 Toru Furukawa
Toru Furukawa- Institute for Integrated Medical Sciences, Tokyo Women’s Medical University, Tokyo, Japan
Pancreatic cancer is characterized by constitutive activation of the mitogen-activated protein kinase (MAPK) pathway. Mutations of KRAS or BRAF and epigenetic abrogation of DUSP6 contribute synergistically to the constitutive activation of MAPK. Active MAPK induces the expression of a variety of genes that are thought to play roles in malignant phenotypes of pancreatic cancer. By blocking the functions of such induced genes, it is possible to attenuate the malignant phenotypes. The development of drugs targeting genes downstream of MAPK may provide a novel therapeutic option for pancreatic cancer.
Introduction
Pancreatic cancer is one of the leading causes of cancer mortality in Japan and many western countries (1, 2). Despite improvements in diagnostic and therapeutic modalities, the 5-year survival rate of patients with pancreatic cancer is still <10% (3). This poor prognosis elicits a pressing need for the development of effective diagnostic and therapeutic measures to improve patient survival. An understanding of the molecular pathobiology involved may provide clues that ultimately lead to the fulfillment of such needs. Pancreatic cancer is characterized by constitutive activation of the mitogen-activated protein kinase (MAPK) pathway. The purpose of this review is to explore the mechanisms and relevance of MAPK activation in the pathobiology of pancreatic cancer.
RAS–MAPK Pathway
The vast majority of pancreatic cancers involve gain-of-function mutations in KRAS (4). KRAS encodes RAS, a guanosine triphosphate (GTP)-binding protein that, when activated by binding with GTP, triggers stimulation of various downstream signaling pathways (5). The activation of RAS is mediated by a guanine nucleotide exchange factor, which is activated by upstream ligand-binding receptor tyrosine kinases (5). Active RAS is self-inactivated by intrinsic GTPase activity, with the help of a GTPase-activating protein (5). Mutations of KRAS observed in pancreatic cancers often occur in codons 12, 13, and 61, at most are G12D or G12R substitutions, which attenuates the intrinsic GTPase activity, thus, such mutations result in prolonged activation of RAS (6). Moreover, some pancreatic cancers harbor activating mutations of BRAF rather than KRAS (7). BRAF encodes B-RAF, a serine/threonine kinase belonging to a family of mitogen-activated protein kinase kinase kinases (MAP3Ks). Mutations in BRAF commonly involve codon 600 and often result in a V600E substitution, which leads to constitutive activation of its kinase function (8). Mutations of KRAS and BRAF are mutually exclusive in pancreatic cancers, which suggest that the activating mutations of these genes can compensate for each other in pancreatic cancer phenotypes (7). B-RAF is an immediately downstream of RAS and activates MAP2K1/MEK, which in turn activates MAPK1/ERK2. Thus, the activating signal is passed on by a chain of kinase reactions, which forms MAPK signaling pathway. Hence, activating mutations of KRAS and BRAF ultimately result in activation of the MAPK signaling pathway, which is crucial for pancreatic cancer (Figure 1). MAPKs are classified into three classes according to their distinctive effectors, ERKs, JNKs, and p38MAPKs (9). Each class of MAPK is involved in different functions, i.e., ERKs are involved mainly in mitosis and proliferation, JNKs in apoptosis and differentiation, and p38MAPKs in stress responses. Activated MAPKs translocate into the nucleus and phosphorylate a variety of transcription factors, altering the expression of various genes, and therefore enacting specific cellular responses (10).
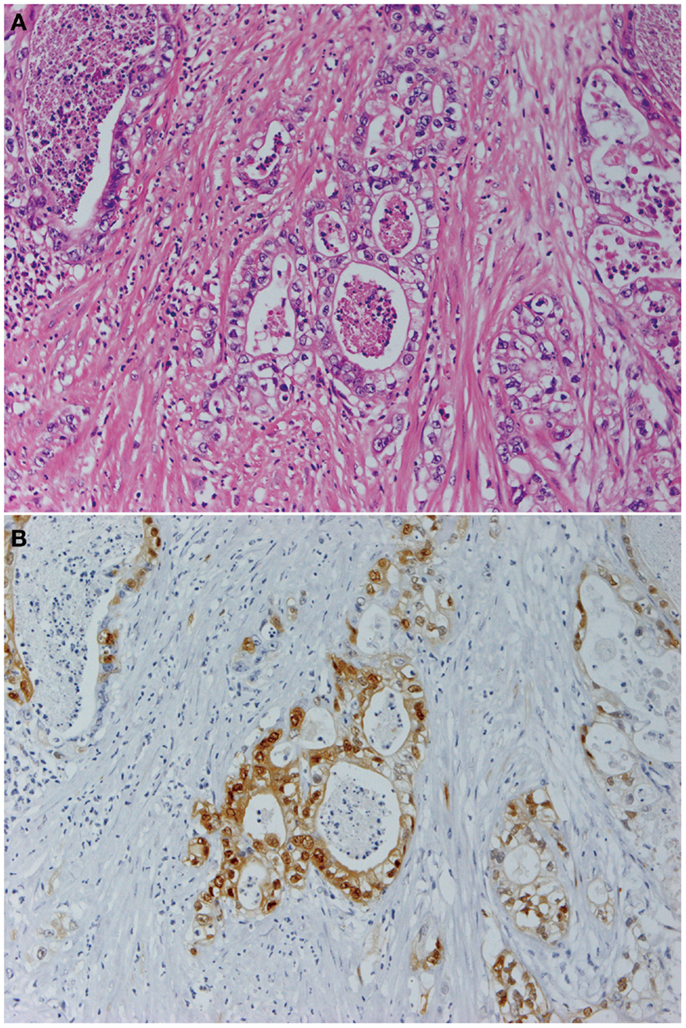
Figure 1. Pancreatic cancer produces constitutively active MAPK. Panels show histological images of invasive ductal adenocarcinoma (A) and its expression of phosphorylated MAPK (B). Staining with hematoxylin and eosin (A) and indirect immunohistochemistry using anti-phosphorylated MAPK antibody (137F5, 1:250; Cell Signaling Technology Inc., Danvers, MA, USA) and a Histofine SAB-PO kit (Nichirei Corp., Tokyo, Japan) according to the manufacturer’s instructions (B). Original magnification was ×200.
Dual-Specificity Protein Phosphatases
Dual-specificity protein phosphatases dephosphorylate phosphorylated serine, threonine, or tyrosine residues of substrates. Dual-specificity protein phosphatases (DUSPs) share a common structure that comprises a carboxyl-terminal catalytic domain and an amino-terminal non-catalytic domain. The catalytic domain contains a sequence homologous to the prototypic VH-1 DUSP encoded by the vaccinia virus (11, 12). The non-catalytic domain contains both a conserved cluster of basic amino acid residues involved in specific recognition of MAPKs, known as the kinase interaction motif, and sequences that determine the subcellular localization of these enzymes (12, 13). MAPKs are prime substrates for DUSPs, i.e., DUSPs primarily dephosphorylate target residues in MAPKs and induce their inactivation. DUSPs are classified into three classes by their subcellular localization and substrate specificities (12). The first class includes DUSP1, DUSP2, DUSP4, and DUSP5, which act in the nucleus and have broad specificities toward various MAPKs. The second class includes DUSP6, DUSP7, and DUSP9, which act in the cytoplasm and strictly on MAPK1/ERK2 (13). The last class includes DUSP8, DUSP10, and DUSP16, which act mainly on MAPK8/JNK1 and MAPK14/p38. DUSPs and MAPKs form feedback loops, each controlling the activities of the other.
DUSP6 in Pancreatic Cancer
Owing to its uniquely strict specificity toward MAPK1/ERK2 whose activation is involved in oncogenesis, an association between DUSP6 and cancer has been a prime focus of study. DUSP6 resides on the chromosome locus 12q21–22, which is commonly deleted in pancreatic cancer (14). In studies of genetic and epigenetic alterations of DUSP6 in pancreatic cancer, mutations of this gene have not been observed, however, marked reduction of its expression has been found (15). This reduced expression is associated with constitutive activation of MAPK1/ERK2 and, despite the presence of mutated KRAS, exogenous overexpression of DUSP6 induces dephosphorylation of MAPK1/ERK2, subsequent suppression of proliferation, and eventual apoptosis of pancreatic cancer cells (16). This indicates that, for their proliferation and survival, pancreatic cancer cells are largely dependent on the activation of MAPK induced as a consequence of the synergistic effect of mutations in KRAS and an abrogation of DUSP6.
While the expression of DUSP6 is markedly reduced in pancreatic cancer cells, the expression of DUSP6 can be restored by treatment with 5-azacytidine, an inhibitor of DNA methyltransferase, which indicates that the reduced expression stems from hypermethylation. A search for possible genomic regions of hypermethylation pertaining to the reduced expression of DUSP6 resulted in the discovery of a cluster of methylated CpG in intron 1 of DUSP6 gene (17). A promoter assay indeed showed promoter activity of the intron 1 (18). Moreover, this activity is shown to be positively associated with activity of MAPK1/ERK2. In other words, active MAPK1/ERK2 induces upregulation of the promoter activity of DUSP6, resulting in an increase in its expression. The increased expression of DUSP6 leads to inactivation of MAPK1/ERK2, hence, which indicates that a negative feedback loop is formed between these two molecules (18).
Pancreatic cancer is hypothesized to develop through multistep progression of dysplastic grades of epithelial cells in the pancreatic duct along with accumulation of genetic abnormalities, which is known as the progression model of pancreatic cancer (19). In this progression model, low-grade dysplasia, termed low-grade pancreatic intraepithelial neoplasia (PanIN), is thought to progress into high-grade dysplasia, or high-grade PanIN, and eventually into invasive carcinoma. The progression is associated with an accumulation of genetic aberrations in KRAS, CDKN2A, SMAD4, and TP53 (19, 20). Therefore, PanIN is regarded as a precursor lesion for pancreatic cancer. The expression of DUSP6 is increased in PanINs while it is decreased in invasive carcinoma, which indicates that DUSP6 may be a gatekeeper preventing progression of the precursor lesion into a fully invasive cancer (21).
DUSP6 is also known to acquire post-translational control of its expression. DUSP6 is degraded by the ubiquitin–proteasome system. For such degradation to take place, serines 159 and 197 of DUSP6 need to be phosphorylated by MAPK1/ERK2 (22). This phosphorylation event is regarded as a positive feedback mechanism for the activation of MAPK1/ERK2 (22). The reduced expression of DUSP6 in cancer cells might be due to the acceleration of this degradation system, which is indeed the case in ovarian cancer cells. The accelerated degradation of DUSP6 in these cells is associated with reactive oxygen species, therefore, blockage of which by an antioxidant increases DUSP6 expression, dephosphorylation of MAPK1/ERK2, and downregulation of cell proliferation (23).
Effects of MAPK Activation
Phosphorylated MAPK1/ERK2 translocates into the nucleus, phosphorylates various transcription factors, and induces expression of downstream genes that are thought to play significant roles in malignant phenotypes of pancreatic cancer (10, 24). To identify such downstream genes, a comparison of the gene expression profiles of pancreatic cancer cells with and without exogenous expression of DUSP6, which dephosphorylates MAPK, was carried out (24). The comparison revealed numerous differentially expressed genes involved in the cell cycle and mitosis, DNA replication, receptor signaling, and transport. These included AURKA, AURKB, TPX2, CENPA, EPHA2, CCNB2, IFITM1, RARRES3, SOX4, and PYCARD (24).
AURKA encodes aurora kinase A, a kinase that phosphorylates molecules involved in spindle formation during mitosis (25). Promoter activity of AURKA is positively associated with MAPK activity, i.e., active MAPK upregulates the promoter activity and induces the expression of AURKA (24). The promoter region of AURKA harbors binding sites for ETS2, a substrate of MAPK (26). Knockdown of ETS2 induces downregulation of AURKA (24). These results indicate that active MAPK induces the expression of AURKA via ETS2. Aurora kinase A is overexpressed in the majority of pancreatic cancers, which is consistent with the observation that most pancreatic cancers produce constitutively active MAPK. Knockdown of AURKA in pancreatic cancer cells results in cell cycle arrest at G2/M and hence, suppression of cellular proliferation (27). Moreover, the knockdown of AURKA enhances the effect of chemotherapy with taxane (27).
To identify genes with an important role in cellular proliferation from among those downstream of MAPK, a knockdown screen was conducted, testing the effect of RNA interference targeting the downstream genes on in vitro proliferation of pancreatic cancer cells (28). The results of this study indicated that AURKB, CENPA, EBNA1BP2, GOLT1A, KIF11, NEDD4L, SON, TPX2, and WDR5 play a crucial role in in vitro proliferation, similarly to AURKA (28). Among them, SON, a gene encoding SON, which is involved in nuclear speckle organization and RNA processing, was the most significant target for suppression of proliferation (28). SON is required for efficient RNA processing of transcripts involved in cell cycle progression (29, 30). Knockdown of SON in cultured pancreatic cancer cells caused G2/M arrest and apoptosis (28). By using a subcutaneous xenograft model in immunocompromised mice, pancreatic cancer cells with knockdown of SON resulted in a significant reduction in in vivo tumorigenesis (28). Moreover, SON is overexpressed in ductal adenocarcinoma of the pancreas, while its expression is lower in PanIN and normal ductal cells (28). The suppressive effect of SON knockdown on in vitro proliferation is unremarkable in normal ductal cells (28). These results indicate that pancreatic cancer cells may depend on SON to maintain their active proliferation and tumorigenicity (28). Effectiveness of these targetable downstream genes for treatment of various cancers has been also reported recently (31–37).
MicroRNAs are endogenous short non-coding RNAs that interact with protein-coding messenger RNAs and regulate translation (38, 39). MiRNAs play important roles in physiological development as well as in pathological conditions, including cancer (40). They are aberrantly expressed in many cancers including pancreatic cancer. Such aberrant expression is thought to be primarily associated with an altered copy number of genomic loci coding for miRNAs. However, signal transduction pathways can also alter the expression of miRNAs, some of which therefore may be affected by active MAPKs in pancreatic cancer. The association between miRNAs and MAPK in pancreatic cancer cells was investigated by comparing miRNA expression in cells with active or attenuated MAPK (41). The results indicated that miR-7, miR-34a, miR-193b, and miR-181d were preferentially associated with MAPK activity, of which, miR-7 was upregulated, while miR-34a, miR-193b, and miR-181d were downregulated. MiR-7 is known to target EGFR, a gene upstream of MAPK. The upregulation of miR-7 by MAPK may induce downregulation of EGFR and subsequent downregulation of MAPK, thus forming a feedback loop. MiR-34a is a highly conserved miRNA and is known to be upregulated by tumor protein 53 (TP53), a tumor suppressor (42, 43). MAPK downregulates miR-34a, therefore, MAPK is thought to counteract TP53’s role in controlling the expression of this miRNA, and can attenuate TP53’s tumor suppressive function. Expression of miR-193b is lowered in various cancers, and its overexpression induces suppression of proliferation of cancer cells, including those of pancreatic cancer. Targets of miR-193b have been shown to be CCND1, NT5E, PLAU, STARD7, STMN1, and YWHAZ, which are involved in the cell cycle, conversion of adenosine monophosphate to adenosine, urokinase-type plasminogen activation, lipid binding, cell cycle progression, and signal transduction, respectively (41). Some of these genes are shown to promote proliferation of pancreatic cancer cells, such that suppression of miR-193b by MAPK would result in accelerated proliferation. Associations between MAPK and miRNAs and their relevance for diseases have been also reported (44–47).
Conclusion and Perspectives
The combination of KRAS mutation and DUSP6 abrogation plays a synergistic role in the constitutive activation of MAPK in pancreatic cancer. Active MAPK induces a variety of genes, including miRNAs involved in malignant cancer phenotypes. By blocking the functions of such induced genes, it is possible to attenuate malignant phenotypes (Figure 2). The development of drugs targeting genes downstream of MAPK may provide a novel therapeutic option for pancreatic cancer.
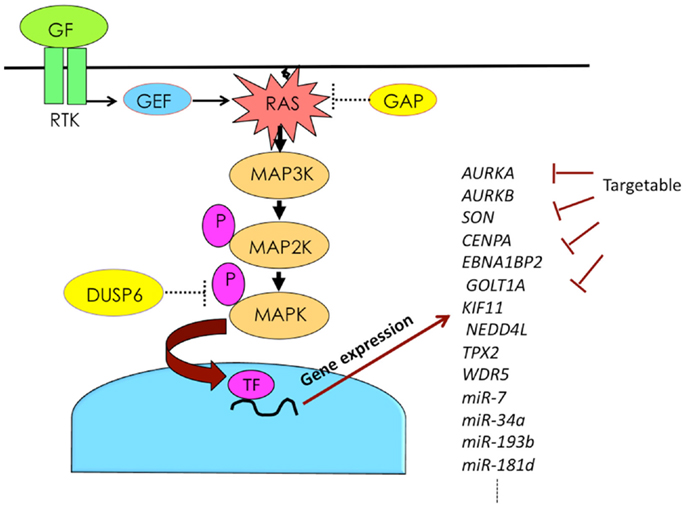
Figure 2. Activation of RAS–MAPK pathway induces expression of numerous genes, some of which can be targeted. Abbreviations are GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GF, growth factor; P, phosphorylation; RTK, receptor tyrosine kinase; and TF, transcription factor.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Matsuda A, Matsuda T, Shibata A, Katanoda K, Sobue T, Nishimoto H. Cancer incidence and incidence rates in Japan in 2008: a study of 25 population-based cancer registries for the monitoring of cancer incidence in Japan (MCIJ) project. Jpn J Clin Oncol (2014) 44(4):388–96. doi:10.1093/jjco/hyu003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin (2009) 59(4):225–49. doi:10.3322/caac.20006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Berrino F, De Angelis R, Sant M, Rosso S, Bielska-Lasota M, Coebergh JW, et al. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995-99: results of the EUROCARE-4 study. Lancet Oncol (2007) 8(9):773–83. doi:10.1016/S1470-2045(07)70245-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell (1988) 53(4):549–54. doi:10.1016/0092-8674(88)90571-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Thatcher JD. The Ras-MAPK signal transduction pathway. Sci Signal (2010) 3(119):tr1. doi:10.1126/scisignal.3119tr1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci U S A (1984) 81(18):5704–8. doi:10.1073/pnas.81.18.5704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol (2003) 163(4):1255–60. doi:10.1016/S0002-9440(10)63485-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417(6892):949–54. doi:10.1038/nature00766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science (2002) 298(5600):1911–2. doi:10.1126/science.1072682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J (1999) 18(3):664–74. doi:10.1093/emboj/18.3.664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Stewart AE, Dowd S, Keyse SM, McDonald NQ. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. Nat Struct Biol (1999) 6(2):174–81. doi:10.1038/5861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev (2008) 27(2):253–61. doi:10.1007/s10555-008-9123-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, Dhalluin C, et al. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell (2001) 7(2):387–99. doi:10.1016/S1097-2765(01)00186-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Kimura M, Furukawa T, Abe T, Yatsuoka T, Youssef EM, Yokoyama T, et al. Identification of two common regions of allelic loss in chromosome arm 12q in human pancreatic cancer. Cancer Res (1998) 58(11):2456–60.
15. Furukawa T, Yatsuoka T, Youssef EM, Abe T, Yokoyama T, Fukushige S, et al. Genomic analysis of DUSP6, a dual specificity MAP kinase phosphatase, in pancreatic cancer. Cytogenet Cell Genet (1998) 82(3–4):156–9. doi:10.1159/000015091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol (2003) 162(6):1807–15. doi:10.1016/S0002-9440(10)64315-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Xu S, Furukawa T, Kanai N, Sunamura M, Horii A. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J Hum Genet (2005) 50(4):159–67. doi:10.1007/s10038-005-0235-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Furukawa T, Tanji E, Xu S, Horii A. Feedback regulation of DUSP6 transcription responding to MAPK1 via ETS2 in human cells. Biochem Biophys Res Commun (2008) 377(1):317–20. doi:10.1016/j.bbrc.2008.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res (2000) 6(8):2969–72.
20. Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol (2001) 25(5):579–86. doi:10.1097/00000478-200105000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Furukawa T, Fujisaki R, Yoshida Y, Kanai N, Sunamura M, Abe T, et al. Distinct progression pathways involving the dysfunction of DUSP6/MKP-3 in pancreatic intraepithelial neoplasia and intraductal papillary-mucinous neoplasms of the pancreas. Mod Pathol (2005) 18(8):1034–42. doi:10.1038/modpathol.3800383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Marchetti S, Gimond C, Chambard JC, Touboul T, Roux D, Pouyssegur J, et al. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol Cell Biol (2005) 25(2):854–64. doi:10.1128/MCB.25.2.854-864.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Chan DW, Liu VW, Tsao GS, Yao KM, Furukawa T, Chan KK, et al. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis (2008) 29(9):1742–50. doi:10.1093/carcin/bgn167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene (2006) 25(35):4831–9. doi:10.1038/sj.onc.1209494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet (1998) 20(2):189–93. doi:10.1038/2496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Foulds CE, Nelson ML, Blaszczak AG, Graves BJ. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol (2004) 24(24):10954–64. doi:10.1128/MCB.24.24.10954-10964.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res (2005) 65(7):2899–905. doi:10.1158/0008-5472.CAN-04-3981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Furukawa T, Tanji E, Kuboki Y, Hatori T, Yamamoto M, Shimizu K, et al. Targeting of MAPK-associated molecules identifies SON as a prime target to attenuate the proliferation and tumorigenicity of pancreatic cancer cells. Mol Cancer (2012) 11:88. doi:10.1186/1476-4598-11-88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A, et al. SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol Cell (2011) 42(2):185–98. doi:10.1016/j.molcel.2011.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sharma A, Takata H, Shibahara K, Bubulya A, Bubulya PA. Son is essential for nuclear speckle organization and cell cycle progression. Mol Biol Cell (2010) 21(4):650–63. doi:10.1091/mbc.E09-02-0126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Teoh PL, Sharrocks AD. WDR5, ASH2L, and RBBP5 control the efficiency of FOS transcript processing. Cell Mol Biol Lett (2014) 19(2):215–32. doi:10.2478/s11658-014-0190-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Boeckx C, Op de Beeck K, Wouters A, Deschoolmeester V, Limame R, Zwaenepoel K, et al. Overcoming cetuximab resistance in HNSCC: the role of AURKB and DUSP proteins. Cancer Lett (2014) 354(2):365–77. doi:10.1016/j.canlet.2014.08.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Zhang C, Han Y, Huang H, Min L, Qu L, Shou C. Integrated analysis of expression profiling data identifies three genes in correlation with poor prognosis of triple-negative breast cancer. Int J Oncol (2014) 44(6):2025–33. doi:10.3892/ijo.2014.2352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Liao P, Wang W, Shen M, Pan W, Zhang K, Wang R, et al. A positive feedback loop between EBP2 and c-Myc regulates rDNA transcription, cell proliferation, and tumorigenesis. Cell Death Dis (2014) 5:e1032. doi:10.1038/cddis.2013.536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Wang F, Lin SL. Knockdown of kinesin KIF11 abrogates directed migration in response to epidermal growth factor-mediated chemotaxis. Biochem Biophys Res Commun (2014) 452(3):642–8. doi:10.1016/j.bbrc.2014.08.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Huerta M, Fernandez-Marquez J, Cabello JL, Medrano A, Querol E, Cedano J. Analysis of gene expression for studying tumor progression: the case of glucocorticoid administration. Gene (2014) 549(1):33–40. doi:10.1016/j.gene.2014.07.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Liu HC, Zhang GH, Liu YH, Wang P, Ma JF, Su LS, et al. TPX2 siRNA regulates growth and invasion of esophageal cancer cells. Biomed Pharmacother (2014) 68(7):833–9. doi:10.1016/j.biopha.2014.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science (2001) 294(5543):853–8. doi:10.1126/science.1064921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature (2010) 466(7308):835–40. doi:10.1038/nature09267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Visone R, Croce CM. MiRNAs and cancer. Am J Pathol (2009) 174(4):1131–8. doi:10.2353/ajpath.2009.080794
41. Ikeda Y, Tanji E, Makino N, Kawata S, Furukawa T. MicroRNAs associated with mitogen-activated protein kinase in human pancreatic cancer. Mol Cancer Res (2012) 10(2):259–69. doi:10.1158/1541-7786.MCR-11-0035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature (2007) 447(7148):1130–4. doi:10.1038/nature05939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell (2007) 26(5):745–52. doi:10.1016/j.molcel.2007.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Tiedje C, Holtmann H, Gaestel M. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J Interferon Cytokine Res (2014) 34(4):220–32. doi:10.1089/jir.2013.0146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Li J, Kong F, Wu K, Song K, He J, Sun W. miR-193b directly targets STMN1 and uPA genes and suppresses tumor growth and metastasis in pancreatic cancer. Mol Med Rep (2014) 10(5):2613–20. doi:10.3892/mmr.2014.2558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Liu SM, Lu J, Lee HC, Chung FH, Ma N. miR-524-5p suppresses the growth of oncogenic BRAF melanoma by targeting BRAF and ERK2. Oncotarget (2014) 5(19):9444–59.
Keywords: pancreatic cancer, mitogen-activated protein kinase, KRAS, BRAF, DUSP6, ERK, MKP-3, microRNA
Citation: Furukawa T (2015) Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front. Oncol. 5:23. doi: 10.3389/fonc.2015.00023
Received: 19 November 2014; Paper pending published: 25 December 2014;
Accepted: 21 January 2015; Published online: 04 February 2015.
Edited by:
Kensuke Adachi, Tokyo Metropolitan Tama Medical Center, JapanReviewed by:
Robert Klein, Icahn School of Medicine at Mount Sinai, USAYong Zhang, AntiCancer Inc., USA
Copyright: © 2015 Furukawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toru Furukawa, Institute for Integrated Medical Sciences, Tokyo Women’s Medical University, 8-1 Kawadacho, Shinjuku, Tokyo 162-8666, Japan e-mail:ZnVydWthd2EudG9ydUB0d211LmFjLmpw