


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 26 December 2012
Sec. Molecular and Cellular Oncology
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00200
This article is part of the Research Topic Oncometabolism View all 11 articles
The Keap1–Nrf2 [Kelch-like ECH-associated protein 1–nuclear factor (erythroid-derived 2)-like 2] pathway plays a central role in the protection of cells against oxidative and xenobiotic stresses. Nrf2 is a potent transcription activator that recognizes a unique DNA sequence known as the antioxidant response element (ARE). Under normal conditions, Nrf2 binds to Keap1 in the cytoplasm, resulting in proteasomal degradation. Following exposure to electrophiles or reactive oxygen species, Nrf2 becomes stabilized, translocates into the nucleus, and activates the transcription of various cytoprotective genes. Increasing attention has been paid to the role of Nrf2 in cancer cells because the constitutive stabilization of Nrf2 has been observed in many human cancers with poor prognosis. Recent studies have shown that the antioxidant and detoxification activities of Nrf2 confer chemo- and radio-resistance to cancer cells. In this review, we provide an overview of the Keap1–Nrf2 system and discuss its role under physiological and pathological conditions, including cancers. We also introduce the results of our recent study describing Nrf2 function in the metabolism of cancer cells. Nrf2 likely confers a growth advantage to cancer cells through enhancing cytoprotection and anabolism. Finally, we discuss the possible impact of Nrf2 inhibitors on cancer therapy.
In our daily lives, we are constantly exposed to miscellaneous chemical and physical insults, including environmental pollutants, food additives, naturally occurring plant alkaloids, ultraviolet and ionizing radiation. In addition to these external stresses, there are many intrinsic toxicants produced during physiological metabolism and pathological processes, including reactive oxygen species (ROS) and proinflammatory cytokines. All aerobic organisms are fundamentally dependent on oxygen, which enables efficient energy production and provokes the oxidative damage of cellular components. To contend with these insults, our bodies are equipped with a cytoprotective mechanism for survival.
The Keap1–Nrf2 [Kelch-like ECH-associated protein 1–nuclear factor (erythroid-derived 2)-like 2] system is one of the most critical cytoprotective mechanisms acquired in vertebrates over the course of evolution (Morita and Motohashi, 2011). The transcription factor Nrf2 is a potent transcriptional activator that plays a central role in the inducible expression of many cytoprotective genes in response to oxidative and electrophilic stresses (Itoh et al., 1997; Ishii et al., 2000). Belonging to the Cap “n” collar (CNC) family of transcription factors, Nrf2 possesses a well-conserved basic region-leucine zipper (bZip) motif and binds to the antioxidant response element (ARE), or electrophile response element (EpRE; TGA(G/C)NNNGC), through heteromerizing with the small Maf protein (Figure 1).
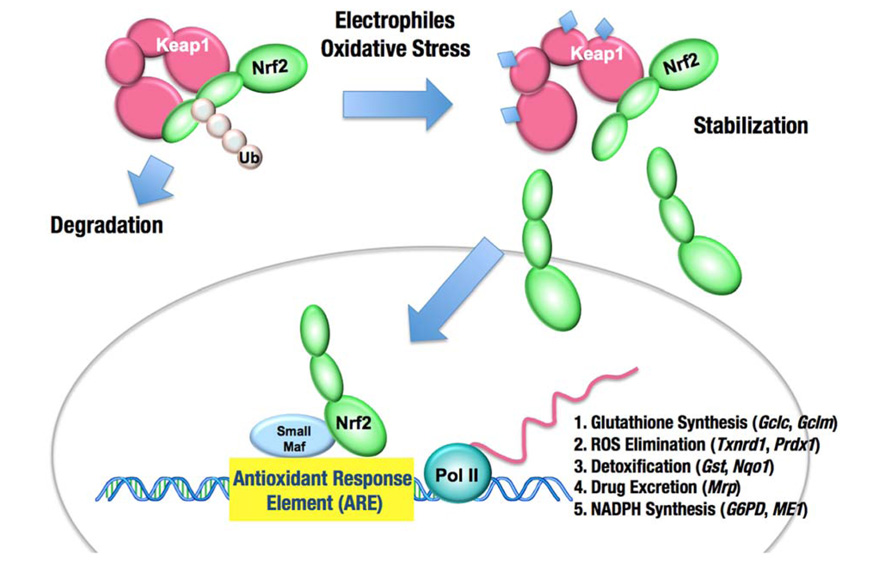
FIGURE 1. The Keap1–Nrf2 system. Under normal conditions, Nrf2 is constantly ubiquitinated through Keap1 and degraded in the proteasome. Following exposure to electrophiles or oxidative stress, Keap1 is inactivated. Stabilized Nrf2 accumulates in the nucleus and activates many cytoprotective genes. Ub, ubiquitin.
Keap1 is essential for the regulation of Nrf2 activity (Itoh et al., 1999). Under normal conditions, Nrf2 is constantly ubiquitinated through Keap1 in the cytoplasm and degraded in the proteasome. Upon exposure to electrophiles or ROS, Keap1 is inactivated and Nrf2 is stabilized. Consequently, Nrf2 translocates into the nucleus and activates the transcription of many cytoprotective genes that encode detoxifying enzymes and antioxidant proteins, including NAD(P)H:quinone oxidoreductase 1, glutathione S-transferase, and heme oxygenase-1. In general, the target genes of Nrf2 are involved in glutathione synthesis, the elimination of ROS, xenobiotic metabolism, and drug transport. Nrf2 coordinately activates these genes and exerts a protective function against xenobiotic and oxidative stresses (Figure 1; Moinova and Mulcahy, 1999; McMahon et al., 2001; Chanas et al., 2002; Solis et al., 2002; Thimmulappa et al., 2002; Bea et al., 2003; Sekhar et al., 2003; MacLeod et al., 2009). Recent studies have shown that Nrf2 is a potent cell survival factor and enhances proliferation of cancers.
Important roles for Nrf2 in the protection from xenobiotic and oxidative stresses have been shown in the analyses of Nrf2-null mice. Nrf2-null mice suffer from acute hepatotoxicity after acetaminophen exposure (Enomoto et al., 2001). The exposure to diesel exhausts increases the formation of DNA adducts in Nrf2-null mice (Aoki et al., 2001). Nrf2-null mice are more susceptible to cigarette smoke-induced emphysema (Rangasamy et al., 2004), bleomycin-induced pulmonary fibrosis (Cho et al., 2004), and hyperoxic lung injury (Cho et al., 2002b). A single-nucleotide polymorphism (SNP) in the promoter region of the mouse Nrf2 gene has been linked to the reduced expression of Nrf2 and subsequent lung damage caused by hyperoxia (Cho et al., 2002a). The human NRF2 gene also harbors SNPs in the promoter region (Yamamoto et al., 2004), which have been linked to the risk of acute lung injury (Marzec et al., 2007). These data demonstrate that Nrf2 significantly contributes to the protection against extrinsic insults.
Nrf2 also plays an important role in the response to intrinsic oxidative stress. Cellular capacities for ROS elimination are limited in Nrf2-null mice (Hirayama et al., 2003). Accordingly, Nrf2-null mice tend to spontaneously develop various inflammatory disorders, including glomerulonephritis, immune-mediated hemolytic anemia, and multi-organ autoimmune inflammation (Yoh et al., 2001; Lee et al., 2004; Ma et al., 2006). The chronic accumulation of intracellular ROS seems to underlie the pathogenesis of these disorders. Thus, Nrf2 also critically contributes to the protection from intrinsic insults.
One of the most important characteristics of Nrf2-mediated transcription is the inducibility in response to xenobiotic and oxidative stresses. Under normal conditions, the activity of the Nrf2-mediated transcription is low, as most of Nrf2 protein is degraded in the proteasome (Itoh et al., 2003). When cells are exposed to electrophiles or ROS, Nrf2 is stabilized and accumulates in the nucleus, which results in the robust activation of Nrf2 target genes. Thus, the mechanism underlying Nrf2 degradation under normal conditions and the stabilization of Nrf2 following exposure to stress are critical clues to the revelation of the molecular basis of our defense system.
Keap1 was identified as a cytoplasmic Nrf2-interacting protein that negatively regulates Nrf2 activity (Itoh et al., 1999). In the absence of Keap1, Nrf2 is constitutively stabilized, and the expression of Nrf2 target genes is maintained at high levels (Wakabayashi et al., 2003). Possessing a BTB domain at the N-terminal region, Keap1 serves as an adaptor for the Cullin 3-based ubiquitin E3 ligase for Nrf2 (Cullinan et al., 2004; Kobayashi et al., 2004; Zhang et al., 2004; Furukawa and Xiong, 2005). Keap1 is a thiol-rich protein that possesses multiple highly reactive cysteine residues (Figure 2). Electrophiles directly modify the cysteine residues, leading to Keap1 inactivation, Nrf2 stabilization, and the induction of many cytoprotective genes (Dinkova-Kostova et al., 2002; Levonen et al., 2004; Sakurai et al., 2006; Rachakonda et al., 2008; Fujii et al., 2010). High concentrations of electrophiles or highly potent electrophiles covalently modify not only Keap1 but also various cellular components, such as nucleic acids, proteins, and lipids, which adversely affects the cellular function. Because of the ultra-sensitive nature of Keap1 cysteine residues, the Keap1–Nrf2 system responds to low levels of electrophiles or less potent electrophiles and induces the cytoprotective machineries for the prevention of cellular damages.

FIGURE 2. Keap1 is a thiol-rich protein that is sensitive to electrophilic covalent modification. Domain structure of Keap1 is shown, and reactive cysteine residues are indicated with brown circles. Direct modification of cysteine residues was demonstrated using various electrophiles. Each electrophile attacks a unique set of cysteines.
The results of biochemical and structural analyses revealed the overall structure of Keap1–Nrf2 complex under the normal condition. Two molecules of Keap1 form a homodimer through the N-terminal BTB domain, and the C-terminal globular domains, called the DC domains, are positioned apart from each other (Ogura et al., 2010; Figures 3A and 4A). Two DC domains of the Keap1 homodimer associate with one molecule of Nrf2 (Tong et al., 2006). The N-terminal region of Nrf2, called the Neh2 domain, bridges the two DC domains at two separate binding sites, namely the ETGE and DLG motifs (Figure 4B). The lysine residues serve as ubiquitination target sites and are clustered in the Neh2 domain between the ETGE and DLG motifs. The two-site binding between Keap1 and Nrf2 appears to be favorable for the efficient ubiquitination of Nrf2.
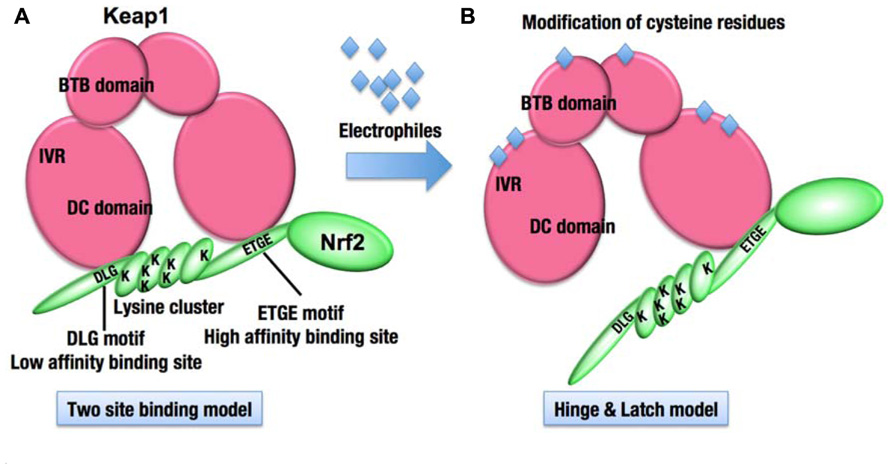
FIGURE 3. Molecular mechanism of stress sensing in the Keap1–Nrf2 system. (A) The Keap1 homodimer binds one molecule of Nrf2. The ETGE and DLG motifs of Nrf2 represent high and low affinity binding sites, respectively. The lysine residues (K) are clustered between the two motifs and represent ubiquitination targets. (B) The modification of cysteine residues in Keap1 with electrophiles is expected to modify the overall conformation of the Keap1 homodimer, resulting in the termination of Nrf2 ubiquitination.
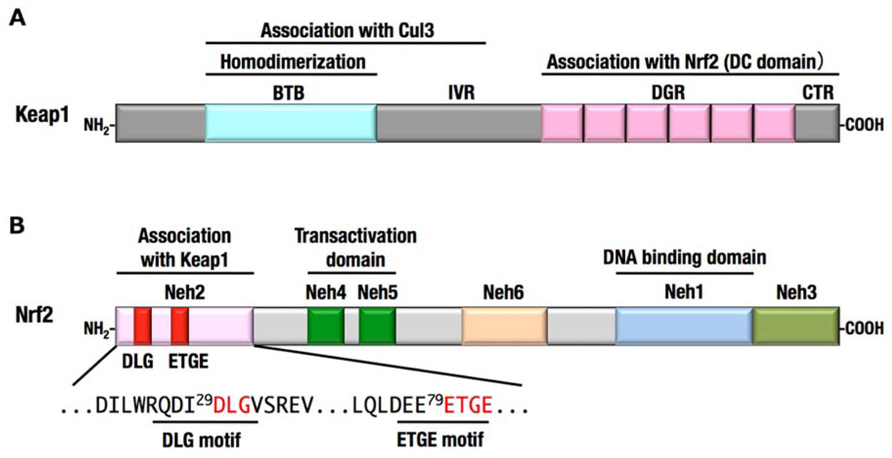
FIGURE 4. Domain structures of Keap1 (A) and Nrf2 (B).(A) The N-terminal region of Keap1 mediates homodimerization and association with Cul3, and the C-terminal region of Keap1 mediates binding with Nrf2. (B) The N-terminal region of Nrf2 is designated Neh2 domain, which contains two motifs, DLG and ETGE, responsible for the interaction with Keap1. Neh4, Neh5, and Neh3 domains are important for the transactivation activity of Nrf2 (Katoh et al., 2001; Nioi et al., 2005). Neh6 domain contains the phosphodegron that is recognized by β-TrCP. Neh1 domain is a basic-region leucine zipper motif for DNA binding and dimerization with small Maf.
Interestingly, the affinity between the ETGE motif and the Keap1 DC domain is much higher than that between the DLG motif and the Keap1 DC domain (Tong et al., 2006). The Keap1 cysteine residue-mutant and electrophile-modified Keap1, both of which have lost the ability to ubiquitinate Nrf2, often retain the capacity to bind with Nrf2. A unique model emerging from these observations is the “hinge and latch” model (Tong et al., 2007; Figure 3B). The cysteine modification is expected to trigger a conformational change of the Keap1 homodimer, which could dissociate the weak binding site, the DLG motif (latch), from the Keap1 DC domain, while the strong binding site, the ETGE motif (hinge), remains attached to the other Keap1 DC domain. The hinge and latch model is one of the attractive hypotheses for the sensing mechanism of the Keap1–Nrf2 system.
Besides the cysteine modification of Keap1, phosphorylation of Nrf2 regulates the activation of Nrf2. Protein kinase C phosphorylates Ser40 of Nrf2, resulting in the activation of Nrf2 (Huang et al., 2002). Nrf2 is also activated by PERK-dependent phosphorylation and promotes cell survival under endoplasmic reticulum stress (Cullinan et al., 2003). In contrast, Nrf2 phosphorylation by GSK-3β at serine residues of Neh6 domain (Figure 4B) inhibits its activity by promoting β-TrCP-mediated ubiquitination and degradation of Nrf2 (Rada et al., 2012). This is a second degradation pathway of Nrf2, which seems to be independent of Keap1.
Because Nrf2 activates many genes encoding detoxification enzymes, Nrf2 deficiency exacerbates the formation of DNA adducts, which increases the risk of carcinogenesis. Conversely, the increased activity of Nrf2 is effective for the prevention of chemical carcinogenesis. Oltipraz (4-methyl-5-[2-pyrazinyl]-1,2-dithiole-3-thione) is an Nrf2 inducer that suppresses the benzo[a]pyrene-induced gastric cancer formation, which is not observed in Nrf2-null mice (Ramos-Gomez et al., 2001). Thus, the antitumor effect of oltipraz requires Nrf2 function. N-nitrosobutyl (4-hydroxybutyl) amine (BBN) causes urinary bladder carcinogenesis, and this effect was also suppressed through oltipraz in an Nrf2-dependent manner (Iida et al., 2004). The results of a field study in China showed that the Nrf2 inducer, sulforaphane, which is contained in broccoli sprouts, is potentially effective for cancer chemoprevention (Wang et al., 1999; Kensler et al., 2005).
Recent studies revealed that Wilms tumor gene on the X chromosome (WTX) and PALB2, a major BRCA2 binding partner known as FANCN, have been shown to interact with the DC domain of KEAP1, inhibit the ubiquitination of Nrf2 and promote NRF2-dependent transcription (Camp et al., 2012; Ma et al., 2012). Functional defects of these gene products enhance the ubiquitination of Nrf2, resulting in the decreased activity of Nrf2. WTX and PALB2 are both considered as tumor suppressor genes since their mutations are often found in kidney tumor (Wilms tumor) and breast and pancreatic cancers, respectively. WTX and PALB2 may suppress carcinogenesis partly through maintaining the Nrf2 activity for cytoprotection.
Intriguingly, various human cancers frequently exhibit increased levels of NRF2 (Singh et al., 2006; Shibata et al., 2008a,b; Wang et al., 2008a; Kim et al., 2010; Solis et al., 2010; Zhang et al., 2010; Taguchi et al., 2011). Highly activated NRF2 target genes, encoding detoxification and antioxidant enzymes, confer a great advantage to cancer cells for survival against anti-cancer drugs and irradiation (Wang et al., 2008b; Singh et al., 2010; Zhang et al., 2010). Constitutively stabilized NRF2 also promotes cell proliferation, as NRF2 knockdown inhibits the proliferation of human lung cancer cell lines (Singh et al., 2008). Cancer cells hijack the KEAP1–NRF2 system, acquiring malignant properties. Indeed, the prognoses of patients carrying NRF2-positve cancers are significantly poor (Shibata et al., 2008b; Solis et al., 2010; Inoue et al., 2012).
Several mechanisms have been reported for the increased activity of NRF2 in cancers (Figure 5): (1) somatic mutations in KEAP1 or NRF2, (2) DNA hypermethylation at the promoter region of KEAP1, (3) the aberrant accumulation of proteins that disrupt the KEAP1–NRF2 interaction, (4) transcriptional up-regulation of NRF2 gene through oncogene-dependent signaling, and (5) the modification of KEAP1 protein through oncometabolites. A detailed description of each mechanism is provided below.
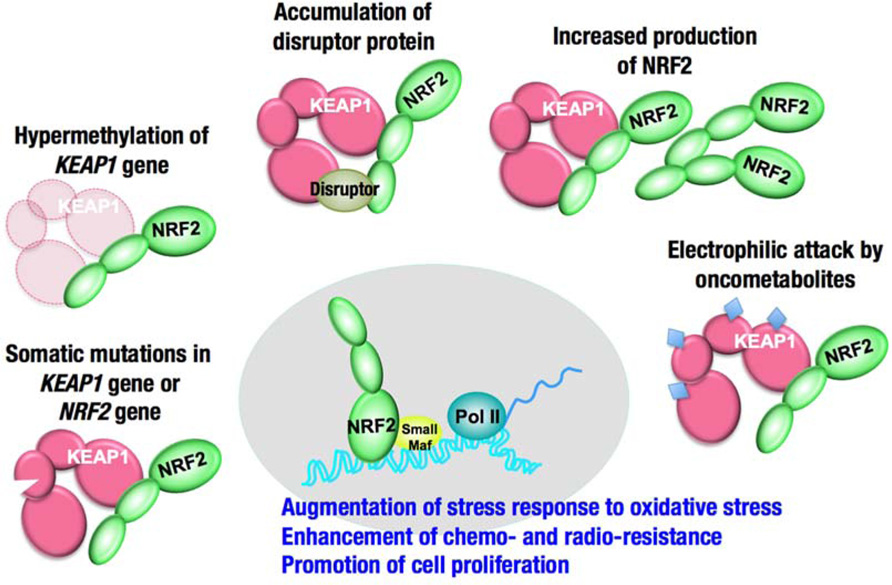
FIGURE 5. Increased activity of NRF2 in cancer cells. The degradation of NRF2 is inhibited in some cases, and the production of NRF2 is increased in other cases.
Missense mutations in the KEAP1 gene have been identified in several human cancers, particularly in solid tumors in the lung, gallbladder and liver (Padmanabhan et al., 2006; Singh et al., 2006; Nioi and Nguyen, 2007; Ohta et al., 2008; Shibata et al., 2008a; Takahashi et al., 2010; Li et al., 2011). Somatic mutations cause amino acid substitutions; thus, the resultant KEAP1 mutant proteins are not able to fulfill the adaptor function of the E3 ubiquitin ligase for NRF2. More than half of the KEAP1 mutations that have been reported so far are distributed in the DC domain, which is essential for association with NRF2 (Taguchi et al., 2011; Figure 6A). Interestingly, heterozygous KEAP1 mutations frequently occur in lung cancers (Padmanabhan et al., 2006; Singh et al., 2006; Ohta et al., 2008; Shibata et al., 2008a). An elegant mouse model demonstrated that a heterozygous mutation in the KEAP1 gene is sufficient to reduce KEAP1 activity and consequently stabilize NRF2 (Suzuki et al., 2011; Figure 7). Based on the observation that Keap1 functions as a homodimer, the heterozygous missense mutation generates three types of Keap1 dimers, i.e., wild-type homodimer, wild-type-mutant heterodimer, and mutant homodimer at a ratio of 1:2:1. Because the hinge and latch hypothesis predicts that the wild-type-mutant heterodimer does not support Nrf2 ubiquitination, a heterozygous missense mutation would result in the 75% reduction of Keap1 activity. The results of a study concerning the graded expression of the Keap1 gene in mice demonstrated that a 50% reduction of Keap1 activity does not induce Nrf2 accumulation, whereas a 75% reduction is enough to elicit this effect (Taguchi et al., 2010). Thus, the heterozygous KEAP1 mutation conferring the growth advantage on cancers is consistent with the two-site binding model and hinge and latch model of the Keap1–Nrf2 system.
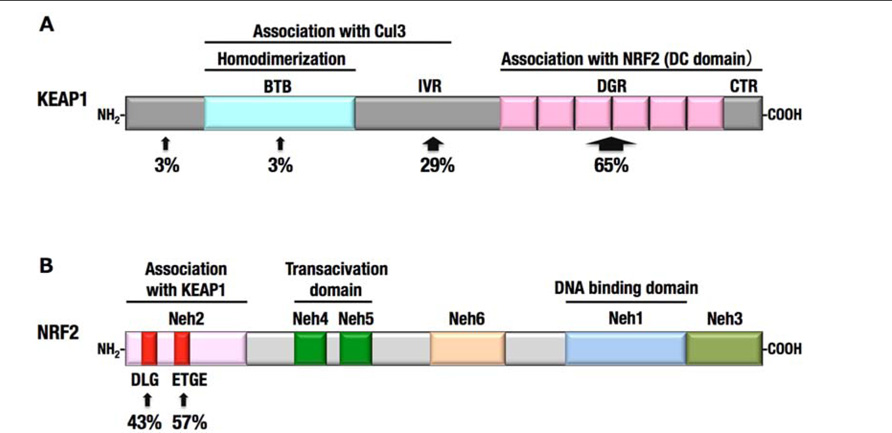
FIGURE 6. Somatic mutations in KEAP1 and NRF2 genes identified in human cancers. (A) More than half of the KEAP1 gene mutations were identified in the DC domain. (B) All NRF2 gene mutations were restricted to the DLG and ETGE motifs.
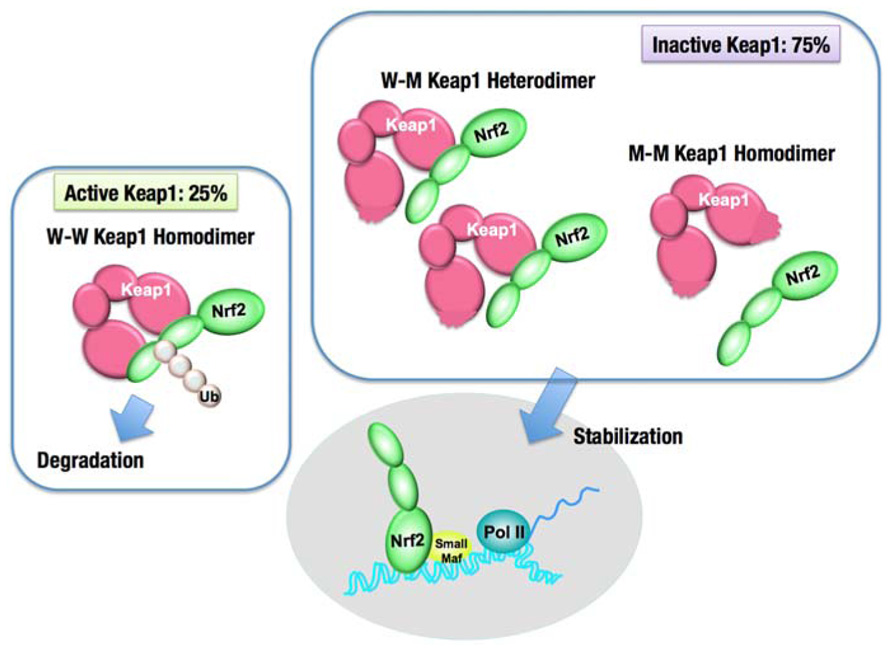
FIGURE 7. Dominant negative effect of the Keap1 gene mutation. The intact Keap1 homodimer ubiquitinates Nrf2, while the Keap1 dimer, containing one or two mutant Keap1 proteins, cannot ubiquitinate Nrf2. A single allele mutation in the Keap1 gene results in the production of an equal molar ratio of the wild-type Keap1 (W) to the mutant Keap1 (M). Keap1 dimerization generates three kinds of dimers, W–W, W–M, and M–M, by 1:2:1 ratio. Since W–M and M–M dimers are incapable of ubiquitinating Nrf2, the overall Keap1 activity is reduced by 75%, and consequently, Nrf2 is stabilized.
Mutations in NRF2 gene were also identified in several cancers, including lung, head and neck, and esophageal cancers (Shibata et al., 2008b, 2011; Kim et al., 2010). Notably, all the mutations in the NRF2 gene are clustered within the DLG (43%) and ETGE (57%) motifs (Figure 6B), which are critical sites for the binding of Nrf2 to the Keap1 DC domain. Mutations in the ETGE motif disrupt the high-affinity binding of Keap1 with Nrf2; thus, ETGE mutant proteins are not ubiquitinated and accumulate in the nucleus (Shibata et al., 2008b). Mutations in the DLG motif disrupt the low-affinity binding of Keap1 with Nrf2, resulting in the stabilization of Nrf2, although the association between Keap1 and Nrf2 through the high-affinity binding site, ETGE, is retained.
Epigenetic alterations also facilitate NRF2 stabilization. The hypermethylation of the promoter region of KEAP1 gene has been identified in cancer cells generated in lung (Wang et al., 2008a; Muscarella et al., 2011b), prostate (Zhang et al., 2010), malignant glioma (Muscarella et al., 2011a), and colorectal cancers (Hanada et al., 2012). The inhibition of KEAP1 gene expression results in NRF2 accumulation, conferring a survival and growth advantage to cancer cells. The epigenetic abnormalities in the KEAP1 gene in lung cancers and malignant gliomas are indeed associated with poor clinical outcomes (Muscarella et al., 2011a,b).
Several non-electrophilic inducers of Nrf2 originating from endogenous stress have been identified. For instance, the cyclin-dependent kinase inhibitor p21, which is a p53-regulated gene product with pro-survival properties, has been shown to associate with the DLG motif of Nrf2 (Chen et al., 2009). Consequently, the two-site binding between Keap1 and Nrf2 is disrupted, and the E3 ligase activity of the Keap1-Cul3 complex is inactivated. Indeed, the activation of cytoprotective genes through Nrf2 is more augmented in the presence than in the absence of p21. As a downstream effector of p53 that mediates cell cycle arrest and apoptosis (Gartel and Tyner, 2002), p21 also promotes cell survival in response to oxidative stress (O’Reilly, 2005). Thus, the cytoprotective function of p21 may be dependent on Nrf2.
Another example of a disruptor protein is SQSTSM1/p62, which is a polyubiquitin binding protein and targets various substrates for autophagy (Komatsu and Ichimura, 2010). The STGE motif within p62 binds to Keap1 DC pockets with a similar affinity as that of the DLG motif of Nrf2 (Komatsu et al., 2010; Lau et al., 2010). When autophagy is impaired, increased p62 competes with the DLG motif for binding to the Keap1 DC pocket and inhibits the ubiquitination of Nrf2, causing Nrf2 stabilization and the increased expression of cytoprotective genes. Importantly, the abnormal accumulation of p62 is often observed in certain cancers, such as hepatocellular carcinoma (Lu et al., 2001; Strnad et al., 2008; Inami et al., 2011), which suggests that the increased activity of NRF2 might contribute to the malignant progression of these cancers.
Because the degradation process primarily regulates the level of Nrf2 protein, there have been a few studies concerning the transcriptional control of the Nrf2 gene. The results of a recent insightful study implicate the transcriptional control of the Nrf2 gene as a determinant of Nrf2 activity (DeNicola et al., 2011). The oncogene K-Ras activates Nrf2 transcription through the Mek-Erk-Jun signaling pathway and reduces the ROS levels in primary fibroblasts. The transcriptional activation of Nrf2 is also suggested as a part of the tumorigenic activity of other oncogene products, such as Braf and c-Myc. Thus, oncogene activation is likely to increase the expression of NRF2, negating the need for mutations in NRF2 or KEAP1. A larger spectrum of cancers might utilize this non-mutational pathway to create a reducing environment that enables tumor promotion.
While unique metabolic activities in cancers, such as aerobic glycolysis, have long been recognized (Koppenol et al., 2011), recent technical advances have accelerated the progress in the field of cancer metabolism. The identification of 2-hydroxyglutarate as an aberrant metabolite produced by mutant IDH1 or IDH2 enzymes in glioma and acute myeloid leukemia established a new concept of oncometabolites (Dang et al., 2009; Ward et al., 2010). Oncometabolites are unique metabolites in cancer cells that are involved in the initiation and/or progression of cancers.
Fumarate, one of the intermediates of the TCA cycle, is considered to be an oncometabolite that promotes cancer based on the human cases carrying heterozygous germline mutations in the fumarate hydratase (FH) gene. These patients exhibit elevated levels of fumarate and develop hereditary leiomyomatosis and renal cell cancer (HLRCC), a syndrome characterized by smooth muscle tumors and papillary renal cell carcinoma type 2 (pRCC-2; Tomlinson et al., 2002). Interestingly, the Nrf2-mediated antioxidant response pathway is highly activated in FH-mutant cells (Adam et al., 2011; Ooi et al., 2011). It was revealed that fumarate, possessing a weak electrophilic property, modifies KEAP1 cysteine residues, resulting in the stabilization of NRF2. Heme oxygenase 1 (HO-1) is an NRF2 target gene that prevents FH deficiency-mediated succinate stagnation through the heme synthesis and degradation pathway (Frezza et al., 2011). The inhibition of HO-1 is effective for the suppression of FH-deficient cancers. The contribution of other NRF2 target gene products to the properties of FH-deficient cancers would be studied in more detail.
In addition to coordinately activating the genes encoding detoxifying enzymes and antioxidant proteins, the constitutive accumulation of Nrf2 confers chemo- and radio-resistance for cancer cell survival. Then, what is the role of Nrf2 in promoting cell proliferation in the absence of external insults? We recently found that Nrf2 redirects glucose and glutamine into anabolic pathways and promotes metabolic activities that are advantageous for proliferation (Mitsuishi et al., 2012). An attempt to identify Nrf2 target genes in cancer cells revealed that Nrf2 directly activates the genes whose products are involved in the pentose phosphate pathway and nicotinamide adenine dinucleotide phosphate (NADPH) production, such as glucose-6-phosphate dehydrogenase (G6PD), phosphogluconate dehydrogenase (PGD), transketolase (TKT) and transaldolase 1 (TALDO1), and malic enzyme 1 (ME1; Figure 8). The metabolite analysis demonstrated that Nrf2 strongly promotes purine nucleotide synthesis, resulting in the increased production of purine nucleotides. Nrf2 also promotes glutamine consumption through enhancing glutaminolysis and glutathione synthesis (Figure 8). Importantly, the effects of Nrf2 on gene expression and metabolic activities are obvious under the sustained activation of PI3K–Akt signaling pathway. The functional expansion of Nrf2 in proliferating cells directs the enhancement of anabolic metabolism, maintains redox homeostasis and further promotes the activation of PI3K–Akt signaling, suggesting the presence of positive feedback between Nrf2 and the PI3K–Akt pathway in proliferating cells (Figure 9).
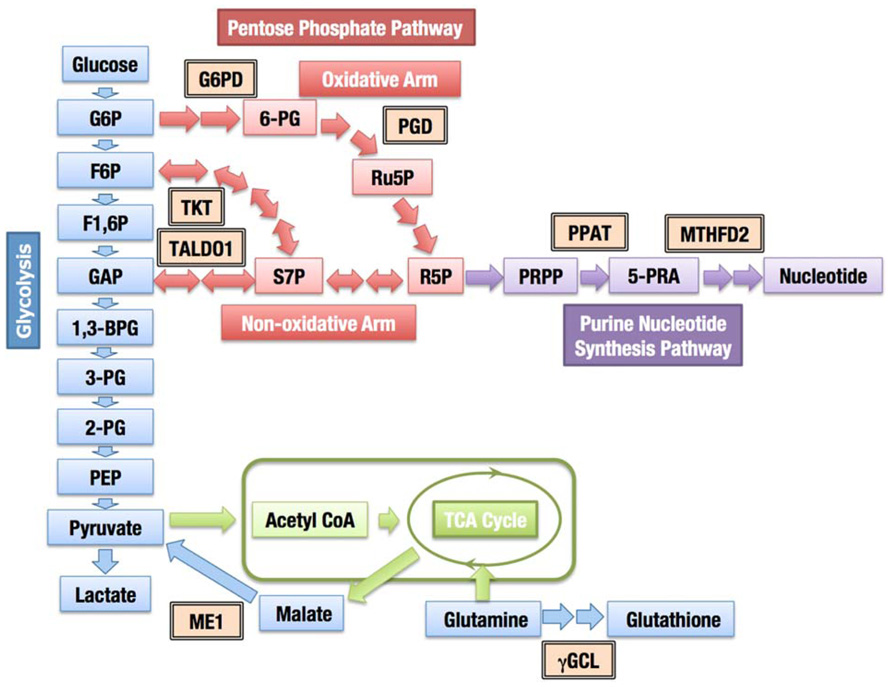
FIGURE 8. Contribution of Nrf2 to cellular metabolism. The enzymes regulated through Nrf2 are indicated with double-framed boxes (G6PD, glucose-6-phosphate dehydrogenase; PGD, phosphogluconate dehydrogenase; TKT, transketolase; TALDO1, transaldolase 1; PPAT, phosphoribosyl pyrophosphate amidotransferase; MTHFD2, methylenetetrahydrofolate dehydrogenase 2; ME1, malic enzyme 1; γGCL, γ-glutamylcysteinyl ligase). Abbreviations of metabolites; 1,3-BPG, 1,3-bisphosphoglycerate; 2-PG, 2-phosphoglycerate; 3-PG, 3-phosphoglycerate; 6-PG, 6-phosphogluconate; 5-PRA, β-5-phosphorybosylamine; F1,6P, fructose 1,6-bis-phosphate; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; GAP, glyceraldehyde 3-phosphate; PEP, phosphoenolpyruvate; PRPP, phosphoribosyl phosphate; R5P, ribose 5-phosphate; Ru5P, ribulose 5-phosphate; S7P, sedoheptulose 7-phosphate.
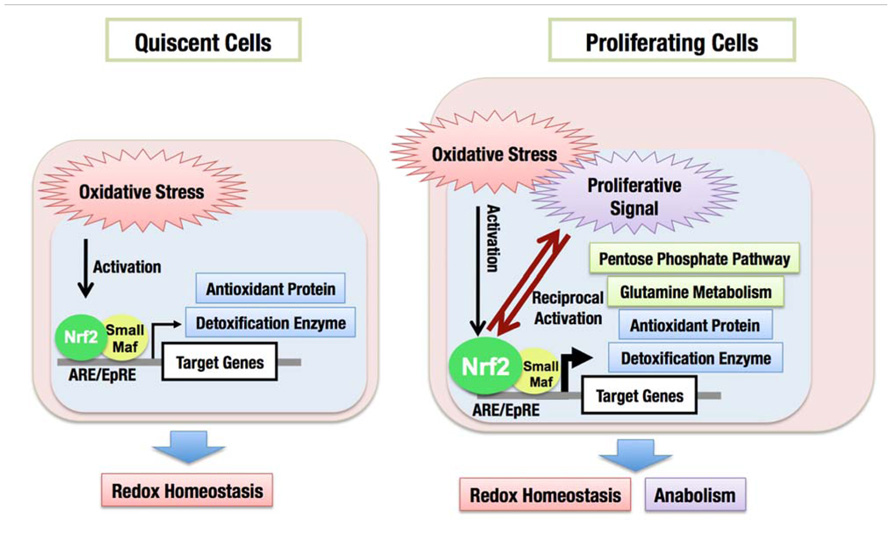
FIGURE 9. Functional expansion of Nrf2 in proliferating cells. In quiescent cells, Nrf2 is activated in response to oxidative stress and induces the expression of cytoprotective genes encoding antioxidant proteins and detoxification enzymes, which maintains the cellular redox homeostasis. In proliferating cells, Nrf2 activity is augmented especially under the sustained activation of PI3K–Akt pathway. Nrf2 activates metabolic genes in addition to cytoprotective genes, resulting in the redirection of glucose and glutamine into anabolic pathway, which is advantageous for cell proliferation.
In good agreement with this observation, a simple accumulation of Nrf2 is not sufficient for the development of spontaneous cancers (Taguchi et al., 2010), although a large number of human cancers depend on Nrf2 activity. In Keap1 knockdown (Keap1KD) mice, the Keap1 mRNA level is reduced to approximately 5% of that in wild-type mice, and constitutive Nrf2 activation is observed in various tissues, such as liver, lung, and kidney. However, Keap1KD mice did not develop any spontaneous cancers. Thus, increased Nrf2 activity does not initiate cancer development but confers advantages in terms of proliferation and stress resistance once a cell acquires uncontrolled proliferative properties. Nrf2 is a critical survival factor for cancer cells, which is best described as a form of non-oncogene addiction.
The pentose phosphate pathway fulfills two important cellular requirements. The first requirement is to generate ribose 5-phosphate for the synthesis of nucleotides, and the other is to provide reducing power in the form of NADPH. Ribose 5-phosphate is a nucleotide precursor, which is indispensable for proliferating cells and generated through two distinct pathways, the oxidative and non-oxidative arms of the pentose phosphate pathway (Figure 8). The oxidative arm is an irreversible mechanism associated with the production of NADPH. The activity of G6PD, one of the enzymes of the oxidative pathway, was associated with thymidine incorporation, indicating a critical role for G6PD in cell growth (Tian et al., 1998). The balance between the need for NADPH or ribose 5-phosphate determines the direction of the non-oxidative arm (Wamelink et al., 2008). When the requirement for NADPH production dominates, pentose phosphates produced from the oxidative arm are recycled back to glycolytic intermediates. When a large quantity of nucleotides is required, such as in cancer cells, both the oxidative and non-oxidative arms are directed toward ribose 5-phosphate production (Boros et al., 2000). The increased expression of one of the enzymes involved in the non-oxidative pathway, TKTL1, was associated with the poor prognosis of colon and urothelial cancers (Langbein et al., 2006), suggesting that the non-oxidative arm is also critical for the malignant growth of some cancers. The inhibition of the TKTL1 activity has been shown to repress the proliferation of hepatoma cells (Zhang et al., 2007).
Nrf2 not only increases the enzyme levels of both the oxidative and non-oxidative arms, but it also facilitates the utilization of ribose 5-phosphate for the purine nucleotide synthesis (Mitsuishi et al., 2012), which appears to maintain the ribose 5-phosphate concentration at a low level and efficiently divert glucose flux into purine nucleotide synthesis through both arms of the pentose phosphate pathway. Although Nrf2 does not directly contribute to aerobic glycolysis, glucose uptake and glycolytic activity are stimulated under the sustained activation of the PI3K–Akt signaling (Elstrom et al., 2004; Wieman et al., 2007), thereby increasing the supply of glycolytic intermediates. Thus, Nrf2 accumulation and activation of PI3K–Akt pathway achieve the efficient synthesis of the purine nucleotides.
The oncoprotein c-Myc regulates nucleotide metabolism (Mannava et al., 2008). c-Myc directly activates the genes involved in nucleotide synthesis, including thymidylate synthase for pyrimidine metabolism, inosine monophosphate dehydrogenase 1 and 2 for purine metabolism, and phosphoribosyl pyrophosphate (PRPP) synthetase 2 for the production of PRPP, which is a common precursor for purine and pyrimidine nucleotides. While purine nucleotide synthesis is selectively affected through Nrf2 activation, c-Myc is involved in the regulation of both purine and pyrimidine nucleotide synthesis.
It has been shown that the activation of mammalian target of rapamycin complex 1 (mTORC1) increases the metabolic flux through both glycolysis and the oxidative arm of the pentose phosphate pathway (Düvel et al., 2010). Sterol regulatory element-binding proteins (SREBP1 and SREBP2) have been suggested as one of the downstream effectors of mTORC1, which are responsible for the regulation of the pentose phosphate pathway enzymes. Nrf2 efficiently activates the pentose phosphate pathway genes in the active PI3K–Akt pathway, where mTORC1 and SREBP are also activated; therefore, Nrf2 and SREBP might synergistically facilitate the oxidative arm of the pentose phosphate pathway.
In contrast, the non-oxidative arm of the pentose phosphate pathway is enhanced in the presence of oncogenic Kras (KrasG12D) in pancreatic tumors (Ying et al., 2012). Myc was suggested as a downstream effector of KrasG12D in this study. Because the KrasG12D-Myc axis has been suggested to induce Nrf2 gene expression and increased the activity of Nrf2 in pancreatic cancers (DeNicola et al., 2011), the activation of the non-oxidative arm of the pentose phosphate pathway might depend on Nrf2.
Another unique regulator of the pentose phosphate pathway is the tumor suppressor p53, which inhibits the pentose phosphate pathway through binding to G6PD and preventing the formation of the active dimer (Jiang et al., 2011). Consequently, wild-type p53 suppresses NADPH production, whereas tumor-associated p53 mutants show almost no activity in inhibiting G6PD, thereby maintaining a high level of NADPH production. The Nrf2-mediated induction of the pentose phosphate pathway at the transcription level would substantially increase the pathway activity in p53-mutated cancers.
The glutathione synthesis pathway is another important anabolic target of Nrf2 (MacLeod et al., 2009). Glutathione is a key molecule for redox homeostasis, and the reduced form of glutathione is essential for the detoxification of ROS and the reduction of oxidized proteins. Considering that the frequently mutated tumor suppressor and oncogenic pathways in cancers commonly lead to the increased accumulation of ROS (Szatrowski and Nathan, 1991; Lee et al., 1999; Vafa et al., 2002; Nogueira et al., 2008), the efficient detoxification of ROS is a requisite for cell proliferation.
Nrf2 directly activates the essential genes for the glutathione synthesis. The genes encoding the regulatory (GCLM) and catalytic (GCLC) subunits of γ-glutamylcysteinyl ligase, a rate-limiting enzyme for glutathione synthesis, are well known targets of Nrf2 (Moinova and Mulcahy, 1999; Solis et al., 2002; Bea et al., 2003; Sekhar et al., 2003). The gene encoding a subunit of the cystine transporter SLC7A11 (xCT) is another Nrf2 target (Sasaki et al., 2002), whose product increases the availability of cysteine for glutathione synthesis. Nrf2 also activates the genes encoding the four major NADPH producing enzymes, G6PD, PGD, ME1, and IDH1, for reducing oxidized glutathione and other cellular components. Thus, Nrf2 induces the production of glutathione and NADPH, conferring a growth advantage to cancer cells.
A recent study has demonstrated that an acute increase in the intracellular concentration of ROS inhibits the glycolytic enzyme pyruvate kinase M2 (PKM2) through the oxidation of Cys358 (Anastasiou et al., 2011). The inhibition of PKM2 redirects glucose into the pentose phosphate pathway and thereby generates NADPH for the detoxification of ROS. An increase in ROS levels also induces Nrf2-dependent transcription, thus the induction of the pentose phosphate pathway enzymes through Nrf2 could contribute to the redirection of glucose and NADPH production.
Due to the multifaceted roles of Nrf2 in cancers, Nrf2 inhibitors could be effective for anticancer therapy. The suppression of antioxidant proteins, glutathione synthesis enzymes, and NADPH-producing enzymes would lead to ROS accumulation in cancer cells and subsequent oxidative damage to various intracellular components, thus compromising the cell viability. The simultaneous inhibition of the oxidative and non-oxidative arms of the pentose phosphate pathway is indeed effective for repressing tumor cell growth (Boros et al., 1997; Ramos-Montoya et al., 2006; Mitsuishi et al., 2012). Nrf2 inhibitors also sensitize cancers to the effects of chemotherapeutic drugs through the down-regulation of detoxification enzymes and drug excretion transporters (Singh et al., 2008; Wang et al., 2008b). While a number of Nrf2 inducers have been developed and tested in clinical trials (Pergola et al., 2011; Gold et al., 2012), few Nrf2 inhibitors have been developed. Brusatol has been purified from a plant extract of Brucea javanica (Simaroubaceae), which is an evergreen shrub grown in Southeast Asia and Northern Australia, and shown to inhibit ARE-luciferase activity and the protein accumulation of Nrf2 (Ren et al., 2011). However, the detailed mechanism by which brusatol enhances Nrf2 degradation and how it selectively inhibits the Keap1–Nrf2 pathway warrants further investigation.
Achieving specificity is the biggest challenge in the development of Nrf2 inhibitors. Nrf2 belongs to the CNC protein family, including NF-E2 p45, Nrf1, Nrf3, Bach1, and Bach2 (Motohashi et al., 2002). All the members form heterodimers with small Maf proteins through leucine zipper motifs and bind to the ARE consensus sequence through basic regions, suggesting that the bZip structure of Nrf2 shares many common properties with that of other CNC members. Thus, one of the targets for Nrf2 inhibitors would be the domain outside the bZip motif, and the other target would be the leucine zipper, as there are substantial variations in the leucine zipper compared with the basic regions in CNC family members.
In addition, achieving delivery specificity is also an important issue. The systemic inhibition of Nrf2 could exacerbate the side effects of chemo- and radiotherapies. Moreover, a recent study showed that Nrf2 deficiency in bone marrow cells aggravates metastasis (Satoh et al., 2010). Thus, a drug delivery method and protocol would need to be developed for the Nrf2 inhibitor to preferentially target cancer cells.
The positive feedback between Nrf2 and active PI3K–Akt signaling, which induces the malignant evolution of cancers, presents another area of interest for therapeutic development. An Nrf2 inhibitor would weaken the PI3K–Akt signaling activity in cancer cells, while inhibitors of PI3K–Akt signaling could antagonize Nrf2 activity. Thus, the disruption of the feedback activation represents an effective anti-cancer therapy. To decipher the functional interactions between Keap1–Nrf2 system and other oncogenic pathways is one of the most important assignments for the conquest of Nrf2-dependent cancers.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported through funding from Grants-in-Aid for Creative Scientific Research (to Masayuki Yamamoto) and Scientific Research (to Masayuki Yamamoto and Hozumi Motohashi) from the JSPS; Grants-in-Aid for Scientific Research on Innovative Areas (to Masayuki Yamamoto and Hozumi Motohashi) from the MEXT; the Tohoku University Global COE for the Conquest of Signal Transduction Diseases with Network Medicine (to Masayuki Yamamoto); the Core Research for Evolutional Science and Technology from the JST (to Masayuki Yamamoto and Hozumi Motohashi); and a research grant from the Princess Takamatsu Cancer Research Fund 09-24118 (to Hozumi Motohashi).
Adam, J., Hatipoglu, E., O’Flaherty, L., Ternette, N., Sahgal, N., Lockstone, H., et al. (2011). Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537.
Anastasiou, D., Poulogiannis, G., Asara, J. M., Boxer, M. B., Jiang, J. K., Shen, M., et al. (2011). Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283.
Aoki, Y., Sato, H., Nishimura, N., Takahashi, S., Itoh, K., and Yamamoto, M. (2001). Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharmacol. 173, 154–160.
Bea, F., Hudson, F. N., Chait, A., Kavanagh, T. J., and Rosenfeld, M. E. (2003). Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ. Res. 92, 386–393.
Boros, L. G., Puigjaner, J., Cascante, M., Lee, W. N., Brandes, J. L., Bassilian, S., et al. (1997). Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 57, 4242–4248.
Boros, L. G., Torday, J. S., Lim, S., Bassilian, S., Cascante, M., and Lee, W. N. (2000). Transforming growth factor beta2 promotes glucose carbon incorporation into nucleic acid ribose through the nonoxidative pentose cycle in lung epithelial carcinoma cells. Cancer Res. 60, 1183–1185.
Camp, N. D., James, R. G., Dawson, D. W., Yan, F., Davison, J. M., Houck, S. A., et al. (2012). Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J. Biol. Chem. 287, 6539–6550.
Chanas, S. A., Jiang, Q., McMahon, M., McWalter, G. K., McLellan, L. I., Elcombe, C. R., et al. (2002). Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3, and Gstm4 genes in the livers of male and female mice. Biochem. J. 365, 405–416.
Chen, W., Sun, Z., Wang, X. J., Jiang, T., Huang, Z., Fang, D., et al. (2009). Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 34, 663–673.
Cho, H. Y., Jedlicka, A. E., Reddy, S. P., Zhang, L. Y., Kensler, T. W., and Kleeberger, S. R. (2002a). Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am. J. Respir. Cell Mol. Biol. 26, 42–51.
Cho, H. Y., Jedlicka, A. E., Reddy, S. P., Kensler, T. W., Yamamoto, M., Zhang, L. Y., et al. (2002b). Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 26, 175–182.
Cho, H. Y., Reddy, S. P., Yamamoto, M., and Kleeberger, S. R. (2004). The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 18, 1258–1260.
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209.
Cullinan, S. B., Gordan, J. D., Jin, J., Harper, J. W., and Diehl, J. A. (2004). The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24, 8477–8486.
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744.
DeNicola, G. M., Karreth, F. A., Humpton, T. J., Gopinathan, A., Wei, C., Frese, K., et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109.
Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., et al. (2002). Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913.
Düvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183.
Elstrom, R. L., Bauer, D. E., Buzzai, M., Karnauskas, R., Harris, M. H., Plas, D. R., et al. (2004). Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64, 3892–3899.
Enomoto, A., Itoh, K., Nagayoshi, E., Haruta, J., Kimura, T., O’Connor, T., et al. (2001). High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 59, 169–177.
Frezza, C., Zheng, L., Folger, O., Rajagopalan, K. N., MacKenzie, E. D., Jerby, L., et al. (2011). Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 477, 225–228.
Fujii, S., Sawa, T., Ihara, H., Tong, K. I., Ida, T., Okamoto, T., et al. (2010). The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J. Biol. Chem. 285, 23970–23984.
Furukawa, M., and Xiong, Y. (2005). BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 25, 162–171.
Gartel, A. L., and Tyner, A. L. (2002). The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 1, 639–649.
Gold, R., Kappos, L., Arnold, D. L., Bar-Or, A., Giovannoni, G., Selmaj, K., et al. (2012). Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 367, 1098–1107.
Hanada, N., Takahata, T., Zhou, Q., Ye, X., Sun, R., Itoh, J., et al. (2012). Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 12:66. doi: 10.1186/1471-2407-12-66
Hirayama, A., Yoh, K., Nagase, S., Ueda, A., Itoh, K., Morito, N., et al. (2003). EPR imaging of reducing activity in Nrf2 transcriptional factor-deficient mice. Free Radic. Biol. Med. 34, 1236–1242.
Huang, H. C., Nguyen, T., and Pickett, C. B. (2002). Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774.
Iida, K., Itoh, K., Kumagai, Y., Oyasu, R., Hattori, K., Kawai, K., et al. (2004). Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 64, 6424–6431.
Inoue, D., Suzuki, T., Mitsuishi, Y., Miki, Y., Suzuki, S., Sugawara, S., et al. (2012). Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 103, 760–766.
Ishii, T., Itoh, K., Takahashi, S., Sato, H., Yanagawa, T., Katoh, Y., et al. (2000). Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 275, 16023–16029.
Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322.
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D., et al. (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86.
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., O’Connor, T., and Yamamoto, M. (2003). Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391.
Inami, Y., Waguri, S., Sakamoto, A., Kouno, T., Nakada, K., Hino, O., et al. (2011). Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193, 275–284.
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., et al. (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 13, 310–316.
Katoh, Y., Itoh, K., Yoshida, E., Miyagishi, M., Fukamizu, A., and Yamamoto, M. (2001). Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6, 857–868.
Kensler, T. W., Chen, J. G., Egner, P. A., Fahey, J. W., Jacobson, L. P., Stephenson, K. K., et al. (2005). Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomarkers Prev. 14, 2605–2613.
Kim, Y. R., Oh, J. E., Kim, M. S., Kang, M. R., Park, S. W., Han, J. Y., et al. (2010). Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 220, 446–451.
Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139.
Komatsu, M., and Ichimura, Y. (2010). Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 584, 1374–1378.
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223.
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337.
Langbein, S., Zerilli, M., Zur Hausen, A., Staiger, W., Rensch-Boschert, K., Lukan, N., et al. (2006). Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br. J. Cancer 94, 578–585.
Lau, A., Wang, X. J., Zhao, F., Villeneuve, N. F., Wu, T., Jiang, T., et al. (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285.
Lee, A. C., Fenster, B. E., Ito, H., Takeda, K., Bae, N. S., Hirai, T., et al. (1999). Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940.
Lee, J. M., Chan, K., Kan, Y. W., and Johnson, J. A. (2004). Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc. Natl. Acad. Sci. U.S.A. 101, 9751–9756.
Levonen, A. L., Landar, A., Ramachandran, A., Ceaser, E. K., Dickinson, D. A., Zanoni, G., et al. (2004). Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 378, 373–382.
Li, Q. K., Singh, A., Biswal, S., Askin, F., and Gabrielson, E. (2011). KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J. Hum. Genet. 56, 230–234.
Lu, M., Nakamura, R. M., Dent, E. D., Zhang, J. Y., Nielsen, F. C., Christiansen, J., et al. (2001). Aberrant expression of fetal RNA-binding protein p62 in liver cancer and liver cirrhosis. Am. J. Pathol. 159, 945–953.
Ma, Q., Battelli, L., and Hubbs, A. F. (2006). Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 168, 1960–1974.
Ma, J., Cai, H., Wu, T., Sobhian, B., Huo, Y., Alcivar, A., et al. (2012). PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 32, 1506–1517.
MacLeod, A. K., McMahon, M., Plummer, S. M., Higgins, L. G., Penning, T. M., Igarashi, K., et al. (2009). Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1–NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 30, 1571–1580.
Mannava, S., Grachtchouk, V., Wheeler, L. J., Im, M., Zhuang, D., Slavina, E. G., et al. (2008). Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7, 2392–2400.
Marzec, J. M., Christie, J. D., Reddy, S. P., Jedlicka, A. E., Vuong, H., Lanken, P. N., et al. (2007). Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 21, 2237–2246.
McMahon, M., Itoh, K., Yamamoto, M., Chanas, S. A., Henderson, C. J., McLellan, L. I., et al. (2001). The Cap‘n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 61, 3299–3307.
Mitsuishi, Y., Taguchi, K., Kawatani, Y., Shibata, T., Nukiwa, T., Aburatani, H., et al. (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79.
Moinova, H. R., and Mulcahy, R. T. (1999). Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 261, 661–668.
Morita, M., and Motohashi, H. (2011). “Survival strategy and disease pathogenesis according to the Nrf2-Small Maf heterodimer,” in Oxidative Stress in Vertebrates and Invertebrates: Molecular Aspects on Cell Signaling, eds T. Farooqui and A. A. Farooqui (Hoboken, NJ: Wiley-Blackwell), 63–82.
Motohashi, H., O’Connor, T., Katsuoka, F., Engel, J. D., and Yamamoto, M. (2002). Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 294, 1–12.
Muscarella, L. A., Barbano, R., D’Angelo, V., Copetti, M., Coco, M., Balsamo, T., et al. (2011a). Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 6, 317–325.
Muscarella, L. A., Parrella, P., D’Alessandro, V., la Torre, A., Barbano, R., Fontana, A., et al. (2011b). Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 6, 710–719.
Nioi, P., Nguyen, T., Sherratt, P. J., and Pickett, C. B. (2005). The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 25, 10895–10906.
Nioi, P., and Nguyen, T. (2007). A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 362, 816–821.
Nogueira, V., Park, Y., Chen, C. C., Xu, P. Z., Chen, M. L., Tonic, I., et al. (2008). Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470.
Ohta, T., Iijima, K., Miyamoto, M., Nakahara, I., Tanaka, H., Ohtsuji, M., et al. (2008). Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 68, 1303–1309.
Ogura, T., Tong, K. I., Mio, K., Maruyama, Y., Kurokawa, H., Sato, C., et al. (2010). Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. U.S.A. 107, 2842–2847.
Ooi, A., Wong, J. C., Petillo, D., Roossien, D., Perrier-Trudova, V., Whitten, D., et al. (2011). An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523.
O’Reilly, M. A. (2005). Redox activation of p21Cip1/WAF1/Sdi1: a multifunctional regulator of cell survival and death. Antioxid. Redox Signal. 7, 108–118.
Padmanabhan, B., Tong, K. I., Ohta, T., Nakamura, Y., Scharlock, M., Ohtsuji, M., et al. (2006). Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 21, 689–700.
Pergola, P. E., Raskin, P., Toto, R. D., Meyer, C. J., Huff, J. W., Grossman, E. B., et al. (2011). Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 365, 327–336.
Rachakonda, G., Xiong, Y., Sekhar, K. R., Stamer, S. L., Liebler, D. C., and Freeman, M. L. (2008). Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 21, 705–710.
Rada, P., Rojo, A. I., Evrard-Todeschi, N., Innamorato, N. G., Cotte, A., Jaworski, T., et al. (2012). Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/(β -TrCP axis. Mol. Cell. Biol. 32, 3486–3499.
Ramos-Gomez, M., Kwak, M. K., Dolan, P. M., Itoh, K., Yamamoto, M., Talalay, P., et al. (2001). Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 3410–3415.
Ramos-Montoya, A., Lee, W. N., Bassilian, S., Lim, S., Trebukhina, R. V., Kazhyna, M. V., et al. (2006). Pentose phosphate cycle oxidative and nonoxidative balance: a new vulnerable target for overcoming drug resistance in cancer. Int. J. Cancer 119, 2733–2741.
Rangasamy, T., Cho, C. Y., Thimmulappa, R. K., Zhen, L., Srisuma, S. S., Kensler, T. W., et al. (2004). Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259.
Ren, D., Villeneuve, N. F., Jiang, T., Wu, T., Lau, A., Toppin, H. A., et al. (2011). Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 108, 1433–1438.
Sakurai, T., Kanayama, M., Shibata, T., Itoh, K., Kobayashi, A., Yamamoto, M., et al. (2006). Ebselen, a seleno-organic antioxidant, as an electrophile. Chem. Res. Toxicol. 19, 1196–1204.
Sasaki, H., Sato, H., Kuriyama-Matsumura, K., Sato, K., Maebara, K., Wang, H., et al. (2002). Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 277, 44765–44771.
Satoh, H., Moriguchi, T., Taguchi, K., Takai, J., Maher, J. M., Suzuki, T., et al. (2010). Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 31, 1833–1843.
Sekhar, K. R., Crooks, P. A., Sonar, V. N., Friedman, D. B., Chan, J. Y., Meredith, M. J., et al. (2003). NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 63, 5636–5645.
Shibata, T., Kokubu, A., Saito, S., Narisawa-Saito, M., Sasaki, H., Aoyagi, K., et al. (2011). NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 13, 864–873.
Shibata, T., Kokubu, A., Gotoh, M., Ojima, H., Ohta, T., Yamamoto, M., et al. (2008a). Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358–1368.
Shibata, T., Ohta, T., Tong, K. I., Kokubu, A., Odogawa, R., Tsuta, K., et al. (2008b). Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. U.S.A. 105, 13568–13573.
Singh, A., Misra, V., Thimmulappa, R. K., Lee, H., Ames, S., Hoque, M. O., et al. (2006). Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Med. 3:e420. doi: 10.1371/journal.pmed.0030420
Singh, A., Boldin-Adamsky, S., Thimmulappa, R. K., Rath, S. K., Ashush, H., Coulter, J., et al. (2008). RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984.
Singh, A., Bodas, M., Wakabayashi, N., Bunz, F., and Biswal, S. (2010). Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal. 13, 1627–1637.
Solis, W. A., Dalton, T. P., Dieter, M. Z., Freshwater, S., Harrer, J. M., He, L., et al. (2002). Glutamate-cysteine ligase modifier subunit: mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 63, 1739–1754.
Solis, L. M., Behrens, C., Dong, W., Suraokar, M., Ozburn, N. C., Moran, C. A., et al. (2010). Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 16, 3743–3753.
Strnad, P., Zatloukal, K., Stumptner, C., Kulaksiz, H., and Denk, H. (2008). Mallory-Denk-bodies: lessons from keratin-containing hepatic inclusion bodies. Biochim. Biophys. Acta 1782, 764–774.
Suzuki, T., Maher, J., and Yamamoto, M. (2011). Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res. 71, 1700–1709.
Szatrowski, T. P., and Nathan, C. F. (1991). Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–798.
Takahashi, T., Sonobe, M., Menju, T., Nakayama, E., Mino, N., Iwakiri, S., et al. (2010). Mutations in Keap1 are a potential prognostic factor in resected non-small cell lung cancer. J. Surg. Oncol. 101, 500–506.
Taguchi, K., Maher, J. M., Suzuki, T., Kawatani, Y., Motohashi, H., and Yamamoto, M. (2010). Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 30, 3016–3026.
Taguchi, K., Motohashi, H., and Yamamoto, M. (2011). Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140.
Thimmulappa, R. K., Mai, K. H., Srisuma, S., Kensler, T. W., Yamamoto, M., and Biswal, S. (2002). Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 62, 5196–5203.
Tian, W. N., Braunstein, L. D., Pang, J., Stuhlmeier, K. M., Xi, Q. C., Tian, X., et al. (1998). Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617.
Tomlinson, I. P., Alam, N. A., Rowan, A. J., Barclay, E., Jaeger, E. E., Kelsell, D., et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410.
Tong, K. I., Katoh, Y., Kusunoki, H., Itoh, K., Tanaka, T., and Yamamoto, M. (2006). Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 26, 2887–2900.
Tong, K. I., Padmanabhan, B., Kobayashi, A., Shang, C., Hirotsu, Y., Yokoyama, S., et al. (2007). Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 27, 7511–7521.
Vafa, O., Wade, M., Kern, S., Beeche, M., Pandita, T. K., Hampton, G. M., et al. (2002). c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044.
Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., et al. (2003). Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245.
Wamelink, M. M. C., Struys, E. A., and Jakobs, C. (2008). The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J. Inherit. Metab. Dis. 31, 703–717.
Wang, J. S., Shen, X., He, X., Zhu, Y. R., Zhang, B. C., Wang, J. B., et al. (1999). Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst. 91, 347–354.
Wang, R., An, J., Ji, F., Jiao, H., Sun, H., and Zhou, D. (2008a). Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 373, 151–154.
Wang, X. J., Sun, Z., Villeneuve, N. F., Zhang, S., Zhao, F., Li, Y., et al. (2008b). Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243.
Ward, P. S., Patel, J., Wise, D. R., Abdel-Wahab, O., Bennett, B. D., Coller, H. A., et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234.
Wieman, H. L., Wofford, J. A., and Rathmell, J. C. (2007). Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 18, 1437–1446.
Yamamoto, T., Yoh, K., Kobayashi, A., Ishii, Y., Kure, S., Koyama, A., et al. (2004). Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 321, 72–79.
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670.
Yoh, K., Itoh, K., Enomoto, A., Hirayama, A., Yamaguchi, N., Kobayashi, M., et al. (2001). Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 60, 1343–1353.
Zhang, D. D., Lo, S. C., Cross, J. V., Templeton, D. J., and Hannink, M. (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941–10953.
Zhang, P., Singh, A., Yegnasubramanian, S., Esopi, D., Kombairaju, P., Bodas, M., et al. (2010). Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 9, 336–346.
Keywords: stress response, redox homeostasis, transcription, purine nucleotide, glutathione
Citation: Mitsuishi Y, Motohashi H and Yamamoto M (2012) The Keap1–Nrf2 system in cancers: stress response and anabolic metabolism. Front. Oncol. 2:200. doi: 10.3389/fonc.2012.00200
Received: 30 August 2012; Accepted: 07 December 2012;
Published online: 26 December 2012.
Edited by:
Lorenzo Galluzzi, Institut National de la Santè et de la Recherche Medicale, FranceReviewed by:
Patrizia Agostinis, Catholic University of Leuven, BelgiumCopyright: © 2012 Mitsuishi, Motohashi and Yamamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Masayuki Yamamoto, Department of Medical Biochemistry, Tohoku University Graduate School of Medicine, 2-1 Seiryo-cho, Aoba-ku, Sendai 980-8575, Japan. e-mail:bWFzaXlhbWFtb3RvQG1lZC50b2hva3UuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.