
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 29 November 2012
Sec. Molecular and Cellular Oncology
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00164
This article is part of the Research Topic Mitochondrial proteins (e.g. VDAC, Bcl-2, HK, ANT2...) as major control points in oncology View all 8 articles
 Varda Shoshan-Barmatz1,2* and Dario Mizrachi1,2
Varda Shoshan-Barmatz1,2* and Dario Mizrachi1,2Here, we review current evidence pointing to the function of VDAC1 in cell life and death, and highlight these functions in relation to cancer. Found at the outer mitochondrial membrane, VDAC1 assumes a crucial position in the cell, controlling the metabolic cross-talk between mitochondria and the rest of the cell. Moreover, its location at the boundary between the mitochondria and the cytosol enables VDAC1 to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities. As a metabolite transporter, VDAC1 contributes to the metabolic phenotype of cancer cells. This is reflected by VDAC1 over-expression in many cancer types, and by inhibition of tumor development upon silencing VDAC1 expression. Along with regulating cellular energy production and metabolism, VDAC1 is also a key protein in mitochondria-mediated apoptosis, participating in the release of apoptotic proteins and interacting with anti-apoptotic proteins. The involvement of VDAC1 in the release of apoptotic proteins located in the inter-membranal space is discussed, as is VDAC1 oligomerization as an important step in apoptosis induction. VDAC also serves as an anchor point for mitochondria-interacting proteins, some of which are also highly expressed in many cancers, such as hexokinase (HK), Bcl2, and Bcl-xL. By binding to VDAC, HK provides both metabolic benefit and apoptosis-suppressive capacity that offers the cell a proliferative advantage and increases its resistance to chemotherapy. VDAC1-based peptides that bind specifically to HK, Bcl2, or Bcl-xL abolished the cell’s abilities to bypass the apoptotic pathway. Moreover, these peptides promote cell death in a panel of genetically characterized cell lines derived from different human cancers. These and other functions point to VDAC1 as a rational target for the development of a new generation of therapeutics.
Research over the past decade has extended the prevailing view of the mitochondrion to include functions well beyond the critical bioenergetics role of supplying ATP to include cell signaling events, inter-organellar communication, aging, cell proliferation, a target in disease, and apoptosis. Mitochondria thus play a central role in the regulation of apoptosis and serve as the venue for cellular decisions leading to cell life or death. One of the mitochondrial proteins controlling cell life and death is the voltage-dependent anion channel (VDAC), also known as mitochondrial porin. VDAC, located in the mitochondrial outer membrane, functions as gatekeeper for the entry and exit of mitochondrial metabolites, thereby controlling cross-talk between mitochondria and the rest of the cell. VDAC is also a key player in mitochondria-mediated apoptosis. Thus, in addition to regulating the metabolic and energetic functions of mitochondria, VDAC appears to act as a convergence point for a variety of cell survival and cell death signals, mediated by its association with various ligands and proteins. The focus of this review will be on the central role of VDAC in cell life and death, addressing VDAC function in the regulation of mitochondria-mediated apoptosis, with an emphasis on structure–function relationships. Understanding VDAC structure–function relationships is critical for deciphering how this channel can perform such a variety of functions, all important for cell life and death. Finally, this review will also provide insight into VDAC function in Ca2+ homeostasis, protection against oxidative stress, regulation of apoptosis, and involvement in several diseases, as well as its role in the action of different drugs.
In mammals, three homologous genes encode three VDAC isoforms, namely VDAC1, VDAC2, and VDAC3 (Shoshan-Barmatz et al., 2010). The three proteins have similar molecular weights (30–35 kDa), each shares approximately 70% identity, and all three can be found in most tissues, albeit in different amounts, with the most abundant sub-type being VDAC1 and the least common form being VDAC3 (Cesar Mde and Wilson, 2004; Yamamoto et al., 2006; De Pinto et al., 2010a; Shoshan-Barmatz et al., 2010; Messina et al., 2012). VDAC1 and VDAC2 but not VDAC3 can form a channel upon reconstitution into an artificial lipid bilayer, with VDAC1 showing voltage-gated high conductance channel properties (see Channel Activity of VDAC1). VDAC2 also presents normal gating activity, while VDAC3 does not insert readily into membranes and generally does not gate well, even at high membrane potentials (up to 80 mV; Sampson et al., 1997; Xu et al., 1999).
The specific role of each VDAC isoform remains unclear, although evidence indicates that they serve different physiologic functions (Sampson et al., 1997; Xu et al., 1999; Wu et al., 1999). For example, the absence of VDAC1 caused multiple defects in respiratory complex activities in both skeletal and cardiac muscle, while in VDAC3-deficient mice, these defects are restricted to the heart (Anflous-Pharayra et al., 2010), suggesting that in vivo, these two isoforms fulfill distinct physiologic roles. In mice, vdac1 and vdac2 deletion reduces respiratory capacity (Wu et al., 1999), the absence of VDAC3 causes male sterility, and a lack of both VDAC1 and VDAC3 causes inhibited growth (Sampson et al., 2001). Furthermore, it was demonstrated that VDAC1- and VDAC3-lacking mice show deficits in learning behavior and synaptic plasticity (Weeber et al., 2002). VDAC3-lacking mice were male-infertile because their mitochondria and the axoneme of their sperm are structurally altered (Sampson et al., 2001). Finally, vdac1- and vdac3-deficient mice are viable, whereas embryos with a homozygous deletion of vdac2 die during development (Cheng et al., 2003).
VDAC1 interacts with different proteins and factors, such as hexokinase (HK; Abu-Hamad et al., 2008) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Tarze et al., 2007), while biochemical data indicate that VDAC1 but not VDAC2 binds HK (Blachly-Dyson et al., 1993). This, however, has been questioned (Azoulay-Zohar and Aflalo, 1999). Lately, it was demonstrated that HK-I and VDAC3 exhibit a higher degree of mitochondrial co-localization than does HK-I with either VDAC1 or VDAC2 (Neumann et al., 2010).
Large proteomic surveys and other studies have shown that all three VDAC isoforms are subject to both phosphorylation and acetylation at multiple sites (Distler et al., 2007; Wang et al., 2008; Choudhary et al., 2009; Gauci et al., 2009; Menzel et al., 2009; Kerner et al., 2012). Analysis of the amino acid sequence of VDAC1 showed that the first methionine is deleted, while the second amino acid, an alanine, is acetylated (Kayser et al., 1989; Gauci et al., 2009). Among the other post-translation modifications VDAC1 undergoes are phosphorylation of serine, threonine, and tyrosine residues (Distler et al., 2007; Kerner et al., 2012) and acetylation of lysines (Kim et al., 2006; Schwer et al., 2009; Zhao et al., 2010; Yang et al., 2011). Recently glycogen synthase kinase 3 (GSK3)-mediated VDAC phosphorylation was reported, allowing for control of outer mitochondrial membrane (OMM) permeabilization in hepatosteatosis (Martel et al., 2012). Currently, the effects of these modifications on VDAC activity are not clear.
VDAC is localized to the OMM of all eukaryotes (Benz, 1994), where it assumes a crucial position in the cell, serving as the main interface between mitochondrial and cellular metabolisms. VDAC is permeable to uncharged molecules up to ∼5,000 Da in the open configuration, mediating the flux of ions, nucleotides and other metabolites across the OMM (Shoshan-Barmatz et al., 2010; Figure 1). In keeping with its two-way trafficking role, VDAC1 enables substrates, including pyruvate, malate, succinate, and NADH, to enter the mitochondria and mediates the exit of newly formed molecules, such as hemes (Shoshan-Barmatz et al., 2010). Hence, down-regulation of VDAC1 expression results in reduced metabolite exchange between mitochondria and the cytosol, making VDAC1 essential for energy production and cell growth (Abu-Hamad et al., 2006). Similarly, alterations in mitochondrial function are linked to VDAC closure, which limits the normal flow of metabolites in and out of mitochondria (Vander Heiden et al., 2000; Holmuhamedov and Lemasters, 2009). VDAC1, at the OMM, is also involved in the entry and exit of Ca2+ (see VDAC1 Transport of Ca2+ and Function in ER-mitochondria Cross-talk). VDAC, furthermore, functions in cholesterol transport across the OMM (Rone et al., 2009). Indeed, VDAC has been proposed to be a necessary component of a protein complex involved in mitochondrial membrane cholesterol distribution and transport and to play an important role in altered cholesterol synthesis and transport in Morris hepatoma cells (Campbell and Chan, 2008).
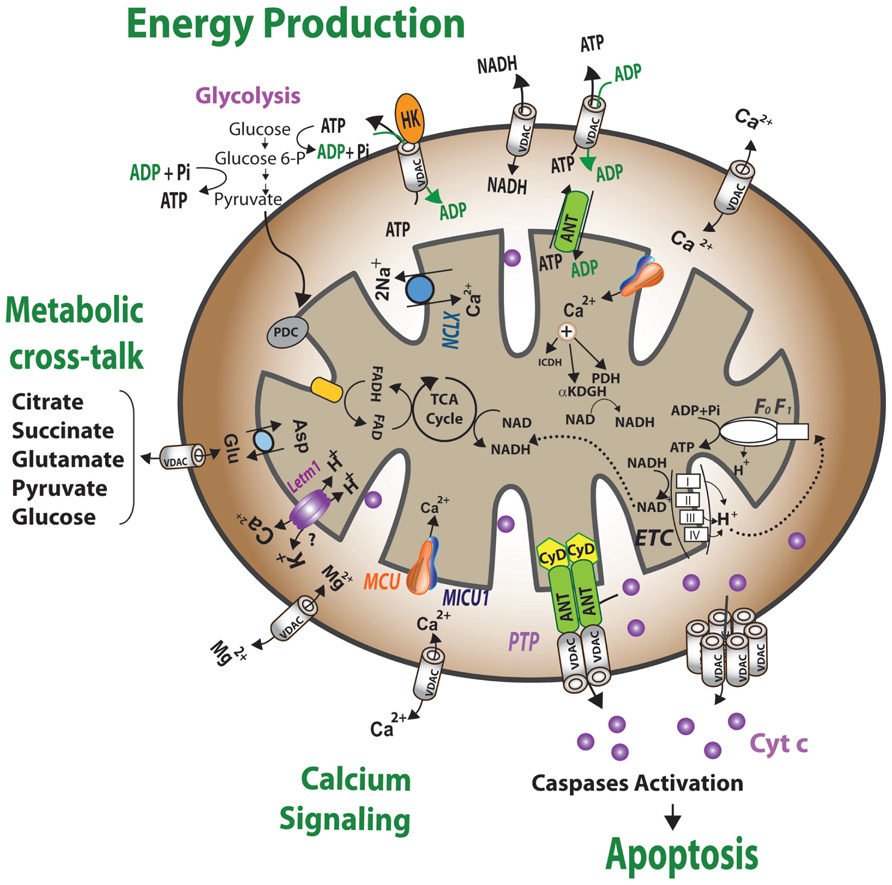
FIGURE 1. Schematic representation of VDAC1 as a multi-functional channel and convergence point for a variety of cell survival and cell death signals. The various functions of VDAC1 include control of the metabolic cross-talk between the mitochondria and the rest of the cell, cellular energy homeostasis by transporting ATP/ADP and NADH between the inter-membrane space and the cytosol and by binding HK, signaling by transporting Ca2+, ROS release to the cytosol and apoptosis, both by binding to the apoptosis regulatory proteins, Bcl-2 family and HK. Also presented are the Ca2+ influx and efflux transport systems in the outer and inner mitochondrial membranes and Ca2+-mediated regulation of the tricarboxylic acid (TCA) cycle. The activation of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH) and α-ketoglutarate dehydrogenase (αKGDH) by intra-mitochondrial Ca2+, leading to enhanced activity of the TCA cycle, is shown. The electron transport chain (ETC) and the ATP synthase (FoF1) are also presented. VDAC in the OMM is presented as a Ca2+ channel. In the IMM, the uptake of Ca2+ into the matrix is mediated by a Ca2+-selective transporter, the mitochondrial Ca2+ uniporter (MCU), regulated by a calcium-sensing accessory subunit (MICU1). Ca2+ efflux is mediated by NCLX, a Na+/Ca2+ exchanger. High levels of matrix Ca2+ accumulation trigger the opening of the PTP, a fast Ca2+ release channel. Molecular fluxes are indicated by arrows.
Given its location at the boundary between the mitochondria and the cytosol, VDAC1 is able to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities. VDAC1 interacts with HK and creatine kinase to convert newly generated ATP into high-energy storage forms, like glucose-6-phosphate (G-6-P) and creatine phosphate, in brain and muscle, respectively. The over-expression of VDAC1 in some cancer cells may be related to its multi-functional activities, as required by high energy-demanding cells (Shoshan-Barmatz and Golan, 2012; see VDAC Expression Levels in Cancers and Enhancement by Pro-apoptotic Drugs).
Thus, VDAC is a dynamic regulator of global mitochondrial function both in health and disease (Lemasters and Holmuhamedov, 2006).
VDAC1 was once thought to be localized solely to the OMM (Yu and Forte, 1996). Indeed, although VDAC is present in abundance in the OMM, various studies have revealed that VDAC is also localized to cell compartments other than mitochondria (Bathori et al., 2000; Shoshan-Barmatz and Israelson, 2005; De Pinto et al., 2010b). VDAC, first isolated from human plasma membrane lymphocytes (Thinnes et al., 1989), was subsequently detected in other cells of various tissues (Bathori et al., 2000; De Pinto et al., 2010b). A splice variant of the mitochondrial VDAC1 gene encoding a leader peptide of 13 amino acids at the NH2 terminus was detected and termed plasmalemmal VDAC1 (pl-VDAC1; Buettner et al., 2000). The protein from this splice variant exhibited extra-mitochondrial trafficking to the endoplasmic reticulum (ER), the Golgi apparatus, and the plasma membrane (Gonzalez-Gronow et al., 2003; De Pinto et al., 2010b). Caveolae and caveolae-like domains were shown to contain VDAC (Bathori et al., 1999). The extra-mitochondrial localization of VDAC in bovine outer dense fibers, a cytoskeletal component of the sperm flagellum, has been reported (Hinsch et al., 2004; Triphan et al., 2008; Cassara et al., 2010). VDAC2 was shown to be present in the membrane components of human spermatozoa (Liu et al., 2011). The presence of VDAC in the sarcoplasmic reticulum (SR) of amphibian and mammalian skeletal muscle and in the ER of rat cerebellum has also been demonstrated (Shoshan-Barmatz et al., 1996, 2004; Shoshan-Barmatz and Israelson, 2005).
The exact functions of the extra-mitochondrial VDAC are as yet unknown. pl-VDAC1 was suggested to facilitate regulatory volume decrease in epithelial cells and to play a role in cellular ATP release and volume control (Okada et al., 2004). The possible functions of VDAC in the SR/ER include providing a pathway for transport of Ca2+, nucleotides and other metabolites across the membrane, and involvement in apoptosis. The participation of VDAC in supra-molecular complexes and intracellular communication, including calcium signal delivery between the ER and mitochondria, has also been postulated (Shoshan-Barmatz and Israelson, 2005; see VDAC1 Transport of Ca2+ and Function in ER-mitochondria Cross-talk).
The channel properties of VDAC1 have been examined following reconstitution of the purified protein into a planar lipid bilayer (PLB). VDAC1 has been purified using various procedures and detergents (Shoshan-Barmatz et al., 2010). Bilayer-reconstituted VDAC1 assumes multiple voltage-dependent conformational states, displaying different selectivities and permeabilities. VDAC1 shows symmetrical bell-shaped voltage-dependent conductance with the highest conductance (4 nS at 1 M KCl) at low potentials of -20 to +20 mV (Colombini, 1989; Benz, 1994; Figure 2). At low potentials, when in the fully open state, VDAC1 selectively conducts small ions (e.g., Cl–, K+, Na+), yet shows a preference for anions, such as phosphate, chloride, adenine nucleotides, glutamate, and other anionic metabolites, and large cations, such as acetylcholine, dopamine, and Tris (Shoshan-Barmatz et al., 2010). At higher positive or negative potentials (>30–60 mV), the channel conductance is reduced and the selectivity shifts to small cations. In this scenario, the channel becomes virtually impermeable to ATP and ADP (Colombini, 1989; Benz, 1994; Shoshan-Barmatz et al., 2010).
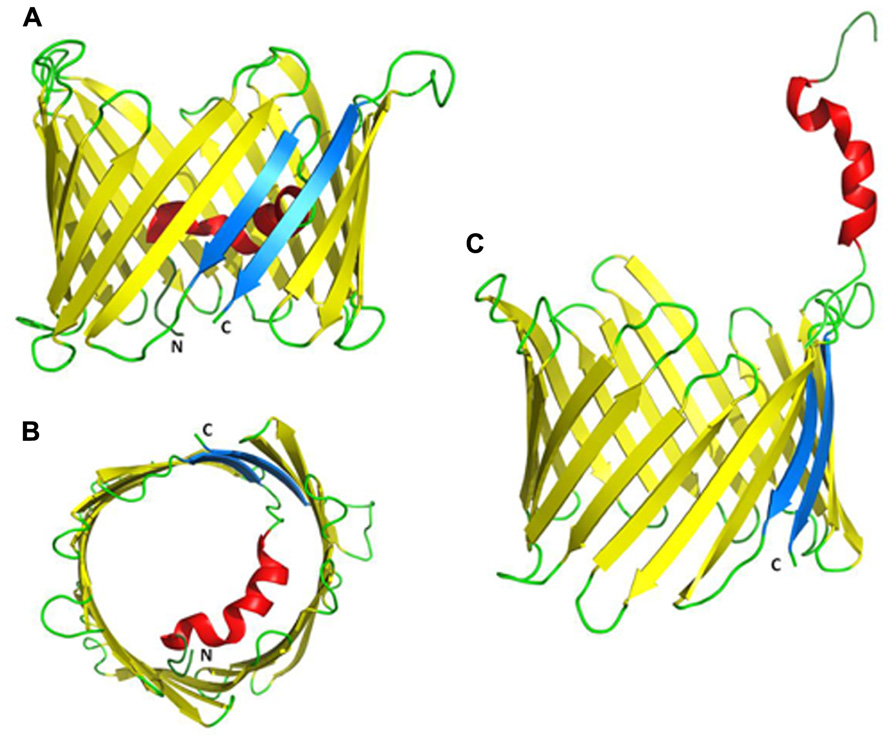
FIGURE 2. Proposed three-dimensional structure of VDAC1. (A) Side-view of the X-ray crystal structure of mouse VDAC1 (Ujwal et al., 2008) in a ribbon representation. The β-barrel is formed by 19 β-strands and the N-terminal helix is folded into the pore interior. β-strands 1 and 19 are parallel and colored blue. The C- and N-termini are annotated as C and N, respectively. Loops and unstructured regions are colored green. (B) Top-view of VDAC1 with the N-terminal inside the pore. (C) VDAC1 in a proposed conformation with the N-terminal outside the channel where it can interact with other proteins (PDB code: 3EMN).
As a voltage-gated channel, VDAC1 must possess a voltage sensor to respond to changes in transmembrane voltage. It is believed that VDAC1 channels rely on two separate gating processes, one that occurs at positive transmembrane potentials and the other at negative potentials (Song et al., 1998). The N-terminal α-helical segment of the channel has been proposed to act as the voltage sensor, gating the pore via conformational changes and/or movements (see The N-terminal Region of VDAC1: Location, Translocation and Channel Gating). Clearly, the molecular nature of VDAC1 gating mechanism has not yet been resolved.
Despite the critical involvement of VDAC1 in various mitochondrial functions, little is known of how VDAC is regulated. Accumulated evidence suggests that VDAC1 function is modulated by various physiological ligands, such as glutamate, adenine nucleotides, NADH, and non-physiological compounds, such as Koenig’s polyanion, ruthenium red (RuR), dicyclohexylcarbodiimide (DCCD), and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS; Shoshan-Barmatz et al., 2006).
NADH was found to regulate the gating of mammalian, fungal, and plant VDAC (Lee et al., 1994). For mammalian VDAC1, molecular and biochemical evidence indicates that the protein possesses one or more nucleotide-binding site(s) (Rostovtseva et al., 2002; Yehezkel et al., 2006, 2007). Glutamate specifically, but not aspartate or GABA, eliminates the bell shape of VDAC voltage-dependence channel conductance (Gincel et al., 2000). Accumulating evidence also suggests that VDAC possesses regulatory binding sites for Ca2+ (reviewed in Shoshan-Barmatz and Gincel, 2003; Shoshan-Barmatz et al., 2006).
Various compounds targeting apoptotic cell death or cell proliferation were found to mediate their activity via interaction with VDAC1, modulating VDAC1 activity (see VDAC as a Pharmacological Target for Compounds Affecting Cell Proliferation or Apoptosis). In addition, VDAC1, acting as a docking site for various proteins, is regulated via protein–protein interactions. Specifically, HK, Bcl2, Bcl-xL, actin, and tubulin were found to interact with VDAC1 and alter its channel conductance (see VDAC1 Association with Proteins and Cancer).
Finally, high cholesterol, known to reduce the activity of membrane-associated proteins, was found to inhibit channel conductance and the metabolic function of VDAC (Campbell and Chan, 2008). It has also been reported that plant VDAC undergoes a reversible regulation of selectivity and voltage-dependence in the presence of sterols (Mlayeh et al., 2010).
Since their discovery in the mid-1970s, great efforts have been devoted to understanding structure–function relationships of VDACs using numerous techniques, including circular dichroism (CD), atomic force microscopy (AFM), electron microscopy, NMR, crystallography, and others.
VDAC1 is a polypeptide of 283 amino acids with Met1 missing in the mature protein (Thinnes et al., 1989; Blachly-Dyson et al., 1993). Various studies have led to the development of models postulating the transmembrane organization of VDAC1, comprising a single α-helix at the N-terminus and 13, 16, or 19 transbilayer β-strands that form a β-barrel (De Pinto et al., 1991; Song et al., 1998; Casadio et al., 2002; Colombini, 2004).
Recently, the 3D structure of recombinant VDAC1 was solved using X-ray crystallography, NMR and a combination of the two. Such studies presented VDAC1 as composed of 19 β-strands arranged as a barrel, with strands β1 and β19 being in parallel conformation (Figure 3; Bayrhuber et al., 2008; Hiller et al., 2008; Ujwal et al., 2008). The N-terminal domain of VDAC1, consisting of 25 amino acids, was shown to be very dynamic and possesses different degrees of α-helical content in each of the three proposed structures (Bayrhuber et al., 2008; Hiller et al., 2008; Ujwal et al., 2008). All three methods employed refolded recombinant VDAC1 expressed in E. coli and purified from inclusion bodies. As such, it has been argued that the refolding conditions employed led to the appearance of non-native structures, as biochemical and biophysical approaches argue for the existence of additional extra-membranal VDAC regions (Colombini, 2009).
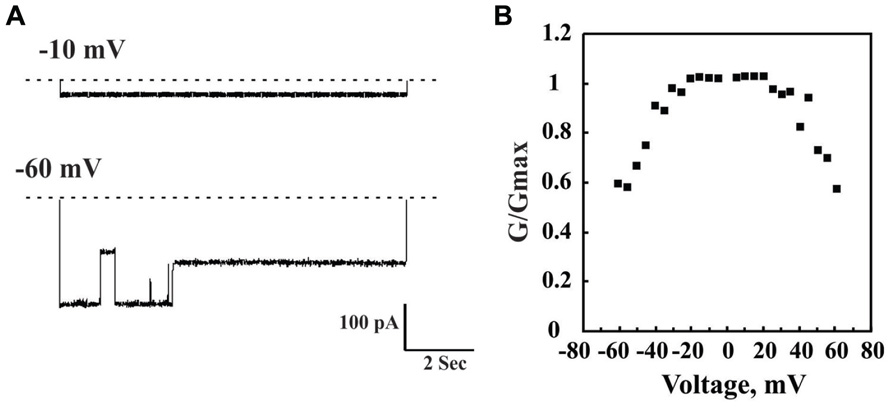
FIGURE 3. Channel properties of bilayer-reconstituted purified VDAC1. Bilayer-reconstituted VDAC single and multi-channel activity was assayed as described previously (Gincel et al., 2000). Purified VDAC (1–5 ng) was reconstituted into a PLB. In (A), a typical activity recording of VDAC incorporated into a PLB is presented as current traces obtained in response to voltage steps from 0 mV to either -10 or -60 mV. In symmetric solution (1 M NaC), when the voltage was changed from -0 to 10 mV, the channel opens and remains stable in this conformation for up to 2 h. However, when the voltage was changed from 0 to -60 mV, the current first increased, due to a greater driving force. However, within less than 1 s, channel conductance decreased and VDAC assumed multiple conductance states. The dashed line indicates the zero current level, while the sub-states of the channel are indicated by arrowheads. In (B), Multi-channel recordings of the average steady-state conductance of VDAC are presented as a function of voltage. The conductance (Go) at a given voltage was normalized to the conductance at -10 mV (Gmax). Each point is the average of three experiments. This voltage-dependent behavior is well known for VDAC.
The pore diameter of the channel has been estimated to be between 3 and 3.8 nm, based on biochemical and structural methods (Colombini, 1980; Goncalves et al., 2007; Bayrhuber et al., 2008) and about 1.5 nm when the N-terminal α-helix is located inside the channel, according to the recent 3D studies (Bayrhuber et al., 2008). Finally, there is evidence that cholesterol is bound to mammalian mitochondrial VDAC and sterols seem to be important for the folding of VDAC (De Pinto et al., 1989; Hiller et al., 2008).
The location of the N-terminal region of VDAC1 with respect to the pore, its role in voltage-gating and its interaction with associated proteins have been the focus of intensive study, as described below.
The VDAC N-terminal region was proposed to lie on the membrane surface (Reymann et al., 1995), to be exposed to the cytoplasm (De Pinto et al., 2003) or to cross the membrane (Colombini, 2004). The 3D structures of recombinant VDAC1, however, revealed that the N-terminal domain is located inside the channel pore, causing a partial obstruction of the wide pore, possibly providing structural reinforcement to the channel walls (Bayrhuber et al., 2008; Hiller et al., 2008; Ujwal et al., 2008; Schneider et al., 2010; Figure 3). Deletion of the N-terminal helical domain did not affect the correct mitochondrial targeting of the protein (De Pinto et al., 2007).
Various studies suggesting that the N-terminal region of VDAC1 constitutes a mobile component of the protein (see VDAC1 N-terminal Domain Function in Voltage-gating and Cell Death). In 2D crystals of Neurospora crassa VDAC1, oblique arrays of the N-terminal domain appear to extend laterally from the barrel, into the aqueous phase (Guo et al., 1995). The N-terminus of the protein was shown to be accessible to anti-VDAC1 antibodies raised against this part of the protein (Guo et al., 1995; Shoshan-Barmatz et al., 2004; Abu-Hamad et al., 2006). The same domain is exposed to kinases, as threonine-13 undergoes phosphorylation (Distler et al., 2007). Additionally, the N-terminal domain of VDAC1 interacts with cytosolic proteins and acts as a recruiting site for HK1, Bcl2, and Bcl-xL and thus is a key structural feature mediating VDAC interaction with anti-apoptotic proteins to enable their function (Shi et al., 2003b; Abu-Hamad et al., 2009; Arzoine et al., 2009; Arbel and Shoshan-Barmatz, 2010), suggesting that this VDAC region is exposed outside the pore. Finally, a recent in silico study supports the role of the N-terminal in controlling shape and permeability of the channel. In the absence of N-terminal, increased overall β-barrel motion, resulting in elliptic, semi-collapsed barrel shapes, is seen (Zachariae et al., 2012).
Taken together, these findings suggest that the N-terminal region of VDAC1 is loosely attached to the barrel wall and can undergo translocation to become exposed at the membrane surface. This movement allows VDAC1 to interact with the anti-apoptotic proteins, HK, Bcl-xL, and Bcl2, and may participate in VDAC1 oligomerization. The signal and nature of conformational changes inducing VDAC1 N-terminal region translocation, both under physiological and apoptotic conditions, await further study.
It was reported that in VDAC1, a glycine-rich sequence (GXXXG), highly conserved in mammals, connects the N-terminal domain to the β-barrel, thus providing the flexibility needed for N-terminal translocation in and out of the pore (Geula et al., 2012a). When this flexible region is mutated by replacing glycines with prolines, the N-terminal region favors a location out of the pore, thereby affecting channel gating. Moreover, in such mutants, the ability to form VDAC1 dimers was highly increased. The GXXXG motif has been linked with dimerization in proteins such as glycophorin A (Gerber and Shai, 2001), human carbonic anhydrase (Whittington et al., 2001), yeast ATP synthase (Saddar and Stuart, 2005), carnitine palmitoyltransferase (Jenei et al., 2009, and others). In VDAC1, this motif is not required for VDAC1 dimerization but may be involved in interaction with VDAC1-associated proteins (Geula et al., 2012a). Interestingly, such a motif in Bcl-xL, interacting with VDAC1 (Arbel et al., 2012), has been recently suggested to mediate VDAC interaction with other proteins (Ospina et al., 2011).
It was reported that during voltage-gating or interaction with molecules, such as cytochrome c (Cyto c), some motion of VDAC occurred (Stanley et al., 1995). N-terminal-truncated mV DAC1 was shown to exhibit high conductance at all tested voltages and was proposed to be a main effector in those apoptotic events dependent on VDAC1 (De Pinto et al., 2008; Abu-Hamad et al., 2009). Using recombinant mutated VDAC1 with the N-terminal cross-linked to channel wall, it was shown that voltage-gating was modified but not completely lost (Teijido et al., 2012). On the other hand, mobility of the N-terminal region and the contribution of this domain to channel gating and interaction with anti-apoptotic proteins were recently demonstrated using site-directed mutagenesis and cysteine substitution, together with a thiol-specific cross-linker (Geula et al., 2012a). Swapping the N-terminal domain of VDAC1 and with that of VDAC3 restores full activity of the channel (Reina et al., 2010).
Different models for the voltage-dependent gating of VDAC1 via the N-terminal region have been proposed: (i) Blockage of the pore by movement of the N-terminal domain within the lumen from the barrel wall toward the center of the channel (Hiller et al., 2008; Ujwal et al., 2008); (ii) transition of the N-terminal region from an α-helical structure that aligns with the barrel wall to a less-structured, unfolded helix element that interacts with the opposing barrel wall (Bayrhuber et al., 2008; Summers and Court, 2010), and (iii) movement of the N-terminal region into and out of the channel lumen (De Pinto et al., 2008; Abu-Hamad et al., 2009; Geula et al., 2012a; Teijido et al., 2012). Calculations using both Poisson–Boltzmann and Poisson–Nernst–Planck electrostatic equations agree with the N-terminal region being involved in gating but not via lateral or horizontal movement of the helix (Choudhary et al., 2010). Thus, the accumulated results and the derived models point to the N-terminal domain as being involved in the channel gating, although the precise mechanism has yet to be described.
In contrast to the extensive studies directed at defining N-terminal domain structure–function, very little research effort has addressed the VDAC1 C-terminal domain. Recently, a frame-shift resulting in appearance of an early stop codon in the VDAC1 gene, leading to the absence of approximately 60% of the C-terminal portion of VDAC1, was noted in gastric and colonic cancers with microsatellite instability (Yoo et al., 2011). Patients with this somatic mutation are heterozygous (Yoo et al., 2011).
A link between the VDAC1 C-terminal region and cancer also comes from studies connecting hypoxia and C-terminal cleavage (Brahimi-Horn et al., 2012). Hypoxic conditions were found to trigger cleavage of the VDAC1 C-terminal to yield a 26-kDa protein (Brahimi-Horn et al., 2012). This C-terminal-cleaved VDAC1 was regulated by HIF-1α and was correlated with hypoxic cell survival and chemotherapy resistance (Brahimi-Horn et al., 2012). Based on the incidence of δC-VDAC1 in lung cancer (about 50% on average and higher in late stage tumors), this form of the protein was proposed to serve as a biomarker to stratify tumor progression in lung cancer patients (Brahimi-Horn et al., 2012). Finally, it was reported that an unidentified mitochondrial calpain cleaves the apoptotic induction factor (AIF) and VDAC1 in a Ca2+-dependent manner, and that such cleavage triggers tVDAC–Bax pores that are associated with the release of tAIF (Ozaki et al., 2009).
Purified and membrane-embedded mammalian VDAC1 were shown to assemble into dimers, trimers, tetramers, and higher oligomers, as revealed by chemical cross-linking and fluorescence resonance energy transfer (FRET) analysis (Zalk et al., 2005). A low-resolution (15 Å) surface structure of VDAC1, obtained by metal shadowing and cryo-electron microscopy of human VDAC1 crystals grown in the presence of phospholipids, showed a dimeric organization of VDAC1 (Dolder et al., 1999). High-resolution AFM of purified native OMM from potato tubers (Hoogenboom et al., 2007) or yeast (Goncalves et al., 2007) showed the distribution of VDAC in an equilibrium ranging from single membrane-embedded pores to hexamers and higher-order oligomers (Hoogenboom et al., 2007), including arrays of up to 20 molecules (Goncalves et al., 2007). In addition, the use of symmetry operators on the NMR-based structure of recombinant hVDAC1 implied that it forms a dimer of monomers arranged in parallel (Bayrhuber et al., 2008), while analysis of the crystal packing of mVDAC1 revealed strong anti-parallel dimers that further assemble into hexamers (Ujwal et al., 2009).
Interestingly, VDAC1 oligomerization is highly increased upon apoptosis induction (Keinan et al., 2010). Structural and computational-based approaches, in combination with site-directed mutagenesis, cysteine replacement, and chemical cross-linking, identified the contact sites between VDAC1 molecules in dimers and higher oligomers (Geula et al., 2012b). Two forms of dimeric VDAC1, one with a contact site involving β-strands 1, 2, and 19 and the second involving β-strands 16 and 17, were identified. Moreover, the results suggest that VDAC1 exists as a dimer that undergoes conformational changes upon apoptosis induction to assemble into higher oligomeric states with contact sites also involving β-strand 8 (Geula et al., 2012b).
The function of VDAC1 oligomers is not known. It was proposed that an organization of VDAC1 beyond the monomeric or dimeric forms may contribute to stabilizing the protein (Ujwal et al., 2009). On the other hand, it was proposed that the oligomeric assembly of VDAC1 offers a platform for other proteins to oligomerize, such as HK (Zalk et al., 2005). HK-I assumes a tetrameric structure that is greatly enhanced when the enzyme is bound to mitochondria (Xie and Wilson, 1990) or when it interacts with the mitochondria to inhibit permeability transition pore (PTP) opening (Azoulay-Zohar et al., 2004). Creatine kinase, when bound to VDAC1 at the inter-membrane space, forms high order oligomers (Brdiczka et al., 1994; Stachowiak et al., 1998), interacting with VDAC1 exclusively in the octameric state, with the dimeric state only showing weak affinity for VDAC1 (Schlattner et al., 2001).
Recently, the function of VDAC1 oligomerization in apoptosis, namely mediating the pathway for the release of Cyto c, was proposed (Zalk et al., 2005; Shoshan-Barmatz et al., 2006, 2008b; Abu-Hamad et al., 2009; Keinan et al., 2010). VDAC1 oligomerization was strongly correlate with apoptosis induction (Shoshan-Barmatz et al., 2008b, 2009; Abu-Hamad et al., 2009; Keinan et al., 2010), while apoptosis induction by various inducers was accompanied by an up to 20-fold increase in VDAC1 oligomerization, indicating a shift in VDAC1 organization toward the oligomeric form under such conditions (Shoshan-Barmatz et al., 2008b; Keinan et al., 2010; see VDAC1 Oligomerization and Release of Cytochrome c).
In apoptosis, a multi-step process that can be initiated by a variety of stimuli, a cascade of cysteine proteases, caspases, are activated, subsequently leading to organized cell demise. Defects in the regulation of apoptosis are often associated with various diseases, with the ability of cells to evade apoptosis being a hallmark of cancer (Hanahan and Weinberg, 2000). Alterations of apoptosis are involved in tumorigenesis, as well as in cellular responses to anti-tumor treatments (Hickman, 2002; Johnstone et al., 2002).
Apoptosis can be activated via the extrinsic or intrinsic pathways. In the extrinsic pathway, apoptosis is induced by extrinsic apoptotic signals initiated by ligand engagement of cell surface receptors, such as Fas and TNF receptors. The intrinsic pathway is initiated in mitochondria in response to different stimuli (Green and Evan, 2002; Johnstone et al., 2002; Chowdhury et al., 2006). Receptor binding by an extrinsic signal typically leads to the recruitment of adapter proteins that promote caspase oligomerization and auto-processing. The extrinsic apoptotic pathway can induce activation of the intrinsic pathway via caspase 8-dependent cleavage of Bid and translocation of the truncated form (tBid) to the mitochondria (Korsmeyer et al., 2000; Yin, 2000). Thus, apoptotic signals initiated by death receptors can be linked to mitochondria-mediated apoptosis.
The intrinsic apoptotic pathway involves mitochondria. Mitochondria contain an arsenal of apoptogenic factors, normally residing in the inter-membranal space (IMS), such as Cyto c, AIF, Smac/DIABLO, and endonuclease G. During transduction of an apoptotic signal into the cell, an alteration in mitochondrial permeability occurs, causing the release of these apoptogenic factors from the IMS (Green and Evan, 2002; Chowdhury et al., 2006; Shoshan-Barmatz et al., 2010). These proteins participate in complex processes resulting in the activation of proteases and nucleases, leading to protein and DNA degradation, and ultimately, cell death. Most notable among the released protein is Cyto c that initiates apoptosis by binding to a central apoptotic regulator, Apaf-1, promoting oligomerization of Apaf-1 and activation of caspase 9, which subsequently activates effector caspases, such as caspases 3, 6, and 7, encouraging execution of cell death. How Cyto c and other apoptogenic factors are released from mitochondria is not clear, and will be discussed below.
Mitochondria-mediated apoptosis can be induced in response to different stimuli (Costantini et al., 2000), including high levels of cytoplasmic Ca2+, reactive oxygen species (ROS), activation of pro-apoptotic Bcl-2 family proteins (Le Bras et al., 2005; Keeble and Gilmore, 2007; Kroemer et al., 2007) or UV damage (Denning et al., 2002). Chemotherapeutic agents act via different mechanisms to induce mitochondria-dependent apoptosis (Costantini et al., 2000). These include betulinic acid (Fulda et al., 1998), PK11195 (Hirsch et al., 1998), diamide (Zamzami et al., 1998), lonidamine (LND; Ravagnan et al., 1999), arsenite (Larochette et al., 1999), 6[3-adamantyl-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD437; Marchetti et al., 1999), 2-chloro-2′-deoxyadenosine, 2-chloro-2′-ara-fluorodeoxy-adenosine (Genini et al., 2000), MT-21 (Watabe et al., 2000), verteporfin (Belzacq et al., 2001), resveratrol (Tinhofer et al., 2001), and paclitaxel (Andre et al., 2000).
As mitochondria represent an appropriate target for therapeutic agents designed to modulate apoptosis, continued research into the mechanisms of mitochondria-mediated apoptotic cell death, coupled with further characterization of the released molecules, will offer new and promising targets for chemotherapeutic intervention in a host of pathologies.
The involvement of VDAC1 in mitochondria-mediated apoptosis has been proposed based on several lines of experimental evidence: (a) Anti-VDAC1 antibodies specifically and effectively prevent As2O3-induced Cyto c release from isolated mitochondria (Zheng et al., 2004) and when microinjected into cells, prevented Bax-induced Cyto c release and subsequent apoptosis, as well as etoposide-, paclitaxel-, and staurosporine (STS)-induced apoptosis (Shimizu et al., 2001). Anti-VDAC1 antibodies also inhibited the interaction of Bax with VDAC and the triggering of cell death (Madesh and Hajnoczky, 2001; Shimizu et al., 2001; Zheng et al., 2004). Microinjection of anti-VDAC antibodies into primary rat hepatocytes effectively prevents apoptosis and Bax–VDAC interactions, as induced by ethanol (Adachi et al., 2004); (b) VDAC1 is the proposed target of pro- and anti-apoptotic members of the Bcl2 family and of HK-I and HK-II (Shimizu et al., 2001; Shi et al., 2003b; Azoulay-Zohar et al., 2004; Zaid et al., 2005; Arbel and Shoshan-Barmatz, 2010; see VDAC1 Transport of Ca2+ and Function in ER-mitochondria Cross-talk); (c) inhibition of Cyto c release and cell death mediated by HK occurred in cells expressing native but not mutated VDAC1 (Zaid et al., 2005; Abu-Hamad et al., 2008; Arzoine et al., 2009); (d) RuR, interacting with Ca2+-binding proteins, interacts with native but not mutated VDAC1 to prevent Cyto c release and cell death (Zaid et al., 2005; Israelson et al., 2008); (e) siRNA-mediated down-expression of VDAC1 prevented cell death and activation of Bax induced by cisplatin and strongly reduced cisplatin-induced release of Cyto c and AIF, as well as the maturation of caspases-3 (Tajeddine et al., 2008). Similarly, reducing VDAC1 expression by siRNA attenuated endostatin-induced apoptosis (Yuan et al., 2008); (f) over-expression of human, murine, yeast, and rice VDAC induce apoptotic cell death (Godbole et al., 2003; Zaid et al., 2005; Abu-Hamad et al., 2008); (g) knockdown of VDAC1 in non-small cell lung cancer (NSCLC) cells inhibited TRAIL-induced activation of caspase-8 and subsequent apoptosis (Chacko et al., 2010); and (h) release of Cyto c was obtained using purified VDAC1 reconstituted into liposomes in which Cyto c had been encapsulated (Madesh and Hajnoczky, 2001; Zalk et al., 2005). Finally, (i) growth factor removal from the medium resulted in reduced nucleotide exchange between mitochondria and cytosol, leading to Cyto c release and apoptosis (Vander Heiden et al., 2000).
In general, there are two views as to the relationship between the conducting state of the VDAC1 channel and cell death. While one view suggests that closure, rather than opening of VDAC leads to OMM permeabilization and apoptosis (Vander Heiden et al., 2000), the second model suggest a distinct VDAC1-based conducting pathways, namely a pore formed in the center of a VDAC1 oligomer (Shimizu et al., 1999; Zalk et al., 2005; Shoshan-Barmatz et al., 2006; see VDAC1 Oligomerization and Function).
Several proposals regarding the mechanism of Cyto c crossing the OMM have been proposed (Figure 4). However, none of the current models of mitochondrial membrane permeabilization can account for all of the experimental data.
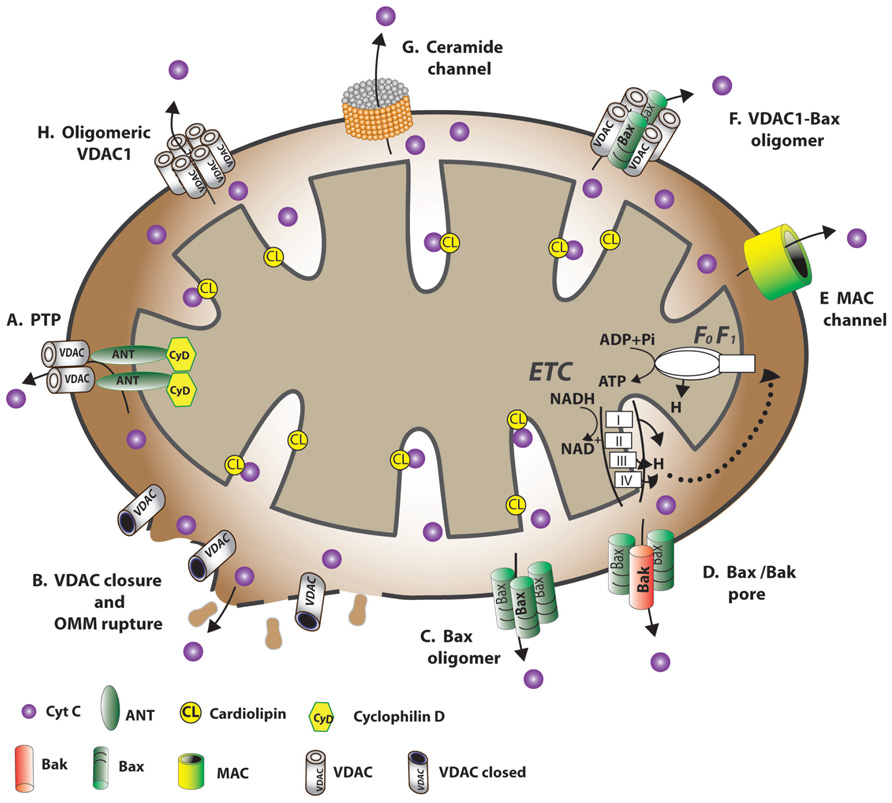
FIGURE 4. Schematic representation of proposed models for the release of apoptogenic proteins from the mitochondrial inter-membrane space mediating the mitochondrial death decision. Different models explaining how OMM permeability changes during apoptosis induction, allowing the release of apoptogenic factors, such as Cyto c. (A) A permeability transition pore (PTP) provides the apoptogenic proteins release pathway. It is proposed that a large conductance pore-forming complex, the PTP, composed of VDAC at the OMM, ANT at the IMM and CypD in the matrix, allows apoptogenic protein release. (B) VDAC closure and OMM rupture serves as the cytochrome c release pathway. Prolonged VDAC closure leads to mitochondrial matrix swelling, OMM rupture, and hence, the appearance of a non-specific release pathway for apoptogenic proteins. (C) Bax activation followed by its oligomerization resulting in OMM permeabilization. Upon apoptosis induction, Bax became associated with mitochondria as a large oligomer/complex, forming a Cyto c-conducting channel in the OMM. (D) A pore formed by oligomerized forms of Bax and Bak after their activation by tBID. BH3-only proteins (e.g., Bid) induce oligomerization of Bax/Bak on the OMM, resulting in Bax activation and OMM permeabilization. (E) MAC as the release pathway. The mitochondrial apoptosis-induced channel, MAC, is a high-conductance channel that forms during early apoptosis and is a putative cytochrome c release channel. MAC formation occurs without loss of outer membrane integrity and depolarization. Members of the Bcl-2 family of proteins regulate apoptosis by controlling the formation of MAC. (F) A Bax- and VDAC-based hetero-oligomer mediates cytochrome c release. The interaction of pro-apoptotic proteins (Bax/Bak) with VDAC forms a cytochrome c release pathway. (G) A lipid channel formed by the lipid, ceramide. Ceramides were shown to induce apoptosis via direct action on mitochondria. A self-assembled ceramide channel is proposed to act as the apoptotic protein release pathway. (H) Oligomeric VDAC1 as a channel for the release of apoptotic proteins. A protein-conducting channel is formed within a VDAC1 homo-oligomer. VDAC1 oligomerization thus functions in mitochondria-mediated apoptosis (see VDAC1 Oligomerization and Release of Cytochrome c). The dynamic equilibrium between VDAC monomeric and oligomeric states can be regulated by various factors, such as Ca2+, oxidative stress and cytochrome c.
In healthy cells, Cyto c is located in the mitochondrial IMS, where it serves as an electron shuttle between complexes III and IV of the respiratory chain, with most Cyto c being bound to cardiolipin (CL). Signals inducing mitochondria-mediated apoptosis result in the release of IMS proteins, including Cyto c (see The Extrinsic and Intrinsic Apoptotic Pathways). The mechanisms by which Cyto c and other pro-apoptotic effector molecules are released have challenged many researchers and several competing models have been proposed to explain the workings of this event (Figure 4; Shoshan-Barmatz et al., 2008a). While some models suggest that the proteins crossing the OMM exclusively involve an increase in OMM permeability due to the formation of a channel large enough to allow for the release of proteins, such as Cyto c, others consider efflux of the proteins to be due to disruption of OMM integrity. The following briefly describes the various proposed models:
One model (Model A, Figure 4) for mitochondrial membrane permeability (MMP) suggests the formation of a PTP, a large high-conductance multi-protein complex comprising several components and spanning both mitochondrial membranes (Bernardi, 1999; Lemasters et al., 1999; Halestrap and Green, 2000; Shoshan-Barmatz and Gincel, 2003; Tsujimoto and Shimizu, 2007). PTP opening is followed by Ca2+ accumulation in the matrix, leading to a sudden increase in permeability to solutes (up to 1,500 Da; Bernardi, 1999; Lemasters et al., 1999; Halestrap and Green, 2000; Shoshan-Barmatz and Gincel, 2003; Tsujimoto and Shimizu, 2007). This Ca2+-dependent increase in MMP leads to loss of membrane potential, mitochondrial swelling, and rupture of the OMM. According to this model, other factors, such as changes in the energetic balance of the mitochondria, anoxia, and ROS can also induce MMP due to PTP opening (reviewed in Tsujimoto and Shimizu, 2007).
The proposed PTP complex components include VDAC1 at the OMM, ANT in the inner mitochondrial membrane (IMM), and cyclophilin D (CypD) in the matrix (Bernardi, 1999; Green and Evan, 2002; Shoshan-Barmatz and Gincel, 2003; Tsujimoto and Shimizu, 2007). Recent studies on the PTP, however, have raised doubts about the proposed members of the complex and the importance of PTP in triggering apoptosis (Kroemer, 1997; Kokoszka et al., 2004; Belizario et al., 2007). Mitochondria isolated from animal models by knockout of genes encoding one or more ANT or VDAC isoforms exhibit Ca2+- and oxidative stress-induced PTP opening, suggesting that the proposed components of the standard model of the PTP complex needs to be reconsidered and further characterized (Kokoszka et al., 2004; Krauskopf et al., 2006; Baines et al., 2007; Berridge et al., 2009). On the other hand, CypD was found to be essential for MMP mediated by Ca2+ overload and that the CypD-dependent MMP regulates some forms of necrotic cell death but not apoptotic death (Belizario et al., 2007).
Although the mechanism(s) responsible for PTP opening and its physiological function have not yet been resolved, a variety of agents were found to promote or inhibit PTP opening, including Ca2+, inorganic phosphate, various oxidizing agents, glutamate, nucleotides, CypD ligands, gelsolin, HK, and proteins of the Bcl-2 family. Some of these compounds have also been shown to interact with VDAC1 directly and modify its channel activity (Shimizu et al., 2000c; Sugiyama et al., 2002; Tsujimoto and Shimizu, 2002; Shoshan-Barmatz and Gincel, 2003; Gincel and Shoshan-Barmatz, 2004; Shoshan-Barmatz et al., 2006). Similarly, the ANT ligands, atractyloside and bongkrekic acid, modulate PTP opening (Haworth and Hunter, 2000). The function of PTP in apoptosis and necrosis (Rasola and Bernardi, 2011) and as a potential therapeutic target for cancer, ischemia-reperfusion injury, and neurodegeneration were proposed (Peixoto et al., 2012).
This model proposes that a sudden increase in IMM permeability to solutes of low molecular weight leads to mitochondria swelling and rupture of the OMM, allowing the efflux of IMS proteins, such as Cyto c, to the cytosol (Feldmann et al., 2000). It has been suggested that matrix swelling and OMM rupture result from a defect in mitochondrial ATP/ADP exchange due to VDAC1 closure, as a result, for example, of removal of a normal growth factor or exposure to G3139 (Model B, Figure 4; Vander Heiden et al., 2000, 2001; Lemasters and Holmuhamedov, 2006). Several studies, however, demonstrated that Cyto c release preceded membrane potential (Δψm) loss in cerebellar granule neurons undergoing apoptotic death and was not accompanied by mitochondrial swelling or OMM rupture (Al-Abdulla et al., 1998; Desagher et al., 1999; Wigdal et al., 2002).
A third proposed mechanism for Cyto c release suggests that oligomeric Bax forms the Cyto c-conducting channel in the OMM (Model C, Figure 4; Antonsson et al., 2000, 2001; Eskes et al., 2000; Zamzami et al., 2000; Kuwana et al., 2002; Reed, 2006). Upon apoptosis induction with STS or UV irradiation, Bax became associated with mitochondria as a large oligomer/complex of 96–260 kDa. While tBid enhances oligomerization of Bax (Lovell et al., 2008), Bcl-2 prevented Bax oligomerization and insertion into the mitochondrial membrane. Several studies, however, showed that apoptosis can be induced in the absence of Bax (Lindenboim et al., 2005; Mizuta et al., 2007; Wan et al., 2008), suggesting that the protein is not obligatory for apoptosis induction and that other mechanisms exist.
The appearance of a channel formed by Bax and Bak hetero-oligomers upon apoptotic insult enabling efflux of pro-apoptotic effectors has been suggested (Model D, Figure 4; Gross et al., 1998; Desagher et al., 1999; Wei et al., 2000, 2001; Antignani and Youle, 2006). tBid was proposed to activate the generation of Bax and Bak complexes up to 500 kDa (Sundararajan et al., 2001). At the same time, anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, were shown to protect cells from apoptosis via a blockage of the Bax–Bak interaction, subsequently preventing Cyto c release (Mikhailov et al., 2003; Dlugosz et al., 2006).
Mitochondrial apoptosis-induced channel (MAC), a supra-molecular high-conductance channel in the OMM, is thought to assemble during early apoptosis and serve as the Cyto c release channel that is regulated by Bcl-2 family members (Guo et al., 2004; Martinez-Caballero et al., 2004, 2005; Dejean et al., 2005, 2006a,b; Model E, Figure 4). The complete molecular identity of MAC is unknown. Recently, it was proposed that Bax is an essential constituent of MAC in some systems, as the electrophysiological characteristics of MAC are very similar to those of Bax channels, while depletion of Bax significantly diminishes MAC activity (Martinez-Caballero et al., 2009).
The formation of hetero-oligomers composed of VDAC1 and Bax were also proposed as a mechanism for Cyto c efflux (Model F, Figure 4; Shimizu et al., 1999; Shimizu and Tsujimoto, 2000; Banerjee and Ghosh, 2004). It was found that recombinant Bax induced permeability in liposomes containing VDAC, implying that VDAC can induce membrane permeability in the presence of Bax (Shimizu and Tsujimoto, 2000). In addition, intracellular microinjection of anti-VDAC antibodies prevented Bax-induced Cyto c release (Shimizu et al., 2001). Electrophysiological studies of Bax and VDAC in PLB revealed that when combined, single-channel conductance rises by factors of 4 and 10 over values attained with VDAC and Bax channels alone, respectively (Banerjee and Ghosh, 2004). Moreover, HK-I and HK-II compete with Bax for interaction with VDAC1 (Pastorino et al., 2002). It was also demonstrated that siRNA-mediated down-expression of VDAC1 strongly suppressed cisplatin-induced activation of Bax (Tajeddine et al., 2008). Finally, another version of this model suggests that oligomeric VDAC is the prime Cyto c release channel and that its pore is regulated by Bax (Debatin et al., 2002).
A self-assembled ceramide-based lipid channel that forms in the OMM was suggested as being the Cyto c release pathway (Model G, Figure 4; Siskind et al., 2006; Stiban et al., 2008). Ceramides were postulated to form a pore in the OMM with a diameter large enough to accommodate Cyto c (Siskind and Colombini, 2000; Siskind et al., 2006; Stiban et al., 2008). An alternative mechanism proposes that ceramides promote the dissociation of Cyto c by altering IMM lipid microdomains (Yuan et al., 2003). Another proposal suggest that ceramides and cholesterol both affect membrane microenvironments so as to favor Bax activation (Birbes et al., 2005; Oh et al., 2006), with translocation to mitochondria fostering propagation of the apoptotic cascade (Martinez-Abundis et al., 2009).
In a model describing Cyto c release developed in our group, it is proposed that mitochondrial pore formation during apoptosis involves the assembly of homo-oligomers of VDAC1 (Model H, Figure 4; Zalk et al., 2005; Shoshan-Barmatz et al., 2006, 2008a; Abu-Hamad et al., 2009). This model is based on a VDAC1 diameter pore (2.5–3.0 nm; Bayrhuber et al., 2008) being large enough for the movement of nucleotides and small molecules but too small to allow passage of a folded protein, like Cyto c. Thus, a model in which Cyto c release takes place through the formation of large protein-conducting channel within a VDAC1 homo-oligomer or in a hetero-oligomer containing VDAC1 and pro-apoptotic proteins formed by oligomerization of VDAC1 is offered (Zalk et al., 2005; Shoshan-Barmatz et al., 2006, 2008a; Abu-Hamad et al., 2009). Substantial evidence for the formation of higher ordered VDAC1-containing complexes exists (see VDAC1 Oligomerization and Function).
Recently, it has been demonstrated that VDAC1 undergoes oligomerization in response to apoptotic stimuli, with VDAC1 oligomerization being enhanced up to 20-fold, as revealed by chemical cross-linking, or directly monitored in living cells using bioluminescence resonance energy transfer (BRET; Shoshan-Barmatz et al., 2008a; Keinan et al., 2010). Enhancement of VDAC oligomerization was obtained regardless of the cell type or apoptosis inducer used, including STS, curcumin, As2O3, etoposide, cisplatin, selenite, TNF-α, H2O2, or UV, all affecting mitochondria yet acting through different mechanisms (Keinan et al., 2010; Figure 5). Conversely, the apoptosis inhibitor, DIDS, prevented STS-induced VDAC1 oligomerization and apoptosis (Shoshan-Barmatz et al., 2008a; Keinan et al., 2010). Moreover, VDAC1 over-expression resulted in VDAC1 oligomerization and apoptosis in the absence of any apoptosis stimuli (Shoshan-Barmatz et al., 2008a). Furthermore, it was demonstrated that in cells expressing a VDAC1 dimeric fusion protein comprising wild type and the RuR-insensitive E72Q-mutated VDAC1 showed no protection against STS-induced apoptosis. This dominant-negative VDAC1 mutant reveals oligomeric VDAC1 to be the active unit in mitochondria-mediated apoptosis (Mader et al., 2010).
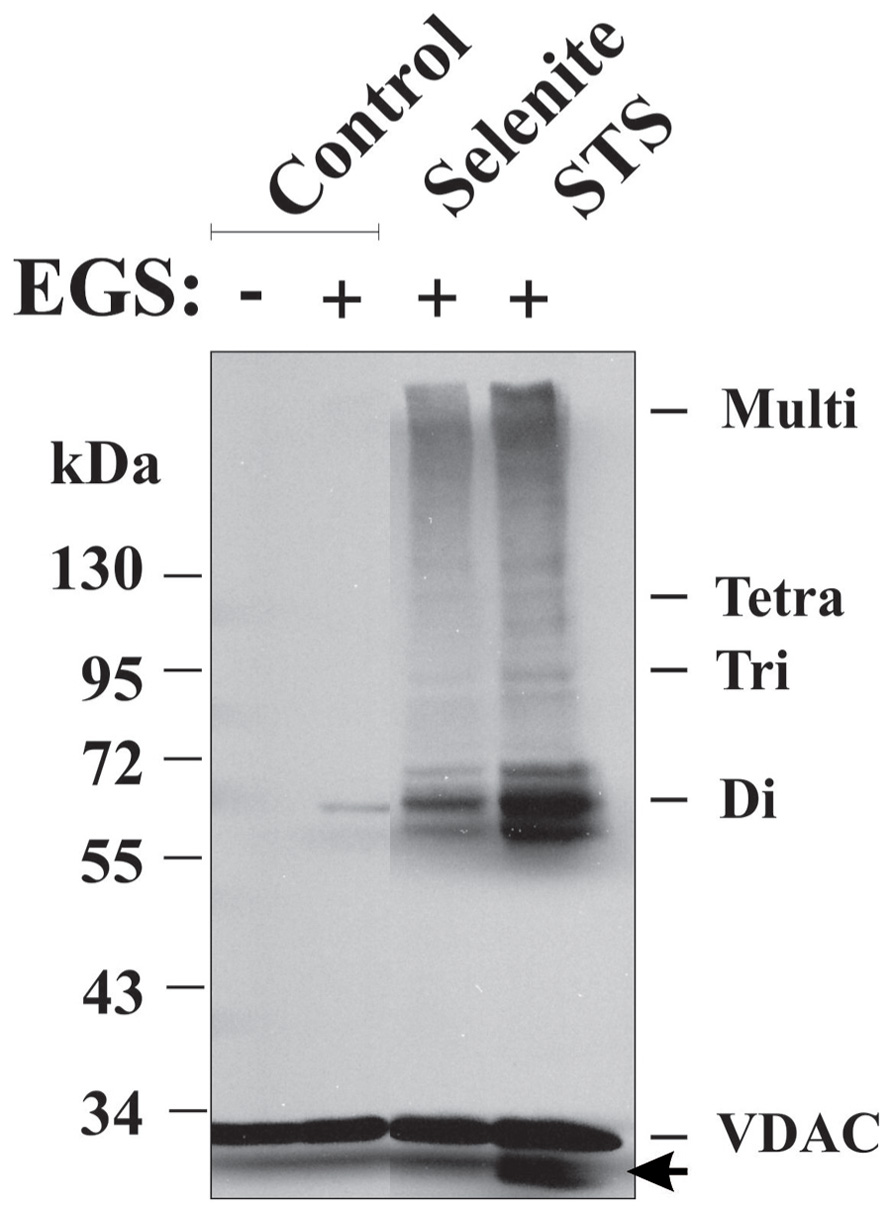
FIGURE 5. Apoptosis induction and VDAC1 oligomerization. VDAC1 oligomerization as induced by apoptosis inducers and revealed by EGS-based cross-linking. HeLa cells were incubated for 16 h with STS (0.2 μM) or selenite (8 μM), as described previously (Keinan et al., 2010), washed with PBS and incubated with EGS (300 μM) at 30°C for 10 min, followed by SDS-PAGE (10% acrylamide) and Western blotting using anti-VDAC1 antibodies. VDAC1 monomers, dimers, trimers, tetramers, and multimers are indicated. The arrow indicates an anti-VDAC1 antibody-labeled protein band migrating below the position of monomeric VDAC1. The positions of molecular weight protein standards are also provided.
The molecular mechanisms triggering VDAC1 oligomerization upon apoptosis induction remain unknown. Post-translational modification of VDAC1, such as phosphorylation (Kerner et al., 2012) or oxidation, may modulate its oligomeric state. The involvement of lipids has been suggested recently (Betaneli et al., 2012). The anionic lipid, phosphatidylglycerol (PG), was found to significantly enhance VDAC1 oligomerization in the membrane, whereas CL disrupts VDAC1 supra-molecular assemblies. Interestingly, during apoptosis, the level of PG in mitochondria increases, whereas that of CL decreases (Betaneli et al., 2012). Another proposed mechanism involves Cyto c detachment from its binding site, namely CL at the IMM. Apoptotic signals leading to the oxidation of CL and dissociation of Cyto c from the lipid (Ott et al., 2002), together with our finding that VDAC1 oligomerization is highly encouraged by Cyto c (Zalk et al., 2005), support a scenario whereby unbound Cyto c promotes VDAC oligomerization, Cyto c release and subsequent apoptosis.
Clearly, a more complete understanding of the mechanisms underlying VDAC1 oligomer assembly and its role in Cyto c release will require additional study. Indeed, the multiple pathways and mechanisms of Cyto c release presented here may co-exist within a single model of cell death, depending on the cell type and the nature of the stimulus (Gogvadze et al., 2006; Galluzzi and Kroemer, 2007).
In recent years, a substantial body of evidence has accumulated indicating a correlation between alterations in cell metabolism and cancer formation. Cancer cells undergo significant metabolic adaptation to fuel cell growth and division (Gatenby and Gillies, 2004; Hanahan and Weinberg, 2011; Koppenol et al., 2011). Malignant cancer cells typically display high rates of glycolysis even when fully oxygenated and are subject to suppressed mitochondrial respiration, despite the fact that glycolysis is a less energy-efficient pathway, a phenomenon known as the “Warburg effect” (Gatenby and Gillies, 2004; Hanahan and Weinberg, 2011; Koppenol et al., 2011). The Warburg effect likely provides the vast majority of cancerous tumors with a number of benefits in the form of precursors for the biosynthesis of nucleic acids, phospholipids, fatty acids, cholesterol, and porphyrins. A second advantage of the Warburg effect is its likely involvement in both tumor protection and invasion. Tumor cells produce lactic acid via glycolysis and transport it out of the cell, leading to increased acidity of the closed microenvironment, generating a low pH “coat.” This is proposed to protect tumors against attack by the immune system while inducing negative effects on normal surrounding cells, aiding in preparing the surrounding tissues for invasion. Additionally, the Warburg effect also assures longer tumor survival time if oxygen becomes limiting (Hanahan and Weinberg, 2011). Moreover, cancer-associated abnormalities in glucose metabolism enhance cellular resistance to apoptosis, with mitochondria playing a key role in this process (Fadeel et al., 2008; Kroemer and Pouyssegur, 2008; Mayevsky, 2009; Fulda et al., 2010; Gogvadze et al., 2010). Finally, mitochondria have been found to playa role in cellular re-programming from the catabolic to the anabolic modes (Fadeel et al., 2008; Kroemer and Pouyssegur, 2008; Mayevsky, 2009; Gogvadze et al., 2010), with such metabolic flexibility and cellular hierarchy being crucial in metastatic cancer (Berridge et al., 2010).
Such metabolic re-programming of cancer cells includes marked over-expression of mitochondrial-bound HK isoforms, considered as the rate-limiting enzyme of glycolysis and serving as the biochemical gate of this pathway (Pedersen et al., 2002; Mathupala et al., 2006; Pedersen, 2007; Shoshan-Barmatz et al., 2010). HK interacts with VDAC1 at the OMM and forms the main interface between mitochondria and the cytosol. VDAC1 contributes to cancer metabolism via transport of various metabolites, mediating ATP/ADP exchange across the OMM and is, therefore, defined as the “food channel.” HK, by association with VDAC1, gains direct access to mitochondrial ATP, reaching VDAC1 via the ANT in the IMM, allowing it to phosphorylate and “trap” any incoming glucose (Pedersen, 2008). With this direct coupling of mitochondrially generated ATP to incoming glucose via VDAC1-bound HK, mitochondria regulate glycolytic flux with that of the tricarboxylic acid (TCA) cycle and ATP synthase to balance the energy requirements of the tumor cell with biochemical requirements for metabolites (i.e., the anaplerotic and cataplerotic pathways, respectively) or metabolite precursors that are required by the tumor (Pedersen et al., 2002; Mathupala et al., 2006; Pedersen, 2007; Shoshan-Barmatz et al., 2010). Thus, both the glycolytic pathway and other seminal metabolic pathways, like the pentose phosphate shunt, are regulated via the energy-coupling resulting from the formation of the VDAC1-bound HK complex. As part of a system that impacts cell growth, the VDAC1–HK complex represents a remarkable target for cancer therapy (see The Interaction of VDAC1 with Hexokinase Regulates Cell Bioenergetics and Apoptosis).
The localization of VDAC1 at the OMM provides structural and functional anchoring sites for a diverse set of cytosolic proteins that together with VDAC1 mediate metabolic and signaling cross-talk between cytosol and mitochondria (Table 1). VDAC1 displays binding sites for glycerol kinase (Adams et al., 1991), HK (Azoulay-Zohar et al., 2004; Pastorino et al., 2005; Abu-Hamad et al., 2008; Goldin et al., 2008; Shoshan-Barmatz et al., 2008b; Arzoine et al., 2009), creatine kinase (Schlattner et al., 2001), C-Raf kinase (Le Mellay et al., 2002), ANT (Vyssokikh and Brdiczka, 2003), the peripheral benzodiazepine receptor [also known as translocator protein (TSPO); Veenman et al., 2007], tubulin (Rostovtseva et al., 2008b), the dynein light chain, mtHSP70, the ORDIC channel, GAPDH (Shoshan-Barmatz and Israelson, 2005), actin (Xu et al., 2001), and gelsolin (Kusano et al., 2000), as well as Bcl-2 family members (Shoshan-Barmatz et al., 2006; Adams and Cory, 2007; Llambi and Green, 2011). Interaction with apoptosis-regulating proteins, such as HK and Bcl-2 family members, make VDAC1 a key protein in apoptosis regulation (Tsujimoto and Shimizu, 2002). Here, we focus on those VDAC1-interacting proteins showing modified levels of expression in cancer cells.
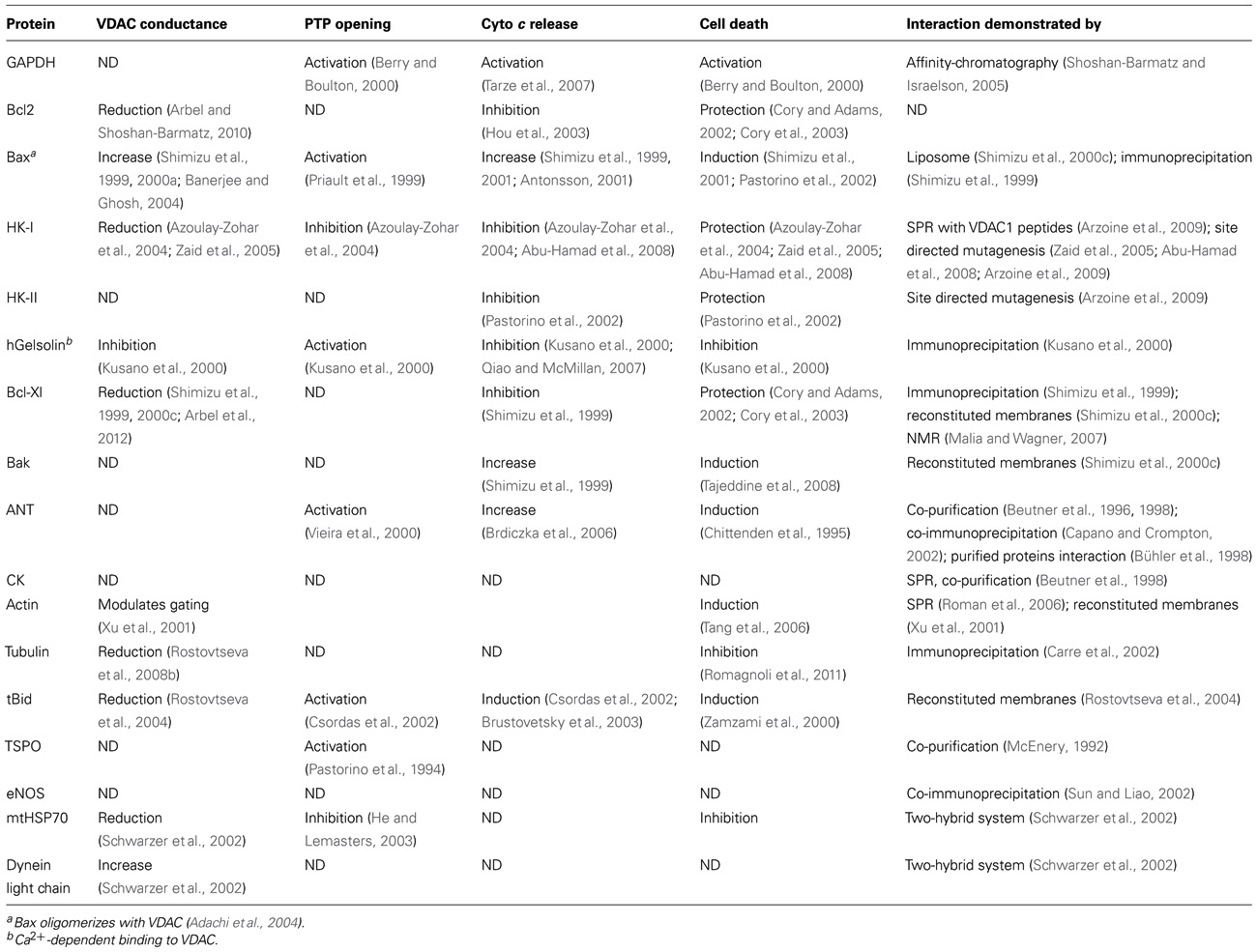
TABLE 1. Modulation of VDAC conductance and mitochondrial-mediated apoptosis by VDAC-associated proteins.
One of the signature phenotypes of highly malignant, poorly differentiated tumors is their high rate of glycolysis, leading to enhanced lactate generation (Brahimi-Horn et al., 2007). This property is frequently dependent on the marked over-expression of VDAC1-bound HK (Rose et al., 1974; Gottlob et al., 2001; Bryson et al., 2002; Pedersen et al., 2002; see Cancer Metabolism, Hexokinase and VDAC). HK thus lies at the apex of the glycolytic pathway that provides the metabolic intermediates required by the biosynthetic pathways on which a transformed cell places such heavy demand (Pedersen, 2007; Mathupala et al., 2010).
Hexokinase catalyzes the rate-limiting step in glycolysis, the ATP-dependent phosphorylation of glucose to G-6-P. The mitochondria bound-isoforms, HK-I and HK-II, were found to be over-expressed in many cancers, including colon, prostate, lymphoma, glioma, gastric adenomas, carcinomas, and breast cancers (Gottlob et al., 2001; Bryson et al., 2002; Mathupala et al., 2010). The elevated levels of mitochondria-bound HK in cancer cells is thus suggested to play a pivotal role in promoting cell growth and survival in rapidly growing, highly glycolytic tumors and in protecting against mitochondria-mediated cell death (Mathupala et al., 2006). The association/dissociation of HK with/from VDAC1 and the switching of VDAC1 between an “open” and a “closed” state regulate cross-talk between mitochondria and the cytosol (Shoshan-Barmatz et al., 2006, 2010). This control is important in maintaining the mitochondria respiration and glycolysis equilibrium at the heart of the energetic and metabolic homeostasis of the cancer cell. Cancer cell, however, possess an escape mechanism that intervenes when G-6-P accumulates and dissociates HK from VDAC (Azoulay-Zohar et al., 2004).
The interaction of HK with the mitochondria, and specifically with VDAC1, has shifted our view of HK as predominantly fulfilling a metabolic role to one of regulation of apoptotic responsiveness of the cell, making the VDAC1–HK complex a target for therapeutic purposes (Pastorino et al., 2002; Azoulay-Zohar et al., 2004; Pastorino et al., 2005; Shoshan-Barmatz et al., 2006, 2008b, 2009; Abu-Hamad et al., 2008; Goldin et al., 2008; Pastorino and Hoek, 2008; Arzoine et al., 2009).
The interaction between HK-I and VDAC1 was first demonstrated in a reconstituted system where HK-I decreased the channel conductance of VDAC1, an effect that was reversed by G-6-P, shown to detach HK from isolated mitochondria (Azoulay-Zohar et al., 2004). The interaction between HK-I and VDAC1 was again demonstrated by co-immunoprecipitation (Shoshan-Barmatz et al., 2008a). The co-localization of HK-I with each of the three isoforms of VDAC1 was demonstrated using two-color stimulated emission depletion (STED) microscopy (Neumann et al., 2010). The HK-I–VDAC1 interaction can be abrogated upon mutagenesis of a single VDAC1 residue (Glu73, Glu202, or Glu65), resulting in an elimination of HK-mediated protection against apoptosis and channel closure (Zaid et al., 2005; Abu-Hamad et al., 2008). N-terminally truncated VDAC1 is incapable of binding HK (Abu-Hamad et al., 2009) or Bcl-xL (Arbel et al., 2012). Moreover, when the N-terminal region α-helix structure in VDAC1 was perturbed, the binding of HK was reduced (Geula et al., 2012a). It was proposed that the N-terminal region of HK-I is inserted into the channel pore, where it interacts with the N-terminal region of VDAC1 (Rosano, 2011). It should be noted that there is strong residue conservation between the HK-I and HK-II N-termini, and that such conservation is not shared by the other two non-bound mitochondrial isoforms, HK-III and HK-IV.
The VDAC1–HK interaction was shown to be negatively regulated by phosphorylation, when GSK3β is up-regulated (Pastorino et al., 2005). Finally, VDAC1-based peptides were found to interact with purified HK and when expressed in cells over-expressing HK, prevented the anti-apoptotic activity of HK (Arzoine et al., 2009).
In vitro and in vivo studies have shown that elevated levels of mitochondria-bound HK in cancer cells also protect against mitochondria-mediated apoptosis via direct interaction with VDAC1.
Mitochondrially associated HK has been shown to protect HeLa and HEK cells from entering apoptosis (Bryson et al., 2002). This protection was related to a blockade of the interaction of the pro-apoptotic protein, Bax, with VDAC1 (Bryson et al., 2002). Moreover, over-expression of HK-I or HK-II in tumor-derived cell lines suppressed STS-induced Cyto c release and apoptosis (Azoulay-Zohar et al., 2004; Zaid et al., 2005; Arzoine et al., 2009). A decrease in apoptosis and an increase in cell proliferation have also been reported to be induced by HK-II expression in the NIH-3T3 (Fanciulli et al., 1994) and rat 1a cell lines (Gottlob et al., 2001). Importantly, mutagenesis studies revealed that single mutations or N-terminal truncation in VDAC1 prevented HK-I-mediated protection against apoptosis and channel closure in a reconstituted membrane system (Zaid et al., 2005; Abu-Hamad et al., 2008, 2009; Shoshan-Barmatz et al., 2009). In addition, binding of HK-II to mitochondria inhibits Bax-induced Cyto c release and apoptosis (Pastorino et al., 2002). HK binding to VDAC1 is regulated by protein kinases, notably GSK-3β (Pastorino et al., 2005) and protein kinase C (PKC; Pastorino and Hoek, 2008), as well as by the cholesterol content of the OMM (Pastorino and Hoek, 2008).
Several mechanisms by which HK binding to VDAC1 protects against apoptosis and promotes cell survival can be considered. These include controlling energy and metabolic homeostasis, as well as preventing VDAC1-mediated Cyto c release and shielding VDAC1 from pro-apoptotic factor binding, thus offering the tumor cell protection from cell death in a synergic manner. The advantages to cancer cells of HK binding to VDAC1 include:
Energy and metabolite production and access. Hexokinase bound to VDAC1 provides cells with metabolic advantages, allowing enhanced cell growth (Pedersen, 2007). Anchoring of HK to VDAC1 offers the enzyme direct access to mitochondrial sources of ATP and greater affinity for Mg2+-ATP (Bustamante and Pedersen, 1980; Pedersen, 2008). HK bound to the cytosolic face of VDAC1 acts as a gate, regulating the traffic of various metabolites through the VDAC1 (Azoulay-Zohar et al., 2004). In addition, VDAC1-bound HK is less sensitive to inhibition by its product, G-6-P (Azoulay-Zohar et al., 2004), thus avoiding product inhibition. The HK–VDAC1 interaction increases energy and metabolite production of the high energy-demanding cancer cells, allowing for maintenance of a high glycolytic flux rate in tumors (Azoulay-Zohar et al., 2004).
VDAC1-bound HK acts as an anti-apoptotic protein. Accumulated evidence demonstrated that HK-I and HK-II also function as anti-apoptotic proteins when bound to VDAC1, while their detachment enabled activation of apoptosis (Pastorino et al., 2002, 2005; Azoulay-Zohar et al., 2004; Shoshan-Barmatz et al., 2006, 2008b, 2009; Abu-Hamad et al., 2008; Goldin et al., 2008; Pastorino and Hoek, 2008; Arzoine et al., 2009). Disruption of HK binding to VDAC1 by mutation in VDAC1 or by addition of VDAC1-based peptides decreased the survival of cancer cells (Arzoine et al., 2009).
Hexokinase interaction with VDAC1 protects against activation of apoptosis by Bax or Bak (Pastorino et al., 2002; Majewski et al., 2004b; Pastorino and Hoek, 2008). The detachment of HK-II from the mitochondria was found to markedly potentiate the onset of caspase-2-induced mitochondrial damage (Shulga et al., 2009).
Regulation of ROS production by HK. Reactive oxygen species act as second messengers in cell signaling and are essential for multiple biological processes in normal cells. However, ROS can also provoke damage to multiple cellular organelles and processes (Auten and Davis, 2009). ROS production is usually increased in cancer cells due to oncogene activation (Zhang et al., 2011; see VDAC1 Function in ROS Release, ROS-mediated Apoptosis and Interaction with NO). Mitochondria-associated HK was shown to reduce mitochondrial ROS generation (da-Silva et al., 2004), with HK-I and HK-II reducing intracellular levels of ROS (Sun et al., 2008). Moreover, expression of both HK-I and HK-II was found to protect against oxidant-induced cell death (Ahmad et al., 2002; Bryson et al., 2002). Thus, detachment of HK from VDAC1 could lead to increased ROS generation and release to the cytoplasm, thereby activating cell death.
Stabilization of both HK and VDAC1. VDAC1, containing an odd number of β-strands, presents conformational intrinsic instability within the first four β-strands (β1–4), relative to other VDAC1 regions (Bayrhuber et al., 2008).
In silico studies of the VDAC1–HK-I interaction predicates that both proteins attain a more stable state through protein–protein interaction (Rosano, 2011). It is proposed that upon binding with the N-terminal helix of HK-I, VDAC1 acquires higher stability via the formation of a network of chemical bonds both due to direct protein–protein contacts and to hydrogen bonds mediated by an ATP molecule and an Mg2+ ion (Rosano, 2011).
Increased synthesis and uptake of cholesterol. Cancer cells have been shown to exhibit a 2- to 10-fold increase in mitochondrial cholesterol content, in comparison to liver mitochondria (Baggetto et al., 1992). It has been proposed that the increased binding of HK to the mitochondria of cancer cells may play a role in mediating increased synthesis and uptake of cholesterol into the mitochondria of cancer cells (Pastorino and Hoek, 2008).
If one considers the metabolic importance of both VDAC1 and HK, the role that HK fulfills in promoting tumor cell survival and the function of the HK–VDAC1 interaction in regulating apoptosis, as well as other functions as presented above, then disruption of the HK–VDAC1 complex represents an attractive target for cancer therapy and may form the basis for novel anti-cancer drugs. Indeed, several different compounds have been employed to disrupt the HK–VDAC1 association, resulting in apoptotic cell death. These include peptides corresponding to the amino terminus of both HK-I (Gelb et al., 1992) and HK-II (Pastorino et al., 2002), clotrimazole (Penso and Beitner, 1998; Pastorino et al., 2002; Shoshan-Barmatz et al., 2008b) and a cell-permeable HK-II-based peptide (Majewski et al., 2004a). Methyl jasmonate (a plant-derived stress hormone) binds to and detaches mitochondria-bound HK from several cancer cell types (Goldin et al., 2008). HK detachment can be visualized using HK–GFP. Cell expression of HK-I–GFP showed punctuated distribution that co-localized with MitoTracker Red (Figure 6), suggesting a mitochondrial localization. The induction of apoptosis by STS resulted in HK–I-GFP detachment, as reflected by the diffuse fluorescence seen (Figure 6).
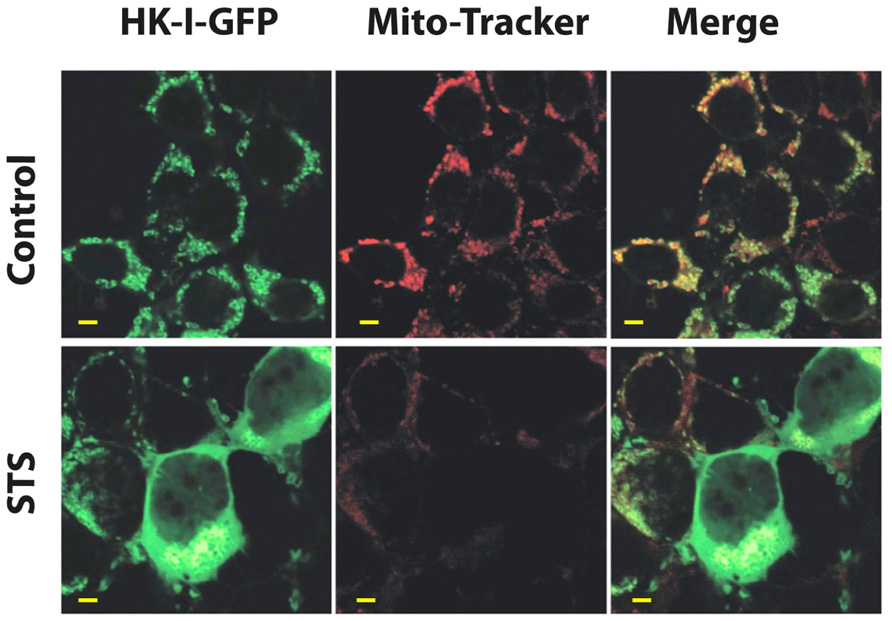
FIGURE 6. Detachment of mitochondrial-bound HK-I-GFP induced by STS. To demonstrate HK-I binding to mitochondria (i.e., VDAC) as well as detachment, a HK-I–GFP fusion protein was expressed in HEK–T cells. Confocal fluorescence microscopy showed that in control cells expressing HK-I–GFP, the fluorescence is punctuated, as expected for mitochondrial distribution. On the other hand, induction of apoptosis by STS detaches mitochondrial-bound HK-I–GFP. HEK–T cells were transfected to express HK-I–GFP and after 48 h were exposed to STS (0.8 μM), stained with 25 nM of the mitochondrial marker MitoTracker Red and visualized with confocal microscopy. The punctuated HK-I–GFP fluorescence, originally co-localized with mitochondria (control), as also reflected in the co-localization with MitoTracker Red (Merge panel), was converted to diffuse labeling of the cytosol after exposure to STS. Images are representative microscopic fields from one of three similar experiments (scale bar = 5 μm).
The small molecule alkylating agent, 3-bromopyruvic acid (3BP) was found to be phosphorylated by HK and to inhibit glycolytic rate in VX2 tumors (Ko et al., 2001). 3BP covalently modifies protein cysteine residues, resulting in rapid activity loss (Meloche and Monti, 1975; Ko and McFadden, 1990; Pereira da Silva et al., 2009). In animal models, 3BP showed high efficacy against advanced stage malignant tumors by inhibiting both glycolysis and mitochondrial energy generation, possibly by interfering with the HK–VDAC1 complex (Berridge et al., 2010). Thus, 3BP is proposed to target metabolism and blocks energy supplies. Other studies demonstrated the potential of 3BP as a potent anti-cancer agent in humans (Ko et al., 2012). Current knowledge related to 3BP and its promise as a future cancer therapeutic are the focus of a special 2012 issue of the Journal of Bioenergetics and Biomembranes (JOBB 44-1, 2012).
In conclusion, since VDAC1-bound HK is essential for tumor cells, conferring several advantages (see HK-linked Protection Against Cell Death is Mediated via Interaction with VDAC1), including protection against apoptotic events and promoting aerobic glycolysis, the detachment of HK from VDAC1 offers a novel therapeutic strategy to augment apoptosis, revert the hyper-glycolytic state and enhance the therapeutic efficacy of conventional chemotherapeutic agents.
The B cell lymphoma/leukemia-2 (Bcl-2) family of proteins plays an essential role in the control of apoptosis (Brown, 1997) at the interface with mitochondria. The Bcl-2 family consists of more than 20 pro-apoptotic (e.g., Bid, Bax, and Bak) and anti-apoptotic (e.g., Bcl-2 and Bcl-xL) members, all characterized by the presence of Bcl2 homology (BH) domains (Adams and Cory, 2007; Youle and Strasser, 2008). Accordingly, these proteins can be sub-divided into three main groups, based on the regions of the BH domain they contain and their function, namely the multi-domain anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1/A1), the multi-domain pro-apoptotic [Bcl-2-associated X protein (Bax) and Bak], and the BH3-only pro-apoptotic (Bid, Bim, Bad, Bik, Noxa, PUMA, Bmf, and Hrk) proteins (Certo et al., 2006). The anti-apoptotic members of the Bcl-2-family contribute to tumor initiation, disease progression, and drug resistance (Miyashita and Reed, 1993; Adams and Cory, 2007). Indeed, enhanced expression of anti-apoptotic Bcl-2 family members was shown to be associated with the resistance of many tumors to chemotherapy (Sentman et al., 1991; Adams and Cory, 2007). For example, in multiple studies, increased levels of Bcl-2, Bcl-xL, and Mcl-1 proteins were linked to survival of multiple myeloma cells and resistance to chemotherapy (Oancea et al., 2004), while in most cases of chronic lymphocytic leukemia (CLL), elevated Bcl-2 mRNA and protein levels are noted (Tsujimoto et al., 1984).
The mechanism whereby Bcl2 family members control apoptosis is still not fully understood. However, it is well established that their activities involve controlling OMM permeability (Scorrano et al., 2003; Adams and Cory, 2007; Youle and Strasser, 2008). Accumulated findings linked the activity of both anti-apoptotic and pro-apoptotic proteins to their association with VDAC (Shimizu and Tsujimoto, 2000; Shimizu et al., 2000b; Sugiyama et al., 2002; Shi et al., 2003a; Tsujimoto, 2003; Malia and Wagner, 2007; Tajeddine et al., 2008; Arbel and Shoshan-Barmatz, 2010). VDAC1 interacts with Bax, Bim, Bcl2, and Bcl-xL in isolated mitochondria and in reconstituted membrane systems (Shimizu et al., 2000b; Rostovtseva et al., 2004; Malia and Wagner, 2007; Arbel et al., 2012). Purified C-terminally truncated Bcl2 and Bcl-xL interact with VDAC1 and reduced the channel conductance of wild type but not of certain mutated forms of VDAC1 (Arbel and Shoshan-Barmatz, 2010; Arbel et al., 2012) or N-terminally truncated VDAC (Abu-Hamad et al., 2009; Arbel et al., 2012). Similarly, both Bcl2 and Bcl-xL prevented apoptosis as induced by various stimuli in cells expressing wild type but not mutated VDAC1 (Abu-Hamad et al., 2009; Arbel and Shoshan-Barmatz, 2010; Arbel et al., 2012). It was proposed that VDAC1 interacts with both Bax and Bcl-xL to form a tertiary complex (Shi et al., 2003a). A direct interaction between Bcl-xL and VDAC1 was demonstrated by NMR (Malia and Wagner, 2007). Interaction of the putative loop region of VDAC1 with Bcl-xL was proposed (Shi et al., 2003a). Bcl-xL was also shown to affect VDAC oligomerization, as revealed by chemical cross-linking of micelle-bound VDAC, shifting the equilibrium of VDAC from the trimeric to the dimeric state (Malia and Wagner, 2007). Furthermore, synthetic peptides corresponding to the VDAC1 N-terminal region and other cytosol-facing VDAC1 sequences bound Bcl2 and Bcl-xL (Shi et al., 2003a; Abu-Hamad et al., 2009; Shoshan-Barmatz et al., 2009; Arbel and Shoshan-Barmatz, 2010; Arbel et al., 2012). Such peptides, in cell-penetrating form or expressed in the cell, were able to suppress the anti-apoptotic activities of Bcl2 and Bcl-xL in cells (Abu-Hamad et al., 2009; Arbel and Shoshan-Barmatz, 2010; Arbel et al., 2012). Thus, VDAC1-based peptides, targeting the anti-apoptotic activity of Bcl-xL and Bcl2, can serve as potential cancer therapeutics.
Bax and VDAC reconstituted into liposomes were shown to form a new channel, with a conductance 4–10 times larger than that of the individual proteins (Shimizu et al., 2000b), and with such increase being prevented by Bcl-xL. The interaction of Bax and Bim with VDAC leads to Cyto c release, with Bcl-xL and anti-VDAC antibodies preventing this release (Shimizu et al., 2001). It has also been shown that Bid but not Bax modulates VDAC channel conductance (Rostovtseva et al., 2004).
In VDAC1-depleted cells, cisplatin-induced activation of Bax was inhibited (Tajeddine et al., 2008), suggesting that VDAC1 is involved in Bax-mediated apoptosis. Furthermore, Bcl-2 and Bcl-xL blocked As2O3-induced VDAC dimerization (Zheng et al., 2004). These results indicate that Bcl-2 family proteins regulate VDAC-mediated apoptosis and hence, the release of apoptogenic proteins from mitochondria.
Translocator protein is an 18-kDa integral membrane protein, originally identified as the peripheral benzodiazepine receptor or isoquinoline-binding protein (Korkhov et al., 2010), that is over-expressed in breast, prostate, colon, and brain cancer, with protein expression linked to cancer progression and poor survival rates (Batarseh and Papadopoulos, 2010). Like VDAC1, TSPO is located in the OMM and was proposed to activate PTP opening, leading to apoptosis (Levin et al., 2005; Veenman et al., 2007; Sileikyte et al., 2011). Interactions between TSPO and VDAC are thought to play a role in apoptosis (Gavish et al., 1999; Levin et al., 2005; Veenman et al., 2007). High-affinity ligand occupancy of TSPO was shown to attenuate anti-neoplastic agent-induced apoptosis (Kugler et al., 2008). The observations that TSPO is involved in the generation of ROS and that TSPO association with VDAC may enhance ROS production prior to apoptotic induction, indirectly links both proteins (Golani et al., 2001; Veenman et al., 2002, 2007). Still, there is no direct evidence for a TSPO–VDAC1 interaction at the molecular level.
In cancer, morphogenic changes are required for high proliferative rates, the acquisition of inappropriate migratory patterns, and invasive characteristics (Hall, 2009). Alterations in the dynamics of the actin and tubulin components of the cytoskeleton are well-known consequences of signaling and the triggering of responses like apoptosis (Franklin-Tong and Gourlay, 2008; Rostovtseva and Bezrukov, 2012). Mitochondria are dynamic organelles that actively travel along the cytoskeleton within the cell. Within this framework, specific association of VDAC1 with tubulin was shown by co-immunoprecipitation (Carre et al., 2002) and by tubulin-induced VDAC1 closure (Rostovtseva et al., 2008a). A model was proposed whereby the negatively charged C-terminus of tubulin penetrates into the positively charged VDAC1 pore, leading to decreased channel conductance (Rostovtseva et al., 2008b). It was proposed that tubulin, VDAC1 and mitochondrial creatine kinase (MtCK) are members of a super complex that is structurally and functionally coupled to the ATP synthasome (Saks et al., 2010). Recent evidence indicates that the lipid composition of the membrane is capable of controlling tubulin–VDAC interactions, thus tuning VDAC sensitivity to blockage by tubulin (Rostovtseva et al., 2012). Similarly, it has been reported that tubulin–VDAC interactions can be regulated by varying the membrane surface charge (Gurnev et al., 2012). In a cellular model of cardiomyocyte physiology, the super complex presents a different composition, namely one in which tubulin is replaced by HK and MtCK is absent, infusing mitochondrially generated ATP into the glycolytic pathway and thereby contributing to the Warburg effect (Saks et al., 2010). Other proteins associated with cytoskeleton dynamics, such as gelsolin (Kusano et al., 2000), adseverin or scinderin (SCIN; Miura et al., 2012), have been reported to associate with VDAC, with concomitant anti-apoptotic effects.
All of these VDAC1–protein interactions establish a new interplay between kinase signaling pathways, mitochondrial respiration, and the highly dynamic microtubule network characteristic of cancerogenesis and cell proliferation. VDAC1, as the common denominator in all the interactions described above, thus presents a single unique target for efforts addressing cell physiological, metabolic, and morphological aspects of cancer.
Many viruses encode for proteins acting on mitochondria, in general, on VDAC1, in particular, or directly targeting proteins in the apoptotic pathway (Boya et al., 2001, 2003; Everett and McFadden, 2001; Irusta et al., 2003; Verrier et al., 2003). Other viruses influence apoptosis by mimicking Bcl-2 family proteins, changing their expression levels or directly interacting with Bcl-2 family proteins (Halestrap et al., 2002; Boya et al., 2004). HIV-1 offers an excellent example of neuronal and immunological-destructive apoptosis involving mitochondria (Sui et al., 2004; Perfettini et al., 2005). HIV-1 envelope proteins can trigger mitochondrial apoptosis, increasing cytosolic Ca2+, ROS, and transcriptional activation of p53, thus increasing the expression of pro-apoptotic proteins, such as Bax (Castedo et al., 2003). The PB1-F2 protein of the influenza A virus, when localized in mitochondria, induces alteration of mitochondrial morphology, dissipation of the mitochondrial membrane potential, and cell death and has been reported to interact directly with VDAC (Zamarin et al., 2005; Danishuddin et al., 2009). Hepatitis B virus (HBV) is another mitochondria-interacting virus (Boya et al., 2003, 2004; Shirakata and Koike, 2003). The HBx protein is highly homologous to VDAC1 and is proposed to “poison” the homo-oligomeric state of VDAC1, creating hetero-oligomeric structures that induce permeability of the mitochondria, allowing Cyto c release (Stanley et al., 1995; Rahmani et al., 2000; Shoshan-Barmatz et al., 2006; Goncalves et al., 2007; Hoogenboom et al., 2007). This action of HBx can result in the development of hepatocellular carcinomas (Rahmani et al., 2000). In infected liver cell lines, hepatic virus (HEV) not only induced up-regulation of VDAC expression in infected human hepatoma cell lines but also encouraged higher levels of VDAC oligomerization, leading to apoptosis (Moin et al., 2007).
In higher eukaryotes, VDAC1 is the most predominant isoform expressed. Several studies have shown that alterations in VDAC1 expression affect both the ability of cells to generate energy and their sensitivity to apoptosis-inducing reagents.
Several studies have demonstrated remarkable differences in the expression levels of VDACs between tumor cells and normal tissues. VDAC1 expression in several cancer cell lines was higher than in the control fibroblast cell line (Simamura et al., 2006, 2008). NSCLC cells specifically exhibited high expression levels of VDAC1. This was shown to directly correlate with poor outcome in early stage NSCLC, thus establishing an association with aggressive tumor behavior (Grills et al., 2011). In addition, in several melanoma and prostate cancer cell lines, a correlation between levels of VDAC1 expression and induction of Cyto c release by G3139 was demonstrated (Lai et al., 2006). In a proteomic analysis of aging-related proteins in human normal colon epithelial tissues, significant up-regulation of 19 proteins was noted, including VDAC1 and VDAC2 (Li et al., 2007).
Several cancer treatments were found to enhance expression of VDAC. Up-regulation of VDAC1 expression was observed in three different acute lymphoblastic leukemia (ALL) cell lines (697, Sup-B15, and RS4;11) following prednisolone treatment, an observation that can be explored for predicting eventual outcome in childhood ALL (Jiang et al., 2011). VDAC1 over-expression was observed in a cisplatin-sensitive cervix squamous cell carcinoma cell line (A431) when exposed to cisplatin, while in a cisplatin-resistant cell line (A431/Pt), the treatment resulted in down-regulation of VDAC1 (Castagna et al., 2004). Up-regulation of VDAC has been reported upon UV irradiation of apoptosis-sensitive cells (Voehringer et al., 2000). When A375 human malignant melanoma cells were treated with the tyrosinase inhibitor, arbutin, a potential anti-tumor agent (Nawarak et al., 2009), VDAC1 expression level was found to be up-regulated (Cheng et al., 2007). In addition, ROS was found to induce up-regulation of VDAC that could be prevented by the ROS chelator, epigallocatechin (Jung et al., 2007).
The relationship between VDAC1 expression levels and sensitivity to various treatments was presented in several studies. The PC3 and DU145 prostate cancer cell lines are relatively resistance to apoptosis as induced by G3139 and were found to express less VDAC than did G3139-sensitive LNCaP cells (Lai et al., 2006). Similarly, reducing VDAC1 expression by siRNA efficiently prevented cisplatin-induced apoptosis and Bax activation in NSCLC (Tajeddine et al., 2008), attenuated endostatin-induced apoptosis (Yuan et al., 2008) and inhibited selenite-induced PTP opening (Tomasello et al., 2009). The anti-cancer activity of furanonaphthoquinones (FNQs) was increased upon VDAC1 over-expression and decreased upon silencing of VDAC1 expression by siRNA (Simamura et al., 2008).
Finally, it has also been shown that the CD45 expression is accompanied by elevated VDAC1 expression in myeloma cells sensitized to a diverse set of apoptotic stimuli via the mitochondrial pathways (Liu et al., 2006).
Among the complex array of genetic changes accompanying cancer development, all cancerous cells develop altered metabolism, with VDAC1 and HK playing important functions in this metabolic reprograming (see The Interaction of VDAC1 with Hexokinase Regulates Cell Bioenergetics and Apoptosis). VDAC1 contributes to cancer metabolism via transport of various metabolites, mediating ATP/ADP exchange across the OMM and by the binding and channeling of mitochondrial ATP directly to HK (see Cancer Metabolism, Hexokinase and VDAC). The importance of VDAC1 for cancer cells is further reflected in the findings that silencing VDAC1 expression reduced cellular ATP levels and cell growth (Abu-Hamad et al., 2006). Furthermore, when HeLa cervical cancer cells stably expressing shRNA directed against hVDAC1 were injected into nude mice, the development of a solid tumor was inhibited (Koren et al., 2010). It has also been shown that VDAC1 silencing potentiates H2O2-induced apoptosis and impairs mitochondrial Ca2+ loading, while silencing VDAC2 had the opposite effects (De Stefani et al., 2012). Together, these findings indicate that over-expression of VDAC1 in cancer cells could contribute to the high glycolytic phenotype seen.
As presented above (see VDAC Expression Levels in Cancers and Enhancement by Pro-apoptotic Drugs), the expression level of VDAC1 serves as a crucial factor in the process of mitochondria-mediated apoptosis as induced by various means. Indeed, over-expression of VDAC from different sources, such as yeast, rice, fish, mice, and humans, was found to induce apoptosis (Godbole et al., 2003; Zaid et al., 2005; Ghosh et al., 2007; Lü et al., 2007). Over-expression of murine (m) VDAC1 or rat (r) VDAC1 in U-937 cells resulted in apoptotic cell death (Zaid et al., 2005). Moreover, over-expression of rice VDAC induced apoptosis (Godbole et al., 2003) that was blocked by Bcl-2 and the VDAC channel inhibitor, DIDS. Similar results were obtained following transfection of fish cells to express Paralichthys olivaceus VDAC (Lü et al., 2007). Over-expression of VDAC1 causes depolarization of the IMM and activation of the PTP (De Pinto et al., 2007; Tomasello et al., 2009).
The mechanism underlying cell death induced by VDAC1 over-expression is not fully understood. However, recent studies demonstrated that VDAC1 over-expression is accompanying by its oligomerization (Zalk et al., 2005; Goncalves et al., 2007; Hoogenboom et al., 2007; Malia and Wagner, 2007; Shoshan-Barmatz et al., 2008a,b; Zeth et al., 2008; Keinan et al., 2010). VDAC1 has been shown to form different oligomeric states, namely monomers, dimers, trimers, tetramers, hexamers, and higher-order oligomers (see VDAC1 Oligomerization and Function). Moreover, the supra-molecular assembly of VDAC1 in cultured cells is highly enhanced upon apoptosis induction, supporting the involvement of VDAC1 oligomerization in Cyto c release and thus, in apoptosis (Shoshan-Barmatz et al., 2008b; Keinan et al., 2010). Moreover, VDAC1 over-expression resulted in its oligomerization in the absence of any apoptosis stimuli (Shoshan-Barmatz et al., 2008b). It should be noted that apoptosis induced by VDAC1 over-expression shares common features with apoptosis induced by other stimuli (e.g., STS, H2O2), as is the case for inhibition by RuR, Bcl2, HK-I over-expression, or DIDS, all agents that interact with VDAC1 (Shoshan-Barmatz et al., 2006). Finally, HEV-infected human hepatoma cells showed enhanced VDAC expression, leading to VDAC oligomerization and apoptosis (Moin et al., 2007).
Thus, the over-expression of VDAC1, as induced by drugs, such as arbutin, prednisolone, or cisplatin, or by viral proteins or UV irradiation, as well as the correlation between drug effectiveness and VDAC expression, suggest that the anti-cancer activities of these drugs is associated with VDAC levels. This is further supported by the finding that cisplatin-induced apoptosis is inhibited in cells silenced for VDAC1 expression (De Pinto et al., 2007; Tomasello et al., 2009). In addition, endostatin-induced apoptosis was decreased upon silencing VDAC1 expression, and enhanced by over-expression of VDAC1 (Yuan et al., 2008). We thus propose that the high levels of VDAC1 promote its oligomerization, leading to apoptosis (Shoshan-Barmatz et al., 2008b; Keinan et al., 2010). Accordingly, anti-cancer strategies that specifically up-regulate VDAC1 levels in cancer cells may activate the mitochondrial apoptotic pathway, with concomitant benefit to the patient.
Reactive oxygen species serve important roles in diverse events, such as cellular proliferation, differentiation, and migration. It is now accepted that cellular “redox” signaling is involved in regulating normal processes and disease progression, including angio- genesis, oxidative stress, aging, and cancer. Cellular levels of ROS are linked to anti-tumor immunity, the oxidative tumor microenvironment, proliferation, and death of cancer cells (Manda et al., 2009).
Cancer cells exhibit high basal levels of oxidative stress due to the activation of oncogenes, loss of tumor suppressors, and the effects of the tumor microenvironment (Cairns et al., 2011). Hypoxia, a characteristic of most solid tumor microenvironments, causes a progressive elevation in mitochondrial ROS production (chronic ROS) which over time leads to the stabilization of cells via increased HIF-2α expression, which serve as signaling molecules that activate the transcription of genes involved in cellular hypoxic adaptation (Hamanaka and Chandel, 2009). It is becoming apparent that hypoxia enables cells to survive with sustained levels of elevated ROS. However, the increased oxidant concentrations associated with cellular transformation promote tumorigenicity through signaling but can also damage DNA, proteins, and lipids. Indeed, promoting oxidative stress in cancer cells selectively kills several cancer cell lines (Trachootham et al., 2006; Nogueira et al., 2008; Ralph et al., 2010).
Reactive oxygen species are mainly produced in the mitochondria as by-products of respiratory chain reactions, with 1–5% of the oxygen consumed by mitochondria in human cells being converted to ROS, such as superoxide anions (), H2O2, and hydroxyl radicals (Murphy, 2009). Oxidative stress results when production of ROS exceeds the capacity of mitochondrial and cellular anti-oxidant defenses to remove these toxic species. Indeed, to combat harmful ROS, cells possess several anti-oxidant defense mechanisms, including catalytic removal of reactive species by enzymes like superoxide dismutase (SOD), catalase, and peroxidase (Barber et al., 2006; Figure 6). However, about 1% of the ROS escapes elimination and cause oxidative cellular damage. The rules governing ROS transport via the OMM and control of this process, as well as the molecular mechanism of ROS activating VDAC to release Cyto c, are not clear.
Since ROS are generated in the mitochondria, they can attack the mitochondria directly and, when released to the cytosol, can attack and modify DNA, lipids, and proteins, thus affecting cell survival (Ott et al., 2007). This requires ROS release from the mitochondria to the cytosol, namely, crossing the OMM. VDAC1 in the OMM may serve as the major gateway for molecules exiting the IMS. Indeed, VDAC1 has been proposed to mediate ROS release from the IMS to the cytosol (Figure 6). Several pieces of evidence support the involvement of VDAC1 in ROS release from the IMS to the cytosol, including:
(i) HK-I and HK-II bound to VDAC1 decrease ROS release from mitochondria when over-expressed in HEK cells (Sun et al., 2008), thereby reducing intracellular levels of ROS (Ahmad et al., 2002; da-Silva et al., 2004). Also, expression of HK-I or HK-II was found to protect against oxidant-induced cell death (Ahmad et al., 2002; Bryson et al., 2002). Thus, detachment of HK from VDAC1 could lead to increased ROS generation and release to the cytoplasm, thereby activating cell death.
(ii) Closure of VDAC appears to impede the efflux of superoxide anions from the IMS, resulting in an increased steady-state level of , causing internal oxidative stress and sensitizing mitochondria toward the Ca2+-induced MPT (Tikunov et al., 2010).
(iii) The VDAC inhibitors, DIDS and dextran sulfate, inhibited release from the mitochondria to the cytosol (Han et al., 2003).
Mitochondria are the major source of ROS that, when released to the cytosol, can damage proteins, lipids and DNA. ROS release is mediated via VDAC1, with its interactors (e.g., HK, DIDS, RuR, AzRu) inhibiting release (Figure 7). Mitochondria also contain several enzymes that can decompose ROS. ROS can promote Cyto c release from mitochondria (Petrosillo et al., 2001, 2003). In fact, it has been shown that apoptosis-inducing agents, such as inorganic arsenic compounds (Shi et al., 2004; Ding et al., 2005) and doxorubicin (Olson et al., 1981; Sokolove, 1994), cause oxidative damage to DNA and protein by inducing ROS generation.
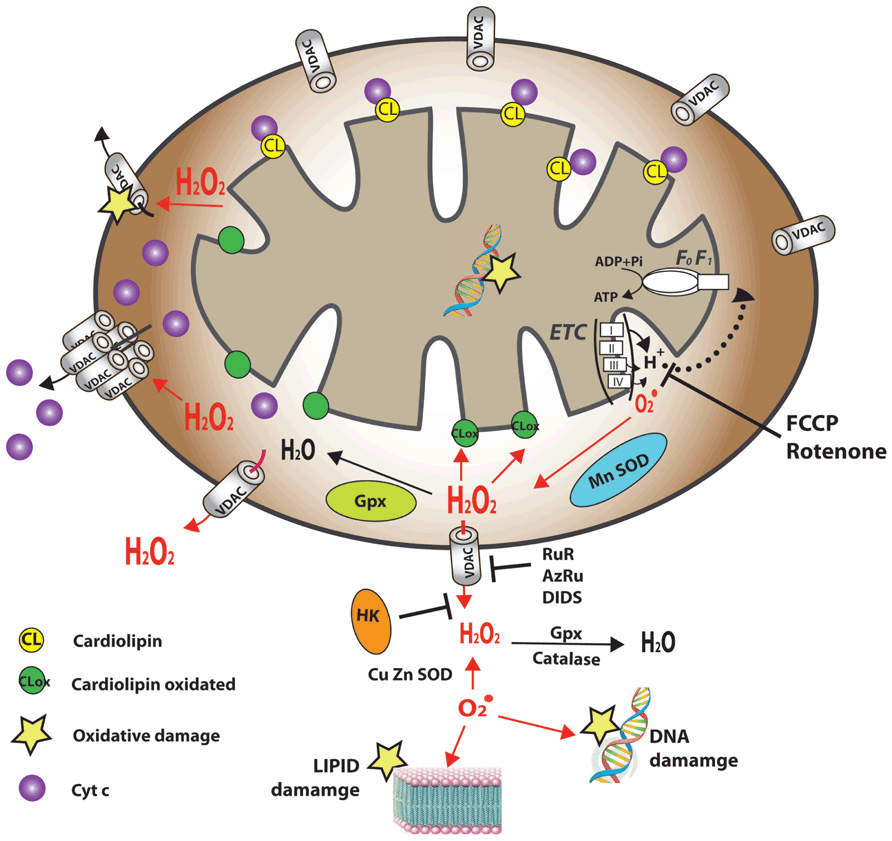
FIGURE 7. Reactive oxygen species occlusion and degradation in mitochondria can prevent cell damage. A schematic presentation summarizing current knowledge and proposed control mechanisms for ROS production, neutralization, and cellular effects (see VDAC1 Function in ROS Release, ROS-mediated Apoptosis and Interaction with NO). Free radicals generated by the electron transport chain are generally regarded as toxic metabolites and, as such, are degraded by specialized enzymes, namely catalases, peroxidases, superoxide dismutases, and glutathione peroxidase. Mitochondrial ROS cross the OMM via VDAC. The ROS that escapes catalytic removal can cause oxidative damage ( ) to mitochondria, and when released from the mitochondria, can damage cellular proteins, lipids, and DNA. In addition, ROS can induce apoptosis. ROS is proposed to induce Cyto c release by oxidizing the mitochondria-specific phospholipid, cardiolipin (CL). ROS-oxidized CL has a markedly lower affinity for Cyto c, thus rendering Cyto c free in the inter-membrane space. ROS can also induce VDAC oligomerization to yield a mega-channel mediating Cyto c release. VDAC inhibitors (e.g., RuR, AzRu, DIDS) and hexokinase (HK) can prevent ROS release to the cytosol. CuZnSOD, cytosolic (copper–zinc-containing) superoxide dismutases; MnSOD, mitochondrial (manganese-containing) superoxide dismutases; GPx, glutathione peroxidase.
) to mitochondria, and when released from the mitochondria, can damage cellular proteins, lipids, and DNA. In addition, ROS can induce apoptosis. ROS is proposed to induce Cyto c release by oxidizing the mitochondria-specific phospholipid, cardiolipin (CL). ROS-oxidized CL has a markedly lower affinity for Cyto c, thus rendering Cyto c free in the inter-membrane space. ROS can also induce VDAC oligomerization to yield a mega-channel mediating Cyto c release. VDAC inhibitors (e.g., RuR, AzRu, DIDS) and hexokinase (HK) can prevent ROS release to the cytosol. CuZnSOD, cytosolic (copper–zinc-containing) superoxide dismutases; MnSOD, mitochondrial (manganese-containing) superoxide dismutases; GPx, glutathione peroxidase.
Inhibition of -induced apoptosis by DIDS, an inhibitor of VDAC channel activity, or anti-VDAC1 antibodies (Shoshan-Barmatz et al., 1996; Madesh and Hajnoczky, 2001; Shimizu et al., 2001; Simamura et al., 2006), suggests that -induces Cyto c release via VDAC-dependent permeabilization of the OMM (Madesh and Hajnoczky, 2001). Moreover, was found to evoke Cyto c release in VDAC-reconstituted liposomes (Madesh and Hajnoczky, 2001). In other studies, it was found that ROS-induced alterations of VDAC1 and/or ANT can induce MMP selective for Cyto c release, without causing further mitochondrial damage (Madesh and Hajnoczky, 2001; Le Bras et al., 2005). In addition, ROS-induced up-regulation of VDAC1 can be prevented by the ROS chelator, epigallocatechin (Jung et al., 2007). It has been suggested that ROS-mediated Cyto c and SOD1 release from mitochondria involves VDAC, leading to increased susceptibility of mitochondria to oxidative stress and apoptosis (Li and Yuan, 2008).
Interactions between TSPO and VDAC1 are proposed to play a role in apoptosis (Gavish et al., 1999; Veenman et al., 2004; Levin et al., 2005; see VDAC and the Translocator Protein). TSPO appears to be involved in the generation of ROS, leading to apoptosis induction (Veenman et al., 2008). It was hypothesized that the close association of TSPO with VDAC may enhance the concentration of ROS generated by TSPO, leading to apoptosis induction (Golani et al., 2001; Veenman et al., 2002, 2007).
Nitric oxide (NO) possesses both pro- and anti-apoptotic effects, depending on both cell type and NO concentration (Brune et al., 1999; Kim et al., 1999). Specific NO molecular targets include inhibition of Bcl-2 cleavage (Kim et al., 1998) and inactivation of caspases by S-nitrosylation (Li et al., 1999). At low physiological release rates, NO was found to inhibit PTP opening in a reversible manner, while at high physiological release rates, NO accelerated PTP opening (Brookes et al., 2000). VDAC1 was found to bind the endothelial NO synthase (eNOS; Sun and Liao, 2002). In vitro binding studies using glutathione S-transferase (GST)-tagged VDAC1 indicated that VDAC1 binds directly to eNOS and that this interaction amplified eNOS activity. Furthermore, the calcium ionophores, A23187 and bradykinin, both known to activate eNOS, increased VDAC1–eNOS complex formation, suggesting a potential role for intracellular Ca2+ in mediating this interaction (Sun and Liao, 2002). The above results indicate that the interaction between VDAC and eNOS may be important for regulating eNOS activity and modulation of VDAC by NO (Sun and Liao, 2002). Recently, it has been reported that exogenous NO directly interacts with purified VDAC from rat heart and inhibited its channel activity in a biphasic manner. This inhibition was prevented by a NO scavenger. Channel activity inhibition paralleled the delay in opening of the cardiac PTP by NO (Cheng et al., 2011).
Ca2+ is a ubiquitous cellular signal, with changes in intracellular Ca2+ concentration not only stimulating a number of intercellular events but also triggering cell death pathways, including apoptosis. Ca2+ signals result in the regulation of Ca2+-dependent enzymes, such as phospholipases, proteases, and nucleases, important during injury or cell death (Berridge et al., 2000). Apart from their metabolic and apoptotic roles, mitochondria are also a major hub of cellular Ca2+ homeostasis. Indeed, mitochondrial Ca2+ homeostasis is fundamental for a wide range of cellular activities, such as control of oxidative phosphorylation (Cox and Matlib, 1993; Maack et al., 2006), modulation of cytosolic calcium signals (Gunter et al., 2000), cell death (Giacomello et al., 2007), and secretion (Maechler et al., 1997; Lee et al., 2003). Intra-mitochondrial Ca2+ controls energy metabolism by enhancing the rate of NADH production by modulating critical enzymes, such as those of the TCA cycle and fatty acid oxidation (Nichols and Denton, 1995; Denton, 2009), linking glycolysis to the TCA cycle (Cárdenas et al., 2010).
Ca2+ transport across the IMM is mediated by several recently identified proteins. The mitochondrial Ca2+ uniporter (MCU; Baughman et al., 2011; De Stefani et al., 2011), as well as its regulatory protein, the EF hand-containing protein termed MICU1 (for mitochondrial calcium uptake 1), acting as a Ca2+ sensor that controls mitochondrial Ca2+ entry mediated by the MCU, were identified (Santo-Domingo and Demaurex, 2010). The high-affinity mitochondrial Ca2+/H+ exchanger, Letm1, is able to import Ca2+ at low (i.e., sub-micromolar) cytosolic concentrations into energized mitochondria (Jiang et al., 2009), while the Na+/Ca2+ exchanger superfamily, NCLX, is the major Ca2+ efflux mechanism (Palty et al., 2010). All the Ca2+ transport systems described above mediate the transport of Ca2+ across the IMM (Figure 1). However, incoming Ca2+ must first traverse the OMM. The only identified protein mediating Ca2+ transport in the OMM is VDAC1. Indeed, VDAC1 is permeable to Ca2+ and possesses Ca2+-binding sites (Gincel et al., 2001; Rapizzi et al., 2002; Tan and Colombini, 2007) and it has been shown that control of OMM Ca2+ permeability is mediated by VDAC (Bathori et al., 2006; Tan and Colombini, 2007).
Although changes in mitochondrial Ca2+ concentration are known to trigger apoptosis, the precise mechanism is not known.
VDAC1 is highly Ca2+-permeable and modulates Ca2+ access to the mitochondrial inter-membrane space (Gincel et al., 2001; Tan and Colombini, 2007). Non-physiological Ca2+ overload depolarizes mitochondria by opening the PTP, with concomitant release of Cyto c and other IMS proteins (Crompton, 1999; Baumgartner et al., 2009) that can lead to both apoptotic and necrotic cell death, associated with disease pathogenesis (Rasola and Bernardi, 2011). Thus, Ca2+ overload disrupts energy production by mitochondria and enables apoptosis and/or necrosis (Shoshan-Barmatz and Gincel, 2003; Kinnally et al., 2011; Rasola and Bernardi, 2011). Ca2+ overload also results in production of ROS. To date, the mitochondrial target for Ca2+ activation of Cyto c release has not been identified. The inhibition of PTP opening by RuR (Gincel et al., 2001), while protection against cell death induced by several apoptotic stimuli (Baek et al., 1997; Ding et al., 2001; Bae et al., 2003) may arise from RuR interaction with VDAC1, inducing channel closure (Gincel et al., 2001). This is supported by the findings that E72Q-VDAC1 did not interact with RuR and cells expressing this mutant were not protected against apoptosis by RuR (Zaid et al., 2005). These observations suggest that Ca2+-binding sites in VDAC1 (Israelson et al., 2008) serve regulatory functions, including those involved in apoptosis induction.
The ER is a dynamic reservoir of Ca2+ ions with electrical or chemical cell stimulation resulting in its activation (Bootman et al., 2002; Verkhratsky and Petersen, 2002), highlighting the role of this organelle as an indispensable component of Ca2+ signaling processes. Ca2+ originating from the ER has been shown to be a potent death-inducing signal (Pinton et al., 2008).
The interplay between ER- and mitochondria-mediated Ca2+ signaling is a determinant of cellular fate through the control of apoptosis and autophagy (Decuypere et al., 2011). Release of Ca2+ from the ER via inositol-1,4,5-trisphosphate receptors (IP3Rs) has been observed in models of apoptosis and has been implicated directly in mitochondrial Ca2+ overload (Hajnoczky et al., 2003). The specific sites of physical association between the ER and mitochondria, known as mitochondria-associated membranes (MAMs), include VDAC among the proteins present at these junctions (Bononi et al., 2012). The mitochondrial chaperone, grp75 (glucose-regulated protein 75), has been proposed to mediate the molecular interaction of VDAC1 with the ER Ca2+-release channel, IP3R, and that chaperone-mediated conformational coupling of these proteins enables better transfer of the Ca2+ ions from the ER to the mitochondria (Szabadkai et al., 2006). In a cell-free system containing purified mitochondria, ER vesicles, IP3R, and VDAC1, PTP activity was identified as being involved in the ER-triggered pro-apoptotic mitochondrial membrane permeabilization process (Deniaud et al., 2008). It has been recently established that VDAC1 (but not isoforms 2 or 3) selectively interacts with IP3Rs and is preferentially involved in the transmission of low-amplitude apoptotic Ca2+ signals to mitochondria (De Stefani et al., 2012). The involvement of VDAC1 in ER–mitochondria Ca2+ cross-talk places VDAC1 at a crucial position on the route transferring Ca2+ signals from the ER to mitochondria, and thus couples ER and mitochondrial functions (De Stefani et al., 2012).
VDAC1, as a multi-functional channel involved both in metabolic homeostasis and apoptosis, can be considered as a prime target for therapeutic agents designed to act via metabolic interference or modulating apoptosis (Shoshan-Barmatz and Ben-Hail, 2011; Shoshan-Barmatz and Golan, 2012). Indeed, several studies identified pharmacological agents that target VDAC as inducing cancer cell death. These agents can be categorized into the following groups:
Reactive oxygen species has been shown to promote Cyto c release from mitochondria, thereby inducing apoptotic cell death (Petrosillo et al., 2001; Petrosillo et al., 2003) (see VDAC1 Function in ROS Release, ROS-mediated Apoptosis and Interaction with NO). Currently known drugs acting via enhancing ROS production include FNQs, erastin, and As2O3.
Furanonaphthoquinones were proposed to induce caspase-dependent apoptosis via the production of NADH-dependent ROS. In HeLa cells, FNQs induce mitochondrial swelling, with subsequent apoptosis, while radical scavengers and anti-VDAC1 antibodies reduced this effect (Simamura et al., 2008), suggesting that FNQ-mediated apoptosis is VDAC-dependent. This claim is further supported by the findings that the ROS production and the anti-cancer activity of FNQs were increased upon VDAC1 over-expression and decreased upon silencing of VDAC1 expression by siRNA (Simamura et al., 2008). FQNs were found to also up-regulate the pro-apoptotic proteins, Bad, Bax, and Cyto c, while down-regulating the anti-apoptotic proteins, Bcl-2, survivin, and XIAP in A549 cells (Su et al., 2010).
Erastin is a cell-permeable piperazinyl–quinazolinone compound that exhibits oncogene-selective lethality toward cells harboring activating mutations in the RAS–RAF–MEK signaling pathway. Erastin treatment in these cells rapidly induces ROS production and a non-apoptotic form of cell death (Yagoda et al., 2007). Knockdown of vdac2 or vdac3 caused resistance to erastin, suggesting their involvement in the mechanism of action of erastin (Yagoda et al., 2007). It was also reported that RSL5, a compound that has increased lethality in the presence of oncogenic RAS, and erastin induced VDAC3-dependent cell death (Yang and Stockwell, 2008). Finally, the erastin analog, PRLX 93936, was found to bind VDAC2 and VDAC3 using a proteomics approach (Sahasrabudhe et al., 2008). PRLX 93936 is currently in clinical Phase I for treatment of solid tumors.
As2O3, a clinically active anti-leukemia agent reported to inhibit mitochondrial respiratory function, increases ROS generation, and enhances the activity of other O2.- producing agents against cultured leukemia cells and primary leukemia cells isolated from patients (Pelicano et al., 2003).
Various compounds interact with VDAC to modify its activity (Table 2). Acrolein (2-propen-1-al), the most reactive of the α,β-unsaturated aldehydes and a toxic compound (Adams and Klaidman, 1993) that was proposed to react with DNA and proteins, preferential binds at p53 mutational hotspots to inhibit DNA repair (Feng et al., 2006). Acrolein is a respiratory irritant that is generated during cooking and is abundant in the environment (Izard and Libermann, 1978). The compound is also endogenously generated by lipid peroxidation at sites of injury (Adams and Klaidman, 1993). In pathological conditions associated with oxidative stress (Voulgaridou et al., 2011), like spinal cord injury (Luo et al., 2005) and Alzheimer’s disease (Dang et al., 2010), levels of acrolein were found to be elevated. Acrolein is reported to enhance carbonylation of VDAC, an event that was found to be significantly increased in the brains of Alzheimer’s disease patients (Mello et al., 2007).
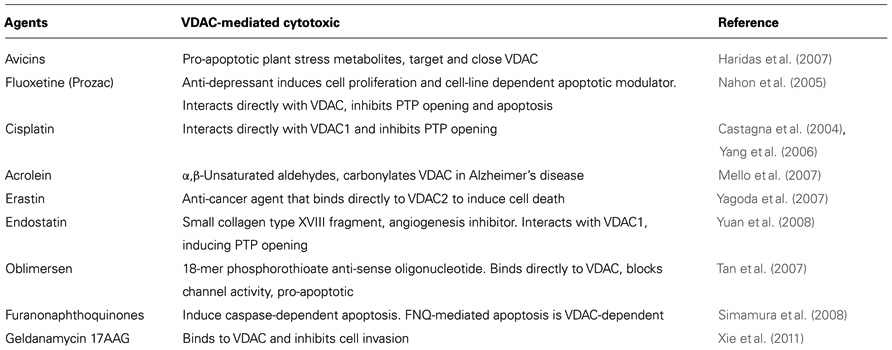
TABLE 2. Reagents proposed to act via VDAC (for details, see VDAC as a Pharmacological Target for Compounds Affecting Cell Proliferation or Apoptosis).
Avicins represents a family of triterpenoid saponins, plant stress metabolites that exhibit cytotoxic activity in tumor cells. These compounds possess anti-inflammatory and anti-oxidant properties capable of perturbing mitochondrial function and initiating apoptosis in tumor cells (Gaikwad et al., 2005; Haridas et al., 2005). Avicin G reduces VDAC1 channel conductance, ATP levels, and respiration rates in Jurkat cells (Lemeshko et al., 2006; Haridas et al., 2007).
Cisplatin (cis-diamminedichloro platinum(II)) is a widely used anti-cancer drug that trigger apoptosis (Santin et al., 2011). Cisplatin acts by forming inter- and intra-strand nuclear DNA cross-links upon binding (Zamarin et al., 2005). Mitochondria, however, have been implicated as a cisplatin target, leading to apoptosis induction (Yang et al., 2006; Cullen et al., 2007). Cisplatin binds to mitochondrial DNA and VDAC (Yang et al., 2006) to regulate VDAC activity (Castagna et al., 2004). Thus, it was proposed that VDAC acts as a cisplatin receptor during apoptosis (Thinnes, 2009). Recently it was suggested that over-expression of SCIN, a calcium-dependent actin-binding protein, is a novel cisplatin-resistant marker in the human bladder cancer cell line, HT1376 (Miura et al., 2012). It was proposed that the high expression of SCIN and its interaction with VDAC mediates resistance to cisplatin (Miura et al., 2012).
Endostatin is a naturally occurring 20 kDa C-terminal fragment derived from type XVIII collagen that may interfere with the pro-angiogenic action of growth factors (Folkman, 2006). Endostatin has a wide range of anti-cancer spectrum targets (Karamouzis and Moschos, 2009). Decreasing VDAC1 levels by siRNA attenuates endostatin-induced apoptosis, while over-expression of VDAC1 enhances the sensitivity of endothelial cells to endostatin (Yuan et al., 2008). Endostatin was found to induce PTP opening through VDAC1 (Yuan et al., 2008). These findings point to VDAC1 as endostatin target.
Fluoxetine (also known as Prozac, Sarafem, Fontex) is an anti-depressant, acting as a selective serotonin re-uptake inhibitor and is clinically used to treat different major depressions, obsessive-compulsive disorder, bulimia nervosa, and panic disorders, among other psychiatric conditions (Wong et al., 1995). The connection between fluoxetine and apoptosis is clouded by a series of contradictory findings. Fluoxetine was reported to enhance apoptosis in different cancer cells and tumors (Cloonan et al., 2010; Lee et al., 2010; Frick et al., 2011), yet was suggested to prevent apoptosis in both cancerous and neuronal cells (Wright et al., 1994; Lee et al., 2001; Chiou et al., 2006; Wang et al., 2011). Fluoxetine was shown to interact with VDAC1 and to increase the voltage-dependence of bilayer-reconstituted VDAC1 (Nahon et al., 2005; Thinnes, 2005). Additionally, fluoxetine was shown to prevent PTP opening and inhibited the release of Cyto c and apoptosis (Nahon et al., 2005). Although its remains unclear as to whether fluoxetine has an anti- or pro-apoptotic effect, it does seem that the compound mediates its effect on apoptosis via interactions with VDAC1.
Oblimersen (G3139) is an 18-mer phosphorothioate anti-sense oligonucleotide being studied as a possible treatment for several types of cancer, including CLL (Advani et al., 2011), B cell lymphoma (Pro et al., 2008), breast cancer (Rom et al., 2009), and melanoma (Lai et al., 2006). Although oblimersen is designed to target the initiation codon region of Bcl-2 mRNA, it was found to directly interact with VDAC1 and reduce its channel activity (Lai et al., 2006; Tan et al., 2007). Oblimersen is reported to induce caspase-dependent apoptosis via the intrinsic mitochondrial pathway in a Bcl-2-independent mechanism (Lai et al., 2006).
Geldanamycin (17AAG) is a derivative of the antibiotic, ansamycin. Geldanamycin binds to a conserved pocket in the amino-terminal portion of the Hsp90 chaperone protein and inhibits its function (Isaacs et al., 2003). In vitro, 17AAG acted as a Ca2+ mitochondrial regulator, increasing intracellular Ca2+ and diminishing the plasma membrane cationic current (Xie et al., 2011). 17AAG was shown to interact with VDAC through a hydrophobic interaction that was independent of Hsp90. This interaction was proposed to mediate inhibition of glioblastoma cell invasion (Xie et al., 2011).
In recent years, we have witnessed a switch in the focus of VDAC research from channel activity properties and regulation at the level of purified protein in yeast and Neurospora to studying its function in mammalian cell cultures and animal models. Indeed, a significant accumulation of knowledge is now available concerning VDAC1 function in cell life and death. Much has been learned about VDAC function in regulating metabolite cross-talk between the mitochondria and rest of the cell, its function in mitochondria–ER Ca2+ signaling, its function in apoptosis and its involvement in various maladies from neurodegenerative diseases to cancer. The studies reviewed above have demonstrated the pivotal roles of mitochondria and VDAC1 in cancer. The dual functions of VDAC1 in both metabolism and apoptosis, as mediated by the protein alone and regulated by associated polypeptides, point to VDAC1 as begin a critical target in addressing cancer therapy.
Although a high-resolution structure was determined for recombinant VDAC1, many questions concerning the architecture of the channel pore, the location of modulator-binding sites, the protein regions involved in the control mechanism of VDAC1 oligomerization and how VDAC is regulated to serve its multiple roles remain unanswered. Likewise, additional studies are required to investigate whether the increased dependence of cancer cells on glycolytic metabolism can be exploited therapeutically.
VDAC1 is integral to mitochondrial ATP production as a metabolite transporter, as the docking site for mitochondrial-bound HK, and is highly expressed in cancers. VDAC1 is associated with the mitochondrial pathway of apoptosis, interacting with over-expressed anti-apoptotic proteins present in cancer and mediating the actions of some anti-cancer drugs. In addition, VDAC1 mediates cholesterol transport and distribution in the mitochondrial membrane, with cancer cells exhibiting several fold higher cholesterol levels than do healthy cells. VDAC1 also functions in ROS production and transport to the cytosol, with elevated ROS generation being seen in cancer cells. These functions, together with VDAC1 being over-expressed in some cancer cells, point to VDAC1 as being a rational target for the development of a new generation of therapeutics.
Delineating the molecular mechanisms underlying the re-programmed metabolism seen in cancers and their cell survival strategies, together with increasing understanding of the difficulty in targeting signal transduction in tumors without significant side-effects, encourages prospects for the development of effective cancer therapies. Thus, the emergence of targeted therapies specifically targeting critical molecules important for cancer cells may provide promising means of selectively killing these malignant cells. VDAC1 is one such a target. Indeed, various strategies involving interference RNA to down-regulate VDAC1 expression levels, VDAC1-based peptides and small molecules, such as methyl jasmonate, are being considered in this capacity. Finally, the discovery that VDAC plays a role at critical control points during energy metabolism and in apoptosis points to VDAC as a novel pharmacologic target for anti-cancer drugs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by grants from the Israel Science Foundation and the Israel Ministry of Health. We thank Danya Ben-Hail and NuritKeinan for providing some of the figures.
AIF, apoptosis-inducing factor; ANT, adenine nucleotide translocase; APAF-1, apoptosis protease-activating factor 1; Bcl-2, B cell lymphoma 2; caspase, cysteinyl/aspartate-specific protease; CLL, chronic lymphocytic leukemia; CyP D, cyclophilin D; Cyto c, cytochrome c; DIDS, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid; ER, endoplasmic reticulum; FNQs, furanonaphthoquinones; G-6-P, glucose-6-phosphate; HK, hexokinase; IMM, inner mitochondrial membrane; IP3Rs, inositol-1,4,5-trisphosphate receptors; LDH, lactate dehydrogenase; MAC, mitochondrial apoptosis-inducing channel; NSCLC, non-small cell lung cancer; OMM, outer mitochondrial membrane; PDH, pyruvate dehydrogenase; PLB, planar lipid bilayer; PT, permeability transition; PTP, permeability transition pore; ROS, reactive oxygen species; RuR, ruthenium red; SR, sarcoplasmic reticulum; STS, staurosporine; TNF-α, tumor necrosis factor alpha; TSPO, translocator protein; UCP, uncoupling protein; VDAC, voltage-dependent anion channel.
Abu-Hamad, S., Arbel, N., Calo, D., Arzoine, L., Israelson, A., Keinan, N., Ben-Romano, R., et al. (2009). The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 122(Pt 11), 1906–1916.
Abu-Hamad, S., Sivan, S., and Shoshan-Barmatz, V. (2006). The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. U.S.A. 103, 5787–5792.
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490.
Adachi, M., Higuchi, H., Miura, S., Azuma, T., Inokuchi, S., Saito, H., et al. (2004). Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G695–G705.
Adams, J. D. Jr., and Klaidman, L. K. (1993). Acrolein-induced oxygen radical formation. Free Radic. Biol. Med. 15, 187–193.
Adams, J. M., and Cory, S. (2007). The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337.
Adams, V., Griffin, L., Towbin, J., Gelb, B., Worley, K., and McCabe, E. R. (1991). Porin interaction with hexokinase and glycerol kinase: metabolic microcompartmentation at the outer mitochondrial membrane. Biochem. Med. Metab. Biol. 45, 271–291.
Advani, P. P., Paulus, A., Masood, A., Sher, T., and Chanan-Khan, A. (2011). Pharmacokinetic evaluation of oblimersen sodium for the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. 7, 765–774.
Ahmad, A., Ahmad, S., Schneider, B. K., Allen, C. B., Chang, L. Y., and White, C. W. (2002). Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L573–L584.
Al-Abdulla, N. A., Portera-Cailliau, C., and Martin, L. J. (1998). Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience 86, 191–209.
Andre, N., Braguer, D., Brasseur, G., Gonçalves, A., Lemesle-Meunier, D., Guise, S., et al. (2000). Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells’. Cancer Res. 60, 5349–5353.
Anflous-Pharayra, K., Lee, N., Armstrong, D. L., and Craigen, W. J. (2010). VDAC3 has differing mitochondrial functions in two types of striated muscles. Biochim. Biophys. Acta 1807, 150–156.
Antignani, A., and Youle, R. J. (2006). How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 18, 685–689.
Antonsson, B. (2001). Bax and other pro-apoptotic Bcl-2 family “killer-proteins” and their victim the mitochondrion. Cell Tissue Res. 306, 347–361.
Antonsson, B., Montessuit, S., Lauper, S., Eskes, R., and Martinou, J. C. (2000). Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 345(Pt 2), 271–278.
Antonsson, B., Montessuit, S., Sanchez, B., and Martinou, J. C. (2001). Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276, 11615–11623.
Arbel, N., Ben-Hail, D., and Shoshan-Barmatz, V. (2012). Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 287, 23152–23161.
Arbel, N., and Shoshan-Barmatz, V. (2010). Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 285, 6053–6062.
Arzoine, L., Zilberberg, N., Ben-Romano, R., and Shoshan-Barmatz, V. (2009). Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 284, 3946–3955.
Auten, R. L., and Davis, J. M. (2009). Oxygen toxicity and reactive oxygen species: the devil is in the details. Pediatr. Res. 66, 121–127.
Azoulay-Zohar, H., and Aflalo, C. (1999). Binding of rat brain hexokinase to recombinant yeast mitochondria: identification of necessary molecular determinants. J. Bioenerg. Biomembr. 31, 569–579.
Azoulay-Zohar, H., Israelson, A., Abu-Hamad, S., and Shoshan-Barmatz, V. (2004). In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 377(Pt 2), 347–355.
Bae, J. H., Park, J. W., and Kwon, T. K. (2003). Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem. Biophys. Res. Commun. 303, 1073–1079.
Baek, J. H., Lee, Y. S., Kang, C. M., Kim, J. A., Kwon, K. S., Son, H. C., et al. (1997). Intracellular Ca2+ release mediates ursolic acid-induced apoptosis in human leukemic HL-60 cells. Int. J. Cancer 73, 725–728.
Baggetto, L. G., Clottes, E., and Vial, C. (1992). Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res. 52, 4935–4941.
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555.
Banerjee, J., and Ghosh, S. (2004). Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 323, 310–314.
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067.
Batarseh, A., and Papadopoulos, V. (2010). Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 327, 1–12.
Bathori, G., Csordás, G., Garcia-Perez, C., Davies, E., and Hajnóczky, G. (2006). Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 281, 17347–17358.
Bathori, G., Parolini, I., Szabó, I., Tombola, F., Messina, A., Oliva, M., et al. (2000). Extramitochondrial porin: facts and hypotheses. J. Bioenerg. Biomembr. 32, 79–89.
Bathori, G., Parolini, I., Tombola, F., Szabò, I., Messina, A., Oliva, M., et al. (1999). Porin is present in the plasma membrane where it is concentrated in caveolae and caveolae-related domains. J. Biol. Chem. 274, 29607–29612.
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345.
Baumgartner, H. K., Gerasimenko, J. V., Thorne, C., Ferdek, P., Pozzan, T., Tepikin, A. V., et al. (2009). Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 284, 20796–20803.
Bayrhuber, M., Meins, T., Habeck, M., Becker, S., Giller, K., Villinger, S., et al. (2008). Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 105, 15370–15375.
Belizario, J. E., Alves, J., Occhiucci, J. M., Garay-Malpartida, M., and Sesso, A. (2007). A mechanistic view of mitochondrial death decision pores. Braz. J. Med. Biol. Res. 40, 1011–1024.
Belzacq, A. S., Jacotot, E., Vieira, H. L., Mistro, D., Granville, D. J., Xie, Z., Reed, J. C., et al. (2001). Apoptosis induction by the photosensitizer verteporfin: identification of mitochondrial adenine nucleotide translocator as a critical target. Cancer Res. 61, 1260–1264.
Benz, R. (1994). Permeation of hydrophilic solutes through mitochondrial outer membranes: review on mitochondrial porins. Biochim. Biophys. Acta 1197, 167–196.
Bernardi, P. (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21.
Berridge, M. V., Herst, P. M., and Lawen, A. (2009). Targeting mitochondrial permeability in cancer drug development. Mol. Nutr. Food Res. 53, 76–86.
Berridge, M. V., Herst, P. M., and Tan, A. S. (2010). Metabolic flexibility and cell hierarchy in metastatic cancer. Mitochondrion 10, 584–588.
Berry, M. D., and Boulton, A. A. (2000). Glyceraldehyde-3-phosphate dehydrogenase and apoptosis. J. Neurosci. Res. 60, 150–154.
Betaneli, V., Petrov, E. P., and Schwille, P. (2012). The role of lipids in VDAC oligomerization. Biophys. J. 102, 523–531.
Beutner, G., Rück, A., Riede, B., and Brdiczka, D. (1998). Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1368, 7–18.
Beutner, G., Ruck, A., Riede, B., Welte, W., and Brdiczka, D. (1996). Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 396, 189–195.
Birbes, H., Luberto, C., Hsu, Y. T., El Bawab, S., Hannun, Y. A., and Obeid, L. M. (2005). A mitochondrial pool of sphingomyelin is involved in TNFalpha-induced Bax translocation to mitochondria. Biochem. J. 386(Pt 3), 445–451.
Blachly-Dyson, E., Zambronicz, E. B., Yu, W. H., Adams, V., McCabe, E. R., Adelman, J., et al. (1993). Cloning and functional expression in yeast of two human isoforms of the outer mitochondrial membrane channel, the voltage-dependent anion channel. J. Biol. Chem. 268, 1835–1841.
Bononi, A., Missiroli, S., Poletti, F., Suski, J. M., Agnoletto, C., Bonora, M., et al. (2012). Mitochondria-associated membranes (MAMs) as hotspot Ca(2+) signaling units. Adv. Exp. Med. Biol. 740, 411–437.
Bootman, M. D., Petersen, O. H., and Verkhratsky, A. (2002). The endoplasmic reticulum is a focal point for co-ordination of cellular activity. Cell Calcium 32, 231–234.
Boya, P., Pauleau, A. L., Poncet, D., Gonzalez-Polo, R. A., Zamzami, N., and Kroemer, G. (2004). Viral proteins targeting mitochondria: controlling cell death. Biochim. Biophys. Acta 1659, 178–189.
Boya, P., Roques, B., and Kroemer, G. (2001). New EMBO members’ review: viral and bacterial proteins regulating apoptosis at the mitochondrial level. EMBO J. 20, 4325–4331.
Boya, P., Roumier, T., Andreau, K., Gonzalez-Polo, R. A., Zamzami, N., Castedo, M., et al. (2003). Mitochondrion-targeted apoptosis regulators of viral origin. Biochem. Biophys. Res. Commun. 304, 575–581.
Brahimi-Horn, M. C., Ben-Hail, D., Ilie, M., Gounon, P., Rouleau, M., Hofman, V., et al. (2012). Expression of a truncated active form of VDAC1 in lung cancer associates with hypoxic cell survival and correlates with progression to chemotherapy resistance. Cancer Res. 72, 2140–2150.
Brahimi-Horn, M. C., Chiche, J., and Pouyssegur, J. (2007). Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229.
Brdiczka, D., Kaldis, P., and Wallimann, T. (1994). In vitro complex formation between the octamer of mitochondrial creatine kinase and porin. J. Biol. Chem. 269, 27640–27644.
Brdiczka, D. G., Zorov, D. B., and Sheu, S. S. (2006). Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim. Biophys. Acta 1762, 148–163.
Brookes, P. S., Salinas, E. P., Darley-Usmar, K., Eiserich, J. P., Freeman, B. A., Darley-Usmar, V. M., et al. (2000). Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J. Biol. Chem. 275, 20474–20479.
Brune, B., von Knethen, A., and Sandau, K. B. (1999). Nitric oxide (NO): an effector of apoptosis. Cell Death Differ. 6, 969–975.
Brustovetsky, N., Dubinsky, J. M., Antonsson, B., and Jemmerson, R. (2003). Two pathways for tBID-induced cytochrome c release from rat brain mitochondria: BAK- versus BAX-dependence. J. Neurochem. 84, 196–207.
Bryson, J. M., Coy, P. E., Gottlob, K., Hay, N., and Robey, R. B. (2002). Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J. Biol. Chem. 277, 11392–11400.
Buettner, R., Papoutsoglou, G., Scemes, E., Spray, D. C., and Dermietzel, R. (2000). Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc. Natl. Acad. Sci. U.S.A. 97, 3201–3206.
Bühler, S., Michels, J., Wendt, S., Rück, A., Brdiczka, D., and Welte, W. (1998). Mass spectrometric mapping of ion channel proteins (porins) and identification of their supramolecular membrane assembly. Proteins 33(Suppl. 2), 63–73.
Bustamante, E., and Pedersen, P. L. (1980). Mitochondrial hexokinase of rat hepatoma cells in culture: solubilization and kinetic properties. Biochemistry 19, 4972–4977.
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95.
Campbell, A. M., and Chan, S. H. (2008). Mitochondrial membrane cholesterol, the voltage dependent anion channel (VDAC), and the Warburg effect. J. Bioenerg. Biomembr. 40, 193–197.
Capano, M., and Crompton, M. (2002). Biphasic translocation of Bax to mitochondria. Biochem. J. 367(Pt 1), 169–178.
Cárdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J., Müller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283.
Carre, M., André, N., Carles, G., Borghi, H., Brichese, L., Briand, C., et al. (2002). Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 277, 33664–33669.
Casadio, R., Jacoboni, I., Messina, A., and De Pinto, V. (2002). A 3D model of the voltage-dependent anion channel (VDAC). FEBS Lett. 520, 1–7.
Cassara, M. C., Menzel, V. A., Hinsch, K. D., Wrenzycki, C., and Hinsch, E. (2010). Voltage-dependent anion channels 1 and 2 are expressed in porcine oocytes. Biosci. Rep. 30, 193–200.
Castagna, A., Antonioli, P., Astner, H., Hamdan, M., Righetti, S. C., Perego, P., et al. (2004). A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics 4, 3246–3267.
Castedo, M., Perfettini, J. L., Andreau, K., Roumier, T., Piacentini, M., and Kroemer, G. (2003). Mitochondrial apoptosis induced by the HIV-1 envelope. Ann. N. Y. Acad. Sci. 1010, 19–28.
Certo, M., Del Gaizo Moore, V., Nishino, M., Wei, G., Korsmeyer, S., Armstrong, S. A., et al. (2006). Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365.
Cesar Mde, C., and Wilson, J. E. (2004). All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2, and VDAC3) are present in mitochondria from bovine, rabbit, and rat brain. Arch. Biochem. Biophys. 422, 191–196.
Chacko, A. D., Liberante, F., Paul, I., Longley, D. B., and Fennell, D. A. (2010). Voltage dependent anion channel-1 regulates death receptor mediated apoptosis by enabling cleavage of caspase-8. BMC Cancer 10, 380. doi: 10.1186/1471-2407-10-380
Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003). VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517.
Cheng, Q., Sedlic, F., Pravdic, D., Bosnjak, Z. J., and Kwok, W. M. (2011). Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 585, 328–334.
Cheng, S. L., Liu, R. H., Sheu, J. N., Chen, S. T., Sinchaikul, S., and Tsay, G. J. (2007). Toxicogenomics of A375 human malignant melanoma cells treated with arbutin. J. Biomed. Sci. 14, 87–105.
Chiou, S. H., Chen, S. J., Peng, C. H., Chang, Y. L., Ku, H. H., Hsu, W. M., et al. (2006). Fluoxetine up-regulates expression of cellular FLICE-inhibitory protein and inhibits LPS-induced apoptosis in hippocampus-derived neural stem cell. Biochem. Biophys. Res. Commun. 343, 391–400.
Chittenden, T., Harrington, E. A., O’Connor, R., Flemington, C., Lutz, R. J., Evan, G. I., et al. (1995). Induction of apoptosis by the Bcl-2 homologue Bak. Nature 374, 733–736.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840.
Choudhary, O. P., Ujwal, R., Kowallis, W., Coalson, R., Abramson, J., and Grabe, M. (2010). The electrostatics of VDAC: implications for selectivity and gating. J. Mol. Biol. 396, 580–592.
Chowdhury, I., Tharakan, B., and Bhat, G. K. (2006). Current concepts in apoptosis: the physiological suicide program revisited. Cell. Mol. Biol. Lett. 11, 506–525.
Cloonan, S. M., Drozgowska, A., Fayne, D., and Williams, D. C. (2010). The antidepressants maprotiline and fluoxetine have potent selective antiproliferative effects against Burkitt lymphoma independently of the norepinephrine and serotonin transporters. Leuk. Lymphoma 51, 523–539.
Colombini, M. (1980). Structure and mode of action of a voltage dependent anion-selective channel (VDAC) located in the outer mitochondrial membrane. Ann. N. Y. Acad. Sci. 341, 552–563.
Colombini, M. (1989). Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 111, 103–111.
Colombini, M. (2004). VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 256–257, 107–115.
Colombini, M. (2009). The published 3D structure of the VDAC channel: native or not? Trends Biochem. Sci. 34, 382–389.
Cory, S., and Adams, J. M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656.
Cory, S., Huang, D. C., and Adams, J. M. (2003). The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22, 8590–8607.
Costantini, P., Jacotot, E., Decaudin, D., and Kroemer, G. (2000). Mitochondrion as a novel target of anticancer chemotherapy. J. Natl. Cancer Inst. 92, 1042–1053.
Cox, D. A., and Matlib, M. A. (1993). A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J. Biol. Chem. 268, 938–947.
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341(Pt 2), 233–249.
Csordas, G., Madesh, M., Antonsson, B., and Hajnóczky, G. (2002). tcBid promotes Ca(2+) signal propagation to the mitochondria: control of Ca(2+) permeation through the outer mitochondrial membrane. EMBO J. 21, 2198–2206.
Cullen, K. J., Yang, Z., Schumaker, L., and Guo, Z. (2007). Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J. Bioenerg. Biomembr. 39, 43–50.
Dang, T. N., Arseneault, M., Murthy, V., and Ramassamy, C. (2010). Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr. Mol. Pharmacol. 3, 66–78.
Danishuddin, M., Khan, S. N., and Khan, A. U. (2009). Molecular interactions between mitochondrial membrane proteins and the C-terminal domain of PB1-F2: an in silico approach. J. Mol. Model. 16, 535–541.
da-Silva, W. S., Gómez-Puyou, A., de Gómez-Puyou, M. T., Moreno-Sanchez, R., De Felice, F. G., de Meis, L., et al. (2004). Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 279, 39846–39855.
Debatin, K. M., Poncet, D., and Kroemer, G. (2002). Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene 21, 8786–8803.
Decuypere, J. P., Monaco, G., Missiaen, L., De Smedt, H., Parys, J. B., and Bultynck, G. (2011). IP(3) receptors, mitochondria, and Ca signaling: implications for aging. J. Aging Res. 2011, 920178.
Dejean, L. M., Martinez-Caballero, S., Guo, L., Hughes, C., Teijido, O., Ducret, T., et al. (2005). Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol. Biol. Cell 16, 2424–2432.
Dejean, L. M., Martinez-Caballero, S., and Kinnally, K. W. (2006a). Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 13, 1387–1395.
Dejean, L. M., Martinez-Caballero, S., Manon, S., and Kinnally, K. W. (2006b). Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim. Biophys. Acta 1762, 191–201.
Deniaud, A., Sharaf el dein, O., Maillier, E., Poncet, D., Kroemer, G., Lemaire, C., et al. (2008). Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299.
Denning, M. F., Wang, Y., Tibudan, S., Alkan, S., Nickoloff, B. J., Qin, J. Z., et al. (2002). Caspase activation and disruption of mitochondrial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ. 9, 40–52.
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316.
De Pinto, V., Benz, R., and Palmieri, F. (1989). Interaction of non-classical detergents with the mitochondrial porin. A new purification procedure and characterization of the pore-forming unit. Eur. J. Biochem. 183, 179–187.
De Pinto, V., Guarino, F., Guarnera, A., Messina, A., Reina, S., Tomasello, F. M., et al. (2010a). Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim. Biophys. Acta 1797, 1268–1275.
De Pinto, V., Messina, A., Lane, D. J., and Lawen, A. (2010b). Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 584, 1793–1799.
De Pinto, V., Messina, A., Accardi, R., Aiello, R., Guarino, F., Tomasello, M. F., et al. (2003). New functions of an old protein: the eukaryotic porin or voltage dependent anion selective channel (VDAC). Ital. J. Biochem. 52, 17–24.
De Pinto, V., Prezioso, G., Thinnes, F., Link, T. A., and Palmieri, F. (1991). Peptide-specific antibodies and proteases as probes of the transmembrane topology of the bovine heart mitochondrial porin. Biochemistry 30, 10191–10200.
De Pinto, V., Reina, S., Guarino, F., and Messina, A. (2008). Structure of the voltage dependent anion channel: state of the art. J. Bioenerg. Biomembr. 40, 139–147.
De Pinto, V., Tomasello, F., Messina, A., Guarino, F., Benz, R., La Mendola, D., et al. (2007). Determination of the conformation of the human VDAC1 N-terminal peptide, a protein moiety essential for the functional properties of the pore. Chembiochem 8, 744–756.
Desagher, S., Osen-Sand, A., Nichols, A., Eskes, R., Montessuit, S., Lauper, S., et al. (1999). Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144, 891–901.
De Stefani, D., Bononi, A., Romagnoli, A., Messina, A., De Pinto, V., Pinton, P., et al. (2012). VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 19, 267–273.
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340.
Ding, W., Hudson, L. G., and Liu, K. J. (2005). Inorganic arsenic compounds cause oxidative damage to DNA and protein by inducing ROS and RNS generation in human keratinocytes. Mol. Cell. Biochem. 279, 105–112.
Ding, W. X., Shen, H. M., and Ong, C. N. (2001). Pivotal role of mitochondrial Ca(2+) in microcystin-induced mitochondrial permeability transition in rat hepatocytes. Biochem. Biophys. Res. Commun. 285, 1155–1161.
Distler, A. M., Kerner, J., and Hoppel, C. L. (2007). Post-translational modifications of rat liver mitochondrial outer membrane proteins identified by mass spectrometry. Biochim. Biophys. Acta 1774, 628–636.
Dlugosz, P. J., Billen, L. P., Annis, M. G., Zhu, W., Zhang, Z., Lin, J., et al. (2006). Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 25, 2287–2296.
Dolder, M., Zeth, K., Tittmann, P., Gross, H., Welte, W., and Wallimann, T. (1999). Crystallization of the human, mitochondrial voltage-dependent anion-selective channel in the presence of phospholipids. J. Struct. Biol. 127, 64–71.
Eskes, R., Desagher, S., Antonsson, B., and Martinou, J. C. (2000). Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 20, 929–935.
Everett, H., and McFadden, G. (2001). Viruses and apoptosis: meddling with mitochondria. Virology 288, 1–7.
Fadeel, B., Ottosson, A., and Pervaiz, S. (2008). Big wheel keeps on turning: apoptosome regulation and its role in chemoresistance. Cell Death Differ. 15, 443–452.
Fanciulli, M., Paggi, M. G., Bruno, T., Del Carlo, C., Bonetto, F., Gentile, F. P., et al. (1994). Glycolysis and growth rate in normal and in hexokinase-transfected NIH-3T3 cells. Oncol. Res. 6, 405–409.
Feldmann, G., Haouzi, D., Moreau, A., Durand-Schneider, A. M., Bringuier, A., Berson, A., et al. (2000). Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology 31, 674–683.
Feng, Z., Hu, W., Hu, Y., and Tang, M. S. (2006). Acrolein is a major cigarette-related lung cancer agent: preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. U.S.A. 103, 15404–15409.
Folkman, J. (2006). Antiangiogenesis in cancer therapy – endostatin and its mechanisms of action. Exp. Cell Res. 312, 594–607.
Franklin-Tong, V. E., and Gourlay, C. W. (2008). A role for actin in regulating apoptosis/programmed cell death: evidence spanning yeast, plants and animals. Biochem. J. 413, 389–404.
Frick, L. R., Rapanelli, M., Arcos, M. L., Cremaschi, G. A., and Genaro, A. M. (2011). Oral administration of fluoxetine alters the proliferation/apoptosis balance of lymphoma cells and up-regulates T cell immunity in tumor-bearing mice. Eur. J. Pharmacol. 659, 265–272.
Fulda, S., Galluzzi, L., and Kroemer, G. (2010). Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464.
Fulda, S., Scaffidi, C., Susin, S. A., Krammer, P. H., Kroemer, G., Peter, M. E., et al. (1998). Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 273, 33942–33948.
Gaikwad, A., Poblenz, A., Haridas, V., Zhang, C., Duvic, M., and Gutterman, J. (2005). Triterpenoid electrophiles (avicins) suppress heat shock protein-70 and x-linked inhibitor of apoptosis proteins in malignant cells by activation of ubiquitin machinery: implications for proapoptotic activity. Clin. Cancer Res. 11, 1953–1962.
Galluzzi, L., and Kroemer, G. (2007). Mitochondrial apoptosis without VDAC. Nat. Cell Biol. 9, 487–489.
Gatenby, R. A., and Gillies, R. J. (2004). Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899.
Gauci, S., Helbig, A. O., Slijper, M., Krijgsveld, J., Heck, A. J., and Mohammed, S. (2009). Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 81, 4493–4501.
Gavish, M., Bachman, I., Shoukrun, R., Katz, Y., Veenman, L., Weisinger, G., et al. (1999). Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 51, 629–650.
Gelb, B. D., Adams, V., Jones, S. N., Griffin, L. D., MacGregor, G. R., and McCabe, E. R. (1992). Targeting of hexokinase 1 to liver and hepatoma mitochondria. Proc. Natl. Acad. Sci. U.S.A. 89, 202–206.
Genini, D., Adachi, S., Chao, Q., Rose, D. W., Carrera, C. J., Cottam, H. B., et al. (2000). Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood 96, 3537–3543.
Gerber, D., and Shai, Y. (2001). In vivo detection of hetero-association of glycophorin-A and its mutants within the membrane. J. Biol. Chem. 276, 31229–31232.
Geula, S., Ben-Hail, D., and Shoshan-Barmatz, V. (2012a). Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 444, 475–485.
Geula, S., Naveed, H., Liang, J., and Shoshan-Barmatz, V. (2012b). Structure-based analysis of VDAC1 protein: defining oligomer contact sites. J. Biol. Chem. 287, 2179–2190.
Ghosh, T., Pandey, N., Maitra, A., Brahmachari, S. K., and Pillai, B. (2007). A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS ONE 2, e1170. doi: 10.1371/journal.pone.0001170
Giacomello, M., Drago, I., Pizzo, P., and Pozzan, T. (2007). Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 14, 1267–1274.
Gincel, D., and Shoshan-Barmatz, V. (2004). Glutamate interacts with VDAC and modulates opening of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 36, 179–186.
Gincel, D., Silberberg, S. D., and Shoshan-Barmatz, V. (2000). Modulation of the voltage-dependent anion channel (VDAC) by glutamate. J. Bioenerg. Biomembr. 32, 571–583.
Gincel, D., Zaid, H., and Shoshan-Barmatz, V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358(Pt 1), 147–155.
Godbole, A., Varghese, J., Sarin, A., and Mathew, M. K. (2003). VDAC is a conserved element of death pathways in plant and animal systems. Biochim. Biophys. Acta 1642, 87–96.
Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2006). Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 1757, 639–647.
Gogvadze, V., Zhivotovsky, B., and Orrenius, S. (2010). The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 31, 60–74.
Golani, I., Weizman, A., Leschiner, S., Spanier, I., Eckstein, N., Limor, R., et al. (2001). Hormonal regulation of peripheral benzodiazepine receptor binding properties is mediated by subunit interaction. Biochemistry 40, 10213–10222.
Goldin, N., Arzoine, L., Heyfets, A., Israelson, A., Zaslavsky, Z., Bravman, T., et al. (2008). Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 27, 4636–4643.
Goncalves, R. P., Buzhynskyy, N., Prima, V., Sturgis, J. N., and Scheuring, S. (2007). Supramolecular assembly of VDAC in native mitochondrial outer membranes. J. Mol. Biol. 369, 413–418.
Gonzalez-Gronow, M., Kalfa, T., Johnson, C. E., Gawdi, G., and Pizzo, S. V. (2003). The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J. Biol. Chem. 278, 27312–27318.
Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R. B., and Hay, N. (2001). Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15, 1406–1418.
Grills, C., Jithesh, P. V., Blayney, J., Zhang, S. D., and Fennell, D. A. (2011). Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS ONE 6, e14635. doi: 10.1371/journal.pone.0014635
Gross, A., Jockel, J., Wei, M. C., and Korsmeyer, S. J. (1998). Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17, 3878–3885.
Gunter, T. E., Buntinas, L., Sparagna, G., Eliseev, R., and Gunter, K. (2000). Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28, 285–296.
Guo, L., Pietkiewicz, D., Pavlov, E. V., Grigoriev, S. M., Kasianowicz, J. J., Dejean, L. M., et al. (2004). Effects of cytochrome c on the mitochondrial apoptosis-induced channel MAC. Am. J. Physiol. Cell Physiol. 286, C1109–C1117.
Guo, X. W., Smith, P. R., Cognon, B., D’Arcangelis, D., Dolginova, E., Mannella, C. A., et al. (1995). Molecular design of the voltage-dependent, anion-selective channel in the mitochondrial outer membrane. J. Struct. Biol. 114, 41–59.
Gurnev, P. A., Queralt-Martin, M., Aguilella, V. M., Rostovtseva, T. K., and Bezrukov, S. M. (2012). Probing tubulin-blocked state of VDAC by varying membrane surface charge. Biophys. J. 102, 2070–2076.
Hajnoczky, G., Davies, E., and Madesh, M. (2003). Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 304, 445–454.
Halestrap, A. P., and Green, D. R. (2000). Mitochondria and cell death. Biochem. Soc. Trans. 28, 170–177.
Halestrap, A. P., McStay, G. P., and Clarke, S. J. (2002). The permeability transition pore complex: another view. Biochimie 84, 153–166.
Hamanaka, R. B., and Chandel, N. S. (2009). Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 21, 894–899.
Han, D., Antunes, F., Canali, R., Rettori, D., and Cadenas, E. (2003). Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Haridas, V., Li, X., Mizumachi, T., Higuchi, M., Lemeshko, V. V., and Colombini, M. (2007). Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion 7, 234–240.
Haridas, V., Sultana, R., Piroddi, M., Cai, J., Pierce, W. M., Klein, J. B., et al. (2005). Avicinylation (thioesterification): a protein modification that can regulate the response to oxidative and nitrosative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 10088–10093.
Haworth, R. A., and Hunter, D. R. (2000). Control of the mitochondrial permeability transition pore by high-affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. J. Bioenerg. Biomembr. 32, 91–96.
He, L., and Lemasters, J. J. (2003). Heat shock suppresses the permeability transition in rat liver mitochondria. J. Biol. Chem. 278, 16755–16760.
Hiller, S., Garces, R. G., Malia, T. J., Orekhov, V. Y., Colombini, M., and Wagner, G. (2008). Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210.
Hinsch, K. D., De Pinto, V., Aires, V. A., Schneider, X., Messina, A., and Hinsch, E. (2004). Voltage-dependent anion-selective channels VDAC2 and VDAC3 are abundant proteins in bovine outer dense fibers, a cytoskeletal component of the sperm flagellum. J. Biol. Chem. 279, 15281–15288.
Hirsch, T., Decaudin, D., Susin, S. A., Marchetti, P., Larochette, N., et al. (1998). PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp. Cell Res. 241, 426–434.
Holmuhamedov, E., and Lemasters, J. J. (2009). Ethanol exposure decreases mitochondrial outer membrane permeability in cultured rat hepatocytes. Arch. Biochem. Biophys. 481, 226–233.
Hoogenboom, B. W., Suda, K., Engel, A., and Fotiadis, D. (2007). The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J. Mol. Biol. 370, 246–255.
Hou, Q., Cymbalyuk, E., Hsu, S. C., Xu, M., and Hsu, Y. T. (2003). Apoptosis modulatory activities of transiently expressed Bcl-2: roles in cytochrome c release and Bax regulation. Apoptosis 8, 617–629.
Irusta, P. M., Chen, Y. B., and Hardwick, J. M. (2003). Viral modulators of cell death provide new links to old pathways. Curr. Opin. Cell Biol. 15, 700–705.
Isaacs, J. S., Xu, W., and Neckers, L. (2003). Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 3, 213–217.
Israelson, A., Zaid, H., Abu-Hamad, S., Nahon, E., and Shoshan-Barmatz, V. (2008). Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1. Cell Calcium 43, 196–204.
Jenei, Z. A., Borthwick, K., Zammit, V. A., and Dixon, A. M. (2009). Self-association of transmembrane domain 2 (TM2), but not TM1, in carnitine palmitoyltransferase 1A: role of GXXXG(A) motifs. J. Biol. Chem. 284, 6988–6997.
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147.
Jiang, N., Kham, S. K., Koh, G. S., Suang Lim, J. Y., Ariffin, H., Chew, F. T., et al. (2011). Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J. Proteomics 74, 843–157.
Johnstone, R. W., Ruefli, A. A., and Lowe, S. W. (2002). Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153–164.
Jung, J. Y., Han, C. R., Jeong, Y. J., Kim, H. J., Lim, H. S., Lee, K. H., et al. (2007). Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci. Lett. 411, 222–227.
Karamouzis, M. V., and Moschos, S. J. (2009). The use of endostatin in the treatment of solid tumors. Expert Opin. Biol. Ther. 9, 641–648.
Kayser, H., Kratzin, H. D., Thinnes, F. P., Götz, H., Schmidt, W. E., Eckart, K., et al. (1989). Identification of human porins. II. Characterization and primary structure of a 31-lDa porin from human B lymphocytes (Porin 31HL). Biol. Chem. Hoppe Seyler 370, 1265–1278.
Keeble, J. A., and Gilmore, A. P. (2007). Apoptosis commitment – translating survival signals into decisions on mitochondria. Cell Res. 17, 976–984.
Keinan, N., Tyomkin, D., and Shoshan-Barmatz, V. (2010). Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 30, 5698–5709.
Kerner, J., Lee, K., Tandler, B., and Hoppel, C. L. (2012). VDAC proteomics: post-translation modifications. Biochim. Biophys. Acta 1818, 1520–1525.
Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618.
Kim, Y. M., Bombeck, C. A., and Billiar, T. R. (1999). Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 84, 253–256.
Kim, Y. M., Kim, T. H., Seol, D. W., Talanian, R. V., and Billiar, T. R. (1998). Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release. J. Biol. Chem. 273, 31437–31441.
Kinnally, K. W., Peixoto, P. M., Ryu, S. Y., and Dejean, L. M. (2011). Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 1813, 616–622.
Ko, Y. H., and McFadden, B. A. (1990). Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate. Arch. Biochem. Biophys. 278, 373–380.
Ko, Y. H., Pedersen, P. L., and Geschwind, J. F. (2001). Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 173, 83–91.
Ko, Y. H., Verhoeven, H. A., Lee, M. J., Corbin, D. J., Vogl, T. J., and Pedersen, P. L. (2012). A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J. Bioenerg. Biomembr. 44, 163–170.
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465.
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337.
Koren, I., Raviv, Z., and Shoshan-Barmatz, V. (2010). Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. Ther. 9, 1046–1052.
Korkhov, V. M., Sachse, C., Short, J. M., and Tate, C. G. (2010). Three-dimensional structure of TspO by electron cryomicroscopy of helical crystals. Structure 18, 677–687.
Korsmeyer, S. J., Wei, M. C., Saito, M., Weiler, S., Oh, K. J., and Schlesinger, P. H. (2000). Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7, 1166–1173.
Krauskopf, A., Eriksson, O., Craigen, W. J., Forte, M. A., and Bernardi, P. (2006). Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim. Biophys. Acta 1757, 590–595.
Kroemer, G. (1997). The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3, 614–620.
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163.
Kroemer, G., and Pouyssegur, J. (2008). Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482.
Kugler, W., Veenman, L., Shandalov, Y., Leschiner, S., Spanier, I., Lakomek, M., et al. (2008). Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell. Oncol. 30, 435–450.
Kusano, H., Shimizu, S., Koya, R. C., Fujita, H., Kamada, S., Kuzumaki, N., et al. (2000). Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 19, 4807–4814.
Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., et al. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342.
Lai, J. C., Tan, W., Benimetskaya, L., Miller, P., Colombini, M., and Stein, C. A. (2006). A pharmacologic target of G3139 in melanoma cells may be the mitochondrial VDAC. Proc. Natl. Acad. Sci. U.S.A. 103, 7494–7499.
Larochette, N., Decaudin, D., Jacotot, E., Brenner, C., Marzo, I., Susin, S. A., et al. (1999). Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp. Cell Res. 249, 413–421.
Le Bras, M., Clément, M. V., Pervaiz, S., and Brenner, C. (2005). Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 20, 205–219.
Lee, A. C., Zizi, M., and Colombini, M. (1994). Beta-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J. Biol. Chem. 269, 30974–30980.
Lee, B., Miles, P. D., Vargas, L., Luan, P., Glasco, S., Kushnareva, Y., et al. (2003). Inhibition of mitochondrial Na+-Ca2+ exchanger increases mitochondrial metabolism and potentiates glucose-stimulated insulin secretion in rat pancreatic islets. Diabetes 52, 965–973.
Lee, C. S., Kim, Y. J., Jang, E. R., Kim, W., and Myung, S. C. (2010). Fluoxetine induces apoptosis in ovarian carcinoma cell line OVCAR-3 through reactive oxygen species-dependent activation of nuclear factor-kappaB. Basic Clin. Pharmacol. Toxicol. 106, 446–453.
Lee, H. J., Kim, J. W., Yim, S. V., Kim, M. J., Kim, S. A., Kim, Y. J., et al. (2001). Fluoxetine enhances cell proliferation and prevents apoptosis in dentate gyrus of maternally separated rats. Mol. Psychiatry 6, 725–728.
Lemasters, J. J., and Holmuhamedov, E. (2006). Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim. Biophys. Acta 1762, 181–190.
Lemasters, J. J., Qian, T., Bradham, C. A., Brenner, D. A., Cascio, W. E., Trost, L. C., et al. (1999). Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J. Bioenerg. Biomembr. 31, 305–319.
Le Mellay, V., Troppmair, J., Benz, R., and Rapp, U. R. (2002). Negative regulation of mitochondrial VDAC channels by C-Raf kinase. BMC Cell Biol. 3, 14. doi: 10.1186/1471-2121-3-14
Lemeshko, V. V., Haridas, V., Quijano Pérez, J. C., and Gutterman, J. U. (2006). Avicins, natural anticancer saponins, permeabilize mitochondrial membranes. Arch. Biochem. Biophys. 454, 114–122.
Levin, E., Premkumar, A., Veenman, L., Kugler, W., Leschiner, S., Spanier, I., et al. (2005). The peripheral-type benzodiazepine receptor and tumorigenicity: isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry 44, 9924–9935.
Li, J., Bombeck, C. A., Yang, S., Kim, Y. M., and Billiar, T. R. (1999). Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 274, 17325–17333.
Li, M., Xiao, Z. Q., Chen, Z. C., Li, J. L., Li, C., and Zhang, P. F., et al. (2007). Proteomic analysis of the aging-related proteins in human normal colon epithelial tissue. J. Biochem. Mol. Biol. 40, 72–81.
Lindenboim, L., Kringel, S., Braun, T., Borner, C., and Stein, R. (2005). Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ. 12, 713–723.
Liu, B., Wang, P., Wang, Z., and Zhang, W. (2011). The use of anti-VDAC2 antibody for the combined assessment of human sperm acrosome integrity and ionophore A23187-induced acrosome reaction. PLoS ONE 6, e16985. doi: 10.1371/journal.pone.0016985
Liu, S., Ishikawa, H., Tsuyama, N., Li, F. J., Abroun, S., Otsuyama, K. I., et al. (2006). Increased susceptibility to apoptosis in CD45(+) myeloma cells accompanied by the increased expression of VDAC1. Oncogene 25, 419–429.
Llambi, F., and Green, D. R. (2011). Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr. Opin. Genet. Dev. 21, 12–20.
Lovell, J. F., Billen, L. P., Bindner, S., Shamas-Din, A., Fradin, C., Leber, B., et al. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084.
Lü, A. J., Dong, C. W., Du, C. S., and Zhang, Q. Y. (2007). Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection. Fish Shellfish Immunol. 23, 601–613.
Luo, J., Uchida, K., and Shi, R. (2005). Accumulation of acrolein-protein adducts after traumatic spinal cord injury. Neurochem. Res. 30, 291–295.
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182.
Mader, A., Abu-Hamad, S., Arbel, N., Gutiérrez-Aguilar, M., and Shoshan-Barmatz, V. (2010). Dominant-negative VDAC1 mutants reveal oligomeric VDAC1 to be the active unit in mitochondria-mediated apoptosis. Biochem. J. 429, 147–155.
Madesh, M., and Hajnoczky, G. (2001). VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015.
Maechler, P., Kennedy, E. D., Pozzan, T., and Wollheim, C. B. (1997). Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO J. 16, 3833–3841.
Majewski, N., Nogueira, V., Bhaskar, P., Coy, P. E., Skeen, J. E., Gottlob, K., et al. (2004a). Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 16, 819–830.
Majewski, N., Nogueira, V., Robey, R. B., and Hay, N. (2004b). Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 24, 730–740.
Malia, T. J., and Wagner, G. (2007). NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry 46, 514–525.
Manda, G., Nechifor, M. T., and Neagu, T.-M. (2009). Reactive oxygen species, cancer and anti-cancer therapies. Curr. Chem. Biol. 3, 342–366.
Marchetti, P., Zamzami, N., Joseph, B., Schraen-Maschke, S., Méreau-Richard, C., Costantini, P., et al. (1999). The novel retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid can trigger apoptosis through a mitochondrial pathway independent of the nucleus. Cancer Res. 59, 6257–6266.
Martel, C., Allouche, M., Esposti, D. D., Fanelli, E., Boursier, C., Henry, C., et al. (2012). GSK3-mediated VDAC phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology. doi: 10.1002/hep.25967 [Epub ahead of print].
Martinez-Abundis, E., Correa, F., Pavón, N., and Zazueta, C. (2009). Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J. 276, 5579–5588.
Martinez-Caballero, S., Dejean, L. M., Jonas, E. A., and Kinnally, K. W. (2005). The role of the mitochondrial apoptosis induced channel MAC in cytochrome c release. J. Bioenerg. Biomembr. 37, 155–164.
Martinez-Caballero, S., Dejean, L. M., and Kinnally, K. W. (2004). Some amphiphilic cations block the mitochondrial apoptosis-induced channel, MAC. FEBS Lett. 568, 35–38.
Martinez-Caballero, S., Dejean, L. M., Kinnally, M. S., Oh, K. J., Mannella, C. A., and Kinnally, K. W. (2009). Assembly of the mitochondrial apoptosis-induced channel, MAC. J. Biol. Chem. 284, 12235–12245.
Mathupala, S. P., Ko, Y. H., and Pedersen, P. L. (2006). Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25, 4777–4786.
Mathupala, S. P., Ko, Y. H., and Pedersen, P. L. (2010). The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 1797, 1225–1230.
Mayevsky, A. (2009). Mitochondrial function and energy metabolism in cancer cells: past overview and future perspectives. Mitochondrion 9, 165–179.
McEnery, M. W. (1992). The mitochondrial benzodiazepine receptor: evidence for association with the voltage-dependent anion channel (VDAC). J. Bioenerg. Biomembr. 24, 63–69.
Mello, C. F., Sultana, R., Piroddi, M., Cai, J., Pierce, W. M., Klein, J. B., et al. (2007). Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience 147, 674–679.
Meloche, H. P., and Monti, C. T. (1975). Bromopyruvate inactivation of 2-keto-3-deoxy-6-phosphogalactonate aldolase of Pseudomonas saccharophila. Kinetics and stereochemistry. Biochemistry 14, 3682–3687.
Menzel, V. A., Cassará, M. C., Benz, R., de Pinto, V., Messina, A., Cunsolo, V., et al. (2009). Molecular and functional characterization of VDAC2 purified from mammal spermatozoa. Biosci. Rep. 29, 351–362.
Messina, A., Reina, S., Guarino, F., and De Pinto, V. (2012). VDAC isoforms in mammals. Biochim. Biophys. Acta 1818, 1466–1476.
Mikhailov, V., Mikhailova, M., Degenhardt, K., Venkatachalam, M. A., White, E., and Saikumar, P. (2003). Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 278, 5367–5376.
Miura, N., Takemori, N., Kikugawa, T., Tanji, N., Higashiyama, S., and Yokoyama, M. (2012). Adseverin: a novel cisplatin-resistant marker in the human bladder cancer cell line HT1376 identified by quantitative proteomic analysis. Mol. Oncol. 6, 311–322.
Miyashita, T., and Reed, J. C. (1993). Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 81, 151–157.
Mizuta, T., Shimizu, S., Matsuoka, Y., Nakagawa, T., and Tsujimoto, Y. (2007). A Bax/Bak-independent mechanism of cytochrome c release. J. Biol. Chem. 282, 16623–16630.
Mlayeh, L., Chatkaew, S., Léonetti, M., and Homblé, F. (2010). Modulation of plant mitochondrial VDAC by phytosterols. Biophys. J. 99, 2097–2106.
Moin, S. M., Panteva, M., and Jameel, S. (2007). The hepatitis E virus Orf3 protein protects cells from mitochondrial depolarization and death. J. Biol. Chem. 282, 21124–21133.
Nahon, E., Israelson, A., Abu-Hamad, S., and Varda, S. B. (2005). Fluoxetine (Prozac) interaction with the mitochondrial voltage-dependent anion channel and protection against apoptotic cell death. FEBS Lett. 579, 5105–5110.
Nawarak, J., Huang-Liu, R., Kao, S. H., Liao, H. H., Sinchaikul, S., Chen, S. T., et al. (2009). Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim. Biophys. Acta 1794, 159–167.
Neumann, D., Bückers, J., Kastrup, L., Hell, S. W., and Jakobs, S. (2010). Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 3, 4.
Nichols, B. J., and Denton, R. M. (1995). Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell. Biochem. 149–150, 203–212.
Nogueira, V., Park, Y., Chen, C. C., Xu, P. Z., Chen, M. L., Tonic, I., et al. (2008). Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470.
Oancea, M., Mani, A., Hussein, M. A., and Almasan, A. (2004). Apoptosis of multiple myeloma. Int. J. Hematol. 80, 224–231.
Oh, H. L., Seok, J. Y., Kwon, C. H., Kang, S. K., and Kim, Y. K. (2006). Role of MAPK in ceramide-induced cell death in primary cultured astrocytes from mouse embryonic brain. Neurotoxicology 27, 31–38.
Okada, S. F., O’Neal, W. K., Huang, P., Nicholas, R. A., Ostrowski, L. E., Craigen, W. J., et al. (2004). Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 124, 513–526.
Olson, R. D., Boerth, R. C., Gerber, J. G., and Nies, A. S. (1981). Mechanism of adriamycin cardiotoxicity: evidence for oxidative stress. Life Sci. 29, 1393–1401.
Ospina, A., Lagunas-Martínez, A., Pardo, J., and Carrodeguas, J. A. (2011). Protein oligomerization mediated by the transmembrane carboxyl terminal domain of Bcl-XL. FEBS Lett. 585, 2935–2942.
Ott, M., Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2007). Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922.
Ott, M., Robertson, J. D., Gogvadze, V., Zhivotovsky, B., and Orrenius, S. (2002). Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. U.S.A. 99, 1259–1263.
Ozaki, T., Yamashita, T., and Ishiguro, S. (2009). Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim. Biophys. Acta 1793, 1848–1859.
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441.
Pastorino, J. G., and Hoek, J. B. (2008). Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182.
Pastorino, J. G., Hoek, J. B., and Shulga, N. (2005). Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554.
Pastorino, J. G., Shulga, N., and Hoek, J. B. (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618.
Pastorino, J. G., Simbula, G., Gilfor, E., Hoek, J. B., and Farber, J. L. (1994). Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem. 269, 31041–31046.
Pedersen, P. L. (2007). Warburg, me and Hexokinase 2: multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 39, 211–222.
Pedersen, P. L. (2008). Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 40, 123–126.
Pedersen, P. L., Mathupala, S., Rempel, A., Geschwind, J. F., and Ko, Y. H. (2002). Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 1555, 14–20.
Peixoto, P. M., Dejean, L. M., and Kinnally, K. W. (2012). The therapeutic potential of mitochondrial channels in cancer, ischemia-reperfusion injury, and neurodegeneration. Mitochondrion 12, 14–23.
Pelicano, H., Feng, L., Zhou, Y., Carew, J. S., Hileman, E. O., Plunkett, W., et al. (2003). Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 278, 37832–37839.
Penso, J., and Beitner, R. (1998). Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur. J. Pharmacol. 342, 113–117.
Pereira da Silva, A. P., El-Bacha, T., Kyaw, N., dos Santos, R. S., da-Silva, W. S., Almeida, F. C., et al. (2009). Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem. J. 417, 717–726.
Perfettini, J. L., Castedo, M., Roumier, T., Andreau, K., Nardacci, R., Piacentini, M., et al. (2005). Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 12(Suppl. 1), 916–923.
Petrosillo, G., Ruggiero, F. M., and Paradies, G. (2003). Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 17, 2202–2208.
Petrosillo, G., Ruggiero, F. M., Pistolese, M., and Paradies, G. (2001). Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 509, 435–438.
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto R. (2008). Calcium and apoptosis: ER–mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418.
Priault, M., Chaudhuri, B., Clow, A., Camougrand, N., and Manon, S. (1999). Investigation of bax-induced release of cytochrome c from yeast mitochondria permeability of mitochondrial membranes, role of VDAC and ATP requirement. Eur. J. Biochem. 260, 684–691.
Pro, B., Leber, B., Smith, M., Fayad, L., Romaguera, J., Hagemeister, F., et al. (2008). Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br. J. Haematol. 143, 355–360.
Qiao, H., and McMillan, J. R. (2007). Gelsolin segment 5 inhibits HIV-induced T-cell apoptosis via Vpr-binding to VDAC. FEBS Lett. 581, 535–540.
Rahmani, Z., Huh, K. W., Lasher, R., and Siddiqui, A. (2000). Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 74, 2840–2846.
Ralph, S. J., Rodríguez-Enríquez, S., Neuzil, J., Saavedra, E., and Moreno-Sánchez, R. (2010). The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation – why mitochondria are targets for cancer therapy. Mol. Aspects Med. 31, 145–170.
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624.
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233.
Ravagnan, L., Marzo, I., Costantini, P., Susin, S. A., Zamzami, N., Petit, P. X., et al. (1999). Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene 18, 2537–2546.
Reed, J. C. (2006). Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 13, 1378–1386.
Reina, S., Palermo, V., Guarnera, A., Guarino, F., Messina, A., Mazzoni, C., et al. (2010). Swapping of the N-terminus of VDAC1 with VDAC3 restores full activity of the channel and confers anti-aging features to the cell. FEBS Lett. 584, 2837–2844.
Reymann, S., Buzhynskyy, N., Prima, V., Sturgis, J. N., and Scheuring, S. (1995). Further evidence for multitopological localization of mammalian porin (VDAC) in the plasmalemma forming part of a chloride channel complex affected in cystic fibrosis and encephalomyopathy. Biochem. Mol. Med. 54, 75–87.
Rom, J., von Minckwitz, G., Marmé, F., Ataseven, B., Kozian, D., and Sievert, M. (2009). Phase I study of apoptosis gene modulation with oblimersen within preoperative chemotherapy in patients with primary breast cancer. Ann. Oncol. 20, 1829–1835.
Romagnoli, R., Baraldi, P. G., Lopez-Cara, C., Cruz-Lopez, O., Carrion, M. D., Kimatrai Salvador, M., et al. (2011). Synthesis and antitumor molecular mechanism of agents based on amino 2-(3′,4′,5′-trimethoxybenzoyl)benzo[b]furan: inhibition of tubulin and induction of apoptosis. ChemMedChem 6, 1841–1853.
Roman, I., Figys, J., Steurs, G., and Zizi, M. (2006). Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 1758, 479–486.
Rone, M. B., Fan, J., and Papadopoulos, V. (2009). Cholesterol transport in steroid biosynthesis: role of protein–protein interactions and implications in disease states. Biochim. Biophys. Acta 1791, 646–658.
Rosano, C. (2011). Molecular model of hexokinase binding to the outer mitochondrial membrane porin (VDAC1): implication for the design of new cancer therapies. Mitochondrion 11, 513–519.
Rose, I. A., O’Connell, E. L., and Litwin, S. (1974). Determination of the rate of hexokinase-glucose dissociation by the isotope-trapping method. J. Biol. Chem. 249, 5163–5168.
Rostovtseva, T. K., Antonsson, B., Suzuki, M., Youle, R. J., Colombini, M., and Bezrukov, S. M. (2004). Bid, but not Bax, regulates VDAC channels. J. Biol. Chem. 279 13575–13583.
Rostovtseva, T. K., and Bezrukov, S. M. (2012). VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 1818, 1526–1535.
Rostovtseva, T. K., Gurnev, P. A., Chen, M. Y., and Bezrukov, S. M. (2012). Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J. Biol. Chem. 287, 29589–29598.
Rostovtseva, T. K., Komarov, A., Bezrukov, S. M., and Colombini, M. (2002). Dynamics of nucleotides in VDAC channels: structure-specific noise generation. Biophys. J. 82(Pt 1), 193–205.
Rostovtseva, T. K., Petrache, H. I., Kazemi, N., Hassanzadeh, E., and Bezrukov, S. M. (2008a). Interfacial polar interactions affect gramicidin channel kinetics. Biophys J. 94, L23–L25.
Rostovtseva, T. K., Sheldon, K. L., Hassanzadeh, E., Monge, C., Saks, V., Bezrukov, S. M., et al. (2008b). Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. U.S.A. 105, 18746–18751.
Saddar, S., and Stuart, R. A. (2005). The yeast F(1)F(0)-ATP synthase: analysis of the molecular organization of subunit g and the importance of a conserved GXXXG motif. J. Biol. Chem. 280, 24435–24442.
Sahasrabudhe, S. R., Lai, S., Pierce, M., Clemens, C., Venkat, R., Rebentisch, M., et al. (2008). Selective in vitro and in vivo anti-tumor activity of PRLX 93936 in biological models of melanoma and ovarian cancer. J. Clin. Oncol. 26(Suppl.), Abstr. 14586.
Saks, V., Guzun, R., Timohhina, N., Tepp, K., Varikmaa, M., Monge, C., et al. (2010). Structure–function relationships in feedback regulation of energy fluxes in vivo in health and disease: mitochondrial interactosome. Biochim. Biophys. Acta 1797, 678–697.
Sampson, M. J., Decker, W. K., Beaudet, A. L., Ruitenbeek, W., Armstrong, D., Hicks, M. J., et al. (2001). Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 276, 39206–39212.
Sampson, M. J., Lovell, R. S., and Craigen, W. J. (1997). The murine voltage-dependent anion channel gene family. Conserved structure and function. J. Biol. Chem. 272, 18966–18973.
Santin, G., Piccolini, V. M., Veneroni, P., Barni, S., Bernocchi, G., and Bottone, M. G. (2011). Different patterns of apoptosis in response to cisplatin in B50 neuroblastoma rat cells. Histol. Histopathol. 26, 831–842.
Santo-Domingo, J., and Demaurex, N. (2010). Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 1797, 907–912.
Schlattner, U., Dolder, M., Wallimann, T., and Tokarska-Schlattner, M. (2001). Mitochondrial creatine kinase and mitochondrial outer membrane porin show a direct interaction that is modulated by calcium. J. Biol. Chem. 276, 48027–48030.
Schneider, R., Etzkorn, M., Giller, K., Daebel, V., Eisfeld, J., Zweckstetter, M., et al. (2010). The native conformation of the human VDAC1 N terminus. Angew. Chem. Int. Ed. Engl. 49, 1882–1885.
Schwarzer, C., Barnikol-Watanabe, S., Thinnes, F. P., and Hilschmann, N. (2002). Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 34, 1059–1070.
Schwer, B., Eckersdorff, M., Li, Y., Silva, J. C., Fermin, D., Kurtev, M. V., et al. (2009). Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606.
Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T., et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139.
Sentman, C. L., Shutter, J. R., Hockenbery, D., Kanagawa, O., and Korsmeyer, S. J. (1991). bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell 67, 879–888.
Shi, H., Hudson, L. G., Ding, W., Wang, S., Cooper, K. L., and Liu, S. (2004). Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chem. Res. Toxicol. 17, 871–878.
Shi, Y., Chen, J., Weng, C., Chen, R., Zheng, Y., Chen, Q., et al. (2003a). Identification of the protein–protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 305, 989–996.
Shi, Y., Jiang, C., Chen, Q., and Tang, H. (2003b). One-step on-column affinity refolding purification and functional analysis of recombinant human VDAC1. Biochem. Biophys. Res. Commun. 303, 475–482.
Shimizu, S., Ide, T., Yanagida, T., and Tsujimoto, Y. (2000a). Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 275, 12321–12325.
Shimizu, S., Konishi, A., Kodama, T., and Tsujimoto, Y. (2000b). BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 3100–3105.
Shimizu, S., Shinohara, Y., and Tsujimoto, Y. (2000c). Bax and Bcl-xL independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene 19, 4309–4318.
Shimizu, S., Matsuoka, Y., Shinohara, Y., Yoneda, Y., and Tsujimoto, Y. (2001). Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 152, 237–250.
Shimizu, S., Narita, M., and Tsujimoto, Y. (1999). Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487.
Shimizu, S., and Tsujimoto, Y. (2000). Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. U.S.A. 97, 577–582.
Shirakata, Y., and Koike, K. (2003). Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J. Biol. Chem. 278, 22071–22078.
Shoshan-Barmatz, V., Arbel, N., and Arzoine, L. (2008a). VDAC, the voltage-dependent anion channel: function, regulation & mitochondrial signaling in cell life and death. Cell Sci. 4, 74–118.
Shoshan-Barmatz, V., Keinan, N., and Zaid, H. (2008b). Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191.
Shoshan-Barmatz, V., and Ben-Hail, D. (2011). VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34.
Shoshan-Barmatz, V., De Pinto, V., Zweckstetter, M., Raviv, Z., Keinan, N., and Arbel, N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285.
Shoshan-Barmatz, V., and Gincel, D. (2003). The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 39, 279–292.
Shoshan-Barmatz, V., and Golan, M. (2012). Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 19, 714–735.
Shoshan-Barmatz, V., Hadad, N., Feng, W., Shafir, I., Orr, I., Varsanyi, M., et al. (1996). VDAC/porin is present in sarcoplasmic reticulum from skeletal muscle. FEBS Lett. 386, 205–210.
Shoshan-Barmatz, V., and Israelson, A. (2005). The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: characterization, modulation and possible function. J. Membr. Biol. 204, 57–66.
Shoshan-Barmatz, V., Israelson, A., Brdiczka, D., and Sheu, S. S. (2006). The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 12, 2249–2270.
Shoshan-Barmatz, V., Zakar, M., Rosenthal, K., and Abu-Hamad, S. (2009). Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 1787, 421–430.
Shoshan-Barmatz, V., Zalk, R., Gincel, D., and Vardi, N. (2004). Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim. Biophys. Acta 1657, 105–114.
Shulga, N., Wilson-Smith, R., and Pastorino, J. G. (2009). Hexokinase II detachment from the mitochondria potentiates cisplatin induced cytotoxicity through a caspase-2 dependent mechanism. Cell Cycle 8, 3355–3364.
Sileikyte, J., Petronilli, V., Zulian, A., Dabbeni-Sala, F., Tognon, G., Nikolov, P., et al. (2011). Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J. Biol. Chem. 286, 1046–1053.
Simamura, E., Hirai, K., Shimada, H., Koyama, J., Niwa, Y., and Shimizu, S. (2006). Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel. Cancer Biol. Ther. 5, 1523–1529.
Simamura, E., Shimada, H., Ishigaki, Y., Hatta, T., Higashi, N., and Hirai, K. (2008). Bioreductive activation of quinone antitumor drugs by mitochondrial voltage-dependent anion channel 1. Anat. Sci. Int. 83, 261–266.
Siskind, L. J., and Colombini, M. (2000). The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 275, 38640–38644.
Siskind, L. J., Kolesnick, R. N., and Colombini, M. (2006). Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion 6, 118–125.
Sokolove, P. M. (1994). Interactions of adriamycin aglycones with mitochondria may mediate adriamycin cardiotoxicity. Int. J. Biochem. 26, 1341–1350.
Song, J., Midson, C., Blachly-Dyson, E., Forte, M., and Colombini, M. (1998). The topology of VDAC as probed by biotin modification. J. Biol. Chem. 273, 24406–24413.
Stachowiak, O., Schlattner, U., Dolder, M., and Wallimann, T. (1998). Oligomeric state and membrane binding behaviour of creatine kinase isoenzymes: implications for cellular function and mitochondrial structure. Mol. Cell. Biochem. 184, 141–151.
Stanley, S., Dias, J. A., D’Arcangelis, D., and Mannella, C. A. (1995). Peptide-specific antibodies as probes of the topography of the voltage-gated channel in the mitochondrial outer membrane of Neurospora crassa. J. Biol. Chem. 270, 16694–16700.
Stiban, J., Caputo, L., and Colombini, M. (2008). Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 49, 625–634.
Su, J. C., Lin, K. L., Chien, C. M., Tseng, C. H., Chen, Y. L., Chang, L. S., et al. (2010). Furano-1,2-naphthoquinone inhibits EGFR signaling associated with G2/M cell cycle arrest and apoptosis in A549 cells. Cell Biochem. Funct. 28, 695–705.
Sugiyama, T., Shimizu, S., Matsuoka, Y., Yoneda, Y., and Tsujimoto, Y. (2002). Activation of mitochondrial voltage-dependent anion channel by apro-apoptotic BH3-only protein Bim. Oncogene 21, 4944–4956.
Sui, Y., Potula, R., Dhillon, N., Pinson, D., Li, S., Nath, A., et al. (2004). Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am. J. Pathol. 164, 1557–1566.
Summers, W. A., and Court, D. A. (2010). Origami in outer membrane mimetics: correlating the first detailed images of refolded VDAC with over 20 years of biochemical data. Biochem. Cell Biol. 88, 425–438.
Sun, J., and Liao, J. K. (2002). Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 99, 13108–13113.
Sun, L., Shukair, S., Naik, T. J., Moazed, F., and Ardehali, H. (2008). Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell. Biol. 28, 1007–1017.
Sundararajan, R., Cuconati, A., Nelson, D., and White, E. (2001). Tumor necrosis factor-alpha induces Bax-Bak interaction and apoptosis, which is inhibited by adenovirus E1B 19K. J. Biol. Chem. 276, 45120–45127.
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911.
Tajeddine, N., Galluzzi, L., Kepp, O., Hangen, E., Morselli, E., Senovilla, L., et al. (2008). Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 10, 4221–4232.
Tan, W., and Colombini, M. (2007). VDAC closure increases calcium ion flux. Biochim. Biophys. Acta 1768, 2510–2515.
Tan, W., Loke, Y. H., Stein, C. A., Miller, P., and Colombini, M. (2007). Phosphorothioate oligonucleotides block the VDAC channel. Biophys. J. 93, 1184–1191.
Tang, H. L., Le, A. H., and Lung, H. L. (2006). The increase in mitochondrial association with actin precedes Bax translocation in apoptosis. Biochem. J. 396, 1–5.
Tarze, A., Deniaud, A., Le Bras, M., Maillier, E., Molle, D., Larochette, N., et al. (2007). GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 26, 2606–2620.
Teijido, O., Ujwal, R., Hillerdal, C. O., Kullman, L., Rostovtseva, T. K., and Abramson, J. (2012). Affixing the N-terminal alpha helix of the voltage dependent anion channel to the channel’s wall does not prevent its voltage gating. J. Biol. Chem. 287, 11437–11445.
Thinnes, F. P. (2005). Does fluoxetine (Prozak) block mitochondrial permeability transition by blocking VDAC as part of permeability transition pores? Mol. Genet. Metab. 84, 378.
Thinnes, F. P. (2009). Human type-1 VDAC, a cisplatin target involved in either apoptotic pathway. Mol. Genet. Metab. 97, 163.
Thinnes, F. P., Götz, H., Kayser, H., Benz, R., Schmidt, W. E., Kratzin, H. D., et al. (1989). Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell. Biol. Chem. Hoppe Seyler 370, 1253–1264.
Tikunov, A., Johnson, C. B., Pediaditakis, P., Markevich, N., Macdonald, J. M., Lemasters, J. J., et al. (2010). Closure of VDAC causes oxidative stress and accelerates the Ca(2+)-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 495, 174–181.
Tinhofer, I., Bernhard, D., Senfter, M., Anether, G., Loeffler, M., Kroemer, G., et al. (2001). Resveratrol, a tumor-suppressive compound from grapes, induces apoptosis via a novel mitochondrial pathway controlled by Bcl-2. FASEB J. 15, 1613–1615.
Tomasello, F., Messina, A., Lartigue, L., Schembri, L., Medina, C., Reina, S., et al. (2009). Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 19, 1363–1376.
Trachootham, D., Zhou, Y., Zhang, H., Demizu, Y., Chen, Z., Pelicano, H., et al. (2006). Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10, 241–252.
Triphan, X., Menzel, V. A., Petrunkina, A. M., Cassará, M. C., Wemheuer, W., Hinsch, K. D., et al. (2008). Localisation and function of voltage-dependent anion channels (VDAC) in bovine spermatozoa. Pflugers Arch. 455, 677–686.
Tsujimoto, Y. (2003). Cell death regulation by the Bcl-2 protein family in the mitochondria. J. Cell Physiol. 195, 158–167.
Tsujimoto, Y., Finger, L. R., Yunis, J., Nowell, P. C., and Croce, C. M. (1984). Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099.
Tsujimoto, Y., and Shimizu, S. (2002). The voltage-dependent anion channel: an essential player in apoptosis. Biochimie 84, 187–193.
Tsujimoto, Y., and Shimizu, S. (2007). Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12, 835–840.
Ujwal, R., Cascio, D., Chaptal, V., Ping, P., and Abramson, J. (2009). Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels (Austin) 3, 167–170.
Ujwal, R., Cascio, D., Colletier, J. P., Faham, S., Zhang, J., Toro, L., et al. (2008). The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U.S.A. 105, 17742–17747.
Vander Heiden, M. G., Chandel, N. S., Li, X. X., Schumacker, P. T., Colombini, M., and Thompson, C. B. (2000). Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. U.S.A. 97, 4666–4671.
Vander Heiden, M. G., Li, X. X., Gottleib, E., Hill, R. B., Thompson, C. B., and Colombini, M. (2001). Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 276, 19414–19419.
Veenman, L., Leschiner, S., Spanier, I., Weisinger, G., Weizman, A., and Gavish, M. (2002). PK 11195 attenuates kainic acid-induced seizures and alterations in peripheral-type benzodiazepine receptor (PBR) protein components in the rat brain. J. Neurochem. 80, 917–927.
Veenman, L., Levin, E., Weisinger, G., Leschiner, S., Spanier, I., Snyder, S. H., et al. (2004). Peripheral-type benzodiazepine receptor density and in vitro tumorigenicity of glioma cell lines. Biochem. Pharmacol. 68, 689–698.
Veenman, L., Papadopoulos, V., and Gavish, M. (2007). Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 13, 2385–2405.
Veenman, L., Shandalov, Y., and Gavish, M. (2008). VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 40, 199–205.
Verkhratsky, A., and Petersen, O. H. (2002). The endoplasmic reticulum as an integrating signalling organelle: from neuronal signalling to neuronal death. Eur. J. Pharmacol. 447, 141–154.
Verrier, F., Mignotte, B., Jan, G., and Brenner, C. (2003). Study of PTPC composition during apoptosis for identification of viral protein target. Ann. N. Y. Acad. Sci. 1010, 126–142.
Vieira, H. L., Haouzi, D., El Hamel, C., Jacotot, E., Belzacq, A. S., and Brenner, C. (2000). Permeabilization of the mitochondrial inner membrane during apoptosis: impact of the adenine nucleotide translocator. Cell Death Differ. 7, 1146–1154.
Voehringer, D. W., Hirschberg, D. L., Xiao, J., Lu, Q., Roederer, M., Lock, C. B., et al. (2000). Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2680–2685.
Voulgaridou, G. P., Anestopoulos, I., Franco, R., Panayiotidis, M. I., and Pappa, A. (2011). DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat. Res. 711, 13–27.
Vyssokikh, M. Y., and Brdiczka, D. (2003). The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol. 50, 389–404.
Wan, K. F., Chan, S. L., Sukumaran, S. K., Lee, M. C., and Yu, V. C. (2008). Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. J. Biol. Chem. 283, 8423–8433.
Wang, B., Malik, R., Nigg, E. A., and Körner, R. (2008). Evaluation of the low-specificity protease elastase for large-scale phosphoproteome analysis. Anal. Chem. 80, 9526–9533.
Wang, Y., Zhang, X. H., and Wang, H. L. (2011). Involvement of BMPR2 in the protective effect of fluoxetine against monocrotaline-induced endothelial apoptosis in rats. Can. J. Physiol. Pharmacol. 89, 345–354.
Watabe, M., Machida, K., and Osada, H. (2000). MT-21 is a synthetic apoptosis inducer that directly induces cytochrome c release from mitochondria. Cancer Res. 60, 5214–5222.
Weeber, E. J., Levy, M., Sampson, M. J., Anflous, K., Armstrong, D. L., Brown, S. E., et al. (2002). The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J. Biol. Chem. 277, 18891–18897.
Wei, M. C., Lindsten, T., Mootha, V. K., Weiler, S., Gross, A., Ashiya, M., et al. (2000). tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14, 2060–2071.
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., et al. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730.
Whittington, D. A., Waheed, A., Ulmasov, B., Shah, G. N., Grubb, J. H., Sly, W. S., et al. (2001). Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. U.S.A. 98, 9545–9550.
Wigdal, S. S., Kirkland, R. A., Franklin, J. L., and Haak-Frendscho, M. (2002). Cytochrome c release precedes mitochondrial membrane potential loss in cerebellar granule neuron apoptosis: lack of mitochondrial swelling. J. Neurochem. 82, 1029–1038.
Wong, D. T., Bymaster, F. P., and Engleman, E. A. (1995). Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci. 57, 411–441.
Wright, S. C., Zhong, J., and Larrick, J. W. (1994). Inhibition of apoptosis as a mechanism of tumor promotion. FASEB J. 8, 654–660.
Wu, S., Sampson, M. J., Decker, W. K., and Craigen, W. J. (1999). Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochim. Biophys. Acta 1452, 68–78.
Xie, G., and Wilson, J. E. (1990). Tetrameric structure of mitochondrially bound rat brain hexokinase: a crosslinking study. Arch. Biochem. Biophys. 276, 285–293.
Xie, Q., Wondergem, R., Shen, Y., Cavey, G., Ke, J., Thompson, R., et al. (2011). Benzoquinone ansamycin 17AAG binds to mitochondrial voltage-dependent anion channel and inhibits cell invasion. Proc. Natl. Acad. Sci. U.S.A. 108, 4105–4110.
Xu, X., Decker, W., Sampson, M. J., Craigen, W. J., and Colombini, M. (1999). Mouse VDAC isoforms expressed in yeast: channel properties and their roles in mitochondrial outer membrane permeability. J. Membr. Biol. 170, 89–102.
Xu, X., Forbes, J. G., and Colombini, M. (2001). Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 180, 73–81.
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868.
Yamamoto, T., Yamada, A., Watanabe, M., Yoshimura, Y., Yamazaki, N., Yoshimura, Y., et al. (2006). VDAC1, having a shorter N-terminus than VDAC2 but showing the same migration in an SDS-polyacrylamide gel, is the predominant form expressed in mitochondria of various tissues. J. Proteome Res. 5, 3336–3344.
Yang, L., Vaitheesvaran, B., Hartil, K., Robinson, A. J., Hoopmann, M. R., Eng, J. K., et al. (2011). The fasted/fed mouse metabolic acetylome: N6-acetylation differences suggest acetylation coordinates organ-specific fuel switching. J. Proteome Res. 10, 4134–4149.
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245.
Yang, Z., Schumaker, L. M., Egorin, M. J., Zuhowski, E. G., Guo, Z., and Cullen, K. J. (2006). Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin. Cancer Res. 12, 5817–5825.
Yehezkel, G., Abu-Hamad, S., and Shoshan-Barmatz, V. (2007). An N-terminal nucleotide-binding site in VDAC1: involvement in regulating mitochondrial function. J. Cell. Physiol. 212, 551–561.
Yehezkel, G., Hadad, N., Zaid, H., Sivan, S., and Shoshan-Barmatz, V. (2006). Nucleotide-binding sites in the voltage-dependent anion channel: characterization and localization. J. Biol. Chem. 281, 5938–5946.
Yin, X. M. (2000). Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 10, 161–167.
Yoo, N. J., Park, S. W., and Lee, S. H. (2011). A frameshift mutation of the pro-apoptotic VDAC1 gene in cancers with microsatellite instability. Gut Liver 5, 548–549.
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59.
Yu, W. H., and Forte, M. (1996). Is there VDAC in cell compartments other than the mitochondria? J. Bioenerg. Biomembr. 28, 93–100.
Yuan, H., Williams, S. D., Adachi, S., Oltersdorf, T., and Gottlieb, R. A. (2003). Cytochrome c dissociation and release from mitochondria by truncated Bid and ceramide. Mitochondrion 2, 237–244.
Yuan, S., Galluzzi, L., Kepp, O., Hangen, E., Morselli, E., Senovilla, L., et al. (2008). Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 22, 2809–2820.
Zachariae, U., Schneider, R., Briones, R., Gattin, Z., Demers, J. P., Giller, K., et al. (2012). beta-Barrel mobility underlies closure of the voltage-dependent anion channel. Structure 20, 1540–1549.
Zaid, H., Abu-Hamad, S., Israelson, A., Nathan, I., Shoshan-Barmatz, V., et al. (2005). The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 12, 751–760.
Zalk, R., Israelson, A., Garty, E. S., Azoulay-Zohar, H., and Shoshan-Barmatz, V. (2005). Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 386(Pt 1), 73–83.
Zamarin, D., García-Sastre, A., Xiao, X., Wang, R., and Palese, P. (2005). Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1, e4. doi: 10.1371/journal.ppat.0010004
Zamzami, N., El Hamel, C., Maisse, C., Brenner, C., Muñoz-Pinedo, C., Belzacq, A. S., et al. (2000). Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 19, 6342–6350.
Zamzami, N., Marzo, I., Susin, S. A., Brenner, C., Larochette, N., Marchetti, P., et al. (1998). The thiol crosslinking agent diamide overcomes the apoptosis-inhibitory effect of Bcl-2 by enforcing mitochondrial permeability transition. Oncogene 16, 1055–1063.
Zeth, K., Meins, T., and Vonrhein, C. (2008). Approaching the structure of human VDAC1, a key molecule in mitochondrial cross-talk. J. Bioenerg. Biomembr. 40, 127–132.
Zhang, T., Tang, S. S., Jin, X., Liu, F. Y., Zhang, C. M., Zhao, W. X., et al. (2011). c-Myc influences olaquindox-induced apoptosis in human hepatoma G2 cells. Mol. Cell. Biochem. 354, 253–261.
Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004.
Keywords: apoptosis, mitochondrial porin, cancer metabolism, VDAC protein, hexokinase, anti-apoptotic Bcl-2
Citation: Shoshan-Barmatz V and Mizrachi D (2012) VDAC1: from structure to cancer therapy. Front. Oncol. 2:164. doi: 10.3389/fonc.2012.00164
Received: 29 July 2012; Accepted: 24 October 2012;
Published online: 29 November 2012.
Edited by:
Catherine Brenner, Institut National de la Santé et de la Recherche Médicale - University of Paris Sud, FranceReviewed by:
Vito De Pinto, University of Catania, ItalyCopyright: © 2012 Shoshan-Barmatz and Mizrachi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Varda Shoshan-Barmatz, Department of Life Sciences, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. e-mail:dmFyZGFzYkBiZ3UuYWMuaWw=.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.