
 Wei Liu
Wei Liu- Metabolism and Cancer Susceptibility Section, Basic Research Laboratory, Center for Cancer Research, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
Studies in metabolism and cancer have characterized changes in core pathways involving glucose and glutamine, emphasizing the provision of substrates for building cell mass. But recent findings suggest that pathways previously considered peripheral may play a critical role providing mechanisms for cell regulation. Several of these mechanisms involve the metabolism of non-essential amino acids, for example, the channeling of glycolytic intermediates into the serine pathway for one-carbon transfers. Historically, we proposed that the proline biosynthetic pathway participated in a metabolic interlock with glucose metabolism. The discovery that proline degradation is activated by p53 directed our attention to the initiation of apoptosis by proline oxidase/dehydrogenase. Now, however, we find that the biosynthetic mechanisms and the metabolic interlock may depend on the pathway from glutamine to proline, and it is markedly activated by the oncogene MYC. These findings add a new dimension to the proline regulatory axis in cancer and present attractive potential targets for cancer treatment.
Introduction
The characterization of metabolic reprogramming in tumor cells has recently progressed rapidly from that first described by Otto Warburg (Vander Heiden et al., 2009). However, the focus remains on core metabolism involving glucose and glutamine (Dang and Semenza, 1999; DeBerardinis et al., 2008; Cairns et al., 2011). The original speculation of a primary defect in mitochondria has not been substantiated, but adaptive changes are prominent in mitochondrial metabolism (Wallace, 2005). The array of changes in reprogramming seems at times disparate and even paradoxical, but they can be understood in a temporo-spatial context (Dang, 2009).
An important consideration is the tumor’s selection of mechanisms to survive hypoxic and nutrient stress, leaving a genetic trail. HIF and AMPK are critical for survival and are frequently elevated in tumors, whereas suppressor mechanisms such as p53 are lost during tumor progression. When the tumor shifts into the proliferative phase and becomes clinically important, it has acquired reprogramming for rapid growth. Oncogenes such as PI3K/AKT, PTEN, RAS, and MYC play an important role during this phase (Vander Heiden et al., 2009; Cairns et al., 2011).
To understand oncometabolism, one must consider the tumor’s requirement for building blocks to incorporate into cell mass (Vander Heiden et al., 2012), i.e., nucleotides for DNA and RNA, amino acids for protein synthesis and fatty acids for lipid and membrane synthesis. That glucose is diverted into these biosynthetic pathways was an important realization, and, indeed, the markedly increased glucose uptake into tumors can be easily seen on PET scans with fluorodeoxyglucose – a feature of tumors not only useful for clinically visualizing and localizing tumors (Visioni and Kim, 2011), but also a hallmark of metabolic reprogramming. The routing of glucose to lactate and its excretion from the cell was seen as a mechanism for regenerating NAD+, a co-factor necessary for the glycolytic pathway at the step of glyceraldehyde-3-P dehydrogenase (Locasale and Cantley, 2011). Thus, the maintenance of flux through the glycolytic pathway requires the sacrifice of pyruvate to regenerate NAD+.
Glutamine was also recognized as playing a critical role in tumor metabolism (Wise et al., 2008; Dang, 2009). Physiologically, this amino acid serves as a conduit for transfer of carbon skeletons and amino groups from one tissue to another. In the postabsorptive state, proteolysis occurs in muscle, and amino groups from released amino acids are transferred to glutamate forming glutamine which circulates to the liver and kidney (Roth, 2008). Also important, the gamma nitrogen of glutamine is used in the initial steps of de novo purine and pyrimidine biosynthesis (Boza et al., 2000). Glutamine is sequentially converted to glutamate and alpha KG which plays an anaplerotic role in the tricarboxylic acid (TCA) cycle as citrate and malate (Le et al., 2012). Glutamine is not the only amino acid which has attracted recent attention in the field of metabolic programming in cancer. In fact, glutamine, arginine, and proline are a metabolic system. This review emphasizes the metabolism of proline which plays a special role in ROS signaling and redox balance (Phang et al., 2008a). Further, recent studies show that the regulation of glutamine and proline metabolism is linked in tumor cells.
The emphasis on “core pathways” during the last 10 years has provided seminal insights (Slaughter and Davies, 1968; Dang, 2009; Locasale and Cantley, 2010; Cairns et al., 2011; Vander Heiden et al., 2012). However, it became clear that core pathways were not the whole story; more “out of the box” thinking was needed. Additionally, these core pathways did not offer useful targets for cancer therapy because pharmacologic blockade often produced severe side effects (Kaufmann et al., 2006; Chang et al., 2007; Brandsma et al., 2010; Kaelin and Thompson, 2012). Thus, we sought evidence for non-core metabolic reprogramming in tumor cells, and fortuitously, insights have derived from several heuristic models. Tumor arrays, metabolomics, screening for targets of oncogenes/suppressor genes, and functional tumor suppression models all have been rewarding. Metabolomics have shown changes in serine and arginine/proline metabolism (Catchpole et al., 2009); p53 target screening has shown that POX/PRODH is a p53-induced gene (Polyak et al., 1997), and that the pyrroline-5-carboxylate (P5C) synthase gene (Aldh 18a1) is a target of MYC in a T-cell activation model (Wang et al., 2011b). Most impressive is the recent systematic screening of anti-tumor effects resulting from knockdown of metabolic genes by their respective siRNAs (Possemato et al., 2011). This study identified five genes when knocked down resulted in robust inhibitory effects on tumors. The authors earmarked 3-phosphoglycerate dehydrogenase (PHGDH), which channels carbons from glycolysis into the serine biosynthetic pathway, as an important enzyme by their criteria and targeted it for a variety of additional studies. Of special interest to us, these authors also identified PYCR1, pyrroline-5-carboxylate reductase 1, the enzyme catalyzing the committed step for proline synthesis, which when knocked down yielded tumor inhibitory effects (Possemato et al., 2011). It was exciting to discover that proline metabolism may play a role not only in suppressing tumors which has been our focus during the last decade, but also in augmenting tumor growth which may make it a possible therapeutic target. But for the latter, there was no convincing evidence that proline biosynthesis was necessary for either bioenergetics or for its direct contributions to supplying building blocks for cell mass. Thus, we felt the need to propose a new regulatory model that explained the observed effects.
Parametabolic Regulation
There are examples of metabolic function for which the term “Parametabolic” might be appropriate. This central idea is that the importance of apparent core products of a given pathway and that of co-factors are switched. The so-called metabolic co-factors are, in fact, the essential products of the metabolic process, and it is this parametabolic regulation which may be critical in the reprogramming of cancer cells (see Figure 1). The conversion of pyruvate to lactate is an example of redox regulation to optimize the flux of glucose through glycolysis (Locasale and Cantley, 2010). Certainly, lactate, itself, is not a physiologically important product. Instead, it is the generation of NAD+ as a co-factor which is the critical metabolic endpoint. Although the role of lactate production has been emphasized for some time, a number of novel parametabolic pathways are beginning to be appreciated. A recently discovered example is the routing of glycolytic intermediates into serine (Locasale et al., 2011; Possemato et al., 2011) not for their contribution to the proteinogenic amino acid pool, no change in serine or glycine concentration was found; more likely, this routing occurred because of the important role of the pathway in 1-carbon transfers which are critical for nucleotide synthesis and for DNA methylation. The role of hexosamines to signal the use of glutamine is another recently discovered example of parametabolic regulation (Wellen et al., 2010). However, the best understood example of parametabolic regulation is that of redox balance to optimize metabolic pathways (Locasale and Cantley, 2011).
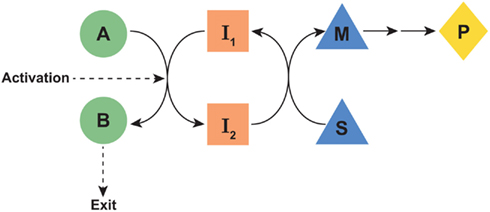
Figure 1. Schematic of proposed parametabolic regulation. The A-B pathway is in a metabolic interlock with the S-M pathway. Genetic or epigenetic upregulation of the A-B pathway would result in an increased production of P. The designated symbols are generic. However, in the context of the proline regulatory axis, A-B would represent the glutamine to proline pathway, I1, and I2 would be pyridine nucleotides, the S-M pathway would represent the oxidative arm of the pentose phosphate pathway and P would be PP-ribose-P. “Exit” may be either excretion, incorporation into protein, or another mechanism for departure from the parametabolic regulatory system.
Redox Regulation
As mentioned above, regulation for redox balance plays an important parametabolic role in cancer metabolism. The conversion of pyruvate to lactate and the excretion of lactate from the cell is well-recognized as a mechanism for regenerating the NAD+ necessary for maintaining the flux catalyzed by glyceraldehyde-3-phosphate dehydrogenase. This redox regeneration has been emphasized by a number of investigators (Vander Heiden et al., 2009; Cairns et al., 2011; Locasale and Cantley, 2011). However, it was also recognized that the diversion of pyruvate to lactate to regenerate NAD+ robbed the metabolic system of an essential precursor for acetyl CoA used for replenishing the TCA cycle as well as serving as a precursor for fatty acid synthesis. Another possible downstream role for the generated NAD+ was demonstrated by the observation that 3-phosphoglycerate is routed into the serine synthetic pathway (Locasale et al., 2011; Possemato et al., 2011). Critically, the step catalyzed by 3-phosphoglycerate dehydrogenase (PHGDH) requires NAD+ (Slaughter and Davies, 1968). Furthermore, providing redox regulation at the oxidative arm of the pentose phosphate pathway (PPP) supplies the PP-ribose-P for nucleotide synthesis (Tian et al., 1998), even though in certain tumors and depending on context, the non-oxidative arm may be important (Tong et al., 2009). The activation of T-cells was accompanied by marked increase in flux through the oxidative arm of the PPP (Wang et al., 2011b), arguing that the contributions of the oxidative versus non-oxidative arm to ribose-5-phosphate may depend on redox balance. Thus, not only must the NAD+/NADH balance be considered, the NADP+/NADPH ratio also may be important. In the 1970s, Eggleston and Krebs emphasized that NADP+/NADPH ratios are the rate-limiting co-factor for the formation of ribose-5-phosphate (Veech et al., 1969). But NADPH has been emphasized in its role for maintenance of the glutathione system used as defense against oxidizing insult, and p53 regulates the PPP to support NADPH formation (Gottlieb and Vousden, 2010). It is significant that MYC activation decreases the defense mechanism against oxidizing insult (Tuttle et al., 1996), a finding consistent with the shift of NADP+/NADPH during proliferation. NADPH is necessary for driving synthetic mechanisms, and it can be produced by isocitrate dehydrogenase 1 in the cytosol and malic enzyme and isocitrate dehydrogenase 2 in mitochondria (Cairns et al., 2011). When NADH is in excess, NADPH can be formed from NADP+ in mitochondria by NADP+ transhydrogenase and the energetics of the proton gradient (Jackson, 2003), and compartmentation of the components of redox balance by either subcellular organelles or protein chaperones may deserve consideration (Arentson et al., 2012).
Amino Acids in Parametabolic Regulation
The non-essential amino acids (NEAA) can play a special role in parametabolic regulation. Of the 20 naturally occurring amino acids, 11 are non-essential. The assignment was made purely on a nutritional basis, i.e., the NEAA are not required in the diet for optimal nutrition. Although some have inferred a less important status for the NEAA, in actuality, the non-essentials provide critical non-proteinogenic functions. The enzymes for their endogenous synthesis were evolutionarily preserved because they contribute special regulatory functions in metabolism. Not surprisingly, metabolism of the NEAA is important in cancer. The role of glutamine has been emphasized (Dang, 2010), and recently the pathway to serine and glycine is a detour from glycolysis; its genomic and functional relevance to cancer has been recognized (Locasale et al., 2011; Possemato et al., 2011). It is well known that arginine and proline are interconvertible; the former is the substrate for nitric oxide (Morris, 2009) and, through ornithine, for polyamine synthesis. In contrast, proline, has not been studied except by a few laboratories. Also worthy of mention, proline is the only secondary proteinogenic amino acid (Adams and Frank, 1980). Because its alpha-amino group is contained within a pyrrolidine ring (see Figure 2), proline cannot be metabolized by the usual amino acid enzymes, i.e., transaminases, decarboxylases, racemases. Instead, a special family of enzymes has evolved and these enzymes have their own regulatory mechanisms as well as their own tissue and subcellular localization. These enzymes mediate the interconversions of proline, glutamate, and arginine, but as we shall see, their functions transcend their contributions to supplying substrates for protein synthesis.
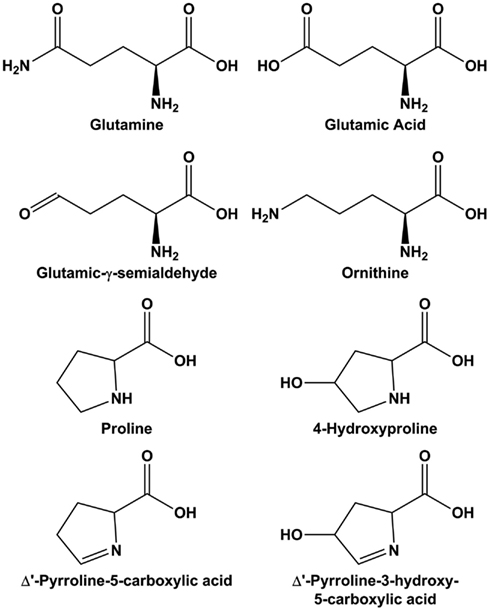
Figure 2. Structure of molecules in the glutamine-arginine-proline pathway. Glutamic-γ-semialdehyde is the open-chain tautomer of Δ1-pyrroline-5-carboxylic acid.
In the setting of interconvertibility of NEAA, the aforementioned pathways involving glutamate, proline, and arginine may be summarized briefly here (see Figure 3). The central metabolite is pyrroline-5-carboxylate (P5C) and its open-chain tautomer, glutamic-γ -semialdehyde. These compounds, in tautomeric equilibrium, are the obligate intermediates for transfer of carbons between the TCA and urea cycles (Phang, 1985; Phang et al., 2001). The fact that P5C is not only the precursor for proline synthesis but also the immediate product of proline degradation aroused our interest, because it is a feature unique in amino acid metabolism.
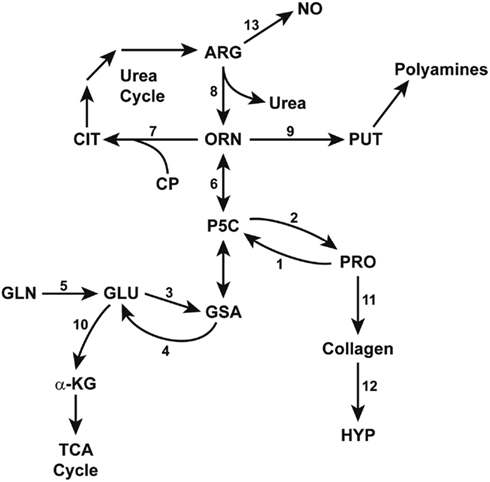
Figure 3. Enzymatic pathways in the interconversions of glutamine, arginine, and proline. GSA (glutamic-γ-semialdehyde) and its tautomer, P5C (Δ1-pyrroline-5-carboxylate) bridge the pathways from glutamine to arginine, thereby linking the TCA and urea cycles. Some reactions are mediated by multiple isozymes with distinct subcellular localizations and functions. The abbreviations are: PRO, proline; HYP, hydroxyproline; GLU, glutamate; GLN, glutamine; a-KG, alphaketoglutarate; ORN, ornithine, CIT, citrulline, ARG, arginine; NO, nitric oxide; PUT, putrescine; CP, carbamyl phosphate. Enzymes catalyzing these reactions are designated by numbers: 1, proline oxidase/dehydrogenase; 2, P5C reductase; 3, P5C synthase; 4, P5C dehydrogenase; 5, glutaminase; 6, ornithine aminotransferases; 7, ornithine transcarbamylase; 8, arginase; 9, ornithine decarboxylase; 10, glutamate dehydrogenase; 11, protein synthesis; 12, prolyl hydroxylase; 13, nitric oxide synthase.
Proline Metabolism and Regulation
The functional role of proline metabolism in redox regulation was first recognized in the 1980s (Phang, 1985). Whether described as a “proline cycle” or “redox shuttle” the interconversions of proline and pyrroline-5-carboxylate, its oxidized congener, transferred redox equivalents between mitochondria and cytosol. The enzymes catalyzing this cycle are proline oxidase (POX), also known as proline dehydrogenase (PRODH) and pyrroline-5-carboxylate reductase (PYCR). Using human erythrocytes which have PYCR but no POX activity, we were able to demonstrate a metabolic interlock (Yeh et al., 1984) with glucose-6-phosphate dehydrogenase (G6PDH) of the PPP. The redox transfer robustly activated the oxidative arm of the PPP and increased the formation of PP-Ribose-P, a critical intermediate for both salvage and de novo pathways for nucleic acids (Yeh and Phang, 1981, 1983). Interestingly, Wu described the flip side of this metabolic interlock. Using porcine enterocytes, he showed that the oxidative arm of the PPP is essential in the synthesis of proline from glutamine (Wu, 1996).
Proline Cycle Revisited
Although the “proline cycle” as a redox regulatory system was proposed almost three decades ago (Hagedorn and Phang, 1983; Phang, 1985), its role in human metabolism was not understood until a decade later (Polyak et al., 1997; Donald et al., 2001; Maxwell and Rivera, 2003). Admittedly, its contributions to bioenergetics and redox regulation were minor under conditions of oxygen and substrate availability with optimal operation of the TCA cycle. Also widely accepted, the optimal functioning of antioxidant enzymes and the glutathione cycle defends against redox toxicity. However, with the recognition that redox signaling is a critical communication system, particularly under conditions of microenvironmental stress, we formulated an overarching concept. A recent review (Cyr and Domann, 2010) referred to our discoveries as the “Proline oxidase (POX) Regulatory Axis” which we have shortened to “proline regulatory axis” (PRA), a designation which includes the protean regulatory effects of proline metabolism. Importantly, the discovery that POX/PRODH is encoded by p53-induced gene 6 (PIG 6) provided persuasive evidence that the PRA provides metabolic responses to stress situations (Polyak et al., 1997). Central to this process is that POX/PRODH is bound to mitochondrial inner membranes and donates electrons to the electron transport chain (ETC; Donald et al., 2001; Liu et al., 2006, 2009; Phang et al., 2008b).
Regulation of POX/PRODH
Not only is POX/PRODH regulated by p53, but also the apoptotic response to p53 is dependent, in large part, on POX/PRODH (Donald et al., 2001; Maxwell and Rivera, 2003; Liu et al., 2005; Hu et al., 2007). In fact, the apoptotic response to p53 is decreased ∼60% when POX/PRODH is knocked down with siRNA (Maxwell and Rivera, 2003). The mechanism of the POX/PRODH pro-apoptotic effect is dependent on the generation of ROS (Donald et al., 2001; Liu et al., 2005). Following this discovery that POX/PRODH is a player in p53-induced apoptosis, we sought to identify additional mechanisms by which POX/PRODH could be induced (see Figure 4). Using a POX promoter/luciferase construct as a target, we co-transfected a variety of transcriptional factors. The usual transcriptional activators (jun/fos, NF-kB) produced a modest effect in activating the POX/PRODH promoter (less than twofold). Strikingly, the expression of peroxisome proliferator-activated receptor gamma (PPARγ robustly activated the POX/PRODH promoter (Pandhare et al., 2006). Furthermore, ligands of PPARγ, whether pharmacologic, e.g., Troglitazone, or physiologic, e.g., oxidized low-density lipoprotein, induced POX/PRODH through a PPARγ-dependent mechanism (Phang et al., 2008c; Zabirnyk et al., 2009). Our discovery that the increase in POX/PRODH induced downstream targets through a ROS-dependent signaling network was subsequently corroborated by several laboratories (Kim et al., 2007; Wang et al., 2011a). Importantly, the apoptotic response to PPARγ and its ligands is blocked by knocking down POX/PRODH. Thus, the regulated activity of POX/PRODH plays a special role in the system for redox signaling for apoptosis, and in other contexts, for autophagy and survival (Phang et al., 2008c; Liu et al., 2012a).
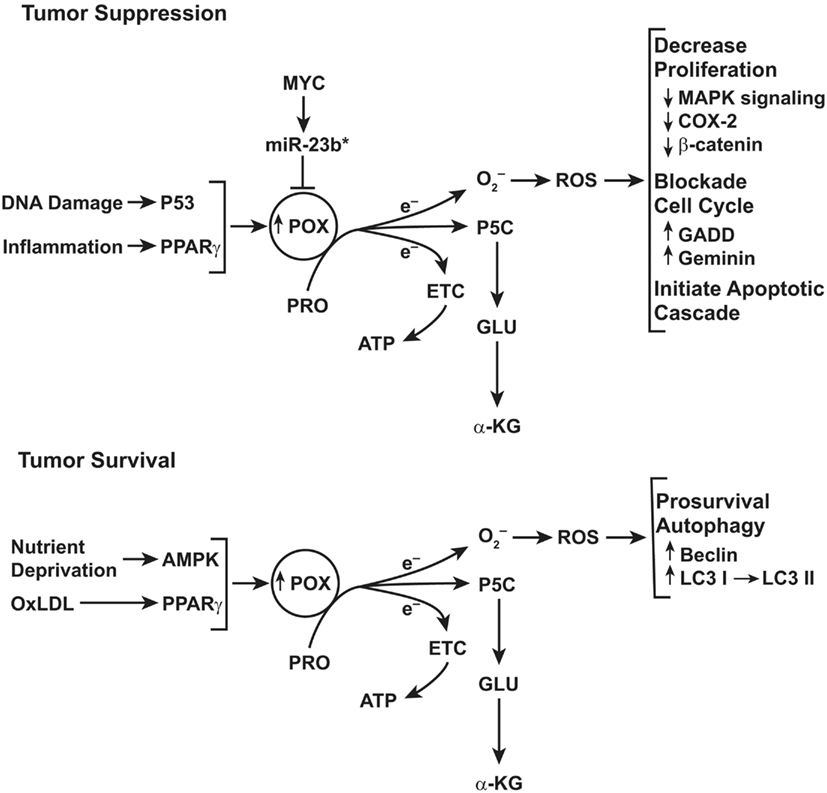
Figure 4. Top: schematic representation of the functions of proline oxidase (POX) in tumor suppression. POX functions as a tumor suppressor even though the gene encoding POX is not a classical tumor suppressor gene. Instead, POX is suppressed by a microRNA, miR-23b* which is over expressed in digestive tract and kidney cancers. For details, see figure in review (Phang and Liu, 2012). Bottom: role of POX signaling under conditions of metabolic stress. The functions of POX depend on metabolic context. Under conditions of glucose or oxygen deprivation or when treated with oxidized LDL, PPARγ, POX expression is upregulated. The proline-dependent ROS generated by POX activate prosurvival autophagy mediated by conversion of LC3 I to LC3 II and by expression of Beclin.
To ascertain the response of the POX/PRODH response to nutrient stress, we studied the effects of low glucose and/or hypoxia (Pandhare et al., 2009; Liu et al., 2012a). We found that POX/PRODH was upregulated by low glucose in a process mediated by AMPK (Pandhare et al., 2009). Similarly, hypoxia increased both HIF1 and AMPK signaling, but the effect on POX/PRODH expression was attenuated by compound C, an inhibitor of AMPK signaling but not by the knockdown of HIF1 (Liu et al., 2012a). The POX/PRODH-mediated response in this context was activation of the pathway for autophagy and cell survival (Figure 4). The response of POX/PRODH to hypoxia was translated to mice with a tumor expressing GFP under the control of a hypoxia response element (HRE). Both GFP and POX could be monitored using immunohistochemistry. Strikingly, the expression of HRE-controlled GFP and POX co-localized in various sections of these xenograft tumors.
POX/PRODH Produces ROS
Increasingly, it is recognized that mitochondria are the source of intracellular ROS signaling. Although other enzymes can be a source of superoxide (NADPH oxidase, xanthine oxidase), they generally function for other purposes, e.g., as a bactericide, and in general, this ROS is excreted extracellularly (Weinberg and Chandel, 2009). Although mitochondria are recognized as an important if not unique source of ROS for signaling, the actual substrate source and molecular mechanism remain uncertain. In general, it has been thought that electrons donated to the ETC can be mobilized for generating ROS. The work of Chandel and others strongly suggested that Complex III is the source of ROS for signaling (Chandel, 2010). They argue that the localization of this complex allows the release of ROS into the intermembrane space rather than into the mitochondrial matrix. Thus, such ROS may be transferred to the cytosol which is the location of many targets for such signaling. As mentioned above, POX/PRODH generates proline-dependent ROS (Donald et al., 2001; Maxwell and Rivera, 2003; Liu et al., 2005; Kim et al., 2007; Wang et al., 2011a). Recently, Chad Hancock in our laboratory has shown that electrons from POX/PRODH are transferred from FAD to ubiquinone, as was first shown by others in Saccharomyces (Wanduragala et al., 2010), and then is transferred further to complex III for proline-dependent ROS (Hancock, C., et al., in preparation; Liu et al., 2012b). Since POX/PRODH can be robustly regulated in response to a variety of stress mechanisms, it seems likely that this is a critical, regulated pathway for the generation of ROS for signaling.
POX/PRODH Functions as a Tumor Suppressor Protein
The regulation of POX/PRODH and its generation of ROS for signaling led us to study its effects on tumorigenesis in a mouse model. Using DLD-tet-off-POX cells injected into immunodeficient mice, the implantation, and growth of tumors was markedly inhibited by the expression of POX/PRODH (Liu et al., 2009). Additionally, using immunohistochemical methods to monitor POX/PRODH levels in a variety of human tumors, we found that POX/PRODH was much lower in tumors as compared to matching normal tissues from the same patient, especially in tumors of the digestive tract and kidney (Liu et al., 2009, 2010). These findings suggested that POX/PRODH may function as a tumor suppressor. However, gene sequencing identified no mutations in the coding or promoter regions. Recently, the criterion for identification of tumor suppressive activity has expanded beyond the classical definition of relying solely upon the mutated tumor suppressor gene. For example, epigenetic mechanisms such as DNA methylation and histone modification, as well as regulation of gene expression by non-coding RNAs, have led to revised concepts of tumor suppressors. Additionally, metabolic enzymes have been identified as mediators of tumor suppression (Thompson, 2009; Luo et al., 2010). Due to these emerging concepts, our characterization of POX/PRODH led us to propose that POX/PRODH functions as a metabolic tumor suppressor protein (Liu et al., 2009; Phang et al., 2010).
POX/PRODH is Regulated by miR-23b*
It was in the context of the above that we sought a mechanism for the downregulation of POX expression in tumors. Wei Liu, in our laboratory, discovered that a specific microRNA, miR-23b*, was increased in tumors compared to normal tissues. POX/PRODH translation was suppressed by mimics of this microRNA, and the POX/PRODH-mediated generation of ROS and induction of apoptosis were inhibited by miR-23b* (Liu et al., 2010). Interestingly, miR-23b, the sibling microRNA processed from the same transcript as miR-23b*, plays an important role in the regulation of glutaminase, and miR-23 a/b is under the regulation of MYC (Gao et al., 2009). In collaboration with Chi Dang and co-workers at Johns Hopkins, we used the P493 cells, cells derived from Burkitt’s lymphoma which carry a tet-off MYC expression vector and showed that MYC expression down regulated POX/PRODH and suppressed both proline-dependent ROS production and POX/PRODH-dependent apoptotic effects (Liu et al., 2012b). Apparently, the suppression of POX/PRODH and its growth-inhibitory and apoptotic effects exercised a permissive role on the proliferative response to MYC.
Δ1-Pyrroline-5-Carboxylate
Another feature of proline metabolism is provided by proline’s oxidized congener, Δ1-pyrroline-5-carboxylate (P5C). This molecule is the product and substrate, respectively, of P5C synthase and pyrroline-5-carboxylate reductase. The cyclized form is in tautomeric equilibrium with its open-chain form, glutamic-γ-semialdehyde (Adams, 1970). We can speculate that this interconvertibility can bestow special properties governed by the intracellular milieu and allowing translocations and interactions not available to other metabolites. We produced [U-14C]-L-pyrroline-5-carboxylate enzymatically with [U-14C]-L-ornithine and partially purified rat liver ornithine aminotransferases (Smith et al., 1977). Using this radiolabeled [U-14C]-L-P5C, we showed that the cellular transport of P5C had special properties (Mixson and Phang, 1988, 1991). Although these were functionally defined over 20 years ago, they shed light on the functional interaction with membranes. Not only was there an energy- and sodium-dependent transport mechanism, but the uptake of P5C was distinct from that for naturally occurring amino acids. Our studies suggested that a transporter or channel existed in close association with P5C reductase. In elegant studies by others, pyrroline-5-carboxylate reductase was shown to exist in the native state as a decamer with a “donut” like, torus structure common to membrane-associated proteins and chaperones (Meng et al., 2006). Future studies will define the structure of the other isozymes, PYCR2, and PYCRL (see below).
We also developed an assay for P5C in biologic fluids using PYCR purified from Escherichia coli (Fleming et al., 1984). With this assay we were able to measure plasma levels and found that it fluctuated with dietary intake (Fleming et al., 1989). Interestingly, it was not related to proline intake. This is an area which could be explored with state-of-the-art analytical methods in metabolomics.
P5C Reductases
Although two PYCRs have been functionally and molecularly recognized for some time (Dougherty et al., 1992; Hu et al., 2008; Wu et al., 2008), a third PYCR (PYCRL or PYCR3) was identified (Suzuki et al., 2001). For mammalian cells, these have been designated PYCR 1/2/L or by some workers, PYCR1/2/3. Recent characterization of these three isozymes suggests that PYCR1 (Reversade et al., 2009) and PYCR2 are associated with mitochondria (De Ingeniis et al., under review); PYCR1 appears to be transmembrane in mitochondria, and its torus structure may be related to its transmembrane localization. PYCR2 is in the mitochondrial matrix whereas PYCRL is in the cytosol (De Ingeniis et al., under review). The preferred reduced pyridine nucleotide for PYCRL is NADPH whereas PYCR1/2 prefer NADH. As mentioned above, earlier work from our lab showed that PYCR activity in human erythrocytes participates in a metabolic interlock with G6PD and 6PGD of the PPP (Yeh et al., 1984). The increased production of phosphoribosyl pyrophosphate was used for the salvage pathway (Yeh and Phang, 1983). Subsequently, the PYCR from erythrocytes (most likely PYCRL) was purified to homogeneity. Not only does the enzyme prefer NADPH, but the affinity for P5C was higher with NADPH than with NADH (Merrill et al., 1989). These and other findings led to the proposal that the erythrocyte enzyme functioned as a P5C-dependent NADPH dehydrogenase. The functional differences in PYCRs were recently reviewed (Hu et al., 2008).
An inborn error with deficiency of PYCR1 has been described associated with skeletal abnormalities, abnormal facial features, and mental retardation (Reversade et al., 2009). Fibroblasts from these patients have increased sensitivity to oxidative challenge. Addition of hydrogen peroxide to normal fibroblasts would cause no obvious damage whereas addition to the PYCR1-deficient fibroblast caused cell death. These findings provide evidence in a genetically derived inborn error that PYCR1 is associated with redox regulation. However, with new data on the three PYCR isozymes and their localizations, it becomes possible to propose new metabolic interlocks from the “proline cycle” proposed over 25 years ago (Hagedorn and Phang, 1983; Phang, 1985). One important feature of P5C reductases should be mentioned here. As mentioned above, crystallographic characterization of PYCR1 shows it to be a decamer forming a torus (Meng et al., 2006). Others have emphasized that such structures frequently are associated with membranes and also may function as a chaperone. One could speculate that such a structure may form protein–protein complexes to facilitate metabolic interlocks.
Since POX/PRODH is upregulated by p53, PPARγ, and AMPK and functionally linked to apoptosis and autophagy, the early proposal that the proline cycle allowed mitochondrial function to be linked to proliferative responses needed revising. The all-important consideration is context. For regulatory purposes, POX/PRODH is generating ROS for signaling, and we have emphasized the importance of the PRA in apoptosis and autophagy, especially for ROS signaling from POX/PRODH. However, the biosynthetic pathway linked to glutamine and MYC does not fit into the aforementioned model. Instead, it may be best explained by a parametabolic redox regulatory pathway.
Myc Regulates Glutamine and Proline Metabolism
Second in importance only to glucose in tumor metabolism, glutamine is used as a source of bioenergetics as well as a donor of nitrogen for the biosynthesis of growth substrates, e.g., nucleotides. Tracing glutamine metabolism with the use of [13C5, 15N2]-glutamine by mass spectrometry and NMR, investigators showed that as glutamate, the carbon skeleton from glutamine is converted to alpha KG to serve an anaplerotic function for the TCA cycle, is oxidized to CO2 and also is converted to lipids (Le et al., 2012). Of special interest to us, in P493 cells expressing MYC controlled by a tet-off promoter, a significant fraction of glutamine was converted to proline. Importantly, suppression of MYC expression markedly decreased the conversion of glutamine to proline (Liu et al., 2012b). It was shown previously that glutaminase, the enzyme catalyzing the first step in glutamine metabolism, was increased by MYC (Gao et al., 2009). With remarkable symmetry, MYC not only downregulated POX/PRODH expression, but markedly increased the enzymes converting glutamate to proline. Both P5C synthase (P5CS) and pyrroline-5-carboxylate reductase 1 (PYCR1) protein levels were markedly increased (Liu et al., 2012b). Importantly, monitoring the flux from labeled glutamine showed that other than CO2, proline was a major end product of glutamine in agreement with the findings of Windmueller and Spaeth in the 1970s (Windmueller and Spaeth, 1974).
Myc in Activated T-Cells
Recently, Wang and co-workers, using a model of T-cell activation, reported that reprogramming of glutamine metabolism played a crucial role, and MYC was the regulator which activated glutamine metabolism (Wang et al., 2011b). Furthermore, they showed that ALDH18A1 was markedly increased. The product of this gene, pyrroline-5-carboylate synthase, plays a critical role in intermediary metabolism synthesizing pyrroline-5-carboxylate (or its open-chain tautomer, glutamic-γ-semialdehyde), the obligate intermediate in the transfer of carbons between the TCA cycle and urea cycle. Activated T-cells showed increased levels of ornithine and putrescine, and this was unaffected by knockout of arginase 1. Also, in studies using 13C-glutamine, these workers found that MYC increased the routing of glutamine into polyamines. In studies of splanchnic metabolism in the 1970s, Windmueller and Spaeth (1974) showed that proline is a significant product of glutamine metabolism, but the current work did not mention the routing of glutamine into proline. Wang et al. emphasized the routing of glutamic-γ-semialdehyde/P5C to polyamines, and they also showed that PRODH was upregulated. The latter was in contrast to our finding where POX/PRODH was down regulated with ectopic over expression of MYC in P493 Burkitt lymphoma cells (Liu et al., 2012b). In Wang’s model, normal T-cells were activated by immune stimulation in mice with and without MYC knockout (Wang et al., 2011b). The disparities in observed PRODH responses to MYC may be due to these contextual differences. Nevertheless, in both models, the emphasis is on the upregulated synthesis of glutamic-γ-semialdehyde/P5C as a central intermediate for MYC-activated proliferation.
Proline Regulatory Axis – A Working Model
In tumor cells, proliferation requires a steady supply of substrates for cell mass. The two main sources are glucose and glutamine. As others have pointed out, the metabolism of glucose through the TCA cycle and oxidative phosphorylation, is an efficient generator of ATP, and glycolysis is a rapid, albeit inefficient generator when glucose is abundant. However, during tumor cell proliferation, the carbons from glucose are required for producing nucleotides, lipids, and other substrates for growth (Vander Heiden et al., 2009; Cairns et al., 2011). Although pyruvate from glucose is an important source of acetyl CoA for lipid biosynthesis, its conversion to lactate is necessary for regeneration of NAD+ (Locasale and Cantley, 2011). Similarly, the critical step for nucleotide synthesis by both de novo and salvage pathways is the oxidative arm of the PPP. Although there are alternative pathways for ribose production, i.e., through the non-oxidative arm of the PPP, rapidly proliferating cells may use either pathway depending on the metabolic context, especially when glucose is readily available (Miccheli et al., 2006; Tong et al., 2009). The critical factor for maintaining ribose formation through the oxidative arm is the availability of NADP+ for the first two steps catalyzed by glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase, respectively (Veech et al., 1969). Thus, we see that two important steps in channeling glucose carbons into cell mass, require the recycling of NAD+ and NADP+ for glycolysis and the oxidative arm of the PPP, respectively. The PRA not only provides p53-induced proline-dependent ROS signaling for metabolic stress, but also the regeneration of oxidizing potential for channeling glucose and glutamine carbons into cell mass for proliferation.
Myc and Parametabolic Regulation
In P493 cells bearing a tet-off MYC construct, MYC induced both P5C synthase and PYCR1. Consistent with these findings, the flux of (U-13C5, 15N2)-glutamine to proline is increased nearly 10-fold with the expression of MYC (Liu et al., 2012b). These findings suggested that the pathway to proline activated a process transcending the proteinogenic function of proline. In fact, in most growing cells, concomitant with the consumption of glutamine, proline is produced in excess of growth requirements and is secreted into the medium (Stoner and Merchant, 1972). The reason for this marked MYC-dependent increase in the flux of glutamine to proline is not fully understood. We are in the process of showing that MYC expression greatly increased glycolysis as well as oxidative phosphorylation, and with knockdown of P5CS or PYCR1, the stimulatory effect of MYC was markedly inhibited (Liu, W., et al., unpublished observations). Additionally, from work done in the 1970s, the exposure of cells to organically synthesized DL-pyrroline-5-carboxylic acid resulted in a large increase in the flux of glucose through the PPP (Phang et al., 1982). This metabolic interlock was thought to be due to the P5C-mediated oxidation of NADPH by P5C reductase (Merrill et al., 1989) and was further substantiated by reconstituted cell-free experiments (Hagedorn and Phang, 1983; Phang, 1985). A number of studies have shown that MYC is associated with increased activity of the oxidative arm of the PPP (Palaskas et al., 2011; Wang et al., 2011b; Manganelli et al., 2012). Our working hypothesis is that the MYC activation of PYCR may be the metabolic link to the activation of the PPP by MYC. From the recent work on isozymes of P5C reductase, a reasonable assumption would be that this was mediated by PYCRL, the isozyme which prefers NADPH as co-factor (De Ingeniis et al., under review). These findings suggest that although glutamine is the source of carbons taken up by the cell, it is the conversion of glutamate to P5C and subsequently to proline which is providing the necessary parametabolic regulation to optimize glucose utilization for cell proliferation. Not only do these findings add to the important functions of glutamine, but suggest that the glutamine-proline pathway is also critical, and it could be targeted selectively for cancer therapy.
These recent developments and new experimental findings indicated that the “Proline cycle” is one component of a more comprehensive “PRA.” With redundant PYCRs, we can hypothesize a number of cycles which can be activated by specific signals for metabolic reprogramming. The data supporting the classical “proline cycle” was from studies using reconstituted cellular components, and the interpretation was that it served as an auxiliary energy-producing mechanism linking the complete oxidation of glucose in the PPP to oxidative phosphorylation with the proline cycle as the shuttle mechanism. We now know that the generation of ROS for signaling is an important function of POX/PRODH, a signaling mechanism responsive to a variety of factors (p53, PPARγ, AMPK). The recent discovery that MYC activates the biosynthetic arm of proline metabolism suggests several novel hypothetical arrangements. First, the marked upregulation of P5CS increases the production of P5C from glutamate. P5C has a number of fates: it can be cycled back to glutamate, it can be converted to ornithine by ornithine aminotransferases or it can be converted to proline by one of the three isozymes of P5C reductase (see Figure 5). Based on their respective subcellular localizations and characteristics, we can speculate that PYCR1 can regenerate NAD+ in the cytosol whereas PYCR2 regenerates NAD+ in the mitochondrial matrix. PYCRL, however, generates NADP+ in the cytosol. One would expect that if these three PYCRs have metabolic interlocks with glucose metabolism, PYCR1/2 would be linked with glycolysis whereas PYCRL would be linked with the PPP. Thus, the biosynthetic arm of the PRA can provide parametabolic redox regulation for glucose metabolism and MYC induction of both P5CS and PYCR may be necessary components of reprogramming by MYC to direct glucose into cell mass.
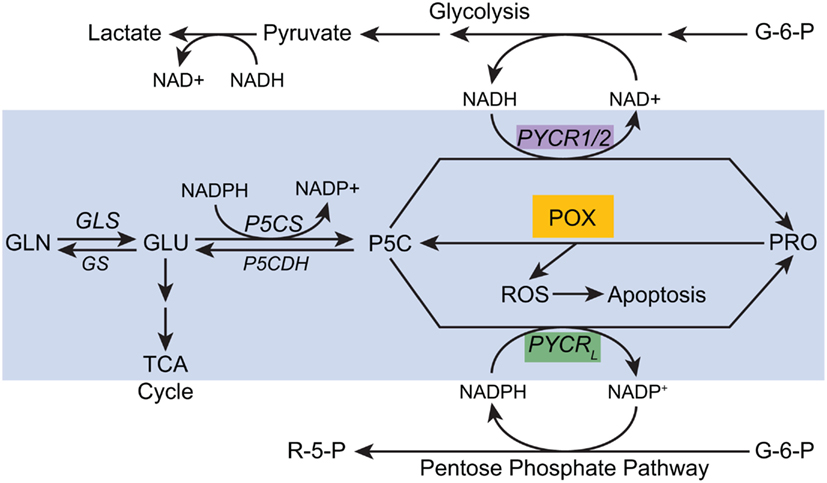
Figure 5. A proposed model for parametabolic redox regulation by the proline regulatory axis. The model is a composite of functions which depend on temporo-spatial context; various activities may not occur simultaneously. For example, MYC inhibits POX but concomitantly activates P5CS and PYCR1 (see text). The enzymes shown are as follows: GLS, glutaminase; GS, glutamine synthase; P5CS, pyrroline-5-carboxylate synthase, P5CDH, pyrroline-5-carboxylate dehydrogenase; POX, proline oxidase/dehydrogenase; PYCR1/2, pyrroline-5-carboxylate reductase 1, and 2; PYCRL, pyrroline-5-carboxylate reductase L. Some of the pathways are simplified. A single arrow may represent multiple steps.
Glutamine to Proline Pathway Mediates Redox Regulation
As mentioned above, a number of reviews have emphasized the role of lactate excretion as a mechanism for regenerating oxidizing potential in the form of NAD+ to maintain increased glucose flux into necessary substrates for cell mass (Cairns et al., 2011; Locasale and Cantley, 2011). Additionally, although the PPP has been primarily considered as a glucose-dependent source of reducing potential to defend against oxidative stress, during reprogramming of cells for growth with the mandatory increase of nucleotides, the oxidative arm of the PPP is activated. Such was the case during activation of T-cells. The flux through the oxidative arm of the PPP is dependent on the level of G6PD and 6PGD, but only in part; it is also regulated by NADP/NADPH ratios (Veech et al., 1969). This is because NADPH is the competitive inhibitor of NADP+ in its binding to G6PD. For most resting cells, NADPH is predominant and there is little flux through the oxidative arm. However, as NADPH is oxidized, the flux through the PPP markedly increases. Thus, for both glycolysis and for the oxidative arm of the PPP, redox regulation is critical.
Depending on which P5C reductase isozyme is used to catalyze the reaction, the pathway from glutamate to proline would oxidize two molecules of NAD(P)+. The fact that there is this metabolic interlock at the PYCR step with the PPP has been shown in a variety of reconstituted systems (Phang et al., 1980; Hagedorn and Phang, 1986). Our ongoing studies using the Seahorse XF flux analyzer has shown that the extracellular acidification rate (ECAR), which may be considered an indicator of lactate formation, is increased with MYC expression, and this effect can be markedly inhibited with the knockdown of either P5CS or PYCR1. Thus, the glutamine to proline pathway is serving as a parametabolic regulator of redox to optimize flux from glucose to substrates for cell mass.
The Fate of Proline
But what happens to the proline? As mentioned above, endogenously synthesized proline can be secreted from the cell (Stoner and Merchant, 1972), or it can be incorporated into collagen (Priest and Davies, 1969) as a sink for proline. When collagen is synthesized, 25% of the amino acids incorporated are proline (Udenfriend, 1966; Phang et al., 2001). Subsequently, about half of these proline residues are hydroxylated forming hydroxyproline. From the perspective of redox balance, collagen can be a sink for reducing potential and be removed from the metabolic pool, but it’s removal from the microenvironment, unlike lactate, is unnecessary. In fact, in many sites with cellular hyperproliferation, collagen synthesis is increased. For example, breast carcinogenesis is often accompanied by collagen formation (Priest and Davies, 1969). A dense, collagen-rich mass co-localizes with the proliferation of tumor cells (Kao et al., 1986; Alowami et al., 2003; Li et al., 2005; Schedin and Keely, 2011), even if the actual production of collagen takes place in stromal cells rather than in tumor cells. Cross-regulation between collagen synthesis and tumor cell proliferation may be mediated by mechanoregulatory (Provenzano et al., 2008) or metabolic reprogramming mechanisms (Walker and Shand, 1972; Savolainen et al., 1984; Anthony et al., 2010). The latter suggestion is plausible because metabolic abnormalities concomitant to inflammatory proliferation is frequently associated with collagen formation, i.e., fibrosis (Davis and Cowie, 1990; Eddy and Giachelli, 1995). This occurs in the lung with chronic infection or autoimmune activation (Verma and Slutsky, 2007).
Collagen requires both proline and hydroxyproline (see Figure 2), but free hydroxyproline is not incorporated into proteins (Stetten, 1949). Instead, proline is post-translationally hydroxylated (Peterkofsky and Udenfriend, 1965). But, there are parallels in proline versus hydroxyproline metabolism (Cooper et al., 2008). Importantly, there are distinct oxidases (dehydrogenases) for the respective imino acids (Adams and Frank, 1980), with little substrate overlap, and hydroyproline oxidase, like proline oxidase, is induced by p53, and can generate ROS (Cooper et al., 2008). However, the respective second step in the degradation, pyrroline-5-carboxyalte dehydrogenase and 3-OH-pyrroline-5-carboxylate dehydrogenase are catalyzed by the same enzyme (Valle et al., 1979; Flynn et al., 1989). Humans with an inherited defect in this enzyme accumulate 3-OH-P5C as well as P5C in blood and excrete large amounts of the two pyrroline carboxylates in the urine. Interestingly, P5C reductase, or at least PYCR1, can use either P5C or 3-OH-P5C as substrate, but this property is not found in enzymes from prokaryotes (Adams, 1970; Steinberg, 1973). Thus, both of these compounds can be used to recycle NAD(P)+. In the 1970s when a number of workers were monitoring the regulation of collagen synthesis, several described a rather interesting phenomenon. As much as 40% of the nascent hydroxylated collagen is not secreted from the cell (Steinberg, 1973). Instead it is degraded to release free hydroxyproline. This apparent wastage of bioenergy could be due to a metabolic function for hydroxyproline and its metabolites which have not yet been identified. Nevertheless, the metabolic pathways for hydroxyproline can act as a backup for those for proline in the PRA.
Conclusion
In recent years, the reprogramming of non-essential amino acid metabolism has attracted attention. We now introduce the concept of “parametabolic regulation” to help understand this reprogramming. Simply stated, it is not the amino acids which are the important products. Rather, their synthesis and degradation provide the scaffold for regenerating or cycling substrates necessary for optimal function of core pathways. The PRA provides a model for such a parametabolic regulatory system.
Since the original formulation of the regulatory functions of proline and its oxidized congener, Δ1-pyrroline-5-carboxylate, the understanding of the physiologic and pathophysiologic functions of the PRA has undergone considerable change. We have been like “blind men and the elephant.” However, the insights provided by a number of laboratories have helped us interpret the new findings.
The interconversions of proline, arginine and glutamate are important under various nutritionally stressed situations, i.e., the production of arginine from proline may augment growth when dietary arginine is deficient, e.g., in weanling pigs (Wu et al., 2008). Additionally, the production of alpha KG from glutamate derived from either proline or arginine may have an anaplerotic role for the TCA cycle. However, there are important linkages with redox which may be the principal regulatory function of the PRA. POX/PRODH is a regulated source of ROS for signaling. The ROS signaling role, described by our laboratory, has been corroborated by other labs. P5C, whether produced from arginine via ornithine, from glutamate by P5CS or from proline by POX/PRODH is a carrier or transfer mechanism for pyridine nucleotide-dependent oxidizing potential. The PRA also plays a role in the reprogramming of cellular metabolism by MYC. This transduction mechanism has been shown to be dependent on P5C reductase and its three isozymes with specific subcellular localizations and individual preferences for NADPH or NADH. Depending on the regulation of this pathway by specific oncogenes, the reprogramming of metabolism may be dependent on the parametabolic regulation provided by the PRA. We are examining the regulation of the specific enzymes by mechanisms from genomic, epigenetic, and non-coding RNA origins. We will verify the metabolic interlocks with important pathways, i.e., glycolysis, PPP, Serine-glycine pathway. The uncovering of these parametabolic mechanisms and the specific reprogramming in various tumors may lead us to the ultimate goal of identifying novel targets for cancer therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project also has been funded in part with Federal funds from the National Cancer Institute, NIH, under contract no. HHSN27612080001. The content of this review does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
References
Adams, E. (1970). Metabolism of proline and of hydroxyproline. Int. Rev. Connect. Tissue Res. 5, 1–91.
Adams, E., and Frank, L. (1980). Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 49, 1005–1061.
Alowami, S., Troup, S., Al-Haddad, S., Kirkpatrick, I., and Watson, P. H. (2003). Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res. 5, R129–R135.
Anthony, B., Allen, J., Li, Y. S., and Mcmanus, D. P. (2010). Schistosoma mansoni egg-induced downregulation of hepatic stellate cell activation and fibrogenesis. J. Gastroenterol. Hepatol. 25, A9–A9.
Arentson, B. W., Sanyal, N., and Becker, D. F. (2012). Substrate channeling in proline metabolism. Front. Biosci. 17, 375–388.
Boza, J. J., Moennoz, D., Bournot, C. E., Blum, S., Zbinden, I., Finot, P. A., and Ballevre, O. (2000). Role of glutamine on the de novo purine nucleotide synthesis in Caco-2 cells. Eur. J. Nutr. 39, 38–46.
Brandsma, D., Dorlo, T. P., Haanen, J. H., Beijnen, J. H., and Boogerd, W. (2010). Severe encephalopathy and polyneuropathy induced by dichloroacetate. J. Neurol. 257, 2099–2100.
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95.
Catchpole, G., Platzer, A., Weikert, C., Kempkensteffen, C., Johannsen, M., Krause, H., Jung, K., Miller, K., Willmitzer, L., Selbig, J., and Weikert, S. (2009). Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell. Mol. Med. 15, 109–118.
Chandel, N. S. (2010). Mitochondrial complex III: an essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 174, 175–181.
Chang, J. M., Chung, J. W., Jae, H. J., Eh, H., Son, K. R., Lee, K. C., and Park, J. H. (2007). Local toxicity of hepatic arterial infusion of hexokinase II inhibitor, 3-bromopyruvate: in vivo investigation in normal rabbit model. Acad. Radiol. 14, 85–92.
Cooper, S. K., Pandhare, J., Donald, S. P., and Phang, J. M. (2008). A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J. Biol. Chem. 283, 10485–10492.
Cyr, A. R., and Domann, F. E. (2010). The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid. Redox Signal. 15, 551–589.
Dang, C. V. (2010). Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 70, 859–862.
Dang, C. V., and Semenza, G. L. (1999). Oncogenic alterations of metabolism. Trends Biochem. Sci. 24, 68–72.
Davis, J. M., and Cowie, H. A. (1990). The relationship between fibrosis and cancer in experimental animals exposed to asbestos and other fibers. Environ. Health Perspect. 88, 305–309.
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G., and Thompson, C. B. (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20.
Donald, S. P., Sun, X. Y., Hu, C. A., Yu, J., Mei, J. M., Valle, D., and Phang, J. M. (2001). Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 61, 1810–1815.
Dougherty, K. M., Brandriss, M. C., and Valle, D. (1992). Cloning human pyrroline-5-carboxylate reductase cDNA by complementation in Saccharomyces cerevisiae. J. Biol. Chem. 267, 871–875.
Eddy, A. A., and Giachelli, C. M. (1995). Renal expression of genes that promote interstitial inflammation and fibrosis in rats with protein-proteinuria. Kidney Int. 47, 1546–1557.
Fleming, G. A., Granger, A., Rogers, Q. R., Prosser, M., Ford, D. B., and Phang, J. M. (1989). Fluctuations in plasma pyrroline-5-carboxylate concentrations during feeding and fasting. J. Clin. Endocrinol. Metab. 69, 448–452.
Fleming, G. A., Hagedorn, C. H., Granger, A. S., and Phang, J. M. (1984). Pyrroline-5-carboxylate in human plasma. Metab. Clin. Exp. 33, 739–742.
Flynn, M. P., Martin, M. C., Moore, P. T., Stafford, J. A., Fleming, G. A., and Phang, J. M. (1989). Type II hyperprolinaemia in a pedigree of Irish travellers (nomads). Arch. Dis. Child. 64, 1699–1707.
Gao, P., Tchernyshyov, I., Chang, T. C., Lee, Y. S., Kita, K., Ochi, T., Zeller, K. I., De Marzo, A. M., Van Eyk, J. E., Mendell, J. T., and Dang, C. V. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765.
Gottlieb, E., and Vousden, K. H. (2010). p53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2, a001040.
Hagedorn, C. H., and Phang, J. M. (1983). Transfer of reducing equivalents into mitochondria by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch. Biochem. Biophys. 225, 95–101.
Hagedorn, C. H., and Phang, J. M. (1986). Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch. Biochem. Biophys. 248, 166–174.
Hu, C. A., Bart Williams, D., Zhaorigetu, S., Khalil, S., Wan, G., and Valle, D. (2008). Functional genomics and SNP analysis of human genes encoding proline metabolic enzymes. Amino Acids 35, 655–664.
Hu, C. A., Donald, S. P., Yu, J., Lin, W. W., Liu, Z., Steel, G., Obie, C., Valle, D., and Phang, J. M. (2007). Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 295, 85–92.
Kaelin, W. G. Jr., and Thompson, C. B. (2012). Q&A: cancer: clues from cell metabolism. Nature 465, 562–564.
Kao, R. T., Hall, J., and Stern, R. (1986). Collagen and elastin synthesis in human stroma and breast carcinoma cell lines: modulation by the extracellular matrix. Connect. Tissue Res. 14, 245–255.
Kaufmann, P., Engelstad, K., Wei, Y., Jhung, S., Sano, M. C., Shungu, D. C., Millar, W. S., Hong, X., Gooch, C. L., Mao, X., Pascual, J. M., Hirano, M., Stacpoole, P. W., Dimauro, S., and De Vivo, D. C. (2006). Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66, 324–330.
Kim, K. Y., Ahn, J. H., and Cheon, H. G. (2007). Apoptotic action of peroxisome proliferator-activated receptor-gamma activation in human non small-cell lung cancer is mediated via proline oxidase-induced reactive oxygen species formation. Mol. Pharmacol. 72, 674–685.
Le, A., Lane, A. N., Hamaker, M., Bose, S., Gouw, A., Barbi, J., Tsukamoto, T., Rojas, C. J., Slusher, B. S., Zhang, H., Zimmerman, L. J., Liebler, D. C., Slebos, R. J., Lorkiewicz, P. K., Higashi, R. M., Fan, T. W., and Dang, C. V. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121.
Li, T., Sun, L., Miller, N., Nicklee, T., Woo, J., Hulse-Smith, L., Tsao, M. S., Khokha, R., Martin, L., and Boyd, N. (2005). The association of measured breast tissue characteristics with mammographic density and other risk factors for breast cancer. Cancer Epidemiol. Biomarkers Prev. 14, 343–349.
Liu, W., Glunde, K., Bhujwalla, Z. M., Raman, V., Sharma, A., and Phang, J. M. (2012a). Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. [Epub ahead of print]. doi: 10.1158/0008-5472.CAN-12-0080.
Liu, W., Le, A., Hancock, C., Lane, A. N., Dang, C. V., Fan, W.-M. F., and Phang, J. M. (2012b). The reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by c-MYC. Proc. Natl. Acad. Sci. U.S.A. 109, 8983–8988.
Liu, Y., Borchert, G. L., Donald, S. P., Diwan, B. A., Anver, M., and Phang, J. M. (2009). Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res. 69, 6414–6422.
Liu, W., Zabirnyk, O., Wang, H., Shiao, Y. H., Nickerson, M. L., Khalil, S., Anderson, L. M., Perantoni, A. O., and Phang, J. M. (2010). miR-23b* targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 29, 4914–4924.
Liu, Y., Borchert, G. L., Donald, S. P., Surazynski, A., Hu, C. A., Weydert, C. J., Oberley, L. W., and Phang, J. M. (2005). MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis 26, 1335–1342.
Liu, Y., Borchert, G. L., Surazynski, A., Hu, C. A., and Phang, J. M. (2006). Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 25, 5640–5647.
Locasale, J. W., and Cantley, L. C. (2010). Altered metabolism in cancer. BMC Biol. 8, 88. doi:10.1186/1741-7007-8-88
Locasale, J. W., and Cantley, L. C. (2011). Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 14, 443–451.
Locasale, J. W., Grassian, A. R., Melman, T., Lyssiotis, C. A., Mattaini, K. R., Bass, A. J., Heffron, G., Metallo, C. M., Muranen, T., Sharfi, H., Sasaki, A. T., Anastasiou, D., Mullarky, E., Vokes, N. I., Sasaki, M., Beroukhim, R., Stephanopoulos, G., Ligon, A. H., Meyerson, M., Richardson, A. L., Chin, L., Wagner, G., Asara, J. M., Brugge, J. S., Cantley, L. C., and Vander Heiden, M. G. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874.
Luo, Z., Zang, M., and Guo, W. (2010). AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future. Oncol. 6, 457–470.
Manganelli, G., Fico, A., Masullo, U., Pizzolongo, F., Cimmino, A., and Filosa, S. (2012). Modulation of the pentose phosphate pathway induces endodermal differentiation in embryonic stem cells. PLoS ONE 7, e29321. doi:10.1371/journal.pone.0029321
Maxwell, S. A., and Rivera, A. (2003). Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J. Biol. Chem. 278, 9784–9789.
Meng, Z., Lou, Z., Liu, Z., Li, M., Zhao, X., Bartlam, M., and Rao, Z. (2006). Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol. 359, 1364–1377.
Merrill, M. J., Yeh, G. C., and Phang, J. M. (1989). Purified human erythrocyte pyrroline-5-carboxylate reductase. Preferential oxidation of NADPH. J. Biol. Chem. 264, 9352–9358.
Miccheli, A., Tomassini, A., Puccetti, C., Valerio, M., Peluso, G., Tuccillo, F., Calvani, M., Manetti, C., and Conti, F. (2006). Metabolic profiling by 13C-NMR spectroscopy: [1,2-13C2]glucose reveals a heterogeneous metabolism in human leukemia T cells. Biochimie 88, 437–448.
Mixson, A. J., and Phang, J. M. (1988). The uptake of pyrroline 5-carboxylate. Group translocation mediating the transfer of reducing-oxidizing potential. J. Biol. Chem. 263, 10720–10724.
Mixson, A. J., and Phang, J. M. (1991). Structural analogues of pyrroline 5-carboxylate specifically inhibit its uptake into cells. J. Membr. Biol. 121, 269–277.
Morris, S. M. Jr. (2009). Recent advances in arginine metabolism: roles and regulation of the arginases. Br. J. Pharmacol. 157, 922–930.
Palaskas, N., Larson, S. M., Schultz, N., Komisopoulou, E., Wong, J., Rohle, D., Campos, C., Yannuzzi, N., Osborne, J. R., Linkov, I., Kastenhuber, E. R., Taschereau, R., Plaisier, S. B., Tran, C., Heguy, A., Wu, H., Sander, C., Phelps, M. E., Brennan, C., Port, E., Huse, J. T., Graeber, T. G., and Mellinghoff, I. K. (2011). 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 71, 5164–5174.
Pandhare, J., Cooper, S. K., and Phang, J. M. (2006). Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J. Biol. Chem. 281, 2044–2052.
Pandhare, J., Donald, S. P., Cooper, S. K., and Phang, J. M. (2009). Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 107, 759–768.
Peterkofsky, B., and Udenfriend, S. (1965). Enzymatic hydroxylation of proline in microsomal polypeptide leading to formation of collagen. Proc. Natl. Acad. Sci. U.S.A. 53, 335–342.
Phang, J. M. (1985). The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr. Top. Cell. Regul. 25, 91–132.
Phang, J. M., Donald, S. P., Pandhare, J., and Liu, Y. (2008a). The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 35, 681–690.
Phang, J. M., Pandhare, J., and Liu, Y. (2008b). The metabolism of proline as microenvironmental stress substrate. J. Nutr. 138, 2008S–2015S.
Phang, J. M., Pandhare, J., Zabirnyk, O., and Liu, Y. (2008c). PPARgamma and proline oxidase in cancer. PPAR Res. 2008, 542694.
Phang, J. M., Downing, S. J., and Yeh, G. C. (1980). Linkage of the HMP pathway to ATP generation by the proline cycle. Biochem. Biophys. Res. Commun. 93, 462–470.
Phang, J. M., Downing, S. J., Yeh, G. C., Smith, R. J., Williams, J. A., and Hagedorn, C. H. (1982). Stimulation of the hexosemonophosphate-pentose pathway by pyrroline-5-carboxylate in cultured cells. J. Cell. Physiol. 110, 255–261.
Phang, J. M., Hu, C. A., and Valle, D. (2001). “Disorders of proline and hydroxyproline metabolism,” in Metabolic and Molecular Basis of Inherited Disease, eds C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York: McGraw-Hill), 1821–1838.
Phang, J. M., Liu, W., and Zabirnyk, O. (2010). Proline metabolism and microenvironmental stress. Annu. Rev. Nutr. 30, 441–463.
Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K. W., and Vogelstein, B. (1997). A model for p53-induced apoptosis. Nature 389, 300–305.
Possemato, R., Marks, K. M., Shaul, Y. D., Pacold, M. E., Kim, D., Birsoy, K., Sethumadhavan, S., Woo, H. K., Jang, H. G., Jha, A. K., Chen, W. W., Barrett, F. G., Stransky, N., Tsun, Z. Y., Cowley, G. S., Barretina, J., Kalaany, N. Y., Hsu, P. P., Ottina, K., Chan, A. M., Yuan, B., Garraway, L. A., Root, D. E., Mino-Kenudson, M., Brachtel, E. F., Driggers, E. M., and Sabatini, D. M. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350.
Priest, R. E., and Davies, L. M. (1969). Cellular proliferation and synthesis of collagen. Lab. Invest. 21, 138–142.
Provenzano, P. P., Inman, D. R., Eliceiri, K. W., Knittel, J. G., Yan, L., Rueden, C. T., White, J. G., and Keely, P. J. (2008). Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 11. doi:10.1186/1741-7015-6-11
Reversade, B., Escande-Beillard, N., Dimopoulou, A., Fischer, B., Chng, S. C., Li, Y., Shboul, M., Tham, P. Y., Kayserili, H., Al-Gazali, L., Shahwan, M., Brancati, F., Lee, H., O’Connor, B. D., Schmidt-Von Kegler, M., Merriman, B., Nelson, S. F., Masri, A., Alkazaleh, F., Guerra, D., Ferrari, P., Nanda, A., Rajab, A., Markie, D., Gray, M., Nelson, J., Grix, A., Sommer, A., Savarirayan, R., Janecke, A. R., Steichen, E., Sillence, D., Hausser, I., Budde, B., Nurnberg, G., Nurnberg, P., Seemann, P., Kunkel, D., Zambruno, G., Dallapiccola, B., Schuelke, M., Robertson, S., Hamamy, H., Wollnik, B., Van Maldergem, L., Mundlos, S., and Kornak, U. (2009). Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 41, 1016–1021.
Savolainen, E. R., Leo, M. A., Timpl, R., and Lieber, C. S. (1984). Acetaldehyde and lactate stimulate collagen synthesis of cultured baboon liver myofibroblasts. Gastroenterology 87, 777–787.
Schedin, P., and Keely, P. J. (2011). Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb, Perspect, Biol. 3, a003228.
Slaughter, J. C., and Davies, D. D. (1968). The isolation and characterization of 3-phosphoglycerate dehydrogenase from peas. Biochem. J. 109, 743–748.
Smith, R. J., Downing, S. J., and Phang, J. M. (1977). Enzymatic synthesis and purification of L-pyrroline-5-carboxylic acid. Anal. Biochem. 82, 170–176.
Stetten, M. R. (1949). Some aspects of the metabolism of hydroxyproline, studied with the aid of isotopic nitrogen. J. Biol. Chem. 181, 31–37.
Stoner, G. D., and Merchant, D. J. (1972). Amino acid utilization by L-M strain mouse cells in a chemically defined medium. In vitro 7, 330–343.
Suzuki, H., Fukunishi, Y., Kagawa, I., Saito, R., Oda, H., Endo, T., Kondo, S., Bono, H., Okazaki, Y., and Hayashizaki, Y. (2001). Protein-protein interaction panel using mouse full-length cDNAs. Genome Res. 11, 1758–1765.
Thompson, C. B. (2009). Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 360, 813–815.
Tian, W. N., Braunstein, L. D., Pang, J., Stuhlmeier, K. M., Xi, Q. C., Tian, X., and Stanton, R. C. (1998). Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617.
Tong, X., Zhao, F., and Thompson, C. B. (2009). The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 19, 32–37.
Tuttle, S., Muschel, R., Bernhard, E., Mckenna, W. G., and Biaglow, J. (1996). Decreased ability of cells overexpressing MYC proteins to reduce peroxide and hydroperoxides. Br. J. Cancer Suppl. 27, S140–S144.
Valle, D., Goodman, S. I., Harris, S. C., and Phang, J. M. (1979). Genetic evidence for a common enzyme catalyzing the second step in the degradation of proline and hydroxyproline. J. Clin. Invest. 64, 1365–1370.
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033.
Vander Heiden, M. G., Lunt, S. Y., Dayton, T. L., Fiske, B. P., Israelsen, W. J., Mattaini, K. R., Vokes, N. I., Stephanopoulos, G., Cantley, L. C., Metallo, C. M., and Locasale, J. W. (2012). Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. [Epub ahead of print]. doi:10.1101/sqb.2012.76.010900.
Veech, R. L., Eggleston, L. V., and Krebs, H. A. (1969). The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 115, 609–619.
Verma, S., and Slutsky, A. S. (2007). Idiopathic pulmonary fibrosis – new insights. N. Engl. J. Med. 356, 1370–1372.
Visioni, A., and Kim, J. (2011). Positron emission tomography for benign and malignant disease. Surg. Clin. North Am. 91, 249–266.
Walker, F., and Shand, J. (1972). Influence of alcohol on collagen synthesis in vitro. Lancet 1, 233–234.
Wallace, D. C. (2005). Mitochondria and cancer: Warburg addressed. Cold Spring Harb. Symp. Quant. Biol. 70, 363–374.
Wanduragala, S., Sanyal, N., Liang, X., and Becker, D. F. (2010). Purification and characterization of Put1p from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 498, 136–142.
Wang, J., Lv, X., Shi, J., Hu, X., and Du, Y. (2011a). Troglitazone induced apoptosis via PPARgamma activated POX-induced ROS formation in HT29 cells. Biomed. Environ. Sci. 24, 391–399.
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., Finkelstein, D., Mccormick, L. L., Fitzgerald, P., Chi, H., Munger, J., and Green, D. R. (2011b). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882.
Weinberg, F., and Chandel, N. S. (2009). Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 66, 3663–3673.
Wellen, K. E., Lu, C., Mancuso, A., Lemons, J. M., Ryczko, M., Dennis, J. W., Rabinowitz, J. D., Coller, H. A., and Thompson, C. B. (2010). The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 24, 2784–2799.
Windmueller, H. G., and Spaeth, A. E. (1974). Uptake and metabolism of plasma glutamine by the small intestine. J. Biol. Chem. 249, 5070–5079.
Wise, D. R., Deberardinis, R. J., Mancuso, A., Sayed, N., Zhang, X. Y., Pfeiffer, H. K., Nissim, I., Daikhin, E., Yudkoff, M., Mcmahon, S. B., and Thompson, C. B. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787.
Wu, G. (1996). An important role for pentose cycle in the synthesis of citrulline and proline from glutamine in porcine enterocytes. Arch. Biochem. Biophys. 336, 224–230.
Wu, G., Bazer, F. W., Datta, S., Johnson, G. A., Li, P., Satterfield, M. C., and Spencer, T. E. (2008). Proline metabolism in the conceptus: implications for fetal growth and development. Amino Acids 35, 691–702.
Yeh, G. C., and Phang, J. M. (1981). The stimulation of purine nucleotide production by pyrroline-5-carboxylic acid in human erythrocytes. Biochem. Biophys. Res. Commun. 103, 118–124.
Yeh, G. C., and Phang, J. M. (1983). Pyrroline-5-carboxylate stimulates the conversion of purine antimetabolites to their nucleotide forms by a redox-dependent mechanism. J. Biol. Chem. 258, 9774–9779.
Yeh, G. C., Roth, E. F. Jr., Phang, J. M., Harris, S. C., Nagel, R. L., and Rinaldi, A. (1984). The effect of pyrroline-5-carboxylic acid on nucleotide metabolism in erythrocytes from normal and glucose-6-phosphate dehydrogenase-deficient subjects. J. Biol. Chem. 259, 5454–5458.
Keywords: proline metabolism, glutamine metabolism, MYC oncogene, tumor suppressor function, redox regulation
Citation: Phang JM, Liu W, Hancock C and Christian KJ (2012) The proline regulatory axis and cancer. Front. Oncol. 2:60. doi: 10.3389/fonc.2012.00060
Received: 30 March 2012; Paper pending published: 23 April 2012;
Accepted: 27 May 2012; Published online: 21 June 2012.
Edited by:
Lorenzo Galluzzi, Institut National de la Santé et de la Recherche Medicale, FranceReviewed by:
Enrico Lugli, National Institutes of Health, USACristina Munoz-Pinedo, Bellvitge Biomedical Research Institute, Spain
Copyright: © 2012 Phang, Liu, Hancock and Christian. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: James Ming Phang, Metabolism and Cancer Susceptibility Section, Basic Research Laboratory, Center for Cancer Research, Frederick National Laboratory for Cancer Research, Building 538, Room 115, Frederick, MD 21702, USA. e-mail:cGhhbmdqQG1haWwubmloLmdvdg==