 David A. Cano
David A. Cano Alfonso Soto-Moreno1,2
Alfonso Soto-Moreno1,2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 August 2014
Sec. Cancer Genetics
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00203
This article is part of the Research Topic Genetically modified mouse models of cancer View all 7 articles
Animal models constitute valuable tools for investigating the pathogenesis of cancer as well as for preclinical testing of novel therapeutics approaches. However, the pathogenic mechanisms of pituitary-tumor formation remain poorly understood, particularly in sporadic adenomas, thus, making it a challenge to model pituitary tumors in mice. Nevertheless, genetically engineered mouse models (GEMMs) of pituitary tumors have provided important insight into pituitary tumor biology. In this paper, we review various GEMMs of pituitary tumors, highlighting their contributions and limitations, and discuss opportunities for research in the field.
Pituitary adenomas occur in 10–15% of the general population as determined by autopsy studies (1, 2). The vast majority are pituitary incidentalomas with no apparent clinical manifestations. However, clinically significant pituitary tumors are associated with significant morbidity and decreased quality of life. Pituitary adenomas may cause symptoms due to abnormal hormone secretion, mass effect, or both. About 50% of pituitary adenomas are prolactinomas that secrete prolactin and cause galactorrhea and hypogonadism. GH-producing somatotropinomas, that cause gigantism and acromegaly, and ACTH-producing corticotropinomas that cause Cushing disease constitute 10 and 5% of pituitary adenomas, respectively. TSH-producing thyrotroph tumors are rare (less that 1%). Clinically non-functioning pituitary adenomas (NFPA) that do not secrete hormones represent about 30% of pituitary adenomas (3, 4).
Pituitary adenomas are usually benign. However, a significant number of pituitary adenomas show an aggressive behavior, with local invasion, increased risk of recurrence after surgery and lack of therapeutic response (5, 6). Malignant pituitary carcinomas are very rare, comprising only 0.2% of all pituitary tumors (7). Non-endocrine tumors can also be found in the pituitary including craniopharyngiomas and blastomas that arise from embryonic remnants of the pituitary (8, 9). Currently available treatment options for pituitary adenomas include drug therapy (mainly dopamine-agonist and somatostatin analogs), surgery, and radiotherapy. However, the currently available therapies fail to control the disease in a significant number of patients emphasizing the need to develop novel therapeutic approaches.
The rarity of pituitary tumors, difficulty to obtain biopsy samples, and lack of human pituitary cell lines pose a significant challenge to pituitary-tumor researchers. Animal models constitute valuable tools for investigating the pathogenesis of pituitary tumors as well as for preclinical testing of novel therapeutics approaches. There are a variety of animal models of pituitary tumors including spontaneous tumors, subcutaneous cell implantation, and genetically engineered mouse models (GEMMs) (transgenic and knockout mice) and each model has its advantages and limitations. In this review, we focus on GEMMs of pituitary tumors (Table 1), highlighting their contributions and limitations, and discuss opportunities for research in the field.
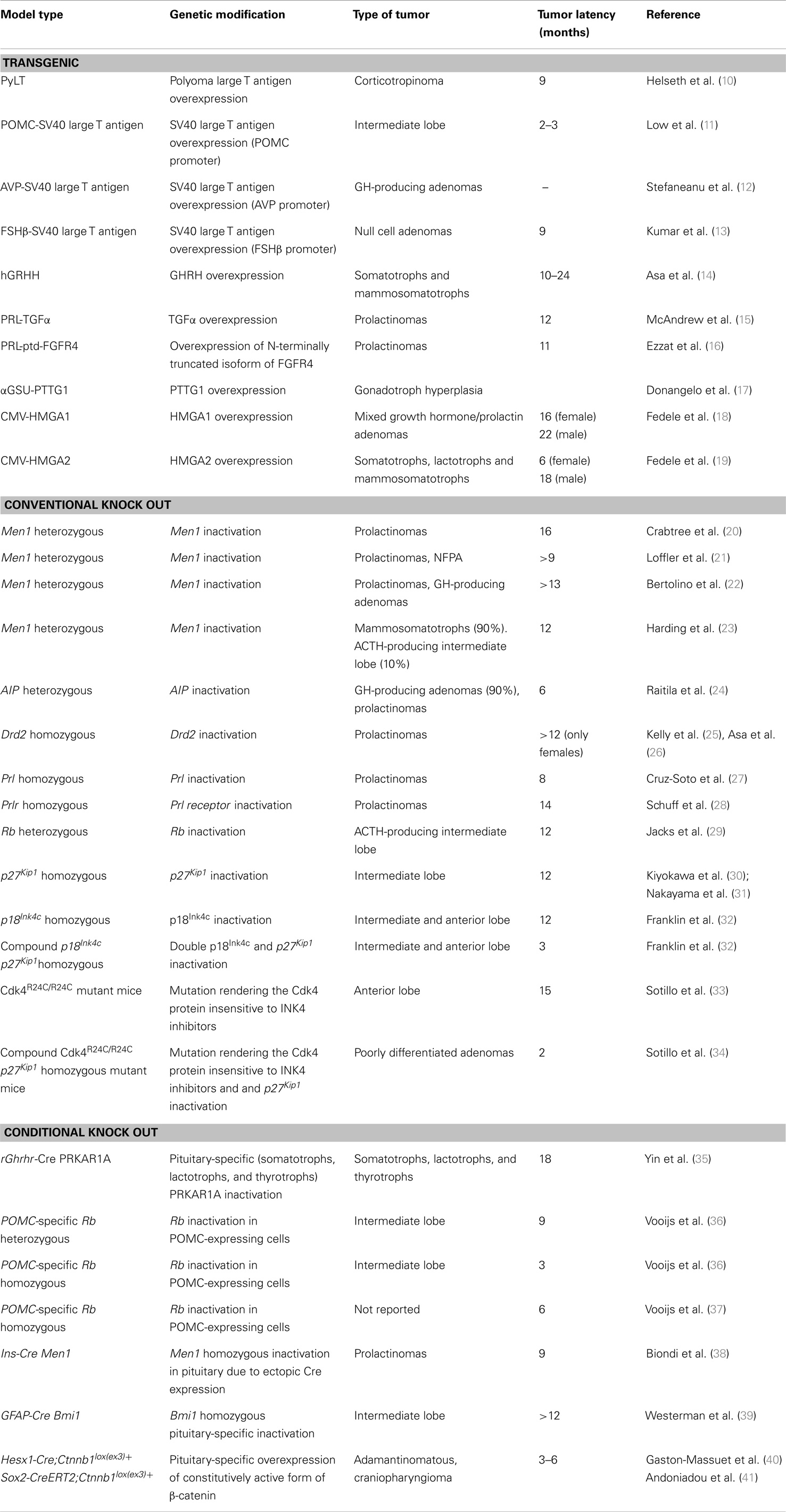
Table 1. Summary of GEMMs for pituitary tumors.
Before we discuss the various GEMMs for pituitary tumors, we will briefly describe the molecular abnormalities in pituitary tumors. For a more comprehensive view of this topic, the reader is directed to several excellent reviews (4, 42–44).
The majority of pituitary adenomas are sporadic. However, approximately 5% of cases arise in the context of familial syndromes (45) and several of the genes involved in these hereditary adenomas have been identified. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant familial disorder with mutations in the tumor suppressor gene MEN1. The cyclin-dependent kinase inhibitor 1B (CDKN1B) is associated with a MEN1-like phenotype now called MEN4. The McCune-Albright syndrome is a rare disease caused by mutations in the GNAS oncogene that encodes the guanine nucleotide-binding protein alpha stimulating activity polypeptide. Inactivating mutations in the PRKAR1A gene that encodes the protein kinase A regulatory subunit-1-alpha are associated with the neoplasia syndrome Carney complex. Familial isolated pituitary adenoma (FIPA) is a rare syndrome defined as familial presentation of pituitary adenomas in the absence of evidence for MEN1 and Carney complex (45). Around 15–40% of FIPA patients harbor germline mutations in the aryl hydrocarbon receptor interacting protein gene (AIP) gene. More recently, it has been suggested that mutations in succinate dehydrogenase genes can predispose to pituitary tumors although the evidence is limited to a single kindred of patients (46).
Pituitary adenomas are thought to be of monoclonal origin according to early studies (47, 48). Despite extensive research on pituitary tumors in the last decade, the molecular mechanisms underlying tumorigenesis in sporadic pituitary tumors remain unclear. There is little evidence to date that the mutations associated to familial pituitary adenomas are involved in most sporadic pituitary adenomas. The GNAS gene is the only gene in which mutations have been found in a significant proportion of sporadic pituitary adenomas (30–40% of GH-producing adenomas) although the oncogenic role of such mutations is still unclear (49, 50). Similarly, mutations in classical oncogenes and tumor suppressor such genes TP53 and MYC are only rarely observed, and restricted to aggressive or pituitary carcinomas. However, a significant number of molecular abnormalities, including cell-cycle deregulation, overexpression of growth factors, hormonal overstimulation, epigenetic silenced tumor suppressor genes, and defective signaling pathways have been found in pituitary adenomas although their causative role in pituitary-tumor formation remains to be firmly established (4, 42–44).
Cell-cycle deregulation is considered the main pathogenic event in the formation of pituitary adenomas. It has been estimated that around 80% of human pituitary adenomas display alterations at least in one cell-cycle regulator (51, 52). These alterations include overexpression of cyclins (mainly D1, D3, and E), as well as to downregulation of the cyclin-dependent kinase inhibitor family (mainly p15Ink4b, p16Ink4a, p18Ink4c, p21Cip1, and p27Kip1) and pRB expression (53). Cyclin D1 and D3 are overexpressed particularly in NFPA while cyclin E is overexpressed mainly in ACTH-producing corticotropinomas. Reduced levels of the INK4 family members of cyclin-dependent kinase inhibitors, p15Ink4b, p16Ink4a, and p18Ink4c in pituitary adenomas appear to be caused mainly by promoter hypermethylation (54–57). Decreased levels of p27Kip1 are common in pituitary carcinomas and ACTH-producing corticotropinomas (58, 59). p21Cip1 is also downregulated in NFPAs but, interestingly, appears to be overexpressed in hormone-secreting pituitary adenomas (60). The retinoblastoma tumor suppressor protein (pRb) plays a critical role in G1-S cell-cycle progression. Loss of pRb expression has been found in human sporadic pituitary adenomas (particularly in invasive types) due to loss of heterozygosity or promoter hypermethylation (61, 62). Pituitary-tumor transforming gene 1 (PTTG1) is a member of the securin family and plays an important role in mitosis by controlling sister chromatid separation. PTTG1 is overexpressed in nearly all pituitary adenomas, especially in invasive hormone-secreting pituitary tumors (63). However, its oncogenic role in human pituitary tumorigenesis is unclear. HMGA2 is another oncogene that has been found to be overexpressed in pituitary adenomas (64). More recently, increased expression of HMGA1 in human pituitary adenomas has also been described. This increased expression correlates with tumor invasiveness and proliferation (65). HMGA1 and 2 are members of the high mobility group A (HMGA) protein family, non-histone chromatin binding proteins that act as modulators of cellular processes such as transcription, replication, and recombination. HMGA proteins appear to induce the development of pituitary adenomas through mechanisms involving cell-cycle deregulation.
Several alterations in the expression of growth factors and/or growth factor receptors are frequently observed in pituitary adenomas, including TGFα, EGF, VEGF, and FGF. FGF is one of the best-studied growth factor family in pituitary tumors. FGF-4 is upregulated in pituitary adenomas and its expression correlates with tumor invasiveness. Expression of a N-terminally truncated isoform of FGFR4 (ptd FGFR4) has been found in pituitary adenomas but not in normal pituitary (16). Experimental evidence suggests a tumorigenic role for this isoform in pituitary (16). In contrast, the FGFR2-IIIb isoform appears to be downregulated in pituitary tumors, due to epigenetic modifications (66).
Deregulation of hypothalamic and pituitary feedback mechanisms have long been proposed to participate in pituitary tumorigenesis based on the defects in hormonal signaling observed in pituitary adenomas, mainly in GH- and ACTH-producing adenomas. Thus, increased GHRH expression has been found in aggressive somatotropinomas (67) and pituitary hyperplasia have been observed in patients with endocrine carcinomas ectopically secreting GHRH (68). An inactivating mutation of the GH receptor has also been described in somatotropinomas (69). Differential expression of vasopressin receptors has been reported in ACTH-producing pituitary tumors (70). Also, mutations in the glucocorticoid receptor that reduce glucocorticoid inhibition have been described in corticotroph tumors (71). In prolactinomas, diminished D2 dopamine receptor activity appears to mediate resistance to dopamine-inhibited hormone secretion (72).
Finally, various signaling pathways have been recently implicated in pituitary tumorigenesis. Akt is overexpressed in pituitary adenomas, particularly NFPAs (73) and mutations and amplifications of the phosphoinositide 3-kinase PIK3CA gene have been found in pituitary adenomas (74) suggesting a role for the PI3K/AKT in pituitary adenoma formation. Activation of the Raf/MEK/ERK signaling pathway has also been observed in pituitary adenomas, as demonstrated by the finding that B-Raf is overexpressed, and its downstream effectors MEK1/2 and ERK1/2 overactivated, in pituitary adenomas (75, 76). Increased levels of the hypoxia-inducible factor HIF1α transcription factor have been found in pituitary tumors but not in the normal pituitary (77, 78). Decreased expression of inhibitors of the canonical Wnt pathway has been reported in pituitary adenomas (79). Although these results might suggest the WNT pathway is overactivated in pituitary adenomas, nuclear accumulation of β-catenin was not observed (79). However, activating mutations have been found in the CTNNB1 gene that encodes β-catenin in around 70% of the pediatric form of human craniopharyngioma (adamantinomatous) (80, 81), a pituitary tumor of embryonic origin. Further mechanistic studies are necessary to fully understand the role of these signaling pathways in pituitary-tumor formation.
A common strategy to generate mouse models of cancer is to express viral oncoproteins in the cell/tissue of interest. These animal models based on viral oncogene overexpression provide limited insight into the mechanisms underlying pituitary-tumor formation. However, they can be considered useful tools to study various aspects of pituitary adenomas such as hormone secretion regulation and pathophysiology of hormone excess.
One of the first mouse models of pituitary tumors was generated by expressing the (PyLT) under the control of the polyoma early region promotor. These transgenic mice developed ACTH-producing microadenomas at 9 months of age, large adenomas at 13–16 months of age, and display features of Cushing’s syndrome (10). Several mouse strains generated using oncogenic viruses were generated specifically targeting the pituitary. Thus, expression of the simian virus 40 (SV40) large T antigen under the control of the proopiomelanocortin (POMC) gene promoter induced the formation of pituitary tumors arising from the intermediate lobe. These tumor cells expressed POMC peptides, but not other pituitary hormones (11). SV40 large T antigen overexpression using another promoter, the arginine vasopressin (AVP) gene promoter, also resulted in pituitary tumors although these tumors were composed of undifferentiated somatotroph cells (12). Transgenic mice carrying a temperature-sensitive mutant of simian virus 40 T antigen driven by regulatory elements of the beta subunit of human follicle-stimulating hormone (FSHβ) gene develop pituitary-tumors reminiscent of human null cell adenomas (13).
As mentioned in the previous section, various hypothalamic hormones and growth factors required for normal pituitary function and formation are overexpressed in pituitary adenomas. Transgenic approaches allow the overexpression of these hormones and factors specifically in the pituitary to evaluate their role in pituitary tumorigenesis. Overexpression of the GH-releasing hormone (GHRH) gene in mice results in hyperplasia of pituitary somatotrophs, lactotrophs, and mammosomatotrophs (cells producing both GH and PRL) by 8 months of age that leads to mixed GH and prolactin secreting pituitary adenomas by 10–24 months of age (14). These results indicate that GHRH might promote proliferation of transformed pituitary cells. However, the extended tumor latency period suggests the requirement for additional molecular alterations in the formation of pituitary tumors in these mice. A transgenic mouse model was generated in which TGFα was specifically overexpressed in the pituitary lactotrophs using the prolactin promoter (15). These transgenic mice developed lactotroph hyperplasia at 6 months of age and prolactin-producing pituitary adenomas with high penetrance by 12 months of age. Pituitary lactotroph-specific overexpression of the N-terminally truncated isoform of FGFR4 (ptd-FGFR4), but not the wild-type FGFR4, in mice leads to the development of tumors reminiscent of human prolactinomas (16). Interestingly, ptd-FGFR4 overexpression causes pituitary adenoma formation without preceding hyperplasia. The mechanism by which ptd-FGFR4 might facilitate pituitary cell transformation appears to involve alterations in neural cell-adhesion molecule/N-cadherin signaling that lead to diminished cell-adhesion (82). The oncogenic role of PTTG1 and HMGA2, two oncogenes previously suggested to play a relevant role in human pituitary adenoma formation, has been analyzed in transgenic mice. Targeted PTTG1 overexpression to gonadotroph cells of the pituitary driven by the alpha-subunit glycoprotein (alphaGSU) promoter results in gonadotroph hyperplasia (17). However, only occasional microadenomas were observed. These observations seem to question the tumorigenic potential of PTTG1 in pituitary. However, when overexpressing-PTTG1 mice were crossed with Rb heterozygous mice, which develop pituitary tumors with high penetrance (see below), increased frequency of pituitary adenomas was observed. On the contrary, PTTG1 deletion diminishes pituitary-tumor development in Rb heterozygous mice (83). Taken together, these results obtained from genetically modified mice indicate that PTTG1 facilitates pituitary adenoma formation. Transgenic mice expressing high levels of the HMGA2 transgene in all tissues (by using the cytomegalovirus promoter) developed mixed growth hormone/prolactin cell pituitary adenomas with high penetrance (85% of female animals) by 6 months of age (19). Of note, transgenic males developed pituitary adenomas with a lower penetrance (40%) and a longer latency period (about 18 months). Interestingly, HMGA1 overexpression also leads to the development of pituitary adenomas secreting prolactin and GH (18). Subsequent studies performed in these transgenic mice have shown that HMGA proteins mediate the formation of these tumors through a mechanism that involves the Rb/E2F1 pathway, thus further supporting a critical role of this pathway in pituitary tumorigenesis (84).
Altogether, transgenic mice overexpressing proteins in the pituitary have provided important insights into pituitary tumorigenesis. One of the major limitations of these mouse models is the regulation of the expression of the transgene that ideally should be limited to adult stages, thus mimicking the human situation.
The use of conventional or whole-body knockout mouse models provides a different approach for analyzing the function of important genes in pituitary tumorigenesis. Thus, conventional knockout mice are a crucial tool to analyze the role of tumor suppressor genes in pituitary tumorigenesis. Conventional knockout mice have been generated to assess the direct effect of several genes linked to familial pituitary tumors such as MEN1, CDKN1B, and AIP in pituitary-tumor development. Four different Men1 knockout mouse strains have been generated (20–23). Homozygous Men1 mice are embryonic lethal due to defects in craniofacial development but heterozygous Men1 mice are viable and have been extensively analyzed for tumor formation. In general, all these Men1 heterozygous mouse models correlate closely with the human MEN1 phenotype regarding tumor incidence and spectrum. These mice develop tumors of various endocrine tissues, including pancreatic islet cell tumors, pituitary and parathyroid adenoma, and adrenocortical tumors. Loss of heterozygosity and menin expression was demonstrated in the tumors, consistent with a tumor suppressor role for the Men1 gene. Furthermore, a higher occurrence of anterior pituitary tumors is observed in female Men1 heterozygous mice, similar to what it is observed in MEN1 patients. However, the different Men1 mouse models also show slight phenotypic differences including the type of pituitary adenomas developed. Thus, only prolactin producing adenomas were observed in the Men1 mouse model generated by Crabtree et al. (20) while prolactinomas and non-secreting adenomas were found in the model generated by Loffler et al. (21). Bertolino et al. (22) reported the formation of prolactinomas and GH-secreting tumors. Finally, Harding et al. (23) reported that the majority the pituitary tumors originated from the pars distalis and contained both prolactin and GH while around 10% of the tumors originated from the pars intermedia and contained ACTH. The reason for these differences in pituitary-tumor formation is unclear but it might be related to the types of Men1 mutations (different exons were targeted) and/or differences in genetic background.
Recently, a conventional knockout mouse deficient in the AIP, a gene linked to familial isolated pituitary adenoma (FIPA), and isolated familial somatotropinomas have been generated. Homozygous knockout Aip mice died during embryogenesis, likely due to cardiovascular abnormalities (24). Heterozygous Aip mice were highly prone to pituitary adenomas, developing tumors as early as at 6 months of age (24). Thus, unlike in humans in whom AIP-associated tumors often develop in childhood or early adulthood (85), heterozygous Aip mice develop adenomas in adulthood. Another major difference between the Aip mouse model and the human disease is the complete penetrance of pituitary tumors in the Aip mouse model (at 15 months of age). The majority of adenomas found in the Aip mouse model were somatotropinomas, although mixed GH/prolactin, prolactinomas, and ACTH-corticotropinomas were also found. Biallelic inactivation of AIP was demonstrated in these tumors.
Analysis of conventional knockout mice has also been useful to analyze the effect of disruption of feedback signaling in pituitary-tumor pathogenesis. Thus, analysis of dopamine receptor 2 (Drd2) knockout mice indicates that functional inactivation of this receptor might contribute to the formation of pituitary adenomas, namely prolactinomas. Female Drd2 knockout mice develop massive lactotroph hyperplasia by 12 months of age and invasive prolactinomas at older ages. (25, 26). Male Drd2 knockout mice develop microadenomas in the absence of hyperplasia. The effects of disrupted prolactin feedback on pituitary tumorigenesis have been demonstrated in knockout mice deficient for prolactin and prolactin receptor (Prlr). Prolactin knockout mice develop lactotroph hyperplasia by 6 weeks of age and prolactinomas by 8 months of age (27). Inactivation of Prlr also results in lactotroph hyperplasia and formation of invasive adenomas (28).
Studies performed in mice with targeted disruption of cell-cycle regulators collectively support the notion that cell-cycle dysregulation is central in pituitary tumorigenesis (51). Mice heterozygous for Rb develop ACTH-producing aggressive tumors arising from the intermediate lobe of the pituitary with high penetrance by 12 months of age (29). Subsequent studies have shown that pituitary-tumor incidence and histological type in Rb heterozygous mice is dependent on the mouse strain suggesting an effect of modifier genes in pituitary adenoma formation (86). Homozygous mice deficient for the cyclin-dependent kinase inhibitors p27Kip1 and p18Ink4c also develop pituitary adenomas. Homozygous p27Kip1mice develop tumors from the intermediate lobe by 12 months of age (30, 31) while p18Ink4c mice develop pituitary adenomas in both the intermediate and anterior lobes (32). Inactivation of other cyclin-dependent kinase inhibitors that are downregulated in human pituitary adenomas, such as p21Cip1, p15Ink4b, and p16Ink4a, does not result in pituitary tumors in mice. The generation of mice with combined inactivation of different cyclin-dependent kinase inhibitors has revealed a cooperation of these cyclin-dependent kinase inhibitors in pituitary-tumor formation. Thus, deletion of p21Cip1, p27Kip1, or p16Ink4a in combination with loss of p18Ink4c results in increased incidence and decreased latency of pituitary tumors compared to single mutants (32, 87, 88). Of note, double p18Ink4c p27Kip1 mutant mice appear to die by 3 months of age due to pituitary adenomas (32). The relevance of INK4 family proteins in pituitary adenoma formation is further supported by the analysis of knock-in mice, which express a CDK4 variant with a mutation that renders the Cdk4 protein insensitive to INK4 inhibitors (Cdk4R24C/R24C mutant mice) (33, 89). These Cdk4R24C/R24C knock-in mice develop multiple tumors including pituitary tumors (25% of penetrance) arising in the anterior pituitary by 15 months of age. A dramatic cooperation in pituitary-tumor formation is observed in double Cdk4R24C/R24C p27Kip1 mutant mice, which develop poorly differentiated pituitary tumors that kill these mutant mice in 8–10 weeks of age (34). Cooperation between cyclin-dependent kinase inhibitors and Rb in pituitary-tumor formation has also been observed. Inactivation of p27Kip1 and p21Cip1in Rb heterozygous mice results in decreased latency of pituitary tumors (90, 91). Similarly, loss of p16Ink4a strongly accelerates intermediate lobe pituitary tumorigenesis in Rb heterozygous mice (92).
The usefulness of conventional knockout mice as models for evaluating therapeutic agents is highlighted by studies performed with bromocriptine, a dopamine agonist used to treat prolactinomas. Thus, it has been reported that bromocriptine decreases prolactin serum levels in Prlr mutant mice harboring tumors (28) and reduces tumor cell proliferation in p27Kip1 deficient mice (93). Bromocriptine does not affect prolactin levels in Drd2 knockout mice, as expected from the absence of the dopamine receptor 2 (28) and thus, these mice can be considered a useful model for dopamine-resistant prolactinomas. The effectiveness of antiangiogenic treatments in dopamine-resistant prolactinomas has been analyzed in Drd2 knockout mice. Anti-VEGF strategies resulted in prolactin inhibition and decreased tumor cell proliferation in these mice (94).
The mouse models discussed above illustrate how conventional knockout mice help to elucidate the causal role of genes that were predicted to be important for human pituitary tumorigenesis. These conventional knockout mice can also be useful as preclinical models for testing of therapeutic agents.
The development of conditional gene targeting has allowed to overcome some of the limitations of conventional knockout mice such as potential embryonic lethality or complications due to systemic effects of gene inactivation. The most widely used conditional system is based on the Cre-loxP technology. Combinations of Cre-loxP systems, specific promoters, and inducible systems (i.e., tamoxifen) allow tissue- and time-specific inactivation of the gene of interest. The number of pituitary-specific Cre lines available is rapidly increasing (35, 95–102). The generation of pituitary cell-specific Cre transgenic lines has been instrumental in advancing our understanding of pituitary cell function and cell lineages. However, to date, relatively few studies have exploited these Cre lines to evaluate the role of genes of interest in pituitary tumorigenesis.
To explore the role of inactivation of the Carney complex gene PRKAR1A on pituitary tumorigenesis, a pituitary-specific knockout of this gene was generated by crossing mice with conditional alleles of Prkar1a with a mouse line expressing the Cre recombinase in pituitary cells using the GHRH receptor promoter (rGhrhr-Cre line) (35). This promoter, drives Cre-mediated recombination in pituitary cells of the Pit1 lineage, including somatotrophs, lactotrophs, and thyrotrophs. Pituitary-specific Prkar1a knockout mice exhibited a moderate increased frequency of pituitary adenomas, although not until advanced age (18 months). These tumors were composed of cells that expressed GH, prolactin, and TSH. Using a different conditional system based on the Flp recombinase, specific inactivation of Rb in pro-opiomelanocortin-expressing cells has been achieved. Mice in which only an allele of Rb was conditionally inactivated in the pituitary displayed atypical lesions in the intermediate lobe of the pituitary by 6–12 weeks of age that resulted in adenoma formation as the mice aged. Accelerated adenoma formation was observed in mice homozygous for the conditional alleles of Rb. These mice developed pituitary adenomas with 100% incidence as early as 12 weeks of age and succumbed to these adenomas by 20 weeks of age (36). The adenomas originated from the intermediate lobe melanotrophs and were indistinguishable from the adenomas observed in conventional Rb heterozygous mice. Pituitary-specific inactivation of Rb has also been achieved using a mouse strain in which Cre expression is under the control of the POMC gene promoter. These mice developed pituitary adenomas with 100% incidence after a relatively short median latency (around 6 months) (37). However, no histological characterization of the adenomas was reported. Interestingly, in this study pituitary adenoma formation in vivo was monitored by crossing conditional Rb knockout mice with transgenic mice that express luciferase under the control of the POMC promoter detecting tumor growth as early as 8 weeks of age.
Pituitary adenomas have been also reported in conditional mouse models using non-pituitary-specific Cre lines. Thus, conditional inactivation of the MEN1 gene using a mouse line in which Cre expression was driven by the insulin promoter resulted in the formation of pituitary adenomas (in addition to islet tumors) by 12 months of age (38). All pituitary adenomas were derived from the pars distalis, as determined by expression of prolactin. The formation of pituitary adenomas in these mice was attributed to aberrant Cre expression in the pituitary. Conditional activation of the polycomb ring finger oncogene using a GFAP-Cre that drives expression in neural and glial lineages developed intermediate and anterior lobe pituitary tumors positive for ACTH and beta-endorphin although with incomplete penetrance and long latency period (1 year) (39). Consistent with these results, Cre expression in the GFAP-Cre line was observed in the pituitary. However, it is unclear whether this Cre activity in the pituitary represents endogenous GFAP expression or ectopic expression of the Cre transgene.
The usefulness of conditional knockout mouse models in the study of pituitary-tumor formation is perhaps best illustrated by two recent studies that analyzed the role of Wnt/β-catenin signaling pathway in pituitary tumorigenesis. Expression of a degradation-resistant mutant form of β-catenin in embryonic pituitary using the Hesx1-Cre line was shown to cause the formation of tumors resembling human adamantinomatous craniopharyngioma (ACP) (40). ACP is a type of pediatric non-hormone-producing tumor derived from pituitary embryonic tissue associated with significant morbidity and mortality (8). Interestingly, expression of mutant β-catenin in differentiated cells in the embryonic or adult pituitary using specific Cre lines (Gh-Cre, Prl-Cre, and Pit1-Cre) did not give rise to tumors (40). Further studies from the same group using specific a tamoxifen-inducible Cre line demonstrated that activation of mutant β-catenin in Sox2-expressing adult pituitary stem cells also result in the formation of ACP-like tumors (41). The molecular characterization of the tumors formed in these mouse models of ACP have uncovered a number of genes and pathways potentially involved in the formation of ACP including the SHH, BMP, and FGF pathways (103). These findings have been validated in human ACP (103) in what is an excellent example of how genetic studies in mice harboring conditional mutations, in addition to providing mechanistic insight into pituitary tumorigenesis, can lead to the generation of new animal models for modeling human disease.
Although current GEMMs of pituitary tumors exhibit, to some degree, biochemical and molecular features of pituitary adenomas, no current mouse model exists that fully recapitulates pituitary-tumor formation. The ideal mouse model of cancer faithfully recapitulates the genetic lesions found in human disease. However, the primary genetic defect/s for pituitary tumorigenesis remains unclear. Furthermore, the pathogenic mechanisms for tumor formation may be specific for the different pituitary adenoma subtypes thus, making it a challenge to model pituitary tumors in mice. Despite these limitations, current GEMMs of pituitary tumors have provided important insight into pituitary-tumor biology. Transgenic and knockout mice have proven to be a powerful tool to examine the role of genes predicted to be important for human pituitary tumorigenesis. Thus, the use of knockout mice has validated the putative oncogenic role of genes linked to familial pituitary adenomas such as MEN1 and AIP. Also, the analysis of knockout mice deficient in cell-cycle regulators such as Rb and cyclin-dependent kinase inhibitors have provided evidence that cell-cycle deregulation may be a critical event in pituitary tumorigenesis. As the number of genes found to be deregulated in pituitary adenomas continues to increase, we can expect to see the generation of new mouse models with deletion or overexpression of those genes in the near future. The recent generation of pituitary-specific Cre-expressing mouse strains will certainly have a significant impact in this area since it will permit the analysis of genes in a cell type-specific and temporal fashion thus circumventing problems often associated to conventional knock out mice such as embryonic lethality. Nevertheless, a more detailed characterization of current GEMMs for pituitary tumors may still provide additional valuable insight into pituitary tumorigenesis. For example, in the vast majority of GEMMs of pituitary tumor, adenomas arise after a long latency period suggesting the requirement of additional genetic events. The identification of such events might uncover underlying mechanisms of adenoma formation. Further studies in existing GEMMs may also help to address important specific questions on pituitary tumors that cannot be easily addressed in human such as the cell of origin for pituitary adenomas, interaction between tumor cells and tumor microenvironment, and mechanisms underlying adenoma invasiveness.
Ultimately, the goal of generating faithful GEMMs of cancer is to evaluate therapeutic strategies. However, there is a scarcity of studies evaluating current available therapies for pituitary tumors in GEMMs. Such studies are fundamental to determine the utility of GEMMs of pituitary tumors for preclinical testing of new therapeutic drugs. In this regard, the use of non-invasive imaging to assess monitoring of tumor growth and response to therapy can enhance the utility of these mouse models for preclinical testing as previously shown in Rb conditional knock out mice (37).
As our understanding of the molecular mechanisms involved in pituitary adenoma formation improves, we can expect the generation of more sophisticated and diverse GEMMs that will undoubtedly facilitate research in pituitary tumorigenesis, the identification of biomarkers of tumor aggressiveness and preclinical testing of novel therapies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Guest Associate Editor Amancio Carnero declares that, despite being affiliated to the same institution as authors David A. Cano, Alfonso Soto-Moreno, and Alfonso Leal-Cerro, the review process was handled objectively and no conflict of interest exists.
David A. Cano was supported by the grants from the Spanish Ministry of Science and Innovation (SAF2011-26805) and Andalusian Regional Ministry of Science and Innovation (CTS-7478). Alfonso Leal-Cerro was supported by a grant from the Spanish Ministry of Health, ISCIII (PI12/0143). Alfonso Soto-Moreno was supported by a grant from the Spanish Ministry of Health, ISCIII (PI12/02043).
1. Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. The prevalence of pituitary adenomas: a systematic review. Cancer (2004) 101:613–9. doi:10.1002/cncr.20412
2. Scheithauer BW, Gaffey TA, Lloyd RV, Sebo TJ, Kovacs KT, Horvath E, et al. Pathobiology of pituitary adenomas and carcinomas. Neurosurgery (2006) 59:341–53. doi:10.1227/01.NEU.0000223437.51435.6E
3. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab (2006) 91:4769–75. doi:10.1210/jc.2006-1668
4. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol (2011) 7:257–66. doi:10.1038/nrendo.2011.40
5. Buchfelder M. Management of aggressive pituitary adenomas: current treatment strategies. Pituitary (2009) 12:256–60. doi:10.1007/s11102-008-0153-z
6. Zada G, Woodmansee WW, Ramkissoon S, Amadio J, Nose V, Laws ER Jr. Atypical pituitary adenomas: incidence, clinical characteristics, and implications. J Neurosurg (2011) 114:336–44. doi:10.3171/2010.8.JNS10290
7. Colao A, Ochoa AS, Auriemma RS, Faggiano A, Pivonello R, Lombardi G. Pituitary carcinomas. Front Horm Res (2010) 38:94–108. doi:10.1159/000318499
8. Muller HL. Childhood craniopharyngioma – current concepts in diagnosis, therapy and follow-up. Nat Rev Endocrinol (2010) 6:609–18. doi:10.1038/nrendo.2010.168
9. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol (2014) 128:111–22. doi:10.1007/s00401-014-1285-z
10. Helseth A, Siegal GP, Haug E, Bautch VL. Transgenic mice that develop pituitary tumors. A model for Cushing’s disease. Am J Pathol (1992) 140:1071–80.
11. Low MJ, Liu B, Hammer GD, Rubinstein M, Allen RG. Post-translational processing of proopiomelanocortin (POMC) in mouse pituitary melanotroph tumors induced by a POMC-simian virus 40 large T antigen transgene. J Biol Chem (1993) 268:24967–75.
12. Stefaneanu L, Rindi G, Horvath E, Murphy D, Polak JM, Kovacs K. Morphology of adenohypophysial tumors in mice transgenic for vasopressin-SV40 hybrid oncogene. Endocrinology (1992) 130:1789–95. doi:10.1210/endo.130.4.1312426
13. Kumar TR, Graham KE, Asa SL, Low MJ. Simian virus 40 T antigen-induced gonadotroph adenomas: a model of human null cell adenomas. Endocrinology (1998) 139:3342–51. doi:10.1210/endo.139.7.6100
14. Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, et al. Pituitary adenomas in mice transgenic for growth hormone-releasing hormone. Endocrinology (1992) 131:2083–9. doi:10.1210/endo.131.5.1425411
15. McAndrew J, Paterson AJ, Asa SL, Mccarthy KJ, Kudlow JE. Targeting of transforming growth factor-alpha expression to pituitary lactotrophs in transgenic mice results in selective lactotroph proliferation and adenomas. Endocrinology (1995) 136:4479–88. doi:10.1210/endo.136.10.7664668
16. Ezzat S, Zheng L, Zhu XF, Wu GE, Asa SL. Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest (2002) 109:69–78. doi:10.1172/JCI14036
17. Donangelo I, Gutman S, Horvath E, Kovacs K, Wawrowsky K, Mount M, et al. Pituitary tumor transforming gene overexpression facilitates pituitary tumor development. Endocrinology (2006) 147:4781–91. doi:10.1210/en.2006-0544
18. Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto AJ, Kenyon L, et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene (2005) 24:3427–35. doi:10.1038/sj.onc.1208501
19. Fedele M, Battista S, Kenyon L, Baldassarre G, Fidanza V, Klein-Szanto AJ, et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene (2002) 21:3190–8. doi:10.1038/sj.onc.1205428
20. Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A (2001) 98:1118–23. doi:10.1073/pnas.98.3.1118
21. Loffler KA, Biondi CA, Gartside M, Waring P, Stark M, Serewko-Auret MM, et al. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. Int J Cancer (2007) 120:259–67. doi:10.1002/ijc.22288
22. Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol (2003) 17:1880–92. doi:10.1210/me.2003-0154
23. Harding B, Lemos MC, Reed AA, Walls GV, Jeyabalan J, Bowl MR, et al. Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia. Endocr Relat Cancer (2009) 16:1313–27. doi:10.1677/ERC-09-0082
24. Raitila A, Lehtonen HJ, Arola J, Heliovaara E, Ahlsten M, Georgitsi M, et al. Mice with inactivation of aryl hydrocarbon receptor-interacting protein (Aip) display complete penetrance of pituitary adenomas with aberrant ARNT expression. Am J Pathol (2010) 177:1969–76. doi:10.2353/ajpath.2010.100138
25. Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, et al. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron (1997) 19:103–13. doi:10.1016/S0896-6273(00)80351-7
26. Asa SL, Kelly MA, Grandy DK, Low MJ. Pituitary lactotroph adenomas develop after prolonged lactotroph hyperplasia in dopamine D2 receptor-deficient mice. Endocrinology (1999) 140:5348–55. doi:10.1210/endo.140.11.7118
27. Cruz-Soto ME, Scheiber MD, Gregerson KA, Boivin GP, Horseman ND. Pituitary tumorigenesis in prolactin gene-disrupted mice. Endocrinology (2002) 143:4429–36. doi:10.1210/en.2002-220173
28. Schuff KG, Hentges ST, Kelly MA, Binart N, Kelly PA, Iuvone PM, et al. Lack of prolactin receptor signaling in mice results in lactotroph proliferation and prolactinomas by dopamine-dependent and -independent mechanisms. J Clin Invest (2002) 110:973–81. doi:10.1172/JCI15912
29. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature (1992) 359:295–300. doi:10.1038/359295a0
30. Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell (1996) 85:721–32.
31. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell (1996) 85:707–20. doi:10.1016/S0092-8674(00)81237-4
32. Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev (1998) 12:2899–911. doi:10.1101/gad.12.18.2899
33. Sotillo R, Dubus P, Martin J, De La Cueva E, Ortega S, Malumbres M, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J (2001) 20:6637–47. doi:10.1093/emboj/20.23.6637
34. Sotillo R, Renner O, Dubus P, Ruiz-Cabello J, Martin-Caballero J, Barbacid M, et al. Cooperation between Cdk4 and p27kip1 in tumor development: a preclinical model to evaluate cell cycle inhibitors with therapeutic activity. Cancer Res (2005) 65:3846–52. doi:10.1158/0008-5472.CAN-04-4195
35. Yin Z, Williams-Simons L, Parlow AF, Asa S, Kirschner LS. Pituitary-specific knockout of the Carney complex gene Prkar1a leads to pituitary tumorigenesis. Mol Endocrinol (2008) 22:380–7. doi:10.1210/me.2006-0428
36. Vooijs M, Van Der Valk M, Te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene (1998) 17:1–12. doi:10.1038/sj.onc.1202169
37. Vooijs M, Jonkers J, Lyons S, Berns A. Noninvasive imaging of spontaneous retinoblastoma pathway-dependent tumors in mice. Cancer Res (2002) 62:1862–7.
38. Biondi CA, Gartside MG, Waring P, Loffler KA, Stark MS, Magnuson MA, et al. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Mol Cell Biol (2004) 24:3125–31. doi:10.1128/MCB.24.8.3125-3131.2004
39. Westerman BA, Blom M, Tanger E, Van Der Valk M, Song JY, Van Santen M, et al. GFAP-Cre-mediated transgenic activation of Bmi1 results in pituitary tumors. PLoS One (2012) 7:e35943. doi:10.1371/journal.pone.0035943
40. Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A (2011) 108:11482–7. doi:10.1073/pnas.1101553108
41. Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, et al. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell (2013) 13:433–45. doi:10.1016/j.stem.2013.07.004
42. Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol (2009) 4:97–126. doi:10.1146/annurev.pathol.4.110807.092259
43. Dworakowska D, Grossman AB. The pathophysiology of pituitary adenomas. Best Pract Res Clin Endocrinol Metab (2009) 23:525–41. doi:10.1016/j.beem.2009.05.004
44. Vandeva S, Jaffrain-Rea ML, Daly AF, Tichomirowa M, Zacharieva S, Beckers A. The genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab (2010) 24:461–76. doi:10.1016/j.beem.2010.03.001
45. Daly AF, Tichomirowa MA, Beckers A. The epidemiology and genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab (2009) 23:543–54. doi:10.1016/j.beem.2009.05.008
46. Xekouki P, Pacak K, Almeida M, Wassif CA, Rustin P, Nesterova M, et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? J Clin Endocrinol Metab (2012) 97:E357–66. doi:10.1210/jc.2011-1179
47. Alexander JM, Biller BM, Bikkal H, Zervas NT, Arnold A, Klibanski A. Clinically nonfunctioning pituitary tumors are monoclonal in origin. J Clin Invest (1990) 86:336–40. doi:10.1172/JCI114705
48. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab (1990) 71:1427–33. doi:10.1210/jcem-71-6-1427
49. Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature (1987) 330:566–8. doi:10.1038/330566a0
50. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature (1989) 340:692–6. doi:10.1038/340692a0
51. Quereda V, Malumbres M. Cell cycle control of pituitary development and disease. J Mol Endocrinol (2009) 42:75–86. doi:10.1677/JME-08-0146
52. Fedele M, Fusco A. Role of the high mobility group A proteins in the regulation of pituitary cell cycle. J Mol Endocrinol (2010) 44:309–18. doi:10.1677/JME-09-0178
53. Farrell WE, Clayton RN. Epigenetic change in pituitary tumorigenesis. Endocr Relat Cancer (2003) 10:323–30. doi:10.1677/erc.0.0100323
54. Jaffrain-Rea ML, Ferretti E, Toniato E, Cannita K, Santoro A, Di Stefano D, et al. p16 (INK4a, MTS-1) gene polymorphism and methylation status in human pituitary tumours. Clin Endocrinol (1999) 51:317–25. doi:10.1046/j.1365-2265.1999.00774.x
55. Ogino A, Yoshino A, Katayama Y, Watanabe T, Ota T, Komine C, et al. The p15(INK4b)/p16(INK4a)/RB1 pathway is frequently deregulated in human pituitary adenomas. J Neuropathol Exp Neurol (2005) 64:398–403.
56. Yoshino A, Katayama Y, Ogino A, Watanabe T, Yachi K, Ohta T, et al. Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J Neurooncol (2007) 83:153–62. doi:10.1007/s11060-006-9316-9
57. Kirsch M, Morz M, Pinzer T, Schackert HK, Schackert G. Frequent loss of the CDKN2C (p18INK4c) gene product in pituitary adenomas. Genes Chromosomes Cancer (2009) 48:143–54. doi:10.1002/gcc.20621
58. Bamberger CM, Fehn M, Bamberger AM, Ludecke DK, Beil FU, Saeger W, et al. Reduced expression levels of the cell-cycle inhibitor p27Kip1 in human pituitary adenomas. Eur J Endocrinol (1999) 140:250–5. doi:10.1530/eje.0.1400250
59. Lidhar K, Korbonits M, Jordan S, Khalimova Z, Kaltsas G, Lu X, et al. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J Clin Endocrinol Metab (1999) 84:3823–30. doi:10.1210/jcem.84.10.6066
60. Neto AG, Mccutcheon IE, Vang R, Spencer ML, Zhang W, Fuller GN. Elevated expression of p21 (WAF1/Cip1) in hormonally active pituitary adenomas. Ann Diagn Pathol (2005) 9:6–10. doi:10.1053/j.anndiagpath.2004.10.002
61. Pei L, Melmed S, Scheithauer B, Kovacs K, Benedict WF, Prager D. Frequent loss of heterozygosity at the retinoblastoma susceptibility gene (RB) locus in aggressive pituitary tumors: evidence for a chromosome 13 tumor suppressor gene other than RB. Cancer Res (1995) 55:1613–6.
62. Simpson DJ, Hibberts NA, Mcnicol AM, Clayton RN, Farrell WE. Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res (2000) 60:1211–6.
63. Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab (1999) 84:761–7. doi:10.1210/jcem.84.2.5432
64. Fedele M, Palmieri D, Fusco A. HMGA2: a pituitary tumour subtype-specific oncogene? Mol Cell Endocrinol (2010) 326:19–24. doi:10.1016/j.mce.2010.03.019
65. Wang EL, Qian ZR, Rahman MM, Yoshimoto K, Yamada S, Kudo E, et al. Increased expression of HMGA1 correlates with tumour invasiveness and proliferation in human pituitary adenomas. Histopathology (2010) 56:501–9. doi:10.1111/j.1365-2559.2010.03495.x
66. Zhu X, Lee K, Asa SL, Ezzat S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol (2007) 170:1618–28. doi:10.2353/ajpath.2007.061111
67. Thapar K, Kovacs K, Stefaneanu L, Scheithauer B, Killinger DW, Lioyd RV, et al. Overexpression of the growth-hormone-releasing hormone gene in acromegaly-associated pituitary tumors. An event associated with neoplastic progression and aggressive behavior. Am J Pathol (1997) 151:769–84.
68. Sano T, Asa SL, Kovacs K. Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev (1988) 9:357–73. doi:10.1210/edrv-9-3-357
69. Asa SL, Digiovanni R, Jiang J, Ward ML, Loesch K, Yamada S, et al. A growth hormone receptor mutation impairs growth hormone autofeedback signaling in pituitary tumors. Cancer Res (2007) 67:7505–11. doi:10.1158/0008-5472.CAN-07-0219
70. Luque RM, Ibanez-Costa A, Lopez-Sanchez LM, Jimenez-Reina L, Venegas-Moreno E, Galvez MA, et al. A cellular and molecular basis for the selective desmopressin-induced ACTH release in Cushing disease patients: key role of AVPR1b receptor and potential therapeutic implications. J Clin Endocrinol Metab (2013) 98:4160–9. doi:10.1210/jc.2013-1992
71. Karl M, Von Wichert G, Kempter E, Katz DA, Reincke M, Monig H, et al. Nelson’s syndrome associated with a somatic frame shift mutation in the glucocorticoid receptor gene. J Clin Endocrinol Metab (1996) 81:124–9. doi:10.1210/jcem.81.1.8550738
72. Caccavelli L, Feron F, Morange I, Rouer E, Benarous R, Dewailly D, et al. Decreased expression of the two D2 dopamine receptor isoforms in bromocriptine-resistant prolactinomas. Neuroendocrinology (1994) 60:314–22. doi:10.1159/000126764
73. Musat M, Korbonits M, Kola B, Borboli N, Hanson MR, Nanzer AM, et al. Enhanced protein kinase B/Akt signalling in pituitary tumours. Endocr Relat Cancer (2005) 12:423–33. doi:10.1677/erc.1.00949
74. Lin Y, Jiang X, Shen Y, Li M, Ma H, Xing M, et al. Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr Relat Cancer (2009) 16:301–10. doi:10.1677/ERC-08-0167
75. Ewing I, Pedder-Smith S, Franchi G, Ruscica M, Emery M, Vax V, et al. A mutation and expression analysis of the oncogene BRAF in pituitary adenomas. Clin Endocrinol (2007) 66:348–52. doi:10.1111/j.1365-2265.2006.02735.x
76. Dworakowska D, Wlodek E, Leontiou CA, Igreja S, Cakir M, Teng M, et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr Relat Cancer (2009) 16:1329–38. doi:10.1677/ERC-09-0101
77. Vidal S, Horvath E, Kovacs K, Kuroki T, Lloyd RV, Scheithauer BW. Expression of hypoxia-inducible factor-1alpha (HIF-1alpha) in pituitary tumours. Histol Histopathol (2003) 18:679–86.
78. Yoshida D, Kim K, Yamazaki M, Teramoto A. Expression of hypoxia-inducible factor 1alpha and cathepsin D in pituitary adenomas. Endocr Pathol (2005) 16:123–31. doi:10.1385/EP:16:2:123
79. Elston MS, Gill AJ, Conaglen JV, Clarkson A, Shaw JM, Law AJ, et al. Wnt pathway inhibitors are strongly down-regulated in pituitary tumors. Endocrinology (2008) 149:1235–42. doi:10.1210/en.2007-0542
80. Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N, et al. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol (2004) 203:814–21. doi:10.1002/path.1562
81. Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrubl F, et al. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol (2005) 109:589–97. doi:10.1007/s00401-005-1004-x
82. Ezzat S, Zheng L, Asa SL. Pituitary tumor-derived fibroblast growth factor receptor 4 isoform disrupts neural cell-adhesion molecule/N-cadherin signaling to diminish cell adhesiveness: a mechanism underlying pituitary neoplasia. Mol Endocrinol (2004) 18:2543–52. doi:10.1210/me.2004-0182
83. Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary hypoplasia in Pttg−/− mice is protective for Rb± pituitary tumorigenesis. Mol Endocrinol (2005) 19:2371–9. doi:10.1210/me.2005-0137
84. Fedele M, Visone R, De Martino I, Troncone G, Palmieri D, Battista S, et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell (2006) 9:459–71. doi:10.1016/j.ccr.2006.04.024
85. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34:239–77. doi:10.1210/er.2012-1013
86. Leung SW, Wloga EH, Castro AF, Nguyen T, Bronson RT, Yamasaki L. A dynamic switch in Rb± mediated neuroendocrine tumorigenesis. Oncogene (2004) 23:3296–307. doi:10.1038/sj.onc.1207457
87. Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol (2000) 20:6147–58. doi:10.1128/MCB.20.16.6147-6158.2000
88. Ramsey MR, Krishnamurthy J, Pei XH, Torrice C, Lin W, Carrasco DR, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res (2007) 67:4732–41. doi:10.1158/0008-5472.CAN-06-3437
89. Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol (2002) 22:644–56. doi:10.1128/MCB.22.2.644-656.2002
90. Brugarolas J, Bronson RT, Jacks T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol (1998) 141:503–14. doi:10.1083/jcb.141.2.503
91. Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci U S A (1999) 96:6382–7. doi:10.1073/pnas.96.11.6382
92. Tsai KY, Macpherson D, Rubinson DA, Nikitin AY, Bronson R, Mercer KL, et al. ARF mutation accelerates pituitary tumor development in Rb± mice. Proc Natl Acad Sci U S A (2002) 99:16865–70. doi:10.1073/pnas.262499599
93. Carneiro C, Jiao MS, Hu M, Shaffer D, Park M, Pandolfi PP, et al. p27 deficiency desensitizes Rb−/− cells to signals that trigger apoptosis during pituitary tumor development. Oncogene (2003) 22:361–9. doi:10.1038/sj.onc.1206163
94. Luque GM, Perez-Millan MI, Ornstein AM, Cristina C, Becu-Villalobos D. Inhibitory effects of antivascular endothelial growth factor strategies in experimental dopamine-resistant prolactinomas. J Pharmacol Exp Ther (2011) 337:766–74. doi:10.1124/jpet.110.177790
95. Cushman LJ, Burrows HL, Seasholtz AF, Lewandoski M, Muzyczka N, Camper SA. Cre-mediated recombination in the pituitary gland. Genesis (2000) 28:167–74. doi:10.1002/1526-968X(200011/12)28:3/4<167::AID-GENE120>3.3.CO;2-E
96. Bingham NC, Verma-Kurvari S, Parada LF, Parker KL. Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis (2006) 44:419–24. doi:10.1002/dvg.20231
97. Charles MA, Saunders TL, Wood WM, Owens K, Parlow AF, Camper SA, et al. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol (2006) 20:1366–77. doi:10.1210/me.2005-0378
98. Jorgez CJ, De Mayo FJ, Matzuk MM. Inhibin alpha-iCre mice: Cre deleter lines for the gonads, pituitary, and adrenal. Genesis (2006) 44:183–8. doi:10.1002/dvg.20198
99. Naik K, Pittman IT, Wolfe A, Miller RS, Radovick S, Wondisford FE. A novel technique for temporally regulated cell type-specific Cre expression and recombination in the pituitary gonadotroph. J Mol Endocrinol (2006) 37:63–9. doi:10.1677/jme.1.02053
100. Luque RM, Amargo G, Ishii S, Lobe C, Franks R, Kiyokawa H, et al. Reporter expression, induced by a growth hormone promoter-driven Cre recombinase (rGHp-Cre) transgene, questions the developmental relationship between somatotropes and lactotropes in the adult mouse pituitary gland. Endocrinology (2007) 148:1946–53. doi:10.1210/en.2006-1542
101. Charles MA, Mortensen AH, Potok MA, Camper SA. Pitx2 deletion in pituitary gonadotropes is compatible with gonadal development, puberty, and fertility. Genesis (2008) 46:507–14. doi:10.1002/dvg.20398
102. Perez-Millan MI, Zeidler MG, Saunders TL, Camper SA, Davis SW. Efficient, specific, developmentally appropriate Cre-mediated recombination in anterior pituitary gonadotropes and thyrotropes. Genesis (2013) 51:785–92. doi:10.1002/dvg.22425
Keywords: pituitary, adenoma, GEMMs, transgenic mice, mouse models
Citation: Cano DA, Soto-Moreno A and Leal-Cerro A (2014) Genetically engineered mouse models of pituitary tumors. Front. Oncol. 4:203. doi: 10.3389/fonc.2014.00203
Received: 18 June 2014; Accepted: 15 July 2014;
Published online: 01 August 2014.
Edited by:
Amancio Carnero, Instituto de Biomedicina de Sevilla, SpainReviewed by:
Cynthia Lilian Andoniadou, King’s College London, UKCopyright: © 2014 Cano, Soto-Moreno and Leal-Cerro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Cano, Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío, Lab110, Avda. Manuel Siurot s/n, 41013 Sevilla, Spain e-mail:ZGNhbm8taWJpc0B1cy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.