 Akari Minami†
Akari Minami† Satoru Matsuda
Satoru Matsuda
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 10 November 2014
Sec. Molecular and Cellular Oncology
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00318
Genomic instability finally induces cell death or apoptosis. The tumor suppressor, phosphatase and tensin homolog on chromosome 10 (PTEN), is a dual-specificity phosphatase, which has protein phosphatase activity and lipid phosphatase activity that antagonizes PI3K activity. Cells that lack PTEN have constitutively higher levels of PIP3 and activated downstream PI3K/AKT targets. BRCA1, a well-known breast cancer tumor suppressor, is to associate with breast cancer risk and genetic susceptibility. Many studies have demonstrated that PTEN, as well as BRCA1, plays a critical role in DNA damage responses. The BRCA1 functionally cooperates with PTEN and might be an essential blockage in the development of several tumors. Actually, the PTEN and BRCA1 genes are recognized as one of the most frequently deleted and/or mutated in many human cancers. The PI3K/AKT pathway is constitutively active in BRCA1-defective human cancer cells. Loss or decrease of these PTEN or BRCA1 function, by either mutation or reduced expression, has a role in various tumor developments. This review summarizes recent findings of the function of BRCA1 and PTEN involved in genomic stability and cancer cell signaling.
Germline mutations in the breast cancer susceptibility gene 1 (BRCA1) extensively increase the risk of breast and ovarian cancers (1, 2). BRCA1-related tumorigenesis may be mainly caused by increased DNA damage and decreased genome stability that is a major hallmark of cancer (3). To maintain genomic integrity, cells are equipped with committed sensors to monitor DNA repair and/or to impose damaged cells into apoptotic cell death (4). Although functional roles of BRCA1 may include the regulation of DNA damage repair, cell cycle progression, and maintenance of genomic integrity, the precise function of the BRCA1 gene as a tumor suppressor is still not clear. It has been shown that BRCA1 deficiency activates the AKT oncogenic signaling pathway (5). Also, activation of the phosphoinositide 3-kinase (PI3K) is often associated with the BRCA1-related breast cancers in clinical sample (6). The PI3K/AKT pathway might have an essential role in the proliferation of malignant tumor cells related to the BRCA1 functions (Figure 1). BRCA1 can downregulate AKT activation via the direct physical interaction (5, 7). In addition, AKT activation inversely correlates with the BRCA1 expression in human breast cancers (8). Moreover, BRCA1 negatively regulates the PI3K/AKT pathway in breast cancer cells (9). Phosphatase and tensin homolog on chromosome 10 (PTEN) is a dual protein/lipid phosphatase that inhibits the PI3K/AKT pathway, whose inhibition eventually reduces cell growth and cell proliferation (10, 11). The PTEN is also a tumor suppressor molecule and seems to protect from bad prognosis of several cancers. In other words, absence of PTEN worsens prognosis in early stages of cancer (12, 13). Furthermore, germ-line mutations of PTEN are the cause of PTEN hamartoma tumor syndromes (Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, PTEN-related Proteus syndrome, Proteus-like syndrome) with increased risk for the development of cancers (14). The PTEN has been shown to be involved in an intricate network of interactions with other molecules (Figure 1). In this review, we summarize the current research and our view of how PTEN and BRCA1 function with their partners to transduce signals downstream and what are the implications for cancer-associated biology.
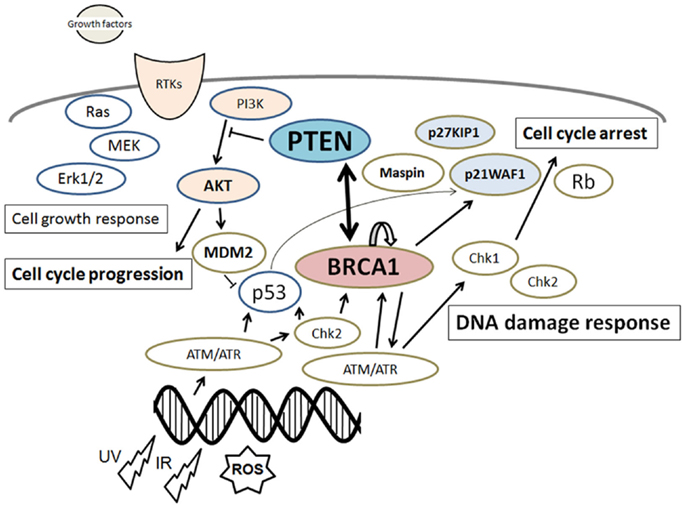
Figure 1. Schematic depiction of the integrative model of tumor suppressors signaling including PTEN and BRCA1. Examples of molecules known to act on DNA damage response, cell proliferation, and cell cycle via the regulatory pathways are shown. Note that some critical pathways have been omitted for clarity.
Because BRCA1 may play an essential role in maintaining genome stability, the mutation of BRCA1 is associated with increased genomic instability in cells (15), which consequently accelerates the mutation rate of other critical genes. Actually, studies have established functional roles for BRCA1 in DNA damage signaling, DNA repair processes, and cell cycle checkpoints (16, 17). In addition, inherited BRCA1 germline mutation revealed a genetic susceptibility leading to high risk of breast and ovarian cancers (18, 19). It has been identified that common variation in BRCA1 gene is also associated with prostate cancer (20). Increased prostate cancer risk and an aggressive clinical course have been reported for BRCA1 mutation (21). Furthermore, several important prostate cancer targets are modulated by BRCA1 (22). BRCA1 cDNA encodes for 1863 amino acids protein with two putative nuclear localization signals and an amino terminal conserved RING finger motif, which is the most common motif present in E3 ubiquitin ligases. The RING finger domain interacts with E2 ubiquitin ligases and exerts maximal E3 ligase activity (23). Knock-in mice with deficient BRCA1 RING finger mutant display diverse genomic instability and tumor-forming phenotypes (24). The carboxyl-terminal domain of BRCA1 is involved in association with specific phosphorylated proteins. BRCA1 itself becomes hyper-phosphorylated after exposure to the DNA damaging agents, and the specific function of BRCA1 seems to be regulated by the phosphorylation (25, 26). Exon 11 encodes a largely unstructured region of the BRCA1 protein that is phosphorylated by the ATM and Chk2 kinases in a DNA damage-dependent manner (27, 28). Principally, the main DNA damage recognition molecule may be the ATM, which is a checkpoint kinase that phosphorylates a number of proteins including BRCA1 and p53 in response to DNA damage (29). Inhibition of DNA repair pathway seems to block the mechanisms that are also required for cell survival in the presence of oncogenic mutations.
Several functions of BRCA1 including roles in the DNA repair may contribute to its tumor suppressor activity. Although BRCA1 gene mutations are rare in sporadic breast and/or ovarian cancers, BRCA1 protein expression is often reduced in the sporadic cancer specimens. The BRCA1 has the important role in concert with Rad50 and Rad51, a DNA recombinase related to the bacterial RecA protein, for the genome stability (30). Phosphorylation status of BRCA1 in response to DNA damage controls the selectivity of DNA repair events, and the function of BRCA1 seems to be regulated by this phosphorylation (30). It has been reported that Chk2 kinase and its downstream target BRCA1 have been functionally linked to the DNA damage response pathway (31). In addition, BRCA1 with the Chk2-mediated phosphorylation is also associated to the cellular spindle formation and chromosomal stability (31). The DNA repair system strictly maintains genomic fidelity through the recognition and repair of the damaged nucleotides. Genetic defects in DNA damage response genes and/or downregulation of the DNA repair mechanism certainly promote genomic instability, which can lead to carcinogenesis. Therefore, cells are equipped with multiple DNA repair mechanisms to the preservation of genomic stability (32). Basically, the role of BRCA1 in cell cycle control has been understood by its ability to interact with various cyclins and various cyclin-dependent kinases (33, 34). The BRCA1 activates the CDK inhibitor p21WAF1 and the p53 tumor suppressor protein, which regulates several genes that control cell cycle checkpoints (33, 34). Given the significant importance of the BRCA1 network in all proliferating cells, insights into the underlying mechanisms of BRCA1 function on chromatin might extend beyond hereditary cancers. Understanding such mechanisms of genome maintenance leads to an improved therapies that target DNA repair deficiency in a variety of malignancies. Hence, the regulation of DNA repair levels may be an innovative therapeutic modality in certain cancers. Either survival or apoptosis, which is determined by the balance between DNA damage and DNA repair levels, may raise the major problems in cancer therapy at that time (35).
Phosphatase and tensin homolog on chromosome 10 tumor suppressor gene is frequently deleted and mutated in various human cancers. Such many somatic PTEN mutations and loss of heterozygosity in cancer at the PTEN locus implicate a key role for PTEN in the etiology of various cancers (36, 37). Human genomic PTEN gene locus on chromosome 10q23.3 contains 9 exons encoding a 5.5 kb mRNA that has a 403 amino-acid open reading frame (38, 39). The PTEN gene is ubiquitously expressed throughout early embryogenesis in almost mammals (40). The PTEN enzyme prefers acidic phospholipid substrates such as PIP3 that is the principal second messenger of the PI3K pathway. The PI3K mediates receptor tyrosine kinase signaling to the survival kinase AKT (Figure 1). PTEN depressingly regulates the activity of PI3K/AKT signaling over converting phosphatidylinositol 3,4,5-triphosphate (PIP3) into phosphatidylinositol 4,5-bisphosphate (PIP2). PTEN might act as a regulator of keeping basal levels of PIP3 below a threshold for the signaling pathway activation. The PTEN inactivation is often involved in the carcinogenesis of some cancers (38), which causes an increase in cellular PIP3 levels. Subsequently, activated PI3K/AKT signaling causes increased expression of several genes for cell growth, cell survival, and cell migration, which are all critical for tumor development (39, 40). Remarkably, some of rosemary extracts may inhibit PTEN expression in K562 culture cells (41). PTEN can be controlled by posttranslational regulation including phosphorylation, acetylation, methylation, oxidation, and so on (42, 43). Because PTEN may be regulated by ubiquitin-mediated proteasomal degradation, a common mechanism to control protein levels, insecurity of PTEN correlated with some of its mutations has been shown to comprise protein interactions (44). Casein kinase 2 mediated phosphorylation stabilizes PTEN protein in an inactive state by inhibiting its proteasomal degradation (45, 46). Therefore, inhibition of the PTEN phosphorylation by the Casein kinase 2 results in enhanced PTEN activity and a subsequent suppression in AKT function (45, 46). Overexpression of PTEN induces growth inhibition by supporting cell cycle arrest, which needs lipid phosphatase activity of PTEN (47, 48). Overexpression of PTEN also correlates with decreased levels and nuclear localization of cyclin D1 (49, 50), a key cell cycle molecule regulated by AKT kinase. One of the mechanisms by which PTEN induces cell cycle arrest is by regulating AKT function so that levels of the cell cycle inhibitor p27KIP1 is increased (51, 52). Despite the central role of PTEN as a negative regulator of the PI3K pathway has been revealed, studies have reported that tumor suppressive activities of PTEN are exerted from within the nucleus, where catalysis of PIP3 does not seem to be present at least a dominant function of the enzyme (53, 54). Nuclear localization of PTEN seems to mediate tumor suppressive activities independent of the AKT pathway through inhibiting anchorage-independent growth (53, 54). The PTEN activities in nucleus may contain the regulation of gene expression and genomic stability (53, 54).
Several growth factor-activated AKT signaling pathway promotes progression of cell cycles by acting on downstream factors involved in controlling the G1/S and/or G2/M transitions (55). Studies have also implicated AKT kinase in modifying the status of genome stability and response for DNA damages (55). In addition, PTEN plays a critical role in damaged DNA repair through its interaction with ATM-p53 pathways in an AKT-independent manner (56). The upregulation of PTEN represses AKT and MDM2 activity, which enhances the level of p53, thereby inducing G2/M arrest and apoptosis (57, 58). In addition, it has been suggested that nuclear PTEN plays a distinctive role to protect cells upon oxidative damage (59). One mechanism by which reactive oxygen species (ROS) are thought to employ its effects may be through the regulation of target molecules including several kinases, PI3K, AKT, and PTEN (60). Actually, the catalytic activity of PTEN can be modulated by the ROS, and cellular PTEN activity is also repressed by the oxidative stress (60, 61). In addition, endogenous oxidant production in macrophages inactivates a fraction of the cellular PTEN (62, 63). It has been reported that ROS levels are increased in the retinal pigment epithelium cells in association with phosphorylation and inactivation of PTEN (64, 65). Phosphorylated inactivation of the PTEN and the consequent AKT activation in cells are withdrawn by antioxidant treatment. ROS mediates PTEN inactivation but ROS does not affect the PTEN expression. Hence, the uncontrolled generation of ROS might contribute to cell proliferation and tumor growth by inhibiting the PTEN function.
Several PI3K inhibitors favorably reduce proliferation of BRCA1-defective breast cancer cells. BEZ235 inhibits not only PI3K/mTOR but also ATM/ATR and some of DNA-dependent protein kinases with similar effectiveness in vitro (66, 67). It is possible that ATM pathways are involved in upregulation of the PI3K/AKT pathway in BRCA1-defective cancer cells. Perifosine, a PI3K/AKT inhibitor, prevents translocation of AKT from the cytoplasm to the plasma membrane by targeting the pleckstrin homology (PH) domain, thereby preventing phosphorylation of AKT by upstream kinases (68). Perifosine prevents proliferation of breast cancer cell lines in a BRCA1-dependent manner (9). Remarkably, combination of PI3K pathway inhibitors with chemotherapeutic drugs such as doxorubicin, cisplatin, or topotecan results in enhancing cancer cell killing properties in BRCA1-defective breast cancer cells (69, 70), suggesting that the PI3K/AKT pathway may be activated in BRCA1-defective breast cancer cells. Hence targeting this PI3K/AKT pathway in combination with chemotherapeutic agents is a plausible strategy for treatment of certain cancer cells. Importantly, it has been shown that depletion of AKT significantly reduces tumor formation induced by Brca1 deficiency in the KO mice (8). On the other hand, AKT activation promotes the expression of BRCA1. In addition, phosphorylation of BRCA1 by AKT increases total BRCA1 protein expression by preventing proteasomal degradation (7). However, it has also been reported that AKT phosphorylation has an inverse correlation with BRCA1 expression in human breast cancers (71, 72). Phosphorylation site in BRCA1 by AKT is at S694 of BRCA1 (7). AKT activation also appears to support nuclear localization of BRCA1, and co-expression of activated AKT with intact BRCA1 decreases radiation sensitivity (7), suggesting this interaction has functional consequences for BRCA1 function in DNA repair.
In contrast, BRCA1 may regulate the PI3K/AKT pathway by acting on upstream kinases of AKT. For example, overexpression of wild-type BRCA1 could further reduce basal phosphorylation (S473/T308) of AKT levels in MCF7 cells. Transient expression of wild-type BRCA1 also abolished the phosphorylation of AKT (S473/T308) in PTEN negative cells (9, 73). Negative mutations and/or decreased expression of the BRCA1 gene may thus activate the PI3K/AKT cancer proliferation pathway (5). In addition, BRCA1 may directly downregulate the AKT protein either by ubiquitin-mediated proteasomal degradation or by activating a protein serine/threonine phosphatase PP2A in breast cancer cells (5). BRCA1 mutant cells accumulate nuclear phosphor-AKT and subsequently inactivate the transcriptional activity of FOXO3a, a central nuclear target of the phosphor-AKT (74, 75). Significantly, some of breast cancers with BRCA1 mutations have high frequencies of PTEN mutations (76), and the resulting PI3K/AKT activation induces the growth of those cancers (77). PTEN loss is highly associated with BRCA1 breast cancers, which could result from genome instability involving homozygous deletions, DNA double-strand breaks and so on (76). Interestingly, PTEN loss is not observed in estrogen hormone receptor-positive BRCA1-associated tumors (76). Loss of PTEN expression might be a starting event in a variety of BRCA1-associated cancers (78). Nuclear PTEN might affect a variety of biological functions and plays a role in DNA repair, cell cycle arrest, and genome stability with BRCA1. In that case, PTEN acts on chromatin and regulates expression of Rad51, which reduces the incidence of spontaneous double-strand breaks (79, 80). Several reports have indicated that reduced levels of PTEN are associated with radioresistance, which can be suppressed by the ectopic PTEN expression (81, 82).
Genome stability might be sustained on several tumor suppressors (Figure 2). Loss of PTEN increases cell survival and reduces DNA repair, which may lead to genomic instability and may enhance radiosensitivity. In case of cancer cells that compromise therapeutic success, targeting inhibition of PTEN-related PI3K/AKT/mTOR pathway has been shown to prevent tumorigenesis and progression. Indeed, for example, rapamycin, an mTOR-specific inhibitor, prevents leukemia development in PTEN-null mouse models (83, 84). However, the effectiveness of rapamycin may require PTEN deletion or genetic loss of PTEN function. The presence of wild-type PTEN may compromise the efficacy of rapamycin (85). In addition, studies show that mTOR inhibition decreases PTEN transcription and subsequently activates AKT (85). Further detailed mechanistic understanding of the roles of PTEN in DNA repair and DNA damage response in different tissues and cell types will help us fully understand the precise molecular mechanisms by which PTEN maintains genomic stability and contributes to tumor suppression and therapeutic efficacy. PTEN and BRCA1 may be regulated and interact each other at multiple levels including transcription, protein modulation, and protein stability. Understanding the connection between tumor suppressor BRCA1 and PTEN would facilitate the development of effective agents and strategies to better treatment against cancer. The PTEN inhibitor has been shown to effectively activate primordial follicles both in neonatal mouse ovaries and in human ovarian cortical tissues (86, 87). It is important to investigate the functional linkage between PTEN and BRCA1 in those ovarian samples, and elucidation of interaction-specific functions may provide insight into regulatory aspects of these tumor suppressors as well as opportunities for therapeutic intervention. Indeed, the regulation is crucial for the effective design of novel ovarian cancer therapeutics. Further mechanistic studies are needed in order to understand the precise molecular mechanisms for the effective treatment of cancers with PTEN/BRCA1 signal alterations. Targets within this pathway could provide strategies for modulation of PTEN/BRCA1 proteins, which may prove therapeutically beneficial for breast, ovarian, and prostate cancer treatment.
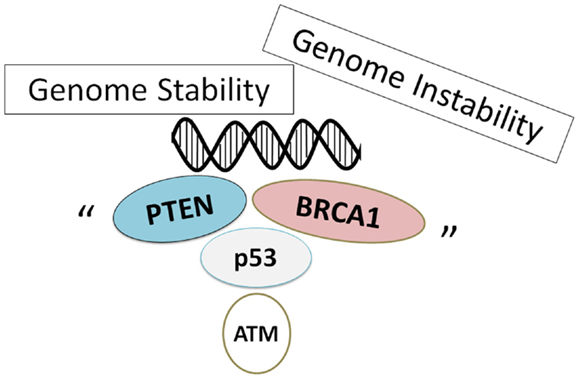
Figure 2. Schematic illustration implying that genome stability is sustained on several tumor suppressors. Note that some critical other functions have been omitted for clarity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by JSPS KAKENHI grant numbers 25560050, 26-12035, and 24240098.
ATM, ataxia telangiectasia mutated; BRCA1, breast cancer susceptibility gene 1; LOH, loss of heterozygosity; MDM2, murine double minute 2; NLS, nuclear localization signal; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PIP3, phosphatidylinositol 3,4,5-triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PI3K, phosphoinositide-3 kinase; RING, really interesting new gene finger domain; ROS, reactive oxygen species.
1. Paul A, Paul S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front Biosci (Landmark Ed) (2014) 19:605–18. doi: 10.2741/4230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. O’Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis (2010) 31:961–7. doi:10.1093/carcin/bgq069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Wang L, Di LJ. BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci (2014) 10:566–75. doi:10.7150/ijbs.8579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Bouafia A, Corre S, Gilot D, Mouchet N, Prince S, Galibert MD. p53 requires the stress sensor USF1 to direct appropriate cell fate decision. PLoS Genet (2014) 10:e1004309. doi:10.1371/journal.pgen.1004309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, Powell SN, et al. Negative regulation of AKT activation by BRCA1. Cancer Res (2008) 68:10040–4. doi:10.1158/0008-5472.CAN-08-3009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. López-Knowles E, O’Toole SA, McNeil CM, Millar EK, Qiu MR, Crea P, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer (2010) 126:1121–31. doi:10.1002/ijc.24831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Nelson AC, Lyons TR, Young CD, Hansen KC, Anderson SM, Holt JT. AKT regulates BRCA1 stability in response to hormone signaling. Mol Cell Endocrinol (2010) 319:129–42. doi:10.1016/j.mce.2010.01.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Xiang T, Jia Y, Sherris D, Li S, Wang H, Lu D, et al. Targeting the Akt/mTOR pathway in Brca1-deficient cancers. Oncogene (2011) 30:2443–50. doi:10.1038/onc.2010.603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Yi YW, Kang HJ, Kim HJ, Hwang JS, Wang A, Bae I. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances antitumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol Carcinog (2013) 52:667–75. doi:10.1002/mc.21905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets (2008) 8:187–98. doi:10.2174/156800908784293659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des (2010) 16:34–44. doi:10.2174/138161210789941865
12. Gao H, Ouyang X, Banach-Petrosky WA, Shen MM, Abate-Shen C. Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Res (2006) 66:7929–33. doi:10.1158/0008-5472.CAN-06-1637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Skírnisdóttir I, Seidal T. Prognostic impact of concomitant p53 and PTEN on outcome in early stage (FIGO I-II) epithelial ovarian cancer. Int J Gynecol Cancer (2011) 21:1024–31. doi:10.1097/IGC.0b013e31821dc906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst (2013) 105:1607–16. doi:10.1093/jnci/djt277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Eyfjord JE, Bodvarsdottir SK. Genomic instability and cancer: networks involved in response to DNA damage. Mutat Res (2005) 592:18–28. doi:10.1016/j.mrfmmm.2005.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res (2010) 108:73–112. doi:10.1016/B978-0-12-380888-2.00003-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene (1999) 18:7883–99. doi:10.1038/sj.onc.1203283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. George SH, Shaw P. BRCA and early events in the development of serous ovarian cancer. Front Oncol (2014) 4:5. doi:10.3389/fonc.2014.00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Moorman PG, Havrilesky LJ, Gierisch JM, Coeytaux RR, Lowery WJ, Peragallo Urrutia R, et al. Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: a systematic review and meta-analysis. J Clin Oncol (2013) 31:4188–98. doi:10.1200/JCO.2013.48.9021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Douglas JA, Levin AM, Zuhlke KA, Ray AM, Johnson GR, Lange EM, et al. Common variation in the BRCA1 gene and prostate cancer risk. Cancer Epidemiol Biomarkers Prev (2007) 16:1510–6. doi:10.1158/1055-9965.EPI-07-0137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, Vora K, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res (2010) 16:2115–21. doi:10.1158/1078-0432.CCR-09-2871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. De Luca P, Moiola CP, Zalazar F, Gardner K, Vazquez ES, De Siervi A. BRCA1 and p53 regulate critical prostate cancer pathways. Prostate Cancer Prostatic Dis (2013) 16:233–8. doi:10.1038/pcan.2013.12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem (2003) 278:5255–63. doi:10.1074/jbc.M204591200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Laufer M, Nandula SV, Modi AP, Wang S, Jasin M, Murty VV, et al. Structural requirements for the BARD1 tumor suppressor in chromosomal stability and homology-directed DNA repair. J Biol Chem (2007) 282:34325–33. doi:10.1074/jbc.M705198200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Thomas JE, Smith M, Tonkinson JL, Rubinfeld B, Polakis P. Induction of phosphorylation on BRCA1 during the cell cycle and after DNA damage. Cell Growth Differ (1997) 8:801–9.
26. Canning MT, Brown DA, Yarosh DB. A bicyclic monoterpene diol and UVB stimulate BRCA1 phosphorylation in human keratinocytes. Photochem Photobiol (2003) 77:46–51. doi:10.1562/0031-8655(2003)0770046ABMDAU2.0.CO2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D. Structure-function of the tumor suppressor BRCA1. Comput Struct Biotechnol J (2012) 1:e201204005. doi:10.5936/csbj.201204005
28. Bowen TJ, Yakushiji H, Montagna C, Jain S, Ried T, Wynshaw-Boris A. Atm heterozygosity cooperates with loss of Brca1 to increase the severity of mammary gland cancer and reduce ductal branching. Cancer Res (2005) 65:8736–46. doi:10.1158/0008-5472.CAN-05-1598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Khanna KK, Chenevix-Trench G. ATM and genome maintenance: defining its role in breast cancer susceptibility. J Mammary Gland Biol Neoplasia (2004) 9:247–62. doi:10.1023/B:JOMG.0000048772.92326.a1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol (2006) 173:195–206. doi:10.1083/jcb.200510130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol (2010) 12:492–9. doi:10.1038/ncb2051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol (2013) 5:a012724. doi:10.1101/cshperspect.a012724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev (2011) 25:2158–72. doi:10.1101/gad.16962311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Jhanwar-Uniyal M. BRCA1 in cancer, cell cycle and genomic stability. Front Biosci (2003) 8:s1107–17. doi:10.2741/1131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, Iseki S, Matsuda S. Against lung cancer cells: to be, or not to be, that is the problem. Lung Cancer Int (2012) 2012:8. doi:10.1155/2012/659365
36. Petrocelli T, Slingerland JM. PTEN deficiency: a role in mammary carcinogenesis. Breast Cancer Res (2001) 3:356–60. doi:10.1186/bcr322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem (2006) 97:433–47. doi:10.1002/jcb.20696
38. Li YL, Tian Z, Wu DY, Fu BY, Xin Y. Loss of heterozygosity on 10q23.3 and mutation of tumor suppressor gene PTEN in gastric cancer and precancerous lesions. World J Gastroenterol (2005) 11:285–8. doi:10.3748/wjg.v11.i2.285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Wang JY, Huang TJ, Chen FM, Hsieh MC, Lin SR, Hou MF, et al. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in advanced gastric carcinomas. Virchows Arch (2003) 442:437–43.
40. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A (2012) 109:3921–6. doi:10.1073/pnas.1117135109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Yoshida H, Okumura N, Kitagishi Y, Nishimura Y, Matsuda S. Ethanol extract of rosemary repressed PTEN expression in K562 culture cells. Int J Appl Biol Pharm Technol (2011) 2:316–22.
42. Zhang P, Chen JH, Guo XL. New insights into PTEN regulation mechanisms and its potential function in targeted therapies. Biomed Pharmacother (2012) 66:485–90. doi:10.1016/j.biopha.2012.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Singh G, Chan AM. Post-translational modifications of PTEN and their potential therapeutic implications. Curr Cancer Drug Targets (2011) 11:536–47. doi:10.2174/156800911795655930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets (2014) 15:65–79. doi:10.2174/1389450114666140106100909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol (2013) 33:3091–8. doi:10.1128/MCB.00319-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Barata JT. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv Enzyme Regul (2011) 51:37–49. doi:10.1016/j.advenzreg.2010.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Leslie NR, Downes CP. PTEN: the down side of PI 3-kinase signalling. Cell Signal (2002) 14:285–95. doi:10.1016/S0898-6568(01)00234-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Chu EC, Tarnawski AS. PTEN regulatory functions in tumor suppression and cell biology. Med Sci Monit (2004) 10:RA235–41.
49. Chung JH, Ostrowski MC, Romigh T, Minaguchi T, Waite KA, Eng C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet (2006) 15:2553–9. doi:10.1093/hmg/ddl177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res (2005) 65:8096–100. doi:10.1158/0008-5472.CAN-05-1888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Qiao L, Paul P, Lee S, Qiao J, Wang Y, Chung DH. Differential regulation of cyclin-dependent kinase inhibitors in neuroblastoma cells. Biochem Biophys Res Commun (2013) 435:295–9. doi:10.1016/j.bbrc.2013.04.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Seminario MC, Precht P, Wersto RP, Gorospe M, Wange RL. PTEN expression in PTEN-null leukaemic T cell lines leads to reduced proliferation via slowed cell cycle progression. Oncogene (2003) 22:8195–204. doi:10.1038/sj.onc.1206872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Jacob AI, Romigh T, Waite KA, Eng C. Nuclear PTEN levels and G2 progression in melanoma cells. Melanoma Res (2009) 19:203–10. doi:10.1097/CMR.0b013e32832ccd6e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci (2008) 121:249–53. doi:10.1242/jcs.022459
55. Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol (2012) 2012:951724. doi:10.1155/2012/951724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Ming M, He YY. PTEN in DNA damage repair. Cancer Lett (2012) 319:125–9. doi:10.1016/j.canlet.2012.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, et al. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem (2007) 282:28609–18. doi:10.1074/jbc.M703796200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Zhou Y, Ho WS. Combination of liquiritin, isoliquiritin and isoliquirigenin induce apoptotic cell death through upregulating p53 and p21 in the A549 non-small cell lung cancer cells. Oncol Rep (2014) 31:298–304. doi:10.3892/or.2013.2849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol (2008) 28:3281–9. doi:10.1128/MCB.00310-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Nakanishi A, Wada Y, Kitagishi Y, Matsuda S. Link between PI3K/AKT/PTEN pathway and NOX proteinin diseases. Aging Dis (2014) 5:203–11. doi:10.14336/AD.2014.0500203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Li ZY, Yang Y, Ming M, Liu B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem Biophys Res Commun (2011) 414:5–8. doi:10.1016/j.bbrc.2011.09.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J (2003) 22:5501–10. doi:10.1093/emboj/cdg513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Simone RE, Russo M, Catalano A, Monego G, Froehlich K, Boehm V, et al. Lycopene inhibits NF-kB-mediated IL-8 expression and changes redox and PPARγ signalling in cigarette smoke-stimulated macrophages. PLoS One (2011) 6:e19652. doi:10.1371/journal.pone.0019652
64. Kim JW, Kang KH, Burrola P, Mak TW, Lemke G. Retinal degeneration triggered by inactivation of PTEN in the retinal pigment epithelium. Genes Dev (2008) 22:3147–57. doi:10.1101/gad.1700108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Kang KH, Lemke G, Kim JW. The PI3K-PTEN tug-of-war, oxidative stress and retinal degeneration. Trends Mol Med (2009) 15:191–8. doi:10.1016/j.molmed.2009.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia (2012) 14:34–43.
67. Li JR, Cheng CL, Yang CR, Ou YC, Wu MJ, Ko JL. Dual inhibitor of phosphoinositide 3-kinase/mammalian target of rapamycin NVP-BEZ235 effectively inhibits cisplatin-resistant urothelial cancer cell growth through autophagic flux. Toxicol Lett (2013) 220:267–76. doi:10.1016/j.toxlet.2013.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Guo JP, Coppola D, Cheng JQ. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem (2011) 286:37389–98. doi:10.1074/jbc.M111.287433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Chock KL, Allison JM, Shimizu Y, ElShamy WM. BRCA1-IRIS overexpression promotes cisplatin resistance in ovarian cancer cells. Cancer Res (2010) 70:8782–91. doi:10.1158/0008-5472.CAN-10-1352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Burstein HJ. Novel agents and future directions for refractory breast cancer. Semin Oncol (2011) 38(Suppl 2):S17–24. doi:10.1053/j.seminoncol.2011.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Ma Y, Hu C, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol Endocrinol (2007) 21:1905–23. doi:10.1210/me.2006-0397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Hinton CV, Fitzgerald LD, Thompson ME. Phosphatidylinositol 3-kinase/Akt signaling enhances nuclear localization and transcriptional activity of BRCA1. Exp Cell Res (2007) 313:1735–44. doi:10.1016/j.yexcr.2007.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Kang HJ, Yi YW, Kim HJ, Hong YB, Seong YS, Bae I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis (2012) 3:e336. doi:10.1038/cddis.2012.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell (1999) 96:857–68. doi:10.1016/S0092-8674(00)80595-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science (2002) 296:530–4. doi:10.1126/science.1068712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Saal LH, Gruvberger-Saal SK, Persson C, Lövgren K, Jumppanen M, Staaf J, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet (2008) 40:102–7. doi:10.1038/ng.2007.39
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Foulkes WD. BRCA1 – sowing the seeds crooked in the furrow. Nat Genet (2008) 40:8–9. doi:10.1038/ng0108-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Martins FC, De S, Almendro V, Gönen M, Park SY, Blum JL, et al. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov (2012) 2:503–11. doi:10.1158/2159-8290.CD-11-0325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell (2007) 128:157–70. doi:10.1016/j.cell.2006.11.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Shen Z, Huhn S. PTEN sumo-wrestles human RAD52 to mystery land. Cell Cycle (2013) 12:3585. doi:10.4161/cc.26820
81. Jung IL, Kang HJ, Kim KC, Kim IG. PTEN/pAkt/p53 signaling pathway correlates with the radioresponse of non-small cell lung cancer. Int J Mol Med (2010) 25:517–23.
82. Li HF, Kim JS, Waldman T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat Oncol (2009) 4:43. doi:10.1186/1748-717X-4-43
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Zhang J, Xiao Y, Guo Y, Breslin P, Zhang S, Wei W, et al. Differential requirements for c-Myc in chronic hematopoietic hyperplasia and acute hematopoietic malignancies in Pten-null mice. Leukemia (2011) 25:1857–68. doi:10.1038/leu.2011.220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Guo W, Schubbert S, Chen JY, Valamehr B, Mosessian S, Shi H, et al. Suppression of leukemia development caused by PTEN loss. Proc Natl Acad Sci U S A (2011) 108:1409–14. doi:10.1073/pnas.1006937108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Chen J, Zhao KN, Li R, Shao R, Chen C. Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Curr Med Chem (2014) 21:3070–80. doi:10.2174/0929867321666140414095605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kim JY. Control of ovarian primordial follicle activation. Clin Exp Reprod Med (2012) 39:10–4. doi:10.5653/cerm.2012.39.1.10
87. Merritt MA, Cramer DW. Molecular pathogenesis of endometrial and ovarian cancer. Cancer Biomark (2010) 9:287–305. doi:10.3233/CBM-2011-0167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: BRCA1, PTEN, genome stability, reactive oxygen species, DNA repair, cell signaling, carcinogenesis
Citation: Minami A, Nakanishi A, Ogura Y, Kitagishi Y and Matsuda S (2014) Connection between tumor suppressor BRCA1 and PTEN in damaged DNA repair. Front. Oncol. 4:318. doi: 10.3389/fonc.2014.00318
Received: 11 September 2014; Accepted: 24 October 2014;
Published online: 10 November 2014.
Edited by:
Masood A. Shammas, Harvard (Dana Farber) Cancer Institute, USAReviewed by:
Alessandro Rimessi, University of Ferrara, ItalyCopyright: © 2014 Minami, Nakanishi, Ogura, Kitagishi and Matsuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoru Matsuda, Department of Food Science and Nutrition, Nara Women’s University, Kita-Uoya Nishimachi, Nara 630-8506, Japan e-mail:c21hdHN1ZGFAY2MubmFyYS13dS5hYy5qcA==
†Akari Minami and Satoru Matsuda have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.