 Marianna Prokopi
Marianna Prokopi Christina A. Kousparou1
Christina A. Kousparou1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 11 February 2015
Sec. Cancer Molecular Targets and Therapeutics
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00389
This article is part of the Research Topic Notch targeting in cancer View all 10 articles
MicroRNAs (miRNAs) have been implicated in the development of some if not all cancer types and have been identified as attractive targets for prognosis, diagnosis, and therapy of the disease. miRNAs are a class of small non-coding RNAs (20–22 nt in length) that bind imperfectly to the 3′-untranslated region of target mRNA regulating gene expression. Aberrantly expressed miRNAs in cancer, sometimes known as oncomiRNAs, have been shown to play a major role in oncogenesis, metastasis, and drug resistance. Amplification of oncomiRNAs during cancer development correlates with the silencing of tumor suppressor genes; on the other hand, down-regulation of miRNAs has also been observed in cancer and cancer stem cells (CSCs). In both cases, miRNA regulation is inversely correlated with cancer progression. Growing evidence indicates that miRNAs are also involved in the metastatic process by either suppressing or promoting metastasis-related genes leading to the reduction or activation of cancer cell migration and invasion processes. In particular, circulating miRNAs (vesicle-encapsulated or non-encapsulated) have significant effects on tumorigenesis: membrane-particles, apoptotic bodies, and exosomes have been described as providers of a cell-to-cell communication system transporting oncogenic miRNAs from tumors to neighboring cells and distant metastatic sites. It is hypothesized that miRNAs control cancer development in a traditional manner, by regulating signaling pathways and factors. In addition, recent developments indicate a non-conventional mechanism of cancer regulation by stem cell reprograming via a regulatory network consisting of miRNAs and Wnt/β-catenin, Notch, and Hedgehog signaling pathways, all of which are involved in controlling stem cell functions of CSCs. In this review, we focus on the role of miRNAs in the Notch-pathway and how they regulate CSC self-renewal, differentiation and tumorigenesis by direct/indirect targeting of the Notch-pathway.
Cancer is a heterogeneous disease with cellular hierarchies and many different phenotypes. Strong evidence points to the fact that the majority of cells in solid tumors are of non-tumorigenic origin. A small population of progenitor cells is thought to be responsible for tumor initiation, growth, metastasis, drug resistance, and recurrence. These cells are known as cancer stem cells (CSCs), since they possess stem cell characteristics such as self-renewal, asymmetric cell division, differentiation, and chemoresistance (1, 2). CSCs act via signaling pathways that mediate self-renewal, including Notch and Wnt. It is becoming widely accepted that irregular stem cell self-renewal is essential for cancer initiation, formation, and relapse and that CSCs play a central role in cancer cell biology. Thus the identification of specific markers of CSCs may be important in the discovery and development of novel oncology therapeutics.
The authors support the hypothesis that cancer initiation and development involve the improper activation of developmental signaling pathways. Normally such pathways control the growth of tissues and organs by maintaining the balance between cell proliferation, differentiation, senescence, and apoptosis. Notch signaling pathway plays a key role in stem cell self-renewal, cell proliferation, and differentiation. Consequently, it has important developmental functions, and its aberrant activation leads to many diseases and cancers (3–5). Notch genes encode large single pass transmembrane proteins that regulate cell fate determination (6). Previous studies in Drosophila, Caenorhabditis elegans, and mammalian cell cultures have shown that Notch act as receptors for the DSL (Delta, Serrate, and Lag-2) family of ligands and signal through two downstream pathways. One of these is via the CSL (CBF1, Suppressor of Hairless, Lag-1) family of transcription factors and the other via the cytoplasmic adapter protein Deltex. In mammals, the Notch signaling pathway includes four receptors (Notch 1–4) and five ligands (Delta-like 1, 3, and 4 & Jagged-1 and 2) (7). Notch signaling is initiated through ligand–receptor binding between two neighboring cells. Upon activation, Notch undergoes cleavage, releasing its intracellular domain NICD and translocates into the nucleus for transcriptional activation of its downstream target genes (8).
Modifications in the Notch-pathway and its associated genes can result in ligand dysregulation having dramatic developmental effects in humans, thus implicating Notch signaling in several inherited diseases such as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, Alagille syndrome, and Spondylocostal dysostosis. Since the discovery of Notch1 gene alterations in T-cell acute lymphoblastic leukemia/lymphoma, deregulated Notch signaling has been connected to many solid tumor pathologies and different cancer types (leukemia, neuroblastomas, skin, cervical, lung, prostate, and breast cancer) (4, 9, 10). The role of Notch signaling in tumorigenesis is thought to be mainly oncogenic, although some observations have suggested an anti-proliferative role in a small number of cancers (hepatocellular carcinoma and skin cancer) (11–13). The oncogenic function of Notch signaling is associated with high levels of Jagged-1, mainly in prostate and breast cancer, as well as with loss of Numb activity – a negative regulator of Notch-pathway – in 40% of breast cancers and 30% of lung cancers (5, 14, 15). Thus, the deregulation of the Notch signaling pathway has so far been linked to metastasis, recurrence, and reduced overall survival. On the other hand, tumor aggressiveness has been linked to the cross-talk between Notch and other oncogenic pathways such as Wnt/β-catenin, NF-kB, Ras, and Akt (16–18).
Although irregular activation of a single pathway may result in tumorigenesis, oncogenic pathways rarely operate in isolation. Cross-talk between signaling pathways adds to the complexity of the disease and are heavily influenced by the microenvironment. Recent studies have revealed that the interaction between the Notch and Wnt/β-catenin signaling pathways drives the CSC uncontrolled self-renewal, resulting in CSC-related tumor recurrence after treatment (19, 20). Notch signaling regulates both the CSC formation and the epithelial-to-mesenchymal-transition (EMT) phenotype during tumor progression. The EMT process, which occurs during tumor progression, drives the CSCs to become metastatic. Indeed, Notch-mediated EMT converts polarized epithelial cells into motile, invasive cells due to loss of E-cadherin – a membrane glycoprotein involved in the adherence of adjacent cells – which results in β-catenin activation and dissemination of cancer cells and CSCs from the primary tumor (21–23). Notch signaling pathway interacts with several oncogenic pathways, transcription, and growth factors (e.g., Snail, Slug, and TGF-β) regulating various biological and pathologic processes during cancer development, progression, and therapy. However, a growing body of evidence indicates that Notch is regulated at molecular level via cross-talk with miRNAs suggesting a critical role for these molecules in tumor biology (24, 25).
MiRNAs are a class of small, non-coding RNAs that regulate mRNA by acting at the post-translational level (26). The interaction between miRNAs and mRNAs is highly complex; in particular each miRNA can control hundreds of gene targets underlining the extraordinary impact of miRNA on protein expression. We are just beginning to understand how this novel class of regulators affects processes, at least, in mammals. Processed from longer primary transcripts by Drosha and Dicer, miRNAs bind through imperfect complementarity to their target genes at the seed sequence (eight-base long), of the 3′ non-coding region leading to degradation of target mRNA due to deadenylation/mRNA cleavage or to repression of mRNA translation initiation (27, 28). MiRNAs have been implicated in a wide range of cell functions – normal or pathological – comprising of cell proliferation, apoptosis, differentiation, and self-renewal (2, 29). Therefore, dysregulation of miRNAs is linked to a range of human pathologies including cancer and its expression is associated with cancer development, progression, and prognosis mainly because of their involvement in cell proliferation and apoptosis (30). MiRNAs were shown to be differentially expressed in cancer forming unique miRNA patterns with some miRNAs to have oncogenic activity while others have tumor suppressor activity (oncosuppressors). Thus, oncogenic miRNAs are upregulated in cancer whereas tumor suppressor miRNAs are downregulated (31). With regards to cancer biology, miRNAs targeting oncogenes are often located in fragile regions with a tension to be downregulated in tumors leading to overexpression of their target oncogenes. A breakthrough study showed that 50% of the annotated human miRNAs are positioned in these unstable sites and are associated with cancer and function in tumor progression (32).
Tissue-specific/tissue-enriched miRNAs, often deregulated, play a major role in cancer progress functioning as both oncosuppressors and oncogenes. For example, upregulated miR-21 is associated with breast and lung cancer, glioblastoma, leukemia, neuroblastoma, and liver metastasis (33, 34). Brain-specific neuromiR-124 is downregulated in glioblastoma resulting in increased CSC numbers and oncogenic capacity (35). On the other hand, let-7, normally expressed in lung, is downregulated in lung cancer and associated with poor survival (36), lung-specific pneumomiR-29 suppresses tumorigenicity in non-small cell lung cancer cells and miR-143 and miR-145 have been shown to be downregulated in breast, cervical, and colorectal cancers (37, 38). Moreover, loss of miR-15 and miR-16 has been related to chronic lymphoid leukemia by negatively regulating the anti-apoptotic gene BCL2, supporting a role in the immune system (39, 40). Similarly, many other miRNAs have been related to Notch signaling pathway, by regulating Notch-associated genes and affecting many types of cancer (Table 1).
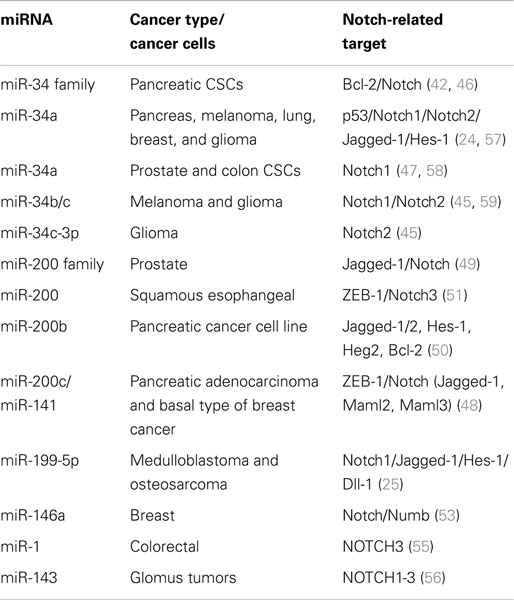
Table 1. Important miRNAs regulating Notch signaling pathway and Notch-associated genes.
MiR-34 family is composed of miR-34a, miR-34b, and miR-34c and has been connected to the regulation of p53 and Notch signaling pathway. Low levels or no expression of miR-34 corresponds to higher probability of breast, brain, pancreatic, and non-small cell lung cancer suggesting a tumor suppressor role (24, 41, 42). Repression of miR-34a was identified in a panel of tumor-derived cancer cell lines such as pancreas, melanoma, lung, breast, and glioma (43, 44). However, miR-34b/c have been identified in malignant melanoma cases and glioma patients’ tissues (45). The involvement of miR-34 family has now been shown to affect major properties of CSCs. For example, it has been revealed that restoration of miR-34 inhibits CD44+/CD133+ pancreatic CSCs self-renewal capacity through direct down-regulation of Bcl-2 and Notch signaling pathways (46). Furthermore, transfection with miR-34c-3p in U251 and U87 glioblastoma cell lines inhibited cell proliferation, induced cell apoptosis, and obstructed glioma cell invasion. Therefore, it was established that miR-34c-3p overexpression reduced the levels of Notch 2 (Figure 1) indicating that cell proliferation inhibition occurred via the Notch signaling pathway (45). On the other hand, it is well recognized that Notch 1 is linked to CSCs “stemness” characteristics and promotes the EMT phenotype, which is closely linked to many types of metastatic cancer. In prostate cancer for instance, upregulated levels of Notch 1 are highly associated with prostate cancer development, metastasis, and progression. Re-expression of miR-34a in C4-2B and CWR22rv1 prostate cancer cells reduced the expression of Notch1, decreased the self-renewal capacity and inhibited the growth of prostate cancer cells (47). Taken together, miR-34 family directs the regulation of Notch 1 and 2 protein expression in glioma cells, pancreatic, and prostate cancer cells mediating the suppression of self-renewal and differentiation properties of CSCs. Restoration of miR-34 levels could be eventually used as cancer therapeutic by down-regulating the Notch family members.
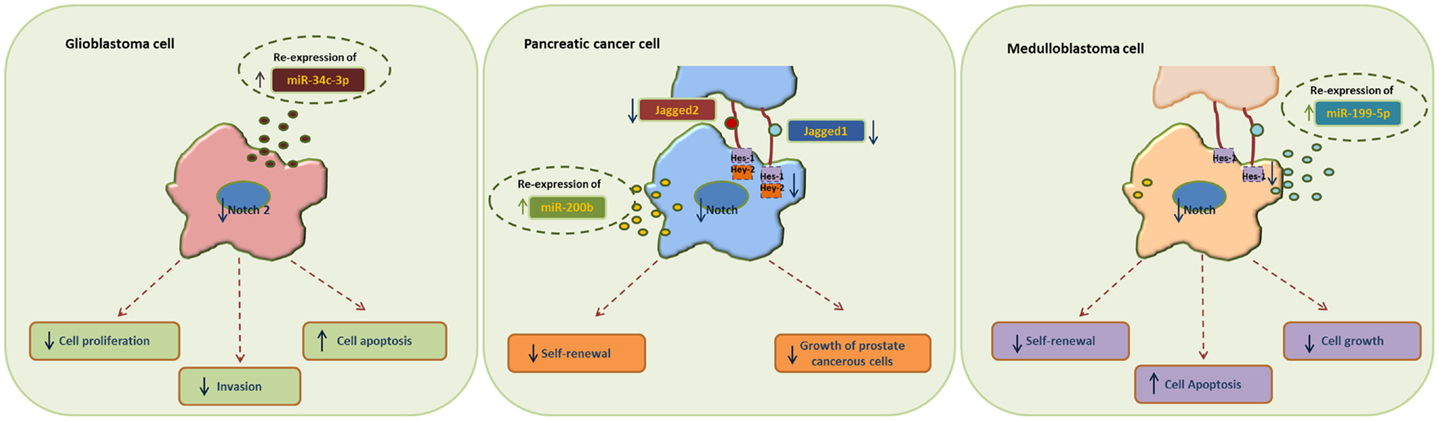
Figure 1. Examples of miRNA re-expression therapy using miR-34, miR-200, and miR-199 family, in glioblastomas, pancreatic, and medulloblastomas cancer cells, all affecting the Notch signaling pathway.
MiR-200 family consists of five members: miR-200a, miR-200b, miR-200c, miR-141, and miR-429. Recent studies have shown that the miR-200 family is highly implicated in the regulation of CSCs with examples involving breast, colorectal, prostate, and brain CSCs. Low levels of miR-200c and miR-141 are correlated with high expression of ZEB-1 – an EMT activator – which in turn activates Notch signaling pathway by targeting the Notch ligand Jagged-1 and Notch co-activators Maml 2 and 3. Reduced expression of miR-141 and miR-200c affects stem cell properties and drug resistance in two human cancer types’ pancreatic adenocarcinoma and basal type of breast cancer (48). Moreover, re-expression of miR-200 family including miR-141 and miR-429 directly inhibits Jagged-1 in human metastatic prostate cancer cells suggesting a new way to control the fate of Notch-pathway (49). Similarly, in another study transfection of miR-200b in Rink-1 cells (pancreatic cell line) have reduced the levels of Jagged-1/2 and these of their target genes Hes-1, Hey-2 (Figure 1), and Bcl-2 leading to cell growth inhibition (50). Another critical study connects NOTCH3 in the regulation of ZEBs and the miR-200 family revealing how critical is Notch in EMT, invasion and tumor formation in squamous esophageal cancers (51). However, more in-depth investigation is required in order to understand how the miR-200 family regulates the Notch signaling pathway.
MiR-199-5p has been linked to the transcription factor Hes-1 in medulloblastoma tumors where it regulates cell growth and several CSC genes via Notch signaling pathway. In metastatic cancer patients’ miR-199-5p expression is lost, yet re-expression of it blocks the Notch signaling pathway (Figure 1) and the population of medulloblastoma stem cell like cells is decreased (25). Zollo’s group went a step further by developing stable nucleic and lipid particles (SNALPs) to deliver miR-199-5p in different tumorigenic cell lines such as colon, breast, glioblastoma, and medulloblastoma. Impairment of cell proliferation and CSC-associated markers was due to effect of miR-199-5p delivery via SNALPs however, different efficacies due to cell type cannot be excluded and the efficiency of these carriers can be limited by their instability and non-specific targeting in vivo (52).
Recently, miR-146a was also found to interact with Notch via regulation of Numb in breast carcinomas (53) and to stimulate NF-κB activity through Notch 1 [reviewed in Ref. (54)]. Another miRNA involved in Notch-pathway is miR-1, which it was reported that directly regulates Notch via Dll-1 protein in mouse embryonic stem cells. MiR-1 has been linked to human hepatocellular carcinoma, lung, prostate, and head and neck cancers; therefore it is only a matter of time for Notch to be associated with the above through miR-1 [reviewed in Ref.(54)]. A recent study by Furukawa (55) demonstrated the association of miR-1 in colorectal tumors and the potential to suppress NOTCH3 expression, which in turn results in reduction of Asef regulating the growth, migration, and invasion potential of cancer cells. Other reports, show an association of MIR143 gene with NOTCH1-3; suggesting that the mechanism of MIR143–NOTCH1-3 tumorigenesis is through oncogenic activation of NOTCH driven by the very strong MIR143 promoter in malignant glomus tumors (56).
The development of therapies against cancer and CSCs has driven toward a new generation of cancer therapeutics. RNA-based approaches promise to be one of the next major classes of cancer therapeutics. Advancements in genetics relating to the role of RNA in an ever-expanding range of cellular pathways and processes have shown that RNA has many of the genetic and regulatory properties formerly attributed only to DNA and proteins. Current work is based on modified mRNA regulating the expression of therapeutic proteins and on RNAi variants such as siRNA and miRNA, which can either block oncogenes or amplify oncosuppressor genes in cancer cells. The success of RNAi therapeutics hinges on their effective and safe delivery to their molecular targets inside cancer cells and tumors. However, targeted delivery remains a major challenge in miRNA therapy because naked ribonucleic acids are subject to rapid degradation by serum nucleases and miRNAs cannot diffuse freely into cells. To deliver the new class of targeted drugs, several technologies are under development including nanocapsules and nanocarriers, micro/nanoparticles, liposomes, and PEGylated vesicles. Delivery of miRNAs or miRNA inhibitors requires a flexible and efficient delivery system, for example, miR-34a has being encapsulated in stable-acid-lipid particles used to target Dll-1 in vitro (52). Other delivery approaches involve the use of agents such as polyethylenimine (PU-PEI) to mediate miRNA delivery. PU-PEI-mediated miR-145 delivery vehicle has been used successfully for miR-145 delivery to glioblastoma cells (60). Another candidate for the successful miRNA therapeutic approach is the vector-mediated overexpression of miRNAs to tumor tissues in vivo using adenoviral or lentiviral delivery. Of note, miR-26a expression in normal liver and liver tumor cells succeeded via adenoviral delivery in order to inhibit tumor cell proliferation and to lead in apoptosis (61). However, the use of this approach can only be employed in systemic local delivery. As the early clinical trial failures in the field have demonstrated, delivery of RNAi therapeutics to their molecular targets inside cancer cells is crucial. The authors have developed a novel system for miRNA delivery applicable for both local and systemic administration with the use of mesenchymal stem cell (MSC)-derived microparticles (MPs). The idea was based on the fact that miRNAs are released in the blood-stream via a controlled and active process through MPs, exosomes, and apoptotic bodies. MiRNAs associated with cellular particles were found to be resistant from nuclease degradation and able to be transferred in a variety of cells and alter the gene expression of the recipient cells (62–64). The novel technology designs for MSC-derived MPs programed to enclose and deliver specific miRNAs that affect the action of genes associated with cancer growth, neovascularization, and metastasis (65). MSC-derived MPs retain the membrane receptors that allow MSCs to home selectively into tumor sites and target malignant cells, thus avoiding the targeting of healthy cells. MSC-derived MPs home and engraft in solid tumors via specific chemokine receptors, fuse to tumor cell membranes, and incorporate miRNA directly into the target cancer cell, thus exerting their therapeutic effect while minimizing side effects associated with conventional therapies (65, 66). MSC-derived MPs offer enhanced therapeutic potential due to their ability to target multiple molecules in malignant cells when compared with approaches targeting single genes and induce immunosuppression through cytokine signaling inhibition making this approach useful in therapy of recurring cancer disease (65, 67).
As miRNA expression seems to be altered in many human diseases, including cancer, the miRNA revolution has already begun and has set the stage for “miRNA re-expression therapy.” Among the many genes that miRNAs can regulate are oncogenes and tumor suppressors, targets of drugs currently used in the clinic. Although a few miRNAs are overexpressed in cancer and seem to function as oncogenes themselves, a greater number of miRNAs have been shown to be downregulated in cancer and have the potential to act as tumor suppressors. MiRNA re-expression and down-regulation have both been shown to have antitumor effects while re-expressing a tumor suppressor miRNA could downregulate multiple oncogenes. Re-expression, to physiological levels, of tissue-specific miRNAs that are lost in cancer can induce the de-differentiation of cancer cells. Re-expressing lost miRNA in a cell can deliver a dramatic effect, because miRNAs regulate a vast number of genes and pathways. However, similar to other RNA-related therapies the key challenge remains the inadequate delivery and stability of the therapeutic agents to the tumor site. MiRNA antagonists and mimics are already available in the market but limited to local administration applicable to only a few target tissues. On the other hand, miRNA therapeutics could follow similar approaches to siRNA chemistry, vector-based systems as in gene therapy, and/or vehicle-based delivery systems. Understanding miRNA biology and how it contributes to cancer development it will provide for new diagnostic and therapeutic tools. MiRNA-based therapy is not a traditional approach and will need further development and improvements in composition, stability, and delivery to the target areas. Nevertheless, miRNA profiling will open a new era in cancer biology providing a new and improved cancer classification system.
The Guest Associate Editor Aleksandra Filipovic declares that, despite having collaborated with author Agamemnon A. Epenetos, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol (2007) 23:675–99. doi: 10.1146/annurev.cellbio.22.010305.104154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Colmont CS, Harding KG, Piguet V, Patel GK. Human skin cancer stem cells: a tale of mice and men. Exp Dermatol (2012) 21:576–80. doi:10.1111/j.1600-0625.2012.01533.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Wang Z, Li Y, Banerjee S, Sarkar FH. Exploitation of the Notch signaling pathway as a novel target for cancer therapy. Anticancer Res (2008) 28:3621–30.
4. Joutel A, Tournier-Lasserve E. Notch signalling pathway and human diseases. Semin Cell Dev Biol (1998) 9:619–25. doi:10.1006/scdb.1998.0261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Stylianou S, Clarke RB, Brennan K. Aberrant activation of Notch signaling in human breast cancer. Cancer Res (2006) 66:1517–25. doi:10.1158/0008-5472.CAN-05-3054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S. Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell (1985) 43:567–81. doi:10.1016/0092-8674(85)90229-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science (1995) 268:225–32. doi:10.1126/science.7716513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi:10.1016/j.cell.2009.03.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer (2011) 11:338–51. doi:10.1038/nrc3035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Reynolds TC, Smith SD, Sklar J. Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the beta T cell receptor gene in human lymphoblastic neoplasms. Cell (1987) 50:107–17. doi:10.1016/0092-8674(87)90667-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett (2009) 279:8–12. doi:10.1016/j.canlet.2008.09.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Dotto GP. Notch tumor suppressor function. Oncogene (2008) 27:5115–23. doi:10.1038/onc.2008.225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet (2003) 33:416–21. doi:10.1038/ng1099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res (2005) 65:8530–7. doi:10.1158/0008-5472.CAN-05-1069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res (2004) 64:6854–7. doi:10.1158/0008-5472.CAN-04-2500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Wang Z, Li Y, Banerjee S, Kong D, Ahmad A, Nogueira V, et al. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J Cell Biochem (2010) 109:726–36. doi:10.1002/jcb.22451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Reedijk M, Odorcic S, Zhang H, Chetty R, Tennert C, Dickson BC, et al. Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol (2008) 33:1223–9. doi:10.3892/ijo_00000112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Wang Z, Banerjee S, Li Y, Rahman KM, Zhang Y, Sarkar FH. Down-regulation of Notch-1 inhibits invasion by inactivation of nuclear factor-kappaB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res (2006) 66:2778–84. doi:10.1158/0008-5472.CAN-05-4281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, et al. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res (2013) 19:1972–80. doi:10.1158/1078-0432.CCR-12-0370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Wang K, Wu X, Wang J, Huang J. Cancer stem cell theory: therapeutic implications for nanomedicine. Int J Nanomedicine (2013) 8:899–908. doi:10.2147/IJN.S38641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Grego-Bessa J, Diez J, Timmerman L, de la Pompa JL. Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle (2004) 3:718–21. doi:10.4161/cc.3.6.949
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med (2007) 204:2935–48. doi:10.1084/jem.20071082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res (2009) 69:7569–76. doi:10.1158/0008-5472.CAN-09-0529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Garzia L, Andolfo I, Cusanelli E, Marino N, Petrosino G, De Martino D, et al. MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS One (2009) 4:e4998. doi:10.1371/journal.pone.0004998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA (2004) 10:1957–66. doi:10.1261/rna.7135204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet (2004) 5:522–31. doi:10.1038/nrg1415
28. Iorio MV, Croce CM. Causes and consequences of microRNA dysregulation. Cancer J (2012) 18:215–22. doi:10.1097/PPO.0b013e318250c001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. O’Hara SP, Mott JL, Splinter PL, Gores GJ, LaRusso NF. MicroRNAs: key modulators of posttranscriptional gene expression. Gastroenterology (2009) 136:17–25. doi:10.1053/j.gastro.2008.11.028
30. Di Leva G, Croce CM. miRNA profiling of cancer. Curr Opin Genet Dev (2013) 23:3–11. doi:10.1016/j.gde.2013.01.004
31. Deng S, Calin GA, Croce CM, Coukos G, Zhang L. Mechanisms of microRNA deregulation in human cancer. Cell Cycle (2008) 7:2643–6. doi:10.4161/cc.7.17.6597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A (2004) 101:2999–3004. doi:10.1073/pnas.0307323101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol (2006) 24:4677–84. doi:10.1200/JCO.2005.05.5194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther (2010) 10:1224–32. doi:10.4161/cbt.10.12.14252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Bian S, Sun T. Functions of noncoding RNAs in neural development and neurological diseases. Mol Neurobiol (2011) 44:359–73. doi:10.1007/s12035-011-8211-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer (2006) 6:857–66. doi:10.1038/nrc1997
37. Hammond SM. RNAi, microRNAs, and human disease. Cancer Chemother Pharmacol (2006) 58(Suppl 1):s63–8. doi:10.1007/s00280-006-0318-2
38. Mishra PJ, Merlino G. MicroRNA reexpression as differentiation therapy in cancer. J Clin Invest (2009) 119:2119–23. doi:10.1172/JCI40107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A (2005) 102:13944–9. doi:10.1073/pnas.0506654102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A (2002) 99:15524–9. doi:10.1073/pnas.242606799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature (2007) 447:1130–4. doi:10.1038/nature05939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One (2009) 4:e6816. doi:10.1371/journal.pone.0006816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. He C, Xiong J, Xu X, Lu W, Liu L, Xiao D, et al. Functional elucidation of MiR-34 in osteosarcoma cells and primary tumor samples. Biochem Biophys Res Commun (2009) 388:35–40. doi:10.1016/j.bbrc.2009.07.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Poellinger L, Lendahl U. Modulating Notch signaling by pathway-intrinsic and pathway-extrinsic mechanisms. Curr Opin Genet Dev (2008) 18:449–54. doi:10.1016/j.gde.2008.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Wu Z, Wu Y, Tian Y, Sun X, Liu J, Ren H, et al. Differential effects of miR-34c-3p and miR-34c-5p on the proliferation, apoptosis and invasion of glioma cells. Oncol Lett (2013) 6:1447–52. doi:10.3892/ol.2013.1579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther (2006) 5:483–93. doi:10.1158/1535-7163.MCT-05-0299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kashat M, Azzouz L, Sarkar SH, Kong D, Li Y, Sarkar FH. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res (2012) 4:432–42.
48. Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J (2011) 30:770–82. doi:10.1038/emboj.2010.349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Vallejo DM, Caparros E, Dominguez M. Targeting Notch signalling by the conserved miR-8/200 microRNA family in development and cancer cells. EMBO J (2011) 30:756–69. doi:10.1038/emboj.2010.358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Wang Z, Banerjee S, Ahmad A, Li Y, Azmi AS, Gunn JR, et al. Activated K-ras and INK4a/Arf deficiency cooperate during the development of pancreatic cancer by activation of Notch and NF-kappaB signaling pathways. PLoS One (2011) 6:e20537. doi:10.1371/journal.pone.0020537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res (2011) 71:6836–47. doi:10.1158/0008-5472.CAN-11-0846
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. de Antonellis P, Liguori L, Falanga A, Carotenuto M, Ferrucci V, Andolfo I, et al. MicroRNA 199b-5p delivery through stable nucleic acid lipid particles (SNALPs) in tumorigenic cell lines. Naunyn Schmiedebergs Arch Pharmacol (2013) 386:287–302. doi:10.1007/s00210-013-0837-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wang X, Lu H, Li T, Yu L, Liu G, Peng X, et al. Kruppel-like factor 8 promotes tumorigenic mammary stem cell induction by targeting miR-146a. Am J Cancer Res (2013) 3:356–73.
54. Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta (2010) 1806:258–67. doi:10.1016/j.bbcan.2010.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Furukawa S, Kawasaki Y, Miyamoto M, Hiyoshi M, Kitayama J, Akiyama T. The miR-1-NOTCH3-Asef pathway is important for colorectal tumor cell migration. PLoS One (2013) 8:e80609. doi:10.1371/journal.pone.0080609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Mosquera JM, Sboner A, Zhang L, Chen CL, Sung YS, Chen HW, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer (2013) 52:1075–87. doi:10.1002/gcc.22102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, et al. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis (2010) 31:1037–44. doi:10.1093/carcin/bgq066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Bu P, Chen KY, Chen JH, Wang L, Walters J, Shin YJ, et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell (2013) 12:602–15. doi:10.1016/j.stem.2013.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A (2008) 105:13556–61. doi:10.1073/pnas.0803055105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials (2012) 33:1462–76. doi:10.1016/j.biomaterials.2011.10.071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell (2009) 137:1005–17. doi:10.1016/j.cell.2009.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia (2006) 20:847–56. doi:10.1038/sj.leu.2404132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Prokopi M, Pula G, Mayr U, Devue C, Gallagher J, Xiao Q, et al. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood (2009) 114:723–32. doi:10.1182/blood-2009-02-205930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Rayner KJ, Hennessy EJ. Extracellular communication via microRNA: lipid particles have a new message. J Lipid Res (2013) 54:1174–81. doi:10.1194/jlr.R034991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Prokopi M, Epenetos A, Anayiotos A, Pitsillides C, Kapnisis K, Kousparou C. Therapeutic miRNAs targeted selectively to tumors by mesenchymal stem cell derived microparticles. Cancer Res (2014) 74:1977. doi:10.1158/1538-7445.AM2014-1977
66. Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat J, Warnock GL. Potential implications of mesenchymal stem cells in cancer therapy. Cancer Lett (2011) 305:8–20. doi:10.1016/j.canlet.2011.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Fritz V, Jorgensen C. Mesenchymal stem cells: an emerging tool for cancer targeting and therapy. Curr Stem Cell Res Ther (2008) 3:32–42. doi:10.2174/157488808783489462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Notch signaling, cancer stem cells, microRNAs, cancer therapeutics, miRNA therapeutics
Citation: Prokopi M, Kousparou CA and Epenetos AA (2015) The secret role of microRNAs in cancer stem cell development and potential therapy: a Notch-pathway approach. Front. Oncol. 4:389. doi: 10.3389/fonc.2014.00389
Received: 07 November 2014; Accepted: 31 December 2014;
Published online: 11 February 2015.
Edited by:
Aleksandra Filipovic, Imperial College London, UKReviewed by:
Giorgio Stassi, University of Palermo, ItalyCopyright: © 2015 Prokopi, Kousparou and Epenetos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianna Prokopi, Trojantec Ltd, The Bank of Cyprus Oncology Centre, 32 Acropoleos Avenue, 2006 Nicosia, Cyprus e-mail:cHJva29waS5tYXJpYW5uYUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.