
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 20 June 2014
Sec. Women's Cancer
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00163
This article is part of the Research Topic Epigenetic and Transcriptional Mechanisms in Gynecologic Cancers Development, Progression, and Resistance to Treatment View all 14 articles
 Brittany Anne Davidson1
Brittany Anne Davidson1 Jennifer M. Rubatt1
Jennifer M. Rubatt1 David L. Corcoran2Deanna K. Teoh1Marcus Q. Bernardini3
David L. Corcoran2Deanna K. Teoh1Marcus Q. Bernardini3 Lisa A. Grace1William John Soper1
Lisa A. Grace1William John Soper1 Andrew Berchuck1
Andrew Berchuck1 Sharareh Siamakpour-Reihani4
Sharareh Siamakpour-Reihani4 Wei Chen5
Wei Chen5 Kouros Owzar5
Kouros Owzar5 Susan K. Murphy1
Susan K. Murphy1 Angeles Alvarez Secord1*
Angeles Alvarez Secord1*Objectives: Underlying mechanisms regulating angiogenesis in ovarian cancer have not been completely elucidated. Evidence suggests that the TP53 tumor suppressor pathway and tumor microenvironment play integral roles. We utilized microarray technology to study the interaction between TP53 mutational status and hypoxia on angiogenic gene expression.
Methods: Affymetrix U133A arrays were analyzed for angiogenic gene expression in 19 ovarian cancer cell lines stratified both by TP53 mutation status and A2780 wild-type (wt) TP53 vs. mutated (m) TP53 cell lines after treatment under hypoxic conditions or with ionizing radiation.
Results: Twenty-eight differentially expressed angiogenic genes were identified in the mTP53 cell lines compared to wtTP53 lines. Five genes were upregulated in mTP53 cells: 40% involved in extracellular matrix (ECM) degradation [matrix metalloproteinase 10 (MMP10)/15] and 60% in angiogenesis (fibroblast growth factor receptor 3/VEGFA/ephrin receptor-B4). Twenty-three genes were upregulated in wtTP53: nearly 22% were ECM constituents or involved in ECM degradation; over 40% were growth factors or mediators of angiogenesis. Five genes were upregulated in the A2780mTP53 cells: 40% involved in ECM remodeling (MMP10, ADAMTS1), 40% with pro-angiogenic activity (EFNB2, factor 2 receptor), and 20% with anti-angiogenic properties (ADAMTS1). Three genes were upregulated in hypoxia treated cells compared to controls: one with anti-angiogenic activity (angiopoietin-like 4) and two with pro-angiogenic activity (VEGFA, EFNA3). No significant gene fold changes were noted after exposure to radiation. Four genes continued to demonstrate significant differential expression (p ≤ 0.05) after adjusting for multiple comparisons. These genes included endoglin upregulation in wt lines (pro-angiogenesis) and upregulation of FGF20 (growth factor), ADAMTS1 (anti-angiogenesis) and MMP10 (ECM degradation) in mTP53 cell lines.
Conclusion: Our exploratory findings indicate that non-overlapping angiogenic pathways may be altered by TP53 mutations and hypoxic conditions in the tumor microenvironment. Further evaluation is needed for confirmation.
The underlying mechanisms that regulate angiogenesis in ovarian cancer have yet to be elucidated but most likely involve interactions controlled by tumors and their microenvironment. Angiogenesis is a complex multistep process that includes increased vascular permeability and dilation followed by extracellular matrix (ECM) degradation; subsequent endothelial cell proliferation and migration; formation of endothelial tubes; and recruitment of pericytes to support the neovasculature. Both tumor and host tissues produce angiogenic factors that influence endothelial cell development and migration (1). The vascular endothelial growth factor (VEGF) family (VEGF-A, -B, -C, FIGF) and fibroblast growth factor 2 (FGF2) are fundamental growth factors in the process of angiogenesis with VEGF-A having the most pro-angiogenic activity. VEGF promoter activity, VEGF mRNA levels, and FGF2 mRNA expression have been shown to be downregulated by wild-type (wt) TP53 (2–4). In mutant (m) TP53 it has been demonstrated that hypoxia inducible factor 1 (HIF1) dependent transcriptional activation of VEGF gene expression has been enhanced (2). TP53 dysfunction has also been associated with increased tumor angiogenesis based on microvessel density (MVD) by immunohistochemistry (IHC) (5, 6). These findings indicate that TP53 may play a role in the regulation of angiogenesis in ovarian cancer.
Hypoxic conditions in the tumor microenvironment have been shown to stimulate angiogenesis in prostate (7), breast (7), melanoma (8), and renal cancer (9). As the distance from tumor to blood supply is increased, the leading edge of the tumor becomes hypoxic, and in turn, induces the expression of the HIF1/hypoxia regulatory element (HRE) complex, a key transcription factor. HIF1α is considered a key regulator of angiogenic factors. HIF1/HRE is responsible for increased gene expression of numerous genes involved in angiogenesis, cell proliferation, and matrix metabolism (10).
In order to investigate the interaction between tumor molecular biology and the microenvironment on the regulation of angiogenesis in ovarian cancer, we utilized genome-scale molecular technology. Our results can enhance our understanding of the molecular profiles of ovarian cancer tumor microenvironment and link crucial processes such as angiogenesis, hypoxia, and perfusion; all of which are established factors in tumor aggressiveness and resistance to therapy. Our primary objective was to determine if angiogenic genes are differentially expressed in ovarian cancer cell lines containing wt vs. mTP53 genes. We also sought to investigate angiogenic gene expression patterns after simulated induction of TP53 and hypoxia-related pathways. Our goal was to identify novel angiogenic targets that may be exploited for therapeutic purposes.
Eighteen immortalized ovarian cancer cell lines maintained by the Duke Gynecologic Oncology research labs (Table 1) were sustained in monolayer culture in RPMI 1640 with 10% fetal bovine serum, sodium pyruvate, glutamine, and non-essential amino acids in 5% CO2 humidified chambers. Cell line authentication was performed using the AmpFlSTR® Identifiler® Plus PCR Amplification Kit (Applied Biosystems, Carlsbad, CA, USA) at the University of Colorado Cancer Center, DNA Sequencing, and Analysis Core (11). The STR genotypes of ovarian cancer cell lines that are available from the American Type Culture Collection or the RIKEN BioResource Center Cell Bank were identical to the source genotypes as reported within their respective STR databases and all other non-commercially available cell lines were shown to be derived from females with unique genotypes. The A2780wtTP53 and A2780mTP53 cell lines were obtained from Professor Robert Brown B.Sc., Ph.D., of the Department of Medical Oncology, University of Glasgow. Protein extractions were performed as previously described (12). All experiments were performed in duplicate or quadruplicate with appropriate controls.

Table 1. Immortalized ovarian cancer cell lines stratified by TP53 status.
A2780 cell lines were grown to 80% confluence in T150 flasks and exposed to hypoxic conditions using 5% O2 in a Bactron Anaerobic Chamber (Sheldon Manufacturing, Cornelius, OH, USA) for 8 or 24 h prior to harvesting. For radiation exposure, the A2780wtTP53 and A2780mTP53 cell lines were plated in 60 mm dishes, exposed to 5 Gy of ionizing radiation for 900 s using the Gammacell 1000 (MDS Nordion, Ottawa, ON, Canada) and harvested at 0, 2, 4, 6, 8, 24, and 48 h. Western blots were performed to evaluate p53 and p21 protein expression per established protocols (12). The following antibodies were used: p53 (sc-126, mouse monoclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p21 (AB-11, mouse monoclonal, NeoMarkers, Fremont, CA, USA), actin (A4700, mouse monoclonal, SIGMA, St. Louis, MO, USA), and goat anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA).
Total RNA was used for probe generation and hybridization to Affymetrix U133A GeneChip arrays as has been described previously in detail (13, 14). The microarray data was screened to select for 378 probe sets belonging to angiogenic candidate genes on the array, based on literature review. Expression patterns were compared between: (1) wtTP53 vs. mTP53 ovarian cancer cell lines; (2) hypoxia treated and untreated controls using the A2780wtTP53 and A2780mTP53 cell lines; (3) radiated A2780wtTP53 after 8 h of exposure and untreated controls. The 8-h sample was chosen because p53 protein expression after radiation exposure was highest between 6 and 8 h (Figure 1).
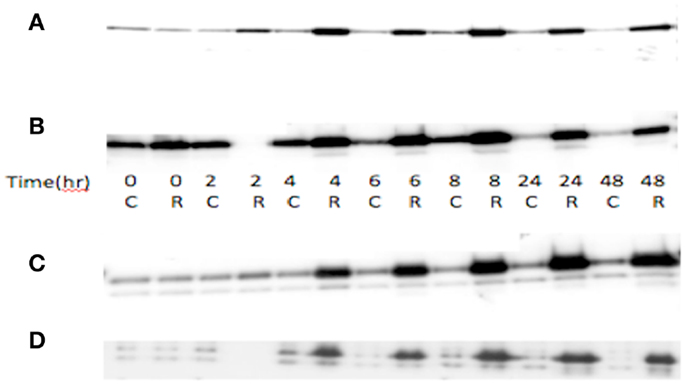
Figure 1. TP53 and p21 protein expression in radiated (R) vs. control (C) in A2780 cell line. (A) Wild-type A2780 cell line shows increased TP53 expression in radiated vs. control samples. (B) TP53 mutant A2780 cell line with induction of TP53 expression in radiated vs. control samples. TP53 protein expression increase seen at 4 h, with greatest increase at 6–8 h, and induction sustained to 48 h. (C) p21 protein expression in wild-type cells. Increased expression noted at 4 h and sustained to 48 h. (D) p21 expression in TP53 mutant cell line. Induction of expression noted at 4 h, sustained to 48 h.
Microarray expression was calculated using the robust multi-array average (RMA) algorithm implemented in the affy package (15) of the Bioconductor (16) extensions to the R statistical programming environment (http://www.R-project.org). RMA generates a background-corrected and quantile-normalized measure of expression on the log2 scale of measurement. The ovarian cancer cell line data, and that generated from the different A2780 conditions, were each analyzed separately in this manner. For each probe set on the array, we used a moderated T-statistic from the limma package (17) to identify differential expression between the wt and mTP53 genotypes. To identify differential expression between the wt and mutant TP53 A2780 cell lines, hypoxia treated cells, and radiated cells, we fit a three way analysis of variance (ANOVA) model with fixed effects for genotype, treatment, and batch for each probe set on the array. Thus genotype effects are treatment and batch corrected, while treatment effects are genotype and batch corrected. The untreated A2780wtTP53 samples served as the baseline for this analysis. The Holm–Bonferroni method was used to correct for multiple hypothesis testing. Candidate genes with an unadjusted p-value <0.05 and an absolute value log2 fold change (L2FC) >1.0 were identified. With these candidate genes, a two-sample t-test was used to analyze the gene expression data available in The Cancer Genome Atlas (TCGA) database. The Benjamini–Hochberg method was used to control the false discovery rate. Clustering of genes for heatmap presentation was done using a correlation distance metric on the z-score normalized expression values.
Eighteen ovarian cancer cell lines with known TP53 genotype (wt vs. m) were analyzed (Table 1). Of the 378 angiogenic candidate gene probes identified during literature review, 28 (7.4%) were found to be differentially expressed in cell lines with wt vs. mTP53 genotype status (Table 2; Figure 2). Five genes (five probe sets) were considered upregulated in the mTP53 cell lines compared to those with a wtTP53 gene given our initial threshold of a p-value ≤0.05 and an absolute value L2FC ≥1. This list includes fibroblast growth factor receptor 3 (FGFR3) (7.0 FC), matrix metalloproteinase 10 (MMP10) (5.7 FC), VEGFA (3.2 FC), MMP15 (2.5 FC), and ephrin receptor-B4 (EPHB4) (2.0 FC). After correcting for multiple hypothesis testing, none of the genes were considered significant at an adjusted p-value <0.05.
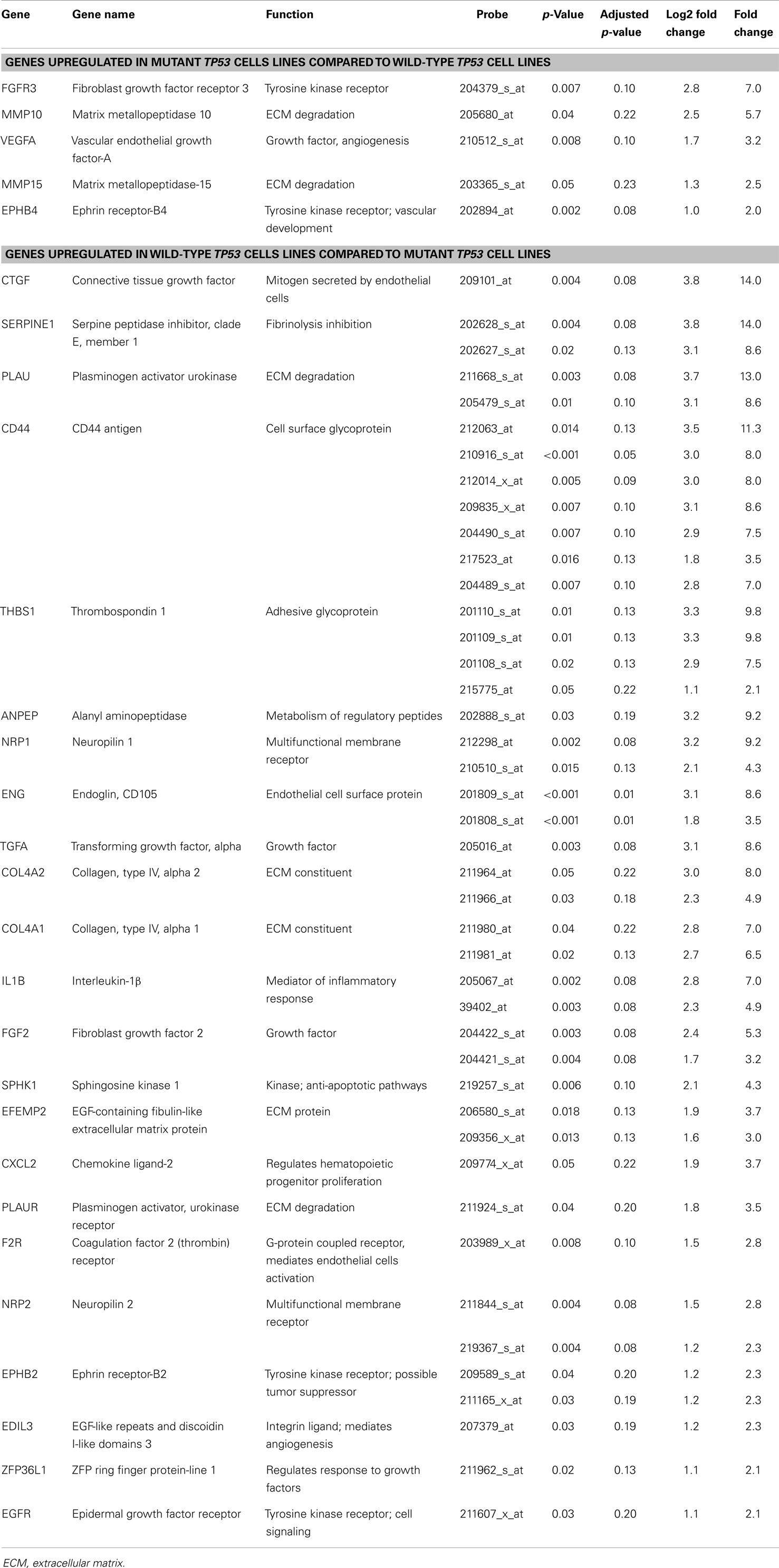
Table 2. Angiogenic genes that are differentially expressed in ovarian cancer cell lines harboring a mutant TP53 gene compared to those with an intact wild-type TP53 gene.
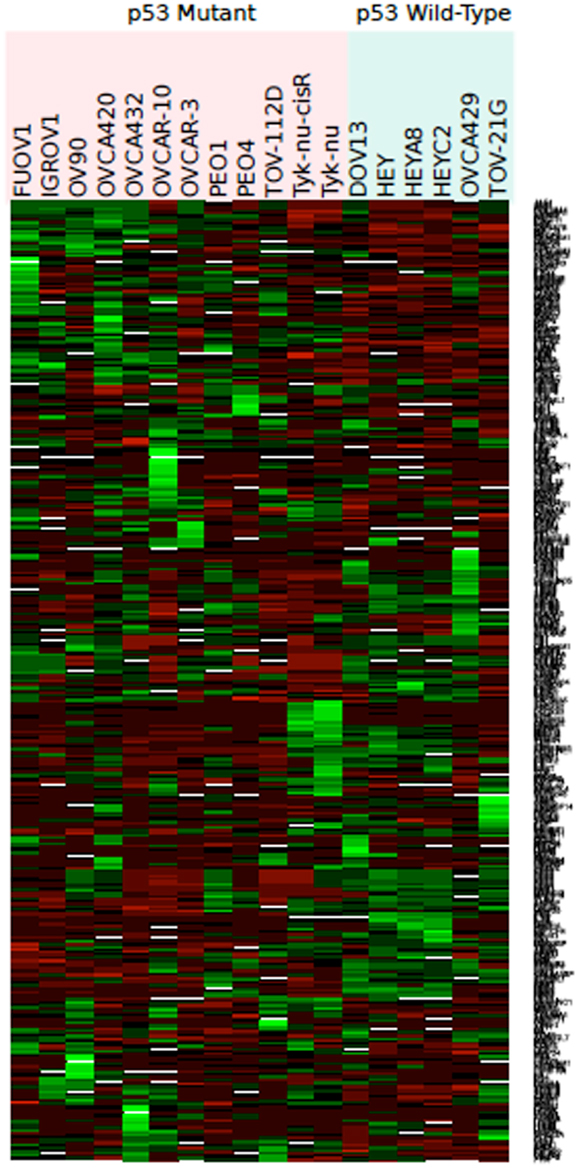
Figure 2. Heat map representing color-coded expression of differentially expressed genes in 18 different wild-type or mutant TP53 cell lines. Twenty-eight genes exhibited statistically significant differential expression by microarray analysis based on TP53 status.
Twenty-three genes (43 probe sets) were upregulated in the wtTP53 relative to the mTP53 cell lines including connective tissue growth factor (CTGF) (14.0 FC), Serpine Peptidase Inhibitor, Clade E, Type 1, Member 1 (SERPINE1) (14.0 FC), plasminogen activator urokinase-type (PLAU) (13.0 FC), CD44 (11.3 FC), thrombospondin 1 (THBS) (9.8 FC), neuropilin 1 (NRP1) (9.2 FC), alanyl aminoopeptidase (ANPEP) (9.2 FC), endoglin (ENG) (8.6 FC), transforming growth factor alpha (TGF-α) (8.6 FC), collagen type IV alpha 2 (COL4A2) (8.0 FC), COL4A1 (7.0 FC), and FGF2 (5.3 FC) (Table 2; Figure 2). Only ENG was significant at a p-value of 0.01 after correcting for multiple hypothesis testing.
The A2780 cell lines differ only by a single TP53 gene mutation and, therefore, allowed for isolation of differences in gene expression related to this TP53 mutation. Five genes were upregulated in the A2780mTP53 compared to A2780wtTP53 cells, including MMP10 (5.3 FC), FGF20 (2.8 FC), A disintegrin-like and metalloprotease with thrombospondin type 1 motif (ADAMTS1) (2.8 FC), ephrin B2 (EPHB2) (2.3 FC), and coagulation F2R (2.0 FC) (Table 3). FGF20 (adjusted p < 0.001), MMP10 (adjusted p = 0.004), and ADAMTS1 (adjusted p < 0.001) were still considered significant after adjusting for multiple hypothesis testing. In contrast, the A2780wtTP53 line demonstrated an upregulation in three genes: VEGFC (2.5 FC), hypoxia inducible factor 1 alpha (HIF1A) (2.3 FC), and angiopoietin-like 4 (ANGPTL4) (2.1 FC) (Table 3).
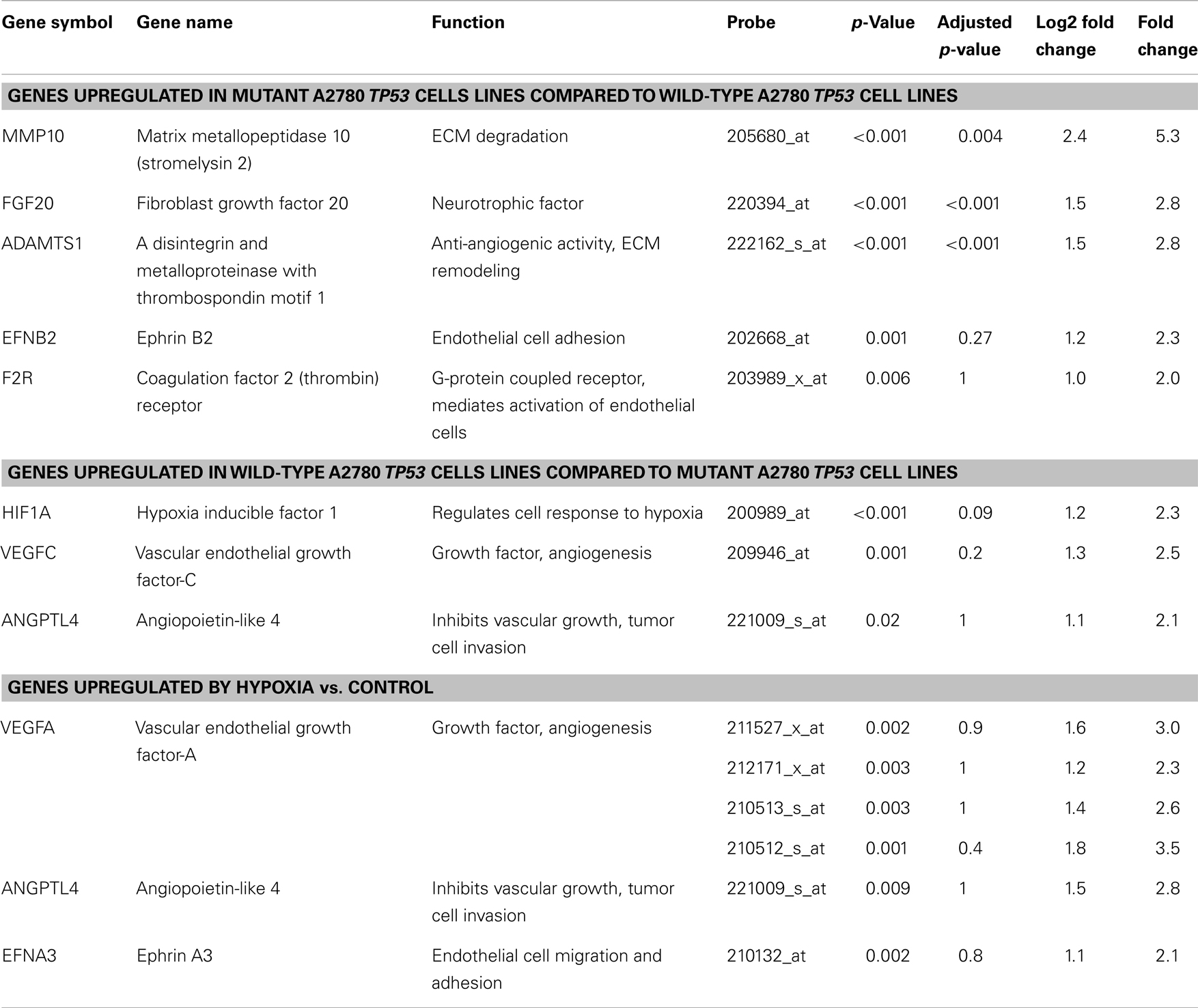
Table 3. Angiogenic genes that are differentially expressed in the A2780 wild-type and mutant TP53 ovarian cancer cell lines and after treatment with hypoxia.
Hypoxia treatment did not increase p53 protein expression (Figure 3). A small increase in p21 protein expression was noted in the TP53 mutant at the 8-h timepoint. This, however, was not sustained at 24 h (Figure 3). Three angiogenic genes were upregulated in hypoxia treated A2780mTP53 cells when compared to A2780wtTP53 controls, including VEGFA (3.5 FC), ANGPTL4 (2.8 FC), and ephrin A3 (EPHA3) (2.1 FC) (Table 3; Figure 4). No genes were identified as upregulated in the A2780wtTP53 when exposed to hypoxia.
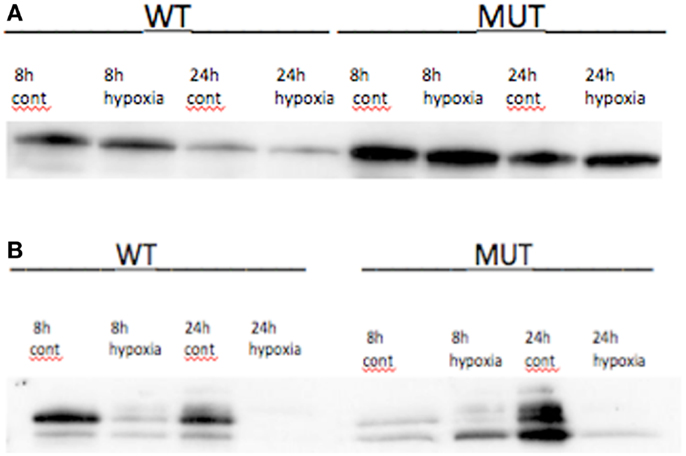
Figure 3. (A) p53 protein expression under hypoxic conditions and control in TP53 wild-type and mutant A2780 cell lines. No changes in protein expression seen at 8 or 24 h. (B) Effects of hypoxia on p21 expression in A2780 cell line. In wtA2780 cell line, no induction of p21 expression was seen under hypoxic conditions. In mA2780 cell line, hypoxia induced a small increase in p21 expression at 8 h but was not present at 24 h.
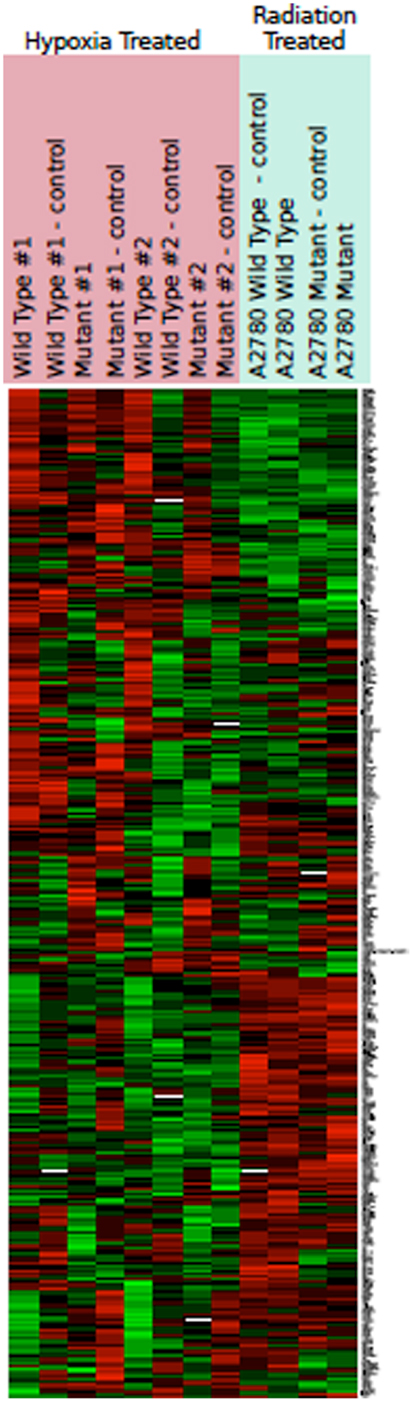
Figure 4. Heat map representing color-coded expression of differentially expressed genes under hypoxic conditions and after radiation. Numbers indicate multiple replicates performed for each sample. Three genes showed statistically significant upregulation by hypoxia. No change in gene expression was noted after radiation.
TP53 protein expression increase seen at 4 h, with greatest increase at 6–8 h (both mutant and wt), and induction sustained to 48 h. p21 showed a similar induction of expression, starting at 4 h and sustained to 48 h, in both wt and mutant cell lines (Figure 1). Exposure to radiation did not yield any significant differentially expressed genes when comparing the A2780mTP53 cells to A2780wtTP53 controls.
The 36 candidate genes were then investigated using cell line data available through the TCGA database. Given that the majority of ovarian cell lines in the database had missing or discordant TP53 status, we looked at cell lines from multiple solid tumor types. Again, after accounting for inconclusive TP53 status and those without gene expression data, 44 cell lines were available for analysis. mRNA expression data between mutant and wt TP53 was then compared. There was not a significant difference in expression between the two groups for any of the candidate genes, though NRP2 (upregulated in wt; p = 0.06) and MMP10 (upregulated in mutant; p = 0.09) trended toward significance.
Understanding the underlying molecular and environmentally responsive pathways driving angiogenesis can provide important insight into the regulation of tumor angiogenesis, development of resistance to VEGF-blocking agents and may assist in the identification of novel targets to exploit in the development of anti-angiogenic therapies. Our exploratory analysis indicates that the regulation of angiogenesis is complex and may be under the control of both TP53-dependent pathways and hypoxic conditions. Furthermore, many of the genes identified are involved in multiple facets of the angiogenic process, such as ECM degradation and remodeling; endothelial cell activation, migration, and adhesion. While our data are exploratory in nature, there were four genes that continued to demonstrate significant differential expression even after adjusting for multiple comparisons. These four differentially regulated genes included ENG (upregulated in wt) and FGF20, ADAMTS1, and MMP10 (upregulated in mutant).
Matrix metalloproteinase 10 encodes a member of the matrix metalloproteinase family of proteins that is responsible for basement membrane degradation. MMPs are cysteine proteases with zinc ion-dependent proteolytic activity that are involved principally in the degradation of the ECM and subsequent tissue remodeling (18–20). Our findings indicate that the intact TP53 tumor suppressor pathway may exert control via the regulation of proteins involved in the ECM. The destruction of the basement membrane and the ECM is a fundamental step in the process of tumor angiogenesis. Forty percent of the upregulated genes identified in the mutated TP53 cell lines were involved in ECM degradation (MMP10 and MMP15). MMPs are also upregulated in response to cytokines, hormone, and growth factors, including VEGF (21–23). Conversely, MMPs can also regulate the activity of various growth factors, again including VEGF (24), as well as chemokines, cytokines, and cell surface adhesion receptors. These components are involved in cell migration and intracellular communication that are directly implicated in wound healing, angiogenesis, tumor progression, and metastasis (18–20, 25, 26). The induction of MMP15 (also known as membrane-type-2 MMP) has been shown to positively correlate with ovarian tumor metastases in murine xenographs (27). MMP15 has also been postulated to have a role in anti-apoptotic pathways, though the precise mechanisms remain unknown (28). MMP10 was also significantly upregulated in mutant A2780 TP53 vs. wt cell lines and marginally upregulated in the TCGA mutant TP53 vs. wt cell lines. Furthermore, MMP10 has been shown to be highly expressed in breast (29), prostate (30), and cervical cancer (31). Compared to epithelial ovarian cancer (EOC), TP53 mutations in these particular malignancies are much less common (32). In a study of head and neck squamous cell malignancies, TP53 mutations were strongly associated with MMP-9 overexpression with a subsequent increase in mean vessel density (33) while elevated p53 levels were associated with decreased MMP-2 levels in patients with invasive breast cancer (34). The notable upregulation of a number of members of the MMP family in mutated TP53 cell lines indicates that deregulation of the TP53 pathway may play an integral role in ECM remodeling during tumor angiogenesis.
We found that several members of the FGF pathway exhibited differential expression. This pathway is comprised of over 20 ligands and 4 tyrosine kinase receptors (FGFRs). Selected FGF growth factors activate the FGFRs in conjunction with heparan sulfate proteoglycans leading to the regulation of cell differentiation, angiogenesis, cell motility, invasion, and survival (35). FGF20 was recently identified and may have a potential role in tumor growth and metastasis (36). Forced expression of FGF20 resulted in increased DNA synthesis, cellular proliferation, in vitro transformation, and in vivo tumor growth (36). There is conflicting data regarding FGF20 expression in ovarian cancer. Our data indicated that FGF20 is expressed in ovarian cancer cell lines and was upregulated in the A2780mTP53 ovarian cancer cells compared to the A2780wtTP53 cells. However, in the study conducted by Jeffers et al. FGF20 mRNA was not expressed in normal ovarian tissue or the six studied ovarian cell lines (OVCAR3, SKOV3, OVCAR4, OVCAR5, IGROV1, OVAR8) (36). In contrast, Chamorro et al. reported that FGF20 is significantly elevated in EOC cells harboring mutations in the WNT/B-catenin signaling pathway (37). FGFR3 expression may also have an important role in cancer progression. Our data show a sevenfold increase in FGFR3 expression in mutant TP53 lines, consistent with Kim et al.’s finding that inhibition of FGFR3 increased target cell apoptosis and decreased resistance to targeted drug therapy (38). FGFR3 overexpression has also been correlated with shorter disease free intervals and overall survival in patients with subtypes of bladder malignancies (39).
In contrast, we found that FGF2 was upregulated in A2780 wt TP53 (5.3 FC) compared to mutant cells. We previously reported that relative high FGF2 protein expression was associated with a significant decreased risk of disease progression and death in women with advanced ovarian cancer treated with taxane and platinum-based therapy. Upon multivariate analysis, however, the association between FGF2 and clinical outcome was no longer significant (40). In addition, we did not detect an association between FGF2 expression and TP53 mutation status or protein expression (40). In our prior study of FGF2, we used immunoblot technology and were unable to assess whether FGF2 is located in the stromal or cellular compartment. Our current study evaluated FGF2 only in cell lines and does not account for stromal expression of FGF2 protein. FGF/FGFR signaling is very complex and function may vary based on the interaction of the specific FGF ligand and the FGFR variant as well as regulatory factors in the tumor and microenvironment (35). Galy et al. elegantly demonstrated that p53 protein directly repressed FGF2 mRNA translation (41). These findings highlight limitations with our microarray analysis. A single microarray analysis cannot capture transcription variances or translational protein alterations. Further exploration is needed to determine if the TP53 pathway is involved in the coordinated regulation of FGF family members.
Our paper is the first to report a potential relationship between TP53 mutation status and ADAMTS1 expression in ovarian cancer. In our study, ADAMTS1 was found to be upregulated in A2780mTP53 cell lines compared to wt. ADAMTS1 is active in ECM degradation and remodeling (42, 43) and has been implicated in normal ovarian follicular development (44). Collagen IV, as well as other basement membrane structural proteins, are poorly organized in ADAMTS1 null ovaries (43). ADAMTS1 may also play a role in ovarian medullary vascular development (44) and its absence leads to a delay in lymphatic development (43). Conflicting evidence exists regarding ADAMTS1 expression in malignancy. A study by Freitas et al. reported decreased ADAMTS1 expression in primary breast malignancies with forced knockdown stimulating migration and invasion of tumor cells in vivo (45). In contrast, others have reported significant upregulation in breast malignancies with subsequent increase in metastatic activity (46, 47). These contrasting findings may result from the auto-proteolytic cleavage of ADAMTS1 with subsequent disparate effects on tumor activity – the full length molecule displays pro-tumor properties while its cleavage products, ADAMTS-1NTF and ADAMTS-1CTF, exhibit anti-tumor activity. This cleavage process is regulated by the rates of production and degradation of heparin sulfate proteoglycans in the tumor microenvironment (48). Our findings suggest that ADAMTS1 regulation may also be controlled via the TP53 pathway.
In contrast to the previous candidate genes with upregulation in mutant lines, ENG was upregulated in the wtTP53 cell lines. ENG (CD105) is a membrane protein overexpressed in tumor-associated endothelial cells and is a marker of proliferating endothelial cells and surrogate for tumor angiogenesis. ENG downregulation in human EOC lines results in decreased vascular proliferation (49). Ziebarth et al. demonstrated that forced ENG inhibition resulted in decreased cell viability, increased apoptosis, induced double-stranded DNA damage, and increased cisplatin sensitivity in ovarian cancer cell lines (50). In our study, ENG was upregulated in the wt TP53 cell lines and was a surprising finding given the association between increased ENG staining in MCV or tumor-associated endothelial cells and advanced stage disease, suboptimal cytoreduction (51), and increased disease progression (52, 53). There are conflicting results regarding the association between ENG and survival (51, 54, 55). High ENG expression in combination with high transforming growth factor B levels prior to chemotherapy have been associated with improved overall survival (55) while others have reported either decrease survival for those with the highest levels of ENG expression (51, 53) or no relationship at all (54). Previously, we used ENG staining to determine MVD. We did not find an association between ENG MVD and TP53 gene mutation status or protein expression (52). The difference between our two studies may be due to the disparate study design (cell lines vs. tumor tissue) and/or methodology (gene vs. protein expression; which may not correlate) as well as the amount of stromal tissue included in the tumor tissues. Our current study evaluating ENG in cell lines alone does not account for stromal expression of ENG protein in the microvasculature. The association between ENG expression and TP53 status in cell lines suggests that ENG may be regulated by the TP53 pathway, though other regulatory mechanisms may also exist.
Our data also demonstrated differential regulation of other well known angiogenic genes in the mTP53 cell lines. Most notably, VEGFA was upregulated in the mTP53 cell lines and under hypoxic conditions demonstrating the convergence of the VEGF pathway by both mechanisms. VEGFA is known to be one of the most potent pro-angiogenic factors. There is conflicting literature regarding TP53 status and association with VEGF protein expression in ovarian cancer specimens. Horiuchi and colleagues reported no association between p53 and VEGF protein expression in ovarian cancers using IHC (56). Previously, we reported an association between p53 protein overexpression and low VEGF protein expression in advanced ovarian cancer specimens, but no association between VEGF and TP53 mutation status (40). Upon further assessment, we found that the association between p53 and VEGF protein expression was limited to the ovarian cancers that contained a wt TP53 gene and lacked p53 protein expression (40). The lack of VEGFA induction with ionizing radiation suggests that higher VEGFA expression may be associated with TP53 mutations, but that TP53 may not be regulating VEGFA expression. In contrast, the VEGF pathway may be primarily regulated via hypoxic conditions in the tumor microenvironment rather than by TP53. It is well established in the literature that hypoxic conditions increase VEGF expression (5, 10, 56, 57) with expression increasing after exposure to hypoxia regardless of TP53 pathway status. This suggests that hypoxia has a dominant role in VEGF regulation (57).
Furthermore, other members of the VEGF family, such as VEGFC and NRP1 and NRP2, were all upregulated in the cell lines harboring wtTP53 compared to those with a mutant gene. NRP2 was also marginally upregulated in the TCGA wt TP53 cell lines compared to mutant cell lines. VEGFC is an integral part of lymphangiogenesis and has been associated with lymph node metastases and prognosis in a variety of malignancies (58–61). NRP1 and NRP2 interact with both VEGF ligands and class 3 semaphorin (SEMA3) ligands in overlapping binding domains (62). While VEGF promotes angiogenesis and interacts with NRP1 to enhance the binding to its receptor, members of the SEMA3 family inhibit angiogenesis (63). Specifically, SEMA3F binding to NRP2 inhibits tumor angiogenesis and metastasis (63). NRP overexpression has been reported in multiple solid malignancies including breast, gastrointestinal, and prostate tumors (63). Conflicting evidence exists regarding NRP expression in ovarian tumors. Bednarek et al. studied NRP1 expression in 50 patients with EOC, the majority of which had weak (n = 13) or no (n = 22) immunohistochemical staining (64). In contrast, Baba et al. reported that 97% of EOC tissue samples and 67% of cell lines stain strongly (65). Recently, Stanton and colleagues reported that the VEGFC–NRP2 axis promoted autophagy, which, in cancer, may represent an adaptive response to promote cell survival (62).
Members of the ephrin family were also differentially expressed in the cell lines and after exposure to hypoxia. The genes encoding the ligand EFNB2 and the receptor EPHB4 were upregulated in mutant TP53 cells; EFNB2 was upregulated in the A2780 line and EPHB4 was upregulated in the larger pool of mutant TP53 lines. In contrast, the ephrin receptor, EPHB2, was upregulated in wt TP53 cell lines while overexpression of the ligand, EFNA3, was induced by hypoxia. Ephrin and the ephrin receptors are a family of membrane-bound tyrosine kinases and receptor tyrosine kinases (RTKs) that are typically highly promiscuous; most of the receptors are capable of binding to numerous ephrin ligands (66, 67). Since both receptors and ligands are membrane-bound, the receptor ligand interactions are capable of bi-directional signaling (67). Members of the ephrin RTK family are expressed on both tumor cells as well as the tumor endothelium and fibroblasts (68, 69). Data from targeted disruption of Eph RTKs and ligands in mice have revealed that the ephrin pathway plays a critical role in embryologic vascular development and tumor angiogenesis (70). EFNA1 and EFNB2 are regulated by TP53 as well as other members of the TP53 family, including p73 and p63 (71, 72). Hypoxia upregulates both mRNA and protein expression of EPHB4, EFNB2, EPHA2, and EFNA1 (73). EPHB4 RTK is expressed in 86% of invasive ovarian cancers and was associated with advanced stage, worse survival (74), and decreased response to chemotherapy (66). EFNB1, an alternate ligand of EPHB4, has been associated with increased MVD in EOC (75). The precise mechanism of ephrin-mediated angiogenesis is unknown, but our data indicate that both the TP53 tumor suppressor pathway and hypoxia may effect ephrin family members.
In addition, we have evaluated our panel of angiogenic genes in women with advanced, high grade serous ovarian carcinoma whose tumors had undergone microarray analysis. We conducted an extreme phenotype study that included women with long survival (>7 years) vs. short survival (<3 years) (76). Thirty-one genes were significantly associated with clinical outcome including several of the genes reported in our current study (CD44−, EPHB2, HIF1A, NRP1, and TGFA). Of these, high CD44 was associated with longer survival in the TCGA database. In contrast, high expression of EPHB2 and NRP1 were associated with shorter survival in an external database (76). Microarray analysis of cell lines may potentially identify genes that have prognostic significance for survival.
Limitations of our study include the use of a simple model of ionizing radiation to simulate TP53 induction (77, 78). To validate our model, we irradiated ovarian cancer cell line OVCA420 to 5 Gy and then subjected cell lysates to immunoblot to assess total p53 expression. When compared to non-irradiated cells, irradiated cells demonstrated a 3.3-fold increase in p53 protein expression 48 h after exposure (Figure 1). We acknowledge that ionizing radiation likely induces other genes in addition to TP53 and these unidentified genes may also play a role in angiogenesis. The in vitro nature of this investigation limits the application of these results to more complex systems such as living organisms. Specifically, cell lines lack the adjacent stroma that is integral to evaluate mesenchymal remodeling, and tumor angiogenesis was not assessed in this model. Another limitation of our study is that while a number of genes showed a significant change in expression, most were no longer significant after correcting the p-values for multiple hypothesis testing. Thus, further verification by another methodology is necessary to establish that these genes are in fact differentially expressed and not the result of statistical error. Our study incorporates data from cell lines of various epithelial ovarian histologies. Though it is well known that an overwhelming majority of serous type tumors possess TP53 mutations, a study by the Gynecologic Oncology Group suggests p53 overexpression may be seen in one-third of mucinous or clear cell histologies while over 70% of tumors deemed “other” also possess p53 overexpression (79).
Despite these limitations the data generated from this study confirmed the complexity of angiogenesis regulation and the presence of convergent and divergent pathways controlled via TP53-dependent and independent mechanisms representing how genetic mechanisms and environmental conditions interact to promote a pro-angiogenic environment. Identification of multiple members of the VEGF, FGF, MMP, and ephrin families as well as other novel genes in our series indicates the existence of multiple regulatory mechanisms involved in tumor angiogenesis. The most differentially expressed genes in our panel represent appealing therapeutic targets that may be exploited to develop anti-angiogenic therapies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research was supported by the Gail Parkins Ovarian Cancer Research Fund as well as anonymous funding.
1. Diaz-Flores L, Gutierrez R, Varela H. Angiogenesis: an update. Histol Histopathol (1994) 9:807–43.
2. Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor expression in tumors. Semin Cancer Biol (2004) 17:123–30. doi: 10.1016/j.semcancer.2003.09.019
3. Mukhopadhyay D, Tsiokas L, Sukhatme VP. Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res (1995) 55:6161–5.
4. Galy B, Créancier L, Zanibellato C, Prats AC, Prats H. Tumour suppressor p53 inhibits human fibroblast growth factor 2 expression by a post-transcriptional mechanism. Oncogene (2001) 20:1669–77. doi:10.1038/sj.onc.1204271
5. Goodheart MJ, Ritchie JM, Rose SL, Fruehauf JP, De Young BR, Buller RE. The relationship of molecular markers of p53 function and angiogenesis to prognosis of stage I epithelial ovarian cancer. Clin Cancer Res (2005) 11:3733–42. doi:10.1158/1078-0432.CCR-04-0056
6. Sood AK, Sorosky JI, Dolan M, Anderson B, Buller RE. Distant metastases in ovarian cancer: association with p53 mutations. Clin Cancer Res (1999) 5:2485–90.
7. Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer (2006) 13:739–49. doi:10.1677/erc.1.00728
8. Michaylira CZ, Nakagawa H. Hypoxic microenvironment as a cradle for melanoma development and progression. Cancer Biol Ther (2006) 5:476–9. doi:10.4161/cbt.5.5.2749
9. Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol (2006) 24:5601–8. doi:10.1200/JCO.2006.08.5415
10. Hewitson K, Schofield CJ. The HIF pathway as a therapeutic target. Drug Discov Today (2004) 9:704–11. doi:10.1016/S1359-6446(04)03202-7
11. Korch C, Spillman MA, Jackson TA, Jacobsen BM, Murphy SK, Lessey BA, et al. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol Oncol (2012) 127(1):241–8. doi:10.1016/j.ygyno.2012.06.017
12. Havrilesky LJ, Alvarez AA, Whitaker RS, Marks JR, Berchuck A. Loss of expression of the p16 tumor suppressor gene is more frequent in advanced ovarian cancers lacking p53 mutations. Gynecol Oncol (2001) 83:491–500. doi:10.1006/gyno.2001.6464
13. Berchuck A, Iversen ES, Lancaster JM, Pittman J, Luo J, Lee P. Patterns of gene expression that characterize long-term survival in advanced stage serous ovarian cancers. Clin Cancer Res (2005) 11(10):3686–96. doi:10.1158/1078-0432.CCR-04-2398
14. Berchuck A, Iversen ES, Luo J, Clarke JP, Horne H, Levine DA, et al. Microarray analysis of early stage serous ovarian cancers shows profiles predictive of favorable outcome. Clin Cancer Res (2009) 15(7):2448–55. doi:10.1158/1078-0432.CCR-08-2430
15. Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (2004) 20(3):307–15. doi:10.1093/bioinformatics/btg405
16. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol (2004) 5(10):R80. doi:10.1186/gb-2004-5-10-r80
17. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol (2004) 3:Article3.
18. Stamenkovic I. Extracellular matrix remodeling: the role of matrix metalloproteinases. J Pathol (2003) 200:448–64. doi:10.1002/path.1400
19. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer (2002) 2:161–74.
20. McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol (2001) 13:534–40. doi:10.1016/S0955-0674(00)00248-9
21. Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem Biol (1996) 3:895–904. doi:10.1016/S1074-5521(96)90178-7
22. Wang FQ, So J, Reierstad S, Fishman DA. Vascular endothelial growth factor-regulated ovarian cancer invasion and migration involves expression and activation of matrix metalloproteinases. Int J Cancer (2006) 118:879–88. doi:10.1002/ijc.21421
23. Yabushita H, Shimazu M, Noguchi M, Kishida T, Narumiya H, Sawaguchi K, et al. Vascular endothelial growth factor activating matrix metalloproteinase in ascitic fluid during peritoneal dissemination of ovarian cancer. Oncol Rep (2003) 10(1):89–95.
24. Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, et al. Matrix metalloproteinases induce the release of vascular endothelial growth factor by ovarian carcinoma cells: implications for ascites formation. Cancer Res (2003) 63(17):5224–9.
25. Matrisian LM. Metalloproteinases and their inhibitors in matrix remodeling. Trends Genet (1990) 6:121–5. doi:10.1016/0168-9525(90)90126-Q
26. Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell (1991) 64:327–36. doi:10.1016/0092-8674(91)90642-C
27. Lin A, Xu HH, Xu DP, Zhang X, Wang Q, Yan WH. Multiple steps of HLA-G in ovarian carcinoma metastasis: alter NK cytotoxicity and induce matrix metalloproteinase-15. Hum Immunol (2013) 74(4):439–46. doi:10.1016/j.humimm.2012.11.021
28. Abraham R, Schäfer J, Rothe M, Bange J, Knyazev P, Ullrich A. Identification of MMP-15 as an apoptotic factor in cancer cells. J Biol Chem (2005) 280:34123–32. doi:10.1074/jbc.M508155200
29. Decock J, Hendrickx W, Drijkoningen M, Wildiers H, Neven P, Smeets A, et al. Matrix metalloproteinase expression patterns in luminal A type breast carcinomas. Dis Markers (2007) 23:189–96. doi:10.1155/2007/281727
30. Riddick AC, Shukla CJ, Pennington CJ, Bass R, Nuttall RK, Hogan A, et al. Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. Br J Cancer (2005) 92:2171–80. doi:10.1038/sj.bjc.6602630
31. Vázquez-Ortíz G, Piña-Sanchez P, Hidalgo A, Lazos M, Moreno J, Alvarado I, et al. Global expression analysis in uterine cervical cancer: metabolic pathways and altered genes. Rev Invest Clin (2005) 57(3):434–41.
32. The TP53 Website. P53 Mutations: All Cancers (2014). Available from: http://p53.free.fr/Database/p53_cancer/all_cancer.html
33. Franchi A, Santucci M, Masini E, Sardi I, Paglierani M, Gallo O. Expression of matrix metalloproteinase 1, matrix metalloproteinase 2 and matrix metalloproteinase 9 in carcinoma of the head and neck. Cancer (2002) 95(9):1902–10. doi:10.1002/cncr.10916
34. Nakopoulou L, Tsirmpa I, Alexandrou P, Louvrou A, Ampela C, Markaki S, et al. MMP-2 protein in invasive breast cancer and the impact of MMP-2/TIMP-2 phenotype on overall survival. Breast Cancer Res Treat (2003) 77(2):145–55. doi:10.1023/A:1021371028777
35. Corn PG, Wang F, McKeehan WL, Navone N. Targeting fibroblast growth factor pathways in prostate cancer. Clin Cancer Res (2013) 19(21):5856–66. doi:10.1158/1078-0432.CCR-13-1550
36. Jeffers M, Shimkets R, Prayaga S, Boldog F, Yang M, Burgess C, et al. Identification of a novel human fibroblast growth factor and characterization of its role in oncogenesis. Cancer Res (2001) 61(7):3131–8.
37. Chamorro MN, Schwartz DR, Vonica A, Brivanlou AH, Cho KR, Varmus HE. FGF-20 and DKK1 are transcriptional targets of beta-catenin and FGF-20 is implicated in cancer and development. EMBO J (2005) 24(1):73–84. doi:10.1038/sj.emboj.7600460
38. Kim B, Wang S, Lee JM, Jeong Y, Ahn T, Son DS, et al. Synthetic lethal screening reveals FGFR as one of the combination targets to overcome resistance to Met-targeted therapy. Oncogene (2014). doi:10.1038/onc.2014.51
39. Sung JY, Sun JM, Chang Jeong B, Il Seo S, Soo Jeon S, Moo Lee H, et al. FGFR3 overexpression is prognostic of adverse outcome for muscle-invasive bladder carcinoma treated with adjuvant chemotherapy. Urol Oncol (2014) 32(1):49.e23–31. doi:10.1016/j.urolonc.2013.07.015
40. Secord AA, Darcy KM, Hutson A, Lee PS, Havrilesky LJ, Grace LA, et al. Co-expression of angiogenic markers and associations with prognosis in advanced epithelial ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol (2007) 106:221–32. doi:10.1016/j.ygyno.2007.03.021
41. Galy B, Créancier L, Prado-Lourenço L, Prats AC, Prats H. p53 Directs conformational change and translation initiation blockade of human fibroblast growth factor 2 mRNA. Oncogene (2001) 20(34):4613–20. doi:10.1038/sj.onc.1204630
42. Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem (2001) 276(16):13372–8. doi:10.1074/jbc.M009737200
43. Brown HM, Dunning KR, Robker RL, Pritchard M, Russell DL. Requirement for ADAMTS-1 in extracellular matrix remodeling during ovarian folliculogenesis and lymphangiogenesis. Dev Biol (2006) 300(2):699–709. doi:10.1016/j.ydbio.2006.10.012
44. Shozu M, Minami N, Yokoyama H, Inoue M, Kurihara H, Matsushima K, et al. ADAMTS-1 is involved in normal follicular development, ovulatory process and organization of the medullary vascular network in the ovary. J Mol Endocrinol (2005) 35(2):343–55. doi:10.1677/jme.1.01735
45. Freitas VM, do Amaral JB, Silva TA, Santos ES, Mangone FR, Pinheiro J, et al. Decreased expression of ADAMTS-1 in human breast tumors stimulates migration and invasion. Mol Cancer (2013) 12:2. doi:10.1186/1476-4598-12-2
46. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3(6):537–49. doi:10.1016/S1535-6108(03)00132-6
47. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest (2005) 115(1):44–55. doi:10.1172/JCI22320
48. Liu YJ, Xu Y, Yu Q. Full-length ADAMTS-1 and the ADAMTS-1 fragments display pro- and antimetastatic activity, respectively. Oncogene (2006) 25(17):2452–67. doi:10.1038/sj.onc.1209287
49. Xu Y, Wang D, Zhao LM, Zhao XL, Shen JJ, Xie Y, et al. Endoglin is necessary for angiogenesis in human ovarian carcinoma-derived primary endothelial cells. Cancer Biol Ther (2013) 14(10):937–48. doi:10.4161/cbt.25940
50. Ziebarth AJ, Nowsheen S, Steg AD, Shah MM, Katre AA, Dobbin ZC, et al. Endoglin contributes to platinum resistance and is a target for tumor-specific therapy in epithelial ovarian cancer. Clin Cancer Res (2013) 19(1):170–82. doi:10.1158/1078-0432.CCR-12-1045
51. Taskiran C, Erdem O, Onan A, Arisoy O, Acar A, Vural C, et al. The prognostic value of endoglin expression in ovarian carcinoma. Int J Gynecol Cancer (2006) 16(5):1789–93. doi:10.1111/j.1525-1438.2006.00658.x
52. Rubatt JM, Darcy KM, Hutson A, Bean SM, Havrilesky LJ, Grace LA, et al. Independent prognostic relevance of microvessel density in advanced epithelial ovarian cancer and associations between CD31, CD105, p53 status, and angiogenic marker expression: a Gynecologic Oncology Group study. Gynecol Oncol (2009) 112(3):469–74. doi:10.1016/j.ygyno.2008.11.030
53. Liu P, Sun YL, Du J, Hou XS, Meng H. CD105/Ki67 coexpression correlates with tumor progression and poor prognosis in epithelial ovarian cancer. Int J Gynecol Cancer (2012) 22(4):586–92. doi:10.1097/IGC.0b013e31823c36b8
54. Bock AJ, Tuft Stavnes H, Kærn J, Berner A, Staff AC, Davidson B. Endoglin expression in ovarian serous carcinoma effusions is related to chemotherapy status. Tumour Biol (2011) 32(3):589–96. doi:10.1007/s13277-011-0157-6
55. Bozas G, Terpos E, Gika D, Karadimou A, Dimopoulos MA, Bamias A. Prechemotherapy serum levels of CD105, transforming growth factor beta2, and vascular endothelial growth factor are associated with prognosis in patients with advanced epithelial ovarian cancer treated with cytoreductive surgery and platinum-based chemotherapy. Int J Gynecol Cancer (2010) 20(2):248–54. doi:10.1111/IGC.0b013e3181cc25c3
56. Horiuchi A, Imai T, Shimizu M, Oka K, Wang C, Nikaido T, et al. Hypoxia-induced changes in the expression of VEGF, HIF-1 alpha and cell cycle-related molecules in ovarian cancer cells. Anticancer Res (2002) 22(5): 2697–702.
57. Laderoute KR, Alarcon RM, Brody MD, Calaoagan JM, Chen EY, Knapp AM, et al. Opposing effects of hypoxia on expression of the angiogenic inhibitor thrombospondin 1 and the angiogenic inducer vascular endothelial growth factor. Clin Cancer Res (2000) 6(7):2941–50.
58. Nakamura Y, Yasuoka H, Tsujimoto M, Imabun S, Nakahara M, Nakao K, et al. Lymph vessel density correlates with nodal status, VEGF-C expression, and prognosis in breast cancer. Breast Cancer Res Treat (2005) 91(2):125–32. doi:10.1007/s10549-004-5783-x
59. Yang J, Wu HF, Qian LX, Zhang W, Hua LX, Yu ML, et al. Increased expressions of vascular endothelial growth factor, VEGF-C and VEGF receptor-3 in prostate cancer tissue are associated with tumor progression. Asian J Androl (2006) 8(2):169–75. doi:10.1111/j.1745-7262.2006.00120.x
60. Gou HF, Chen XC, Zhu J, Jiang M, Yang Y, Cao D, et al. Expressions of COX-2 and VEGF-C in gastric cancer: correlations with lymphangiogenesis and prognostic implications. J Exp Clin Cancer Res (2011) 28(30):14. doi:10.1186/1756-9966-30-14
61. Guo X, Chen Y, Xu Z, Xu Z, Qian Y, Yu X. Prognostic significance of VEGF-C expression in correlation with COX-2, lymphatic microvessel density, and clinicopathologic characteristics in human non-small cell lung cancer. Acta Biochim Biophys Sin (2009) 41(3):217–22. doi:10.1093/abbs/gmp004
62. Stanton MJ, Dutta S, Zhang H, Polavaram NS, Leontovich AA, Hönscheid P, et al. Autophagy control by the VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer Res (2013) 73(1):160–71. doi:10.1158/0008-5472.CAN-11-3635
63. Bielenberg DR, Pettaway CA, Takashima S, Klagsbrun M. Neuropilins in neoplasms: expression, regulation, and function. Exp Cell Res (2006) 312(5):584–93. doi:10.1016/j.yexcr.2005.11.024
64. Bednarek W, Mazurek-Kociubowska M, Sobstyl M, Wertel I, Czekierdowski A. Expression of lymphangiogenesis marker neuropilin-1 in different types of ovarian cancer. Ginekol Pol (2010) 81(3):176–82.
65. Baba T, Kariya M, Higuchi T, Mandai M, Matsumura N, Kondoh E, et al. Neuropilin-1 promotes unlimited growth of ovarian cancer by evading contact inhibition. Gynecol Oncol (2007) 105(3):703–11. doi:10.1016/j.ygyno.2007.02.005
66. Wu Q, Suo Z, Kristensen GB, Baekelandt M, Nesland JM. The prognostic impact of EphB2/B4 expression on patients with advanced ovarian carcinoma. Gynecol Oncol (2006) 102:15–21. doi:10.1016/j.ygyno.2005.11.034
67. Haramis APG, Perrakis A. Selectivity and promiscuity in Eph receptors. Structure (2006) 14:169–71. doi:10.1016/j.str.2006.01.005
68. Brantley DM, Cheng N, Thompson EJ, Lin Q, Brekken RA, Thorpe PE, et al. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene (2002) 21:7011–26. doi:10.1038/sj.onc.1205679
69. Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res (2001) 61:2301–6.
70. Brantley-Sieders DM, Chen J. Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis (2004) 7:17–28. doi:10.1023/B:AGEN.0000037340.33788.87
71. Dohn M, Jiang J, Chen X. Receptor tyrosine kinase EphA2 is regulated by p53-family proteins and induced apoptosis. Oncogene (2001) 20:6503–15. doi:10.1038/sj.onc.1204816
72. Maxwell SA, Davis GE. Biological and molecular characterization of an ECV-304-derived cell line resistant to p53-mediated apoptosis. Apoptosis (2000) 5:277–90. doi:10.1023/A:1009660714216
73. Vihanto MM, Plock J, Erni D, Frey BM, Frey FJ, Huynh-Do U. Hypoxia up-regulates expression of Eph receptors and ephrins in mouse skin. FASEB J (2005) 19:1689–91. doi:10.1096/fj.04-3647fje
74. Kumar SR, Masood R, Spannuth WA, Singh J, Scehnet J, Kleiber G, et al. The receptor tyrosine kinase EphB4 is overexpressed in ovarian cancer, provides survival signals and predicts poor outcome. Br J Cancer (2007) 96:1083–91. doi:10.1038/sj.bjc.6603642
75. Castellvi J, Garcia A, de la Torre J, Hernandez J, Gil A, Xercavins J, et al. Ephrin B expression in epithelial ovarian neoplasms correlates with tumor differentiation and angiogenesis. Hum Pathol (2006) 37:883–9. doi:10.1016/j.humpath.2006.02.021
76. Siamakpour-Reihani S, Owzar K, Jiang C, et al. Prognostic significance of differential expression of angiogenic genes in women with invasive high-grade serous ovarian carcinoma. Presented at American Society of Clinical Oncology 49th Annual Meeting. Chicago, IL: (2013).
77. Siddik ZH, Mims B, Lozano G, Thai G. Independent pathways of p53 induction by cisplatin an X-rays in a cisplatin-resistant ovarian tumor cell line. Cancer Res (1998) 58:698–703.
78. Hill DA, de la Serna IL, Veal TM, Imbalzano AN. BRCA1 interacts with dominant negative SWI/SNF enzymes without affecting homologous recombination of radiation-induced gene activation of p21 or Mdm2. J Cell Biochem (2004) 91:987–98. doi:10.1002/jcb.20003
Keywords: angiogenesis, TP53, ovarian carcinoma, hypoxia, VEGF
Citation: Davidson BA, Rubatt JM, Corcoran DL, Teoh DK, Bernardini MQ, Grace LA, Soper WJ, Berchuck A, Siamakpour-Reihani S, Chen W, Owzar K, Murphy SK and Secord AA (2014) Differential angiogenic gene expression in TP53 wild-type and mutant ovarian cancer cell lines. Front. Oncol. 4:163. doi: 10.3389/fonc.2014.00163
Received: 10 February 2014; Accepted: 06 June 2014;
Published online: 20 June 2014.
Edited by:
Ivan Garcia-Bassets, University of California San Diego, USAReviewed by:
Paul M. Hwang, National Institutes of Health, USACopyright: © 2014 Davidson, Rubatt, Corcoran, Teoh, Bernardini, Grace, Soper, Berchuck, Siamakpour-Reihani, Chen, Owzar, Murphy and Secord. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angeles Alvarez Secord, DUMC 3079, Department of Obstetrics and Gynecology, Division of Gynecology Oncology, Duke University Medical Center, Durham, NC 27710, USA e-mail:c2Vjb3IwMDJAbWMuZHVrZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.