 Emily Padfield
Emily Padfield Hayley P. Ellis
Hayley P. Ellis Kathreena M. Kurian
Kathreena M. Kurian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 29 January 2015
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00005
Epidermal growth factor receptor (EGFR) and EGFRvIII analysis is of current interest in glioblastoma – the most common malignant primary CNS tumor, because of new EGFRvIII vaccine trials underway. EGFR activation in glioblastoma promotes cellular proliferation via activation of MAPK and PI3K–Akt pathways, and EGFRvIII is the most common variant, leading to constitutively active EGFR. This review explains EGFR and EGFRvIII signaling in GBM; describes targeted therapy approaches to date including tyrosine kinase inhibitor, antibody-based therapies, vaccines and pre-clinical RNA-based therapies, and discusses the difficulties encountered with these approaches including pathway redundancy and intratumoral heterogeneity.
There is an urgent need for new molecular targeted therapies for newly diagnosed GBM (1–4). Recent data from The Cancer Genome Atlas project has proposed various subtypes of GBM, each with distinct molecular properties and genetic aberrations (5), although there is increasing recognition that there is molecular heterogeneity within individual tumors (6–8). Primary GBM is frequently associated with epidermal growth factor receptor (EGFR) amplifications compared with secondary GBM, which may arise from lower grade precursors (5, 9).
Overall, aberrant amplification, deletion, or mutation of at least one receptor tyrosine kinase (RTK) has been found in 67.3% of GBM, with EGFR accounting for 57.4% (10) (as shown in Figure 1). In addition, around 50% of patients with EGFR amplification harbor a specific mutation – known as EGFRvIII – which results from an in-frame deletion of exons 2–7 (11, 12). EGFRvIII can also be present independently of EGFR amplification (13).
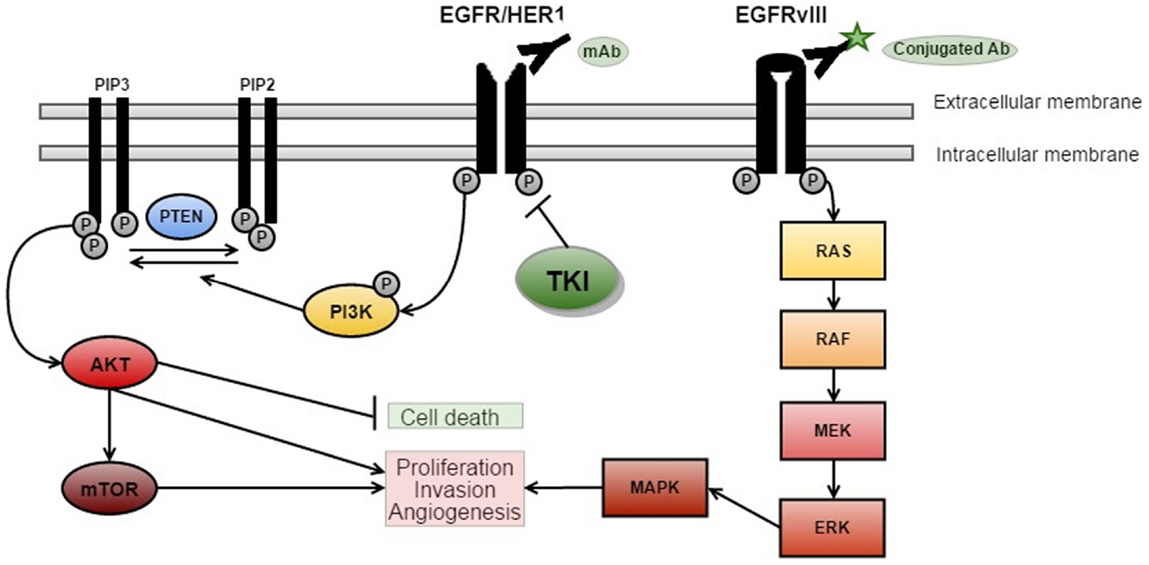
Figure 1. EGFR signaling and targeted therapies. There are three key signaling pathways in GBM. The RTK/RAS/PI(3)K pathway is involved in cell growth, apoptosis resistance, invasion, and migration (pictured above with targeted therapies). The other critically important pathways that regulate cell proliferation and survival are p53 and RB signaling (5) [adapted from Ref. (14)].
Due to the frequency of EGFR aberrations, many EGFR-targeted therapies are in development or clinical trials (14). Although EGFR kinase inhibitor therapy has shown initial success in other cancers such non-small cell lung cancer (15), previous trials in glioblastoma have been unsuccessful to date (16–18). More recently, interest has focused on an anti-EGFRvIII vaccine (known as rindopepimut), which has entered clinical trials (19).
Epidermal growth factor receptor signaling has an important role in many cancers, as cellular proliferation is mainly controlled by growth factors and their receptors (Figure 1). EGFR (also known as HER1 or ERBB1) is a receptor belonging to the ERBB family of the RTKs (20). Ligand-binding by EGF results in the activation of the RTK/RAS/PI(3)K pathway (21) via receptor phosphorylation, ultimately resulting in cellular proliferation, angiogenesis, and increased local tissue invasion as well as resistance to apoptosis (21–23).
Ligand-binding to RTKs simultaneously activates PI3K (see Figure 1) (22). PI3K phosphorylates phosphatidylinositol 4,5-biphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3), which results in further activation of AKT (22). AKT then promotes activation of mTOR – a protein, which exists as two complexes; mTORC1 and mTORC2 (24). Activation of mTORC1 promotes cellular growth by biosynthesis of proteins, lipids, and organelles in addition to inhibition of catabolic activity (22). mTORC2 activation results in phosphorylation and subsequent activation of several molecules (including AKT) that are involved in cell survival, metabolism, and proliferation (22). PTEN is a major inhibitor of this pathway by preventing phosphorylation of PIP2 to PIP3 (22). Loss of PTEN, such as the homozygous deletions observed in around 36% of gliomas, results in dramatic up-regulation of this pathway, and may be a major source of resistance to EGFR therapies (5, 22, 23).
In vitro studies suggest that the signaling mechanism of EGFRvIII cells can confer resistance to EGFR inhibitors (25), because EGFRvIII signals via an mTOR2 pathway whereas wtEGFR uses mTOR1 (24).
EGFRvIII is often co-expressed with wtEGFR, typically in tumors with EGFR over-expression, which complicates our understanding of its contribution to tumorigenesis (13, 26, 27). It has been suggested that the transformation happens intracellularly, with EGFRvIII being phosphorylated in an EGFR-dependent manner leading to increased downstream STAT signaling (27). There is also evidence in GBM of EGFRvIII internalization to form an oncogenic complex with STAT3 (27).
A retrospective analysis of clinical trials found that of 40 patients with EGFR amplification, those also expressing EGFRvIII had significantly shorter survival (0.839 years) than patients without (1.374 years), P = 0.0031 (13). Considerable growth advantage has been observed in EGFRvIII transfected GBM cell lines (U87MG.EGFRvIII), when compared to wtEGFR cell lines (28–30). This growth advantage is thought to result from an elevated proliferation rate coupled with a reduction in apoptosis (28). Similar to EGFR signaling, EGFRvIII activates the RTK/RAS/PI3K pathway as a result of EGFRvIII expression (31, 32). This results in increased levels of phosphorylated AKT and reduced levels of P27KIP1, a cell cycle regulator that inhibits G1–S phase transition in cell lines (33). Furthermore, abnormal spindle-like microcephaly-associated (ASPM) protein expression has been described in the U87MG.EGFRvIII cells compared to parental U87MG cells (34). ASPM promotes neural stem cell self-proliferation and hence it has been postulated the increased expression results in enhanced GBM progression by promoting cancer stem cell self-renewal (34).
The proliferative effects of EGFRvIII may be potentiated by the anti-apoptotic nature of brain tissue through up-regulation of Bcl-XL, which is a potent inhibitor of apoptosis (35). EGFRvIII has also been shown to have a role in tumor invasiveness, for example, in vitro studies demonstrated that U87MG.EGFRvIII cells displayed up-regulation of genes that promote an invasive phenotype such as matrix metalloproteinase (MMP)-13 (36). In both scratch tests and Matrigel Invasion Chamber assays, the cells also showed greater ability for migration and local tissue invasion than wtEGFR cells (36).
In the presence of amplified EGFR, it has been found that regulation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway through IκBαM gene transfer could play a role in glioma angiogenesis by regulating the expression of vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) (37).
Four modes of targeted therapies have been used to target EGFR including tyrosine kinase inhibitors (TKIs), antibody-based therapy, immunotherapy, and pre-clinical trials of RNA therapies. TKIs are small-molecule inhibitors, which bind to the ligand-binding site on the extracellular domain, and are the most clinically advanced EGFR-targeting therapy to date (14). Antibody-based therapy uses monoclonal antibodies that correspond to the receptor landscape to inhibit signaling, and can also use conjugated antibodies that allow toxins or radioactive isotopes to be targeted to specific cells (38). The current immunotherapy for EGFRvIII can be administered in the form of an intradermal vaccine CDX-110 and granulocyte macrophage-colony stimulating factor (GM-CSF) (39). RNA therapies will also be discussed, which involve creating antisense oligonucleotides or siRNA complementary to the regions that it would be clinically beneficial to silence (40).
Epidermal growth factor receptor TKIs gefitinib and erlotinib (see Table 1) have been found to significantly increase progression-free survival in non-small cell lung carcinoma (NSCLC) patients, with one meta-analysis reporting 42.9% of patients receiving TKI therapy reaching at least 1 year of progression-free survival compared to 9.7% with chemotherapy (41). A study of gefitinib as palliative therapy for patients with brain metastases from NSCLC found that 45% of patients experienced symptom improvement, with the experimental group maintaining progression-free survival for 6 months longer than the control group (42). As an initial therapy for asymptomatic brain metastases in never-smokers with adenocarcinoma of the lung, the combination of gefitinib and erlotinib has shown response rates of up to 70% (43). Lapatanib is another tyrosine kinase inhibitor used in treatment of HER2+ breast cancer, which when used in combination with capecitabine was found to increase progression-free survival to 8.4 months compared to 4.4 months receiving capecitabine monotherapy at the primary endpoint of a clinical trial of metastatic breast cancer patients (44). In the monotherapy group, 11 patients had CNS metastases compared with 4 in the combination therapy group (44), though lapatinib has not yet been shown to have activity against recurrent GBM in clinical trials (45).
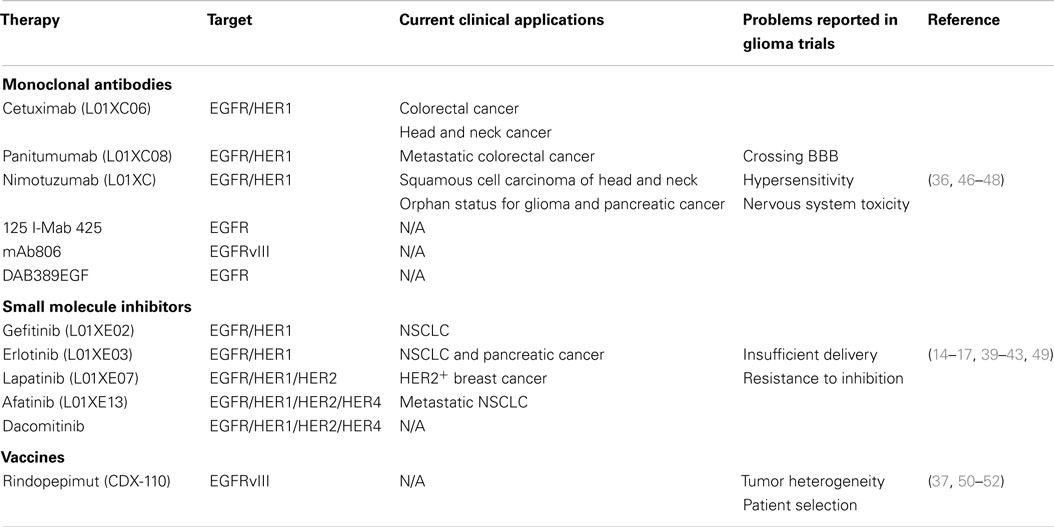
Table 1. A summary of therapies targeting EGFR and EGFRvIII.
Pre-clinical results demonstrate the ability of TKIs to inhibit tumor cell growth, angiogenesis, survival, and proliferation in several different EGFR transfected GBM cell lines (36, 49, 53, 54). However, these results do not appear to be clinically translatable, as response rates in GBM patients are disappointing for many inhibitors including gefitinib and erlotinib (55, 56). One explanation of this could be that TKIs are most efficacious when targeting tumor cells that express mutations in exons 19 and 21 of the EGFR kinase domain, which has been identified in various cancer types but has not yet been elucidated in GBM (14).
Despite the success of antibody-based therapy in the treatment of renal cell carcinoma, melanoma, and hematologic cancers, these results have not been replicated in GBM (46, 57, 58). Conjugated and unconjugated antibodies have been developed to target both wtEGFR and EGFRvIII, the most successful so far being cetuximab, panitumumab, and nimotuzumab (41, 47). The unconjugated antibodies bind the extracellular domain of EGFR, and they are also suggested to cause internalization of EGFRvIII, though clinical trials have had varying results (48).
Treatment of EGFR-amplified GBM cells with cetuximab in subcutaneous and intracranial mouse xenografts has been found to result in a significant decrease in proliferation, and an increase in overall survival as well as apoptosis (59). A decrease in the expression of VEGF in cell supernatant was observed using an enzyme-linked immunosorbent assay, suggesting further potential for application in GBM, as this signaling pathway also contributes to tumor maintenance and angiogenesis (59).
A Phase II study stratified patients depending on their EGFR gene amplification status and both groups were administered cetuximab intravenously (60). Cetuximab had little effect in both study groups and the median overall survival was 5 months, eliciting no significant correlation between EGFR status and response or overall survival (60). Other clinical trials involving similar antibody-based therapies have been equally unsuccessful, though a decrease in skin toxicity has been reported with use of nimotuzumab, which could increase its viability as an adjuvant therapy in GBM (47).
In a Phase III study, patients were administered nimotuzumab with concurrent radiotherapy (47). Although there was no statistically significant difference in overall survival of patients, the patients with the greatest median overall survival had molecular markers of EGFR amplification and unmethylated MGMT (47).
Antibodies, which utilize toxins or radioisotopes, could provide a potent adjuvant therapy for GBM as they enhance cell killing by the immune system in addition to inhibition of EGFR signaling (61). Various early clinical trials report that administering the radiolabeled antibody 125I-MAb 425 intravenously, either alone or with standard of care treatment, significantly improves median survival (38, 50, 61). In the largest Phase II trial to date combined treatment of 125I-mAb 425 and TMZ provided the greatest survival benefit with a median survival of 20.4 months, compared to treatment of 125I-mAb 425 alone, which was 14.5 months (50). Antibodies conjugated to death receptor agonists have been reported to induce apoptosis in GBM cell lines, for example, the scFvM58–sTRAIL fusion, which has been shown to selectively target GBM cells that express multidrug resistance protein 3 (62).
In initial Phase I trials, vaccinations comprising dendritic cells (DCs) primed with EGFRvIII peptides were found to be safe, with only grade I and II skin reactions at the vaccine injection site reported (51). Patients were also found to be immunologically responsive when their cellular immune responses were tested regularly using skin tests (51). Histological analysis in recurrent GBM patients who received the same vaccine showed no residual expression of EGFRvIII, demonstrating that the vaccine can effectively eliminate EGFRvIII cells, though all other cell types remain intact (63).
The Phase II trial “ACTIVATE” included 19 patients with newly diagnosed GBM received vaccines comprising PEPvIII-KLH (the EGFRvIII peptide coupled keyhole limpet hemocyanin to illicit both humoral and cellular immune responses) and GM-CSF (64). Progression-free survival was 12 months and patients who demonstrated immune sensitization to EGFRvIII had an overall survival of 47.7 months in comparison to 22.8 months for those who did not (64). It is important to note that positive results in these clinical trials could be due at least in part to the use of GM-CSF, as its use in cancer immunotherapy is enhancement of immunotherapeutic mechanisms of tumor destruction (52).
Another arm of this trial “ACT II” compares rindopepimut/GM-CSF concurrently with either standard or dose-intensive adjuvant TMZ (19). All patients were found to have an immune response to EGFRvIII; however, patients in the dose-intensive cohort had an even greater serum response, which may be partly due to a decrease in regulatory T cells (19). Most importantly, overall survival was greatly improved (23.6 months) in comparison to historical case-matched controls (19).
The larger “ACT III” trial sought to evaluate the clinical efficacy of the peptide vaccine CDX-110 in addition to radiotherapy and TMZ and produced similar results (65). The study was initially a randomized Phase II/III study, but patients belonging to the non-vaccine group withdrew from the trial (66). The median OS was 24.6 months compared to 15.2 months for matched EGFRvIII-positive controls (66). In addition, the results appeared to show a benefit in patients with methylated and unmethylated MGMT promoters (66).
A Phase III clinical trial, “ACT IV,” which compares rindopepimut plus GM-CSF and TMZ to current standard of care alone with a control (keyhole limpet hemocyanin) has been undertaken. Screening closed for this study on 30/9/14 with around 700 patients enrolled, and primary data collection is expected to take place in late 2016 (NCT01480479). A further Phase II trial (ReACT) is also underway involving patients with recurring EGFR-positive GBM receiving the EGFRvIII vaccine in addition to bevacizumab (NCT01498328). Also, at Stanford University a Phase I trial is underway utilizing the EGFRvIII vaccine in children with diffuse intrinsic pontine gliomas, as EGFR expression has been found to occur in ~50% of the tumors studied (67).
The use of antisense oligonucleotides to inhibit translation of mRNA has already yielded good results in pre-clinical studies for NSCLC and prostate cancer (68, 69). Following this, several experimental RNA methods targeting EGFR and EGFRvIII have been developed, including antisense oligonucleotides, RNA interference (RNAi), and ribozymes (40, 70, 71).
Antisense RNA appears to be efficacious in targeting EGFR expressing cells in vitro (70, 72, 73). Injection of vectors containing antisense RNA to target EGFRvIII into intracranial glioblastoma xenografts were found to reduce tumor volume by >40-fold compared with controls (74). In addition, in a U251 subcutaneous mouse model treated with antisense RNA and siRNA had significantly smaller tumor volumes by 29 and 19%, respectively, when compared to controls, further demonstrating efficacy in vivo (40).
Therapy with siRNA leads to post-transcriptional gene silencing that results in the destruction of the target mRNA (75). siRNA against EGFR has caused up to 90% knockdown of EGFR mRNA in U251 glioma cells (40). These results were reproduced using an intracranial xenograft mouse model, where median overall survival increased by almost 90% (40).
In pre-clinical studies, ribozymes targeting EGFRvIII were shown to inhibit ERM5–1 and U87MG glioblastoma cells (71, 76). In U87MG.EGFRvIII cells, anti-EGFRvIII hairpin ribozymes resulted in >90% reduction of EGFRvIII mRNA and a reduction in proliferation (71).
There may also be future potential for adjuvant miRNA-based therapies, as miR-7 has been shown to be an efficacious inhibitor of the EGFR signaling pathway in glioblastoma cell lines in vitro by direct inhibition of the EGFR receptor and further independent down-regulation of AKT, leading to a decrease in cell invasiveness (77). An increase in the radio-sensitivity of resistant cancer cells has also been described following miR-7 (78). The first miRNA-based cancer therapy (MRX34) has recently entered a Phase I clinical trial to evaluate its safety for use against primary liver cancer and liver metastases (NCT01829971). However, development of miRNA-based therapies against glioma may be considerably more difficult due to the lack of a delivery system sufficient to bypass the blood–brain barrier (79).
Limitations using TKIs, such as erlotinib may have an inability to pass the BBB due to the presence of efflux transporters on the endothelial cells associated with the BBB (65). Additionally, at present very little is known about the long-term adverse effects of non-specifically inhibiting EGFR signaling using TKIs, and knowledge of the biological effects of EGFR inhibitors on GBM cells is still incomplete (14).
Drawbacks described using antibody therapy may relate to local compared with systemic administration, for example, in mouse models, systemic administration of an antibody directed to EGFRvIII had no improvement in survival compared to controls, but intratumoral injection of the antibody resulted in an increase of median overall survival of 286% (64). A further study in rats demonstrated that cetuximab applied by an osmotic mini-pump significantly reduced tumor growth in the brain versus systemic application – which failed to block tumor growth (52). This could be due to the large molecular weight of the antibodies reducing their ability to traverse the BBB without assisted transport vectors (80).
Cautious interpretation of the utility of the ACT IV immunotherapy trial is needed because patients eligible to enroll in the trial were very highly selected, for example, they were newly diagnosed with complete tumor resection and no evidence of progressive disease (NCT01480479). This is because it has been shown that in patients with tumor resection of <95% neither over-expression of wtEGFR nor the presence of EGFRvIII can be used independently to predict patient survival (81). One hypothesis is that a small tumor may still be relatively immunoprivileged; thus, the immune system may be relatively naïve to tumor antigens (39).
In RNA studies, AAV/shRNA vectors were found to be severely toxic, and caused fatality in 64% of mice due to oversaturation of RNAi pathways (82, 83). Furthermore, RNA entities are anionic, hydrophilic, and unable to enter cell by passive diffusion mechanisms, so the BBB is essentially impenetrable to any potentially therapeutic RNA molecules (84).
Moreover, intratumoral heterogeneity may be a complicating factor. A study cohort of 57 glioma cases, examining tumoral heterogeneity via immunohistochemistry found that in one case the over-expression and amplification were localized to one half of the glioma, with the other half demonstrating normal levels of EGFR expression (85). Furthermore, not all of the samples with genetic amplifications also had increased levels of RNA, which further complicates the assessment of the viability of using EGFR-targeted therapies (85).
It has been previously shown that targeting of the EGFR receptor can lead to selection pressure for somatic mutations at other points in the pathway, such as inactivating phosphorylation of PTEN, leading to resistance to EGFR inhibitors (86), which could present a further problem for the ACT IV trial. Loss of PTEN has previously been found to be strongly correlated with treatment failure in GBM (87), to the extent that the analysis of EGFRvIII and PTEN levels may be used to predict tumor response to TKI therapy (88).
Additionally, in GBM there is a redundancy in activation of PI3K due to the availability of several types of tyrosine kinases upstream (86), including GFR1, MET, PDGFRα/β, and uPAR (89, 90). Increased activation of other members of the ERBB family of tyrosine kinases has also been described, as compensatory activation of ERBB2 and ERBB3 was noted after EGFR withdrawal in GBM cancer-stem-cell lines (91). This means that even after totally depleting EGFRvIII expressing populations of cells specifically, other tumor sub-clones with alternative mutations could be selected for, maintaining the tumor population and allowing functional resistance to EGFR-targeting therapies, and inhibitors of other ERBB family members may also be required for down-regulation of downstream elements (86, 91).
The loss of PTEN and increase in expression of other ERBB receptors may render EGFR signaling dispensable in the tumor, allowing growth and survival without EGFR signaling and thus negating the therapeutic viability of EGFR-targeting therapies in these cases (87, 88, 91). This uncoupling of the downstream pathways from EGFR signaling could be a possible explanation for the poor clinical responses exhibited with TKIs such as erlotinib (88).
Additionally, studies examining intratumoral heterogeneity found that it can be maintained by interactions between tumor cells, including the up-regulation of IL-6 production in EGFRvIII cells to activate neighboring wtEGFR cells, enhancing tumor growth and resistance to therapy (7). The mechanisms by which intratumoral heterogeneity arises are poorly understood; however, tumor cells have been found to reversibly increase or decrease levels of EGFRvIII expression in order to maximize their growth potential (92). Erlotinib resistance appears to be linked to EGFRvIII suppression in extrachromosomal DNA in order to successfully evade therapeutic mechanisms that target extrachromosomal oncogenes (92). Surprisingly, in this study, Nathanson et al. also described a reversal of erlotinib resistance within 72 h upon withdrawal of the drug, where extrachromosomal EGFRvIII DNA was dramatically upregulated, and restored sensitivity to TKI-induced cell death (92).
A study comparing the efficacy of lapatinib in lung cancer and GBM found that the lack of response to therapy in GBM could be due to the location of the mutation. Lung cancer EGFR mutations tend to occur in the kinase domain, whereas GBM EGFR mutations are mainly in the extracellular domain, which could allow the GBM mutant receptors sufficient flexibility within the kinase domain to accommodate lapatinib and other type II EGFR kinase inhibitors (93).
The intratumoral heterogeneity of EGFR expression in GBM may ultimately limit the clinical utility of therapies such as TKIs, antibody-based therapies, and RNA-based therapies, because it will not be possible to target every single neoplastic cell in the tumor population. Moreover, it is likely that other sub-clones of tumor cells will arise or other pathways may be upregulated as a mechanism of resistance to EGFR-targeted therapies. Although problems with EGFR intratumoral heterogeneity and pathway redundancy will also apply to immunotherapy approaches, vaccines may ultimately be more attractive because they alert the immune system to the presence of tumor and may trigger a more non-specific tumoricidal immune response, which may potentially eradicate all tumor cells.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G; ESMO Guidelines Working Group. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol (2014) 25(Suppl 3):iii93–101. doi:10.1093/annonc/mdu050
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi:10.1056/NEJMoa043330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev (2007) 21:2683–710. doi:10.1101/gad.1596707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol (2012) 107:359–64. doi:10.1007/s11060-011-0749-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi:10.1038/nature07385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res (2011) 71:4055–60. doi:10.1158/0008-5472.CAN-11-0153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev (2010) 24:1731–45. doi:10.1101/gad.1890510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344:1396–401. doi:10.1126/science.1254257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi:10.1016/j.ccr.2009.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155:462–77. doi:10.1016/j.cell.2013.09.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol (2007) 25:2288–94. doi:10.1200/JCO.2006.08.0705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Del Vecchio CA, Giacomini CP, Vogel H, Jensen KC, Florio T, Merlo A, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene (2013) 32:2670–81. doi:10.1038/onc.2012.280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res (2003) 63:6962–70.
14. Karpel-Massler G, Schmidt U, Unterberg A, Halatsch ME. Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: where do we stand? Mol Cancer Res (2009) 7:1000–12. doi:10.1158/1541-7786.MCR-08-0479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Yang X, Yang K, Kuang K. The efficacy and safety of EGFR inhibitor monotherapy in non-small cell lung cancer: a systematic review. Curr Oncol Rep (2014) 16:390. doi:10.1007/s11912-014-0390-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev (2008) 18:73–9. doi:10.1016/j.gde.2008.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Wykosky J, Mukasa A, Furnari F, Cavenee WK. Escape from targeted inhibition: the dark side of kinase inhibitor therapy. Cell Cycle (2010) 9:1661–2. doi:10.4161/cc.9.9.11592
18. Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res (2008) 14:2895–9. doi:10.1158/1078-0432.CCR-07-2248
19. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol (2011) 13:324–33. doi:10.1093/neuonc/noq157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Ren H, Yang BF, Rainov NG. Receptor tyrosine kinases as therapeutic targets in malignant glioma. Rev Recent Clin Trials (2007) 2:87–101. doi:10.2174/157488707780599384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Patel R, Leung HY. Targeting the EGFR-family for therapy: biological challenges and clinical perspective. Curr Pharm Des (2012) 18:2672–9. doi:10.2174/138161212800626148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, et al. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol (2004) 22:1926–33. doi:10.1200/JCO.2004.07.193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi:10.1126/science.1164382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov (2011) 1:524–38. doi:10.1158/2159-8290.CD-11-0124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Huang PH, Cavenee WK, Furnari FB, White FM. Uncovering therapeutic targets for glioblastoma: a systems biology approach. Cell Cycle (2007) 6:2750–4. doi:10.4161/cc.6.22.4922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res (2005) 11:1462–6. doi:10.1158/1078-0432.CCR-04-1737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Fan Q-W, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIll driving STAT3/5 and progression in glioblastoma. Cancer Cell (2013) 24:438–49. doi:10.1016/j.ccr.2013.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Nagane M, Coufal F, Lin H, Bögler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res (1996) 56:5079–86.
29. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A (1994) 91:7727–31. doi:10.1073/pnas.91.16.7727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sugawa N, Yamamoto K, Ueda S, Morita N, Kita M, Nishino H, et al. Function of aberrant EGFR in malignant gliomas. Brain Tumor Pathol (1998) 15:53–7. doi:10.1007/BF02482101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol (2006) 65:1181–8. doi:10.1097/01.jnen.0000248549.14962.b2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A (2007) 104:12867–72. doi:10.1073/pnas.0705158104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Narita Y, Nagane M, Mishima K, Huang HJ, Furnari FB, Cavenee WK. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res (2002) 62:6764–9.
34. Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A (2006) 103:17402–7. doi:10.1073/pnas.0608396103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell (1993) 74:597–608. doi:10.1016/0092-8674(93)90508-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Lal A, Glazer CA, Martinson HM, Friedman HS, Archer GE, Sampson JH, et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res (2002) 62:3335–9.
37. Wu JL, Abe T, Inoue R, Fujiki M, Kobayashi H. IkappaBalphaM suppresses angiogenesis and tumorigenesis promoted by a constitutively active mutant EGFR in human glioma cells. Neurol Res (2004) 26:785–91. doi:10.1179/016164104225014139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Emrich JG, Brady LW, Quang TS, Class R, Miyamoto C, Black P, et al. Radioiodinated (I-125) monoclonal antibody 425 in the treatment of high grade glioma patients: ten-year synopsis of a novel treatment. Am J Clin Oncol (2002) 25:541–6. doi:10.1097/00000421-200212000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol (2009) 19:713–23. doi:10.1111/j.1750-3639.2009.00318.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kang CS, Zhang ZY, Jia ZF, Wang GX, Qiu MZ, Zhou HX, et al. Suppression of EGFR expression by antisense or small interference RNA inhibits U251 glioma cell growth in vitro and in vivo. Cancer Gene Ther (2006) 13:530–8. doi:10.1038/sj.cgt.7700932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Liang W, Wu X, Fang W, Zhao Y, Yang Y, Hu Z, et al. Network meta-analysis of erlotinib, gefitinib, afatinib and icotinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. PLoS One (2014) 9:e85245. doi:10.1371/journal.pone.0085245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Wu C, Li YL, Wang ZM, Li Z, Zhang TX, Wei Z. Gefitinib as palliative therapy for lung adenocarcinoma metastatic to the brain. Lung Cancer (2007) 57:359–64. doi:10.1016/j.lungcan.2007.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kim JE, Lee DH, Choi Y, Yoon DH, Kim SW, Suh C, et al. Epidermal growth factor receptor tyrosine kinase inhibitors as a first-line therapy for never-smokers with adenocarcinoma of the lung having asymptomatic synchronous brain metastasis. Lung Cancer (2009) 65:351–4. doi:10.1016/j.lungcan.2008.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med (2006) 355:2733–43. doi:10.1056/NEJMoa064320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol (2010) 65:353–61. doi:10.1007/s00280-009-1041-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Capietto A-H, Keirallah S, Gross E, Dauguet N, Laprevotte E, Jean C, et al. Emerging concepts for the treatment of hematological malignancies with therapeutic monoclonal antibodies. Curr Drug Targets (2010) 11:790–800. doi:10.2174/138945010791320845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bode U, Massimino M, Bach F, Zimmermann M, Khuhlaeva E, Westphal M, et al. Nimotuzumab treatment of malignant gliomas. Expert Opin Biol Ther (2012) 12:1649–59. doi:10.1517/14712598.2012.733367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ, et al. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res (2007) 27:3355–66.
49. Knight LA, Di Nicolantonio F, Whitehouse P, Mercer S, Sharma S, Glaysher S, et al. The in vitro effect of gefitinib (‘Iressa’) alone and in combination with cytotoxic chemotherapy on human solid tumours. BMC Cancer (2004) 4:83. doi:10.1186/1471-2407-4-83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Li L, Quang TS, Gracely EJ, Kim JH, Emrich JG, Yaeger TE, et al. A Phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J Neurosurg (2010) 113:192–8. doi:10.3171/2010.2.JNS091211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther (2009) 8:2773–9. doi:10.1158/1535-7163.MCT-09-0124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Thorne SH. The role of GM-CSF in enhancing immunotherapy of cancer. Immunotherapy (2013) 5:817–9. doi:10.2217/imt.13.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Halatsch ME, Gehrke EE, Vougioukas VI, Bötefür IC, A-Borhani F, Efferth T, et al. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J Neurosurg (2004) 100:523–33. doi:10.3171/jns.2004.100.3.0523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Quatrale AE, Porcelli L, Silvestris N, Colucci G, Angelo A, Azzariti A. EGFR tyrosine kinases inhibitors in cancer treatment: in vitro and in vivo evidence. Front Biosci (Landmark Ed) (2011) 16:1962–72. doi:10.2741/3834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci (2009) 16:748–54. doi:10.1016/j.jocn.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Rich JN, Rasheed BK, Yan H. EGFR mutations and sensitivity to gefitinib. N Engl J Med (2004) 351:1260–1. doi:10.1056/NEJM200409163511221
57. Escudier B, Albiges L. Pazopanib for the treatment of advanced renal cell cancer. Expert Opin Orphan Drugs (2014) 2:605–16. doi:10.1517/21678707.2014.912579
58. Gyorki DE, Spillane J, Speakman D, Shackleton M, Henderson MA. Current management of advanced melanoma: a transformed landscape. ANZ J Surg (2014) 84:612–7. doi:10.1111/ans.12673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Eller JL, Longo SL, Hicklin DJ, Canute GW. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery (2002) 51:1005–13. doi:10.1227/00006123-200210000-00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol (2009) 20:1596–603. doi:10.1093/annonc/mdp032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Quang TS, Brady LW. Radioimmunotherapy as a novel treatment regimen: 125I-labeled monoclonal antibody 425 in the treatment of high-grade brain gliomas. Int J Radiat Oncol Biol Phys (2004) 58:972–5. doi:10.1016/j.ijrobp.2003.09.096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Wang LH, Ni CW, Lin YZ, Yin L, Jiang CB, Lv CT, et al. Targeted induction of apoptosis in glioblastoma multiforme cells by an MRP3-specific TRAIL fusion protein in vitro. Tumor Biol (2014) 35:1157–68. doi:10.1007/s13277-013-1155-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol (2008) 20:267–75. doi:10.1016/j.smim.2008.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Heimberger AB, Hussain SF, Aldape K, Sawaya R, Archer GA, Friedman H, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol (2006) 24:107S–107S.
65. Sampson JH, Aldape KD, Gilbert MR, Hassenbusch SJ, Sawaya R, Schmittling B, et al. Temozolomide as a vaccine adjuvant in GBM. J Clin Oncol (2007) 25:18S.
66. Lai RK, Recht LD, Reardon DA, Paleologos N, Groves M, Rosenfeld MR, et al. Long-term follow-up of act III: a phase II trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma. Neuro Oncol (2011) 13:34–34.
67. Li G, Mitra SS, Monje M, Henrich KN, Bangs CD, Nitta RT, et al. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol (2012) 108:395–402. doi:10.1007/s11060-012-0842-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Laskin JJ, Nicholas G, Lee C, Gitlitz B, Vincent M, Cormier Y, et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J Thorac Oncol (2012) 7:579–86. doi:10.1097/JTO.0b013e31823f459c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Tannock IF, Fizazi K, Ivanov S, Karlsson CT, Fléchon A, Skoneczna I, et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): a phase 3, double-blind randomised trial. Lancet Oncol (2013) 14:760–8. doi:10.1016/S1470-2045(13)70184-0
70. Liu A-X, Liu X-W, Pu P-Y, Peng Q, Cheng A-G. Induction of apoptosis and inhibition of growth in glioblastoma cell line by EGFR antisense RNA. J Neurochem (1996) 67:S37.
71. Halatsch ME, Schmidt U, Bötefür IC, Holland JF, Ohnuma T. Marked inhibition of glioblastoma target cell tumorigenicity in vitro by retrovirus-mediated transfer of a hairpin ribozyme against deletion-mutant epidermal growth factor receptor messenger RNA. J Neurosurg (2000) 92:297–305. doi:10.3171/jns.2000.92.2.0297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Petch AK, Sohail M, Southern E, Akhtar S. Combination therapy using two antisense oligonucleotides against EGFr mRNA. J Pharm Pharmacol (1999) 51:125.
73. Liu X, Liu A, Pu P. Inhibitory effect of EGFR antisense RNA on growth of glioma cells in vitro. Chin Clin Oncol (1998) 25:869–73.
74. Shir A, Levitzki A. Inhibition of glioma growth by tumor-specific activation of double-stranded RNA-dependent protein kinase PKR. Nat Biotechnol (2002) 20:895–900. doi:10.1038/nbt730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Guo D, Wang B, Han F, Lei T. RNA interference therapy for glioblastoma. Expert Opin Biol Ther (2010) 10:927–36. doi:10.1517/14712598.2010.481667
76. Yamazaki H, Kijima H, Ohnishi Y, Abe Y, Oshika Y, Tsuchida T, et al. Inhibition of tumor growth by ribozyme-mediated suppression of aberrant epidermal growth factor receptor gene expression. J Natl Cancer Inst (1998) 90:581–7. doi:10.1093/jnci/90.8.581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res (2008) 68:3566–72. doi:10.1158/0008-5472.CAN-07-6639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Lee KM, Choi EJ, Kim IA. microRNA-7 increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Radiother Oncol (2011) 101:171–6. doi:10.1016/j.radonc.2011.05.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Li M, Li J, Liu L, Li W, Yang Y, Yuan J. MicroRNA in human glioma. Cancers (Basel) (2013) 5:1306–31. doi:10.3390/cancers5041306
80. Dennis MS, Watts RJ. Transferrin antibodies into the brain. Neuropsychopharmacology (2012) 37:302–3. doi:10.1038/npp.2011.196
81. Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med (2005) 3:38. doi:10.1186/1479-5876-3-38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature (2009) 457:426–33. doi:10.1038/nature07758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature (2006) 441:537–41. doi:10.1038/nature04791
84. Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol (2009) 9(Suppl 1):S3. doi:10.1186/1471-2377-9-S1-S3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Strommer K, Hamou MF, Diggelmann H, de Tribolet N. Cellular and tumoural heterogeneity of EGFR gene amplification in human malignant gliomas. Acta Neurochir (Wien) (1990) 107:82–7. doi:10.1007/BF01405784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Fenton TR, Nathanson D, Kuga A, Iwanami J, Dang H, Yang K, et al. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc Natl Acad Sci U S A (2012) 109:14164–9. doi:10.1073/pnas.1211962109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Mellinghoff IK, Cloughesy TF, Mischel PS. PTEN-mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res (2007) 13:378–81. doi:10.1158/1078-0432.CCR-06-1992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med (2005) 353:2012–24. doi:10.1056/NEJMoa051918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science (2007) 318:287–90. doi:10.1126/science.1142946
90. Hu J, Jo M, Cavenee WK, Furnari F, VandenBerg SR, Gonias SL. Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc Natl Acad Sci U S A (2011) 108:15984–9. doi:10.1073/pnas.1113416108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Clark PA, Iida M, Treisman DM, Kalluri H, Ezhilan S, Zorniak M, et al. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia (2012) 14:420–8.
92. Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science (2014) 343:72–6. doi:10.1126/science.1241328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Differential sensitivity of glioma-versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov (2012) 2:458–71. doi:10.1158/2159-8290.CD-11-0284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: EGFR, EGFRvIII, EFGR inhibitors, glioblastoma multiforme, molecular marker
Citation: Padfield E, Ellis HP and Kurian KM (2015) Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 5:5. doi: 10.3389/fonc.2015.00005
Received: 05 November 2014; Accepted: 09 January 2015;
Published online: 29 January 2015.
Edited by:
Kerrie Leanne McDonald, University of NSW, AustraliaReviewed by:
Keith Giles, Western Australian Institute for Medical Research, AustraliaCopyright: © 2015 Padfield, Ellis and Kurian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hayley P. Ellis, Brain Tumour Research Group, Institute of Clinical Sciences, Southmead Hospital, University of Bristol, Level 1, Learning and Research Building, Bristol BS10 5NB, UK e-mail:aGUxNTE5QGJyaXN0b2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.