pathway activation in lymphoid cells under oxidative stress (55).
CBP/p300 is an essential co-activator for the transforming capacity of c-Myb (56). CBP/p300 is required for the ability of c-Myb to repress several key target genes involved in myeloid differentiation and p300 is recruited to c-Myb-binding sites close to c-Myb target genes (57). The interaction of p300/CBP with c-Myb is essential for leukemic transformation by the myeloid leukemia oncogenes AE, MLL-ENL, and MLL-AF9 (58). The p300–c-Myb interaction is essential for the ability of AE, MLL-ENL, and MLL-AF9 fusion proteins to confer self-renewal properties on myeloid progenitor cells. In the absence of this interaction, these fusion oncoproteins are unable to impose a block of differentiation, leading instead to terminal differentiation. Myeloid progenitors from Plt6 mice, which have a mutation in p300, are also refractory to transformation by the AE and MLL fusion proteins. Taken together, the specific interaction between p300 and c-Myb is required to control a transcriptional program, which is essential for the acquisition of self-renewal and possibly other leukemogenic properties upon expression of fusion oncoproteins. Thus, disruption of the p300–c-Myb interaction could be a potential therapeutic strategy for acute myeloid leukemia (AML) (58).
Notch3 is acetylated at lysine 1692 and lysine 1731 by p300, which can be deacetylated by HDAC1. The acetylation of Notch3 by p300 is able to promote its ubiquitination and protein degradation through proteasome system. Consequently, the expression level of Notch3 and its transcriptional activity are decreased in the non-acetylatable Notch3 mutant transgenic mice, which leads to defects in the Notch3 downstream signaling. The non-acetylatable Notch3 mutant can enhance Notch3-induced growth of T cell leukemia proliferation, which can be blocked by a histone deacetylases (HDAC) inhibitor. In the Notch3 transgenic mouse model, HDAC inhibitor-mediated hyperacetylation of Notch3 inhibits the proliferation of T cell leukemia/lymphoma cells. Altogether, targeting Notch3 deacetylation could be a promising therapeutic strategy for T-cell leukemia (25).
The CBP gene is fused with the MLL gene in patients with t(11;16) MDS; the MLL–CBP fusion contains a mostly intact CBP, suggesting involvement of CBP in leukemogenesis (59). Both the CBP HAT domain and bromodomain are required for MLL–CBP-induced transformation in AML, which is usually preceded by an MDS phase. The replacement of the MLL–CBP HAT domain with the PCAF/GCN5 HAT domain enhanced the proliferation of hematopoietic progenitor cells and led to the loss of myeloid cell surface markers in these cells. These phenotypes were not observed when the CBP bomodomain of MLL–CBP was replaced by the PCAF/TAFII250 bromodomain. The recipient mice transplanted with domain-swapped hematopoietic progenitors developed lymphoid disease or had low-frequency MDS that progressed to AML. Thus, the CBP HAT domain and bromodomain have different functions but play important roles in the pathogenesis of MLL–CBP-positive leukemias (60).
We have shown that transcriptional activation by AE is crucial for leukemogenesis and that AE interacts with the transcriptional co-activator, p300. The important function of the AML1–ETO/p300 interaction is that AE can be acetylated, as acetylation of AE is essential for its self-renewal promoting effects. The acetylation of AE9a by p300 at a specific lysine residue (K43) is required for its ability to induce leukemia in mice. Pharmacological and RNA interference-mediated inhibition of p300 specifically impairs AE/AE9a-induced transcriptional activation and leukemogenesis, but does not affect the development of MLL–AF9-induced leukemia. Acetyl-AEK43 is present in blast cells isolated from t(8;21) leukemia patients and these leukemia cells, but not normal human CD34+ cells, are sensitive to growth inhibition by p300 inhibitors. AE and p300 co-localize at the regulatory regions of many AE upregulated genes, which includes the regulators of self-renewal (e.g., Id1, p21, and Egr1). AE and p300 can cooperate in the transcriptional regulation of these target genes. Several TFIID subunits that specifically bind to a K43 acetylated AML1 peptide but not to the identical non-acetylated peptide. Furthermore, these results establish a novel link between post-translational modification of a non-histone protein (by a “histone modifying enzyme”) and transcriptional regulation, which has crucial implications for the study of cancer and the regulation of gene expression. As ETO is thought to be a key component of co-repressor complexes, its interactions with the co-activator p300 in cells is surprising and raises a question about whether “bipotential” complexes, like bivalent histone marks, may allow cells to turn on or off a given set of genes in response to certain internal or external signals. Thus, acetylation of an oncogenic fusion transcription factor itself can promote gene activation independent of any effect on histone acetylation, and can be essential for leukemia stem cell to self-renewal and malignant transformation (4, 61).
Tip60 regulates several transcription factors, which can promote or suppress carcinogenesis (e.g., p53 and Myc). Tip60 regulates DNA-damage response (DDR) signaling induced by oncogenes, which can prevent cancer progression. Loss of one allele of Tip60 inhibited Myc-induced DDR but had no effect on DDR in normal B cells. In the Tip60 heterozygous knockout mice, Tip60 inhibits Myc-induced lymphomagenesis at the pre-tumoral stage. The mono-allelic loss of Tip60 occurs often in lymphomas with concomitant reduction in mRNA levels; this often coexists with p53 mutations and is related to the disease grade. Thus, Tip60 functions as a tumor suppressor in a haplo-insufficient manner, and Tip60 is essential for controlling Myc-induced DDR in cancer cells (62).
TIP60 interacts with c-Myb, which requires the transactivation domain of c-Myb and the HAT domain of TIP60. Coexpression of TIP60 impairs c-Myb-induced transcription activation. TIP60 can bind to the promoters of c-Myb target genes, which is dependent on c-Myb. Furthermore, c-Myb interacts with HDAC1 and HDAC2, which are associated with TIP60 and cause transcriptional repression. TIP60 negatively regulates the transcription activity of c-Myb through interacting with HDACs in human hematopoietic cells. Consistently, knockdown of Tip60 increased the expression level of c-Myc. It has been found that the expression level of Tip60 is much (~60%) lower in the patients with AML. These findings suggest that TIP60 regulates the function of c-Myb and that dysregulated TIP60 could be involved in c-Myb-driven leukemogenesis (63).
MOZ–TIF2 is associated with AML chromosomal abnormalities at inv(8)(p11q13). MOZ–TIF2 contains the CBP interaction domain (CID) of TIF2 and the HAT domains of MOZ and TIF2 (64–67). In a murine bone marrow transplant assay, MOZ–TIF2 causes AML that could be serially transplanted (67). It has been found that the interaction between MOZ–TIF2 and CBP through the CID domain and the C2HC nucleosome recognition motif in MOZ are essential for transformation (68). MOZ–TIF2 dominantly inhibits the transcription activity of CBP-dependent activation (e.g., p53 and nuclear receptors), which requires the CID domain. The nuclear localization of MOZ–TIF2 is abnormal, which is dependent on the MOZ portion of this fusion protein. CBP expression is decreased in the cells expressing MOZ–TIF2, which results in the depletion of CBP from PML bodies. The critical characteristics of MOZ–TIF2 are to disrupt the normal activity of CBP/CBP-dependent activators in acute myeloid leukemia (69). MOZ–TIF2 binds to the promoter of RARβ2, leading to the dissociation of CBP/p300, the abnormal histone acetylation and the downregulation of RARβ2. MOZ–TIF2 was recruited to AML1 target promoters and upregulated transcription mediated by AML1. Both MOZ and MOZ–TIF2 were found to co-localize with AML1 (70). MOZ–TIF2 impaired retinoic acid-mediated transcription activation of C/EBPβ/CD11b, and inhibited nuclear receptor-induced gene activation through aberrant recruitment of CBP, which required both the MOZ and TIF2 domains of this fusion protein (71).
In a transgenic zebrafish model, spi1 promoter-driven MOZ–TIF2 expression induced the development of AML in 2 of 180 embryos expressing MYST3/NCOA2, in which kidney invasion by myeloid blast cells was observed (72). MOZ–TIF2 interacted with PU.1 to stimulate the expression of M-CSFR, and PU.1 is required for the establishment and maintenance of LSCs by MOZ–TIF2. Loss of CSF1R impairs MOZ–TIF2-induced leuekmogenesis in mouse models and CSF1R inhibitors delay the development of MOZ–TIF2-induced AML (73). MOZ–TIF2 can cooperate with FLT3–ITD to transform hematopoietic cells. STAT5 signaling is required for MOZ–TIF2-induced leukemogenesis. Deletion of STAT5 led to the differentiation of MOZ–TIF2-transduced fetal liver cells, and these cells lost their replating ability. The recipient mice transplanted with Stat5−/− MOZ–TIF2 leukemia cells have longer latency and incomplete penetrance. STAT5 is essential for the self-renewal of leukemia stem cells in MOZ–TIF2 driven leukemia (74). Overexpression of HOXA9, HOXA10, and MEIS1 was observed in AML patients with MOZ fusions. MOZ–TIF2 forms a stable complex with bromodomain-PHD finger protein 1 (BRPF1), and MOZ–TIF2/BRPF1 associate with Hox genes in the MOZ–TIF2 driven leukemia cells. BRPF1 depletion disrupts the localization of MOZ on the Hox genes, which led to the inhibition of MOZ–TIF2-induced transformation. Moreover, mutant MOZ–TIF2 lacking HAT activity could not deregulate HOX genes or initiate AML. Thus, MOZ–TIF2/BRPF1 induces leukemogenesis through the upregulation of HOX genes, which is regulated by MOZ-dependent histone acetylation (75).
The recurrent translocation t(8;16)(p11;p13) leads to the MOZ–CBP fusion gene, which contains the MOZ finger motifs and HAT domain and a mostly intact CBP (76). This t(8;16)(p11;p13) translocation occurs in the M5 subtype AML, which is characterized by erythrophagocytosis and a poor prognosis. The CBP–MOZ mRNA is not in-frame, which suggested that MOZ–CBP is the critical fusion for the initiation of leukemia (77, 78). MOZ, CBP, and MOZ–CBP are all able to acetylate the transcription factor AML1. The level of MOZ–AML1 complex upregulates when M1 myeloid cells differentiate into macrophages/monocytes. This finding suggests that the MOZ–AML1 complex could have important functions in the differentiation of myeloid cells. MOZ–CBP inhibits the differentiation of M1 myeloid cells, and could induce the development of leukemia through impairing AML1-induced transcription activation (79). MOZ–CBP cooperates with steroid receptor co-activator-1 to activate transcription, and the CBP portion of MOZ–CBP is required for the transcription activity of this fusion protein. It has been found that the interaction between MOZ–CBP and NF-κB could also have critical functions in leukemogenesis (80). MOZ–CBP inhibits p53-mediated transcription, and the impairment of MOZ/p53-induced transcription contributes to the development of leukemia (81).
The translocation t(8;22)(p11;q13) in acute myeloid leukemia generates the fusion gene MOZ–p300, and the MOZ zinc finger/HAT domain are fused to a mostly intact p300. Thus, MOZ–p300 has two HAT domains from MOZ and p300 portions, and may play an important role in the development of leukemia through deregulation of histone acetylation (82). It has been found that MOZ is fused with an unknown partner at 2p23 in one patient with tMDS. Therefore, MOZ might lead to the abnormal histone acetylation and promote the pathogenesis of myeloid malignancies (83).
The t(1;19) translocation was found in pediatric pre-B cell ALL, which leads to the fusion of E2A and PBX1 and the generation of a E2A–PBX1 fusion protein. E2A–PBX1 is able to induce the transformation of hematopoietic cells. It has been found that SPT3–TAFII31–GCN5L acetylase (STAGA) and its HAT subunit GCN5 directly binds to the E2A portion of E2A–PBX1. GCN5 can acetylate and stabilize the E2A–PBX1 fusion protein (23). AML1/MDS1/EVI1 (AME), a transcription repressor generated by translocation t(3;21) in human leukemia, binds to P/CAF and GCN5 through two binding sites, with one of the binding sites being in the Runt domain. GCN5 and P/CAF are able to acetylate AME, and either P/CAF or GCN5 can cooperate with AME to impair the repression of AML1-dependent transcriptional activation (84, 85).
The transcription factor p53 was the first non-histone substrate discovered to be acetylated by HATs (5). Levels of p53 acetylation are associated with the activation and stabilization of p53 (86–93); acetylation of p53 also stimulates its sequence-specific DNA-binding (94–97). The seven different lysine (K164, K305, K370, K372, K373, K381, and K382) in the C-terminus of p53 are acetylated by CBP and PCAF (98). Acetylation of p53 is crucial for the recruitment of CBP/p300 to the promoters of its target genes. CBP and p300 are able to promote p53-mediated transcription activation (99). TIP60/hMOF acetylates p53 at position 120 (100, 101), which can be induced by DNA damage or oncogenic stress-mediated p19ARF activation (102). Mutation of p53 lysine 120 to arginine inhibited p53-induced transcription activation. p53 can also be acetylated at one lysine outside its C-terminus, which is critical for the activation of the proapoptotic genes PUMA and BAX. In the p53 acetylation-deficient knockin mouse, the expression of p53 target genes is decreased after DNA damage (103). The deletion of the lysine residue at position 164 and other acetylation sites in p53 blocked p53-mediated transactivation of p21 and inhibition of cell growth (104). These findings suggest that p53 acetylation may play a critical role in the pathogenesis of hematological malignancies.
The FDA approved drug, Beleodaq, is used for the therapy of patients with peripheral T cell lymphomas (PTCL), an aggressive disease, which accounts for ~15% of all non-Hodgkin lymphomas. Beleodaq inhibits HDAC and it is the third drug to receive FDA approval for PTCL. In the trial that led to the FDA approval, Beleodaq’s overall response rates were comparable to those of Folotyn and Istodax; 10.8% of patients experienced a complete response and 15% had a partial response. The response rate was even higher in patients with angioimmunoblastic T cell lymphoma, which suggests that targeting acetylation is a promising therapeutic strategy for the therapy of hematological malignancies.
The HAT inhibitor, Garcinol, is derived from plants. It has been identified as a stimulator of human HSPCs expansion in the screening of natural products. During a 7-day culture of CD34+CD38− HSCs or CD34+ HSPCs, Garcinol was able to induce the expansion of HSPCs, and this ability is associated with its inhibitory effect on HATs. The derivatives of Garcinol, which can expand HSPCs, are also able to inhibit HAT activity and histone acetylation. Altogether, the Garcinol effects suggest that targeting HATs could be a promising strategy for expanding HSPCs (105).
Foxp3+ Treg cells can not only regulate immune homeostasis/autoimmunity but also limit the immune response of hosts to tumors. Thus, targeting Foxp3+ Treg cells could be a promising strategy to improve antitumor immunity. Conditional deletion or pharmacological inhibition of p300 was able to increase T cell receptor-induced apoptosis in Foxp3+ Treg cells and abrogate the suppressive functions of Treg cells. Inhibition of p300 can also impair the induction of peripheral Treg cells and tumor growth in the immunocompetent mouse model. Collectively, p300 is critical for the homeostasis and function of Foxp3+ Treg cells, and targeting p300 could be a new approach for cancer immunotherapy (54).
The HAT p300 can enhance the self-renewal ability of leukemia stem cells through acetylating AE and activating the target genes of AE, which indicated that p300 could be a promising drug target for t(8;21) leukemia. C646, a selective and competitive p300 inhibitor, can inhibit the proliferation and colony formation of t(8;21) leukemia cells, and induce apoptosis and G1 phase cell cycle arrest in t(8;21) leukemia cells and primary cells isolated from patients with t(8;21) leukemia. C646 does not significantly affect the normal HSPCs mobilized by GCSF. In particular, AML1−ETO+ AML cells are more sensitive to C646 compared with AML1−ETO− AML cells. The growth inhibition of AML1−ETO+ leukemia cells induced by C646 are associated with the decreased acetylation of histone H3 and downregulation of Bcl2/C-kit, suggesting that C646 could be a promising drug candidate for the treatment of AML1−ETO+ leukemia (4, 106).
Epigallocatechin-3-gallate (EGCG) has been found as a HAT inhibitor in natural compound screening. EGCG can block p300-mediated acetylation of p65, impairing its translocation to the nucleus, and it can upregulate the amount of IκBα in the cytoplasm, thus inhibiting NF-κB activity in several ways, and decreasing the expression of NF-κB target genes. EGCG impairs B cell transformation by EBV, perhaps via suppression of NF-κB acetylation (107), and it can inhibit the binding of p300 to the IL-6 promoter and block cytokine gene expression. Thus, EGCG could be a potential therapy for B cell malignancies.
p300 plays an important role in signal transduction pathways that promote the proliferation and survival of malignant cells. Therefore, p300 represents a promising drug target for hematological malignancies, and libraries of compounds have been screened for p300 inhibitors. One candidate, L002, inhibits p300 in vitro, with an IC50 of ~2 μM. L002 can block histone acetylation and p53 acetylation, and can inhibit the activation of STAT3. Biochemical testing of a series of related compounds revealed functional groups that may impact the inhibitory potency of L002 against p300. Interestingly, these analogs show inhibitory activities against CBP, PCAF, and GCN5, but against several other acetyltransferases (KAT5, KAT6B, and KAT7), HDACs and HMTases. Among the NCI-60 panel of cancer cell lines, leukemia, and lymphoma cell lines were extremely sensitive to L002. Thus, this new acetyltransferase inhibitor, L002, is a potential anticancer agent (108).
The different acetylation of proteins correlates with the development of BCR–ABL-positive leukemia. A derivative of anacardic acid – small molecule MG153, which is developed to have stronger HAT inhibitory ability, is a potent inhibitor of PCAF. The inhibition of PCAF decreases proliferation and induces apoptosis, which correlates with loss of the mitochondrial membrane potential and DNA fragmentation. Importantly, cells expressing BCR–ABL are more sensitive to PCAF inhibition compared to parental cells without BCR–ABL. Moreover, inhibition of PCAF in BCR–ABL-expressing cells breaks their resistance to DNA damage-induced cell death. Targeting the PCAF alone or in combination with DNA-damaging drugs shows cytotoxic effects and should be considered as a prospective therapeutic strategy in chronic myeloid leukemia (CML) cells. Moreover, anacardic acid derivative MG153 is a valuable agent and further studies validating its therapeutic relevance should be performed (109).
The a-methylene-g-butyrolactone 3 (MB-3) is a cell-permeable inhibitor against GCN5, and is able to decrease the levels of histone H3 acetylation and non-histone substrate (a-tubulin) acetylation. GCN5 acetylates E2A–PBX1, and MB-3 reduces the levels of E2A–PBX1 acetylation and E2A–PBX1 protein in a dose-dependent manner. RCH–ACV cells are derived from the bone marrow cells of a patient with pre-B cell acute lymphoid leukemia, and has t(1;19) translocation, which generates the E2A–PBX1 fusion gene. E2A–PBX1 acetylation was inhibited by MB-3, and the level of E2A–PBX1 and GCN5 protein was decreased when RCH–ACV cells were treated with MB-3. The E2A–PBX1 half-life was shorter in the cells treated with MB-3, indicating GCN5-dependent acetylation can affect the stability of E2A–PBX1 protein. A reduction in Wnt16, an E2A–PBX1 target gene were also observed in RCH–ACV cells cultured with MB-3, indicating the importance for the pathogenesis of t(1;19)-positive pre-B cell leukemia. Additionally, the expression of E2A–PBX1, E2A, and Wnt16 were significantly decreased in the primary t(1;19) pre-B ALL cells treated with MB-3. MB-3 does not affect the expression of Pol II or Tubulin, which suggests that MB-3 can destabilize certain proteins probably through the inhibition of GCN5-dependent histone or non-histone substrate acetylation. These findings indicate that GCN5 inhibitors have potential value as therapeutic agents for ALL (23). Some recently identified GCN5 inhibitors, such as (thiazol-2-yl)hydrizones (110, 111), might also be able to target hematological malignancies.
Lysine acetylation acetylation occurs not only at the histone tails but also in the non-histone proteins. LATs are catalytic subunits of multiprotein complexes, whose biochemical and molecular characterization have yielded much important information about the function and regulation of acetyltransferase activity. Importantly, HATs have the catalytic/non-catalytic and histone/non-histone effects on the hematopoietic cells, which confer HAT the ability to control a variety of cellular events in normal and malignant hematopoiesis. Genetic approaches are very useful to study how protein acetylation controls a variety of cellular events in normal and malignant hematopoiesis. The study on the gene knockout mouse models shows that p300 and CBP play distinct roles in hematopoiesis; GCN5 but not PCAF is essential in early embryonic development. Thus, such an in vivo approach will generate the new findings on the role and mechanism of LATs. One major finding in the mechanism study on the function of protein acetylation is that bromodomain can bind to the acetylated lysine. Since a lot of proteins have bromodomain, it would be interesting to understand whether all bromodomains can recognize the acetylated lysine and how bromodomains specifically recognize the acetylated lysine. MOZ, MORF, p300, and CBP are involved in the leukemia-associated chromosomal translocation, which can generate leukemogenic fusion genes. Thus, LATs have critical functions in the pathogenesis of hematological malignancies. The direct involvement of LATs in hematological malignancies indicates that compounds with the ability to regulate the activity of LATs are the potential drugs for the treatment of hematological malignancies. Thus, the studies on the function and mechanism of histone/non-histone proteins acetylation will not only shed new light on how lysine acetylation controls a variety of cellular events in normal and malignant hematopoiesis but also provide critical insights into the development of new therapeutic strategies for the therapy of hematological malignancies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by funding from an Institutional Research Grant (IRG-98-277-13) from the American Cancer Society (LW), Gabrielle’s Angel Foundation for Cancer Research (LW), the Women’s Cancer Association of the University of Miami (LW), Leukemia Research Foundation (LW), and a grant from the National Institutes of Health R01CA166835 (SN).
1. Furdas SD, Kannan S, Sippl W, Jung M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch Pharm (2012) 345(1):7–21. doi: 10.1002/ardp.201100209
2. Friedmann DR, Marmorstein R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J (2013) 280(22):5570–81. doi:10.1111/febs.12373
3. Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell (2007) 131(4):633–6. doi:10.1016/j.cell.2007.10.039
4. Yamaguchi Y, Kurokawa M, Imai Y, Izutsu K, Asai T, Ichikawa M, et al. AML1 is functionally regulated through p300-mediated acetylation on specific lysine residues. J Biol Chem (2004) 279(15):15630–8. doi:10.1074/jbc.M400355200
5. Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science (2011) 333(6043):765–9. doi:10.1126/science.1201662
6. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell (1997) 90(4):595–606. doi:10.1016/S0092-8674(00)80521-8
7. Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep (2003) 4(5):484–90. doi:10.1038/sj.embor.embor821
8. Santander VS, Bisig CG, Purro SA, Casale CH, Arce CA, Barra HS. Tubulin must be acetylated in order to form a complex with membrane Na(+), K(+)-ATPase and to inhibit its enzyme activity. Mol Cell Biochem (2006) 291(1–2):167–74. doi:10.1007/s11010-006-9212-9
9. Ivanov D, Schleiffer A, Eisenhaber F, Mechtler K, Haering CH, Nasmyth K. Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr Biol (2002) 12(4):323–8. doi:10.1016/S0960-9822(02)00681-4
10. Buerki C, Rothgiesser KM, Valovka T, Owen HR, Rehrauer H, Fey M, et al. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res (2008) 36(5):1665–80. doi:10.1093/nar/gkn003
11. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem (2001) 70:81–120. doi:10.1146/annurev.biochem.70.1.81
12. Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol (2010) 17(5):471–82. doi:10.1016/j.chembiol.2010.03.006
13. Voss AK, Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays (2009) 31(10):1050–61. doi:10.1002/bies.200900051
14. Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, et al. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell (1997) 90(3):569–80. doi:10.1016/S0092-8674(00)80516-4
15. Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature (1997) 389(6647):194–8. doi:10.1038/38304
16. Blanco-Garcia N, Asensio-Juan E, de la Cruz X, Martinez-Balbas MA. Autoacetylation regulates P/CAF nuclear localization. J Biol Chem (2009) 284(3):1343–52. doi:10.1074/jbc.M806075200
17. Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood (2011) 118(9):2443–53. doi:10.1182/blood-2011-01-331892
18. Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood (2007) 110(8):2864–71. doi:10.1182/blood-2007-01-065201
19. Bruserud O, Stapnes C, Tronstad KJ, Ryningen A, Anensen N, Gjertsen BT. Protein lysine acetylation in normal and leukaemic haematopoiesis: HDACs as possible therapeutic targets in adult AML. Expert Opin Ther Targets (2006) 10(1):51–68. doi:10.1517/14728222.10.1.51
20. Huang HT, Kathrein KL, Barton A, Gitlin Z, Huang YH, Ward TP, et al. A network of epigenetic regulators guides developmental haematopoiesis in vivo. Nat Cell Biol (2013) 15(12):1516–25. doi:10.1038/ncb2870
21. Shen H, Chen Z, Ding X, Qi X, Cen J, Wang Y, et al. BMI1 reprogrammes histone acetylation and enhances c-fos pathway via directly binding to Zmym3 in malignant myeloid progression. J Cell Mol Med (2014) 18(6):1004–17. doi:10.1111/jcmm.12246
22. Kosan C, Ginter T, Heinzel T, Kramer OH. STAT5 acetylation: mechanisms and consequences for immunological control and leukemogenesis. JAKSTAT (2013) 2(4):e26102. doi:10.4161/jkst.26102
23. Holmlund T, Lindberg MJ, Grander D, Wallberg AE. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia (2013) 27(3):578–85. doi:10.1038/leu.2012.265
24. Joosten M, Seitz V, Zimmermann K, Sommerfeld A, Berg E, Lenze D, et al. Histone acetylation and DNA demethylation of T cells result in an anaplastic large cell lymphoma-like phenotype. Haematologica (2013) 98(2):247–54. doi:10.3324/haematol.2011.054619
25. Palermo R, Checquolo S, Giovenco A, Grazioli P, Kumar V, Campese AF, et al. Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene (2012) 31(33):3807–17. doi:10.1038/onc.2011.533
26. Perez-Campo FM, Costa G, Lie-a-Ling M, Kouskoff V, Lacaud G. The MYSTerious MOZ, a histone acetyltransferase with a key role in haematopoiesis. Immunology (2013) 139(2):161–5. doi:10.1111/imm.12072
27. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov (2014) 13(5):337–56. doi:10.1038/nrd4286
28. Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem (2012) 287(51):43137–55. doi:10.1074/jbc.M112.413047
29. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell (2013) 24(6):777–90. doi:10.1016/j.ccr.2013.11.003
30. Tolani B, Gopalakrishnan R, Punj V, Matta H, Chaudhary PM. Targeting Myc in KSHV-associated primary effusion lymphoma with BET bromodomain inhibitors. Oncogene (2014) 33(22):2928–37. doi:10.1038/onc.2013.242
31. Mottok A, Gascoyne RD. Bromodomain inhibition in diffuse large B-cell lymphoma – giving MYC a brake. Clin Cancer Res (2015) 21(1):4–6. doi:10.1158/1078-0432.CCR-14-1651
32. Hnisz D, Abraham BJ, Lau A, Saint-André V, Sigova AA, Hoke HA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155:934–47. doi:10.1016/j.cell.2013.09.053
33. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153:320–34. doi:10.1016/j.cell.2013.03.036
34. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science (2014) 346(6215):1373–7. doi:10.1126/science.1259037
35. Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia (2014) 28(8):1725–35. doi:10.1038/leu.2014.70
36. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell (2014) 159(7):1524–37. doi:10.1016/j.cell.2014.11.013
37. Marushige K. Activation of chromatin by acetylation of histone side chains. Proc Natl Acad Sci U S A (1976) 73(11):3937–41. doi:10.1073/pnas.73.11.3937
38. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res (2010) 38(15):4958–69. doi:10.1093/nar/gkq244
39. Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, et al. Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood (1999) 93(9):2771–9.
40. Chan WI, Hannah RL, Dawson MA, Pridans C, Foster D, Joshi A, et al. The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol Cell Biol (2011) 31(24):5046–60. doi:10.1128/MCB.05830-11
41. Kung AL, Rebel VI, Bronson RT, Ch’ng LE, Sieff CA, Livingston DM, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev (2000) 14(3):272–7. doi:10.1101/gad.14.3.272
42. Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci U S A (2002) 99(23):14789–94. doi:10.1073/pnas.232568499
43. Thomas T, Corcoran LM, Gugasyan R, Dixon MP, Brodnicki T, Nutt SL, et al. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev (2006) 20(9):1175–86. doi:10.1101/gad.1382606
44. Perez-Campo FM, Borrow J, Kouskoff V, Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood (2009) 113(20):4866–74. doi:10.1182/blood-2008-04-152017
45. Perez-Campo FM, Costa G, Lie ALM, Stifani S, Kouskoff V, Lacaud G. MOZ-mediated repression of p16 is critical for the self-renewal of neural and hematopoietic stem cells. Stem Cells (2014) 32(6):1591–601. doi:10.1002/stem.1606
46. Kimbrel EA, Lemieux ME, Xia X, Davis TN, Rebel VI, Kung AL. Systematic in vivo structure-function analysis of p300 in hematopoiesis. Blood (2009) 114(23):4804–12. doi:10.1182/blood-2009-04-217794
47. Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, et al. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature (2002) 419(6908):738–43. doi:10.1038/nature01062
48. Kasper LH, Fukuyama T, Lerach S, Chang Y, Xu W, Wu S, et al. Genetic interaction between mutations in c-Myb and the KIX domains of CBP and p300 affects multiple blood cell lineages and influences both gene activation and repression. PLoS One (2013) 8(12):e82684. doi:10.1371/journal.pone.0082684
49. Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang HY, et al. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell (2009) 33(2):248–56. doi:10.1016/j.molcel.2008.12.016
50. Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, et al. HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol Cell (2009) 33(2):257–65. doi:10.1016/j.molcel.2009.01.007
51. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi:10.1182/blood-2009-02-207118
52. Du T, Nagai Y, Xiao Y, Greene MI, Zhang H. Lysosome-dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp Mol Pathol (2013) 95(1):38–45. doi:10.1016/j.yexmp.2013.04.003
53. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
54. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med (2013) 19(9):1173–7. doi:10.1038/nm.3286
55. Kikuchi H, Kuribayashi F, Takami Y, Imajoh-Ohmi S, Nakayama T. GCN5 regulates the activation of PI3K/Akt survival pathway in B cells exposed to oxidative stress via controlling gene expressions of Syk and Btk. Biochem Biophys Res Commun (2011) 405(4):657–61. doi:10.1016/j.bbrc.2011.01.088
56. Pattabiraman DR, Sun J, Dowhan DH, Ishii S, Gonda TJ. Mutations in multiple domains of c-Myb disrupt interaction with CBP/p300 and abrogate myeloid transforming ability. Mol Cancer Res (2009) 7(9):1477–86. doi:10.1158/1541-7786.MCR-09-0070
57. Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res (2011) 39(11):4664–79. doi:10.1093/nar/gkr024
58. Pattabiraman DR, McGirr C, Shakhbazov K, Barbier V, Krishnan K, Mukhopadhyay P, et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood (2014) 123(17):2682–90. doi:10.1182/blood-2012-02-413187
59. Taki T, Sako M, Tsuchida M, Hayashi Y. The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood (1997) 89(11):3945–50.
60. Santillan DA, Theisler CM, Ryan AS, Popovic R, Stuart T, Zhou MM, et al. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer Res (2006) 66(20):10032–9. doi:10.1158/0008-5472.CAN-06-2597
61. Sun XJ, Wang Z, Wang L, Jiang Y, Kost N, Soong TD, et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature (2013) 500(7460):93–7. doi:10.1038/nature12287
62. Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature (2007) 448(7157):1063–7. doi:10.1038/nature06055
63. Zhao H, Jin S, Gewirtz AM. The histone acetyltransferase TIP60 interacts with c-Myb and inactivates its transcriptional activity in human leukemia. J Biol Chem (2012) 287(2):925–34. doi:10.1074/jbc.M111.279950
64. Carapeti M, Aguiar RC, Goldman JM, Cross NC. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood (1998) 91(9):3127–33.
65. Liang J, Prouty L, Williams BJ, Dayton MA, Blanchard KL. Acute mixed lineage leukemia with an inv(8)(p11q13) resulting in fusion of the genes for MOZ and TIF2. Blood (1998) 92(6):2118–22.
66. Carapeti M, Aguiar RC, Watmore AE, Goldman JM, Cross NC. Consistent fusion of MOZ and TIF2 in AML with inv(8)(p11q13). Cancer Genet Cytogenet (1999) 113(1):70–2. doi:10.1016/S0165-4608(99)00007-2
67. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell (2004) 6(6):587–96. doi:10.1016/j.ccr.2004.10.015
68. Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell (2003) 3(3):259–71. doi:10.1016/S1535-6108(03)00051-5
69. Kindle KB, Troke PJ, Collins HM, Matsuda S, Bossi D, Bellodi C, et al. MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol Cell Biol (2005) 25(3):988–1002. doi:10.1128/MCB.25.3.988-1002.2005
70. Collins HM, Kindle KB, Matsuda S, Ryan C, Troke PJ, Kalkhoven E, et al. MOZ-TIF2 alters cofactor recruitment and histone modification at the RARbeta2 promoter: differential effects of MOZ fusion proteins on CBP- and MOZ-dependent activators. J Biol Chem (2006) 281(25):17124–33. doi:10.1074/jbc.M602633200
71. Yin H, Glass J, Blanchard KL. MOZ-TIF2 repression of nuclear receptor-mediated transcription requires multiple domains in MOZ and in the CID domain of TIF2. Mol Cancer (2007) 6:51. doi:10.1186/1476-4598-6-51
72. Zhuravleva J, Paggetti J, Martin L, Hammann A, Solary E, Bastie JN, et al. MOZ/TIF2-induced acute myeloid leukaemia in transgenic fish. Br J Haematol (2008) 143(3):378–82. doi:10.1111/j.1365-2141.2008.07362.x
73. Aikawa Y, Katsumoto T, Zhang P, Shima H, Shino M, Terui K, et al. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat Med (2010) 16(5):580–5. doi:10.1038/nm.2122
74. Tam WF, Hahnel PS, Schuler A, Lee BH, Okabe R, Zhu N, et al. STAT5 is crucial to maintain leukemic stem cells in acute myelogenous leukemias induced by MOZ-TIF2. Cancer Res (2013) 73(1):373–84. doi:10.1158/0008-5472.CAN-12-0255
75. Shima H, Yamagata K, Aikawa Y, Shino M, Koseki H, Shimada H, et al. Bromodomain-PHD finger protein 1 is critical for leukemogenesis associated with MOZ-TIF2 fusion. Int J Hematol (2014) 99(1):21–31. doi:10.1007/s12185-013-1466-x
76. Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet (1996) 14(1):33–41. doi:10.1038/ng0996-33
77. Panagopoulos I, Isaksson M, Lindvall C, Bjorkholm M, Ahlgren T, Fioretos T, et al. RT-PCR analysis of the MOZ-CBP and CBP-MOZ chimeric transcripts in acute myeloid leukemias with t(8;16)(p11;p13). Genes Chromosomes Cancer (2000) 28(4):415–24. doi:10.1002/1098-2264(200008)28:4<415::AID-GCC7>3.0.CO;2-I
78. Rozman M, Camos M, Colomer D, Villamor N, Esteve J, Costa D, et al. Type I MOZ/CBP (MYST3/CREBBP) is the most common chimeric transcript in acute myeloid leukemia with t(8;16)(p11;p13) translocation. Genes Chromosomes Cancer (2004) 40(2):140–5. doi:10.1002/gcc.20022
79. Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A, Ohki M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J (2001) 20(24):7184–96. doi:10.1093/emboj/20.24.7184
80. Chan EM, Chan RJ, Comer EM, Goulet RJ III, Crean CD, Brown ZD, et al. MOZ and MOZ-CBP cooperate with NF-kappaB to activate transcription from NF-kappaB-dependent promoters. Exp Hematol (2007) 35(12):1782–92. doi:10.1016/j.exphem.2007.07.015
81. Rokudai S, Aikawa Y, Tagata Y, Tsuchida N, Taya Y, Kitabayashi I. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J Biol Chem (2009) 284(1):237–44. doi:10.1074/jbc.M805101200
82. Kitabayashi I, Aikawa Y, Yokoyama A, Hosoda F, Nagai M, Kakazu N, et al. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia (2001) 15(1):89–94. doi:10.1038/sj.leu.2401983
83. Imamura T, Kakazu N, Hibi S, Morimoto A, Fukushima Y, Ijuin I, et al. Rearrangement of the MOZ gene in pediatric therapy-related myelodysplastic syndrome with a novel chromosomal translocation t(2;8)(p23;p11). Genes Chromosomes Cancer (2003) 36(4):413–9. doi:10.1002/gcc.10172
84. Senyuk V, Sinha KK, Chakraborty S, Buonamici S, Nucifora G. P/CAF and GCN5 acetylate the AML1/MDS1/EVI1 fusion oncoprotein. Biochem Biophys Res Commun (2003) 307(4):980–6. doi:10.1016/S0006-291X(03)01288-9
85. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res (2004) 32(3):959–76. doi:10.1093/nar/gkh252
86. Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature (2000) 408(6810):377–81. doi:10.1038/35042612
87. Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol (2000) 20(22):8458–67. doi:10.1128/MCB.20.22.8458-8467.2000
88. Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell (2001) 8(6):1243–54. doi:10.1016/S1097-2765(01)00414-2
89. Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J (2001) 20(6):1331–40. doi:10.1093/emboj/20.6.1331
90. Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol (2006) 173(4):533–44. doi:10.1083/jcb.200512059
91. Li AG, Piluso LG, Cai X, Gadd BJ, Ladurner AG, Liu X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol Cell (2007) 28(3):408–21. doi:10.1016/j.molcel.2007.09.006
92. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature (2008) 451(7178):583–6. doi:10.1038/nature06500
93. Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature (2008) 451(7178):587–90. doi:10.1038/nature06515
94. Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol (1999) 19(2):1202–9.
95. Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev (1998) 12(18):2831–41. doi:10.1101/gad.12.18.2831
96. Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A (2004) 101(8):2259–64. doi:10.1073/pnas.0308762101
97. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol (2006) 26(7):2782–90. doi:10.1128/MCB.26.7.2782-2790.2006
98. Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem (2003) 278(28):25568–76. doi:10.1074/jbc.M212574200
99. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev (2000) 14(13):1553–77. doi:10.1101/gad.14.13.1553
100. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell (2006) 24(6):841–51. doi:10.1016/j.molcel.2006.11.026
101. Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell (2006) 24(6):827–39. doi:10.1016/j.molcel.2006.11.021
102. Mellert H, Sykes SM, Murphy ME, McMahon SB. The ARF/oncogene pathway activates p53 acetylation within the DNA binding domain. Cell Cycle (2007) 6(11):1304–6. doi:10.4161/cc.6.11.4343
103. Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol (2005) 25(13):5389–95. doi:10.1128/MCB.25.13.5389-5395.2005
104. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell (2008) 133(4):612–26. doi:10.1016/j.cell.2008.06.005
105. Nishino T, Wang C, Mochizuki-Kashio M, Osawa M, Nakauchi H, Iwama A. Ex vivo expansion of human hematopoietic stem cells by garcinol, a potent inhibitor of histone acetyltransferase. PLoS One (2011) 6(9):e24298. doi:10.1371/journal.pone.0024298
106. Gao XN, Lin J, Ning QY, Gao L, Yao YS, Zhou JH, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One (2013) 8(2):e55481. doi:10.1371/journal.pone.0055481
107. Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res (2009) 69(2):583–92. doi:10.1158/0008-5472.CAN-08-2442
108. Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol Cancer Ther (2013) 12(5):610–20. doi:10.1158/1535-7163.MCT-12-0930
109. Kusio-Kobialka M, Dudka-Ruszkowska W, Ghizzoni M, Dekker FJ, Piwocka K. Inhibition of PCAF by anacardic acid derivative leads to apoptosis and breaks resistance to DNA damage in BCR-ABL-expressing cells. Anticancer Agents Med Chem (2013) 13(5):762–7. doi:10.2174/1871520611313050010
110. Secci D, Carradori S, Bizzarri B, Bolasco A, Ballario P, Patramani Z, et al. Synthesis of a novel series of thiazole-based histone acetyltransferase inhibitors. Bioorg Med Chem (2014) 22(5):1680–9. doi:10.1016/j.bmc.2014.01.022
Keywords: histone acetyltransferases, hematopoiesis, transcriptional regulation, hematopoietic stem cells, hematological malignancies
Citation: Sun X-J, Man N, Tan Y, Nimer SD and Wang L (2015) The role of histone acetyltransferases in normal and malignant hematopoiesis. Front. Oncol. 5:108. doi: 10.3389/fonc.2015.00108
Received: 27 February 2015; Accepted: 25 April 2015;
Published: 26 May 2015
Edited by:
Wen Zhou, Columbia University, USAReviewed by:
Hao Jiang, University of Alabama at Birmingham, USACopyright: © 2015 Sun, Man, Tan, Nimer and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Wang, Department of Biochemistry and Molecular Biology, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, 1501 NW 10th Avenue, Biochemical Research Building, 701 Miami, FL 33136, USA,bC53YW5nQG1lZC5taWFtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.