
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 November 2013
Sec. Cancer Molecular Targets and Therapeutics
Volume 3 - 2013 | https://doi.org/10.3389/fonc.2013.00290
This article is part of the Research TopicInhibiting PARP as a strategic target in cancerView all 10 articles
 Alice N. Weaver1
Alice N. Weaver1 Eddy S. Yang1,2,3*
Eddy S. Yang1,2,3*Poly(ADP-ribose) polymerases (PARPs) are DNA-dependent nuclear enzymes that transfer negatively charged ADP-ribose moieties from cellular nicotinamide-adenine-dinucleotide (NAD+) to a variety of protein substrates, altering protein–protein and protein-DNA interactions. The most studied of these enzymes is poly(ADP-ribose) polymerase-1 (PARP-1), which is an excellent therapeutic target in cancer due to its pivotal role in the DNA damage response. Clinical studies have shown susceptibility to PARP inhibitors in DNA repair defective cancers with only mild adverse side effects. Interestingly, additional studies are emerging which demonstrate a role for this therapy in DNA repair proficient tumors through a variety of mechanisms. In this review, we will discuss additional functions of PARP-1 – including regulation of inflammatory mediators, cellular energetics and death pathways, gene transcription, sex hormone- and ERK-mediated signaling, and mitosis – and the role these PARP-1-mediated processes play in oncogenesis, cancer progression, and the development of therapeutic resistance. As PARP-1 can act in both a pro- and anti-tumor manner depending on the context, it is important to consider the global effects of this protein in determining when, and how, to best use PARP inhibitors in anticancer therapy.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme which binds DNA via two zinc finger motifs and transfers chains of ADP-ribosyl moieties (PARs) from nicotinamide-adenine-dinucleotide (NAD+) to chromatin-associated acceptor proteins, including PARP-1 itself. This post-translational modification plays an important role in promoting DNA repair by releasing PARP-1 from DNA and allowing for recruitment of proteins involved in both base excisional repair (BER) and homologous recombination (HR) (1). Accordingly, PARP-1 is an attractive anticancer target, and poly(ADP-ribose) polymerase (PARP) inhibitors have been identified as chemo- and radiation-sensitizing agents in an array of cancers (2–5), including our report on the sensitization of head and neck cancer to radiotherapy following PARP inhibition (6). Perhaps the most well-known tumoricidal effects of PARP inhibitors are in BRCA-mutated cancers, which harbor DNA repair defects and become dependent on PARP-1-mediated repair for survival. Two landmark studies (7, 8) found inhibition of PARP-1 in cells containing BRCA mutations resulted in the generation of chromatid breaks, G2 cell cycle arrest, and enhancement of apoptosis, results which have been confirmed in early phase clinical trials (9, 10).
Interestingly, recent studies also show potential efficacy of PARP inhibition in sporadic tumors lacking DNA repair defects. A clinical study of the PARP inhibitor olaparib in women with heavily pretreated high-grade serous ovarian cancer without germline BRCA1/2 mutations resulted in objective responses in 11/46 (24%) (11), indicating there may be additional determinants of sensitivity to PARP inhibition. Pre-clinical studies have identified susceptibility to PARP inhibition alone in HR-proficient HER2-positive breast cancer, pancreatic cancer, prostate cancer, Ewing’s sarcoma, small cell lung carcinoma, and neuroblastoma, among others (12–17). These reports demonstrate the existence of non-DNA repair functions of PARP-1 that may be targetable for cancer treatment. It is thus becoming increasingly apparent that a number of PARP-1-mediated cellular processes influence characteristics of tumor development, progression, and treatment response, including several of the eight “hallmarks of cancer” proposed by Hanahan and Weinberg (18) (Figure 1). In this review, we will discuss cancer-related functions of PARP-1 – including regulation of inflammatory mediators through NF-κB, cell death and energetics, ERK-mediated tumor progression and invasion, mitosis, gene transcription, and sex hormone signaling – and examples of how these functions may be exploited to expand the patient population potentially benefiting from treatment with PARP inhibitors.
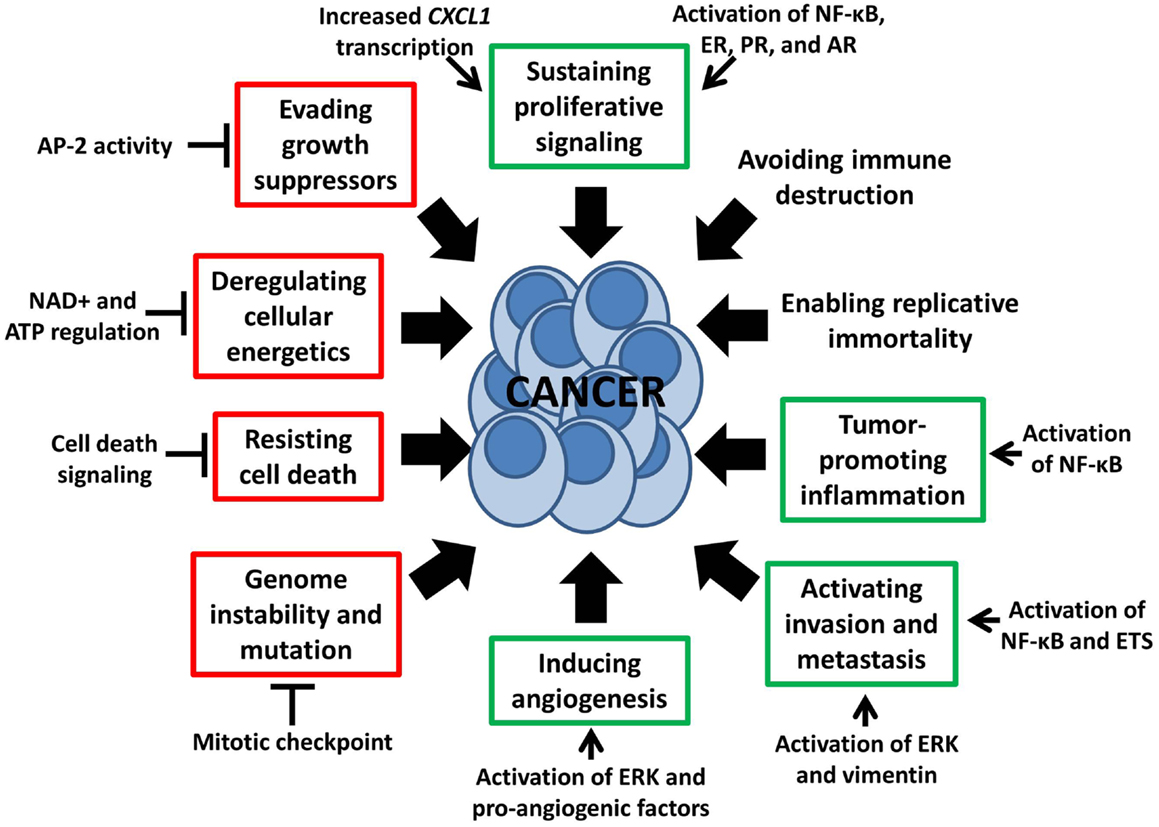
Figure 1. Non-DNA repair functions of PARP-1 influence the “hallmarks of cancer” (18). This schematic depicts multiple PARP-1-mediated processes which either stimulate or inhibit six of the eight “hallmarks of cancer,” as indicated by green and red boxes respectively. These hallmarks, proposed by Hanahan and Weinberg, are malignant characteristics that provide a framework for understanding the biology of cancer.
In multiple cancers, including breast, prostate, and head and neck among others, the NF-κB signaling pathway undergoes a loss of regulation resulting in constitutive activation (19). Briefly, NF-κB is a family of transcription factors including RelA/p65, RelB, c-Rel, p50, and p52, which exist as homo- and hetero-dimers. DNA-binding affinity and DNA sequence specificity is dependent on the composition of the dimer. Inhibitory proteins bind NF-κB dimers and sequester them in the cytosol in the absence of a stimulus; pathway activation causes proteasomal degradation of inhibitors, allowing the dimer to translocate to the nucleus and activate pro-inflammatory transcription programs. Although NF-κB signaling mediates the acute immune response responsible for targeting and eliminating cancerous cells, chronic inflammation mediated by this “hallmark” pathway can lead to the malignant phenotype (Figure 1), facilitating escape from immune surveillance, cancer survival, metastasis, and angiogenesis (20).
Activation of NF-κB can be regulated by PARP-1 via multiple mechanisms (Figure 2). First, PARP-1 directly interacts with histone acetyl-transferases p300 and CREB-binding protein (CBP) to synergistically co-activate NF-κB-dependent gene expression. In response to inflammatory stimuli, p300/CBP acetylates PARP-1 at specific lysine residues. This modification is necessary for PARP-1-p50 interaction, enhancement of p300–p50 interaction, and co-activation of NF-κB-mediated transcription programs (21, 22). Co-activation is negatively regulated by the activity of class I histone deacetylases (HDACs) (22) and SUMO1/3-mediated SUMOylation of the automodification domain of PARP-1 (23). Second, enzymatic activation of PARP-1 variably affects NF-κB, with outcomes dependent on the identity of the PAR acceptor protein. AutoPARylation of PARP-1 following detection of DNA strand breaks promotes the formation of a “signalosome” containing IKKγ (NEMO), the regulatory subunit of a NF-κB inhibitory complex, along with PIASγ, and ATM. Chains of PAR on activated PARP-1 provide the scaffold needed for SUMOylation of IKKγ by the PIASγ PAR binding motif, leading to activation of IKK and NF-κB (24). The effects of PARylation on NF-κB itself are less clear, with different sources reporting decreased, increased, or unaffected DNA-binding activity (25–27). Taken together, these studies demonstrate a strong role for PARP-1 in regulating NF-κB activity.
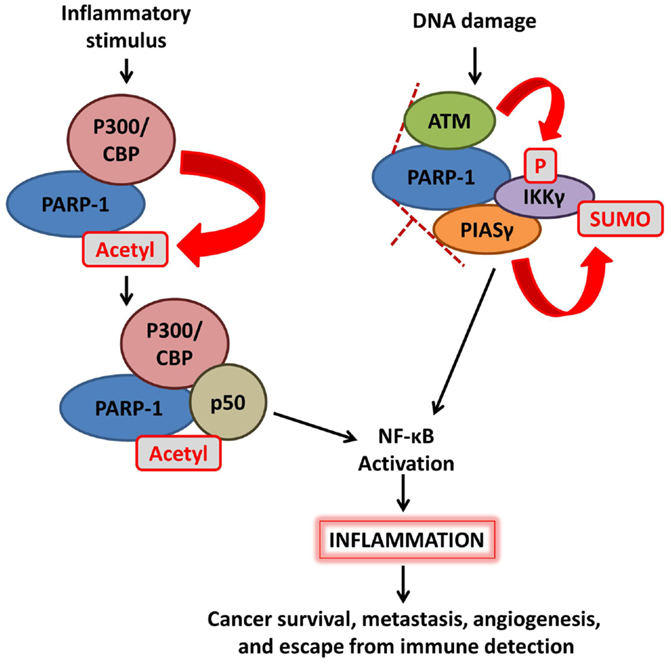
Figure 2. Poly(ADP-ribose) polymerase-1 mediates activation of NF-κB signaling. (Left) inflammatory stimulation triggers p300/CBP acetylation of PARP-1, enhancing the interaction between p50 and PARP-1 as well as the p50–p300 interaction; this ultimately leads to activation of NF-κB. (Right) DNA damage detection promotes the formation of a complex including PARP-1, ATM, PIASγ, and IKKγ (NEMO); chains of PAR on PARP-1 provide a structure upon which PIASγ SUMOylates IKKγ, leading to NF-κB activation.
The interaction between PARP-1 and the NF-κB pathway promotes production of pro-inflammatory cytokines such as TNFα, IL-6, INFγ, E-selectin, and ICAM-1, as well as expression of nitric oxide synthase (28–30); PARP inhibition has been shown to attenuate upregulation of these factors in response to inflammatory stimuli (28, 29). Furthermore, PARP inhibition may also prevent inflammation-associated adverse side effects of traditional chemotherapeutics (31), supporting the use of PARP inhibitors in multidrug regimens. Loss of PARP-1 activity not only decreases pro-tumor inflammation, but also inhibits two related hallmarks of cancer through anti-inflammatory mechanisms: proliferative signaling (32) and metastasis (33, 34) (Figure 1).
Recently, we discovered an unexpected sensitivity to PARP inhibition in DNA repair proficient HER2-positive breast cancer cells through attenuation of NF-κB-mediated signaling (13). HER2 over-expressing cancers have activated NF-κB, which acts to block apoptosis and possibly mediate resistance to HER2-targeted drugs (35). In HER2-positive breast cancer cells, treatment with PARP inhibitor significantly reduced the expression of NF-κB activator IKKα and phosphorylated p65 while increasing inhibitory IkBα. These events resulted in decreased NF-κB transcriptional activity in HER2-positive, but not HER2-negative, breast cancer cells (13). Furthermore, overexpression of HER2 alone was sufficient to confer sensitivity to PARP inhibitor, suggesting synthetic lethality with PARP inhibition in tumors that are oncogene-addicted to HER2 signaling through NF-κB. This study represents a specific application of PARP-1-regulated NF-κB signaling to cancer therapy, one that may soon be expanded into a clinical trial.
Cancer cells are characterized by excessive proliferation, impaired cell death signaling, and deregulated metabolism (Figure 1). These features are often mediated by altered mitochondrial activity coupled with inactivation of apoptotic signaling through decreased expression of pro-apoptotic factors like p53 or overexpression of anti-apoptotic factors like Bcl-x. Integrity of regulatory pathways for cell death and metabolism is important for response to many cancer treatment modalities, as well as in cancer imaging and diagnostics. Cellular energetics and death signaling are heavily regulated by PARP-1, allowing activity of this protein to serve as a switch between cell fates and to affect both tumor proliferation and therapeutic response.
In response to damage stimuli, activated PARP-1 acts early in the apoptosis initiation pathway to stabilize p53 and facilitate its function (36). If damage is excessive, high levels of PAR synthesis by PARP-1 deplete its NAD+ substrate; additional interactions between PARP-1 and NMNAT-1, a NAD+ synthase, and SIRT1, a NAD+-dependent protein deacetylase, further contribute to PARP-1 as a controller of NAD+ availability and, thus, NAD-dependent metabolic reactions. ATP-dependent NAD+ salvage saps cellular ATP stores, resulting in energy deprivation and, eventually, energy crisis-induced necrosis (Figure 3). Furthermore, PARP-1-mediated PARylation may inactivate caspase-8 and reduce caspase-mediated apoptotic signaling (37). Hyperactivation of PARP-1 and accumulation of PAR can also cause translocation of PAR to the cytosol, where it interacts with the outer mitochondrial surface. Here it binds apoptosis inducing factor (AIF) and induces its release and translocation to the nucleus, ultimately resulting in large-scale DNA fragmentation and a novel PARP-1-dependent cell death mechanism known as “parthanatos” (38). To prevent these events, activated caspases cleave PARP-1 into two fragments: an 89-kDa C-terminal fragment with low levels of catalytic activity and a 24-kDa N-terminal peptide which inhibits the catalytic activity of uncleaved nuclear PARP-1. Conservation of NAD+ and, thus, ATP allows the cell to undergo programed cell death (39–41). Accordingly, inhibition of PARP-1 preserves ATP levels, improves antioxidant status, and normalizes anti-apoptotic Bcl-x levels in the kidney following chemotherapy-induced injury (42, 43).
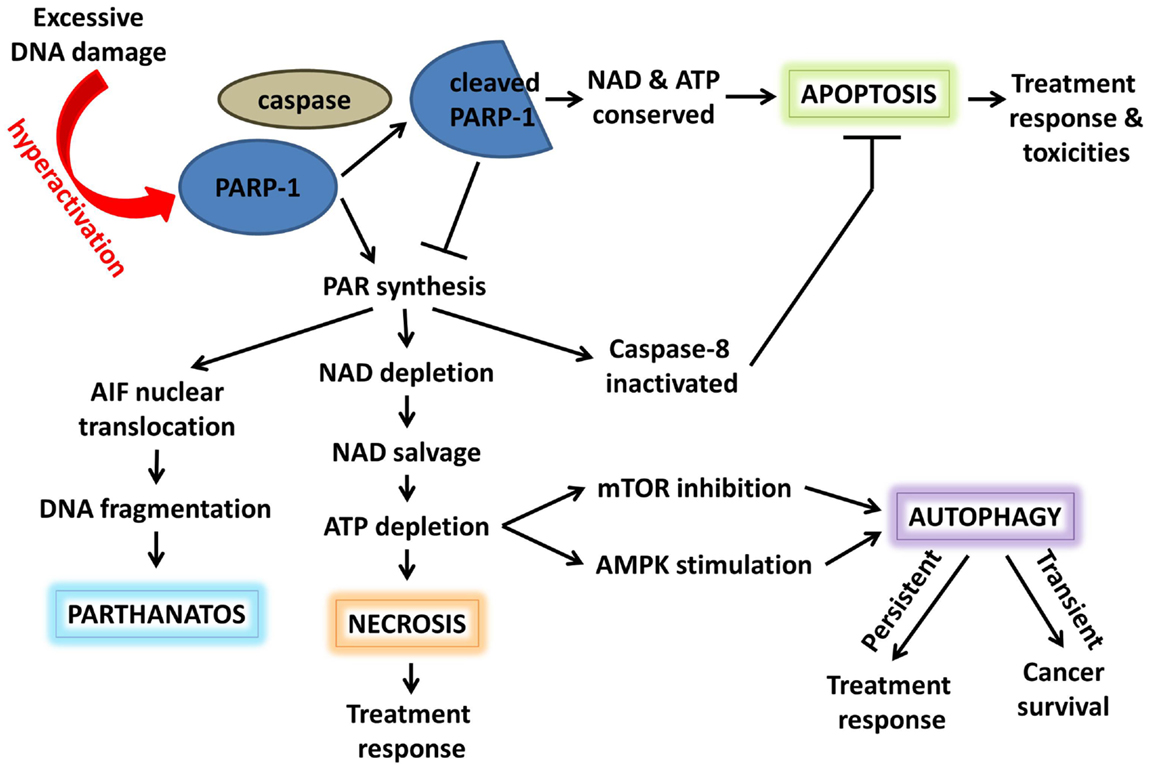
Figure 3. Poly(ADP-ribose) polymerase-1 acts as a switch between cell fates. Hyperactivation of PARP-1 and PAR synthesis depletes NAD and, subsequently, ATP. Elevated PAR can promote necrosis, autophagy, or AIF-induced parthanatos. In addition, PARylation inactivates caspase-8, inhibiting apoptotic signaling. Alternatively, activated caspases can cleave PARP-1; the resulting cleavage product inhibits uncleaved PARP-1, conserving NAD/ATP, and promoting apoptosis. These cell death pathways play a role in both cancer survival and response to anticancer therapy.
Poly(ADP-ribose) polymerase-1 also regulates the classical necroptotic pathway mediated by the death promoting MAP kinase, c-Jun N-terminal kinase (JNK). This signaling network is activated in many cancers and has been implicated as a driver of both tumor development and treatment response (44, 45). PARP-1 downregulates MAP kinase phosphatase MKP-1 expression and inhibits the survival kinase Akt, both of which activate JNK (46, 47), suggesting potential benefit for PARP inhibition in tumors with elevated JNK activity. JNK1 mediates phosphorylation and sustained activation of PARP-1, creating a feed-forward regulatory loop (48). In conjunction, PARP-1-induced depletion of ATP stimulates AMP-activated protein kinase (AMPK) while inhibiting mTOR to promote autophagy, yet another cell death pathway important in cancer survival and treatment response (49). Pharmacologic inhibition of PARP-1 promotes Akt activity and mTOR signaling resulting in decreased cell death (50), although these results are contradicted by a recent report showing PHLPP1-mediated downregulation of Akt activity and increased cell death following PARP inhibition (51).
Clinically, targeting the role of PARP-1 in cell death pathways appears to be complex. PARP-1 inhibition may reduce PAR-mediated inactivation of caspase-8, sensitizing cancer cells to tumor necrosis factor-related apoptosis-induced ligand (TRAIL) therapy (37). Additionally, inhibition of PARP-1 prevented cisplatin- and methotrexate-induced ATP depletion and nephrotoxicity (42, 43), as well as imatinib (Gleevec)-induced JNK activation and cardiotoxicity (52), without significantly affecting the anticancer activity of these agents. However, activation of the Akt survival pathway may counteract the cytotoxic effects of PARP inhibition and cause resistance to therapy (47), suggesting Akt pathway inhibition may enhance PARP inhibition in anti-tumor therapy. Despite these complexities, the influence of PARP-1 on metabolic co-factors and cell death signaling is significant, and further studies examining the role of PARP inhibition in manipulating these processes is warranted.
In addition to the JNK-mediated signaling described previously, a second family of MAP kinases known as extracellular signal-regulated kinases or ERKs is involved not only in cell death determination but also in tumor progression, angiogenesis, and metastasis. ERK activation is pivotal in cancer cell survival through upregulation of anti-apoptotic proteins and inhibition of caspase activity (53). Inhibition of this pathway by targeting ERK or MEK, which is immediately upstream of ERK in signaling, has been associated with suppression of ovarian tumor growth (54), reduced metastatic potential of melanoma cells (55), and increased sensitivity to cytotoxic agents (56). Recent studies indicate an important role for PARP-1 in promoting ERK signaling.
Poly(ADP-ribose) polymerase-1 is activated and autoPARylated by a direct interaction with phosphorylated ERK2 (pERK2), resulting in enhanced pERK2-catalyzed phosphorylation of target transcription factors and increased gene expression (57). Furthermore, PARP inhibition causes loss of ERK2 stimulation by decreasing the activity of critical pro-angiogenic factors including vascular endothelial growth factor (VEGF), transmembrane signaling protein syndecan-4 (SDC-4), platelet/endothelial cell adhesion molecule (PECAM1/CD31), and hypoxia inducible factor (HIF). This ultimately results in reduced angiogenesis and inflammation (58–62). The effects of PARP-1 on ERK signaling are further enhanced by PARP-1-mediated transcription of vimentin, an intermediary angiogenic filament upregulated in tumor vasculature and pivotal for the endothelial-to-mesenchymal transition characteristic of metastasis (63). Pharmacologic inhibition of PARP reverted this transition, correlating with a reduction in the number and size of metastatic melanoma foci in a mouse model (63).
Collectively, these studies indicate PARP-1 directly fosters ERK signaling in addition to mediating separate but parallel signaling pathways reinforcing the same end result of increased angiogenesis and metastasis, two tumor-promoting features (Figure 1). As such, PARP inhibition may be effective in blocking the ERK signaling network or increasing activity of ERK/MEK inhibitors, agents already shown to be efficacious in acute myeloid leukemia, multiple myeloma, melanoma, colorectal, breast, lung, and pancreatic cancers (64–68). Furthermore, selective ERK inhibition induces tumor regression in MEK inhibitor-resistant models (67), raising the question of whether PARP inhibition could be similarly effective in either MEK or ERK-resistant tumors due to its proximity in the signaling pathway. As MEK, ERK, and PARP inhibitors have only recently entered early phase clinical trials, it will be some time before we know which patients benefit most from these drugs, either alone or in combination, but their interaction warrants further investigation.
The high proliferation rate of cancer cells is a result not only of decreased cell death but also of improperly regulated cell cycling, allowing evasion of growth suppressing signals. Although multiple cell cycle checkpoints can be impaired in cancer, the mitotic or spindle assembly checkpoint is of great importance both in tumorigenesis and as an anticancer target. This point of regulation, which is responsible for ensuring appropriate chromosome segregation, is required for cell viability. Cells with a weakened mitotic checkpoint are capable of survival but do not maintain proper chromosome segregation, resulting in genomic instability and aneuploidy. These are common features of tumor cells and may even act as drivers in cancer development (Figure 1). PARP-1 can act on many mediators of cell cycle progression through its effects on gene expression (68), which will be detailed in a later section. However, direct regulation of the mitotic checkpoint by PARP-1 is another important factor that may be targetable in cancer treatment.
Recent reports suggest multiple roles for PARP-1 in the structural machinery of mitosis. First, PAR, which is primarily synthesized by PARP-1, is required for assembly and function of the bipolar spindle (69). In addition, PARP-1 both localizes to and PARylates proteins at centromeres and centrosomes during mitosis (70, 71). PARP-1 also mediates PARylation of p53, which is responsible for regulating centrosome duplication and monitoring chromosomal stability (71). Loss of PARP-1activity is associated with mislocalization of centromeric and centrosomal proteins, resulting in incomplete synapsis of homologous chromosomes, defective chromatin modifications, and failure to maintain metaphase arrest, indicating loss of mitotic checkpoint integrity (71, 72). Similarly, inhibition of PARP-1 is associated with genomic instability characterized by reduced stringency of mitotic checkpoints, centrosome hyperamplification, and chromosomal aneuploidy, the most common characteristic of solid tumors (71, 73, 74).
Furthermore, PARP-1 has been shown to interact with the E3 ubiquitin ligase, CHFR, a tumor suppressor with an important role in the early mitotic checkpoint. Binding of these two proteins results in degradation of PARP-1 and cell cycle arrest in prophase, an effect stimulated by the microtubule inhibitor docetaxel resulting in resistance to this drug in CHFR-over-expressing cancer cells. Concomitant use of a PARP inhibitor with docetaxel significantly increased apoptosis in these cells, suggesting a role for PARP inhibition in sensitizing cancers with high CHFR activity to microtubule inhibitors (75).
The clinical characteristics of cancer, including growth, metastatic potential, and response to treatment, are greatly influenced by dysregulation of gene transcription. Gene expression profiles are currently being utilized as tumor biomarkers, indicators of treatment sensitivity or resistance, and prognostic predictors. In the future, there may even be a role for therapeutic agents that reactivate a silenced tumor suppressor or silence an activated oncogene. In total, 3.5% of the transcriptome is regulated by PARP-1 with 60–70% positively regulated (76), including genes involved in tumor promotion such as JUND, MDM2, HGF, FLT1 (VEGFR1), EGFR, HIF2A (EPAS1), SPP1 (OPN), MMP28, ANGPT2, and PDGF (77). As discussed below and shown in Figure 4, this regulation can occur broadly through interactions with nucleosomes and modification of chromatin, can be gene specific through interactions with promoters and binding factors, or can result as a combination of the two, as binding of PARP-1 to nucleosomes mediates its localization to specific target gene promoters (78, 79).
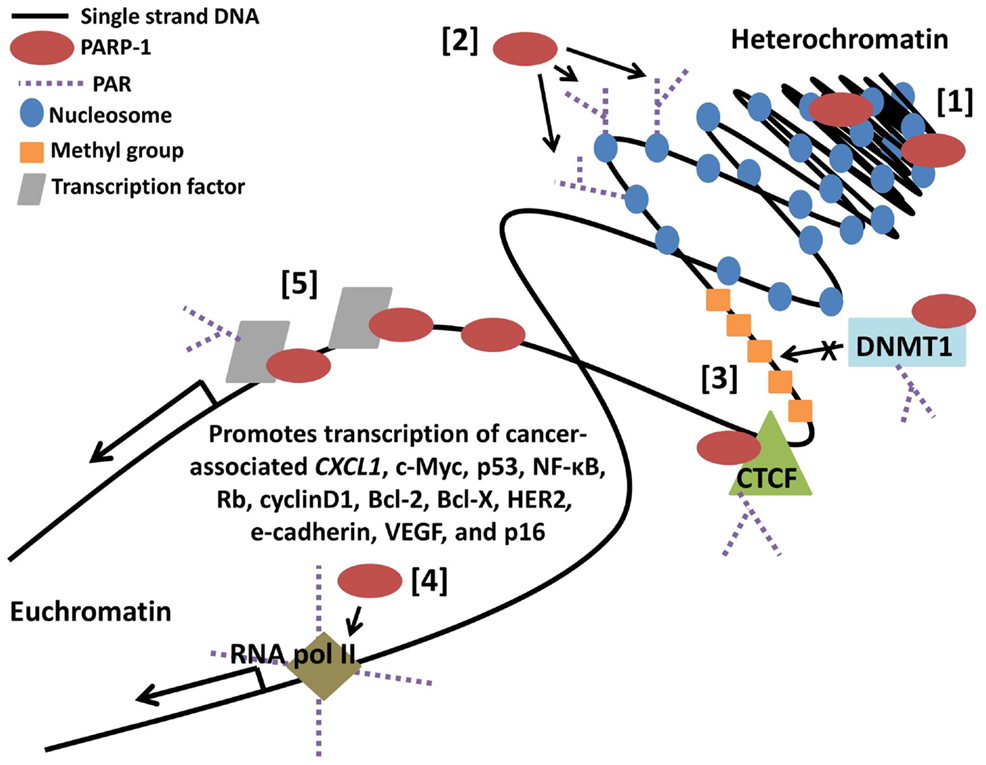
Figure 4. Poly(ADP-ribose) polymerase-1-regulates gene transcription through multiple mechanisms. [1] PARP-1 binds neighboring nucleosomes resulting in chromatin compaction. [2] PARP-1 PARylation of core histones mediates chromatin relaxation. [3] PARP-1 promotes hypomethylation of DNA by enhancing the chromatin insulator activity of CCCTC-binding factor (CTCF) while inhibiting methyltransferase activity of DNMT1. [4] PARP-1 promotes loading and retention of RNA polymerase II at active promoters. [5] PARP-1 binds regulatory DNA sequences and transcription factors, PARylates transcription factors, and recruits additional regulatory binding proteins in a target gene specific manner.
One mechanism by which PARP-1 alters gene expression is through regulation of chromatin structure and, thus, DNA accessibility. Simultaneous binding of multiple neighboring nucleosomes by PARP-1 compacts chromatin into a supranucleosomal structure, repressing gene transcription (79). This structural change is further stimulated by histone deacetylation mediated by a complex consisting of PARP-1, ATP-dependent helicase Brg1 (SmarcA4), and HDACs (80). Conversely, PARylation of core histones promotes charge repulsion-induced relaxation of chromatin and recruitment of transcription machinery (81–83). PARP-1-mediated PARylation also results in disassociation of linker histone H1, a repressor of RNA polymerase II-mediated transcription; accordingly, higher proportions of PARP-1:H1 indicate active promoters (84), suggesting potential utility of PARP-1 as a biomarker for actively transcribed genes. Although these outcomes can be separated by PARP-1 activity (protein binding versus enzymatic function), pharmacologic inhibition of PARP affect both actions, indicating manipulation of chromatin accessibility through PARP-1 is not currently an option for cancer therapy.
Along with chromatin structure, methylation patterns also play a large role in determining DNA accessibility. Alterations in DNA methylation are commonly found in many cancers and serve as a functional equivalent to a gene mutation in the process of tumorigenesis. Inhibition of PARP-1 is associated with transcriptional silencing through accumulation of DNA methylation and CpG island hypermethylation throughout the genome (85). This effect may be mediated by dimerization of PARP-1 with CCCTC-binding factor (CTCF), a chromatin insulator which binds to hypomethylated DNA regions. As the CTCF-PARP-1 interaction is PAR-dependent, decreased PAR following PARP inhibition abrogates this function (86, 87). Loss of CTCF-PARP-1 complex activity results in transcriptional silencing of multiple loci including tumor suppressors CDKN2A-INK4 (p16), CDH1 (e-cadherin), and P19ARF (88, 89).
Poly(ADP-ribose) polymerase-1 can also hinder DNA methylation by dimerization with DNA (cytosine-5-)-methyltransferase 1 (DNMT1), a methyltransferase found overexpressed in gastrointestinal tract carcinomas, resulting in inhibition of its methyltransferase activity (85, 90). In contrast, PARP-1 binding and PARylation of the Dnmt1 promoter actually enhances its transcription by preventing methylation-induced silencing (91). The reduced catalytic efficiency of PARylated DNMT1 may come as a result of negatively charged PARylated PARP-1 out-competing DNA for binding with DNMT1 (92). Interestingly, PARP-1-DNMT1 can form a ternary complex with CTCF at unmethylated CTCF-target sites in a PAR-dependent manner. Loss of PAR from this complex causes dissociation of PARP-1 and CTCF, allowing the still-bound DNMT1 to methylate the site and inhibit transcription (92).
Although some specific tumor suppressors are mentioned above as being affected by PARP-1-mediated chromatin insulation, the activity of PARP-1 in regulating DNA methylation patterns at specific genes or genic regions is largely unknown. As such, it is difficult to predict the effect of PARP inhibition on cancer growth and progression through this mechanism. However, with the advent of genomic profiling, it has recently become possible to identify methylation changes specific to certain cancer subtypes. Anticancer agents with epigenetic modifying activity, such as DNA methyltransferase inhibitors, are being investigated in these cancers and show promising results, especially in hematologic malignancies (93). The effect of PARP inhibition on epimutations has not been studied, but the reports described above suggest PARP inhibitors could have similar applicability.
Poly(ADP-ribose) polymerase-1 can also promote transcription in a more sequence-specific manner by positively regulating RNA polymerase II activity at active promoters. This occurs through: (1) PARylation-induced exclusion of histone demethylase KDM5B, maintaining levels of activating histone mark K3K4me3 (82), (2) PARylation-induced dissociation of the DEK repressor, promoting loading of the RNA polymerase II mediator complex (94), and (3) creation of a PAR scaffold for retention of RNA polymerase II (95). Surprisingly, a recent report showed that inhibition of PARP-1 enzymatic activity was associated with increased H3K4me3, resulting in upregulation of sodium iodide symporter transcription and elevated radio-iodine uptake in thyroid cancer cell lines (96). This contradictory work may result from target gene specific functions of PARP-1, as the previously cited studies were focused on genes known to be positively regulated by PARP-1. However, it does illustrate the need for greater understanding of PARP-1 involvement at active gene promoters, as well as the potential for manipulating PARP-1-mediated transcription to enhance efficacy of cancer therapy.
Gene expression can be further regulated by direct interactions between PARP-1 and DNA elements or binding factors. PARP-1 acts as a promoter-specific switch at target genes, facilitating the release of inhibitory co-regulators and recruitment of stimulatory co-regulators (97, 98). PARP-1 binding of the NF-κB immediate upstream region (IUR) element activates transcription of CXCL1, which encodes melanoma growth stimulatory activity protein and is overexpressed in the progression of malignant melanoma (99). Binding of PARP-1 to the transcription factor E2F-1 increases E2F-1 promoter activity and expression of the E2F-1-responsive oncogene Myc (c-Myc) (100). PARP-1 expression and activity are also required for cancer cell invasion (Figure 1) mediated by ETS transcription factors – whose fusion products drive Ewing’s sarcoma, acute myeloid leukemia, and prostate cancer – and the Ewing’s sarcoma fusion protein EWS-FLI (14, 15). While PARP-1 interaction with these factors promotes pro-tumor signaling, other interactions have the opposite effect. PARP-1 suppresses self-inhibition of AP-2, a transcription factor that negatively regulates cell cycle and proliferation (101). Increased AP-2 expression suppresses cancer cell growth (102) and may inhibit ras oncogene-mediated transformation (101), effects likely diminished by PARP inhibition (Figure 1). PARP-1 has also been shown to bind the inhibitory element of COX-2, which mediates inflammation and promotes VEGF-mediated pro-angiogenesis pathways activated in cancer cells (103, 104).
Instances of PARP-1-mediated enzymatic activity affecting specific transcription factors or genes often translate to a clear role for PARP-1 inhibitors as anticancer agents, even in monotherapy. For example, ETS-positive prostate tumors and EWS-FLI-positive Ewing’s sarcomas are highly sensitive to PARP inhibitors (14, 15). However, PARP-1 has multiple and diverse functions involving both PARylation activity and DNA-binding capability. Enzymatic inhibition, which decreases PARP-1 self PARylation, actually increases DNA binding and may be detrimental in some cancers, such as the malignant melanoma example given above. A greater understanding of the relative effects of PARP-1 on transcriptional activity is needed in order to select tumors with a molecular profile conducive to pharmacologic inhibition through this mechanism.
Sex hormones have been implicated in development, progression, and treatment sensitivity of prostate, breast, gynecologic, and colon cancers. Sex steroid effects are mediated through their receptors, which act as transcription factors in steroid-responsive tissues. Any of the multiple levels of regulation controlling these signaling pathways can become impaired, leading to abnormal proliferative responses characteristic of cancer progression (Figure 1). Similar to PARP-1-mediated regulation of transcription factor activity, PARP-1 plays a role in regulating three of the sex hormone receptors most commonly linked to cancer: estrogen receptor (ER), progesterone receptor (PR), and androgen receptor (AR).
Approximately 80% of breast carcinomas are positive for ER, identifying ER-targeted therapies as excellent, although not un-failable, treatment options in these cancers (105). PARP-1 interacts with the ERa isoform both directly and through estradiol-induced PARylation to enhance binding of ERa and other activating factors to target gene promoters (106, 107), suggesting PARP inhibition may enhance the activity of ER-targeted agents. A similar interaction occurs between PARP-1 and PR: PARP-1 binding of PR, as well as hormone-activated CDK2-induced PR PARylation, acts to stimulate cancer cell proliferation (108). PARP-1 regulation of PR activity is of great interest in endometrioid carcinomas specifically, as expression of PARP-1 and PR is positively correlated at each pathologic stage of this cancer (109). However, the effects of PARP inhibition in endometrial cancer have yet to be determined.
Recently, a report detailing the strong interaction between PARP-1 and AR has generated much excitement over the potential for PARP inhibitors in prostate cancer treatment. Human prostatic adenocarcinoma, a cancer highly resistant to standard therapies, is reliant on AR activity for growth and survival. Accordingly, AR-targeted therapies are the primary treatment for these patients. Unfortunately, there are multiple mechanisms for AR reactivation leading to tumor recurrence, a lethal phenotype known as castration-resistant prostate cancer. PARP-1 enzymatic activity, which is significantly upregulated in castration-resistant prostate cancer, promotes both AR chromatin binding and transcription factor functions. Although PARP-1 does localize with AR to regulatory sites of AR-target genes, the two proteins appear to be members of separate complexes at these loci. Inhibition of PARP-1 in vivo: (1) depletes both PARP-1 and AR at target genes, (2) significantly reduces expression of target genes, including pro-tumorigenic ets genes referenced previously, (3) sensitizes both castration-resistant and castration-sensitive prostate cancer cells to genotoxic insult and androgen depletion, (4) enhances the anti-tumor effects of anti-androgen therapy, and (5) delays onset of resistance to anti-androgen therapy. Ex vivo studies of castration resistance prostate tumors displayed a significant anti-tumor response to both veliparib and olaparib, two well-known PARP inhibitors, that correlates with reduced AR activity (110). These results suggest PARP inhibitors have the potential to significantly enhance existing prostate cancer therapy and improve outcomes for patients with castration-resistant tumors.
Poly(ADP-ribose) polymerase inhibitors are exciting new drugs that are easily delivered, can be highly efficacious, and are associated with few side effects. Mild nausea is commonly reported, with rare instances of more serious symptoms such as temporary cognitive deficits and myelosuppression. While ongoing clinical trials are focused on exploiting the role of PARP-1 in DNA repair, we have identified in this review multiple targetable functions of PARP-1 that are not dependent on HR defects (Figures 1–4; Table 1). One of the challenges in broadening the use of PARP inhibitors in anticancer therapy is more efficient identification of patients who may respond to these drugs. Some ongoing clinical trials include analysis of protein expression – including HR proteins, NF-κB, and PARP-1 itself – in relation to clinical response in search for potential biomarkers of sensitivity. However, the list of candidates is extensive and will continue to grow as additional functions of PARP-1 are discovered. Banking tumor biopsies from patients enrolled in PARP-1 clinical trials will greatly expedite the development of a panel of biomarkers, as will increased use of cancer genome sequencing and microarray technologies. Another challenge will be in identifying and overcoming mechanisms of resistance to PARP inhibition. For example, a second BRCA mutation or a deletion of the original mutation can cause reversion to HR-proficiency and resistance to PARP inhibitors in BRCA-mutated cancers (111). As the majority of clinical applications proposed here are theoretical or in pre-clinical development, associated mechanisms of resistance are entirely unknown, although development of such resistance is practically assured. Thirdly, many of the functions discussed here are effected by PARP-1 binding rather than enzymatic activity. Currently available PARP inhibitors act at the catalytic site of PARP-1, which does result in some degree of altered binding capacity via changes in autoPARylation status. However, treatment with PARP inhibitors may not effectively inhibit specific PARP-1 interactions, or may require different dosing. It will be important to study the various clinically available agents to determine if, and to what extent, binding domains are affected. Despite these obstacles, PARP inhibition is an extremely promising anticancer strategy and, as the first agents near completion of phase III trials, it will be exciting to see the magnitude of impact PARP inhibitors will have in clinical practice.
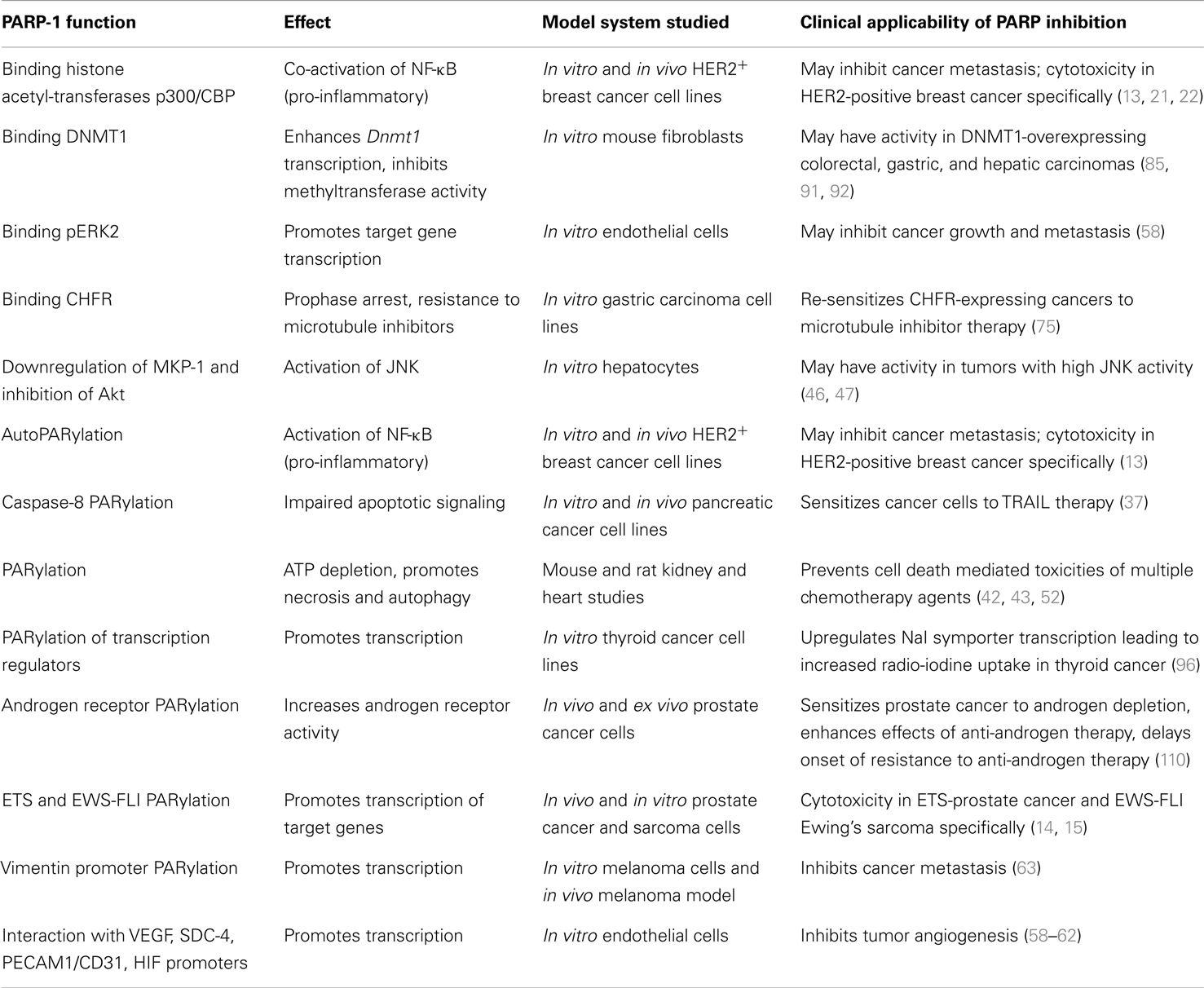
Table 1. Summary of reported non-DNA repair functions of PARP-1 with potential clinical correlations.
Alice N. Weaver and Eddy S. Yang conceptualized the topic. Alice N. Weaver conducted the literature review and wrote the article. Eddy S. Yang critically revised the article and provided guidance and supervision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose work was not cited due to space constraints. We would like to thank Amanda Swindall, Jennifer Stanley, Monica Wielgos, and Tiffiny Cooper for assistance in preparing the manuscript. This work is supported by grants from the Department of Defense (Eddy S. Yang), the American Association of Cancer Research (Eddy S. Yang), and the 5T32GM008361-21 grant from the NIH-National Institute of General Medical Sciences (Alice N. Weaver).
1. Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med (2006) 7(3):165–72. doi: 10.1016/j.carrev.2006.02.002
2. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst (2004) 96(1):56–67. doi:10.1093/jnci/djh005
3. Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res (2008) 14(23):7917–23. doi:10.1158/1078-0432.CCR-08-1223
4. Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer (2011) 104(5):750–5. doi:10.1038/bjc.2011.8
5. Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res (2012) 18(6):1726–34. doi:10.1158/1078-0432.CCR-11-2821
6. Nowsheen S, Bonner JA, Yang ES. The poly(ADP-ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol (2011) 99(3):331–8. doi:10.1016/j.radonc.2011.05.084
7. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434(7035):913–7. doi:10.1038/nature03443
8. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi:10.1038/nature03445
9. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi:10.1056/NEJMoa0900212
10. Tutt A, Robson M, Garber JE, Domchek S, Audeh MW, Weitzel JN, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J Clin Oncol (2009) 27(185):CRA501. doi:10.1016/S0140-6736(10)60892-6
11. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol (2011) 12(9):852–61. doi:10.1016/S1470-2045(11)70214-5
12. Melisi D, Ossovskaya V, Zhu C, Rosa R, Ling J, Dougherty PM, et al. Oral poly(ADP-ribose) polymerase-1 inhibitor BSI-401 has antitumor activity and synergizes with oxaliplatin against pancreatic cancer, preventing acute neurotoxicity. Clin Cancer Res (2009) 15(20):6367–77. doi:10.1158/1078-0432.ccr-09-0910.
13. Nowsheen S, Cooper T, Bonner JA, LoBuglio AF, Yang ES. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res (2012) 72(18):4796–806. doi:10.1158/0008-5472.CAN-12-1287
14. Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell (2011) 19(5):664–78. doi:10.1016/j.ccr.2011.04.010
15. Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res (2012) 72(7):1608–13. doi:10.1158/0008-5472.CAN-11-3648
16. Byers LA, Nilsson MB, Masrorpour F, Wang J, Diao L, Bhardwaj V, et al. Investigation of PARP1 as a therapeutic target in small cell lung cancer. J Clin Oncol (2012) 30(Suppl):7096. doi:10.1371/journal.pone.0056284
17. Addioui A, Belounis A, Cournoyer S, Nyalendo C, Brito R-M, Beaunoyer M, et al. Preclinical study of a PARP inhibitor in neuroblastoma. J Clin Oncol (2012) 30(Suppl):9570.
18. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
19. Gilmore T, Gapuzan ME, Kalaitzidis D, Starczynowski D. Rel/NF-kappa B/I kappa B signal transduction in the generation and treatment of human cancer. Cancer Lett (2002) 181(1):1–9. doi:10.1016/S0304-3835(01)00795-9
20. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi:10.1186/1476-4598-12-86
21. Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem (2003) 278(46):45145–53. doi:10.1074/jbc.M307957200
22. Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem (2005) 280(49):40450–64. doi:10.1074/jbc.M507553200
23. Messner S, Schuermann D, Altmeyer M, Kassner I, Schmidt D, Schar P, et al. Sumoylation of poly(ADP-ribose) polymerase 1 inhibits its acetylation and restrains transcriptional coactivator function. FASEB J (2009) 23(11):3978–89. doi:10.1096/fj.09-137695
24. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell (2009) 36(3):365–78. doi:10.1016/j.molcel.2009.09.032
25. Chang WJ, Alvarez-Gonzalez R. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem (2001) 276(50):47664–70. doi:10.1074/jbc.M104666200
26. Zerfaoui M, Errami Y, Naura AS, Suzuki Y, Kim H, Ju J, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol (2010) 185(3):1894–902. doi:10.4049/jimmunol.1000646
27. Veuger SJ, Hunter JE, Durkacz BW. Ionizing radiation-induced NF-kappaB activation requires PARP-1 function to confer radioresistance. Oncogene (2009) 28(6):832–42. doi:10.1038/onc.2008.439
28. Haddad M, Rhinn H, Bloquel C, Coqueran B, Szabo C, Plotkine M, et al. Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br J Pharmacol (2006) 149(1):23–30. doi:10.1038/sj.bjp.0706837
29. Jijon HB, Churchill T, Malfair D, Wessler A, Jewell LD, Parsons HG, et al. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am J Physiol Gastrointest Liver Physiol (2000) 279(3):G641–51.
30. Naura AS, Datta R, Hans CP, Zerfaoui M, Rezk BM, Errami Y, et al. Reciprocal regulation of iNOS and PARP-1 during allergen-induced eosinophilia. Eur Respir J (2009) 33(2):252–62. doi:10.1183/09031936.00089008
31. Korkmaz A, Kurt B, Yildirim I, Basal S, Topal T, Sadir S, et al. Effects of poly(ADP-ribose) polymerase inhibition in bladder damage caused by cyclophosphamide in rats. Exp Biol Med (Maywood) (2008) 233(3):338–43. doi:10.3181/0706-RM-151
32. Martin-Oliva D, O’Valle F, Munoz-Gamez JA, Valenzuela MT, Nunez MI, Aguilar M, et al. Crosstalk between PARP-1 and NF-kappaB modulates the promotion of skin neoplasia. Oncogene (2004) 23(31):5275–83. doi:10.1038/sj.onc.1207696
33. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene (2001) 20(31):4188–97. doi:10.1038/sj.onc.1204535
34. Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev (2011) 25(12):1245–61. doi:10.1101/gad.625811
35. Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A (2007) 104(40):15852–7. doi:10.1073/pnas.0706728104
36. Wesierska-Gadek J, Ranftler C, Schmid G. Physiological ageing: role of p53 and PARP-1 tumor suppressors in the regulation of terminal senescence. J Physiol Pharmacol (2005) 56(Suppl 2):77–88.
37. Yuan K, Sun Y, Zhou T, McDonald JM, Chen Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin Cancer Res (2013) 19(17):4750–9. doi:10.1158/1078-0432.CCR-13-0516
38. Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal (2011) 4(167):ra20. doi:10.1126/scisignal.2000902
39. Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis (2002) 7(4):321–8. doi:10.1023/A:1016119328968
40. Ivana Scovassi A, Diederich M. Modulation of poly(ADP-ribosylation) in apoptotic cells. Biochem Pharmacol (2004) 68(6):1041–7. doi:10.1016/j.bcp.2004.04.023
41. Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem (1998) 273(22):13703–12. doi:10.1074/jbc.273.22.13703
42. Racz I, Tory K, Gallyas F Jr, Berente Z, Osz E, Jaszlits L, et al. BGP-15 – a novel poly(ADP-ribose) polymerase inhibitor – protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem Pharmacol (2002) 63(6):1099–111. doi:10.1016/S0006-2952(01)00935-2
43. Dalaklioglu S, Sahin P, Ordueri EG, Celik-Ozenci C, Tasatargil A. Potential role of poly(ADP-ribose) polymerase (PARP) activation in methotrexate-induced nephrotoxicity and tubular apoptosis. Int J Toxicol (2012) 31(5):430–40. doi:10.1177/1091581812457430
44. Chen N, Nomura M, She QB, Ma WY, Bode AM, Wang L, et al. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res (2001) 61(10):3908–12.
45. Yang Y, Ikezoe T, Saito T, Kobayashi M, Koeffler HP, Taguchi H. Proteasome inhibitor PS-341 induces growth arrest and apoptosis of non-small cell lung cancer cells via the JNK/c-Jun/AP-1 signaling. Cancer Sci (2004) 95(2):176–80. doi:10.1111/j.1349-7006.2004.tb03200.x
46. Racz B, Hanto K, Tapodi A, Solti I, Kalman N, Jakus P, et al. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: a new mechanism for the cytoplasmic effect of PARP-1 activation. Free Radic Biol Med (2010) 49(12):1978–88. doi:10.1016/j.freeradbiomed.2010.09.026
47. Szanto A, Hellebrand EE, Bognar Z, Tucsek Z, Szabo A, Gallyas F Jr, et al. PARP-1 inhibition-induced activation of PI-3-kinase-Akt pathway promotes resistance to taxol. Biochem Pharmacol (2009) 77(8):1348–57. doi:10.1016/j.bcp.2009.01.008
48. Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ (2007) 14(5):1001–10. doi:10.1038/sj.cdd.4402088
49. Virag L, Robaszkiewicz A, Vargas JM, Javier Oliver F. Poly(ADP-ribose) signaling in cell death. Mol Aspects Med (2013) 34(6):1153–67. doi:10.1016/j.mam.2013.01.007
50. Ethier C, Tardif M, Arul L, Poirier GG. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS One (2012) 7(10):e47978. doi:10.1371/journal.pone.0047978
51. Wang S, Wang H, Davis BC, Liang J, Cui R, Chen SJ, et al. PARP1 inhibitors attenuate AKT phosphorylation via the upregulation of PHLPP1. Biochem Biophys Res Commun (2011) 412(2):379–84. doi:10.1016/j.bbrc.2011.07.107
52. Sarszegi Z, Bognar E, Gaszner B, Konyi A, Gallyas F Jr, Sumegi B, et al. BGP-15, a PARP-inhibitor, prevents imatinib-induced cardiotoxicity by activating Akt and suppressing JNK and p38 MAP kinases. Mol Cell Biochem (2012) 365(1–2):129–37. doi:10.1007/s11010-012-1252-8
53. Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem (2000) 79(3):355–69. doi:10.1002/1097-4644(20001201)79:3<355::AID-JCB20>3.0.CO;2-0
54. Kandala PK, Wright SE, Srivastava SK. Blocking epidermal growth factor receptor activation by 3,3’-diindolylmethane suppresses ovarian tumor growth in vitro and in vivo. J Pharmacol Exp Ther (2012) 341(1):24–32. doi:10.1124/jpet.111.188706
55. Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS, Lim JS. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol (2012) 83(3):385–94. doi:10.1016/j.bcp.2011.11.008
56. Kohno M, Tanimura S, Ozaki K. Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biol Pharm Bull (2011) 34(12):1781–4. doi:10.1248/bpb.34.1781
57. Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell (2007) 25(2):297–308. doi:10.1016/j.molcel.2006.12.012
58. Pyriochou A, Olah G, Deitch EA, Szabo C, Papapetropoulos A. Inhibition of angiogenesis by the poly(ADP-ribose) polymerase inhibitor PJ-34. Int J Mol Med (2008) 22(1):113–8.
59. Lacal PM, Tentori L, Muzi A, Ruffini F, Dorio AS, Xu W, et al. Pharmacological inhibition of poly(ADP-ribose) polymerase activity down-regulates the expression of syndecan-4 and Id-1 in endothelial cells. Int J Oncol (2009) 34(3):861–72. doi:10.3892/ijo_00000213
60. Tentori L, Muzi A, Dorio AS, Bultrini S, Mazzon E, Lacal PM, et al. Stable depletion of poly (ADP-ribose) polymerase-1 reduces in vivo melanoma growth and increases chemosensitivity. Eur J Cancer (2008) 44(9):1302–14. doi:10.1016/j.ejca.2008.03.019
61. Martin-Oliva D, Aguilar-Quesada R, O’Valle F, Munoz-Gamez JA, Martinez-Romero R, Garcia Del Moral R, et al. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res (2006) 66(11):5744–56. doi:10.1158/0008-5472.CAN-05-3050
62. Rajesh M, Mukhopadhyay P, Godlewski G, Batkai S, Hasko G, Liaudet L, et al. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem Biophys Res Commun (2006) 350(4):1056–62. doi:10.1016/j.bbrc.2006.09.160
63. Rodriguez MI, Peralta-Leal A, O’Valle F, Rodriguez-Vargas JM, Gonzalez-Flores A, Majuelos-Melguizo J, et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet (2013) 9(6):e1003531. doi:10.1371/journal.pgen.1003531
64. Kerr AHJ, James JA, Smith MA, Willson C, Court EL, Smith JG. An investigation of the MEK/ERK inhibitor U0126 in acute myeloid leukemia. Ann N Y Acad Sci (2003) 1010(1):86–9. doi:10.1196/annals.1299.013
65. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res (2007) 13(5):1576–83. doi:10.1158/1078-0432.CCR-06-1150
66. Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF, et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood (2007) 110(5):1656–63. doi:10.1182/blood-2007-03-081240
67. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov (2013) 3(7):742–50. doi:10.1158/2159-8290.CD-13-0070
68. Lee P, Wallace E, Yeh T, Poch G, Litwiler K, Pheneger T, et al. ARRY-142886, a potent and selective MEK inhibitor: III) efficacy in murine xenograft models correlates with decreased ERK phosphorylation. Proc Am Assoc Cancer Res (2004) 45:3890.
69. Chang P, Jacobson MK, Mitchison TJ. Poly(ADP-ribose) is required for spindle assembly and structure. Nature (2004) 432(7017):645–9. doi:10.1038/nature03061
70. Saxena A, Saffery R, Wong LH, Kalitsis P, Choo KH. Centromere proteins cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated. J Biol Chem (2002) 277(30):26921–6. doi:10.1074/jbc.M200620200
71. Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M. Involvement of poly(ADP-ribose) polymerase 1 and poly(ADP-ribosyl)ation in regulation of centrosome function. Mol Cell Biol (2003) 23(7):2451–62. doi:10.1128/MCB.23.7.2451-2462.2003
72. Yang F, Baumann C, De La Fuente R. Persistence of histone H2AX phosphorylation after meiotic chromosome synapsis and abnormal centromere cohesion in poly (ADP-ribose) polymerase (Parp-1) null oocytes. Dev Biol (2009) 331(2):326–38. doi:10.1016/j.ydbio.2009.05.550
73. Halappanavar SS, Shah GM. Defective control of mitotic and post-mitotic checkpoints in poly(ADP-ribose) polymerase-1(-/-) fibroblasts after mitotic spindle disruption. Cell Cycle (2004) 3(3):335–42. doi:10.4161/cc.3.3.670
74. Tong WM, Yang YG, Cao WH, Galendo D, Frappart L, Shen Y, et al. Poly(ADP-ribose) polymerase-1 plays a role in suppressing mammary tumourigenesis in mice. Oncogene (2007) 26(26):3857–67. doi:10.1038/sj.onc.1210156
75. Kashima L, Idogawa M, Mita H, Shitashige M, Yamada T, Ogi K, et al. CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J Biol Chem (2012) 287(16):12975–84. doi:10.1074/jbc.M111.321828
76. Ogino H, Nozaki T, Gunji A, Maeda M, Suzuki H, Ohta T, et al. Loss of Parp-1 affects gene expression profile in a genome-wide manner in ES cells and liver cells. BMC Genomics (2007) 8:41. doi:10.1186/1471-2164-8-41
77. Quiles-Perez R, Munoz-Gamez JA, Ruiz-Extremera A, O’Valle F, Sanjuan-Nunez L, Martin-Alvarez AB, et al. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology (2010) 51(1):255–66. doi:10.1002/hep.23249
78. Kotova E, Lodhi N, Jarnik M, Pinnola AD, Ji Y, Tulin AV. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc Natl Acad Sci U S A (2011) 108(15):6205–10. doi:10.1073/pnas.1019644108
79. Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell (2004) 119(6):803–14. doi:10.1016/j.cell.2004.11.002
80. Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature (2010) 466(7302):62–7. doi:10.1038/nature09130
81. Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A (1982) 79(11):3423–7. doi:10.1073/pnas.79.11.3423
82. Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell (2010) 39(5):736–49. doi:10.1016/j.molcel.2010.08.014
83. Kraus WL, Lis JT. PARP goes transcription. Cell (2003) 113(6):677–83. doi:10.1016/S0092-8674(03)00433-1
84. Frizzell KM, Gamble MJ, Berrocal JG, Zhang T, Krishnakumar R, Cen Y, et al. Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J Biol Chem (2009) 284(49):33926–38. doi:10.1074/jbc.M109.023879
85. Reale A, Matteis GD, Galleazzi G, Zampieri M, Caiafa P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene (2005) 24(1):13–9. doi:10.1038/sj.onc.1208005
86. Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet (2004) 36(10):1105–10. doi:10.1038/ng1426
87. Ong CT, Van Bortle K, Ramos E, Corces VG. Poly(ADP-ribosyl)ation regulates insulator function and intrachromosomal interactions in Drosophila. Cell (2013) 155(1):148–59. doi:10.1016/j.cell.2013.08.052
88. Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell (2009) 34(3):271–84. doi:10.1016/j.molcel.2009.04.001
89. Farrar D, Rai S, Chernukhin I, Jagodic M, Ito Y, Yammine S, et al. Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl)ation. Mol Cell Biol (2010) 30(5):1199–216. doi:10.1128/MCB.00827-09
90. Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer (2001) 91(2):205–12. doi:10.1002/1097-0215(200002)9999:9999<::AID-IJC1040>3.0.CO;2-2
91. Zampieri M, Passananti C, Calabrese R, Perilli M, Corbi N, De Cave F, et al. Parp1 localizes within the Dnmt1 promoter and protects its unmethylated state by its enzymatic activity. PLoS One (2009) 4(3):e4717. doi:10.1371/journal.pone.0004717
92. Zampieri M, Guastafierro T, Calabrese R, Ciccarone F, Bacalini MG, Reale A, et al. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem J (2012) 441(2):645–52. doi:10.1042/BJ20111417
93. Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst (2005) 97(20):1498–506. doi:10.1093/jnci/dji311
94. Gamble MJ, Fisher RP. SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol (2007) 14(6):548–55. doi:10.1038/nsmb1248
95. Zobeck KL, Buckley MS, Zipfel WR, Lis JT. Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol Cell (2010) 40(6):965–75. doi:10.1016/j.molcel.2010.11.022
96. Lavarone E, Puppin C, Passon N, Filetti S, Russo D, Damante G. The PARP inhibitor PJ34 modifies proliferation, NIS expression and epigenetic marks in thyroid cancer cell lines. Mol Cell Endocrinol (2013) 365(1):1–10. doi:10.1016/j.mce.2012.08.019
97. Ju BG, Solum D, Song EJ, Lee KJ, Rose DW, Glass CK, et al. Activating the PARP-1 sensor component of the groucho/TLE1 corepressor complex mediates a CaMKinase IIdelta-dependent neurogenic gene activation pathway. Cell (2004) 119(6):815–29. doi:10.1016/j.cell.2004.11.017
98. Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, et al. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell (2005) 18(1):83–96. doi:10.1016/j.molcel.2005.02.034
99. Nirodi C, NagDas S, Gygi SP, Olson G, Aebersold R, Richmond A. A role for poly(ADP-ribose) polymerase in the transcriptional regulation of the melanoma growth stimulatory activity (CXCL1) gene expression. J Biol Chem (2001) 276(12):9366–74. doi:10.1074/jbc.M009897200
100. Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, et al. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene (2003) 22(52):8460–71. doi:10.1038/sj.onc.1206897
101. Kannan P, Yu Y, Wankhade S, Tainsky MA. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res (1999) 27(3):866–74. doi:10.1093/nar/27.3.866
102. Zeng Y-X, Somasundaram K, El-Deiry WS. AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nat Genet (1997) 15(1):78–82. doi:10.1038/ng0197-78
103. Lin Y, Tang X, Zhu Y, Shu T, Han X. Identification of PARP-1 as one of the transcription factors binding to the repressor element in the promoter region of COX-2. Arch Biochem Biophys (2011) 505(1):123–9. doi:10.1016/j.abb.2010.09.016
104. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer (2011) 11(11):761–74. doi:10.1038/nrc3106
105. Folkerd EJ, Dowsett M. Influence of sex hormones on cancer progression. J Clin Oncol (2010) 28(26):4038–44. doi:10.1200/JCO.2009.27.4290
106. Zhang F, Wang Y, Wang L, Luo X, Huang K, Wang C, et al. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor alpha-dependent gene transcription. J Biol Chem (2013) 288(16):11348–57. doi:10.1074/jbc.M112.429134
107. Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science (2006) 312(5781):1798–802. doi:10.1126/science.1127196
108. Wright RH, Castellano G, Bonet J, Le Dily F, Font-Mateu J, Ballare C, et al. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev (2012) 26(17):1972–83. doi:10.1101/gad.193193.112
109. Ghabreau L, Roux JP, Frappart PO, Mathevet P, Patricot LM, Mokni M, et al. Poly(ADP-ribose) polymerase-1, a novel partner of progesterone receptors in endometrial cancer and its precursors. Int J Cancer (2004) 109(3):317–21. doi:10.1002/ijc.11731
110. Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov (2012) 2(12):1134–49. doi:10.1158/2159-8290.CD-12-0120
Keywords: PARP-1, PARP inhibitors, NF-κB, genetic transcription, sex hormone signaling, ERK signaling, angiogenesis, mitotic spindle
Citation: Weaver AN and Yang ES (2013) Beyond DNA repair: additional functions of PARP-1 in cancer. Front. Oncol. 3:290. doi: 10.3389/fonc.2013.00290
Received: 20 September 2013; Accepted: 13 November 2013;
Published online: 27 November 2013.
Edited by:
Christina Annunziata, National Cancer Institute, USAReviewed by:
Xu-Dong Zhu, McMaster University, CanadaCopyright: © 2013 Weaver and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eddy S. Yang, Department of Radiation Oncology, School of Medicine, The University of Alabama at Birmingham, 176F HSROC Suite 2232N, 1700 6th Avenue South Birmingham, AL 35249-6832, USA e-mail:ZXlhbmdAdWFiLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.