 Sameera Kongara1,2 Vassiliki Karantza1,2,3*
Sameera Kongara1,2 Vassiliki Karantza1,2,3*- 1Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, Piscataway NJ, USA
- 2The Cancer Institute of New Jersey, New Brunswick, NJ, USA
- 3Division of Medical Oncology, Department of Internal Medicine, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, Piscataway, NJ, USA
Reactive oxygen species (ROS) at physiological levels are important cell signaling molecules. However, aberrantly high ROS are intimately associated with disease and commonly observed in cancer. Mitochondria are primary sources of intracellular ROS, and their maintenance is essential to cellular health. Autophagy, an evolutionarily conserved process whereby cytoplasmic components are delivered to lysosomes for degradation, is responsible for mitochondrial turnover and removal of damaged mitochondria. Impaired autophagy is implicated in many pathological conditions, including neurological disorders, inflammatory bowel disease, diabetes, aging, and cancer. The first reports connecting autophagy to cancer showed that allelic loss of the essential autophagy gene BECLIN1 (BECN1) is prevalent in human breast, ovarian, and prostate cancers and that Becn1+/- mice develop mammary gland hyperplasias, lymphomas, lung and liver tumors. Subsequent studies demonstrated that Atg5-/- and Atg7-/- livers give rise to adenomas, Atg4C-/- mice are susceptible to chemical carcinogenesis, and Bif1-/- mice are prone to spontaneous tumors, indicating that autophagy defects promote tumorigenesis. Due to defective mitophagy, autophagy-deficient cells accumulate damaged mitochondria and deregulated ROS levels, which likely contribute to their tumor-initiating capacity. However, the role of autophagy in tumorigenesis is complex, as more recent work also revealed tumor dependence on autophagy: autophagy-competent mutant-Ras-expressing cells form tumors more efficiently than their autophagy-deficient counterparts; similarly, FIP200 deficiency suppresses PyMT-driven mammary tumorigenesis. These latter findings are attributed to the fact that tumors driven by powerful oncogenes have high metabolic demands catered to by autophagy. In this review, we discuss the relationship between ROS and autophagy and summarize our current knowledge on their functional interactions in tumorigenesis.
Introduction
Reactive oxygen species (ROS) are a family of molecules that include highly reactive free oxygen radicals, such as the superoxide anion ( ) and the hydroxyl radical (OH. ), and stable non-radical oxidants, such as hydrogen peroxide (H2O2). ROS are produced in cells through metabolism of oxygen and, when at high levels, can function as destructive molecules, actively participating in microbial elimination within phagocytes and also contributing to genomic instability, thereby resulting in cell death and/or tumorigenesis. In more recent years, however, ROS at lower physiological levels have been recognized as intracellular signal transduction molecules that regulate kinase-driven pathways, in turn mediating cellular responses to external stimuli or challenges, such as growth factors, nutrient deprivation, or hypoxia (Gough and Cotter, 2011). Mitochondria are a major source of intracellular ROS, as leaky electrons from oxidative phosphorylation [1–5% of electrons escape from complexes I and III along the electron transport chain (ETC)] and enzymatic complexes, such as α-glycerophosphate dehydrogenase and α-ketoglutarate dehydrogenase, generate short-lived superoxide anions in the presence of oxygen, which are rapidly converted into relatively stable and membrane diffusible hydrogen peroxide by superoxide dismutase (Mn-SOD), a resident protein of the mitochondrial matrix (Karihtala and Soini, 2007; Miao and St Clair, 2009; Marchi et al., 2012). Other important intracellular sources of ROS include enzymes, such as the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, (XO), the ER luminal thiol oxidase Ero1, and myeloperoxidase (Arnhold and Flemmig, 2010; Tavender and Bulleid, 2010; Dikalov, 2011; Fransen et al., 2012), as well as other organelles, such as peroxisomes (Fransen et al., 2012). Reactive nitrogen species (RNS), such as nitric oxide (NO) and peroxynitrite (ONOO), also act as signaling agents regulating numerous physiological and pathological conditions (Fukuto et al., 2012; Lee et al., 2012); however, in this XO, xanthine oxidase. review we will only focus on ROS.
At physiological levels, ROS function as signaling molecules through oxidation of redox-sensitive cysteine residues and resultant inhibition of phosphatases that negatively regulate signaling cascades (Meng et al., 2002), whereas excessive ROS create an imbalance between the oxidant and antioxidant responses in cells, thus resulting in oxidative stress, which has been etiologically implicated in the pathogenesis of several malignancies, including breast, lung, liver, and prostate cancers (Moller et al., 2008; Nowsheen et al., 2009; Khandrika et al., 2009; Severi et al., 2010; Adcock et al., 2011; Sakaguchi et al., 2011; Vera-Ramirez et al., 2011; Thapa and Ghosh, 2012). Abnormally high ROS levels result in oxidation of nuclear and mitochondrial DNA, proteins and lipids, thus impairing their structure and function and causing aberrant expression and regulation of tumor-promoting proteins and/or pathways (Valko et al., 2006). Indeed, high levels of malondialdehyde, a marker of lipid peroxidation, are observed in plasma samples from patients with breast cancer as compared to normal controls (Huang et al., 1999; Gonenc et al., 2001; Kumaraguruparan et al., 2002; Polat et al., 2002; Yeh et al., 2005; Sener et al., 2007). Conversely, overexpression of the antioxidant enzymes copper/zinc superoxide dismutase (SOD1) and manganese superoxide dismutase (SOD2) in human breast cancer cell lines suppresses xenograft tumor growth (Weydert et al., 2006), and transgenic overexpression of catalase in PyMT mice diminishes tumor invasiveness and metastasis (Goh et al., 2011).
Autophagy, derived from the Greek words for self (auto) and eating (phagy), is an evolutionarily conserved catabolic process that targets proteins, cytoplasm and organelles to lysosomes for degradation, followed by recycling of free amino acids and ATP into the cytoplasm for biomolecule synthesis within the same cell (He and Klionsky, 2009; Yang and Klionsky, 2010). Three modes of autophagy exist, namely macroautophagy, microautophagy, and chaperone-mediated autophagy. Microautophagy and chaperone-mediated autophagy are characterized by the direct delivery of cargo into lysosomes, while macroautophagy (hereafter referred to as autophagy) is marked by the formation of double membrane vesicles, which capture cytoplasmic material for delivery to lysosomes. While autophagy occurs at a basal level in most tissue types and promotes cellular homeostasis by recycling proteins and organelles, it is up-regulated in response to cellular insults, such as glucose and amino acid deprivation, hypoxia, oxidative stress, and chemotherapeutic drugs. Impaired autophagy has been implicated in the pathogenesis of diverse diseases, including cancer, diabetes, Crohn’s disease, and neurodegenerative conditions, like Alzheimer’s, Parkinson’s, and Huntington’s disease (Sridhar et al., 2012; Quan et al., 2012).
The role of autophagy in tumorigenesis is complex. Allelic loss of the essential autophagy regulator BECLIN1 (BECN1) is observed in over 50% of human breast, ovarian, and prostate cancers (Aita et al., 1999), and Becn1+/- mice are susceptible to hepatocellular carcinomas, lung adenocarcinomas, lymphomas, and mammary gland hyperplasias (Qu et al., 2003; Yue et al., 2003). Furthermore, autophagy-compromised, apoptosis-defective immortalized mouse mammary epithelial cells (iMMECs) and baby mouse kidney (iBMK) cells generate allograft tumors in nude mice more efficiently than their wild-type counterparts (Karantza-Wadsworth et al., 2007; Mathew et al., 2007). Mice lacking Bif1, a positive regulator of autophagy, are prone to spontaneous lymphomas, sarcomas, duodenal adenocarcinomas, small cell lung carcinomas, esophageal squamous cell carcinomas, and hepatocellular carcinomas (Takahashi et al., 2007), whereas Atg4C-/- mice are susceptible to chemical-induced fibrosarcomas (Marino et al., 2007), and Atg5 or Atg7 deletion in the liver results in hepatocellular adenomas (Takamura et al., 2011; Inami et al., 2011). Paradoxically though, autophagy is required for the robust progression of tumors (Guo et al., 2011; Lock et al., 2011; Kim et al., 2011; Yang et al., 2011) and also supports PyMT-driven mammary tumorigenesis, as loss of FIP200, a positive regulator of autophagy, inhibits primary tumor formation and metastasis (Wei et al., 2011). This conundrum can be, at least partially, attributed to the important roles of autophagy in both maintenance of cellular homeostasis under regular growth conditions and protection of cell viability under stress. As a housekeeping pathway, autophagy degrades damaged or aggregated proteins and organelles, which are known sources of ROS, thereby preventing tumor initiation via suppression of oxidative and genotoxic stress (Karantza-Wadsworth et al., 2007; Mathew et al., 2007). On the other hand, oncogene activation increases the metabolic demands of transformed cells and intact autophagy supports tumor cell survival by providing substrates for mitochondrial metabolism (Guo et al., 2011). How the functional status of autophagy impacts tumorigenesis is still under investigation, and tissue- and context-specific roles are likely to be revealed.
This review will focus on the interplay between ROS and autophagy and their functional interactions in the context of cancer.
Autophagic Machinery
Early work led to the identification of several Atg (Autophagy-related) genes, which are indispensable for the functional execution of the autophagic cascade in yeast. Most yeast Atg proteins have mammalian homologs participating in a similarly ordered process. Initiation of autophagy is characterized by the formation of nascent double membrane structures called phagophores, which elongate and enclose cytoplasmic material in vesicles or autophagosomes. Mature autophagosomes subsequently fuse with lysosomes to form autolysosomes, thereby leading to the degradation of cargo and the release of amino acids and ATP into the cytoplasm (He and Klionsky, 2009; Yang and Klionsky, 2010).
In mammalian cells, under nutrient-rich conditions, the nutrient sensor and serine/threonine kinase mammalian target of rapamycin complex 1 (mTORC1) phosphorylates ULK1/2, and mAtg13, preventing the assembly of a multi-protein complex, which is required for autophagosome formation. Under starvation, mTORC1 is inactivated, ULK1/2 undergo autophosphorylation, and then phosphorylate mAtg13 and FIP200 to form an ULK1/2–mAtg13–FIP200 complex that is stabilized by Atg10 (Hara et al., 2008; Ganley et al., 2009; Hosokawa et al., 2009; Jung et al., 2009; Mercer et al., 2009). The nucleation and formation of the phagophore also requires the Class III PI3K/Vps34 complex, which consists of PI3K/Vps34, the serine/threonine kinase p150 and Beclin1; this complex is positively regulated by Atg14L, the ultraviolet irradiation resistant-associated gene (UVRAG), the Bax-interacting factor 1 (Bif-1), and the activating molecule in Beclin 1-regulated autophagy (AMBRA), and negatively regulated by the RUN domain Beclin 1-interacting cysteine-rich-containing protein (RUBICON; Kihara et al., 2001; Liang et al., 2006; Fimia et al., 2007; Takahashi et al., 2007; Sun et al., 2008; reviewed in Yang and Klionsky, 2010). The ULK and the Class III PI3K complexes recruit two ubiquitin-like conjugation systems for expansion of the autophagosome membrane. Following activation by Atg7 (E1-like enzyme), Atg12 is transferred to Atg10 (E2-like enzyme) and becomes covalently bound to Atg5 via a lysine residue. Atg16L combines with Atg5–Atg12 to form the Atg5–Atg12–Atg16L complex that self-oligomerizes and binds to the phagophore (Mizushima et al., 1998, 2001, 2003; Tanida et al., 2001; reviewed in He and Klionsky, 2009). Similarly, following cleavage at its C-terminus by the cysteine protease Atg4 and activation by Atg7 (E1-like enzyme), Atg8 (or LC3) is transferred to Atg3 (E2-like enzyme), conjugated to phosphatidylethanolamine (PE) and incorporated into the autophagosome membrane, in a process facilitated by – but not exclusively dependent on – the Atg5–Atg12 complex (Kabeya et al., 2000; Tanida et al., 2002; Nishida et al., 2009; reviewed in He and Klionsky, 2009). Atg4 can also delipidate LC3-PE to release LC3 from autophagosomes and, thus, increase the free LC3 pool (Kabeya et al., 2004; Tanida et al., 2004). The protein p62/SQSTM1, which accumulates under conditions of oxidative stress, binds to ubiquitinated proteins via its ubiquitin-associated (UBA) domain, sequesters them by self-oligomerization, and delivers them to autophagosomes via its LC3 binding domain (Bjorkoy et al., 2005; Komatsu et al., 2007; Pankiv et al., 2007). Finally, the autophagosomal cargo, including p62, is degraded in lysosomes (Bjorkoy et al., 2005; Komatsu et al., 2007; Pankiv et al., 2007).
Crosstalk Between ROS and Autophagy: ROS Modulate Autophagy
Autophagy Induction in Response to ROS
Direct effect on Atg proteins
Amino acid deprivation induces the formation of H2O2 in mitochondria in a Class III PI3K-dependent manner, and this ROS is essential for the induction of autophagy in response to starvation (Scherz-Shouval et al., 2007). Specifically, the Cys81 residue near the catalytic site of Atg4 is a direct oxidation target by H2O2; its oxidized form inactivates the protease activity of Atg4 and prevents the delipidation of LC3 without affecting the C-terminal processing of LC3 by Atg4, thus leading to increased autophagosome formation. In a reducing environment, the Atg4 protease remains active and delipidates LC3, thereby suppressing autophagosome membrane formation and resulting in autophagy inhibition.
Indirect induction of autophagy
The energy-dependent activity of mTOR is regulated by the heterotrimeric enzyme AMPK, which is in turn activated by phosphorylation in response to high intracellular AMP/ATP ratios. Under nutrient-depleted conditions, active AMPK phosphorylates TSC2, as well as the mTORC1 subunit Raptor, effectively preventing the activation of mTORC1 and, therefore, inducing autophagy (Inoki et al., 2003; Gwinn et al., 2008). More recent studies demonstrate a direct role for AMPK in the induction of autophagy. Under glucose deprivation, AMPK directly activates ULK1 by phosphorylation at Ser317 and Ser777 and results in the induction of autophagy (Kim et al., 2011). In another study, phosphorylated ULK1 (at Ser555) is detected in wild-type hepatocytes, but not in hepatocytes from AMPK-/- mice treated with metformin, an activator of AMPK (Egan et al., 2011). Reconstitution of ULK1 in ULK1-/- MEFs, but not a mutated ULK1 construct lacking the AMPK phosphorylation site, rescues the viability of cells under starvation by the induction of mitochondrial-specific autophagy (Egan et al., 2011). AMPK is a major mediator of indirect activation of autophagy by ROS, as it is sensitive to oxidative stress induced by H2O2 and gets phosphorylated at Thr172 of its α1 catalytic subunit (Choi et al., 2001). AMPK also induces autophagy in Atg5- and LKB1-, but not AMP/ATP ratio-, dependent manners in response to hypoxia-associated mitochondrial ROS production (Papandreou et al., 2008; Emerling et al., 2009).
Reactive oxygen species-associated autophagy induction is also mediated by ataxia-telangiectasia mutated (ATM), a kinase that is critical for orchestrating cellular responses to DNA damage and, in parallel to its nuclear functions, increases TSC2 activity in the cytoplasm via the LKB1/AMPK metabolic pathway, thus resulting in mTORC1 inactivation (Alexander et al., 2010). Similarly, ROS-induced and p53-activated sestrins (Sesns) have been shown to suppress oxidative stress by promoting autophagy via the AMPK-mTOR axis in Drosophila (Lee et al., 2010; Budanov, 2011)
High mobility group box 1 (HMGB1) is a nuclear protein that is released extracellularly in response to cytokines, trauma, and cell death, and can mediate inflammation by binding to receptors, such as the receptor for advanced glycation end products (RAGE) and Toll-like receptors (TLRs; Lotze and Tracey, 2005; Sims et al., 2010). Recent studies demonstrated that loss of HMGB1 attenuates autophagy (Tang et al., 2010a,b). Furthermore, HMGB1 translocates from the nucleus to the cytoplasm in response to autophagy-inducing stimuli in a ROS-dependent manner, since treatment with the antioxidant N-acetyl cysteine (NAC) dampens, while loss of the antioxidant enzymes SOD1 and SOD2 promotes, autophagy induction and HMGB1 cytoplasmic translocation (Tang et al., 2010b). HMGB1, in turn, promotes autophagy by disrupting the Beclin1–Bcl-2 interaction and enhancing ERK1/2 activity (Tang et al., 2010b). The redox sensitivity of HMGB1 is critical for autophagy induction, as oxidation of cysteine residues at positions 23 and 45 is essential for the ability of HMGB1 to disrupt the Beclin1–Bcl-2 interaction, while Cysteine 106 is important for the cytoplasmic localization of HMGB1 (Tang et al., 2010b). Similarly, the HMGB1 receptor RAGE positively regulates autophagy and promotes survival of pancreatic cancer cells upon treatment with hydrogen peroxide (Kang et al., 2011a,b).
Hypoxia triggers an acute induction of ROS over a short period of time, while prolonged hypoxia results in decreased intracellular ROS levels as compared to normoxia (Semenza, 2011). Mitochondrial ROS generated in response to hypoxia prevent degradation of the hypoxia-inducible factor-1 alpha (HIF-1α; Chandel et al., 2000; Guzy et al., 2005). Hypoxia-induced stabilization of HIF-1α leads to transcription of several target genes, including BNIP3 and BNIP3L, which induce autophagy in response to low oxygen conditions (Bruick, 2000; Sowter et al., 2001; Zhang et al., 2008; Bellot et al., 2009). In addition to hypoxia, BNIP3 mediates autophagy induction in response to H2O2 and during myocardial ischemia/reperfusion injury (Hamacher-Brady et al., 2007; Byun et al., 2009). H2O2-responsive BNIP3-mediated induction of autophagy occurs via dephosphorylation of mTOR at the S2481 residue, resulting in its inactivation (Byun et al., 2009), while hypoxia-driven autophagy induction relies on the ability of BNIP3 to bind to and interfere with the activity of the GTPase Rheb, a positive regulator of mTORC1, thereby preventing TORC1 activation (Li et al., 2007). Additionally, mutating a redox-sensitive cysteine residue at position 64 diminishes the ability of BNIP3 to undergo homodimerization in response to oxidative stress, such as that caused by ischemia and reperfusion (Kubli et al., 2008), and this homodimerization is critical for the interaction of BNIP3 with LC3 and the induction of autophagy (Hanna et al., 2012).
Other mediators of autophagy induction in response to ROS and oxidative stress include the forkhead transcriptional regulator FOXO1 and FOXO3 (Sengupta et al., 2011), the p38 mitogen-activated protein kinase (MAPK; Cheng et al., 2009; McClung et al., 2010), the extracellular regulated kinase (ERK) and the c-Jun N-terminal kinase (JNK; Wu et al., 2009; Wong et al., 2010), and the endoplasmic reticulum kinase PERK (Avivar-Valderas et al., 2011).
Inhibitory Effect of ROS on Positive Regulators of Autophagy
Direct effect on Atg proteins
Another point of direct autophagy regulation by ROS may be at the two ubiquitin-like conjugation systems described earlier. While direct evidence is lacking in this regard, it has been speculated that the sulfhydryl group-containing ubiquitin-like enzymes Atg3, Atg7, and Atg10 are redox-sensitive in a manner similar to the E1 and E2 enzymes of the ubiquitin system, which possess catalytic cysteines and can undergo reversible oxidation and inactivation in the presence of ROS (Filomeni et al., 2010).
Indirect effect on other positive autophagy regulators
Reactive oxygen species can also modulate autophagy indirectly by affecting the activity of the master autophagy regulator mTORC1 rather than the Atg proteins themselves. mTORC1, a multi-protein complex consisting of the conserved serine/threonine kinase mTOR, Raptor, mLST8, PRAS40, and Deptor, is sensitive to the availability of amino acids, growth factors, hormones, and energy, and subsequently regulates translation, cell growth, and autophagy (Zoncu et al., 2011). The GTPases Rheb and Rag regulate the activity and the spatial localization of mTORC1, respectively, in response to amino acid and growth factor availability. Rheb is in turn regulated by the GTP activating proteins (GAP) TSC1/TSC2: upon amino acid and growth factor stimulation, PKB/Akt inactivates TSC2 by phosphorylation, resulting in Rheb and mTORC1 activation and concomitant suppression of autophagy (Zoncu et al., 2011). An earlier study indicated that mTORC1 activity increases in the presence of oxidizing agents, such as diamide or the cysteine oxidant phenylarsine oxide (PAO; Sarbassov and Sabatini, 2005), and more recent work demonstrated that PAO activates mTORC1 via Rheb (Yoshida et al., 2011). Interestingly, Tsc1-/- and Tsc2-/- MEFs exhibit constitutively high, rapamycin-sensitive mTOR activity regardless of redox modulation by an oxidant (PAO) or a reducing (British anti-Lewisite) agent, thus indicating that TSC1/2 maybe redox-sensitive proteins and potentially regulated by ROS (Yoshida et al., 2011).
mTOR activation due to growth factor stimulation is mediated by a number of signaling pathways, including the PI3K–Akt axis. For instance, binding of insulin to its receptor recruits PI3K to the cell membrane, where it phosphorylates phosphatidylinositol-4,5-phosphate (PIP2) to generate phosphatidylinositol-3,4,5-phosphate (PIP3) in a reaction that is reversibly catalyzed by the phosphatase-and-PI3K antagonist PTEN (Hernandez-Aya and Gonzalez-Angulo, 2011; Zoncu et al., 2011). Formation of PIP3 leads to recruitment and activation of PH-domain proteins, such as PDK1 and PKB/Akt, leading to further downstream signaling and mTORC1 activation. Activation of the PI3K axis in response to growth factors, such as EGF, insulin, and PDGF, is facilitated by concomitant production of H2O2, which inactivates PTEN by oxidizing a cysteine residue (Cys124) at its catalytic site and reversibly cross-linking it to Cys71 via a disulfide bond (Lee et al., 2002; Kwon et al., 2004). Although not formally demonstrated, ROS-induced inactivation of PTEN can result in increased mTORC1 activity via the PI3K–Akt pathway, thus leading to suppression of autophagy. An antioxidant buffer ameliorating growth factor-associated ROS production, and thus preserving autophagy in the context of PI3K axis activation, is provided by peroxiredoxins, particularly peroxiredoxin I (Prdx1) and peroxiredoxin II (Prdx2), which modulate PTEN oxidation (Kwon et al., 2004; Cao et al., 2009). Interestingly, MMTV-v-H-Ras;Prdx1-/- mice develop mammary tumors at a higher frequency compared to MMTV-v-H-Ras;Prdx1+/+ mice, possibly in association with increased PTEN oxidation and resultant PI3K/Akt/mTOR axis activation, as mammary epithelial cells isolated from MMTV-v-H-Ras; Prdx1-/- mice demonstrate higher levels of oxidized PTEN compared to their wild-type counterparts (Cao et al., 2009). These studies indicate that ROS may negatively regulate autophagy by mTORC1 induction via oxidation and inactivation of PTEN and TSC1/2, and suggest that other mechanism(s) protective against mTORC1 activation must be simultaneously active, so that autophagy is allowed to proceed as a prosurvival cell function under oxidative stress.
Crosstalk Between ROS and Autophagy: Autophagy Modulates Mitochondrial ROS
While the discussion has thus far focused on the impact of ROS on the functional status of autophagy in response to various cellular stressors, the autophagic pathway in turn modulates the cellular levels of ROS. Autophagy is essential for the turnover of normal mitochondria, as well as the removal of damaged mitochondria, which are primary sources of intracellular ROS. This specific form of autophagy is called mitophagy (Lemasters, 2005; Youle and Narendra, 2011). Autophagy-deficient cells have significantly higher ROS levels compared to their wild-type counterparts (Mathew et al., 2009; Kongara et al., 2010). The ROS scavenger NAC improves autophagy-deficient cell survival under metabolic stress in association with attenuation of p62 accumulation, indicating that excessive ROS results in p62 up-regulation and impairment of cell survival (Mathew et al., 2009; Kongara et al., 2010). Mammary tumors from FIP200f/f;MMTV-Cre;MMTV-PyMT mice are characterized by the presence of abnormal mitochondria, as well as accumulation of both healthy and dysfunctional mitochondria compared to the wild-type controls (Wei et al., 2011), and conditional deletion of FIP200 in hematopoietic stem cells and neural cells results in mitochondria accumulation and increased ROS levels (Liang et al., 2010; Liu et al., 2010). Morphologically abnormal mitochondria are also observed in Atg7-/- livers (Komatsu et al., 2005; Inami et al., 2011) and pancreatic β cells (Ebato et al., 2008; Jung et al., 2008), Atg5-/- cardiac tissue (Nakai et al., 2007), and Paneth cells with a hypomorphic allele of Atg16L1 (Cadwell et al., 2008). Furthermore, increased ROS levels are associated with increased mitochondrial content in Atg5-/- MEFs (Tal et al., 2009) and Atg7-/- T cells (Pua et al., 2009). Taken together, all these studies indicate that mitophagy plays an essential role in recycling of functional and removal of damaged mitochondria and clearly demonstrate that defective autophagy-associated blocks in mitophagy lead to increased intracellular levels of ROS, which in turn may be etiologically linked to tumorigenesis.
Mitophagy has been extensively examined in normal development, such as erythrocyte maturation, which involves developmental elimination of mitochondria from red blood cells, as well as in the context of mitochondrial depolarization induced by chemicals, such as CCCP (carbonyl cyanide m-chlorophenylhydrazone). Mitochondrial elimination during erythrocyte maturation depends upon the outer mitochondrial membrane protein NIP3-like protein X (Nix, also known as BNIP3L), since Nix-/- reticulocytes retain their mitochondria (Schweers et al., 2007; Sandoval et al., 2008). Similarly, ULK1-/- and Atg7-/- reticulocytes exhibit delayed mitochondrial clearance (Kundu et al., 2008; Zhang et al., 2009). Atg7-/- reticulocytes also accumulate damaged mitochondria, accompanied by increased ROS (Mortensen et al., 2010). Although Nix has been shown to directly interact with LC3 via its WXXL motif (Novak et al., 2010), its precise role in mitochondrial clearance remains unclear.
While Nix is involved in removal of functional mitochondria during normal red blood cell development, the serine/threonine kinase PTEN-induced kinase1 (PINK1) and the E3 ubiquitin ligase Parkin are essential for the elimination of damaged or dysfunctional mitochondria. PINK1 is constitutively synthesized and proteolytically degraded in normal mitochondria. Loss of membrane potential and depolarization of damaged mitochondria results in stabilization and accumulation of PINK1, which in turn recruits cytoplasmic Parkin to abnormal mitochondria and triggers mitophagy (Matsuda et al., 2010; Narendra et al., 2008, 2010a; Vives-Bauza et al., 2010). Parkin ubiquitinates mitochondrial proteins involved in mitochondrial fusion, such as mitofuscin 1 and 2 (MFN1 and 2), and results in their degradation (Gegg et al., 2010; Poole et al., 2010; Rakovic et al., 2011). The precise function of this degradation remains unknown, as are the details of how Parkin stimulates mitophagy. p62/SQSTM1 is recruited to damaged mitochondria by Parkin, but there are conflicting reports regarding its requirement for mitophagy. Reduced mitochondrial clearance is observed in HeLa cells with a transient p62 knockdown upon treatment with the mitochondrial uncoupler CCCP (Geisler et al., 2010). On the other hand, p62-/- MEFs do not have a defect in mitochondrial clearance upon CCCP treatment, despite the finding that p62 causes perinuclear, and thus autophagosome-like, mitochondrial clustering (Okatsu et al., 2010; Narendra et al., 2010b).
Functional Interactions Between Autophagy and ROS Intumorigenesis
DNA Damage and Genomic Instability
As mentioned above, Becn1+/- iMMECs (Kongara et al., 2010) and iBMK cells (Mathew et al., 2009) exhibit elevated intracellular ROS levels compared to their wild-type counterparts and, although not directly demonstrated, oxidative stress due to excess ROS is likely contributing to the DNA damage and genomic instability observed in autophagy-defective iMMECs and iBMK cells in vitro and in their resultant allograft tumors in nude mice in vivo (Karantza-Wadsworth et al., 2007; Mathew et al., 2007; Mathew et al., 2009), thus providing a plausible link between autophagy defects, ROS accumulation, and enhanced tumorigenic potential, especially in the background of impaired apoptosis and cell cycle check-point defects (Karantza-Wadsworth et al., 2007).
Role of p62
Loss of Atg5 and Atg7 in mouse livers results in accumulation of abnormal mitochondria, formation of inclusion bodies, liver injury, and development of benign hepatocellular adenomas (Komatsu et al., 2007; Inami et al., 2011; Takamura et al., 2011). p62 is a component of these inclusion bodies, which are cytoplasmic aggregates of ubiquitinated proteins commonly associated with pathological conditions, such as Alzheimer’s (Kuusisto et al., 2002), Huntington’s (Nagaoka et al., 2004), and Parkinson’s (Kuusisto et al., 2001) diseases, as well as alcoholic and non-alcoholic steatohepatitis (Zatloukal et al., 2002) and hepatocellular carcinoma (Zatloukal et al., 2002). Combined loss of p62 and autophagy in Atg7-/-; p62-/- livers abrogates inclusion body formation, alleviates hepatic injury and retards tumor progression compared to Atg7-/- deficiency alone (Komatsu et al., 2007; Takamura et al., 2011). On the contrary, p62 overexpression increases ROS and accelerates tumorigenesis in an autophagy-defective background, but not in an autophagy-competent one (Mathew et al., 2009), suggesting that the tumor-forming abilities of autophagy-deficient cells may be dependent on persistent and abnormal p62 accumulation, which is the result of combined p62 induction in response to defective autophagy-associated oxidative stress and impaired p62 elimination, again due to deficient autophagy. As explained below, p62 in turn activates prosurvival antioxidant cell responses, including the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway, but in aggregate form also leads to further ROS production, thereby maintaining a potentially tumorigenic vicious cycle.
p62/SQSTM1 activates the Nrf2 pathway in a non-canonical manner and likely modulates ROS levels as a consequence. Nrf2 is a transcription factor, which under normal growth conditions is constitutively targeted for degradation by the Kelch-like ECH-associated protein 1 (Keap1) complex (Taguchi et al., 2011). Upon induction of oxidative stress, Keap1 undergoes conformational changes leading to the release, stabilization, and translocation of Nrf2 into the nucleus, where it activates the transcription of genes under control of the antioxidant responsive element (ARE), which in turn function to restore physiological ROS levels (Taguchi et al., 2011). p62 competes with Nrf2 for binding sites on Keap1, resulting in Nrf2 stabilization and enabling its nuclear translocation and activation of target genes, including p62, thus resulting in a positive feedback loop (Jain et al., 2010; Komatsu et al., 2010; Lau et al., 2010). Indeed, under autophagy-deficient conditions, Keap1 is a component of inclusion bodies together with p62 and is, similar to p62, also degraded via autophagy (Jain et al., 2010; Komatsu et al., 2010). As expected, liver-specific Atg7 loss activates the Nrf2 pathway, while simultaneous p62 loss abrogates this activation (Komatsu et al., 2007; Inami et al., 2011). Importantly, similar to concurrent p62 and Atg7 loss, combined Nrf2 and Atg7 deficiency alleviates the cellular injury observed in Atg7-/- livers, whereas combined loss of Keap1 and Atg7 exacerbates hepatocyte damage (Komatsu et al., 2010). Taken together, these results indicate that liver injury due to autophagy defects is driven by p62 accumulation and its resultant activation of the Nrf2 pathway. In support of this hypothesis, p62 deletion in the human hepatocellular carcinoma cell line Jhh-5 diminishes its colony-forming ability in soft agar in conjunction with failure to activate Nrf2 (Inami et al., 2011). Whether Nrf2 activity is required for the formation of tumors in the context of autophagy deficiency and p62 accumulation in vivo remains to be investigated.
In addition to up-regulating the Nrf2 antioxidant response, p62 modulates ROS levels by also activating the nuclear factor kappa B (NF-κB), which is known to be induced by and also regulate ROS (Duran et al., 2008; Morgan and Liu, 2011). The absence of p62 delays tumor formation and reduces tumor burden in a tetracycline-inducible K-Ras model of lung tumorigenesis, and this effect is attributed to loss of NF-κB activity in the p62-/- background (Duran et al., 2008). Similarly, H-RasV12-transformed p62-/- MEFs, when compared to their wild-type counterparts, exhibit higher levels of apoptosis in association with increased JNK activity, which in turn is caused by elevated ROS levels (Duran et al., 2008). Treatment of H-RasV12-transformed p62-/- MEFs with the antioxidant butylated hydroxyanisole (BHA) restores cell viability, indicating that excessive ROS is responsible for the increase in cell death (Duran et al., 2008). Intriguingly, in an autophagy-deficient background, p62 aggregation and oligomerization impair its ability to activate the NF-κB pathway, thereby mimicking a functional loss of p62 with respect to this particular pathway, and may partly explain the increased ROS associated with autophagy deficiency (Mathew et al., 2009). Thus, p62 accumulation accompanying impaired autophagy fails to activate the NF-κB pathway, leading to increased ROS production, while it simultaneously activates the antioxidant Nrf2 pathway via Keap1 sequestration. The balance between increased ROS production and ROS tempering via the Nrf2 pathway may be an important player in tumor progression in the context of autophagy deficiency.
Inflammasome Activation
Inflammasomes are multi-protein complexes responsible for the processing of caspase-1 and the subsequent maturation and secretion of the proinflammatory cytokines interleukin IL-1β and IL-18 (Martinon et al., 2009). The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, pyrin domain-containing 3 (NLRP3) inflammasome consists of the NLR family member NLRP3, the adaptor apoptosis-associated speck-like (ASC) protein and pro-caspase-1, and is activated in response to diverse stimuli, such as bacteria, viruses, silica, asbestos, uric acid crystals, ATP, lysosomal damage, and impaired phagocytosis (Martinon et al., 2009; Gross et al., 2011). Proteolytic activity of the NLRP3 inflammasome requires two steps: (1) priming, which is characterized by the transcriptional up-regulation of NLRP3 and pro-IL-1β, and (2) activation, which involves the oligomerization of NLRP3 in response to various stimuli (Chen and Nunez, 2011; Gross et al., 2011). Interestingly, ROS are critical mediators of inflammasome activity, most likely at the priming step (Bauernfeind et al., 2011), since antioxidants attenuate IL-1β maturation in response to uric acid crystals (Petrilli et al., 2007; Dostert et al., 2008), asbestos (Dostert et al., 2008), and ATP (Cruz et al., 2007; Dostert et al., 2008). In agreement with the above findings, defective autophagy-induced mitochondrial ROS also lead to inflammasome activation (Nakahira et al., 2011; Zhou et al., 2011), and macrophages isolated from Atg7-/-, Atg16L1-/-, LC3B-/-, and Becn1+/- mice secrete elevated levels of IL-1β and IL-18 upon stimulation (Saitoh et al., 2008; Nakahira et al., 2011), thus intimately linking autophagy impairment to inflammasome activation and its resultant inflammatory response.
The connection between inflammasome-associated cytokine production and tumorigenesis is complex, as IL-1β and IL-18 have both pro- and anti-tumorigenic effects. In support of a tumor-promoting role, loss of IL-1β slows the progression of tumors in a model of chemical-induced carcinogenesis (Krelin et al., 2007), implantation of B16 melanoma cells in IL-1β-null mice prevents the formation of tumors as well as lung metastases (Vidal-Vanaclocha et al., 2000; Voronov et al., 2003), Lewis lung carcinoma cells overexpressing IL-1β exhibit accelerated rate of tumor formation accompanied by increased vasculature (Saijo et al., 2002), and IL-1β-overexpressing A459 cells form lung metastasis more efficiently than control cells, in a process inhibited by an antibody to IL-1β (Yano et al., 2003). Furthermore, implantation of mammary carcinoma 4T1 cells in IL-1 receptor-null (IL-1R-/-) mice slows tumor progression in association with immune suppression (Bunt et al., 2007), and fibroblast growth factor receptor-1 (FGFR-1)-mediated mammary tumorigenesis is dependent on the induction of IL-1β and inhibited by administration of anti-IL-1β antibody (Reed et al., 2009). Taken together, these studies implicate IL-1β in promotion of tumor cell proliferation, migration, angiogenesis, invasion, and inhibition of immune surveillance. Studies conducted on IL-18 indicate that IL-18 increases apoptosis and decreases metastasis in experimental models of bone metastasis (Ohtsuki et al., 1997; Nakata et al., 1999; Iwasaki et al., 2002); however, IL-18 has also been reported to increase angiogenesis (Park et al., 2001) and suppress immune surveillance (Terme et al., 2011).
While earlier studies indicated that IL-1β and IL-18, together with other components of the inflammasome, exacerbate colitis and promote colitis-associated cancer (Siegmund et al., 2001a,b; Sivakumar et al., 2002; Ishikura et al., 2003; Loher et al., 2004; Bauer et al., 2007), more recent evidence suggests that these molecules play a protective role by impeding colitis and progression to cancer (Takagi et al., 2003; Allen et al., 2010; Dupaul-Chicoine et al., 2010; Salcedo et al., 2010; Zaki et al., 2010a,b). The reason for this discrepancy is unclear, although it can be partly attributed to differences in gut-residing microflora and in experimental design (Zaki et al., 2011). Furthermore, components of the inflammasome, and in particular IL-1β, are required for the efficient recruitment of the immune system to attack tumor cells upon chemotherapeutic treatment (Ghiringhelli et al., 2009). In conclusion, modulation of tumor growth and survival by IL-1β and IL-18 is complex and likely context-dependent. Similarly, how inflammasome activation due to autophagy defects impacts tumorigenesis remains unclear and has understandably become a vigorously investigated subject.
Summary
The functional interactions between autophagy and ROS are elaborate (Figure 1) and their role in tumorigenesis is complex (Figure 2), and likely tissue- and context-dependent. Excess ROS up-regulate autophagy by multiple and at times counteracting mechanisms, and in turn this catabolic cellular process restores physiological ROS levels. Defective autophagy promotes the accumulation of damaged mitochondria and results in oxidative stress, which may be partly responsible for the tumor-forming abilities of autophagy-deficient cells by functioning as a genotoxic and mutagenic agent and by triggering secondary, non-cell autonomous effects, such as inflammation.
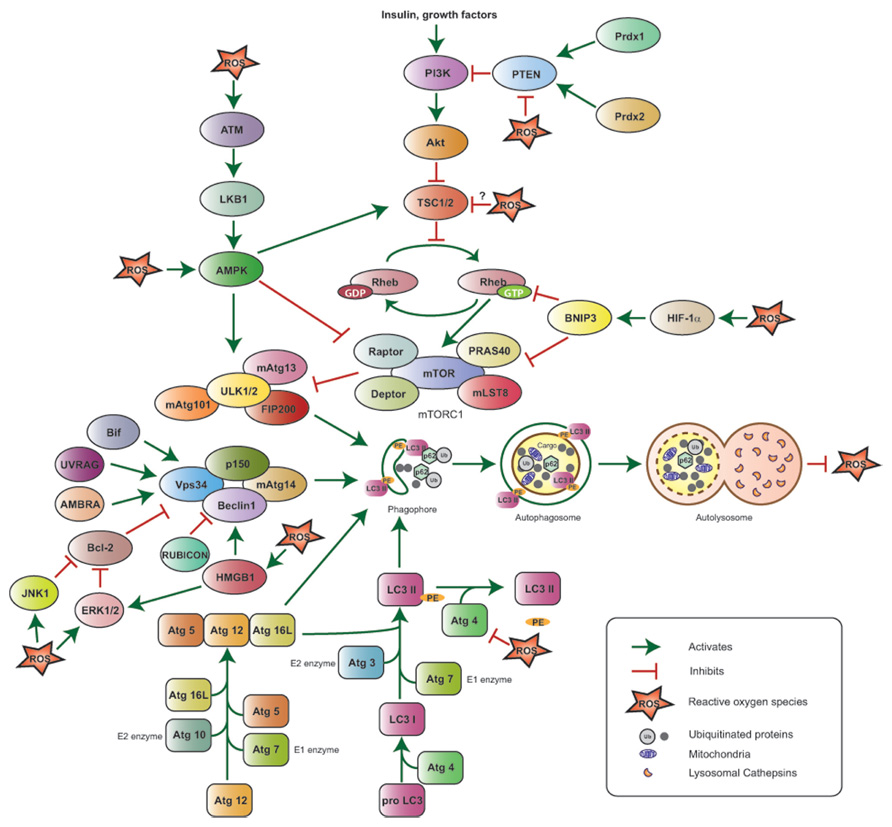
FIGURE 1. Reactive oxygen species-mediated autophagy regulation. The ULK1/2–FIP200–mAtg13 and Beclin1–Vps34–p150 protein complexes together with the two ubiquitin-like proteins Atg12 and LC3 (or Atg8) and their conjugating proteins, including Atg7 (E1-like enzyme), Atg10 (E2-like enzyme), and Atg3 (E2-like enzyme), are the primary regulators of autophagosome formation and, thus, autophagy induction. Atg4 cleaves Pro-LC3 at its C-terminal-end, Atg7 and Atg3 add phosphatidyl ethanolamine (PE), and LC3-PE is then incorporated into the autophagosome membrane; Atg4 can also cleave LC3-PE to release free LC3 and negatively impact autophagy. The PI3K–Akt–mTORC1 pathway suppresses autophagy by inhibiting the ULK1/2–FIP200–mAtg13 complex. PI3K is activated in response to hormones and growth factors and suppresses TSC1/2 activity, thus resulting in Akt and mTORC1 activation; the phosphatase PTEN antagonizes PI3K, while the peroxiredoxins Prdx1 and 2 maintain PTEN function by preventing its oxidation. mTORC1 is also negatively regulated by HIF-1α via BNIP3 induction and by ATM via LKB1 and AMPK. The Beclin1–Vps34–p150 complex is positively regulated by Bif-1, UVRAG, AMBRA, and HMGB1, and negatively regulated by Bcl-2 and RUBICON. ROS stimulate autophagy by activating HIF-1α, AMPK, ATM, HMGB1, JNK1, ERK1/2 and inhibiting the PE-cleaving activity of Atg4, without affecting its ability to process the C-terminal end of LC3. ROS inhibit PTEN and are hypothesized to negatively affect TSC1/2 activity, potentially inhibiting autophagy. Upon its induction, autophagy results in ROS suppression.
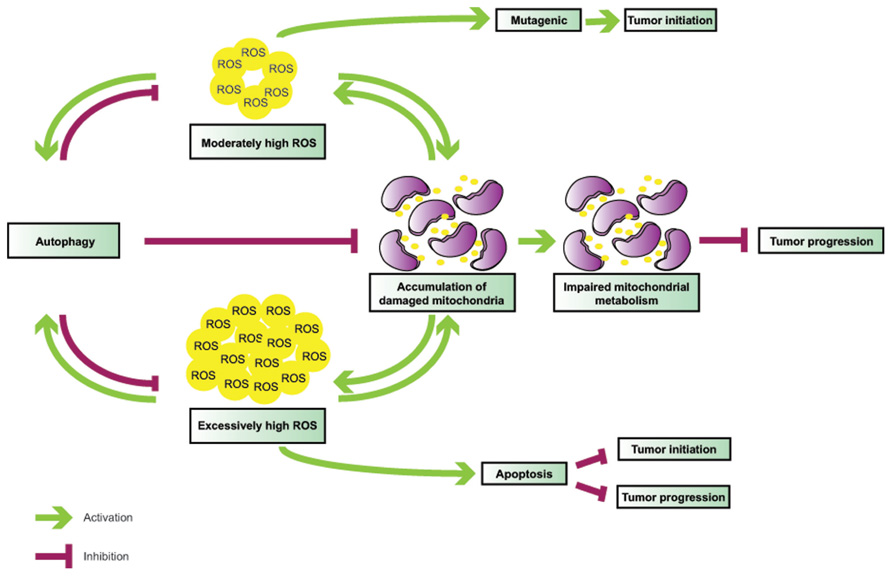
FIGURE 2. Interplay between ROS and autophagy in tumorigenesis. Autophagy is induced by ROS and functions to mitigate oxidative stress. Defective autophagy leads to ROS accumulation and mitochondrial damage; in turn, malfunctioning mitochondria become additional sources of ROS, thereby creating a vicious cycle. While moderately high ROS levels are mutagenic and likely mediate tumor initiation in association with autophagy defects, very high ROS levels may result in apoptosis, and, thus, inhibit both tumor initiation and progression.
The finding that Atg5-/- or Atg7-/- mouse livers give rise to benign adenomas, but not malignant hepatocellular carcinomas, is likely indicative of the opposing roles that autophagy may plays at different stages of tumorigenesis. Chronic impairment of autophagy may be detrimental to cellular health due to accumulation of aggregated proteins and damaged organelles, such as aging mitochondria, thus leading to steadily rising, protumorigenic levels of ROS over time. Since autophagy is one of the primary mechanisms by which cells moderate ROS, autophagy impairment can create a vicious cycle, where inability to temper slightly elevated ROS levels leads to further ROS production, which, if it exceeds a critical threshold, may trigger cell death, and thus, impede tumor progression. Accumulating evidence indicates that tumors driven by metabolically demanding oncogenes, such as mutant Ras and PyMT, also depend on autophagy. Indeed, oncogenic Ras-driven tumors rely on mitochondrial oxidative phosphorylation to meet their metabolic needs, and autophagy is critical for the supply of essential metabolic intermediates and the maintenance of mitochondrial health. In parallel to increased metabolic demands, potent oncogene activation is also associated with excess ROS production and, thus, autophagy may not only support cancer metabolism, but also function as a toxic ROS scavenger to again promote tumor cell survival and enable cancer progression.
In conclusion, the functional status of autophagy is intricately connected to intracellular ROS levels in complex ways, and these elaborate interactions likely impact tumorigenesis in context-specific manners that are worthy of further investigation and may provide novel insights into how to best pharmacologically modulate autophagy for cancer prevention and treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMBRA, activating molecule in beclin 1-regulated autophagy; AMP, adenosine monophosphate; AMPK, AMP activated protein kinase; ARE, antioxidant response element; ASC, apoptosis-associated speck-like protein containing a CARD; Atg 3, autophagy-related 3; Atg4C, autophagy-related 4C; Atg5, autophagy-related 5; Atg7, autophagy-related 7; Atg8/LC3, autophagy-related 8/microtubule-associated protein light chain 3; Atg10, autophagy-related 10; Atg12, autophagy-related 12; mAtg13, mammalian autophagy-related 13; Atg16L, autophagy-related 16-like; ATM, ataxia-telangiectasia mutated; ATP, adenosine triphosphate; Bcl-2, B cell CLL/lymphoma 2; Beclin1, coiled-coil myosin-like BCL2-interacting protein; BHA, butylated hydroxyanisole; Bif1, Bax interacting factor 1; BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3; BNIP3L/Nix, BCL2/adenovirus E1B interacting protein 3-like/NIP3-like protein X; CCCP, carbonyl cyanidem-chlorophenylhydrazone; Deptor, DEP domain containing MTOR-interacting protein; E1, ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; EGF, epidermal growth factor; ER, endoplasmic reticulum; ERK1/2, extracellular regulated kinase 1/2; Ero1, endoplasmic reticulum oxidoreductin-1; ETC, electron transport chain; FGFR-1, fibroblast growth factor receptor-1; FIP200, FAK family kinase-interacting protein of 200 kDa; FOXO1/O3, forkhead box protein O1/O3; GAP, GTPase activating protein; HIF-1α, hypoxia-inducible factor-1 alpha; HMGB1, high mobility group box 1; H2O2, hydrogen peroxide; iBMKs, immortalized baby mouse kidney cells; IL-1β, interleukin 1 beta; IL-18, interleukin 18; IL-1R, interleukin 1 receptor; iMMECs, immortalized mouse mammary epithelial cells; JNK, c-Jun N-terminal kinase 1; Keap1, Kelch-like ECH-associated protein 1; LKB1, liver kinase B1; MAPK, mitogen-activated protein kinase; MEFs, mouse embryonicfibroblasts; MFN1/2, mitofuscin 1/2; mLST8, MTOR-associated protein, LST8 homolog; MMTV, mouse mammary tumor virus; Mn-SOD, manganese superoxide dismutase; mTORC1, mammalian target of rapamycin 1; NAC, N-acetyl cysteine; NADPH oxidase, nicotinamide adenine dinucleotide phosphate-oxidase; NF-κB, nuclear factor kappa B; NLR, Nod-like receptor; NLRP3, NLR family, pyrin domain containing 3; NO, nitric oxide; Nrf2, nuclear factor erythroid 2-related factor 2; , superoxide anion; OH., hydroxyl radical; ONOO, peroxynitrite; p62/SQSTM1, sequestosome 1; p150, phosphatidylinositol 3-kinase-associated p150; PAO, phenylarsine oxide; Parkin, Parkinson protein, E3 ubiquitin protein ligase; PDGF, platelet-derived growth factor; PDK1, phosphoinositide-dependent kinase 1; PE, phosphatidyl ethanolamine; PERK, PRKR-like endoplasmic reticulum kinase; PH, Pleckstrin homology; PI3K/Vps34, phosphatidylinositol 3-kinase/vacuolar protein sorting 34; PINK1, PTEN-induced putative kinase 1; PIP2, phosphatidylinositol-4,5-phosphate; PIP3, phosphatidylinositol-3,4,5-phosphate; PKB/Akt, protein kinase B-alpha; PRAS40, proline-rich AKT substrate, 40 kDa; Prdx1/2, peroxiredoxin 1/2; PTEN, phosphatase and tensin homolog; PyMT, polyoma middle T; Rag, Ras-related GTP-binding protein; RAGE, receptor for advanced glycation end products; Raptor, regulatory associated protein of MTOR, complex 1; Rheb, Ras homolog enriched in brain; RNS, reactive nitrogen species; ROS, reactive oxygen species; RUBICON, RUN domain Beclin 1-interacting cysteine-rich-containing protein; Sesns, sestrins; SOD1, copper/zinc superoxide dismutase; SOD2, manganese superoxide dismutase; TLRs, Toll-like receptors; TSC 1/2, tuberous sclerosis 1/2; UBA, ubiquitin-associated; ULK1/2, Unc-51-like kinase 1/2; UVRAG, ultraviolet irradiation resistant-associated gene; xanthine oxidase.
References
Aita, V. M., Liang, X. H., Murty, V. V., Pincus, D. L., Yu, W., Cayanis, E., et al. (1999). Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59, 59–65.
Adcock, I. M., Caramori, G., and Barnes, P. J. (2011). Chronic obstructive pulmonary disease and lung cancer: new molecular insights. Respiration 81, 265–284.
Alexander, A., Cai, S. L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., et al. (2010). ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U.S.A. 107, 4153–4158.
Allen, I. C., TeKippe, E. M., Woodford, R. M., Uronis, J. M., Holl, E. K., Rogers, A. B., et al. (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056.
Arnhold, J., and Flemmig, J. (2010). Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 500, 92–106.
Avivar-Valderas, A., Salas, E., Bobrovnikova-Marjon, E., Diehl, J. A., Nagi, C., Debnath, J., et al. (2011). PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 31, 3616–3629.
Bauer, C., Loher, F., Dauer, M., Mayer, C., Lehr, H. A., Schonharting, M., et al. (2007). The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig. Dis. Sci. 52, 1642–1652.
Bauernfeind, F., Bartok, E., Rieger, A., Franchi, L., Nunez, G., and Hornung, V. (2011). Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617.
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouyssegur, J., et al. (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581.
Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614.
Bruick, R. K. (2000). Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. U.S.A. 97, 9082–9087.
Budanov, A. V. (2011). Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 15, 1679–1690.
Bunt, S. K., Yang, L., Sinha, P., Clements, V. K., Leips, J., and Ostrand-Rosenberg, S. (2007). Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 67, 10019–10026.
Byun, Y. J., Kim, S. K., Kim, Y. M., Chae, G. T., Jeong, S. W., and Lee, S. B. (2009). Hydrogen peroxide induces autophagic cell death in C6 glioma cells via BNIP3-mediated suppression of the mTOR pathway. Neurosci. Lett. 461, 131–135.
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
Cao, J., Schulte, J., Knight, A., Leslie, N. R., Zagozdzon, A., Bronson, R., et al. (2009). Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 28, 1505–1517.
Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M., et al. (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138.
Chen, G. Y., and Nunez, G. (2011). Inflammasomes in intestinal inflammation and cancer. Gastroenterology 141, 1986–1999.
Cheng, Y., Qiu, F., Ye, Y. C., Guo, Z. M., Tashiro, S., Onodera, S., et al. (2009). Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J. 276, 1291–1306.
Choi, S. L., Kim, S. J., Lee, K. T., Kim, J., Mu, J., Birnbaum, M. J., et al. (2001). The regulation of AMP-activated protein kinase by H(2)O(2). Biochem. Biophys. Res. Commun. 287, 92–97.
Cruz, C. M., Rinna, A., Forman, H. J., Ventura, A. L., Persechini, P. M., and Ojcius, D. M. (2007). ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 282, 2871–2879.
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301.
Dupaul-Chicoine, J., Yeretssian, G., Doiron, K., Bergstrom, K. S., McIntire, C. R., LeBlanc, P. M., et al. (2010). Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378.
Duran, A., Linares, J. F., Galvez, A. S., Wikenheiser, K., Flores, J. M., Diaz-Meco, M. T., et al. (2008). The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 13, 343–354.
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677.
Ebato, C., Uchida, T., Arakawa, M., Komatsu, M., Ueno, T., Komiya, K., et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332.
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461.
Emerling, B. M., Weinberg, F., Snyder, C., Burgess, Z., Mutlu, G. M., Viollet, B., et al. (2009). Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 46, 1386–1391.
Filomeni, G., Desideri, E., Cardaci, S., Rotilio, G., and Ciriolo, M. R. (2010). Under the ROS...thiol network is the principal suspect for autophagy commitment. Autophagy 6, 999–1005.
Fimia, G. M., Stoykova, A., Romagnoli, A., Giunta, L., Di Bartolomeo, S., Nardacci, R., et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125.
Fransen, M., Nordgren, M., Wang, B., and Apanasets, O. (2012). Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim. Biophys. Acta 1822, 1363–1373.
Fukuto, J. M., Carrington, S. J., Tantillo, D. J., Harrison, J. G., Ignarro, L. J., Freeman, B. A., et al. (2012). Small molecule signaling agents: the integrated chemistry, and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 25, 769–793.
Ganley, I. G., Lam du, H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009). ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305.
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870.
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131.
Ghiringhelli, F., Apetoh, L., Tesniere, A., Aymeric, L., Ma, Y., Ortiz, C., et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178.
Goh, J., Enns, L., Fatemie, S., Hopkins, H., Morton, J., Pettan-Brewer, C., et al. (2011). Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 11, 191. doi: 10.1186/1471-2407-11-191
Gonenc, A., Ozkan, Y., Torun, M., and Simsek, B. (2001). Plasma malondialdehyde (MDA) levels in breast and lung cancer patients. J. Clin. Pharm. Ther. 26, 141–144.
Gough, D. R., and Cotter, T. G. (2011). Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2, e213.
Gross, O., Thomas, C. J., Guarda, G., and Tschopp, J. (2011). The inflammasome: an integrated view. Immunol. Rev. 243, 136–151.
Guo, J. Y., Chen, H. Y., Mathew, R., Fan, J., Strohecker, A. M., Karsli-Uzunbas, G., et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470.
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., et al. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408.
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226.
Hamacher-Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., et al. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 14, 146–157.
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, A. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104.
Hara, T., Takamura, A., Kishi, C., Iemura, S., Natsume, T., Guan, J. L., et al. (2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
Hernandez-Aya, L. F., and Gonzalez-Angulo, A. M. (2011). Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist 16, 404–414.
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991.
Huang, Y. L., Sheu, J. Y., and Lin, T. H. (1999). Association between oxidative stress and changes of trace elements in patients with breast cancer. Clin. Biochem. 32, 131–136.
Inami, Y., Waguri, S., Sakamoto, A., Kouno, T., Nakada, K., Hino, O., c (2011). Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193, 275–284.
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590.
Ishikura, T., Kanai, T., Uraushihara, K., Iiyama, R., Makita, S., Totsuka, T., et al. (2003). Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 18, 960–969.
Iwasaki, T., Yamashita, K., Tsujimura, T., Kashiwamura, S., Tsutsui, H., Kaisho, T., et al. (2002). Interleukin-18 inhibits osteolytic bone metastasis by human lung cancer cells possibly through suppression of osteoclastic bone-resorption in nude mice. J. Immunother. 25(Suppl. 1), S52–S60.
Jain, A., Lamark, T., Sjottem, E., Larsen, K. B., Awuh, J. A., Overvatn, A., et al. (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 285, 22576–22591.
Jung, H. S., Chung, K. W., Won Kim, J., Kim, J., Komatsu, M., Tanaka, K., et al. (2008). Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 8, 318–324.
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003.
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728.
Kabeya, Y., Mizushima, N., Yamamoto, A., Oshitani-Okamoto, S., Ohsumi, Y., and Yoshimori, T. (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812.
Kang, R., Tang, D., Livesey, K. M., Schapiro, N. E., Lotze, M. T., and Zeh, H. J. III. (2011a). The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 15, 2175–2184.
Kang, R., Tang, D., Lotze, M. T., and Zeh, H. J. III. (2011b). RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy 7, 442–444.
Karantza-Wadsworth, V., Patel, S., Kravchuk, O., Chen, G., Mathew, R., Jin, S., et al. (2007). Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21, 1621–1635.
Karihtala, P., and Soini, Y. (2007). Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 115, 81–103.
Khandrika, L., Kumar, B., Koul, S., Maroni, P., and Koul, H. K. (2009). Oxidative stress in prostate cancer. Cancer Lett. 282, 125–136.
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335.
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141.
Kim, M. J., Woo, S. J., Yoon, C. H., Lee, J. S., An, S., Choi, Y. H., et al. (2011). Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286, 12924–12932.
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434.
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163.
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223.
Kongara, S., Kravchuk, O., Teplova, I., Lozy, F., Schulte, J., Moore, D., et al. (2010). Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol. Cancer Res. 8, 873–884.
Krelin, Y., Voronov, E., Dotan, S., Elkabets, M., Reich, E., Fogel, M., et al. (2007). Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67, 1062–1071.
Kubli, D. A., Quinsay, M. N., Huang, C., Lee, Y., and Gustafsson, A. B. (2008). Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 295, H2025–H2031.
Kumaraguruparan, R., Subapriya, R., Viswanathan, P., and Nagini, S. (2002). Tissue lipid peroxidation and antioxidant status in patients with adenocarcinoma of the breast. Clin. Chim. Acta 325, 165–170.
Kundu, M., Lindsten, T., Yang, C. Y., Wu, J., Zhao, F., Zhang, J., et al. (2008). Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112, 1493–1502.
Kuusisto, E., Salminen, A., and Alafuzoff, I. (2001). Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12, 2085–2090.
Kuusisto, E., Salminen, A., and Alafuzoff, I. (2002). Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol. Appl. Neurobiol. 28, 228–237.
Kwon, J., Lee, S. R., Yang, K. S., Ahn, Y., Kim, Y. J., Stadtman, E. R., et al. (2004). Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. U.S.A. 101, 16419–16424.
Lau, A., Wang, X. J., Zhao, F., Villeneuve, N. F., Wu, T., Jiang, T., et al. (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285.
Lee, S. R., Yang, K. S., Kwon, J., Lee, C., Jeong, W., and Rhee, S. G. (2002). Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342.
Lee, J., Giordano, S., and Zhang, J. (2012). Autophagy, mitochondria, and oxidative stress: cross-talk, and redox signalling. Biochem. J. 441, 523–540.
Lee, J. H., Budanov, A. V., Park, E. J., Birse, R., Kim, T. E., Perkins, G. A., et al. (2010). Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327, 1223–1228.
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5.
Li, Y., Wang, Y., Kim, E., Beemiller, P., Wang, C. Y., Swanson, J., et al. (2007). Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 282, 35803–35813.
Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., et al. (2006). Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699.
Liang, C. C., Wang, C., Peng, X., Gan, B., and Guan, J. L. (2010). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 285, 3499–3509.
Liu, F., Lee, J. Y., Wei, H., Tanabe, O., Engel, J. D., Morrison, S. J., et al. (2010). FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 116, 4806–4814.
Lock, R., Roy, S., Kenific, C. M., Su, J. S., Salas, E., Ronen, S. M., et al. (2011). Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178.
Loher, F., Bauer, C., Landauer, N., Schmall, K., Siegmund, B., Lehr, H. A., et al. (2004). The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharmacol. Exp. Ther. 308, 583–590.
Lotze, M. T., and Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342.
Marchi, S., Giorgi, C., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., et al. (2012). Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 329635.
Marino, G., Salvador-Montoliu, N., Fueyo, A., Knecht, E., Mizushima, N., and Lopez-Otin, C. (2007). Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 282, 18573–18583.
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265.
Mathew, R., Kongara, S., Beaudoin, B., Karp, C. M., Bray, K., Degenhardt, K., et al. (2007). Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381.
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075.
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221.
McClung, J. M., Judge, A. R., Powers, S. K., and Yan, Z. (2010). p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 298, C542-549.
Meng, T. C., Fukada, T., and Tonks, N. K. (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9, 387–399.
Mercer, C. A., Kaliappan, A., and Dennis, P. B. (2009). A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5, 649–662.
Miao, L., and St Clair, D. K. (2009). Regulation of superoxide dismutase genes: implications in disease. Free Radic. Biol. Med. 47, 344–356.
Mizushima, N., Sugita, H., Yoshimori, T., and Ohsumi, Y. (1998). A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892.
Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., et al. (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668.
Mizushima, N., Kuma, A., Kobayashi, Y., Yamamoto, A., Matsubae, M., Takao, T., et al. (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679–1688.
Moller, P., Folkmann, J. K., Forchhammer, L., Brauner, E. V., Danielsen, P. H., Risom, L., et al. (2008). Air pollution, oxidative damage to DNA, and carcinogenesis. Cancer Lett. 266, 84–97.
Morgan, M. J., and Liu, Z. G. (2011). Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 21, 103–115.
Mortensen, M., Ferguson, D. J., Edelmann, M., Kessler, B., Morten, K. J., Komatsu, M., et al. (2010). Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 832–837.
Nagaoka, U., Kim, K., Jana, N. R., Doi, H., Maruyama, M., Mitsui, K., et al. (2004). Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 91, 57–68.
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230.
Nakai, A., Yamaguchi, O., Takeda, T., Higuchi, Y., Hikoso, S., Taniike, M., et al. (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624.
Nakata, A., Tsujimura, T., Sugihara, A., Okamura, H., Iwasaki, T., Shinkai, K., et al. (1999). Inhibition by interleukin 18 of osteolytic bone metastasis by human breast cancer cells. Anticancer Res. 19, 4131–4138.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803.
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al,. (2010a). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. doi: 10.1371/journal.pbio.1000298
Narendra, D., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010b). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106.
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658.
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51.
Nowsheen, S., Wukovich, R. L., Aziz, K., Kalogerinis, P. T., Richardson, C. C., Panayiotidis, M. I., et al. (2009). Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 674, 131–136.
Ohtsuki, T., Micallef, M. J., Kohno, K., Tanimoto, T., Ikeda, M., and Kurimoto, M. (1997). Interleukin 18 enhances Fas ligand expression and induces apoptosis in Fas-expressing human myelomonocytic KG-1 cells. Anticancer Res. 17, 3253–3258.
Okatsu, K., Saisho, K., Shimanuki, M., Nakada, K., Shitara, H., Sou, Y. S., et al. (2010). p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900.
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145.
Papandreou, I., Lim, A. L., Laderoute, K., and Denko, N. C. (2008). Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 15, 1572–1581.
Park, C. C., Morel, J. C., Amin, M. A., Connors, M. A., Harlow, L. A., and Koch, A. E. (2001). Evidence of IL-18 as a novel angiogenic mediator. J. Immunol. 167, 1644–1653.
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589.
Polat, M. F., Taysi, S., Gul, M., Cikman, O., Yilmaz, I., Bakan, E., et al. (2002). Oxidant/antioxidant status in blood of patients with malignant breast tumour and benign breast disease. Cell Biochem. Funct. 20, 327–331.
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5, e10054. doi: x 10.1371/journal.pone.0010054
Pua, H. H., Guo, J., Komatsu, M., and He, Y. W. (2009). Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 182, 4046–4055.
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, 1809–1820.
Quan, W., Lim, Y. M., and Lee, M. S. (2012). Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic beta-cells. Exp. Mol. Med. 44, 81–88.
Rakovic, A., Grunewald, A., Kottwitz, J., Bruggemann, N., Pramstaller, P. P., Lohmann, K., et al. (2011). Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE 6, e16746. doi: 10.1371/journal.pone.0016746
Reed, J. R., Leon, R. P., Hall, M. K., and Schwertfeger, K. L. (2009). Interleukin-1beta and fibroblast growth factor receptor 1 cooperate to induce cyclooxygenase-2 during early mammary tumourigenesis. Breast Cancer Res. 11, R21.
Saijo, Y., Tanaka, M., Miki, M., Usui, K., Suzuki, T., Maemondo, M., et al. (2002). Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J. Immunol. 169, 469–475.
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268.
Sakaguchi, S., Takahashi, S., Sasaki, T., Kumagai, T., and Nagata, K. (2011). Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab. Pharmacokinet. 26, 30–46.
Salcedo, R., Worschech, A., Cardone, M., Jones, Y., Gyulai, Z., Dai, R. M., et al. (2010). MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 207, 1625–1636.
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235.
Sarbassov, D. D., and Sabatini, D. M. (2005). Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 280, 39505–39509.
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760.
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505.
Semenza, G. L. (2011). Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 1813, 1263–1268.
Sener, D. E., Gonenc, A., Akinci, M., and Torun, M. (2007). Lipid peroxidation and total antioxidant status in patients with breast cancer. Cell Biochem. Funct. 25, 377–382.
Sengupta, A., Molkentin, J. D., Paik, J. H., DePinho, R. A., and Yutzey, K. E. (2011). FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 286, 7468–7478.
Severi, T., van Malenstein, H., Verslype, C., and van Pelt, J. F. (2010). Tumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 31, 1409–1420.
Siegmund, B., Fantuzzi, G., Rieder, F., Gamboni-Robertson, F., Lehr, H. A., Hartmann, G., et al. (2001a). Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1264–R1273.
Siegmund, B., Lehr, H. A., Fantuzzi, G., and Dinarello, C. A. (2001b). IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 98, 13249–13254.
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388.
Sivakumar, P. V., Westrich, G. M., Kanaly, S., Garka, K., Born, T. L., Derry, J. M., et al. (2002). Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 50, 812–820.
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Sridhar, S., Botbol, Y., Macian, F., and Cuervo, A. M. (2012). Autophagy and disease: always two sides to a problem. J. Pathol. 226, 255–273.
Sun, Q., Fan, W., Chen, K., Ding, X., Chen, S., and Zhong, Q. (2008). Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 105, 19211–19216.
Taguchi, K., Motohashi, H., and Yamamoto, M. (2011). Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140.
Takagi, H., Kanai, T., Okazawa, A., Kishi, Y., Sato, T., Takaishi, H., et al. (2003). Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 38, 837–844.
Takahashi, Y., Coppola, D., Matsushita, N., Cualing, H. D., Sun, M., Sato, Y., et al. (2007). Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151.
Takamura, A., Komatsu, M., Hara, T., Sakamoto, A., Kishi, C., Waguri, S., et al. (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800.
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775.
Tang, D., Kang, R., Cheh, C. W., Livesey, K. M., Liang, X., Schapiro, N. E., et al. (2010a). HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310.
Tang, D., Kang, R., Livesey, K. M., Cheh, C. W., Farkas, A., Loughran, P., et al. (2010b). Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892.
Tanida, I., Tanida-Miyake, E., Ueno, T., and Kominami, E. (2001). The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 276, 1701–1706.
Tanida, I., Tanida-Miyake, E., Komatsu, M., Ueno, T., and Kominami, E. (2002). Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 277, 13739–13744.
Tanida, I., Sou, Y. S., Ezaki, J., Minematsu-Ikeguchi, N., Ueno, T., and Kominami, E. (2004). HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-asso-ciated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276.
Tavender, T. J., and Bulleid, N. J. (2010). Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox Signal. 13, 1177–1187.
Terme, M., Ullrich, E., Aymeric, L., Meinhardt, K., Desbois, M., Delahaye, N., et al. (2011). IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 71, 5393–5399.
Thapa, D., and Ghosh, R. (2012). Antioxidants for prostate cancer chemoprevention: challenges and opportunities. Biochem. Pharmacol. 83, 1319–1330.
Vera-Ramirez, L., Sanchez-Rovira, P., Ramirez-Tortosa, M. C., Ramirez-Tortosa, C. L., Granados-Principal, S., Lorente, J. A., et al. (2011). Free radicals in breast carcinogenesis, breast cancer progression and cancer stem cells. Biological bases to develop oxidative-based therapies. Crit. Rev. Oncol. Hematol. 80, 347–368.
Valko, M., Rhodes, C. J., Moncol, J., Izakovic, M., and Mazur, M. (2006). Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 160, 1–40.
Vidal-Vanaclocha, F., Fantuzzi, G., Mendoza, L., Fuentes, A. M., Anasagasti, M. J., Martin, J., et al. (2000). IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl. Acad. Sci. U.S.A. 97, 734–739.
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383.
Voronov, E., Shouval, D. S., Krelin, Y., Cagnano, E., Benharroch, D., Iwakura, Y., et al. (2003). IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 2645–2650.
Wei, H., Wei, S., Gan, B., Peng, X., Zou, W., and Guan, J. L. (2011). Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 25, 1510–1527.
Weydert, C. J., Waugh, T. A., Ritchie, J. M., Iyer, K. S., Smith, J. L., Li, L., et al. (2006). Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic. Biol. Med. 41, 226–237.
Wong, C. H., Iskandar, K. B., Yadav, S. K., Hirpara, J. L., Loh, T., and Pervaiz, S. (2010). Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 5, e9996. doi: 10.1371/journal.pone.0009996
Wu, H., Wang, M. C., and Bohmann, D. (2009). JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy. Mech. Dev. 126, 624–637.
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131.
Yang, S., Wang, X., Contino, G., Liesa, M., Sahin, E., Ying, H., et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729.
Yano, S., Nokihara, H., Yamamoto, A., Goto, H., Ogawa, H., Kanematsu, T., et al. (2003). Multifunctional interleukin-1beta promotes metastasis of human lung cancer cells in SCID mice via enhanced expression of adhesion-, invasion- and angiogenesis-related molecules. Cancer Sci. 94, 244–252.
Yeh, C. C., Hou, M. F., Tsai, S. M., Lin, S. K., Hsiao, J. K., Huang, J. C., et al. (2005). Superoxide anion radical, lipid peroxides and antioxidant status in the blood of patients with breast cancer. Clin. Chim. Acta 361, 104–111.
Yoshida, S., Hong, S., Suzuki, T., Nada, S., Mannan, A. M., Wang, J., et al. (2011). Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 286, 32651–32660.
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14.
Yue, Z., Jin, S., Yang, C., Levine, A. J., and Heintz, N. (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U.S.A. 100, 15077–15082.
Zaki, M. H., Boyd, K. L., Vogel, P., Kastan, M. B., Lamkanfi, M., and Kanneganti, T. D. (2010a). The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391.
Zaki, M. H., Vogel, P., Body-Malapel, M., Lamkanfi, M., and Kanneganti, T. D. (2010b). IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920.
Zaki, M. H., Lamkanfi, M., and Kanneganti, T. D. (2011). The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179.
Zatloukal, K., Stumptner, C., Fuchsbichler, A., Heid, H., Schnoelzer, M., Kenner, L., et al. (2002). p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160, 255–263.
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903.
Zhang, J., Randall, M. S., Loyd, M. R., Dorsey, F. C., Kundu, M., Cleveland, J. L., et al. (2009). Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 114, 157–164.
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225.
Keywords: autophagy, ROS, oxidative stress, cancer, p62, inflammation
Citation: Kongara S and Karantza V (2012) The interplay between autophagy and ROS in tumorigenesis. Front. Oncol. 2:171. doi: 10.3389/fonc.2012.00171
Received: 23 August 2012; Accepted: 30 October 2012;
Published online: 21 November 2012.
Edited by:
Carola A. Neumann, Medical University of South Carolina, USAReviewed by:
Daniel C. Hoessli, International Center for Chemical and Biological Sciences, SwitzerlandCatherine Brenner, Institut National de la Santé et de la Recherche Médicale - University of Paris Sud, France
Copyright: © 2012 Kongara and Karantza. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Vassiliki Karantza, The Cancer Institute of New Jersey, 195 Little Albany Street, Room 3550, New Brunswick, NJ 08903, USA. e-mail:karantva@umdnj.edu