 Joshua Oaks
Joshua Oaks Besim Ogretmen
Besim Ogretmen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 15 January 2015
Sec. Hematologic Malignancies
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00388
This article is part of the Research Topic The Emerging Role for PP2A in Hematologic Malignancies View all 8 articles
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that is a primary regulator of cellular proliferation through targeting of proliferative kinases, cell cycle regulators, and apoptosis inhibitors. It is through the regulation of these regulatory elements that gives PP2A tumor suppressor functions. In addition to mutations on the regulatory subunits, the phosphatase/tumor suppressing activity of PP2A is also inhibited in several cancer types due to overexpression or modification of the endogenous PP2A inhibitors such as SET/I2PP2A. This review focuses on the current literature regarding the interactions between the lipid signaling molecules, selectively sphingolipids, and the PP2A inhibitor SET for the regulation of PP2A, and the therapeutic potential of sphingolipids as PP2A activators for tumor suppression via targeting SET oncoprotein.
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase, which serves as a regulator of cell death and/or cell division, functions performed by targeting kinases and other effectors of these processes including Bcl-2 and the Aurora kinases (1–3). In non-malignant cells, PP2A restrains cell division to occur only when necessary stimuli is present, and induces cell death by triggering extrinsic or intrinsic death signals.
Structurally, PP2A is a holoenzyme, which typically contains a catalytic “C” subunit, coupled with an “A” scaffolding subunit, and a regulatory “B” subunit (Figure 1A) (4). Some form of PP2A holoenzyme is found in all tissues with variations of the subunit isoforms and/or post-translational modification of the subunits accounting for tissue specificity, subcellular distribution, and substrate selection. In this article, we focused on one of the major cancer-related PP2A inhibitors, SET, and its regulation by sphingolipids, primarily ceramide for PP2A reactivation and tumor suppression.
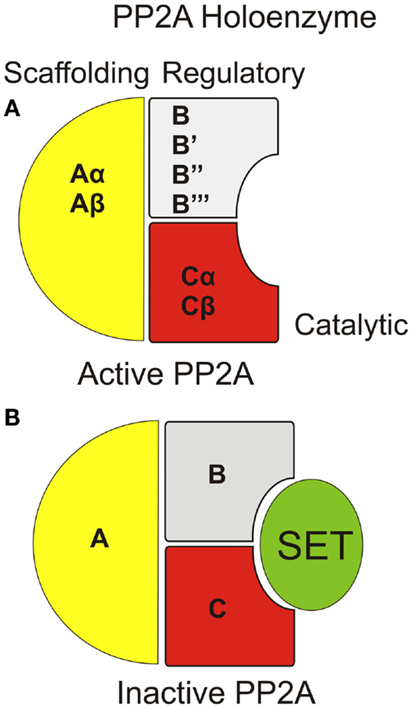
Figure 1. Organization of PP2A holoenzyme with/without SET/I2PP2A interaction. (A) The PP2A holoenzyme consists of a scaffolding “A,” regulatory “B,” and catalytic “C” subunit. Each subunit type can include any one of multiple species for each subunit. (B) SET inhibits PP2A activity through direct interaction within the catalytic domain of PP2A.
Protein phosphatase 2A was first identified as a tumor suppressor when it was discovered that the known carcinogen okadaic acid is a specific PP2A inhibitor (5–7). Similarly, the transforming (oncogenic) simian vacuolating virus 40 (SV40) small T-antigen is believed to be tumorigenic due to PP2A inhibition (8). Likewise, PP2A activity was inhibited in a variety of cancers including chronic myeloid leukemia (CML), acute myeloid leukemia (AML), polycythemia vera (PV), and lung cancer (9–12). Importantly, reactivation of PP2A through genetic or pharmacologic means induces cell death in various cancer cells in culture and in animal models.
Protein phosphatase 2A can be regulated through changes in phosphatase activity as well as changes in substrate specificity, the latter of which is primarily controlled by the regulatory B subunits. The B subunits are often tissue-specific and mediate subcellular localization of the holoenzyme. The catalytic activity is controlled by post-translational modification of the C-subunit and through endogenous inhibitory proteins. Although the specific interplay between each of these inhibitory proteins and the PP2A holoenzyme is not fully understood, they are typically found together in a complex suggesting a role for direct inhibition.
One of these endogenous inhibitors is Inhibitor 1 of PP2A (I1PP2A, also known as acidic leucine-rich nuclear phosphoprotein 32 A or ANP32A) (13). I1PP2A (ANP32A) is a sphingosine/dimethylsphingosine-sensitive PP2A inhibitor that regulates PP2A function in human umbilical vein endothelia cells (HUVEC) (14). I1PP2A also functions as part of the histone acetyltransferase regulatory INHAT complex. Another member of the ANP32 family, ANP32e (formerly cerebellar developmental-regulated protein 1, CPD1), is a PP2A inhibitor and also plays a role in neuronal development (15, 16).
Cancerous Inhibitor of PP2A (CIP2A) was first described as a fusion protein detected in a leukemia patient (17), which is an endogenous PP2A inhibitor that has been shown to have both proto-oncogenic and prognostic value in cancer. CIP2A is overexpressed in several different cancer types including head and neck squamous cell carcinoma (HNSCC), colon-cancer and CML (18), a subset of myleodysplasic syndromes (19), and osteosarcoma (20) as well as others (21). CIP2A also serves as a prognostic marker in cutaneous malignant melanoma (CMM) (22), cholangiocarcinoma (23), pancreatic ductal adenocarcinoma (PDA) (24), aggressive astrocytoma (25), and others (26, 27). Physiologically, CIP2A is particularly important for shielding c-Myc from dephosphorylation by PP2A, leading to protection of c-Myc from proteasomal degradation (28).
Inhibitor 2 of PP2A (I2PP2A/SET also known as TAF-1β) is an endogenous PP2A inhibitor and a well-described proto-oncoprotein. Although the specific mechanism by which SET inhibits PP2A is believed to be through direct binding to the catalytic domain of PP2A (Figure 1B), the details of this interaction have yet to described. Similar to CIP2A, SET plays a role in a variety of cancers (29, 30) and was originally discovered as a chimeric protein (31). A fusion protein consisting of the entire SET protein sequence coupled with the carboxyl-terminal end of the CAN nuclear pore protein was found in a single acute lymphocytic leukemia patient sample and postulated to be an oncogene (32, 33). Mechanistically, SET inhibits PP2A in cell-free assays at low (nanometer) concentrations while not inhibiting related phosphatases (34). Since its discovery, SET has been found to be involved in a variety of cancers, primarily as an inhibitor of PP2A. Specifically, there are three described mechanisms by which SET has been shown to become an active PP2A inhibitor: (i) overexpression of SET is detected in several leukemias, Wilms tumors (9, 29), epithelial ovarian cancer (35), prostate cancer (36), and lung cancer (12); (ii) altered phosphorylation of SET was observed in prostate cancer, PV, Alzheimer’s disease, and in activated T-cells with the suspected kinases defined as PKC, casein kinase II, and PKD, respectively. PI3K is also suspected of phosphorylating SET for activation (11, 36–39); and (iii) changes in endogenous ceramide (an inhibitor of SET, discussed below) are also common in several cancers, reviewed in Ref. (40). Independent of PP2A inhibitory function, SET also functions to regulate gene expression through alteration of histone acetylation (41, 42).
Several of the domains of the SET protein have been attributed to specific functions. An internal span close to the N-terminus (residues 36–124) has been found to be critical for PP2A inhibitory function (43). Conversely, the C-terminal end is responsible for histone binding (44). Additional published data demonstrate that both the N- and C-terminus segments of granzyme A (45) cleaved SET at N175 to bind and inhibit PP2A (46). The coiled-coil domain (E25-Q65) is critical for SET dimerization, which modulates some, but not all, of the nuclear functions of SET. Dimerization is not necessary for SET to inhibit PP2A (47). Interestingly, the structural details of specific interactions between SET and the PP2A holoenzyme with specific A, B, and C subunits with tumor suppressor functions have not yet been described.
Overexpression of SET is found in CML, Wilms tumors, and in lung cancers. In CML, an increase in heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) leads to an enhanced mRNA stability, which produces an increase in SET protein levels (9). Knock-down of SET in both CML cell lines as well as CML CD34+ primary patient samples restored PP2A activity indicating a causal role for SET in PP2A inhibition in these cells. Additionally, CML cells expressing SET knock-down had reduced BCR/ABL1 phosphorylation (activity) as well as reduced clonogenic potential. SET was also found to be overexpressed in lung tumors when compared to normal lung tissue with an accompanying increase in PP2A phosphorylation (inhibition) (12). Knock-down of SET in A549 lung cancer cells produced an increase in PP2A activity and a reduction in tumor proliferation in situ and in SCID mice. SET was found overexpressed in B-CLL cells as well as in the immortal, non-CLL B-cell cell lines, Raji and Ramos (48). Reactivation of PP2A through SET knock-down using shRNAs or a SET-targeting peptide induced cytotoxicity in both Raji and Ramos cells (48).
Ceramides are a class of sphingolipids consisting of a sphingosine backbone, which is N-acylated and joined to a fatty acid moiety (Figure 2). Ceramide can be generated de novo through a series of steps starting with serine palmitoyltransferase (SPT), which uses acetyl-CoA and serine to form 3-keto-dihydrosphingosine. This product is desaturated by dihydrosphingosine desaturase (DES) to produce dihydrosphingosine, which is the acylated by ceramide synthase (CerS1-6) to form ceramide with the desaturation reaction by dihydroceramide desaturase. Alternative pathways of synthesis of ceramide are through the breakdown of sphingomyelin by sphingomyelinases or originating directly from sphingosine produced by the dephosphorylation of sphingosine-1-phosphate (S1P) via the salvage pathway involving CerS function (Figure 2).
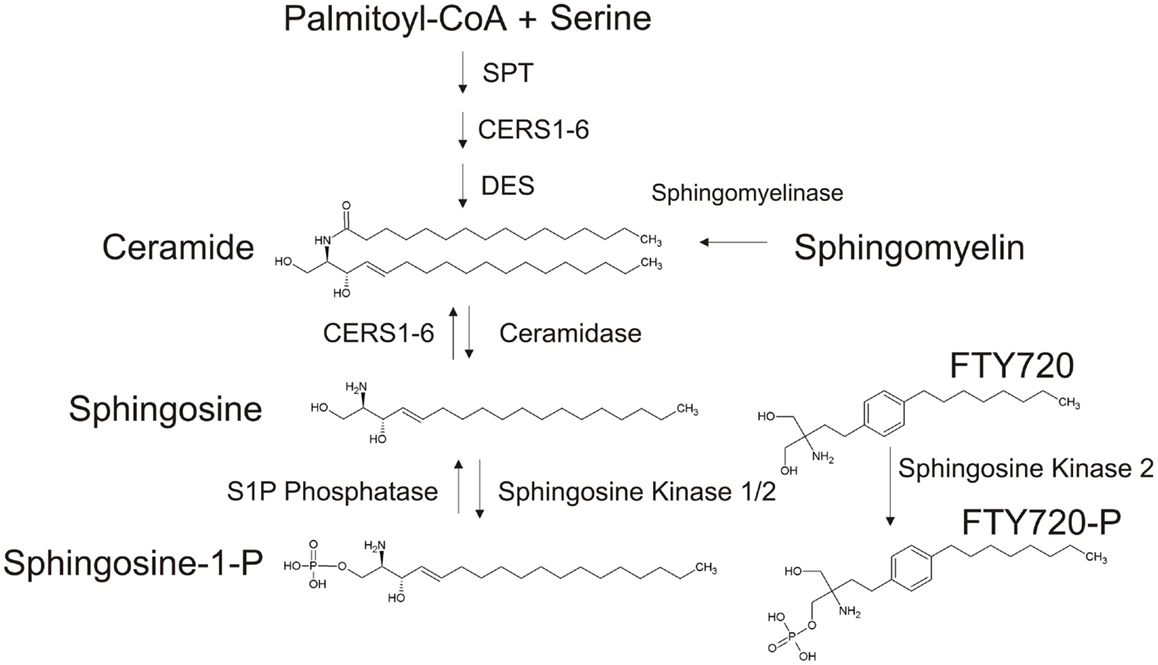
Figure 2. Generation of ceramide and FTY720-P. Ceramide can be generated through de novo synthesis originating with palmitoyl-CoA and serine, though the hydrolysis of sphingomyelin or through the “salvage pathway” via the acetylation of sphingosine. FTY720, a sphingosine (myriocin) analog, is phosphorylated by SphK (mainly by Sphk2 in the nucleus).
Ceramide was shown to activate PP2A (49–51), as well as the related phosphatase PP2C (52), without any mechanistic information. Recent data provided a unique mechanism for ceramide-mediated PP2A activation, revealing that ceramide directly binds SET, relieving PP2A from SET, increasing PP2A activity, leading to tumor suppression, consistent with anti-proliferative roles of ceramide (48–52).
FTY720 (fingolimod, Gilenya, Novartis) is a synthetic sphingosine (myriocin) analog currently approved for the treatment of multiple sclerosis. Originally designed as an immunosuppressant, FTY720 is given orally as a pro-drug. Once internalized within a target cell, FTY720 is mainly phosphorylated by sphingosine kinase 2 (SphK2), producing FTY720-P. Following phosphorylation, FTY720-P is exported out of the cell, where it functions in an autocrine/paracrine manner by binding to the sphingosine-1-phosphate receptor 1 (S1PR1). Following binding to S1PR (except S1PR2), the receptor is internalized and degraded, leading to immune suppression. Thus FTY720-P is a functional antagonist for S1PR1. FTY720 was found to activate PP2A by Nagahara et al. in HL-60RG and Jurkat cells (53). As a sphingosine analog, FTY720 retains many of the functional characteristics of natural sphingolipids, but it has a good bioavailability and a protracted half-life (54).
While SET can be regulated through expression levels or phosphorylation changes, the inhibitory properties of SET are also controlled through sphingolipid signaling. While sphingolipids have been shown to bind another PP2A inhibitor, I1PP2A/ANP32A, SET is unique in that it contains a binding “pocket,” which affords high affinity and substrate selectivity for ceramide.
Ceramide was described as a PP2A activator in 1993 (49) and, although there has been evidence of an interaction between PP2Ac and ceramide (55), an alternative mechanism by which ceramide activates PP2A is likely through direct binding to SET.
In fact, SET was found to be a direct target of ceramide by Mukhopadhyay et al. by employing biotin-labeled C6-ceramide to pull-down ceramide associated proteins found in A549 lung carcinoma cell extracts using avidin-conjugated columns (56). The prominent protein found through SDS-PAGE was excised and identified though LC/MS as SET. Further examination helped identify that different species of ceramide had vastly different binding affinities for SET. First, using recombinant SET protein, our laboratory found that only the natural isomer, D-e-C6-ceramide, and not L-e-C6-ceramide binds SET. Additionally, de novo-generated biotin-labeled C18-ceramide, but not C16-ceramide, bound to SET following an avidin pull-down. Moreover, purified trimeric PP2A supplemented with recombinant SET was activated by the addition of water-soluble pyridinium (Pyr) D-e-C6-ceramide, while its isomers Pyr-dihydro-C6-ceramide, Pyr-L-e-C6-ceramide, Pyr-D-theo-C6-ceramide, or Pyr-D-theo-C6-ceramide produced no PP2A activation. Taken together, these data indicate that ceramide functions as a PP2A activator through, at least in part, the binding to SET.
This attribute of ceramide selectivity for SET binding led to the construction of a molecular model wherein ceramide could be docked and specific interactions were predicted. Using computational modeling, a hydrophobic pocket within the SET protein was identified as the expected site for ceramide binding. This pocket includes the residue K209 on helix 7, which was predicted through in silico modeling to be significant for ceramide binding. When mutated to K209D (replacing the positively charged R-group of lysine with the negatively charged group of aspartate), the ability of ceramide to bind SET was decreased by >75%. Moreover, ceramide-mediated PP2A activation induced proteasomal degradation of c-Myc, and tumor suppression (56).
Additional work, which demonstrates the binding of ceramide to SET, expanded on the interactions between ceramide and SET but also ascertained the mechanism of action of the PP2A activator/synthetic sphingolipid analog FTY720 (12). Data indicated preferential binding of C18-ceramide over C16-ceramide to SET. Indeed, C18-ceramide had the highest affinity for wild-type SET, although lesser affinity ceramides were also found to bind SET including C20-, C22-, C26-ceramides, and minute binding of C24-ceramide. Moreover, in addition to the K209 residue (56), Y122 was suspected to interact with K209 via a hydrophobic/ionic interaction. The Y122C mutant of SET was employed to determine the potential role of opening the “gate” produced through the K209/Y122 interaction with ceramide. The Y122C mutant bound to all ceramides. More importantly, by using a SET mutant, which is retained within the endoplasmic reticulum (ER) rather than the nucleus, the ER-targeted SET demonstrated high affinity for C16- rather than C18-ceramide despite the higher concentration of C16-ceramide in both compartments (12).
When compared to normal lung tissue, an increase in SET protein levels as well as a corresponding decrease in C18-ceramide were detected, indicating a two-pronged reduction in PP2A activity through both an increase in SET and a decrease in the SET inhibitor C18-ceramide. Moreover, mRNA levels of ceramide synthase 1 (CerS1), which is responsible for the generation of C18-ceramide were decreased in lung tumor tissue compared to normal lung tissue. Interestingly, the mRNA of CerS6 and its product, C16-ceramide were both increased in lung tumor tissue (12).
The structural and functional similarities of the synthetic sphingolipid analog FTY720 (myriocin analog) suggest that it too, may be a ligand for SET. In silico, molecular docking experiments indicated that the binding of FTY720, but not FTY720-P, to SET is favorable and employs the same K209 residue as ceramide. Indeed, using a combination of LC/MS, surface plasma resonance (SPR) and biotin-tagged FTY720 pull-downs FTY720, but not immunosuppressive FTY720-P, was found to bind directly to SET (12). Similar to the findings with ceramide, the K209D mutant SET did not bind FTY720 with the high affinity when compared to wild-type SET (12). These studies suggested that FTY720 and not immune suppressive FTY720-P exerts anti-cancer activities against various cancer types. Interestingly, FTY720-P was shown to inhibit colitis-mediated colon-cancer growth/proliferation (57). However, whether PP2A reactivation was involved in the suppression of colitis-mediated colon tumor growth/proliferation by FTY720-P, which appeared to be phosphorylated by tumor SphK1 in SphK2−/− mice, has not been examined.
FTY720 has favorable pharmacokinetic properties compared to natural ceramides, therefore Saddoughi et al. used FTY720 as a therapeutic drug for the treatment of A549 lung cancer cell-derived tumors. Interestingly, both FTY720 and SET knock-down prevented the growth of A549 in situ and in vivo. Surprisingly, through the use of caspase 3/7 knockout MEFs and the caspase inhibitor Z-VAD, FTY720-induced cell death was found to be caspase-independent (12). Further experiments using receptor-interacting serine/threonine-protein kinase 1 (RIP1) knockout cells, siRNA, or chemical inhibitor of RIPK1 (necrostatin) helped determine the cell death induced by FTY720 in A549 cells to be a caspase-independent, RIP1-dependent programed necrosis known as necroptosis (12).
Prior to the quantitation of SET levels in CLL by Christensen et al. (48), Liu et al. found therapeutic potential in administering FTY720 to primary CD19+ lymphocytes from B-CLL patients (58). Likewise, several cell lines were found to be sensitive to FTY720; MEC-1 B-CLL derived cells, Ramos and Raji cells (described above), and the acute lymphoblastic leukemia cell lines 697 and RS4;11. While SET was not examined in this study, PP2A was found to be activated by FTY720 and re-inhibition of PP2A with okadaic acid rescued cells from FTY720-mediated cytotoxicity. While the cytotoxic effects of FTY720 were found to be PP2A-dependent, the primary receptor involved in FTY720-induced immunosuppression, the S1PR1, was found to be not involved (48).
Similarly, Oaks et al. found that targeting SET with FTY720 was able to selectively kill hematopoietic progenitor cells expressing the oncogenic Jak2V617F tyrosine kinase (11). Again, FTY720-P was found to be ineffective in activating PP2A. While cells expressing Jak2V617F did not exhibit increased SET expression, serine phosphorylation of SET was found to require Jak2 activity. Indeed, phosphorylation of SET on the serine 9 residue was necessary for complete PP2A inhibition, consistent with data obtained in other models (37).
Mukhopadhyay et al. have found that synthetic C6-ceramide is a viable treatment for prostate cancer cells, which overexpress the SET protein (59). When compared to normal prostate epithelium, the androgen-resistant prostate cancer cell lines PC-3 and DU145, as well as the androgen-sensitive LNCaP cell line were all found to have elevated SET levels. Likewise, c-Myc was also overexpressed in all of the prostate cancer cells compared to the normal prostate tissue. While total PP2Ac levels were consistent between the cancer and non-cancerous cell lines, PP2Ac phosphorylation at tyrosine 307 was higher in cancer cell lines indicating that the PP2A was inactive. Ceramide levels were examined in these cells, and it was found to induce apoptosis (60), a known activator of PP2A (49) and a direct inhibitor of SET (56). Therefore, endogenous ceramide levels were measured in cancerous PC-3 and LNCaP cells and compared to normal prostate epithelium. The PC-3 cells were found to have elevated C20-ceramide and a decrease in C14-, C16-, C18-, C22-, C24-, and C26-ceramides. The LNCaP cells had an elevated C16-, C18-, and C20- and lower levels of C14, C22-, C24-, and C26-ceramides.
To increase ceramide levels, synthetic C6-ceramide was given at concentrations ranging from 5 to 40 μM and cell viability assayed 24 and 48-h post-treatment (59). While normal prostate epithelium as unaffected at any dose or time point, all three prostate cancer cell lines showed a dose- and time-dependent cell death. Importantly, treatment with C6-ceramide (10 μM, 48 h) reduced c-Myc levels in each of the cancer cell lines. Moreover, pretreatment with the PP2A inhibitor okadaic acid (10 nM) increased basal c-Myc levels and prevented a decrease following treatment with C6-ceramide (10 μM, 48 h).
Recently, Chen et al. found that treating AML-ETO+ cell lines with FTY720-induced caspase-depended apoptosis through a PP2A-dependent pathway (61). To unravel the mechanisms involved in FTY720-induced cell death in these cells, the authors performed gene microarray analyses. Among the genes found to be upregulated by FTY720 were acid sphingomyelinase, acid beta-glucosidase and sphingolipid delta(4)-desaturase, all of which are involved in the regulation of ceramide metabolism. Further study using qPCR found another ceramide-generating gene, neutral sphingomyelinase 2 (nSmase2) to be upregulated with FTY720 treatment. Accordingly, using high-performance liquid chromatography–electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS), the authors found an increase in ceramide levels in FTY720-treated Kasumi-1 cells. In particular, C18-, C20-, and C22-ceramides were increased in a time (4–6 h) and dose (7.5–10 μM)-dependent manner. Particularly, these same ceramides were enriched in the mitochondrial fraction of FTY720-treated cells with C22-ceramide showing a dominant increase. Intriguingly, sphingosine-1-phosphate (S1P), a known inhibitor of PP2A activity (11), was found to be decreased following FTY720 treatment. Accordingly, ceramide synthase inhibitor fumonisin B1 (FB1) and the neutral sphingomyelinase inhibitor GW4869 partially protected Kasumi-1 cells from apoptosis following FTY720 treatment (61).
Protein phosphatase 2A is commonly deregulated in cancers, typically due to inhibition by SET. The protein level of SET can be increased or SET can be subject to post-translational modification through a variety of oncogenic pathways, each resulting in the inhibition of PP2A activity (Figure 3). The commonality of PP2A inhibition by SET oncoprotein, which is overexpressed in various tumor tissues compared to non-cancerous tissue counterparts, makes SET a promising target for cancer therapeutics. Previously, most of the research on PP2A used ceramide as tool for activation. The recent data (12) provide us with the groundwork for understanding the biological properties of both ceramide/FTY720 and PP2A. Our knowledge of the relationship between sphingolipids and PP2A has been greatly enhanced through the employment of mass spectrometry, which allows for identification of specific lipids and lipid species, as well as computational modeling of SET to estimate the interactions between ceramide/FTY720 and SET. The use of sphingolipids and, synthetic sphingolipid analogs, or newly developed SET inhibitors, as cancer therapeutics is likely to increase as we are better able to target SET to reactivate PP2A, leading to tumor suppression. We believe that structural studies coupled with studies to identify the mechanism by which ceramide/SET binding regulates tumor suppressor PP2A will lead to the development of novel SET inhibitors with improved anti-cancer activity without immune suppression and other toxic effects.
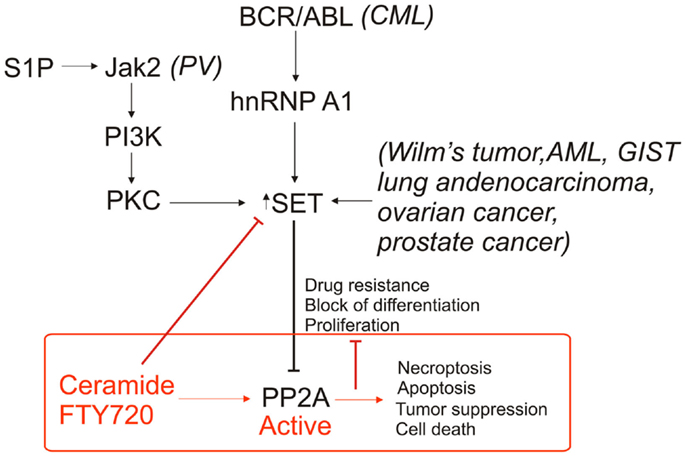
Figure 3. Roles of SET/I2PP2A and ceramide/FTY720 on PP2A-dependent regulation of cell death. PP2A inhibition is inhibited through various mechanisms in cancer via an increase in SET expression as well as altered phosphorylation of SET. Targeting SET by ceramide and/or FTY720 inhibits SET, activates PP2A, and induces cell death in various cancer cells.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J (2001) 353(Pt 3):417–39. doi: 10.1042/0264-6021:3530417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Horn V, Thelu J, Garcia A, Albiges-Rizo C, Block MR, Viallet J. Functional interaction of aurora-A and PP2A during mitosis. Mol Biol Cell (2007) 18(4):1233–41. doi:10.1091/mbc.E06-12-1152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Low IC, Loh T, Huang Y, Virshup DM, Pervaiz S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood (2014) 124(14):2223–34. doi:10.1182/blood-2014-03-563296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Kremmer E, Ohst K, Kiefer J, Brewis N, Walter G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol Cell Biol (1997) 17(3):1692–701.
5. Saydam G, Aydin HH, Sahin F, Selvi N, Oktem G, Terzioglu E, et al. Involvement of protein phosphatase 2A in interferon-alpha-2b-induced apoptosis in K562 human chronic myelogenous leukaemia cells. Leuk Res (2003) 27(8):709–17. doi:10.1016/S0145-2126(02)00347-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J (1988) 256(1):283–90.
7. Suganuma M, Fujiki H, Suguri H, Yoshizawa S, Hirota M, Nakayasu M, et al. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci U S A (1988) 85(6):1768–71. doi:10.1073/pnas.85.6.1768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Mungre S, Enderle K, Turk B, Porras A, Wu YQ, Mumby MC, et al. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-t antigen in vitro decrease viral transformation. J Virol (1994) 68(3):1675–81.
9. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell (2005) 8(5):355–68. doi:10.1016/j.ccr.2005.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Roberts KG, Smith AM, McDougall F, Carpenter H, Horan M, Neviani P, et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res (2010) 70(13):5438–47. doi:10.1158/0008-5472.CAN-09-2544
11. Oaks JJ, Santhanam R, Walker CJ, Roof S, Harb JG, Ferenchak G, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood (2013) 122(11):1923–34. doi:10.1182/blood-2013-03-492181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med (2013) 5(1):105–21. doi:10.1002/emmm.201201283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell (2001) 104(1):119–30. doi:10.1016/S0092-8674(01)00196-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Habrukowich C, Han DK, Le A, Rezaul K, Pan W, Ghosh M, et al. Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J Biol Chem (2010) 285(35):26825–31. doi:10.1074/jbc.M110.147058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Hoffarth S, Zitzer A, Wiewrodt R, Hahnel PS, Beyer V, Kreft A, et al. pp32/PHAPI determines the apoptosis response of non-small-cell lung cancer. Cell Death Differ (2008) 15(1):161–70. doi:10.1038/sj.cdd.4402256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Hayashi E, Kuramitsu Y, Okada F, Fujimoto M, Zhang X, Kobayashi M, et al. Proteomic profiling for cancer progression: differential display analysis for the expression of intracellular proteins between regressive and progressive cancer cell lines. Proteomics (2005) 5(4):1024–32. doi:10.1002/pmic.200401132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Coenen EA, Zwaan CM, Meyer C, Marschalek R, Pieters R, van der Veken LT, et al. KIAA1524: a novel MLL translocation partner in acute myeloid leukemia. Leuk Res (2011) 35(1):133–5. doi:10.1016/j.leukres.2010.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell (2007) 130(1):51–62. doi:10.1016/j.cell.2007.04.044
19. Li N, Abe S, Kurata M, Abe-Suzuki S, Onishi I, Kirimura S, et al. Over-expression of cancerous inhibitor of PP2A (CIP2A) in bone marrow cells from patients with a group of high-risk myelodysplastic syndromes. Pathol Oncol Res (2014) 20(2):399–407. doi:10.1007/s12253-013-9709-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Zhai M, Cong L, Han Y, Tu G. CIP2A is overexpressed in osteosarcoma and regulates cell proliferation and invasion. Tumour Biol (2014) 35(2):1123–8. doi:10.1007/s13277-013-1150-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Rantanen T, Kauttu T, Akerla J, Honkanen T, Krogerus L, Salo J, et al. CIP2A expression and prognostic role in patients with esophageal adenocarcinoma. Med Oncol (2013) 30(3):684. doi:10.1007/s12032-013-0684-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shi F, Ding Y, Ju S, Wu X, Cao S. Expression and prognostic significance of CIP2A in cutaneous malignant melanoma. Biomarkers (2014) 19(1):70–6. doi:10.3109/1354750X.2013.871752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Xu P, Huang Q, Xie F, Xu XL, Shao F. Increased expression of CIP2A in cholangiocarcinoma and correlation with poor prognosis. Hepatogastroenterology (2013) 60(124):669–72. doi:10.5754/hge11139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Wang L, Gu F, Ma N, Zhang L, Bian JM, Cao HY. CIP2A expression is associated with altered expression of epithelial-mesenchymal transition markers and predictive of poor prognosis in pancreatic ductal adenocarcinoma. Tumour Biol (2013) 34(4):2309–13. doi:10.1007/s13277-013-0775-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Yi F, Ni W, Liu W, Bai J, Li W. Expression and biological role of CIP2A in human astrocytoma. Mol Med Rep (2013) 7(5):1376–80. doi:10.3892/mmr.2013.1357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Yu G, Liu G, Dong J, Jin Y. Clinical implications of CIP2A protein expression in breast cancer. Med Oncol (2013) 30(2):524. doi:10.1007/s12032-013-0524-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood (2011) 117(24):6660–8. doi:10.1182/blood-2010-08-304477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Wiegering A, Pfann C, Uthe FW, Otto C, Rycak L, Mader U, et al. CIP2A influences survival in colon cancer and is critical for maintaining Myc expression. PLoS One (2013) 8(10):e75292. doi:10.1371/journal.pone.0075292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Carlson SG, Eng E, Kim EG, Perlman EJ, Copeland TD, Ballermann BJ. Expression of SET, an inhibitor of protein phosphatase 2A, in renal development and Wilms’ tumor. J Am Soc Nephrol (1998) 9(10):1873–80.
30. Fornerod M, Boer J, van Baal S, Jaegle M, von Lindern M, Murti KG, et al. Relocation of the carboxyterminal part of CAN from the nuclear envelope to the nucleus as a result of leukemia-specific chromosome rearrangements. Oncogene (1995) 10(9):1739–48.
31. Adachi Y, Pavlakis GN, Copeland TD. Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J Biol Chem (1994) 269(3):2258–62.
32. von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3’ half to different genes: characterization of the set gene. Mol Cell Biol (1992) 12(8):3346–55.
33. Saito S, Miyaji-Yamaguchi M, Nagata K. Aberrant intracellular localization of SET-CAN fusion protein, associated with a leukemia, disorganizes nuclear export. Int J Cancer (2004) 111(4):501–7. doi:10.1002/ijc.20296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem (1996) 271(19):11059–62. doi:10.1074/jbc.271.19.11059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ouellet V, Le Page C, Guyot MC, Lussier C, Tonin PN, Provencher DM, et al. SET complex in serous epithelial ovarian cancer. Int J Cancer (2006) 119(9):2119–26. doi:10.1002/ijc.22054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Anazawa Y, Nakagawa H, Furihara M, Ashida S, Tamura K, Yoshioka H, et al. PCOTH, a novel gene overexpressed in prostate cancers, promotes prostate cancer cell growth through phosphorylation of oncoprotein TAF-Ibeta/SET. Cancer Res (2005) 65(11):4578–86. doi:10.1158/0008-5472.CAN-04-4564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Yu G, Yan T, Feng Y, Liu X, Xia Y, Luo H, et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol Aging (2013) 34(7):1748–58. doi:10.1016/j.neurobiolaging.2012.12.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Irie A, Harada K, Araki N, Nishimura Y. Phosphorylation of SET protein at Ser171 by protein kinase D2 diminishes its inhibitory effect on protein phosphatase 2A. PLoS One (2012) 7(12):e51242. doi:10.1371/journal.pone.0051242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Prasad SV. Inhibition of protein phosphatase 2A activity by PI3Kgamma regulates beta-adrenergic receptor function. Mol Cell (2011) 41(6):636–48. doi:10.1016/j.molcel.2011.02.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Furuya H, Shimizu Y, Kawamori T. Sphingolipids in cancer. Cancer Metastasis Rev (2011) 30(3–4):567–76. doi:10.1007/s10555-011-9304-1
41. Kim JY, Lee KS, Seol JE, Yu K, Chakravarti D, Seo SB. Inhibition of p53 acetylation by INHAT subunit SET/TAF-Ibeta represses p53 activity. Nucleic Acids Res (2012) 40(1):75–87. doi:10.1093/nar/gkr614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kim JY, Kim KB, Son HJ, Chae YC, Oh ST, Kim DW, et al. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Ibeta. FEBS Lett (2012) 586(19):3159–65. doi:10.1016/j.febslet.2012.06.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Saito S, Miyaji-Yamaguchi M, Shimoyama T, Nagata K. Functional domains of template-activating factor-I as a protein phosphatase 2A inhibitor. Biochem Biophys Res Commun (1999) 259(2):471–5. doi:10.1006/bbrc.1999.0790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Karetsou Z, Emmanouilidou A, Sanidas I, Liokatis S, Nikolakaki E, Politou AS, et al. Identification of distinct SET/TAF-Ibeta domains required for core histone binding and quantitative characterisation of the interaction. BMC Biochem (2009) 10:10. doi:10.1186/1471-2091-10-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Beresford PJ, Kam CM, Powers JC, Lieberman J. Recombinant human granzyme A binds to two putative HLA-associated proteins and cleaves one of them. Proc Natl Acad Sci U S A (1997) 94(17):9285–90. doi:10.1073/pnas.94.17.9285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett (2011) 585(17):2653–9. doi:10.1016/j.febslet.2011.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Miyaji-Yamaguchi M, Okuwaki M, Nagata K. Coiled-coil structure-mediated dimerization of template activating factor-I is critical for its chromatin remodeling activity. J Mol Biol (1999) 290(2):547–57. doi:10.1006/jmbi.1999.2898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Christensen DJ, Chen Y, Oddo J, Matta KM, Neil J, Davis ED, et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood (2011) 118(15):4150–8. doi:10.1182/blood-2011-04-351072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem (1993) 268(21):15523–30.
50. Kowluru A, Metz SA. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett (1997) 418(1–2):179–82. doi:10.1016/S0014-5793(97)01379-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem (1999) 274(29):20313–7. doi:10.1074/jbc.274.29.20313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Perry DM, Kitatani K, Roddy P, El-Osta M, Hannun YA. Identification and characterization of protein phosphatase 2C activation by ceramide. J Lipid Res (2012) 53(8):1513–21. doi:10.1194/jlr.M025395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Nagahara Y, Matsuoka Y, Saito K, Ikekita M, Higuchi S, Shinomiya T. Coordinate involvement of cell cycle arrest and apoptosis strengthen the effect of FTY720. Jpn J Cancer Res (2001) 92(6):680–7. doi:10.1111/j.1349-7006.2001.tb01148.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Single-dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol (2004) 57(5):586–91. doi:10.1111/j.1365-2125.2003.02065.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res (2004) 45(3):496–506. doi:10.1194/jlr.M300347-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J (2009) 23(3):751–63. doi:10.1096/fj.08-120550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell (2013) 23(1):107–20. doi:10.1016/j.ccr.2012.11.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Liu Q, Zhao X, Frissora F, Ma Y, Santhanam R, Jarjoura D, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood (2008) 111(1):275–84. doi:10.1182/blood-2006-10-053884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Mukhopadhyay A, Tabanor K, Chaguturu R, Aldrich JV. Targeting inhibitor 2 of protein phosphatase 2A as a therapeutic strategy for prostate cancer treatment. Cancer Biol Ther (2013) 14(10):962–72. doi:10.4161/cbt.25943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science (1993) 259(5102):1769–71. doi:10.1126/science.8456305
61. Chen L, Luo LF, Lu J, Li L, Liu YF, Wang J, et al. FTY720 induces apoptosis of M2 subtype acute myeloid leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS One (2014) 9(7):e103033. doi:10.1371/journal.pone.0103033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ceramide, sphingolipids, FTY720, PP2A, tumor suppression
Citation: Oaks J and Ogretmen B (2015) Regulation of PP2A by sphingolipid metabolism and signaling. Front. Oncol. 4:388. doi: 10.3389/fonc.2014.00388
Received: 19 November 2014; Paper pending published: 18 December 2014;
Accepted: 27 December 2014; Published online: 15 January 2015.
Edited by:
Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Husheng Ding, Mayo Clinic, USACopyright: © 2015 Oaks and Ogretmen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Besim Ogretmen, Department of Biochemistry and Molecular Biology, Hollings Cancer Center, Medical University of South Carolina, 86 Jonathan Lucas Street, Charleston, SC 29425, USA e-mail:b2dyZXRtZW5AbXVzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.