
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 16 November 2012
Sec. Cancer Molecular Targets and Therapeutics
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00162
This article is part of the Research Topic Targeting MYCN in paediatric cancers View all 9 articles
 Laura D. Gamble1
Laura D. Gamble1 Michael D. Hogarty2
Michael D. Hogarty2 Xueyuan Liu2
Xueyuan Liu2 David S. Ziegler1,3 Glenn Marshall1,3 Murray D. Norris1 Michelle Haber1*
David S. Ziegler1,3 Glenn Marshall1,3 Murray D. Norris1 Michelle Haber1*Polyamines are highly regulated essential cations that are elevated in rapidly proliferating tissues, including diverse cancers. Expression analyses in neuroblastomas suggest that up-regulation of polyamine pro-synthetic enzymes and down-regulation of catabolic enzymes is associated with poor prognosis. Polyamine sufficiency may be required for MYCN oncogenicity in MYCN amplified neuroblastoma, and targeting polyamine homeostasis may therefore provide an attractive therapeutic approach. ODC1, an oncogenic MYCN target, is rate-limiting for polyamine synthesis, and is overexpressed in many cancers including neuroblastoma. Inhibition of ODC1 by difluoromethylornithine (DFMO) decreased tumor penetrance in TH-MYCN mice treated pre-emptively, and extended survival and synergized with chemotherapy in treating established tumors in both TH-MYCN and xenograft models. Efforts to augment DFMO activity, or otherwise maximally reduce polyamine levels, are focused on antagonizing polyamine uptake or augmenting polyamine export or catabolism. Since polyamine inhibition appears to be clinically well tolerated, these approaches, particularly when combined with chemotherapy, have great potential for improving neuroblastoma outcome in both MYCN amplified and non-MYCN amplified neuroblastomas.
Neuroblastoma originates from the primitive cells of the sympathetic nervous system and is the most common solid tumor of early childhood. It is an aggressive cancer that often presents with high risk clinical and genetic features. In these cases, despite the use of intense multimodal therapies, long-term survival rates remain below 50% (Maris et al., 2007). Current treatment regimens are also associated with substantial morbidity, so novel therapeutic strategies are urgently needed. MYCN amplification, identified in up to 30% of neuroblastomas, is a powerful and reliable marker of aggressive disease and is strongly prognostic of poor outcome (Cohn and Tweddle, 2004). As a transcription factor, MYCN induces and represses a large number of genes involved in multiple biological processes including cell growth and differentiation. However, the genes necessary or sufficient to initiate neuroblastoma tumorigenesis downstream of MYCN remain to be established.
The polyamine pathway is frequently deregulated in neuroblastoma, and a number of genes involved in polyamine homeostasis are known to be MYCN or c-MYC targets (Bello-Fernandez et al., 1993; Lutz et al., 1996; Fernandez et al., 2003; Li et al., 2003; Forshell et al., 2010), while the expression of others is linked to MYCN status (Hogarty et al., 2008; Rounbehler et al., 2009). This suggests a mechanism by which MYCN may contribute to the malignant phenotype of neuroblastoma. Therapeutic approaches targeting the polyamine pathway may therefore provide an effective strategy for the treatment of high risk neuroblastoma, particularly in tumors dependent on deregulated Myc activity, such as those with MYCN amplification.
Polyamines are positively charged multifunctional polycations derived from amino acids and found in all living organisms. They are indispensable for cell growth, differentiation, and cell survival and function by forming electrostatic bonds with negatively charged macromolecules to mediate a number of biological processes. These include DNA synthesis and stability, replication, transcription and translation, ribosome biogenesis, modulation of ion channels and receptors, and protein phosphorylation (Pegg, 1988; Panagiotidis et al., 1995; Johnson, 1996; Igarashi and Kashiwagi, 2000; Childs et al., 2003; Gerner and Meyskens, 2004; Pegg, 2006). Polyamines are also required for covalent activation of eIF5A, a major protein translation factor, via hypusination, a polyamine-dependent modification (Cooper et al., 1983). Whereas polyamine depletion leads to growth arrest, overexpression of these essential cations is cytotoxic (Poulin et al., 1993; Tobias and Kahana, 1995; Ray et al., 2001; Li et al., 2002). Therefore, tight regulation of intracellular polyamine levels is critical and is dependent on the proliferative state of the cell. Regulatory mechanisms include de novo synthesis, recycling via a back converting catabolic pathway and through transmembrane import and efflux (Gerner and Meyskens, 2004; Casero and Marton, 2007). An overview of the polyamine pathway is shown in Figure 1.
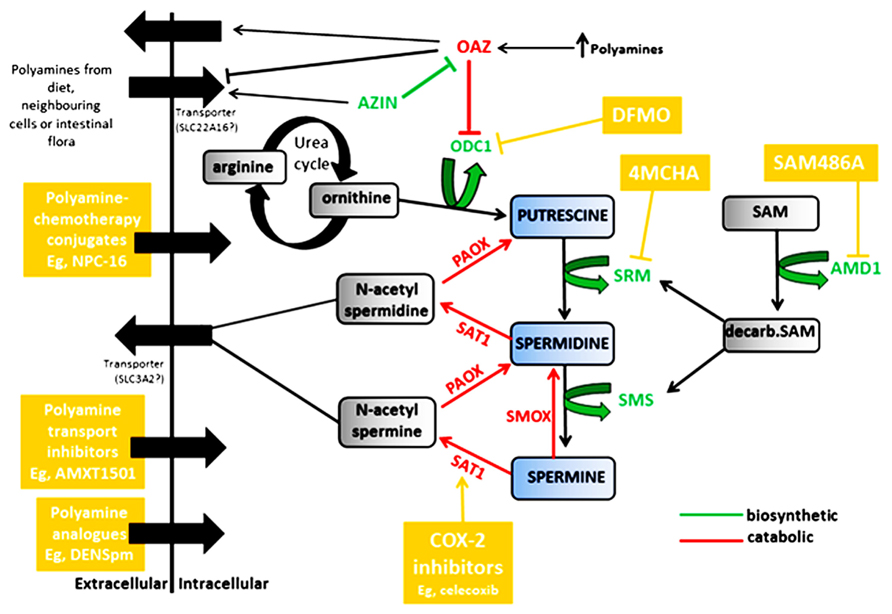
FIGURE 1. Regulation of the polyamines putrescine, spermidine and spermine by biosynthetic enzymes (shown in green) and catabolic enzymes (shown in red). Compounds and classes of compounds that target various aspects of polyamine regulation are shown in yellow. ODC1, ornithine decarboxylase; OAZ, antizyme; AZIN, antizyme inhibitor; SRM, spermidine synthase; SMS, spermine synthase; AMD1, adenosylmethionine decarboxylase; SAT1, spermine/spermidine N1-acetyltransferase; PAOX, polyamine oxidase; SMOX, spermine oxidase.
The first rate-limiting enzyme in the polyamine pathway is ornithine decarboxylase (ODC1), which catalyzes the decarboxylation and conversion of ornithine, a product of the urea cycle, to the primary polyamine putrescine (Pegg, 2006). Putrescine is the precursor for spermidine and spermine synthesis, and is further processed into these more abundant polyamines by two aminopropyltransferases, spermidine synthase (SRM) and spermine synthase (SMS). The second rate-limiting enzyme, adenosylmethionine decarboxylase (AMD1), decarboxylates S-adenosylmethionine (SAM) to provide the aminopropyl donor for the conversions to spermidine and spermine. Both ODC1 and AMD1 are highly controlled at the transcriptional and post-transcriptional levels, and have among the shortest half-lives of any mammalian enzymes. In addition, ODC1 turnover is regulated by antizymes (OAZ1, OAZ2, and OAZ3) which in turn are controlled by antizyme inhibitors (AZIN1 and AZIN2). Antizymes initiate ODC1 degradation by binding the ODC monomer, inhibiting its activity and shunting ODC1 to the 26S proteasome for degradation (Li and Coffino, 1992; Murakami et al., 1992). Of the three antizymes, OAZ1 is the most effective at stimulating ODC1 degradation. Antizyme expression is also induced by a feedback mechanism. An increase in intracellular polyamine levels stimulates a +1 frame-shift by the ribosomes during translation of antizyme mRNA, increasing expression of the full-length protein (Matsufuji et al., 1995). In response to increased intracellular polyamines, antizymes negatively regulate polyamine transport by promoting polyamine secretion and inhibiting uptake, while antizyme degradation by the ubiquitin pathway is also inhibited (Mitchell et al., 1994; Suzuki et al., 1994; Palanimurugan et al., 2004).
Antizyme inhibitors antagonize the function of antizymes by mimicking ODC1 (Koguchi et al., 1997; Kanerva et al., 2008). They are highly homologous to ODC1, but lack enzymatic activity due to critical amino acid substitutions and bind antizymes with greater affinity than ODC1 (Albeck et al., 2008). Increased antizyme inhibitor activity therefore results in the release of ODC1 from the inactive ODC1-antizyme complex, which in turn increases the production of polyamines (Murakami et al., 1996; Mangold, 2006; Pegg, 2006). In addition, forced induction of AZIN1 in cell cultures has also been shown to increase polyamine uptake (Keren-Paz et al., 2006). Polyamine levels themselves act as down-regulators of both ODC1 and AMD1 and as up-regulators of antizymes by a feedback homeostasis mechanism.
Polyamine catabolism allows for the re-utilization of polyamines as spermine is converted back to spermidine and spermidine back to putrescine. A number of key enzymes are involved in this process as shown in Figure 1. The degradation of polyamines depends on three enzymes; spermine/spermidine N1-acetyltransferase (SAT1), polyamine oxidase (PAOX), and spermine oxidase (SMOX). SAT1, a highly inducible cytosolic enzyme, acetylates spermine and spermidine (Casero and Pegg, 1993), which are then either exported from the cell, or oxidized by the peroxisomal enzyme PAOX, resulting in conversion to spermidine or putrescine, H2O2 and 3-aminopropanol (Seiler, 1995). PAOX preferentially catalyzes the oxidation of the N1-acetylspermine/spermidine produced by SAT1 activity, rather than spermine or spermidine, whereas SMOX is a cytosolic enzyme which catalyzes the oxidation of spermine directly to spermidine, without acetylation and produces H2O2 and 2 aminopropanol (Vujcic et al., 2002; Wang et al., 2003; Casero and Pegg, 2009; Pegg, 2009). Mostly, PAOX is constitutively expressed and dependent on SAT1 as it is rate-limited by the availability of the acetylated spermidine/spermine (Casero and Pegg, 1993; Vujcic et al., 2002). SAT1, the rate limiting enzyme in polyamine catabolism, is therefore extensively regulated at transcriptional and post-transcriptional levels (Fogel-Petrovic et al., 1993; Coleman et al., 1996), and is a gatekeeper regulating flux through the polyamine pathway (Kramer et al., 2008).
Cellular polyamine levels are also regulated by transmembrane transport where cells can take up polyamines from their surroundings and also export them to the extracellular space, and this can make a significant contribution to cellular polyamine levels. Known polyamine transporters include SLC3A2 (Uemura et al., 2008) and SLC22A16 (Aouida et al., 2010). SAT1 is co-localized with the SLC3A2 transporter and catalyzes the export of acetylated polyamines via a polyamine/arginine exchange reaction, suggesting a role for acetylation in polyamine efflux (Uemura et al., 2008). SLC22A16 has also been identified as a high affinity transporter directing polyamine import in mammalian cells (Aouida et al., 2010). Polyamine uptake by caveolae-dependent endocytosis has also been reported (Roy et al., 2008). Polyamines are present in the extracellular space from dietary intake, export from neighboring cells and synthesis by intestinal bacteria. Such microenvironment polyamines provide a reservoir whereby polyamine antagonized cancer cells can circumvent biosynthetic blockade through augmented uptake.
Polyamines are elevated in rapidly proliferating cells, including cancer cells, and substantial evidence suggests cancer development is associated with altered polyamine regulation. The biological association between increased polyamines and tumor formation is well established in numerous cancers including breast, prostate, colon, skin carcinoma and neuroblastoma (Cipolla et al., 1993; Leveque et al., 2000; Thomas and Thomas, 2003; Gerner and Meyskens, 2004; Casero and Marton, 2007). There is also evidence that increased polyamine biosynthesis is not just a consequence of increased proliferation in these cells, but may be necessary for the development of specific cancers (Gerner and Meyskens, 2004; Casero and Marton, 2007). The mechanism by which MYCN amplification results in such a poor prognosis has yet to be fully elucidated, and recent evidence suggests that its effect on the polyamine pathway may play a critical role. A number of polyamine genes have been shown to be c-MYC target genes (ODC1, AMD1, and SRM) whereas others appear to be regulated by MYC/MYCN (Bello-Fernandez et al., 1993; Fernandez et al., 2003; Hogarty et al., 2008; Rounbehler et al., 2009; Forshell et al., 2010). However, with the exception of ODC1 (Lutz et al., 1996), the polyamine genes that are direct transcriptional targets of MYCN remain to be established. It is highly likely that polyamine synthesis may be specifically required to support downstream MYCN-governed functions.
ODC1 is a well-established oncogene in its own right (Auvinen et al., 1992), with high ODC1 activity associated with tumor growth in several human cancers, including neuroblastoma (O’Brien et al., 1975; Janne et al., 1978; Scalabrino and Ferioli, 1981; Crozat et al., 1992; Mohan et al., 1999; Wallace and Caslake, 2001; Hogarty et al., 2008). The contribution of ODC1 activity to MYC-induced lymphomagenesis was examined in a mouse model of B-cell lymphoma, the Eμ-Myc transgenic mouse. In this model, ODC1 ablation inhibited lymphomagenesis, but subsequent restoration of ODC1 activity promoted tumor onset (Nilsson et al., 2005). In addition, enforced expression of ODC1 in the skin of transgenic mice led to increased tumor incidence (O’Brien et al., 1997; Chen et al., 2000). In neuroblastoma there is significant evidence that ODC1 is overexpressed in high risk disease. It is often co-amplified with MYCN or overexpressed, and is associated with poor prognosis in both MYCN amplified and non-MYCN amplified tumors (Hogarty et al., 2008; Rounbehler et al., 2009; Geerts et al., 2010).
Evaluation of several polyamine genes included in the Neuroblastoma Prognosis Database (publically available at http://home.ccr.cancer.gov/oncology/oncogenomics/) revealed that increased expression of biosynthetic SMS, AMD1, and AZIN, and decreased expression of catabolic OAZ2 was associated with decreased survival and poor prognosis as shown in Figure 2A. The levels of SAT1 or SRM expression on the other hand, were not prognostic of survival. However, all of these genes, including SAT1 and SRM, were associated either positively or negatively with MYCN amplification dependent on their biosynthetic or catabolic role (Figure 2B). Since MYCN is upstream of the polyamine biosynthesis pathway, this suggests a major role for MYCN in regulating polyamine biosynthesis, and a mechanism by which MYCN contributes to neuroblastoma development. Several studies support these findings. Geerts et al. (2010) found increased ODC1 and reduced OAZ2 expression to be excellent predictors of survival and poor prognosis in both MYCN amplified and non-amplified neuroblastomas. OAZ1 and OAZ3 on the other hand played no role in predicting survival. Transcriptome analysis of 101 primary neuroblastomas found several polyamine biosynthetic genes, including ODC1, AMD1, SRM, and SMS, to be up-regulated in the MYCN amplified high risk cohort (and again ODC1 expression was elevated in non-MYCN amplified high risk group; Hogarty et al., 2008). OAZ2 was expressed at lower levels in high risk MYCN amplified tumors but also significantly reduced in non-MYCN amplified high risk tumors. In addition catabolic SMOX was decreased, while the level of SAT1 expression was not associated with any particular risk group (Hogarty et al., 2008). These studies suggest a role for ODC1, and OAZ2, independent of MYCN, in promoting an aggressive phenotype. Further evidence supporting this conclusion comes from the finding that ODC1 is not always co-amplified with MYCN in neuroblastomas, while copy number gain of ODC1 has been reported in half of high risk neuroblastomas without MYCN amplification, suggesting a mechanism by which the polyamine pathway is up-regulated in this subset (George et al., 1997; Mosse et al., 2007; Hogarty et al., 2008).
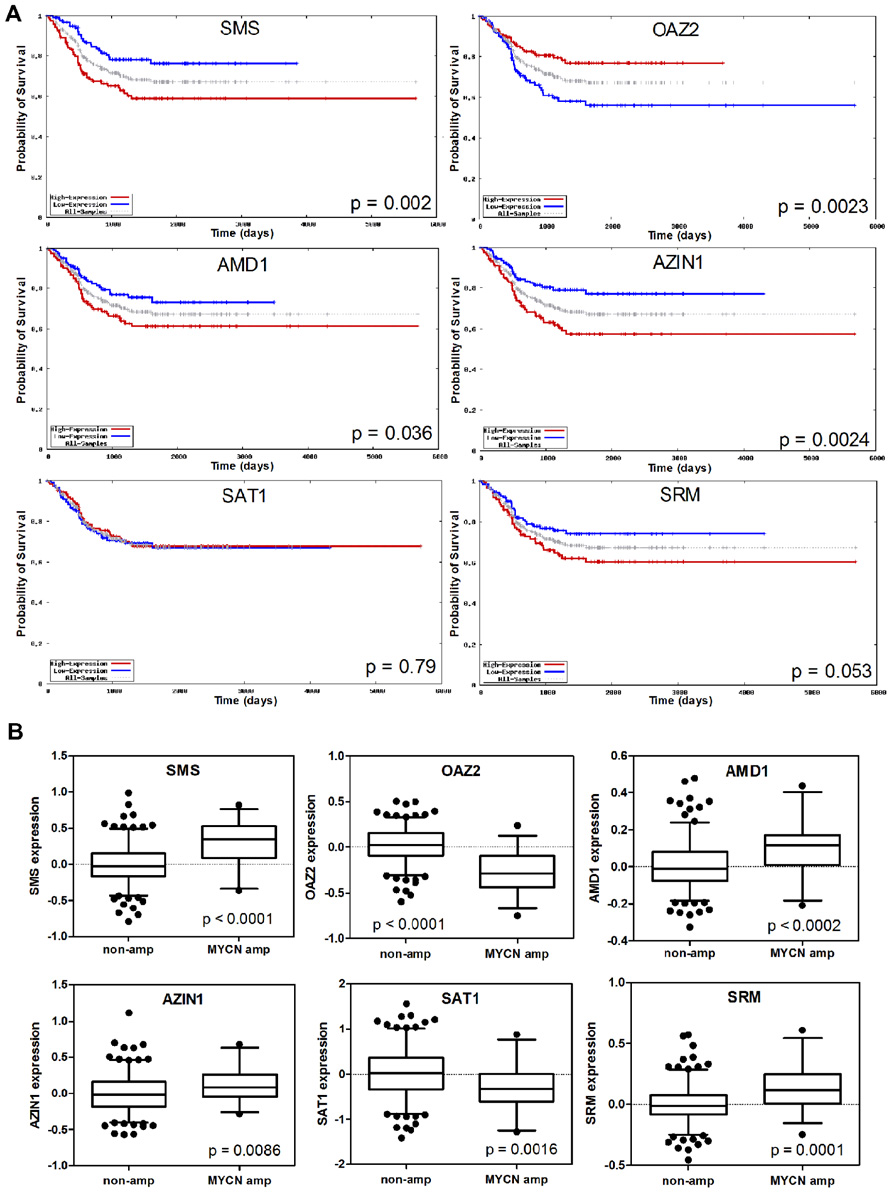
FIGURE 2. Analysis of expression of the polyamine pathway regulators SMS, OAZ2, AMD1, AZIN1, SAT1, and SRM, and their association with neuroblastoma outcome. (A) Kaplan–Meier survival curves in the overall neuroblastoma cohort with dichotomization for high/low expression around the median. (B) Expression of polyamine pathway genes in the subsets of tumors with and without MYCN amplification. Data was obtained from the Neuroblastoma Prognosis Database (publically available at http://home.ccr.cancer.gov/oncology/oncogenomics/).
These data suggest that systemic alterations in polyamine metabolism correlate with MYCN amplification, but that polyamine enhancement in non-MYCN amplified tumors is also associated with high risk disease. Polyamine depletion strategies may be broadly effective against high risk tumors, rather than just MYCN amplified tumors.
Since elevated polyamines are sustained in rapidly proliferating cells and levels are increased in cancer tissues compared to surrounding tissues, suppression of polyamine biosynthesis provides an attractive therapeutic approach for many cancers. Inhibitors of the rate-limiting enzymes in polyamine biosynthesis, ODC1 and AMD1, have been developed and extensively tested in preclinical and clinical trials. α-difluoromethylornithine (DFMO) acts as a specific suicide inhibitor of ODC1 and is the most widely studied inhibitor of polyamine metabolism both as a chemotherapeutic and a chemopreventive agent (Meyskens and Gerner, 1999; Levin et al., 2000; Takahashi et al., 2000; Fabian et al., 2002; Levin et al., 2003). Exposure of a number of cancer cell lines, tumors and tissues to DFMO has shown a considerable decrease in intracellular putrescine concentrations, subsequent decreases in spermidine levels, and growth inhibition as a result of impaired synthesis of RNA, DNA, and proteins (Mamont et al., 1982; Sunkara and Rosenberger, 1987). Despite promising preclinical results, the anti-tumor activity of DFMO has to date failed to translate to the clinic. However, further investigations have shown additive and synergistic activities when used in combination therapies for the treatment of specific cancers in animal models (Bartholeyns and Koch-Weser, 1981; Marton, 1987; Quemener et al., 1992) and the potential for synthetic-lethal interactions in MYC-driven cancers specifically has yet to be directly tested.
It has previously been shown that ODC1 heterozygosity or treatment with DFMO impairs MYC induced lymphomagenesis in the Eμ-Myc transgenic mice, and that this was achieved though impairment of MYC’s ability to suppress p27KIP1, a CDK inhibitor (Nilsson et al., 2005). A number of recent studies have investigated the effect of DFMO in neuroblastoma cell lines and animal models. A MYCN transgenic mouse model (TH-MYCN mice), which faithfully recapitulates the features of human neuroblastoma with similar biochemical features and syntenic chromosomal rearrangements to human neuroblastoma, was used (Weiss et al., 1997). Tumor formation is dependent on the level of MYCN gene dosage, with homozygous mice developing tumors with a short latency and 100% tumor penetrance, and hemizygous mice displaying longer tumor latency and only 20–30% penetrance. In this model TH-MYCN tumors have up-regulated ODC1, AZIN, AMD1, SRM, and SMS, and down-regulated OAZ2, SMOX, and SAT1 compared with sympathetic ganglia (Evageliou and Hogarty, 2009). Since a similar pattern of polyamine deregulation is seen in primary human neuroblastoma, this suggests that results using this model are likely to be translatable to the human disease. DFMO treatment in neuroblastoma cell lines inhibited proliferation, and when extended to in vivo studies using the TH-MYCN transgenic mouse, DFMO treatment from birth increased tumor latency and overall survival (Hogarty et al., 2008). Interestingly, no tumors developed after DFMO withdrawal, suggesting a finite period during which embryonic tumors can develop and also supporting a role for DFMO as a chemopreventive agent for neuroblastoma. Giving hemizygous mice DFMO from birth resulted in reduced tumor initiation. DFMO treatment of mice with detectable tumors delayed tumor progression and extended survival time (Hogarty et al., 2008). Similarly, Rounbehler et al. (2009) found that DFMO selectively impaired the proliferative response of MYCN amplified neuroblastomas, and delayed tumor incidence and onset in vivo. However, once a tumor developed it had a similar aggressive phenotype as tumours in mice that had not received DFMO, and whilst there was a reduction in putrescine, spermidine was only moderately reduced and spermine levels remained unchanged (Hogarty et al., 2008). Importantly, DFMO enhanced the effect of the anticancer drugs, cyclophosphamide and cisplatin in vivo. Tumor-free survival after cyclophosphamide treatment in combination with DFMO was increased to 80% compared to 20% for cyclophosphamide alone, and DFMO significantly increased the survival time of mice treated with cisplatin, although all of these mice did eventually succumb to the disease (Hogarty et al., 2008).
SAM486A is a derivative of the first generation AMD1 inhibitor mitoguazone (MGBG), and exerts potent and specific inhibition of AMD1 (Regenass et al., 1992, 1994). Its efficacy has been assessed in a number of cancer cells and animal systems, and has been tested in phase I and II clinical trials in adult cancers. However, like DFMO, when used as a single agent, results have been disappointing. In neuroblastoma, in vitro studies found p53 wild-type cells to be highly sensitive to SAM486A independent of their MYCN status (Koomoa et al., 2009). In these cells SAM486A functions by inducing p53, possibly through DNA damage induced by ATM, and by reducing Akt/PKB expression to induce apoptosis and inhibit cell proliferation (Koomoa et al., 2009). In addition, large increases in intracellular putrescine levels correlated with increased p53. SAM486A treatment of p53 mutant neuroblastoma cells inhibited polyamine-dependent cell growth and caused a G1 arrest, which was further enhanced upon combination with DFMO. Neither compound, either alone or in combination, induced apoptosis (Wallick et al., 2005). Following removal of these inhibitors in the p53 mutant cells, the proliferative capacity of the cells was slow and only partially restored, but this was shown to be largely due to DFMO and not SAM486A. DFMO has been shown to induce cell cycle arrest in a p53 mutant neuroblastoma cell line via induction of two contradictory pathways; cell survival via PI3K/PKB signaling, and cell cycle arrest through p27KIP1 phosphorylation (Wallick et al., 2005; Koomoa et al., 2008).
The disappointing clinical trials with either DFMO or SAM486A as single agents are likely due to activation of compensatory mechanisms following DFMO or SAM486A exposure. This allows intracellular polyamine levels to be maintained in a cell upon loss of a single biosynthetic enzyme activity. Polyamines may be imported from extracellular pools, and compensatory induction of other biosynthetic enzymes or reduced polyamine catabolism may be involved.
AMD1 has been shown to be up-regulated following ODC1 inhibition (Wallick et al., 2005) and it has been reported that combined DFMO and SAM486A therapy is synergistic in neuroblastoma (Evageliou and Hogarty, 2009). This indicates that attacking the polyamine synthesis pathway with multiple compounds may be a more effective approach, particularly if the two rate-limiting enzymes are simultaneously inhibited. DFMO and SAM486A are of particular interest because clinical trials in other cancer types have shown both inhibitors to be well tolerated, even at high doses, with only the occasional occurrence of reversible ototoxicity (DFMO only), nausea and mild neutropenia. In addition, DFMO is already FDA approved as it is used in the treatment of African trypanosomiasis (Van Nieuwenhove et al., 1985; Sjoerdsma and Schechter, 1999).
As well as being tested as a chemotherapeutic agent, DFMO has demonstrated promising results in human trials as a chemopreventive agent. This polyamine inhibitor has been shown to suppress skin carcinogenesis in patients with moderate to severe actinic keratosis (Alberts et al., 2000), and also slowed prostate cancer growth in men with a family history of prostate cancer (Simoneau et al., 2008). In addition, DFMO in combination with sulindac, a SAT1 inducing COX2 inhibitor, resulted in a remarkable decrease in colon adenomas in patients with previous disease (Meyskens et al., 2008). There were no significant toxicities in any of these studies. Whilst chemopreventive approaches are not currently practical in neuroblastoma, the use of polyamine antagonists could prove useful in managing minimal residual disease (MRD) post-autologous stem cell transplantation in order to reduce the risk of relapse.
Another compound that targets polyamine biosynthesis is the SRM inhibitor, trans-4-methylcyclohexamide (4MCHA). This inhibitor has been tested in a B-cell lymphoma mouse model where 4MCHA had chemopreventive effects in vivo, but was not effective against established lymphomas (Shirahata et al., 1993; Forshell et al., 2010). SRM is a MYC target and interestingly, it was found to be more potently induced by MYC than ODC1 suggesting it may be important in MYC-induced oncogenesis.
Polyamine analogs have a multistep role in depleting polyamine pools. They function by mimicking natural polyamines and subsequently lowering intracellular polyamine levels by feedback inhibition. The result is down-regulation of synthetic enzymes such as ODC1 and AMD1, but also induction of catabolic enzymes such as SAT1 and SMOX. Elevated levels of SAT1 increases export of acetyl-polyamines due to co-localization with a polyamine transporter (such as SLC3A2), and induction of SMOX results in a subsequent production of H2O2 (Wang et al., 2001; Casero et al., 2003; Pledgie-Tracy et al., 2009). The resulting accumulation of non-functional analogs competitively inhibits polyamine function, and the depleted intracellular polyamine pool reduces cell proliferation and induces growth inhibition (Reddy et al., 1998; Casero and Marton, 2007). A number of these compounds have been developed and have shown promising results in in vitro and in vivo models.
N1, N11-diethylnorspermine (DENSpm) is a spermine analog that induces growth inhibition in a number of cancer cell lines (Kramer et al., 1997; Gabrielson et al., 1999; Schipper et al., 2000; Myhre et al., 2008). When tested in neuroblastoma cell lines, the compound induced G1 arrest and apoptosis in p53 wild-type cells, but similar to SAM486A treatment, only induced growth inhibition in p53 mutant cells (Soderstjerna et al., 2010). DENSpm, as well as the related compound, BENSpm, and the second-generation CPENSpm, have all been tested in phase I and II clinical studies but were not successful as single agents (Creaven et al., 1997; Hahm et al., 2002; Wolff et al., 2003). BENSpm has been shown to synergistically induce growth inhibition in combination with standard chemotherapy agents in cell lines (Davidson et al., 1993; Pledgie-Tracy et al., 2009). In addition BENSpm combined with cisplatin produced synergistic cell death in cisplatin resistant ovarian carcinoma cells (Marverti et al., 1998). This resulted in a synergistic increase in SAT1 activity, and the subsequent polyamine pool depletion by this combination was significantly greater than either agent alone (Tummala et al., 2011). However, in vivo studies using breast cancer cell xenografts found that BENSpm in combination with paclitaxel did not further reduce tumor growth compared to either agent alone. In vitro, synergy between BENSpm and various chemotherapeutic agents was dependent on the cell line and the combined chemotherapeutic used (Pledgie-Tracy et al., 2009). Surprisingly, whereas some studies report induction of SMOX by polyamine analogs, BENSpm and CPENSpm have been shown to bind the catalytic site of SMOX, inhibiting activity and may explain why SMOX levels appear to be induced, as a result of stabilization (Cervelli et al., 2010). The subsequent reduction in H2O2 in the tumor mass may partly explain the lack of activity observed in clinical trials.
PG11047 is a novel conformationally restricted analog of spermine that competitively inhibits spermine function (Reddy et al., 1998), and has recently been studied in phase I clinical trials for adult cancers both as a single agent and in combination with chemotherapy (Clinical Trial Identifier: NCT00705874). PG11047 inhibits cell proliferation in a range of cell lines, and inhibits tumor growth in vivo in prostate and NSCLC xenografts (Hacker et al., 2008; Dredge et al., 2009). However, whilst Ewings sarcoma cell lines were particularly sensitive to PG11047-mediated growth inhibition, neuroblastoma cells were less sensitive and in vivo, a cytostatic effect similar to that seen with DFMO was reported, with only a modest delay in tumor onset after treatment (Smith et al., 2011).
Since tumor cells exhibit enhanced polyamine transport activity by comparison with normal cells, and since the pharmacological inhibition of polyamine biosynthesis leads to a compensatory increase in polyamine salvaging activity (Pegg, 1988; Seiler et al., 1996), another mechanism of inhibiting this pathway includes antagonizing polyamine uptake. A number of compounds are under preclinical development, including D-lysine spermine (MQT-1426), N1-spermyl-L-lysinamide (OR1202), and a spermine analog D-Lys(C16acyl)-Spm (AMXT1501; Weeks et al., 2000; Chen et al., 2006; Burns et al., 2009). Despite a poor response to D-Lys(C16acyl)-Spm alone, combination with DFMO had remarkable efficacy against cutaneous squamous cell carcinomas (SCC) in a transgenic ODC1 mouse model of skin cancer. The majority of large aggressive SCCs underwent complete or near-complete remission, even in the presence of extracellular spermidine, indicating that D-Lys(C16acyl)-Spm in combination with DFMO is successful in reducing intracellular polyamines (Burns et al., 2009). Available evidence indicates that high levels of expression of SLC22A16, a polyamine transporter, are prognostic of poor outcome in MYCN non-amplified metastatic neuroblastoma (http://pob.abcc.ncifcrf.gov/cgi-bin/JK). Targeting this polyamine transporter may be required to effectively reduce intracellular polyamine levels.
The use of NSAIDs such as celecoxib and sulindac, has also been investigated, which function by influencing polyamine acetylation and export through up-regulation of SAT1 (Babbar et al., 2003). Celecoxib combined with anticancer agents induces synergistic and anti-proliferative effects (Shirode and Sylvester, 2010), exerting their chemopreventive action by affecting SAT1. DFMO in combination with NSAIDs has been shown to suppress colorectal carcinogenesis in murine models and in phase II clinical trials (Fischer et al., 2003; Gerner et al., 2007).
Polyamines conjugated to cytotoxic drugs, such as naphthalimide, anthracene, or anthraquinone, can be transported into cancer cells via the polyamine transporter system, and have been shown to exert potent anti-tumor effects (Tian et al., 2009; Xie et al., 2011a). Since the polyamine transporter is up-regulated in many tumor cells, these compounds may provide a targeted therapy, inhibiting cell proliferation through simultaneously delivering a cytotoxic drug, and also depleting intracellular polyamine content. A number of preclinical studies have shown promising results in a variety of cancers, although no studies have been carried out in neuroblastoma. In colorectal cancer cell lines, a naphthalimide-polyamine conjugate (NPC-16) in combination with celecoxib produced enhanced apoptosis as a result of elevated NPC-16 uptake due to up-regulated SAT1 activity, and decreased intracellular polyamine levels (Xie et al., 2011b). A putrescine conjugated with anthracene, Ant 4, was shown to induce cytotoxicity and subsequent apoptosis in a promyelogenous leukemia cell line (Palmer et al., 2009). Putrescine uptake was significantly reduced, demonstrating that this conjugate could successfully compete with its native polyamine for uptake. The spermine-podophyllotoxin conjugate F14512 has shown exceptional cytotoxicity in cells with enhanced polyamine uptake in vitro, as well as inhibiting breast carcinoma in a xenograft model (Barret et al., 2008). Whilst preclinical data using this class of compound look promising, to date no clinical trials have taken place. It is attractive to speculate that combining a polyamine-chemotherapy conjugate with other polyamine depleting agents will facilitate the uptake of these conjugates and provide a more active targeted approach in reducing polyamines.
Many compounds targeting the polyamine pathway have been developed or are under development. However, those that have made it to clinical trials have produced limited effects, most likely as a result of compensatory mechanisms that allow a cell to circumvent polyamine depletion. Polyamine depletion compounds have been well tolerated clinically, and in combination with chemotherapeutic agents have produced synergistic effects. It is likely that optimization of polyamine depletion, by using compounds that decrease polyamine synthesis and eradicate compensatory mechanisms, in combination with chemotherapeutic agents may have significant clinical potential in improving the outcome of patients with aggressive neuroblastoma. Furthermore, these compounds are likely to be effective in both MYCN amplified and non-MYCN amplified patients since polyamine deregulation has been observed in both tumor groups. A phase I clinical trial, coordinated by the New Approaches to Neuroblastoma Therapy (NANT) consortium, for the treatment of refractory neuroblastoma using high dose DFMO and celecoxib in combination with standard chemotherapy (cyclophosphamide and topotecan) is in development, and results from this study will be invaluable in determining the potential use of polyamine depletion for the treatment of neuroblastoma.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Albeck, S., Dym, O., Unger, T., Snapir, Z., Bercovich, Z., and Kahana, C. (2008). Crystallographic and biochemical studies revealing the structural basis for antizyme inhibitor function. Protein Sci. 17, 793–802.
Alberts, D. S., Dorr, R. T., Einspahr, J. G., Aickin, M., Saboda, K., Xu, M. J., et al. (2000). Chemoprevention of human actinic keratoses by topical 2-(difluoromethyl)-dl-ornithine. Cancer Epidemiol. Biomarkers Prev. 9, 1281–1286.
Aouida, M., Poulin, R., and Ramotar, D. (2010). The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J. Biol. Chem. 285, 6275–6284.
Auvinen, M., Paasinen, A., Andersson, L. C., and Holtta, E. (1992). Ornithine decarboxylase activity is critical for cell transformation. Nature 360, 355–358.
Babbar, N., Ignatenko, N. A., Casero, R. A. Jr., and Gerner, E. W. (2003). Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J. Biol. Chem. 278, 47762–47775.
Barret, J. M., Kruczynski, A., Vispe, S., Annereau, J. P., Brel, V., Guminski, Y., et al. (2008). F14512, a potent antitumor agent targeting topoisomerase II vectored into cancer cells via the polyamine transport system. Cancer Res. 68, 9845–9853.
Bartholeyns, J., and Koch-Weser, J. (1981). Effects of alpha-difluoromethylornithine alone and combined with adriamycin or vindesine on L1210 leukemia in mice, EMT6 solid tumors in mice, and solid tumors induced by injection of hepatoma tissue culture cells in rats. Cancer Res. 41, 5158–5161.
Bello-Fernandez, C., Packham, G., and Cleveland, J. L. (1993). The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 90, 7804–7808.
Burns, M. R., Graminski, G. F., Weeks, R. S., Chen, Y., and O’Brien, T. G. (2009). Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a polyamine biosynthesis inhibitor. J. Med. Chem. 52, 1983–1993.
Casero, R. A. Jr., and Marton, L. J. (2007). Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 6, 373–390.
Casero, R. A. Jr., and Pegg, A. E. (1993). Spermidine/spermine N1-acetyltransferase-the turning point in polyamine metabolism. FASEB J. 7, 653–661.
Casero, R. A. Jr., and Pegg, A. E. (2009). Polyamine catabolism and disease. Biochem. J. 421, 323–338.
Casero, R. A. Jr., Wang, Y., Stewart, T. M., Devereux, W., Hacker, A., Smith, R., et al. (2003). The role of polyamine catabolism in anti-tumour drug response. Biochem. Soc. Trans. 31, 361–365.
Cervelli, M., Bellavia, G., Fratini, E., Amendola, R., Polticelli, F., Barba, M., et al. (2010). Spermine oxidase (SMO) activity in breast tumor tissues and biochemical analysis of the anticancer spermine analogues BENSpm and CPENSpm. BMC Cancer 10, 555. doi: 10.1186/1471-2407-10-555
Chen, Y., Megosh, L. C., Gilmour, S. K., Sawicki, J. A., and O’Brien, T. G. (2000). K6/ODC transgenic mice as a sensitive model for carcinogen identification. Toxicol. Lett. 116, 27–35.
Chen, Y., Weeks, R. S., Burns, M. R., Boorman, D. W., Klein-Szanto, A., and O’Brien, T. G. (2006). Combination therapy with 2-fluoromethylornithine and a polyamine transport inhibitor against murine squamous cell carcinoma. Int. J. Cancer 118, 2344–2349.
Childs, A. C., Mehta, D. J., and Gerner, E. W. (2003). Polyamine-dependent gene expression. Cell Mol. Life Sci. 60, 1394–1406.
Cipolla, B., Guille, F., Moulinoux, J. P., Quemener, V., Staerman, F., Corbel, L., et al. (1993). Polyamines and prostatic carcinoma: clinical and therapeutic implications. Eur. Urol. 24, 124–131.
Cohn, S. L., and Tweddle, D. A. (2004). MYCN amplification remains prognostically strong 20 years after its “clinical debut.” Eur. J. Cancer 40, 2639–2642.
Coleman, C. S., Huang, H., and Pegg, A. E. (1996). Structure and critical residues at the active site of spermidine/spermine-N1-acetyltransferase. Biochem. J. 316(Pt 3), 697–701.
Cooper, H. L., Park, M. H., Folk, J. E., Safer, B., and Braverman, R. (1983). Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc. Natl. Acad. Sci. U.S.A. 80, 1854–1857.
Creaven, P. J., Perez, R., Pendyala, L., Meropol, N. J., Loewen, G., Levine, E., et al. (1997). Unusual central nervous system toxicity in a phase I study of N1N11 diethylnorspermine in patients with advanced malignancy. Invest. New Drugs 15, 227–234.
Crozat, A., Palvimo, J. J., Julkunen, M., and Janne, O. A. (1992). Comparison of androgen regulation of ornithine decarboxylase and S-adenosylmethionine decarboxylase gene expression in rodent kidney and accessory sex organs. Endocrinology 130, 1131–1144.
Davidson, N. E., Mank, A. R., Prestigiacomo, L. J., Bergeron, R. J., and Casero, R. A. Jr. (1993). Growth inhibition of hormone-responsive and -resistant human breast cancer cells in culture by N1, N12-bis(ethyl)spermine. Cancer Res. 53, 2071–2075.
Dredge, K., Kink, J. A., Johnson, R. M., Bytheway, I., and Marton, L. J. (2009). The polyamine analog PG11047 potentiates the antitumor activity of cisplatin and bevacizumab in preclinical models of lung and prostate cancer. Cancer Chemother. Pharmacol. 65, 191–195.
Evageliou, N. F., and Hogarty, M. D. (2009). Disrupting polyamine homeostasis as a therapeutic strategy for neuroblastoma. Clin. Cancer Res. 15, 5956–5961.
Fabian, C. J., Kimler, B. F., Brady, D. A., Mayo, M. S., Chang, C. H., Ferraro, J. A., et al. (2002). A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: breast tissue, imaging, and serum and urine biomarkers. Clin. Cancer Res. 8, 3105–3117.
Fernandez, P. C., Frank, S. R., Wang, L., Schroeder, M., Liu, S., Greene, J., et al. (2003). Genomic targets of the human c-Myc protein. Genes Dev. 17, 1115–1129.
Fischer, S. M., Conti, C. J., Viner, J., Aldaz, C. M., and Lubet, R. A. (2003). Celecoxib and difluoromethylornithine in combination have strong therapeutic activity against UV-induced skin tumors in mice. Carcinogenesis 24, 945–952.
Fogel-Petrovic, M., Shappell, N. W., Bergeron, R. J., and Porter, C. W. (1993). Polyamine and polyamine analog regulation of spermidine/spermine N1-acetyltransferase in MALME-3M human melanoma cells. J. Biol. Chem. 268, 19118–19125.
Forshell, T. P., Rimpi, S., and Nilsson, J. A. (2010). Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. (Phila) 3, 140–147.
Gabrielson, E. W., Pegg, A. E., and Casero, R. A. Jr. (1999). The induction of spermidine/spermine N1-acetyltransferase (SSAT) is a common event in the response of human primary non-small cell lung carcinomas to exposure to the new antitumor polyamine analogue N1,N11-bis(ethyl)norspermine. Clin. Cancer Res. 5, 1638–1641.
Geerts, D., Koster, J., Albert, D., Koomoa, D. L., Feith, D. J., Pegg, A. E., et al. (2010). The polyamine metabolism genes ornithine decarboxylase and antizyme 2 predict aggressive behavior in neuroblastomas with and without MYCN amplification. Int. J. Cancer 126, 2012–2024.
George, R. E., Kenyon, R., Mcguckin, A. G., Kohl, N., Kogner, P., Christiansen, H., et al. (1997). Analysis of candidate gene co-amplification with MYCN in neuroblastoma. Eur. J. Cancer 33, 2037–2042.
Gerner, E. W., and Meyskens, F. L. Jr. (2004). Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 4, 781–792.
Gerner, E. W., Meyskens, F. L. Jr., Goldschmid, S., Lance, P., and Pelot, D. (2007). Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids 33, 189–195.
Hacker, A., Marton, L. J., Sobolewski, M., and Casero, R. A. Jr. (2008). In vitro and in vivo effects of the conformationally restricted polyamine analogue CGC-11047 on small cell and non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 63, 45–53.
Hahm, H. A., Ettinger, D. S., Bowling, K., Hoker, B., Chen, T. L., Zabelina, Y., et al. (2002). Phase I study of N(1),N(11)-diethylnorspermine in patients with non-small cell lung cancer. Clin. Cancer Res. 8, 684–690.
Hogarty, M. D., Norris, M. D., Davis, K., Liu, X., Evageliou, N. F., Hayes, C. S., et al. (2008). ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 68, 9735–9745.
Igarashi, K., and Kashiwagi, K. (2000). Polyamines: mysterious modulators of cellular functions. Biochem. Biophys. Res. Commun. 271, 559–564.
Janne, J., Poso, H., and Raina, A. (1978). Polyamines in rapid growth and cancer. Biochim. Biophys. Acta 473, 241–293.
Johnson, T. D. (1996). Modulation of channel function by polyamines. Trends Pharmacol. Sci. 17, 22–27.
Kanerva, K., Makitie, L. T., Pelander, A., Heiskala, M., and Andersson, L. C. (2008). Human ornithine decarboxylase paralogue (ODCp) is an antizyme inhibitor but not an arginine decarboxylase. Biochem. J. 409, 187–192.
Keren-Paz, A., Bercovich, Z., Porat, Z., Erez, O., Brener, O., and Kahana, C. (2006). Overexpression of antizyme-inhibitor in NIH3T3 fibroblasts provides growth advantage through neutralization of antizyme functions. Oncogene 25, 5163–5172.
Koguchi, K., Kobayashi, S., Hayashi, T., Matsufuji, S., Murakami, Y., and Hayashi, S. (1997). Cloning and sequencing of a human cDNA encoding ornithine decarboxylase antizyme inhibitor. Biochim. Biophys. Acta 1353, 209–216.
Koomoa, D. L., Borsics, T., Feith, D. J., Coleman, C. C., Wallick, C. J., Gamper, I., et al. (2009). Inhibition of S-adenosylmethionine decarboxylase by inhibitor SAM486A connects polyamine metabolism with p53-Mdm2-Akt/protein kinase B regulation and apoptosis in neuroblastoma. Mol. Cancer Ther. 8, 2067–2075.
Koomoa, D. L., Yco, L. P., Borsics, T., Wallick, C. J., and Bachmann, A. S. (2008). Ornithine decarboxylase inhibition by alpha-difluoromethylornithine activates opposing signaling pathways via phosphorylation of both Akt/protein kinase B and p27Kip1 in neuroblastoma. Cancer Res. 68, 9825–9831.
Kramer, D. L., Diegelman, P., Jell, J., Vujcic, S., Merali, S., and Porter, C. W. (2008). Polyamine acetylation modulates polyamine metabolic flux, a prelude to broader metabolic consequences. J. Biol. Chem. 283, 4241–4251.
Kramer, D. L., Fogel-Petrovic, M., Diegelman, P., Cooley, J. M., Bernacki, R. J., Mcmanis, J. S., et al. (1997). Effects of novel spermine analogues on cell cycle progression and apoptosis in MALME-3M human melanoma cells. Cancer Res. 57, 5521–5527.
Leveque, J., Foucher, F., Bansard, J. Y., Havouis, R., Grall, J. Y., and Moulinoux, J. P. (2000). Polyamine profiles in tumor, normal tissue of the homologous breast, blood, and urine of breast cancer sufferers. Breast Cancer Res. Treat. 60, 99–105.
Levin, V. A., Hess, K. R., Choucair, A., Flynn, P. J., Jaeckle, K. A., Kyritsis, A. P., et al. (2003). Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin. Cancer Res. 9, 981–990.
Levin, V. A., Uhm, J. H., Jaeckle, K. A., Choucair, A., Flynn, P. J., Yung, W. K. A., et al. (2000). Phase III randomized study of postradiotherapy chemotherapy with alpha-difluoromethylornithine-procarbazi-ne, N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosurea, vincristine (DFMO-PCV) versus PCV for glioblastoma multiforme. Clin. Cancer Res. 6, 3878–3884.
Li, L., Liu, L., Rao, J. N., Esmaili, A., Strauch, E. D., Bass, B. L., et al. (2002). JunD stabilization results in inhibition of normal intestinal epithelial cell growth through P21 after polyamine depletion. Gastroenterology 123, 764–779.
Li, X., and Coffino, P. (1992). Regulated degradation of ornithine decarboxylase requires interaction with the polyamine-inducible protein antizyme. Mol. Cell. Biol. 12, 3556–3562.
Li, Z., Van Calcar, S., Qu, C., Cavenee, W. K., Zhang, M. Q., and Ren, B. (2003). A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 8164–8169.
Lutz, W., Stohr, M., Schurmann, J., Wenzel, A., Lohr, A., and Schwab, M. (1996). Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 13, 803–812.
Mamont, P. S., Danzin, C., Wagner, J., Siat, M., Joder-Ohlenbusch, A. M., and Claverie, N. (1982). Accumulation of decarboxylated S-adenosyl-L-methionine in mammalian cells as a consequence of the inhibition of putrescine biosynthesis. Eur. J. Biochem. 123, 499–504.
Mangold, U. (2006). Antizyme inhibitor: mysterious modulator of cell proliferation. Cell Mol. Life Sci. 63, 2095–2101.
Maris, J. M., Hogarty, M. D., Bagatell, R., and Cohn, S. L. (2007). Neuroblastoma. Lancet 369, 2106–2120.
Marton, L. J. (1987). Effects of treatment with DNA-directed cancer chemotherapeutic agents after polyamine depletion. Pharmacol. Ther. 32, 183–190.
Marverti, G., Piccinini, G., Ghiaroni, S., Barbieri, D., Quaglino, D., and Moruzzi, M. S. (1998). N1,N12-bis(ethyl)spermine effect on growth of cis-diamminedichloroplatinum(II)-sensitive and -resistant human ovarian-carcinoma cell lines. Int. J. Cancer 78, 33–40.
Matsufuji, S., Matsufuji, T., Miyazaki, Y., Murakami, Y., Atkins, J. F., Gesteland, R. F., et al. (1995). Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 80, 51–60.
Meyskens, F. L. Jr., and Gerner, E. W. (1999). Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 5, 945–951.
Meyskens, F. L. Jr., Mclaren, C. E., Pelot, D., Fujikawa-Brooks, S., Carpenter, P. M., Hawk, E., et al. (2008). Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev. Res. (Phila) 1, 32–38.
Mitchell, J. L., Judd, G. G., Bareyal-Leyser, A., and Ling, S. Y. (1994). Feedback repression of polyamine transport is mediated by antizyme in mammalian tissue-culture cells. Biochem. J. 299(Pt 1), 19–22.
Mohan, R. R., Challa, A., Gupta, S., Bostwick, D. G., Ahmad, N., Agarwal, R., et al. (1999). Overexpression of ornithine decarboxylase in prostate cancer and prostatic fluid in humans. Clin. Cancer Res. 5, 143–147.
Mosse, Y. P., Diskin, S. J., Wasserman, N., Rinaldi, K., Attiyeh, E. F., Cole, K., et al. (2007). Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression. Genes Chromosomes Cancer 46, 936–949.
Murakami, Y., Ichiba, T., Matsufuji, S., and Hayashi, S. (1996). Cloning of antizyme inhibitor, a highly homologous protein to ornithine decarboxylase. J. Biol. Chem. 271, 3340–3342.
Murakami, Y., Matsufuji, S., Kameji, T., Hayashi, S., Igarashi, K., Tamura, T., et al. (1992). Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360, 597–599.
Myhre, L., Alm, K., Hegardt, C., Staaf, J., Jonsson, G., Larsson, S., et al. (2008). Different cell cycle kinetic effects of N1,N11-diethylnorspermine-induced polyamine depletion in four human breast cancer cell lines. Anticancer Drugs 19, 359–368.
Nilsson, J. A., Keller, U. B., Baudino, T. A., Yang, C., Norton, S., Old, J. A., et al. (2005). Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell 7, 433–444.
O’Brien, T. G., Megosh, L. C., Gilliard, G., and Soler, A. P. (1997). Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 57, 2630–2637.
O’Brien, T. G., Simsiman, R. C., and Boutwell, R. K. (1975). Induction of the polyamine-biosynthetic enzymes in mouse epidermis and their specificity for tumor promotion. Cancer Res. 35, 2426–2433.
Palanimurugan, R., Scheel, H., Hofmann, K., and Dohmen, R. J. (2004). Polyamines regulate their synthesis by inducing expression and blocking degradation of ODC antizyme. EMBO J. 23, 4857–4867.
Palmer, A. J., Ghani, R. A., Kaur, N., Phanstiel, O., and Wallace, H. M. (2009). A putrescine-anthracene conjugate: a paradigm for selective drug delivery. Biochem. J. 424, 431–438.
Panagiotidis, C. A., Artandi, S., Calame, K., and Silverstein, S. J. (1995). Polyamines alter sequence-specific DNA-protein interactions. Nucleic Acids Res. 23, 1800–1809.
Pegg, A. E. (1988). Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 48, 759–774.
Pledgie-Tracy, A., Billam, M., Hacker, A., Sobolewski, M. D., Woster, P. M., Zhang, Z., et al. (2009). The role of the polyamine catabolic enzymes SSAT and SMO in the synergistic effects of standard chemotherapeutic agents with a polyamine analogue in human breast cancer cell lines. Cancer Chemother. Pharmacol. 65, 1067–1081.
Poulin, R., Coward, J. K., Lakanen, J. R., and Pegg, A. E. (1993). Enhancement of the spermidine uptake system and lethal effects of spermidine overaccumulation in ornithine decarboxylase-overproducing L1210 cells under hyposmotic stress. J. Biol. Chem. 268, 4690–4698.
Quemener, V., Moulinoux, J. P., Havouis, R., and Seiler, N. (1992). Polyamine deprivation enhances antitumoral efficacy of chemotherapy. Anticancer Res. 12, 1447–1453.
Ray, R. M., Mccormack, S. A., and Johnson, L. R. (2001). Polyamine depletion arrests growth of IEC-6 and Caco-2 cells by different mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G37–G43.
Reddy, V. K., Valasinas, A., Sarkar, A., Basu, H. S., Marton, L. J., and Frydman, B. (1998). Conformationally restricted analogues of 1N,12N-bisethylspermine: synthesis and growth inhibitory effects on human tumor cell lines. J. Med. Chem. 41, 4723–4732.
Regenass, U., Caravatti, G., Mett, H., Stanek, J., Schneider, P., Muller, M., et al. (1992). New S-adenosylmethionine decarboxylase inhibitors with potent antitumor activity. Cancer Res. 52, 4712–4718.
Regenass, U., Mett, H., Stanek, J., Mueller, M., Kramer, D., and Porter, C. W. (1994). CGP 48664, a new S-adenosylmethionine decarboxylase inhibitor with broad spectrum antiproliferative and antitumor activity. Cancer Res. 54, 3210–3217.
Rounbehler, R. J., Li, W., Hall, M. A., Yang, C., Fallahi, M., and Cleveland, J. L. (2009). Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 69, 547–553.
Roy, U. K., Rial, N. S., Kachel, K. L., and Gerner, E. W. (2008). Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol. Carcinog. 47, 538–553.
Scalabrino, G., and Ferioli, M. E. (1981). Polyamines in mammalian tumors. Part I. Adv. Cancer Res. 35, 151–268.
Schipper, R. G., Deli, G., Deloyer, P., Lange, W. P., Schalken, J. A., and Verhofstad, A. A. (2000). Antitumor activity of the polyamine analog N(1), N(11)-diethylnorspermine against human prostate carcinoma cells. Prostate 44, 313–321.
Seiler, N., Delcros, J. G., and Moulinoux, J. P. (1996). Polyamine transport in mammalian cells. An update. Int. J. Biochem. Cell Biol. 28, 843–861.
Shirahata, A., Takahashi, N., Beppu, T., Hosoda, H., and Samejima, K. (1993). Effects of inhibitors of spermidine synthase and spermine synthase on polyamine synthesis in rat tissues. Biochem. Pharmacol. 45, 1897–1903.
Shirode, A. B., and Sylvester, P. W. (2010). Synergistic anticancer effects of combined gamma-tocotrienol and celecoxib treatment are associated with suppression in Akt and NFkappaB signaling. Biomed. Pharmacother. 64, 327–332.
Simoneau, A. R., Gerner, E. W., Nagle, R., Ziogas, A., Fujikawa-Brooks, S., Yerushalmi, H., et al. (2008). The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: results of a year-long phase IIb randomized placebo-controlled chemoprevention trial. Cancer Epidemiol. Biomarkers Prev. 17, 292–299.
Sjoerdsma, A., and Schechter, P. J. (1999). Eflornithine for African sleeping sickness. Lancet 354, 254.
Smith, M. A., Maris, J. M., Lock, R., Kolb, E. A., Gorlick, R., Keir, S. T., et al. (2011). Initial testing (stage 1) of the polyamine analog PG11047 by the pediatric preclinical testing program. Pediatr. Blood Cancer 57, 268–274.
Soderstjerna, E., Holst, C. M., Alm, K., and Oredsson, S. M. (2010). Apoptosis induced by the potential chemotherapeutic drug N1, N11-diethylnorspermine in a neuroblastoma cell line. Anticancer Drugs 21, 917–926.
Sunkara, P. S., and Rosenberger, A. L. (1987). Antimetastatic activity of DL-alpha-difluoromethylornithine, an inhibitor of polyamine biosynthesis, in mice. Cancer Res. 47, 933–935.
Suzuki, T., He, Y., Kashiwagi, K., Murakami, Y., Hayashi, S., and Igarashi, K. (1994). Antizyme protects against abnormal accumulation and toxicity of polyamines in ornithine decarboxylase-overproducing cells. Proc. Natl. Acad. Sci. U.S.A. 91, 8930–8934.
Takahashi, Y., Mai, M., and Nishioka, K. (2000). alpha-difluoromethylornithine induces apoptosis as well as anti-angiogenesis in the inhibition of tumor growth and metastasis in a human gastric cancer model. Int. J. Cancer 85, 243–247.
Thomas, T., and Thomas, T. J. (2003). Polyamine metabolism and cancer. J. Cell Mol. Med. 7, 113–126.
Tian, Z. Y., Xie, S. Q., Du, Y. W., Ma, Y. F., Zhao, J., Gao, W. Y., et al. (2009). Synthesis, cytotoxicity and apoptosis of naphthalimide polyamine conjugates as antitumor agents. Eur. J. Med. Chem. 44, 393–399.
Tobias, K. E., and Kahana, C. (1995). Exposure to ornithine results in excessive accumulation of putrescine and apoptotic cell death in ornithine decarboxylase overproducing mouse myeloma cells. Cell Growth Differ. 6, 1279–1285.
Tummala, R., Diegelman, P., Hector, S., Kramer, D. L., Clark, K., Zagst, P., et al. (2011). Combination effects of platinum drugs and N1, N11 diethylnorspermine on spermidine/spermine N1-acetyltransferase, polyamines and growth inhibition in A2780 human ovarian carcinoma cells and their oxaliplatin and cisplatin-resistant variants. Cancer Chemother. Pharmacol. 67, 401–414.
Uemura, T., Yerushalmi, H. F., Tsaprailis, G., Stringer, D. E., Pastorian, K. E., Hawel, L. III, et al. (2008). Identification and characterization of a diamine exporter in colon epithelial cells. J. Biol. Chem. 283, 26428–26435.
Van Nieuwenhove, S., Schechter, P. J., Declercq, J., Bone, G., Burke, J., and Sjoerdsma, A. (1985). Treatment of gambiense sleeping sickness in the Sudan with oral DFMO (DL-alpha-difluoromethylornithine), an inhibitor of ornithine decarboxylase; first field trial. Trans. R. Soc. Trop. Med. Hyg. 79, 692–698.
Vujcic, S., Diegelman, P., Bacchi, C. J., Kramer, D. L., and Porter, C. W. (2002). Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem. J. 367, 665–675.
Wallace, H. M., and Caslake, R. (2001). Polyamines and colon cancer. Eur. J. Gastroenterol. Hepatol. 13, 1033–1039.
Wallick, C. J., Gamper, I., Thorne, M., Feith, D. J., Takasaki, K. Y., Wilson, S. M., et al. (2005). Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene 24, 5606–5618.
Wang, Y., Devereux, W., Woster, P. M., Stewart, T. M., Hacker, A., and Casero, R. A. Jr. (2001). Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 61, 5370–5373.
Wang, Y., Murray-Stewart, T., Devereux, W., Hacker, A., Frydman, B., Woster, P. M., et al. (2003). Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem. Biophys. Res. Commun. 304, 605–611.
Weeks, R. S., Vanderwerf, S. M., Carlson, C. L., Burns, M. R., O’Day, C. L., Cai, F., et al. (2000). Novel lysine-spermine conjugate inhibits polyamine transport and inhibits cell growth when given with DFMO. Exp. Cell Res. 261, 293–302.
Weiss, W. A., Aldape, K., Mohapatra, G., Feuerstein, B. G., and Bishop, J. M. (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995.
Wolff, A. C., Armstrong, D. K., Fetting, J. H., Carducci, M. K., Riley, C. D., Bender, J. F., et al. (2003). A Phase II study of the polyamine analog N1,N11-diethylnorspermine (DENSpm) daily for five days every 21 days in patients with previously treated metastatic breast cancer. Clin. Cancer Res. 9, 5922–5928.
Xie, S. Q., Li, Q., Zhang, Y. H., Wang, J. H., Mei, Z. H., Zhao, J., et al. (2011a). NPC-16, a novel naphthalimide-polyamine conjugate, induced apoptosis and autophagy in human hepatoma HepG2 cells and Bel-7402 cells. Apoptosis 16, 27–34.
Keywords: polyamines, MYCN, neuroblastoma, ODC1, DFMO
Citation: Gamble LD, Hogarty MD, Liu X, Ziegler DS, Marshall G, Norris MD and Haber M (2012) Polyamine pathway inhibition as a novel therapeutic approach to treating neuroblastoma. Front. Oncol. 2:162. doi: 10.3389/fonc.2012.00162
Received: 16 August 2012; Paper pending published: 10 September 2012;
Accepted: 24 October 2012; Published online: 16 November 2012.
Edited by:
Arturo Sala, Brunel University/University College London Institute of Child Health, UKReviewed by:
Hongying Wang, Cancer Institute and Hospital, Chinese Academy of Medical Sciences (Peking Union Medical College), ChinaCopyright: © 2012 Gamble, Hogarty, Liu, Ziegler, Marshall, Norris and Haber. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Michelle Haber, Children’s Cancer Institute Australia for Medical Research, Lowy Cancer Research Centre, P.O. Box 81, Randwick, Sydney, NSW 2031, Australia. e-mail:bWhhYmVyQGNjaWEudW5zdy5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.