
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 13 June 2012
Sec. Molecular and Cellular Oncology
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00059
This article is part of the Research TopicYeast programmed cell death and ageingView all 16 articles
 Caitlin Clapp1† Liam Portt1† Chamel Khoury2 Sara Sheibani1,3 Rawan Eid1 Matthew Greenwood1 Hojatollah Vali3 Craig A. Mandato3
Caitlin Clapp1† Liam Portt1† Chamel Khoury2 Sara Sheibani1,3 Rawan Eid1 Matthew Greenwood1 Hojatollah Vali3 Craig A. Mandato3 Michael T. Greenwood1*
Michael T. Greenwood1*Genetically programmed cell death (PCD) mechanisms, including apoptosis, are important for the survival of metazoans since it allows, among things, the removal of damaged cells that interfere with normal function. Cell death due to PCD is observed in normal processes such as aging and in a number of pathophysiologies including hypoxia (common causes of heart attacks and strokes) and subsequent tissue reperfusion. Conversely, the loss of normal apoptotic responses is associated with the development of tumors. So far, limited success in preventing unwanted PCD has been reported with current therapeutic approaches despite the fact that inhibitors of key apoptotic inducers such as caspases have been developed. Alternative approaches have focused on mimicking anti-apoptotic processes observed in cells displaying increased resistance to apoptotic stimuli. Hormesis and pre-conditioning are commonly observed cellular strategies where sub-lethal levels of pro-apoptotic stimuli lead to increased resistance to higher or lethal levels of stress. Increased expression of anti-apoptotic sequences is a common mechanism mediating these protective effects. The relevance of the latter observation is exemplified by the observation that transgenic mice overexpressing anti-apoptotic genes show significant reductions in tissue damage following ischemia. Thus strategies aimed at increasing the levels of anti-apoptotic proteins, using gene therapy or cell penetrating recombinant proteins are being evaluated as novel therapeutics to decrease cell death following acute periods of cell death inducing stress. In spite of its functional and therapeutic importance, more is known regarding the processes involved in apoptosis than anti-apoptosis. The genetically tractable yeast Saccharomyces cerevisiae has emerged as an exceptional model to study multiple aspects of PCD including the mitochondrial mediated apoptosis observed in metazoans. To increase our knowledge of the process of anti-apoptosis, we screened a human heart cDNA expression library in yeast cells undergoing PCD due to the conditional expression of a mammalian pro-apoptotic Bax cDNA. Analysis of the multiple Bax suppressors identified revealed several previously known as well as a large number of clones representing potential novel anti-apoptotic sequences. The focus of this review is to report on recent achievements in the use of humanized yeast in genetic screens to identify novel stress-induced PCD suppressors, supporting the use of yeast as a unicellular model organism to elucidate anti-apoptotic and cell survival mechanisms.
Getting rid of unwanted or potentially damaging cells is critical for the normal functioning of metazoans (Wyllie, 2010; Portt et al., 2011; Ulukaya et al., 2011). More recently, genetically encoded mechanisms have been discovered to be of equal importance in regulating cell death and cell survival in all eukaryotes including single cell microbes such as the yeast Saccharomyces cerevisiae (Carmona-Gutierrez and Madeo, 2009; Shemarova, 2010; Kaczanowski et al., 2011). This has led to dramatic changes in how programmed cell death (PCD) is perceived and it has opened up a number of important avenues of research that allows genetic approaches to the study of death inducing and cell survival strategies. It has long been established that the functional expression of human genes in yeast has facilitated the study of individual members of complex gene family in an isolated but functional cell system. The use of humanized yeast cells for the study of apoptosis started many years ago when key regulators of mammalian apoptosis were found to retain their pro- and anti-apoptotic functions when expressed in yeast (Manon et al., 1997; Ligr et al., 1998; Lisa-Santamaria et al., 2009). Today humanized yeast cells are commonly used as a means of identifying and characterizing novel apoptotic regulators and processes (Greenwood and Ludovico, 2010; Silva et al., 2011a). As a prelude to a detailed discussion of the use of humanized yeast, it is necessary to introduce a few related topics. A general discussion will begin with the fact that although a multitude of specialized sub-forms have been described there are three main types of PCDs that receive the most attention (Hotchkiss et al., 2009; Orrenius et al., 2011; Portt et al., 2011; Galluzzi et al., 2012b).
Type I PCD or apoptosis has long been recognized as containing two distinct types called the extrinsic and intrinsic forms (Kroemer et al., 2009; Wyllie, 2010). The extrinsic form is largely due to the activation of cell surface death receptors such as TNFα and is more studied in the context of the immune cells. The intrinsic form is centered on the mitochondria and is activated by a variety of stresses including a number of chemicals (pesticides, cancer therapeutics), physical agents (high osmolarity, change in temperature or pH), and intracellular stresses such as DNA damage and accumulation of misfolded proteins especially in the endoplasmic reticulum (ER; Carmona-Gutierrez et al., 2010; Orrenius et al., 2011). These stresses lead to the activation of intracellular pathways and processes that cause alterations in mitochondrial membrane permeability and the release of pro-apoptogenic factors including cytochrome c, AIF, and Endo G. The effects of cytochrome c are mediated by its ability to form an active apoptosome complex with the Apoptosis Protease Activating Factor 1 (APAF-1) that serves to activate procaspase 9. This in turn leads to cleavage mediated activation of executioner caspases such as caspase 3. There are number of other interrelated pathways associated with the intrinsic pathway which involves a large variety of pro-apoptotic proteins. Many of these stresses can be shown to induce cell death when overexpressed and to reduce stress mediated cell death when their genes are knocked out or down regulated by siRNA based strategies. The reader is referred to a number of recent reviews for more detailed accounts of these processes (Orrenius et al., 2011; Shamas-Din et al., 2011; Ulukaya et al., 2011; Galluzzi et al., 2012b).
Type II PCD or autophagic cell death may not be more complex, but at the moment, it certainly is a lot more confusing (Denton et al., 2012; Galluzzi et al., 2012a,b; Shen et al., 2012). This is because autophagy (Greek, self-eating) is in itself a cellular process that serves to protect the cell from stress (Moreau et al., 2009). Although there are multiple different forms, the most understood form is called macro-autophagy (He and Klionsky, 2009). Here the autophagic machinery is activated in response to nutritional stress where it earmarks cellular constituents that are expendable (i.e., material required for growth). These are then broken down and serve as building blocks for the synthesis of molecules and the expression of genes that can serve to prevent premature cell death. The autophagic machinery gets activated in the absence of nutrient by a well studied complex process involving a variety of regulatory proteins including TOR (Loewith and Hall, 2011). The ability to genetically identify a large number of autophagic (ATG) genes in yeast was instrumental in developing our understanding of autophagy (He and Klionsky, 2009). The availability of mutants lacking ATG genes due to knock out or knock down have shown that the process of autophagy is critical for cellular survival in response to a variety of stresses including amino acid and glucose starvation as well chemical inducers of apoptosis (Pan et al., 2009). Other forms of autophagy that carry out specialized functions such as the selective removal of certain cellular constituents including the specific removal of damaged mitochondria by mitophagy may also be critical for cellular survival under some conditions (Kissova and Camougrand, 2010). On the other hand low level removal of damaged cellular material by constitutive autophagy is thought to have housekeeping functions that are required to maintain proper order in a cell (Gottlieb et al., 2009; Marino et al., 2011).
In contrast to the large body of knowledge about the protective effects and processes associated with the pro-survival functions of autophagy (see also below) even the bare essential framework for autophagic cell death is lacking (Chen and Klionsky, 2011). Autophagic cell death was originally defined as a form of cell death that is associated with autophagosomes that is likely non-apoptotic (caspase, Bax independent; Marino et al., 2011; Meschini et al., 2011). Autophagosomes are intracellular vesicles that are the site of the autophagic cargo destined for degradation that consist of the membrane engulfed cargo fused to lysosomes. The connection between autophagy and autophagic cell death still remains obscure. In fact there is quite a strong and growing conviction by many that autophagic cell death would be better labeled as cell death associated with autophagosomes (Denton et al., 2012; Shen et al., 2012). Thus autophagy is often labeled as an “innocent bystander” in the process of an ongoing death (Rami, 2009). It is argued that conditions that serve to initiate cell death, will lead to stress mediated activation of autophagy as a sort of cellular defense mechanism (Schleicher et al., 2010). The increased autophagy is thus a way for the cell to try and mitigate and resist undergoing inappropriate cell death (Meschini et al., 2011). The up-regulation of such cellular survival processes have been identified in response to numerous stresses (Fulda et al., 2010; Portt et al., 2011). The inability to prevent cell deaths that are thought to be occurring by autophagy, by inhibiting the autophagic process is regarded as strong evidence that autophagic cell death is likely over diagnosed (Galluzzi et al., 2012b; Shen et al., 2012). Strong guidelines or recommendations by a number of leaders in the field have been proposed to try and demystify the process (Galluzzi et al., 2012b). It nevertheless remains technically challenging to identify cell death by autophagy. This is partially due to the fact that small chemical inhibitors of autophagy used are non-specific. The ability to develop specific chemical inhibitors or genes encoding negative regulators, as was recently done for necrosis, would be required to clearly identify and characterize autophagic death (Wu et al., 2012).
Nevertheless, autophagic cell death has been acknowledged to exist, even by the most ardent anti-autophagic death crowd, under certain circumstances, and in some organisms (Shen et al., 2012). For example apoptotic inducing stimuli leads to autophagic cell death in mouse embryonic fibroblasts (MEFs) cells that are unable to carry out apoptosis due to knock outs of both pro-apoptotic Bcl-2 family members, Bax and Bak (Shimizu et al., 2004). Others have argued that autophagic cell death may simply be a sort of death of last resort that occurs after prolonged autophagy where the cell simply becomes exhausted and has no more resources to combat prolonged stresses (Rami, 2009). This could occur by a process of autophagosome lysis and subsequent death by the released catalytically active enzymes in a process analogous to lysosomal rupture that occurs in some forms of ER stress or necrosis (Orrenius et al., 2011). The application of proteomic approaches to the study of autophagy may be useful in further delineating the role of autophagy in cell death (Dengjel et al., 2012).
Type III PCD is also known as necrosis. This cell death differs from apoptosis in a number of readily detectable key features including several biochemical, cellular, and morphological differences (Berghe et al., 2010; Eisenberg et al., 2010; Fulda et al., 2010; Galluzzi et al., 2012b). The early loss of membrane integrity observed in necrotic cells coupled to the fact that membrane externalization occurs in apoptosis allows the use to use fluorescently labeled nuclear vital dyes and differentially labeled protein capable of binding inner membranes (annexin V) to discriminate between these forms of death. Also key among the differences is the blebbing of apoptotic cells followed by the engulfment of the debris by immune cells. Necrotic cells on other hand remain within the tissue and lead to inflammation as they decay. Necrosis is the form of cell death that occurs in response to severe catastrophic stress that leads to deleterious and irreversible cellular injury. More recent studies suggest that there is a genetically programmed form of necrosis, called necroptosis, which can be triggered by severe stresses including some forms of DNA damage (Weinlich et al., 2011; Wu et al., 2012). A biochemical pathway involving the death type receptor (i.e., TNF-α) mediated formation of a necrosome type complex that leads to activation of Receptor Interacting Protein Kinase 1 and 3 (RIPK-1 and 3) can lead to both apoptotic and necrotic cell death (Green et al., 2011). Compounds capable of inhibiting the RIPKs as well as potential intrinsically expressed proteins such as the prepro region of cathepsin D have been identified as inhibitors of the process of necroptosis (Carmona-Gutierrez et al., 2011a). These and other studies suggest that clinical strategies aimed at preventing some forms of necrosis may be possible (Christofferson and Yuan, 2010).
Alternative forms of apoptosis have been known for a long time (Orrenius et al., 2011). There are non-traditional forms of PCD that diverge due to a number of differences such as being caspase or Bax independent (Kroemer et al., 2009). Other forms include infections with organisms such as Salmonella that initiate a caspase 1 dependent cell death called pyroptosis. A caspase 12 dependent process appears to be involved in certain forms of cell death such as the pro-inflammatory condition that is seen in sepsis. Anoikis refers to the PCD that occurs in response to cell detachment from its neighbors. Increased resistance to this form of cell death may lead to the ability to grow in an anchorage independent manner that is often seen in cancer cells (Sakamoto and Kyprianou, 2010). Many of these PCDs appear to have characteristics of apoptosis such as similar morphological changes indicating that many may simply represent variants on apoptosis. Thus despite differences in triggering the many forms of PCDs, the mitochondria still plays a central role in mediating many of these varied forms of cell death. Even the PCD that occurs in aging, which shows a great deal of dependence on glucose metabolism, appears to be dependent on the mitochondria (Laun et al., 2008). Understanding how a cell integrates all information to come up with the appropriate cell death response will require more knowledge about the regulation of cell death.
All the different forms of PCD are not always stand-alone processes. There is a great deal of reported cross-talk between the different forms of cell death (Amelio et al., 2011; Giansanti et al., 2011; Orrenius et al., 2011; Shen and Codogno, 2012). On the large scale the three types of PCDs can be shown to influence one another such that the process of autophagy can serve to inhibit apoptosis and necrosis while cells unable to undergo apoptosis will undergo autophagy or necrotic cell death (Zhivotovsky and Orrenius, 2010; Shen and Codogno, 2012). The most highly reported of such an example is the early experiment using MEFs cultured from embryos having a double knock out (DKO) of the two genes encoding the pro-apoptotic Bcl-2 proteins Bax and Bak (Shimizu et al., 2004). The DKO embryos are lethal which likely reflects the importance of these two proteins in regulating developmental apoptosis. When challenged with apoptotic stimuli, DKO MEFs appear to undergo a cell death that has the hallmarks not of apoptosis but of autophagic cell death. On the other hand, there are a number of other cases where the inhibition of caspase can effectively prevent apoptotic cell death without sparring the cell’s life since it induces necrosis (Rami et al., 2008). Alternatively autophagy may be co-activated with apoptosis as a defense mechanism that can serve to prevent premature execution of apoptosis or necrosis (Amelio et al., 2011; Shen and Codogno, 2012). These and multiple other forms of cross-talk have been observed in a number of different ways including the fact that many individual proteins play roles in more than one cell death modality. For example, the BH3 containing Bcl-2 family member Beclin 1 is a key regulator of autophagy that can be inhibited by Bcl-2 (Kang et al., 2011). Thus Bcl-2 mediated inhibition of Beclin and of Bax allows it to have direct roles in preventing autophagy and apoptosis induction. On the other hand caspase mediated cleavage of Beclin can serve as a mechanism by which the activation of apoptotic machinery can inhibit autophagy (Djavaheri-Mergny et al., 2010). The regulation does not seem to be reciprocal since the overexpression does not appear to inhibit the anti-apoptotic effects of Bcl-2 (Kang et al., 2011). Other well-known examples of proteins with dual functions include FLIP and the autophagic specific gene ATG5 (Giansanti et al., 2011). FLIP is a well-known inhibitor of the extrinsic apoptotic pathway that can also interact with ATG3 and serve as a negative regulator of autophagy (Lee et al., 2009). Less is known about possible cross-talk in unicellular eukaryotes such as yeast. So although the three functional PCDs exist in yeast, there will be a number of strategic differences by which cross-talk between the pathways is used. For example, the ATG6 gene serves the same autophagic regulatory function as Beclin 1 but the yeast ortholog does not appear to have a BH3 domain.
The most commonly examined form of cell death is the intrinsic form of apoptosis that, as mentioned above, involves the stress mediated release of pro-apoptogenic factors from the mitochondria. This pathway mediates the effects of many of the stresses a cell encounters including a wide range of chemicals and physical agents as well as a myriad of others including intracellular stresses such as DNA damage, ER stress as well as the stimuli that occurs in a large number of pathophysiological conditions (see below). The proportion of cells that undergo apoptosis in a given population of cells is directly related to the intensity of the stress encountered (Figure 1; Orrenius et al., 2011; Qi et al., 2012). The intensity of the stress is directly related to a combination of both the dose encountered and the time of exposure to any given stress. For example, keeping the time of exposure constant, we can decrease the percentage of viable cells in a population by increasing the concentration of the stress inducing agent. Thus the average cell population under normal type conditions will respond in a dose dependent manner to a stress and give a standard profile of intensity of stress vs. percentage viability that can be experimentally determined (Figure 1). This profile can shifted to the right or to left by respectively increasing or reducing the expression of a number of pro-apoptotic genes (Figure 1). Thus the normal response to a given stress can be changed by altering the apoptotic machinery. If the stress is too intense, accidental death will occur since the cells will undergo necrosis.
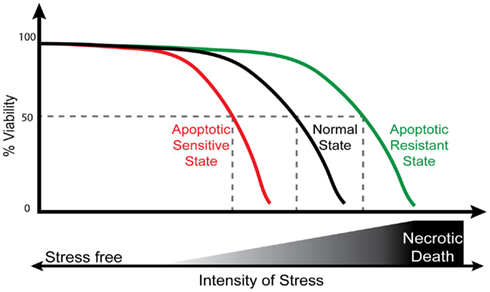
Figure 1. Stress is a dose dependent mediator of cell death. A theoretical and graphical depiction of the effects of increasing the intensity of stress on cellular viability. In a stress free environment cellular viability is maintained close to 100% with only a basal level of cell death. In the normal state (depicted in black), the percentage of cell viability begins to decreases when a specific threshold of stress intensity is encountered. Intensity of stress reflects a combination of time of exposure to the stress and to its intensity. Cell death inducing stresses include a variety of different physical or chemical agents or pathophysiological stresses as discussed in the text. Once the threshold level of stress is encountered, the % viability further decreases in dose dependent manner with respect to the increase in the intensity of stress. In an apoptotic sensitive state (depicted in red), the minimal threshold of the intensity of stress that is required in order to observe a decrease in viability is reduced. Such an apoptotic sensitive state can be observed in cells that are lacking mechanism that are involved in apoptotic resistance such as the loss of an anti-apoptotic resistant gene or a mutation leading to defects in the induction of autophagy. Alternatively, overexpression of a pro-apoptotic gene can lead to a similar phenotype. The opposite phenotype, that is the state of apoptotic resistance (depicted in green), occurs in cells that are lacking some pro-apoptotic genes, that are overexpressing an anti-apoptotic gene or that have increased activation of other pro-survival processes such as autophagy. The net effect of altering a cell’s sensitivity to apoptosis can be observed in the different intensity of stress that is required to give rise to half maximal viability (50% viability, depicted by dashed lines). Thus the threshold of sensitivity to death inducing stress is variable and is established by complex regulatory processes that involve both pro- and anti-apoptotic processes. It should be noted that the processes that trigger apoptosis and that serve to induce cell death are not altered by the apoptotic resistant or sensitive states.
The mechanism by which stress leads to the activation of apoptotic response involves the increases in the levels of two well characterized intermediates, activated Bax, and Reactive Oxygen Species (ROS). Stress also results in an increases in the levels of a number of other pro-apoptotic second messengers including calcium, cAMP, the nucleotide dUTP, and the sphingolipid ceramide (Ozbayraktar and Ulgen, 2009; Vertessy and Toth, 2009; Circu and Aw, 2010; Ganesan and Colombini, 2010; Pourova et al., 2010; Insel et al., 2011; Li et al., 2011; Orrenius et al., 2011; Wilson et al., 2012). Increases in the level of these pro-apoptotic second messengers appear to be tightly coordinated and to be synergistic in their effects (Portt et al., 2011; Ray et al., 2012). For example, increasing activated Bax leads to increased ROS and ceramide while increases in ROS and ceramide can serve to activate Bax (Kumar and Jugdutt, 2003; Lecour et al., 2006; Ganesan et al., 2010). Functionally, one can manipulate a decrease in the levels of ROS or ceramide and this can serve to prevent Bax mediated cell death (see also below; Moon et al., 2002; Yang et al., 2006; Separovic et al., 2007). This suggests that stress mediated cell death involves the cooperative response of numerous cellular pathways and that this cross-talk may serve as redundant, additive, or even synergistic processes to induce cell death (Portt et al., 2011). Specific recent examples of additive effects include Bax mediated permeabilization of the outer mitochondrial membrane serving to increase the release of ROS from the mitochondria. Increased levels of sphingolipids such as ceramide in the mitochondrial membrane may in turn serve to facilitate the insertion of Bax into the mitochondria (Ganesan et al., 2010; Chipuk et al., 2012).
In order to avoid triggering inappropriate PCD, cells have developed a large repertoire of pro-survival tools and strategies that play critical roles in the balance between life and death (Owsianowski et al., 2008; Busca et al., 2009; Fulda et al., 2010; Ashida et al., 2011; Portt et al., 2011; Rodrigues et al., 2012). Anti-apoptotic responses are not simply the inverse of apoptosis instead it is more like a regulatory network that serves to prevent cell death when it is not warranted. As we shall describe in the following few sections, this network also includes and cross-talks extensively with the pro-survival component of autophagy. Of further importance is the fact that this network may be pre-activated in order to create a state of increased resistance to future death inducing stimuli. Examples of the importance of the modulatory nature of apoptotic responses are well illustrated by a recent paper by Shlezinger et al. (2011). They examined the role that apoptosis and anti-apoptosis may play in the ability of the necrotrophic fungus Botrytis cinerea to infect plants. Using genetically altered organisms, they were able to show that plants send out signals to try and induce apoptosis in the invader while an increase in the expression of anti-apoptotic sequences in the fungus increased its ability to infect the plant. Thus the ability to modulate responses to apoptotic stimuli can be a useful tool in organisms with complex life cycles (Rodrigues et al., 2012). Here we will focus on the major regulatory processes that serve to negatively regulate PCD.
Anti-apoptotic genes can be defined as sequences that confer an apoptotic resistant state to a cell while its knock out or a knock down of its normal expression levels leads to a apoptotic sensitive state (Figure 1). Exceptions may exist especially if the anti-apoptotic gene functions in a parallel pathway to another similar genes or processes. The most common anti-apoptotic gene, Bcl-2, functions at least in part by antagonizing the pro-apoptotic Bcl-2 protein Bax (Khoury and Greenwood, 2008). Other very well characterized anti-apoptotic proteins include the IAPs (Inhibitors of Apoptosis), HSPs (Heat Shock Proteins), and ROS scavengers. IAPs function to inhibit caspase activation; HSPs act as chaperones that help in stabilizing protein structure while ROS scavengers serve in inactivating damage inducing ROS (Beere, 2004; Busca et al., 2009; Circu and Aw, 2010). The functional analysis of new anti-apoptotic genes often serves to reinforce the importance of some apoptotic pathways or they may serve to uncover novel ones. Thus the importance of ROS and misfolded/denatured proteins in the induction of apoptosis from multiple stresses is reinforced by the classical observation that overexpression of genes encoding ROS scavengers and chaperonin type HSPs serve to decrease cell death (Garrido et al., 2003; Kim et al., 2006; Guaragnella et al., 2008). Uncovering unexpected cell survival sequences such as the Metalloprotease 15 (MMP15) suggests a novel role for extracellular matrix or alternative functions to well-known proteins (Abraham et al., 2005). The ability to increase cell survival by increasing the expression of the gene encoding dUTPase supports the emerging consensus that nucleotides such as dUTP serve as stress-induced pro-apoptotic second messengers (Williams et al., 2011; Wilson et al., 2012). On the other hand, the cytoprotective effects of the ceramide utilizing enzyme sphingomyelin synthase is consistent with the long standing role of ceramide as a stress-induced pro-apoptotic second messenger (Yang et al., 2006; Separovic et al., 2007, 2008). The diversity in the function of anti-apoptotic sequences exemplifies the diverse roles played by these sequences.
Sequences capable of specifically preventing other forms of PCD without effecting apoptotic cell death have been reported. This is especially true for the ability of genes capable of preventing necrotic cell death (Weinlich et al., 2011). For example death receptor mediated activation of receptor interacting protein kinases-1 and 3 (RIPK-1 and 3) is one of the early steps in the activation of necroptosis. Proteins like FLIP are capable of inhibiting RIPKs and preventing necroptosis and are thus functionally similar to anti-apoptotic sequences (Green et al., 2011). Other examples of such necroptotic regulators, is the ability of Pep4 (encoding the yeast ortholog of the mammalian cathepsin D) to decrease aging induced necrotic death (Carmona-Gutierrez et al., 2011a).
On the other hand there are a number of genes that can be considered as conditional anti-apoptotic genes. For example, there are genes that serve to repair DNA damage in response to stresses like UV light and thus can prevent cell death in response to DNA damage. Most of these sequences are unable to prevent PCD in response to most other stresses. So these are pro-survival genes that represent stimulus dependent pro-survival anti-apoptotic genes. Finally, there are a large number of genes that have been identified that can prevent autophagy (Kroemer et al., 2009; Galluzzi et al., 2012b). These genes are essential for autophagy and they function in the process of inducing autophagy, so their removal does not in fact prevent autophagic cell death (He and Klionsky, 2009). The ability to prevent autophagic cell death is limited to a number of non-specific pharmacological agents that mostly serve to prevent acidification of lysosomes. These are thought to function by preventing similar processes in autophagolysosomes.
As mentioned above, basal autophagy serves as a housekeeping process that can get rid of damaged cellular constituents that accumulate while running the basic processes of cellular life. When activated in response to stress, it can increase the removal of damaged material and serve as a mechanism to prevent premature or inappropriate cell death (Shen and Codogno, 2012). Thus the removal of damaged mitochondria can decrease stress mediated increases in ROS levels while the removal of ER clogged with denatured proteins can prevent unwanted ER stress mediated PCD (Gottlieb et al., 2009). Thus a number of other more specialized sub-forms of autophagy such as mitophagy may also be operational in preventing PCD (Kissova and Camougrand, 2010; Marino et al., 2011).
Other cellular processes, analogous to autophagy, can also have specific pro-survival functions in response to specific types of stresses and are thus not always considered in general discussions of cellular survival (Schonthal, 2009; Taylor and Rutter, 2011). A common example is the ER stress response (Schonthal, 2009). This pathway is activated in response to stresses such as the accumulation of misfolded proteins in the ER. Thus defects in this pathway may account for pathological situations like Parkinson’s disease (Nagley et al., 2010). Activating the ER stress response can serve to prevent PCD is response to ER stress but it will be unable to prevent general forms of PCD. Similar to ERAD, a stress inducible mitochondrial quality control system (Mitochondrial-associated degradation, MAD) has recently been described (Heo et al., 2010). This process involves Cdc48p and a newly identified Cdc48p interacting protein called VSM1. This process is thought to be able to clean up damaged mitochondria and prevent cell death (Taylor and Rutter, 2011).
Just as the regulation of cell death is a balance between pro- and anti-apoptotic machineries, it is of interest that cells produce both anti- and pro- survival intracellular second messengers. ROS represents the classical second messenger regulator of apoptosis since it can switch from being anti- to pro- apoptotic depending on its levels (Ray et al., 2012). Similarly, cAMP also has the ability to be both pro- and anti-apoptotic but that may be more dependent on cell type (Insel et al., 2011). One of the most interesting but less studied intracellular secondary messenger is spermidine. Spermidine along with putrescine and spermine make up a family of polyamine compounds that are ubiquitous and present in high amounts in cells from bacteria, plants, and other eukaryotes including man and yeast (Igarashi and Kashiwagi, 2010; Minois et al., 2011). Although their exact function remains largely enigmatic they are involved in a number of different cellular processes including apoptosis (Igarashi and Kashiwagi, 2010; Minois et al., 2011). In spite of the existence of a few reports demonstrating a pro-apoptotic effect of polyamines, their ability to serve as anti-apoptotic agents appears to be more common (Igarashi and Kashiwagi, 2010; Gill and Tuteja, 2011). The potential importance of spermidine as a pro-survival and protective effector has been increased by the recent report showing that it has protective effects in a number of different aging models including yeast, worm, fly, and mouse (Eisenberg et al., 2009). Of interest, the effects of spermidine appear to function by activating autophagy. The ability to activate the protective effects of autophagy has been reported for a number of compounds such as the anti-aging compound resveratrol. This is widely attributed to be the anti-aging mediator that is responsible for the reported beneficial effects of red wine (Rockenfeller and Madeo, 2010). This up-regulating of autophagy may be a common phenomenon as it is seen in the apoptotic resistance associated with many pathophysiological conditions such as cancer (see also below) and may also account for the pro-survival effects of a wide range of other conditions (Periyasamy-Thandavan et al., 2009; Wilkinson and Ryan, 2010). Clues as to the mechanisms involved in regulating the levels of protective spermidine come from further studies of Madeo’s group who show the ability of cathepsin D to prevent aging mediated necroptosis is associated with an increase in polyamines (Carmona-Gutierrez et al., 2011a).
Adaptation to stresses that do not lead to cell death induces a transient condition where the cell shows an enhanced resistance to later exposure of otherwise lethal stresses (Balakumar et al., 2008; Le Bourg, 2009; Lehotsky et al., 2009; Calabrese et al., 2011a). A series of experiments using yeast cells reported by Davies et al. (1995) represent a simple but elegant demonstration of the processes of pre-conditioning. They were able to show that a 2-h exposure to 3.2 mM H2O2 was sufficient to kill greater than 99.9% of yeast cells. Cells that were pre-treated with a sub-lethal dose of H2O2, say 0.1 mM H2O2 for 45 min, showed enhanced viability with 5% of cells surviving in response to a later exposure 2 h exposure to 3.2 mM H2O2. This response was dose dependent with increasing concentration of H2O2 used as pretreatment with 0.4 mM leading to a greater than 30% viability following the 2-h exposure to 3.2 mM H2O2. This and numerous other studies have shown that there is a window of opportunity in which the protection occurs and that the protection that is elicited in response to a sub-lethal stress may lead to cross-protection and confer resistance to the lethal effects of a different stress (Berry and Gasch, 2008; Calabrese et al., 2011a). A similar process is seen in all cell types including mammalian and bacterial cells. The diagram in Figure 1 illustrates in a simple schematic form, the differences that exist between the responses of a cell in a “Normal State” compared to an adapted cell that now has entered an “Apoptotic Resistant State.” The adapted cell shows an enhanced resistance to the cell death inducing effects of stress and this is demonstrated by the observation that they require a greater dose of stress in order to kill off 50% of the population of cells (Figure 1). This process of adaptation induced by mild stress that serves to increase resistance to stronger stress is often referred to as a form of hormesis. As we will discuss later on, hormesis is also likely to play an important role in our evolving concept of the role ROS stress plays in the cell death that occurs in processes such as aging.
An increase in the levels of ROS, albeit not as intense as observed for cells undergoing apoptosis, plays a role in inducing the process by which cellular adaptation occurs in response to mild stress seen in hormesis and pre-conditioning (Martins et al., 2011; Ristow and Schmeisser, 2011; Ristow and Zarse, 2011). As in apoptosis, the mitochondria appear to be the major source of ROS for hormesis (Pan, 2011). In contrast when ROS is present at physiologically relevant levels such as observed during normal or mildly stressed conditions, ROS functions as an intracellular second messenger (Ray et al., 2012). The moderate increase in ROS levels can have a number of effects on proteins for example, it can react with cysteine residues and alter structure and the function of several proteins (Ray et al., 2012). Thus ROS has been shown to be able to alter a number of cellular responses by affecting a variety of different pathways such as MAP and PI3 kinase cascades. The role of ROS in mediating hormetic effects have been reported in all organisms including bacteria. For example, the hormetic effect of sub-lethal low levels of bactericidal antibiotic has been reported to be mediated by increased levels of ROS (Belenky and Collins, 2011).
The free radical theory of aging is a long standing hypothesis that has been used to explain aging (Harman, 1956). At its simplest, the theory posits that free radicals accumulate during the aging process and this accounts for the increased damage that accumulates at the cellular, tissue, and organismal level (Ristow and Zarse, 2011). First proposed in the 1950s the hypothesis gained a lot of momentum as it made a lot of inherent sense and it was consistent with many observations. It is so widely accepted, even by the general public, that anti-oxidants such as vitamin C and E are commonly accepted anti-aging components of most anti-aging creams. The theory of increased free radicals was also commonly adopted as a mechanism to explain the damage that occurs in response to numerous pathophysiological stresses including many that lead to cell death (Kumar et al., 2002; Misra et al., 2009). Thus anti-oxidants were examined for their potential to prevent cellular death in multiple diseases including numerous forms of cardiomyopathies. Numerous discrepancies uncovered over the years have served to question the validity of the model. Of importance to this skepticism was the effect of calorie restriction (CR) on aging. CR, without malnutrition, had been widely reported to slow down aging and lead to increased longevity in all species examined including yeast, flies, worms, and mammals (Ristow and Zarse, 2011). An excess production of ROS has not been able to fully explain the effects of CR. Instead, CR appears to act like a mild stress leading to moderate increases in ROS levels and subsequent anti-aging effects that are mediated by hormesis (Martins et al., 2011; Ristow and Schmeisser, 2011; Ristow and Zarse, 2011). The similarities between CR and hormesis include up-regulating the same pro-survival strategies including increases in the expression of anti-apoptotic sequences and activation of autophagy (Calabrese et al., 2011a; Martins et al., 2011). In support of this, are the widespread observations that the anti-aging effects mediated by other processes such as the removal of specific genes and the effects of compounds like resveratrol and spermidine function, at least in part, by increasing stress response and autophagy (Calabrese et al., 2011a). For example the loss of the CTA1 catalase encoding gene in yeast serves to extend chronological lifespan by increasing the levels of ROS in a manner analogous to CR (Mesquita et al., 2010).
Other stress inducible pro-apoptotic second messengers such as ceramide may also have dual functions in regulating cell death responses (Lecour et al., 2005). Of interest, sub-lethal increases in ROS may also account for the increased autophagy that is observed in many scenarios involving mildly stressed cells. This suggests that there is a mechanism for the direct cross-talk between different forms of pro-survival processes. Other evidence for such cross-talk comes from the recent observations that some anti-apoptotic genes may function by activating autophagy (Gurusamy et al., 2009; Qian et al., 2009). An alternative scenario to explain the later observation is based on the fact that the overexpression of a number of genes is mildly toxic and they serve to stress the cell (Liu et al., 1992). For example, it is well-known that cells used in the biotechnology industry that are forced by genetic manipulations to overexpress different gene products are often stressed to the point where they undergo apoptotic like cell death (Krampe and Al-Rubeai, 2010; Mokdad-Gargouri et al., 2012). It could thus be envisioned that some anti-apoptotic genes prevent cell death due the fact that their overexpression leads to mild stress and subsequent induction of the pro-survival machinery including activation of autophagy and hormesis. Other scenarios that serve to induce a protective pre-condition or hormetic phenotype include the loss by knockout of some genes as well as number of different compounds including many toxic chemicals administered at sub-lethal levels (Kharade et al., 2005; Wang et al., 2009; Mesquita et al., 2010; Martins et al., 2011; Orrenius et al., 2011). This indicates that there are multitudes of ways of eliciting a protective phenotype. This serves to increase the repertoire of strategies that can be clinically used to combat unwanted cell death (Fleming et al., 2011).
Analysis of the expression of numerous genes, and more recently the analysis of global gene expression profiles, reveal that cellular adaptation (pre-conditioning and hormesis) is associated with alterations in the expression of many genes including the increased expression of a number of anti-apoptotic genes (Wu et al., 2004; Coles et al., 2005; Balakumar et al., 2008; Fulda, 2009b). In spite of the fact that that the expression of so many different genes are observed, a stress resistant phenotype can be recapitulated by the simple overexpression of a multitude of different but individual anti-apoptotic genes (Gil-Gómez and Brady, 1998; Yenari et al., 2005; Khan et al., 2006; Nakka et al., 2008). The fact that a single gene is sufficient for this phenotype suggests that there is a strong redundancy in the numerous anti-apoptotic processes that are invoked in the process of adaptation. Increased autophagy, probably macro-autophagy, is also a response to mild stress. Similarly to what is observed with the increased expression of anti-apoptotic genes, increased autophagy on its own is capable of increasing pro-survival phenotypes. This can be demonstrated by using specific macro-autophagy activating drugs like rapamycin (Galluzzi et al., 2012a). Such cells show increased resistance to numerous stresses including many that would be sufficient to induce apoptosis. One of the questions that remain is why there is so much apparent redundancy and co-activation of anti-apoptotic and pro-survival responses.
In eukaryotic microbes, pre-conditioning is a process that has numerous similarities to hormesis. Classical pre-conditioning can be demonstrated by temporally decreasing blood supply to a tissue leading to mild ischemia like conditions (Balakumar et al., 2008; Porter et al., 2012). As in hormesis, the mildly stressed tissue can be subsequently shown to have acquired an increased resistance to more intense levels of stress including longer periods of ischemia. The normal form of pre-conditioning called late onset pre-conditioning involves an increase in the expression of anti-apoptotic genes and is likely to be similar if not identical to what occurs during hormesis (as described above). There is also an early form of cellular protection that occurs soon after the pre-conditioning stimuli. This process is much quicker than the late onset form of pre-conditioning and usually involves post-translational mechanisms that serve to activate pro-survival proteins and cascades such as MAP kinases (Hausenloy et al., 2005; Balakumar et al., 2008). The question as to whether such an early form of hormesis exists in yeast has not been fully addressed.
There a plethora of diseases and pathophysiologies that are associated with defective apoptotic responses (Krakstad and Chekenya, 2010; Whelan et al., 2010; Zhivotovsky and Orrenius, 2010; Ashida et al., 2011; Lavu et al., 2011; Liu et al., 2011; Orrenius et al., 2011; Strasser et al., 2011; Ulukaya et al., 2011; Oerlemans et al., 2012; Porter et al., 2012). Some such as cancer and some virally infected cells have decreased apoptotic responses that lead to the accumulation of unwanted cells. These conditionally apoptotic resistant cells have been useful in identifying genes whose up-regulation can confer anti-apoptotic phenotypes (Busca et al., 2009; Fulda, 2009b). In effect, the most common anti-apoptotic gene, Bcl-2, was first identified as a gene that is up-regulated in cancer cells (Vaux et al., 1988). Altered Bcl-2 levels are now known to occur in a large proportion of cancers. Developing combinational chemotherapeutic strategies aimed at killing cancer cells combined with inhibitors of anti-apoptotic genes or of their protein product are being developed as useful clinical processes (Strasser et al., 2011). Cancer cells develop in stressful micro-environments that can include low nutrient and oxygen levels (Fulda, 2010). Strategies aimed at preventing new blood vessel formation (angiogenesis) are based on preventing the establishment of these tumor cells (Galluzzi et al., 2010b). These types of stresses faced by cancer cells is thought to lead to pre-condition or hormesis like responses that serve to increase apoptotic resistance (Fulda, 2009b; Fulda et al., 2010). Thus there is also an increase in other cell survival strategies in these cells including an increase in autophagy that accounts for some of the anti-apoptotic responses (Liu et al., 2011; Meschini et al., 2011). These observations have led to the current interest in the development of autophagy inhibitors to be used as chemotherapeutic adjuvants (Wilkinson and Ryan, 2010; Meschini et al., 2011).
On the other hand, there are a large number of other pathophysiological conditions where an increase in cell death occurs (Fulda et al., 2010; Whelan et al., 2010; Ulukaya et al., 2011). These are seen in response to stresses such as ischemia/reperfusion that occur due to heart attacks and strokes, in autoimmune diseases like Multiple Sclerosis (MS), and in diseases associated with defects of protein folding and quality control leading to the accumulation of misfolded proteins such as occurs in Alzheimer’s disease. In unicellular eukaryotes such as yeast, exposure to excess mating pheromone in the absence of an appropriate mating partner or the heterologous expression of disease causing human genes like α-synuclein can be seen as analogous to pathological situations that lead to premature death (Buttner et al., 2006; Franssens et al., 2010; Khurana and Lindquist, 2010). In more practical situations, inappropriate, or unwanted cell death occurs in industrially used cells including yeast and cultured mammalian cells that undergo apoptosis due to the stresses imposed by the forced overproduction of biotechnologically important compounds and therapeutics (Krampe and Al-Rubeai, 2010; Mokdad-Gargouri et al., 2012). In many pathologies, there are very limited therapies given that the underlying causes of many of these diseases are not known. Increased apoptosis is not usually the cause since it occurs as a result of the stress a cell encounters due to the pathophysiological condition. Nevertheless, the ability to prevent PCD following the onset of the disease process could be of tremendous clinical value. This is apparent for the PCD that occurs after acute conditions like a heart attack following an ischemic/reperfusion event (Oerlemans et al., 2012). The most common form of a heart attack involves a blockage of a blood vessel leading to ischemic event that leads to nutrients and oxygen deprivation stress of the cells downstream of the obstruction (Ong and Gustafsson, 2012; Figure 2). The first priority is to unblock the blood vessel and allow tissue reperfusion (Porter et al., 2012). The level of stress is graded with the most severe stress occurring in the cells that are most deprived of blood (Figure 2). In addition, there is a great deal of stress that occurs in the cells due to the effects of reperfusion (Ong and Gustafsson, 2012). Necrosis is likely to occur in the part of the tissue that is severely deprived, while apoptosis is triggered in some other areas having less stress (Ong and Gustafsson, 2012). Thus there is a zone of necrotic death that likely occurs soon after a heart attack and these cells are likely beyond therapeutic rescue (Figure 2). Conversely, cell death due to PCD (apoptotic and possibly autophagic death), will occur over the next few days (Ong and Gustafsson, 2012). Thus there is a therapeutic window in which the prevention of PCD would be a great benefit in limiting the infarct size, increasing survivability and decreasing morbidity, following ischemia/reperfusion events. Thus the process of post-conditioning can be used to prevent some of the post-infarct induced PCD (Balakumar et al., 2008; Lehotsky et al., 2009). Post-conditioning refers to the ability to stimulate a hormetic like response in cells using chemical agents that can induce pre-conditioning even after the apoptosis inducing event has occurred (Balakumar et al., 2008; Lehotsky et al., 2009). Further, there is a great deal of evidence that serves to convince that therapeutic interference in the process of PCD post-infarct would serve to limit cellular demise (Oerlemans et al., 2012). Of importance here is the observation that transgenic animals that overexpress anti-apoptotic genes in a cardiac or brain specific manner, have significantly decreased zones of death following ischemic events (Figure 2; Yenari et al., 2005; Khan et al., 2006; Nakka et al., 2008). These effects are widespread since they are observed with a number of different anti-apoptotic genes and it demonstrates the potential clinical usefulness of anti-apoptosis (Rami et al., 2008; Fulda, 2009a; Krakstad and Chekenya, 2010; Dietz, 2011; Lavu et al., 2011).
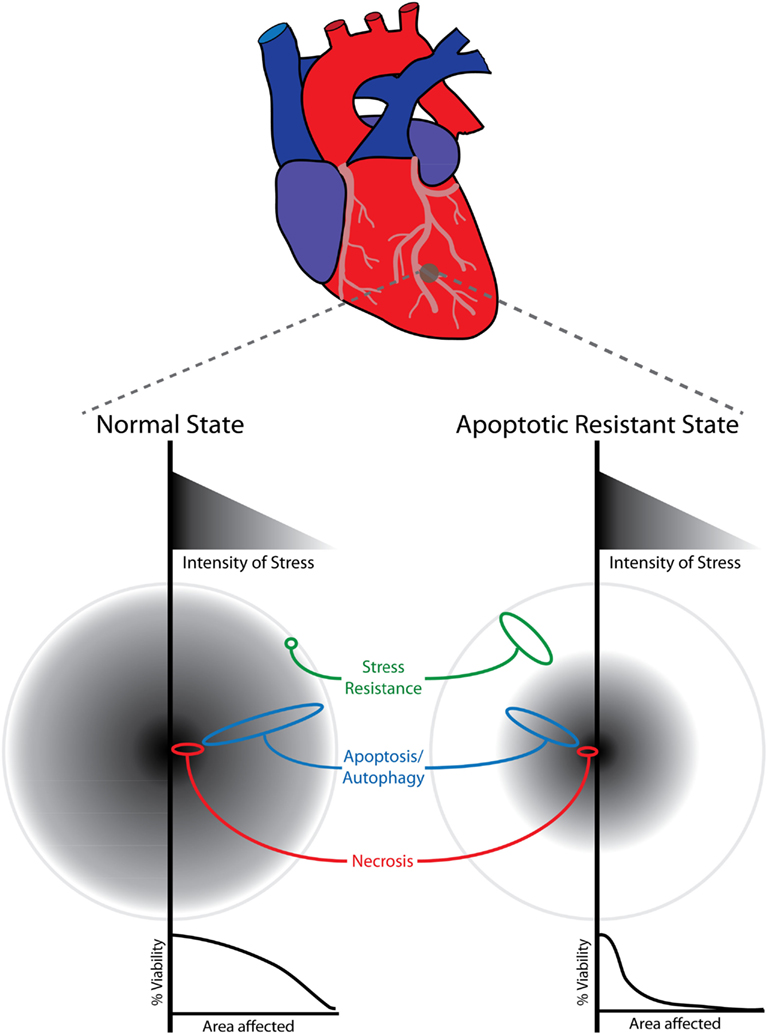
Figure 2. Schematic depiction of the stress generated and subsequent cell death mediated by cardiac stress events. Zones of cardiac cell death can be induced experimentally including the generation of myocardial infarction (MI) by ligation of left descending artery or by direct cardiac ischemia reperfusion (Tarnavski et al., 2004). In the normal state (left side), a gradient of stress intensity occurs in the deprived tissue where the most severely deprived cells undergo necrosis while other stressed cells undergo cell death that appears to include apoptosis and possibly autophagic death. Loss of viability is more pronounced as we move closer to the site of the most intense stress. Transgenic animals that overexpress an anti-apoptotic gene in a cardiac specific manner represent an apoptotic resistant state (right side). Identical levels of stress generated in the hearts of these animals leads to significant reduction in the zone of dead cells.
Numerous strategies have been developed in order to try and overcome the apoptotic resistance encountered in cancer cells (Strasser et al., 2011; Porter et al., 2012). Most chemotherapeutics as well as radiation therapies appear to function by targeting the rapidly dividing phenotype associated with these cells in order to induce a specific apoptotic response. As mentioned above, strategies such as the inhibition of autophagy, are being developed to render cancer cells more sensitive to apoptotic inducing stimuli. In the case of cells that are more resistant to apoptosis due to an infection by an infective agent, investigations are underway to understand the life cycle of these organisms to target the factors that increase apoptotic resistance (Galluzzi et al., 2010a; Ashida et al., 2011; Rodrigues et al., 2012).
On the other hand, given the success achieved by overexpressing anti-apoptotic genes in transgenic animals to prevent ischemic damage, there is intense investigation to try and develop strategies that can serve to inhibit apoptosis (Rami et al., 2008; Yacoubian and Standaert, 2009; Dietz, 2011; Oerlemans et al., 2012). Thus the development of small molecule inhibitors of proteins involved in mediating apoptotic death was examined by many (Oerlemans et al., 2012). Given their central role in apoptosis, caspases were identified as ideal targets given that their inhibition is predicted to serve to prevent at least some forms of PCD. Caspase inhibitors with a good deal of specificity and effectiveness were developed and shown to be effective in vitro cell cultures (Oerlemans et al., 2012). Many of the inhibitors were indeed effective at blocking caspase and caspase mediated cell death. In contrast these were of limited effectiveness at preventing cell death in vivo (Oerlemans et al., 2012). It appears that cells that are stimulated with appropriate stress will undergo alternate modes of PCD if their apoptotic machinery is impaired. This situation is commonly observed and is reminiscent of the autophagic cell death that occurs in apoptotically stimulated cells that are unable to undergo apoptosis due to a double KO of the pro-apoptotic Bcl-2 family members Bax and Bak (Shimizu et al., 2004).
The limited effectiveness encountered with these caspase inhibitors suggests that targeting a single protein may not be able to inhibit apoptosis. Thus a great deal of effort is being made into understanding the processes that occur during apoptosis and anti-apoptosis in order to develop strategies aimed at tapping into the natural anti-apoptotic network (Balakumar et al., 2008; Lehotsky et al., 2009; Lochner et al., 2009; Yacoubian and Standaert, 2009; Boll et al., 2011). This concept is in line with the fact that agents causing up-regulation of endogenous defense mechanisms by hormesis or by pre-conditioning are effective at preventing or decreasing cell death (Balakumar et al., 2008; Rami et al., 2008; Calabrese et al., 2011b; Martins et al., 2011; Porter et al., 2012). Thus, anti-apoptotic genes, which are part of the in vivo regulatory responses to counteract stress mediated cell death, are readily effective when overexpressed in vivo (via transgenics; Gil-Gómez and Brady, 1998; Yenari et al., 2005; Khan et al., 2006). Strategies are currently being developed to increase the levels of anti-apoptotic genes in tissues. Approaches include delivery of expressible gene constructs by gene therapy or the use of lipophilically modified recombinant proteins that could diffuse into apoptotically challenged cells (Dietz, 2011; Lavu et al., 2011). Such approaches could only serve to give rise to a short term increase in the levels of anti-apoptotic proteins, but this may be sufficient to prevent cell death in the short term after an ischemic/reperfusion event in tissues like the heart.
In addition to its long history in the food and biotechnology industries, the yeast S. cerevisiae may be the most widely used model organism (Botstein and Fink, 2011). As a eukaryotic cell system it has numerous advantages over other model cell types including the ability to use genetic approaches. In addition, its basic cellular processes, such as cell cycle control, DNA replication and repair, mitochondrial based respiration, and autophagy are carried out by simplified cellular process that are virtually interchangeable with the same but more complex processes of metazoan eukaryotes including humans (Perocchi et al., 2008). Thus it comes as no surprise that the description of a PCD in S. cerevisiae in 1997 has led to an explosion of research into the study of these processes in yeast (Madeo et al., 1997; Carmona-Gutierrez et al., 2010). Although the utility of a PCD process in unicellular eukaryotes was at first difficult to imagine, it is now widely accepted and has in fact been extensively documented to occur in a wide variety of different unicellular eukaryotes (Shemarova, 2010). Over the last few years, a large variety of studies have been instrumental in documenting many of the processes and in identifying many of the proteins involved in mediating yeast PCD (Frohlich et al., 2007; Carmona-Gutierrez et al., 2010). Although there are some differences, it would appear that yeast PCD is very similar to the mitochondrial or intrinsic apoptosis seen in metazoans (Eisenberg et al., 2007; Pereira et al., 2008). The complexity is decreased compared to mammalian apoptosis which is exemplified by the fact that most apoptotic regulators are encoded by multiple copies in humans but by single copy genes in yeast. So, yeast has single functional counterparts to many mammalian apoptotic regulators including a caspase (YCA1), a BH3 containing Bcl-2 family like protein (yBH3), an AIF (AIF), an OMI serine protease (NMA111), and an Endonuclease G (NUC1; Madeo et al., 2002; Fahrenkrog et al., 2004; Wissing et al., 2004; Buttner et al., 2007, 2011). As in mammalian cells, yeast will undergo apoptosis in response to a variety of different stresses including many chemicals, physical process such as altered osmolarity and pH, as well as the overexpression of genes encoding pro-apoptotic proteins including mammalian Bax and caspases (Manon et al., 1997; Lisa-Santamaria et al., 2009; Sharon et al., 2009; Carmona-Gutierrez et al., 2010; Orrenius et al., 2011). The intracellular events mediating yeast PCD also show a great deal of similarity with a number of other processes such as an increase in mitochondrial outer membrane permeability, increase in pro-apoptotic second messengers like ROS and ceramide, the release of apoptogenic factors including cytochrome c, DNA fragmentation as well as the externalization of phosphatidylserine (Manon et al., 1997; Ligr et al., 1998; Madeo et al., 1999; Yang et al., 2006; Ganesan et al., 2010; Carmona-Gutierrez et al., 2011b). In addition, naturally occurring physiological processes including ER stress and chronological aging will lead to mitochondrial and necrotic like cell death as it does in mammalian cells (Burhans et al., 2003; Fabrizio and Longo, 2008; Winderickx et al., 2008; Eisenberg et al., 2010; Madeo et al., 2010; Pan, 2011). On the other hand, ER mediated cell death in yeast is reminiscent of what occurs in a number of diseases that involves the accumulation of misfolded proteins such as Parkinson’s and Huntington’s (Winderickx et al., 2008; Khurana and Lindquist, 2010).
Of importance to our interests is the fact that regulated cell death, including PCD and necrosis, in yeast is also under negative regulation (Owsianowski et al., 2008; Carmona-Gutierrez et al., 2011a; Zdralevic et al., 2012). In fact many of the proteins that are known to prevent cell death in mammalian cells, such as free radical scavengers, chaperonin type proteins like HSPs, the yeast IAP (Inhibitor of Apoptosis Protein) protein (Bir1p) as well as numerous heterologous mammalian proteins such as Bcl-2 also prevent cell death (Manon et al., 1997; Moon et al., 2002; Flower et al., 2005; Walter et al., 2006). Yeast also has a well-defined stress activated autophagic process that has long been known to promote cellular longevity in response to stresses such as amino acid or glucose starvation and aging (Eisenberg et al., 2009; He and Klionsky, 2009). In spite of this, there have been very relatively few studies directed toward examining the possible role of autophagic cell death in yeast (Camougrand et al., 2003; Kissova et al., 2006; Thevissen et al., 2010; Sampaio-Marques et al., 2011).
Observed differences between yeast and mammalian cells are also of interest. For example, in a screen for yeast mutants showing increased resistance to the heterologous expression of the pro-apoptotic Bax, the UTH1 gene was identified (Camougrand et al., 2004). Although the exact function of Uth1p has yet to be defined, deletion of this gene leads to a number of interesting pleiotropic effects including increasing replicative lifespan, increasing resistance to the autophagic inducing drug rapamycin and ER inducing stresses (Kissova and Camougrand, 2010; Ritch et al., 2010). Surprisingly, a direct mammalian ortholog of UTH1 has yet to be found but it would be of interest to set up a suicide screen of UTH1Δ cells in order to identify potential functional homologs. Such screens have proven useful in the past for the identification and characterization of a number different human orthologs that can functionally complement phenotypes of a variety of different yeast mutants (Perier et al., 1994; Sato et al., 2006; Osborn and Miller, 2007). The study of yeast specific apoptotic regulators open up possibilities of increasing our understanding of the basic ancestral processes involved in regulating PCD. More recent developments including systems biology and other global approaches are being applied to understanding the processes of PCD in yeast (Munoz et al., 2012). For example a global search for yeast genes that are potential regulators of PCD was carried out by evaluating all yeast gene knockout strains for increased susceptibility to stress mediated cell death (Teng et al., 2011). Over 800 of the 6000 or so yeast genes were thus identified as potential pro-apoptotic genes. This suggests that many more genes are involved in the process of PCD than we know of now and these types of approaches in yeast may actually serve to identify all players in the process of PCD. Other such global strategies are now being combined with the power of yeast genetics to characterize other processes involved in PCD such as identifying the metabolic products (metabolome) that accumulate in different process associated with different forms of PCD (Ring et al., 2012).
Humanized yeast has been extensively used to study the structure and function of human genes in an uncluttered cellular environment (Osborn and Miller, 2007; Greenwood and Ludovico, 2010; Silva et al., 2011a). For example, all mammalian cells express a multitude of G-Protein Coupled Receptors (GPCRs) and studies focused on individual members of this large gene family are further encumbered by the fact that different members of the same family are capable of functionally interacting with each other (Panetta and Greenwood, 2008). Thus the ability to functionally replace the yeast sex pheromone GPCRs and their regulatory proteins with human orthologs allows the study of individual human genes uncluttered by the presence of other members (Kong et al., 2002; Ladds et al., 2005). Given the similarity between the processes of metazoan mitochondrially induced apoptosis and yeast apoptosis (Cheng et al., 2008) it is not surprising that humanized yeast have been successfully used in the pursuit of knowledge regarding the genes involved and the processes regulating PCD (Greenwood and Ludovico, 2010; Khurana and Lindquist, 2010; Silva et al., 2011a; Zdralevic et al., 2012).
Early studies by a number of yeast researchers lead to the discovery that mammalian regulators of apoptosis including various Bcl-2 family members including pro-apoptotic Bax and anti-apoptotic Bcl-2 could be functionally expressed in yeast (Greenhalf et al., 1996; Manon et al., 1997; Tao et al., 1997). Thus Bax mediated PCD in yeast was extensively studied and all indications suggested it induced apoptosis, at least in some cells, since it lead to apoptotic hallmark events similar to what was observed in mammalian cells (Priault et al., 2003; Khoury et al., 2008). Bcl-2 was also able to prevent Bax as well as other forms of stress mediated PCD in yeast. Some of these early studies had noticed that Bax expression not only lead to typical apoptotic phenotypes but it also lead to the activation of autophagy (Camougrand et al., 2003; Kissova et al., 2006). This is indicative of some form of cross-talk and is suggestive of some of the useful avenues of knowledge that are uncovered using humanized yeast in the study of PCD. Not surprisingly other pro-apoptotic human proteins such as caspases can trigger the same types of effects in yeast (Lisa-Santamaria et al., 2009). The functional expression of Bax and of other mammalian apoptotic regulators continues to serve to uncover insights into PCD (Greenwood and Ludovico, 2010). In the case of Bax, recent work has been useful in characterizing the roles of a variety of different proteins including protein kinase C and the mitochondrial receptor Tom22 in the functional activation of Bax (Renault et al., 2011; Silva et al., 2011b). Up until recently it was thought that the yeast genome did not code for any BH3 containing proteins, so the effects of heterologously expressed Bcl-2 proteins was thought to reflect intrinsic activities of these proteins or it was thought that yeast must contain a functional that is a not structural homolog of human Bax (Priault et al., 2003; Khoury et al., 2008). More recently, an endogenously encoded BH3 domain containing protein, called yBH3p, was identified as a pro-apoptotic protein in yeast (Buttner et al., 2011). Whether it has all the functions of mammalian Bax remains to be determined.
Bax mediated lethality in yeast was exploited in other ways. Notably, yeast cells conditionally expressing Bax, most often under a galactose inducible promoter, were used in numerous suicide screens (Figure 3; Liu et al., 1992; Sato et al., 2006; Osborn and Miller, 2007; Greenwood and Ludovico, 2010). Copy DNA (cDNA) libraries obtained from a range of different species including bacteria, plants, and different tissues were screened in order to identify sequences that permitted yeast to grow in the presence of Bax (Greenwood and Ludovico, 2010; Laloux et al., 2010). Many of the Bax suppressors identified corresponded to obvious anti-apoptotic sequences including numerous free radical scavenging proteins (Kampranis et al., 2000; Moon et al., 2002; Camougrand et al., 2004). This is consistent with the notion that Bax is indeed inducing a PCD that has similarities to apoptosis. Other proteins identified represent a challenge since they are clearly able to prevent PCD from a number of different stresses in yeast, but the mechanism by which they carry out this function remains unknown. This class of protein includes some with well-known functions such as Vacuolar Protein Sorting 24 (VPS24) and the small GTP binding protein Ran as well as proteins that are essentially functional orphans including TMEM85 and TMEM14 (Khoury et al., 2007; Ring et al., 2008; Woo et al., 2008, 2011). Other Bax suppressors identified include the dUTP hydrolyzing enzyme dUTPase and the ceramide utilizing enzyme sphingomyelin synthase (Yang et al., 2006; Williams et al., 2011). This suggests that stress mediated cell death in yeast, like observed in mammalian cells, involves an up-regulation of the levels of apoptosis inducing second messengers including ROS, ceramide, and dUTP (Portt et al., 2011; Wilson et al., 2012). Many of the screens have reported the identification of multiple Bax suppressors but the identity of many of these have yet to be reported. For example screening of a human T cell library lead to the identification or 24 Bax suppressors but the group has only reported a detailed analysis of four of these (Eun et al., 2008; Woo et al., 2008, 2009, 2011). Similarly, we have isolated over 60 different Bax suppressors from a screen of a human cardiac library and we have since published a detailed characterization of five of these (Yang et al., 2006; Khoury et al., 2007, 2008; Ring et al., 2008; Williams et al., 2011). Parallel or subsequent analysis of a number of these Bax suppressors reveal that they are bone-fide cell survival genes since they also function as anti-apoptotic genes in mammalian cells (Fiol et al., 2007; Separovic et al., 2007; Eun et al., 2008; Woo et al., 2008, 2009, 2011; Greenwood and Ludovico, 2010; Williams et al., 2011; Wilson et al., 2012). Taken together these screens suggests that there is a lot more that remains to be discovered on the anti-apoptotic and cell survival mechanisms used by a cell and that yeast is an ideal system for this task.
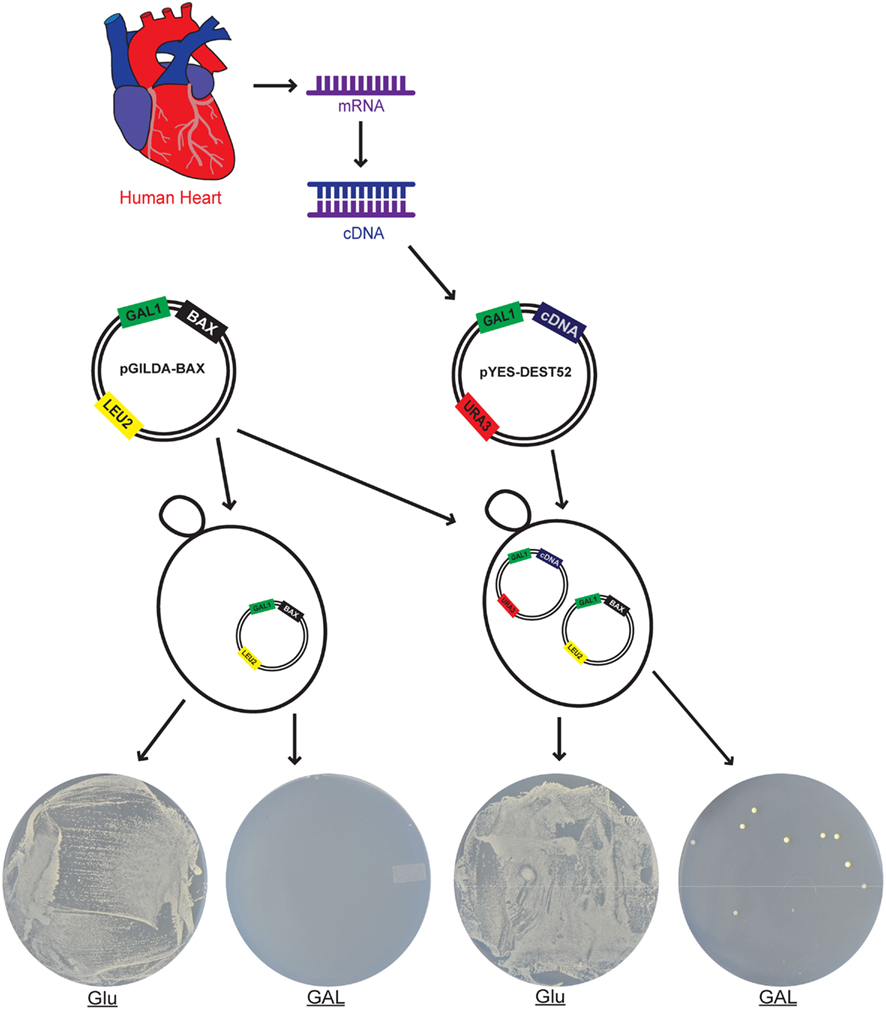
Figure 3. Schematic depiction of a yeast based functional strategy to screen for and identify cardiac specific sequences capable of preventing Bax mediated cell death. Poly A+ RNA isolated from human cardiac tissue was used to generate a cDNA library in the URA3 selectable marker yeast expression plasmid pYES-DEST52 (Sato et al., 2006). This places the expression of the human cardiac cDNAs under the control of the galactose inducible GAL1 promoter. Plasmid pGILDA-Bax was used to express an activated mouse Bax in yeast under the control of galactose inducible GAL1 promoter. Yeast cells harboring pGILDA-Bax alone show normal growth when grown on glucose media but undergo cell death due to the expression of Bax when grown on galactose media. Close to 106 yeast transformants harboring both pGILDA-Bax as well as different cDNAs from the pYES-DEST52 cardiac cDNA library were grown on galactose media. A total of 72 different colonies containing over 60 different putative Bax suppressors were identified when the pYES-DEST52 cardiac cDNAs were isolated and their nucleotide sequence determined (Yang et al., 2006). Similar apoptotic resistant phenotypes can be achieved in the heart by subjecting the tissue to a period of mild stress in order to induce pre-conditioning. Glu, glucose; GAL, galactose.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Space prevented us from citing the work from all the many laboratories that have made significant contributions to the field of anti-apoptosis and cell survival. Work in MTG’s laboratory is supported by NSERC, the ARP program at the Royal Military College, and a joint grant with CAM from the Heart and Stroke Foundation of Canada.
Abraham, R., Schafer, J., Rothe, M., Bange, J., Knyazev, P., and Ullrich, A. (2005). Identification of MMP-15 as an anti-apoptotic factor in cancer cells. J. Biol. Chem. 280, 34123–34132.
Amelio, I., Melino, G., and Knight, R. A. (2011). Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem. Biophys. Res. Commun. 414, 277–281.
Ashida, H., Mimuro, H., Ogawa, M., Kobayashi, T., Sanada, T., Kim, M., and Sasakawa, C. (2011). Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942.
Balakumar, P., Rohilla, A., and Singh, M. (2008). Pre-conditioning and postconditioning to limit ischemia-reperfusion-induced myocardial injury: what could be the next footstep? Pharmacol. Res. 57, 403–412.
Beere, H. M. (2004). “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 117, 2641–2651.
Belenky, P., and Collins, J. J. (2011). Microbiology. Antioxidant strategies to tolerate antibiotics. Science 334, 915–916.
Berghe, T. V., Vanlangenakker, N., Parthoens, E., Deckers, W., Devos, M., Festjens, N., Guerin, C. J., Brunk, U. T., Declercq, W., and Vandenabeele, P. (2010). Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17, 922–930.
Berry, D. B., and Gasch, A. P. (2008). Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol. Biol. Cell 19, 4580–4587.
Boll, M. C., Alcaraz-Zubeldia, M., and Rios, C. (2011). Medical management of Parkinson’s disease: focus on neuroprotection. Curr. Neuropharmacol. 9, 350–359.
Botstein, D., and Fink, G. R. (2011). Yeast: an experimental organism for 21st Century biology. Genetics 189, 695–704.
Burhans, W. C., Weinberger, M., Marchetti, M. A., Ramachandran, L., D’Urso, G., and Huberman, J. A. (2003). Apoptosis-like yeast cell death in response to DNA damage and replication defects. Mutat. Res. 532, 227–243.
Busca, A., Saxena, M., Kryworuchko, M., and Kumar, A. (2009). Anti-apoptotic genes in the survival of monocytic cells during infection. Curr. Genomics 10, 306–317.
Buttner, S., Eisenberg, T., Carmona-Gutierrez, D., Ruli, D., Knauer, H., Ruckenstuhl, C., Sigrist, C., Wissing, S., Kollroser, M., Frohlich, K. U., Sigrist, S., and Madeo, F. (2007). Endonuclease G regulates budding yeast life and death. Mol. Cell 25, 233–246.
Buttner, S., Eisenberg, T., Herker, E., Carmona-Gutierrez, D., Kroemer, G., and Madeo, F. (2006). Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J. Cell Biol. 175, 521–525.
Buttner, S., Ruli, D., Vogtle, F. N., Galluzzi, L., Moitzi, B., Eisenberg, T., Kepp, O., Habernig, L., Carmona-Gutierrez, D., Rockenfeller, P., Laun, P., Breitenbach, M., Khoury, C., Frohlich, K. U., Rechberger, G., Meisinger, C., Kroemer, G., and Madeo, F. (2011). A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J. 30, 2779–2792.
Calabrese, V., Cornelius, C., Cuzzocrea, S., Iavicoli, I., Rizzarelli, E., and Calabrese, E. J. (2011a). Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Aspects Med. 32, 279–304.
Calabrese, V., Cornelius, C., Dinkova-Kostova, A. T., Calabrese, E. J., and Mattson, M. P. (2011b). Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 13, 1763–1811.
Camougrand, N., Grelaud-Coq, A., Marza, E., Priault, M., Bessoule, J. J., and Manon, S. (2003). The product of the UTH1 gene, required for Bax-induced cell death in yeast, is involved in the response to rapamycin. Mol. Microbiol. 47, 495–506.
Camougrand, N., Kissova, I., Velours, G., and Manon, S. (2004). Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res. 5, 133–140.
Carmona-Gutierrez, D., Bauer, M. A., Ring, J., Knauer, H., Eisenberg, T., Buttner, S., Ruckenstuhl, C., Reisenbichler, A., Magnes, C., Rechberger, G. N., Birner-Gruenberger, R., Jungwirth, H., Frohlich, K. U., Sinner, F., Kroemer, G., and Madeo, F. (2011a). The propeptide of yeast cathepsin D inhibits programmed necrosis. Cell Death Dis. 2, e161.
Carmona-Gutierrez, D., Reisenbichler, A., Heimbucher, P., Bauer, M. A., Braun, R. J., Ruckenstuhl, C., Buttner, S., Eisenberg, T., Rockenfeller, P., Frohlich, K. U., Kroemer, G., and Madeo, F. (2011b). Ceramide triggers metacaspase-independent mitochondrial cell death in yeast. Cell Cycle 10, 3973–3978.
Carmona-Gutierrez, D., Eisenberg, T., Buttner, S., Meisinger, C., Kroemer, G., and Madeo, F. (2010). Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 17, 763–773.
Carmona-Gutierrez, D., and Madeo, F. (2009). “Tracing the roots of death: apoptosis in Saccharomyces cerevisiae,” in Essentials of Apoptosis, 2nd Edn, eds Z. Dong and X. M. Yin (Dusseldorf: Springer), 325–354.
Chen, Y., and Klionsky, D. J. (2011). The regulation of autophagy – unanswered questions. J. Cell Sci. 124, 161–170.
Cheng, W. C., Leach, K. M., and Hardwick, J. M. (2008). Mitochondrial death pathways in yeast and mammalian cells. Biochim. Biophys. Acta 1783, 1272–1279.
Chipuk, J. E., McStay, G. P., Bharti, A., Kuwana, T., Clarke, C. J., Siskind, L. J., Obeid, L.M., and Green, D. R. (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000.
Christofferson, D. E., and Yuan, J. (2010). Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 22, 263–268.
Circu, M. L., and Aw, T. Y. (2010). Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 48, 749–762.
Coles, J. G., Boscarino, C., Takahashi, M., Grant, D., Chang, A., Ritter, J., Dai, X., Du, C., Musso, G., Yamabi, H., Goncalves, J., Kumar, A. S., Woodgett, J., Lu, H., and Hannigan, G. (2005). Cardioprotective stress response in the human fetal heart. J. Thorac. Cardiovasc. Surg. 129, 1128–1136.
Davies, J. M., Lowry, C. V., and Davies, K. J. (1995). Transient adaptation to oxidative stress in yeast. Arch. Biochem. Biophys. 317, 1–6.
Dengjel, J., Hoyer-Hansen, M., Nielsen, M. O., Eisenberg, T., Harder, L. M., Schandorff, S., Farkas, T., Kirkegaard, T., Becker, A. C., Schroeder, S., Vanselow, K., Lundberg, E., Nielsen, M. M., Kristensen, A. R., Akimov, V., Bunkenborg, J., Madeo, F., Jaattela, M., and Andersen, J. S. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell. Proteomics 11, M111 014035.
Denton, D., Nicolson, S., and Kumar, S. (2012). Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 19, 87–95.
Dietz, G. P. (2011). Protection by neuroglobin and cell-penetrating peptide-mediated delivery in vivo: a decade of research. Comment on Cai et al: TAT-mediated delivery of neuroglobin protects against focal cerebral ischemia in mice. Exp. Neurol. 227, 224–231. [Exp. Neurol. 231, 1–10].
Djavaheri-Mergny, M., Maiuri, M. C., and Kroemer, G. (2010). Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29, 1717–1719.
Eisenberg, T., Buttner, S., Kroemer, G., and Madeo, F. (2007). The mitochondrial pathway in yeast apoptosis. Apoptosis 12, 1011–1023.
Eisenberg, T., Carmona-Gutierrez, D., Buttner, S., Tavernarakis, N., and Madeo, F. (2010). Necrosis in yeast. Apoptosis 15, 257–268.
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., Ring, J., Schroeder, S., Magnes, C., Antonacci, L., Fussi, H., Deszcz, L., Hartl, R., Schraml, E., Criollo, A., Megalou, E., Weiskopf, D., Laun, P., Heeren, G., Breitenbach, M., Grubeck-Loebenstein, B., Herker, E., Fahrenkrog, B., Frohlich, K. U., Sinner, F., Tavernarakis, N., Minois, N., Kroemer, G., and Madeo, F. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314.
Eun, S. Y., Woo, I. S., Jang, H. S., Jin, H., Kim, M. Y., Kim, H. J., Lee, J. H., Chang, K. C., Kim, J. H., and Seo, H. G. (2008). Identification of cytochrome c oxidase subunit 6A1 as a suppressor of Bax-induced cell death by yeast-based functional screening. Biochem. Biophys. Res. Commun. 373, 58–63.
Fabrizio, P., and Longo, V. D. (2008). Chronological aging-induced apoptosis in yeast. Biochim. Biophys. Acta 1783, 1280–1285.
Fahrenkrog, B., Sauder, U., and Aebi, U. (2004). The S. cerevisiae HtrA-like protein Nma111p is a nuclear serine protease that mediates yeast apoptosis. J. Cell Sci. 117, 115–126.
Fiol, D. F., Mak, S. K., and Kultz, D. (2007). Specific TSC22 domain transcripts are hypertonically induced and alternatively spliced to protect mouse kidney cells during osmotic stress. FEBS J. 274, 109–124.
Fleming, A., Noda, T., Yoshimori, T., and Rubinsztein, D. C. (2011). Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 7, 9–17.
Flower, T. R., Chesnokova, L. S., Froelich, C. A., Dixon, C., and Witt, S. N. (2005). Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J. Mol. Biol. 351, 1081–1100.
Franssens, V., Boelen, E., Anandhakumar, J., Vanhelmont, T., Buttner, S., and Winderickx, J. (2010). Yeast unfolds the road map toward alpha-synuclein-induced cell death. Cell Death Differ. 17, 746–753.
Frohlich, K. U., Fussi, H., and Ruckenstuhl, C. (2007). Yeast apoptosis – from genes to pathways. Semin. Cancer Biol. 17, 112–121.
Fulda, S. (2009b). Exploiting apoptosis pathways for the treatment of pediatric cancers. Pediatr. Blood Cancer 53, 533–536.
Fulda, S. (2010). Evasion of apoptosis as a cellular stress response in cancer. Int. J. Cell Biol. 2010, 370835.
Fulda, S., Gorman, A. M., Hori, O., and Samali, A. (2010). Cellular stress responses: cell survival and cell death. Int. J. Cell Biol. 2010, 214074.
Galluzzi, L., Kepp, O., Morselli, E., Vitale, I., Senovilla, L., Pinti, M., Zitvogel, L., and Kroemer, G. (2010a). Viral strategies for the evasion of immunogenic cell death. J. Intern. Med. 267, 526–542.
Galluzzi, L., Morselli, E., Kepp, O., Vitale, I., Rigoni, A., Vacchelli, E., Michaud, M., Zischka, H., Castedo, M., and Kroemer, G. (2010b). Mitochondrial gateways to cancer. Mol. Aspects Med. 31, 1–20.
Galluzzi, L., Morselli, E., Kepp, O., Vitale, I., Younes, A. B., Maiuri, M. C., and Kroemer, G. (2012a). Evaluation of rapamycin-induced cell death. Methods Mol. Biol. 821, 125–169.
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., Dawson, T. M., Dawson, V. L., El-Deiry, W. S., Fulda, S., Gottlieb, E., Green, D. R., Hengartner, M. O., Kepp, O., Knight, R. A., Kumar, S., Lipton, S. A., Lu, X., Madeo, F., Malorni, W., Mehlen, P., Nunez, G., Peter, M. E., Piacentini, M., Rubinsztein, D. C., Shi, Y., Simon, H. U., Vandenabeele, P., White, E., Yuan, J., Zhivotovsky, B., Melino, G., and Kroemer, G. (2012b). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on cell death 2012. Cell Death Differ. 19, 107–120.
Ganesan, V., and Colombini, M. (2010). Regulation of ceramide channels by Bcl-2 family proteins. FEBS Lett. 584, 2128–2134.
Ganesan, V., Perera, M. N., Colombini, D., Datskovskiy, D., Chadha, K., and Colombini, M. (2010). Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 15, 553–562.
Garrido, C., Schmitt, E., Cande, C., Vahsen, N., Parcellier, A., and Kroemer, G. (2003). HSP27 and HSP70: potentially oncogenic apoptosis inhibitors. Cell Cycle 2, 579–584.
Giansanti, V., Torriglia, A., and Scovassi, A. I. (2011). Conversation between apoptosis and autophagy: “Is it your turn or mine?” Apoptosis 16, 321–333.
Gil-Gómez, G., and Brady, H. J. (1998). Transgenic mice in apoptosis research. Apoptosis 3, 215–228.
Gill, S. S., and Tuteja, N. (2011). Polyamines and abiotic stress tolerance in plants. Plant Signal. Behav. 5, 26–33.
Gottlieb, R. A., Finley, K. D., and Mentzer, R. M. Jr. (2009). Cardioprotection requires taking out the trash. Basic Res. Cardiol. 104, 169–180.
Green, D. R., Oberst, A., Dillon, C. P., Weinlich, R., and Salvesen, G. S. (2011). RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol. Cell 44, 9–16.
Greenhalf, W., Stephan, C., and Chaudhuri, B. (1996). Role of mitochondria and C-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in Saccharomyces cerevisiae. FEBS Lett. 380, 169–175.
Greenwood, M. T., and Ludovico, P. (2010). Expressing and functional analysis of mammalian apoptotic regulators in yeast. Cell Death Differ. 17, 737–745.
Guaragnella, N., Antonacci, L., Giannattasio, S., Marra, E., and Passarella, S. (2008). Catalase T and Cu,Zn-superoxide dismutase in the acetic acid-induced programmed cell death in Saccharomyces cerevisiae. FEBS Lett. 582, 210–214.
Gurusamy, N., Lekli, I., Gherghiceanu, M., Popescu, L. M., and Das, D. K. (2009). BAG-1 induces autophagy for cardiac cell survival. Autophagy 5, 120–121.
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300.
Hausenloy, D. J., Tsang, A., Mocanu, M. M., and Yellon, D. M. (2005). Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 288, H971–H976.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
Heo, J. M., Livnat-Levanon, N., Taylor, E. B., Jones, K. T., Dephoure, N., Ring, J., Xie, J., Brodsky, J. L., Madeo, F., Gygi, S. P., Ashrafi, K., Glickman, M. H., and Rutter, J. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480.
Hotchkiss, R. S., Strasser, A., McDunn, J. E., and Swanson, P. E. (2009). Cell death. N. Engl. J. Med. 361, 1570–1583.
Igarashi, K., and Kashiwagi, K. (2010). Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 42, 39–51.
Insel, P. A., Zhang, L., Murray, F., Yokouchi, H., and Zambon, A. C. (2011). Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol (Oxf.) 204, 277–287.
Kaczanowski, S., Sajid, M., and Reece, S. E. (2011). Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasit. Vectors 4, 44.
Kampranis, S. C., Damianova, R., Atallah, M., Toby, G., Kondi, G., Tsichlis, P. N., and Makris, A. M. (2000). A novel plant glutathione S-transferase/peroxidase suppresses Bax lethality in yeast. J. Biol. Chem. 275, 29207–29216.
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580.
Khan, A. A., Wang, Y., Sun, Y., Mao, X. O., Xie, L., Miles, E., Graboski, J., Chen, S., Ellerby, L. M., Jin, K., and Greenberg, D. A. (2006). Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc. Natl. Acad. Sci. U.S.A. 103, 17944–17948.
Kharade, S. V., Mittal, N., Das, S. P., Sinha, P., and Roy, N. (2005). Mrg19 depletion increases S. cerevisiae lifespan by augmenting ROS defence. FEBS Lett. 579, 6809–6813.
Khoury, C. M., and Greenwood, M. T. (2008). The pleiotropic effects of heterologous Bax expression in yeast. Biochim. Biophys. Acta 1783, 1449–1465.
Khoury, C. M., Yang, Z., Ismail, S., and Greenwood, M. T. (2007). Characterization of a novel alternatively spliced human transcript encoding an N-terminally truncated Vps24 protein that suppresses the effects of Bax in an ESCRT independent manner in yeast. Gene 391, 233–241.
Khoury, C. M., Yang, Z., Li, X. Y., Vignali, M., Fields, S., and Greenwood, M. T. (2008). A TSC22-like motif defines a novel antiapoptotic protein family. FEMS Yeast Res. 8, 540–563.
Khurana, V., and Lindquist, S. (2010). Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker’s yeast? Nat. Rev. Neurosci. 11, 436–449.
Kim, H. J., So, H. S., Lee, J. H., Park, C., Park, S. Y., Kim, Y. H., Youn, M. J., Kim, S. J., Chung, S. Y., Lee, K. M., and Park, R. (2006). Heme oxygenase-1 attenuates the cisplatin-induced apoptosis of auditory cells via down-regulation of reactive oxygen species generation. Free Radic. Biol. Med. 40, 1810–1819.
Kissova, I., Plamondon, L. T., Brisson, L., Priault, M., Renouf, V., Schaeffer, J., Camougrand, N., and Manon, S. (2006). Evaluation of the roles of apoptosis, autophagy and mitophagy in the loss of plating efficiency induced by Bax-expression in yeast. J. Biol. Chem. 281, 36187–36197.
Kissova, I. S., and Camougrand, N. (2010). Mitophagy in yeast: actors and physiological roles. FEMS Yeast Res. 10, 1023–1034.
Kong, J. L., Panetta, R., Song, W., Somerville, W., and Greenwood, M. T. (2002). Inhibition of somatostatin receptor 5-signaling by mammalian Regulators of G-protein Signaling (RGS) in yeast. Biochim. Biophys. Acta 1542, 95–105.
Krakstad, C., and Chekenya, M. (2010). Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol. Cancer 9, 135.
Krampe, B., and Al-Rubeai, M. (2010). Cell death in mammalian cell culture: molecular mechanisms and cell line engineering strategies. Cytotechnology 62, 175–188.
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., El-Deiry, W. S., Golstein, P., Green, D. R., Hengartner, M., Knight, R. A., Kumar, S., Lipton, S. A., Malorni, W., Nunez, G., Peter, M. E., Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky, B., and Melino, G. (2009). Classification of cell death: recommendations of the Nomenclature Committee on cell death 2009. Cell Death Differ. 16, 3–11.
Kumar, D., and Jugdutt, B. I. (2003). Apoptosis and oxidants in the heart. J. Lab. Clin. Med. 142, 288–297.
Kumar, D., Lou, H., and Singal, P. K. (2002). Oxidative stress and apoptosis in heart dysfunction. Herz 27, 662–668.
Ladds, G., Goddard, A., and Davey, J. (2005). Functional analysis of heterologous GPCR signalling pathways in yeast. Trends Biotechnol. 23, 367–373.
Laloux, G., Deghelt, M., de Barsy, M., Letesson, J. J., and De Bolle, X. (2010). Identification of the essential Brucella melitensis porin Omp2b as a suppressor of Bax-induced cell death in yeast in a genome-wide screening. PLoS ONE 5, e13274. doi:10.1371/journal.pone.0013274
Laun, P., Heeren, G., Rinnerthaler, M., Rid, R., Kossler, S., Koller, L., and Breitenbach, M. (2008). Senescence and apoptosis in yeast mother cell-specific aging and in higher cells: a short review. Biochim. Biophys. Acta 1783, 1328–1334.
Lavu, M., Gundewar, S., and Lefer, D. J. (2011). Gene therapy for ischemic heart disease. J. Mol. Cell. Cardiol. 50, 742–750.
Lecour, S., Owira, P., and Opie, L. H. (2005). Ceramide-induced preconditioning involves reactive oxygen species. Life Sci. 78, 1702–1706.
Lecour, S., Van der Merwe, E., Opie, L. H., and Sack, M. N. (2006). Ceramide attenuates hypoxic cell death via reactive oxygen species signaling. J. Cardiovasc. Pharmacol. 47, 158–163.
Lee, J. S., Li, Q., Lee, J. Y., Lee, S. H., Jeong, J. H., Lee, H. R., Chang, H., Zhou, F. C., Gao, S. J., Liang, C., and Jung, J. U. (2009). FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362.
Lehotsky, J., Burda, J., Danielisova, V., Gottlieb, M., Kaplan, P., and Saniova, B. (2009). Ischemic tolerance: the mechanisms of neuroprotective strategy. Anat. Rec. (Hoboken) 292, 2002–2012.
Li, Z. Y., Yang, Y., Ming, M., and Liu, B. (2011). Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 414, 5–8.
Ligr, M., Madeo, F., Frohlich, E., Hilt, W., Frohlich, K. U., and Wolf, D. H. (1998). Mammalian Bax triggers apoptotic changes in yeast. FEBS Lett. 438, 61–65.
Lisa-Santamaria, P., Neiman, A. M., Cuesta-Marban, A., Mollinedo, F., Revuelta, J. L., and Jimenez, A. (2009). Human initiator caspases trigger apoptotic and autophagic phenotypes in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1793, 561–571.
Liu, H., Krizek, J., and Bretscher, A. (1992). Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics 132, 665–673.
Liu, J. J., Lin, M., Yu, J. Y., Liu, B., and Bao, J. K. (2011). Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett. 300, 105–114.
Lochner, A., Marais, E., Genade, S., Huisamen, B., du Toit, E. F., and Moolman, J. A. (2009). Protection of the ischaemic heart: investigations into the phenomenon of ischaemic preconditioning. Cardiovasc. J. Afr. 20, 43–51.
Loewith, R., and Hall, M. N. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177–1201.
Madeo, F., Eisenberg, T., Buttner, S., Ruckenstuhl, C., and Kroemer, G. (2010). Spermidine: a novel autophagy inducer and longevity elixir. Autophagy 6, 160–162.
Madeo, F., Frohlich, E., and Frohlich, K. U. (1997). A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 139, 729–734.
Madeo, F., Frohlich, E., Ligr, M., Grey, M., Sigrist, S. J., Wolf, D. H., and Frohlich, K. U. (1999). Oxygen stress: a regulator of apoptosis in yeast. J. Cell Biol. 145, 757–767.
Madeo, F., Herker, E., Maldener, C., Wissing, S., Lachelt, S., Herlan, M., Fehr, M., Lauber, K., Sigrist, S. J., Wesselborg, S., and Frohlich, K. U. (2002). A caspase-related protease regulates apoptosis in yeast. Mol. Cell 9, 911–917.
Manon, S., Chaudhuri, B., and Guerin, M. (1997). Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 415, 29–32.
Marino, G., Madeo, F., and Kroemer, G. (2011). Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 23, 198–206.
Martins, I., Galluzzi, L., and Kroemer, G. (2011). Hormesis, cell death and aging. Aging (Albany N.Y.) 3, 821–828.
Meschini, S., Condello, M., Lista, P., and Arancia, G. (2011). Autophagy: molecular mechanisms and their implications for anticancer therapies. Curr Cancer Drug Targets 11, 357–379.
Mesquita, A., Weinberger, M., Silva, A., Sampaio-Marques, B., Almeida, B., Leao, C., Costa, V., Rodrigues, F., Burhans, W. C., and Ludovico, P. (2010). Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. U.S.A. 107, 15123–15128.
Minois, N., Carmona-Gutierrez, D., and Madeo, F. (2011). Polyamines in aging and disease. Aging (Albany N.Y.) 3, 716–732.
Misra, M. K., Sarwat, M., Bhakuni, P., Tuteja, R., and Tuteja, N. (2009). Oxidative stress and ischemic myocardial syndromes. Med. Sci. Monit. 15, RA209–RA219.
Mokdad-Gargouri, R., Abdelmoula-Soussi, S., Hadiji-Abbès, N., Amor, I. Y., Borchani-Chabchoub, I., and Gargouri, A. (2012). Yeasts as a tool for heterologous gene expression. Methods Mol. Biol. 824, 359–370.
Moon, H., Baek, D., Lee, B., Prasad, D. T., Lee, S. Y., Cho, M. J., Lim, C. O., Choi, M. S., Bahk, J., Kim, M. O., Hong, J. C., and Yun, D. J. (2002). Soybean ascorbate peroxidase suppresses Bax-induced apoptosis in yeast by inhibiting oxygen radical generation. Biochem. Biophys. Res. Commun. 290, 457–462.
Moreau, K., Luo, S., and Rubinsztein, D. C. (2009). Cytoprotective roles for autophagy. Curr. Opin. Cell Biol. 22, 206–211.
Munoz, A. J., Wanichthanarak, K., Meza, E., and Petranovic, D. (2012). Systems biology of yeast cell death. FEMS Yeast Res. 12, 249–265.
Nagley, P., Higgins, G. C., Atkin, J. D., and Beart, P. M. (2010). Multifaceted deaths orchestrated by mitochondria in neurones. Biochim. Biophys. Acta 1802, 167–185.
Nakka, V. P., Gusain, A., Mehta, S. L., and Raghubir, R. (2008). Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol. Neurobiol. 37, 7–38.
Oerlemans, M. I., Koudstaal, S., Chamuleau, S. A., de Kleijn, D. P., Doevendans, P. A., and Sluijter, J. P. (2012). Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int. J. Cardiol. doi:10.1016/j.ijcard.2012.03.055
Ong, S. B., and Gustafsson, A. B. (2012). New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 94, 190–196.
Orrenius, S., Nicotera, P., and Zhivotovsky, B. (2011). Cell death mechanisms and their implications in toxicology. Toxicol. Sci. 119, 3–19.
Osborn, M. J., and Miller, J. R. (2007). Rescuing yeast mutants with human genes. Brief. Funct. Genomic. Proteomic. 6, 104–111.
Owsianowski, E., Walter, D., and Fahrenkrog, B. (2008). Negative regulation of apoptosis in yeast. Biochim. Biophys. Acta 1783, 1303–1310.
Ozbayraktar, F. B., and Ulgen, K. O. (2009). Molecular facets of sphingolipids: mediators of diseases. Biotechnol. J. 4, 1028–1041.
Pan, T., Rawal, P., Wu, Y., Xie, W., Jankovic, J., and Le, W. (2009). Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 164, 541–551.
Pan, Y. (2011). Mitochondria, reactive oxygen species, and chronological aging: a message from yeast. Exp. Gerontol. 46, 847–852.
Panetta, R., and Greenwood, M. T. (2008). Physiological relevance of GPCR oligomerization and its impact on drug discovery. Drug Discov. Today 13, 1059–1066.
Pereira, C., Silva, R. D., Saraiva, L., Johansson, B., Sousa, M. J., and Corte-Real, M. (2008). Mitochondria-dependent apoptosis in yeast. Biochim. Biophys. Acta 1783, 1286–1302.
Perier, F., Coulter, K. L., Liang, H., Radeke, C. M., Gaber, R. F., and Vandenberg, C. A. (1994). Identification of a novel mammalian member of the NSF/CDC48p/Pas1p/TBP-1 family through heterologous expression in yeast. FEBS Lett. 351, 286–290.
Periyasamy-Thandavan, S., Jiang, M., Schoenlein, P., and Dong, Z. (2009). Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Renal Physiol. 297, F244–F256.
Perocchi, F., Mancera, E., and Steinmetz, L. M. (2008). Systematic screens for human disease genes, from yeast to human and back. Mol. Biosyst. 4, 18–19.
Porter, K., Medford, H. M., McIntosh, C. M., and Marsh, S. A. (2012). Cardioprotection requires flipping the ‘posttranslational modification’ switch. Life Sci. 90, 89–98.
Portt, L., Norman, G., Clapp, C., Greenwood, M., and Greenwood, M. T. (2011). Anti-apoptosis and cell survival: a review. Biochim. Biophys. Acta 1813, 238–259.
Pourova, J., Kottova, M., Voprsalova, M., and Pour, M. (2010). Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiol. (Oxf) 198, 15–35.
Priault, M., Camougrand, N., Kinnally, K. W., Vallette, F. M., and Manon, S. (2003). Yeast as a tool to study Bax/mitochondrial interactions in cell death. FEMS Yeast Res. 4, 15–27.
Qi, H., Han, Y., and Rong, J. (2012). Potential roles of PI3K/Akt and Nrf2-Keap1 pathways in regulating hormesis of Z-ligustilide in PC12 cells against oxygen and glucose deprivation. Neuropharmacology 62, 1659–1670.
Qian, J., Ren, X., Wang, X., Zhang, P., Jones, W. K., Molkentin, J. D., Fan, G. C., and Kranias, E. G. (2009). Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 105, 1223–1231.
Rami, A. (2009). Review: autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol. Appl. Neurobiol. 35, 449–461.
Rami, A., Bechmann, I., and Stehle, J. H. (2008). Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol. 85, 273–296.
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990.
Renault, T. T., Grandier-Vazeille, X., Arokium, H., Velours, G., Camougrand, N., Priault, M., Teijido, O., Dejean, L. M., and Manon, S. (2011). The cytosolic domain of human Tom22 modulates human Bax mitochondrial translocation and conformation in yeast. FEBS Lett. 586, 116–121.
Ring, G., Khoury, C. M., Solar, A. J., Yang, Z., Mandato, C. A., and Greenwood, M. T. (2008). Transmembrane protein 85 from both human (TMEM85) and yeast (YGL231c) inhibit hydrogen peroxide mediated cell death in yeast. FEBS Lett. 582, 2637–2642.
Ring, J., Sommer, C., Carmona-Gutierrez, D., Ruckenstuhl, C., Eisenberg, T., and Madeo, F. (2012). The metabolism beyond programmed cell death in yeast. Exp. Cell Res. 318, 1193–1200.
Ristow, M., and Schmeisser, S. (2011). Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 51, 327–336.
Ristow, M., and Zarse, K. (2011). How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 45, 410–418.
Ritch, J. J., Davidson, S. M., Sheehan, J. J., and Austriaco, N. (2010). The Saccharomyces SUN gene, UTH1, is involved in cell wall biogenesis. FEMS Yeast Res. 10, 168–176.
Rodrigues, V., Cordeiro-da-Silva, A., Laforge, M., Ouaissi, A., Silvestre, R., and Estaquier, J. (2012). Modulation of mammalian apoptotic pathways by intracellular protozoan parasites. Cell. Microbiol. 14, 325–333.
Sakamoto, S., and Kyprianou, N. (2010). Targeting anoikis resistance in prostate cancer metastasis. Mol. Aspects Med. 31, 205–214.
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, F., and Ludovico, P. (2011). Yeast chronological lifespan and proteotoxic stress: is autophagy good or bad? Biochem. Soc. Trans. 39, 1466–1470.
Sato, M., Cismowski, M. J., Toyota, E., Smrcka, A. V., Lucchesi, P. A., Chilian, W. M., and Lanier, S. M. (2006). Identification of a receptor-independent activator of G protein signaling (AGS8) in ischemic heart and its interaction with Gbetagamma. Proc. Natl. Acad. Sci. U.S.A. 103, 797–802.
Schleicher, S. M., Moretti, L., Varki, V., and Lu, B. (2010). Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist. Updat. 13, 79–86.
Schonthal, A. H. (2009). Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett. 275, 163–169.
Separovic, D., Hanada, K., Maitah, M. Y., Nagy, B., Hang, I., Tainsky, M. A., Kraniak, J. M., and Bielawski, J. (2007). Sphingomyelin synthase 1 suppresses ceramide production and apoptosis post-photodamage. Biochem. Biophys. Res. Commun. 358, 196–202.
Separovic, D., Semaan, L., Tarca, A. L., Awad Maitah, M. Y., Hanada, K., Bielawski, J., Villani, M., and Luberto, C. (2008). Suppression of sphingomyelin synthase 1 by small interference RNA is associated with enhanced ceramide production and apoptosis after photodamage. Exp. Cell Res. 314, 1860–1868.
Shamas-Din, A., Brahmbhatt, H., Leber, B., and Andrews, D. W. (2011). BH3-only proteins: orchestrators of apoptosis. Biochim. Biophys. Acta 1813, 508–520.
Sharon, A., Finkelstein, A., Shlezinger, N., and Hatam, I. (2009). Fungal apoptosis: function, genes and gene function. FEMS Microbiol. Rev. 33, 833–854.
Shemarova, I. V. (2010). Signaling mechanisms of apoptosis-like programmed cell death in unicellular eukaryotes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 155, 341–353.
Shen, H. M., and Codogno, P. (2012). Autophagy is a survival force via suppression of necrotic cell death. Exp. Cell Res. Available at: http://dx.doi.org/10.1016/j.yexcr.2012.02.006
Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228.
Shlezinger, N., Minz, A., Gur, Y., Hatam, I., Dagdas, Y. F., Talbot, N. J., and Sharon, A. (2011). Anti-apoptotic machinery protects the necrotrophic fungus Botrytis cinerea from host-induced apoptotic-like cell death during plant infection. PLoS Pathog. 7, e1002185. doi:10.1371/journal.ppat.1002185
Silva, R. D., Manon, S., Goncalves, J., Saraiva, L., and Corte-Real, M. (2011a). The importance of humanized yeast to better understand the role of bcl-2 family in apoptosis: finding of novel therapeutic opportunities. Curr. Pharm. Des. 17, 246–255.
Silva, R. D., Manon, S., Goncalves, J., Saraiva, L., and Corte-Real, M. (2011b). Modulation of Bax mitochondrial insertion and induced cell death in yeast by mammalian protein kinase C alpha. Exp. Cell Res. 317, 781–790.
Strasser, A., Cory, S., and Adams, J. M. (2011). Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 30, 3667–3683.
Tao, W., Kurschner, C., and Morgan, J. I. (1997). Modulation of cell death in yeast by the Bcl-2 family of proteins. J. Biol. Chem. 272, 15547–15552.
Tarnavski, O., McMullen, J. R., Schinke, M., Nie, Q., Kong, S., and Izumo, S. (2004). Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol. Genomics 16, 349–360.
Taylor, E. B., and Rutter, J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513.
Teng, X., Cheng, W. C., Qi, B., Yu, T. X., Ramachandran, K., Boersma, M. D., Hattier, T., Lehmann, P. V., Pineda, F. J., and Hardwick, J. M. (2011). Gene-dependent cell death in yeast. Cell Death Dis. 2, e188.
Thevissen, K., Yen, W. L., Carmona-Gutierrez, D., Idkowiak-Baldys, J., Aerts, A. M., Francois, I. E., Madeo, F., Klionsky, D. J., Hannun, Y. A., and Cammue, B. P. (2010). Skn1 and Ipt1 negatively regulate autophagy in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 303, 163–168.
Ulukaya, E., Acilan, C., and Yilmaz, Y. (2011). Apoptosis: why and how does it occur in biology? Cell Biochem. Funct. 29, 468–480.
Vaux, D. L., Cory, S., and Adams, J. M. (1988). Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442.
Vertessy, B. G., and Toth, J. (2009). Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42, 97–106.
Walter, D., Wissing, S., Madeo, F., and Fahrenkrog, B. (2006). The inhibitor-of-apoptosis protein Bir1p protects against apoptosis in S. cerevisiae and is a substrate for the yeast homologue of Omi/HtrA2. J. Cell. Sci. 119, 1843–1851.
Wang, C., Skinner, C., Easlon, E., and Lin, S. J. (2009). Deleting the 14-3-3 protein Bmh1 extends life span in Saccharomyces cerevisiae by increasing stress response. Genetics 183, 1373–1384.
Weinlich, R., Dillon, C. P., and Green, D. R. (2011). Ripped to death. Trends Cell Biol. 21, 630–637.
Whelan, R. S., Kaplinskiy, V., and Kitsis, R. N. (2010). Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 72, 19–44.
Wilkinson, S., and Ryan, K. M. (2010). Autophagy: an adaptable modifier of tumourigenesis. Curr. Opin. Genet. Dev. 20, 57–64.
Williams, D., Norman, G., Khoury, C., Metcalfe, N., Briard, J., Laporte, A., Sheibani, S., Portt, L., Mandato, C. A., and Greenwood, M. T. (2011). Evidence for a second messenger function of dUTP during Bax mediated apoptosis of yeast and mammalian cells. Biochim. Biophys. Acta 1813, 315–321.
Wilson, P. M., Labonte, M. J., Lenz, H. J., Mack, P. C., and Ladner, R. D. (2012). Inhibition of dUTPase induces synthetic lethality with thymidylate synthase-targeted therapies in non-small cell lung cancer. Mol. Cancer Ther. 11, 616–628.
Winderickx, J., Delay, C., De Vos, A., Klinger, H., Pellens, K., Vanhelmont, T., Van Leuven, F., and Zabrocki, P. (2008). Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim. Biophys. Acta 1783, 1381–1395.
Wissing, S., Ludovico, P., Herker, E., Buttner, S., Engelhardt, S. M., Decker, T., Link, A., Proksch, A., Rodrigues, F., Corte-Real, M., Frohlich, K. U., Manns, J., Cande, C., Sigrist, S. J., Kroemer, G., and Madeo, F. (2004). An AIF orthologue regulates apoptosis in yeast. J. Cell Biol. 166, 969–974.
Woo, I. S., Eun, S. Y., Jang, H. S., Kang, E. S., Kim, G. H., Kim, H. J., Lee, J. H., Chang, K. C., Kim, J. H., Han, C. W., and Seo, H. G. (2009). Identification of ADP-ribosylation factor 4 as a suppressor of N-(4-hydroxyphenyl)retinamide-induced cell death. Cancer Lett. 276, 53–60.
Woo, I. S., Jang, H. S., Eun, S. Y., Kim, H. J., Ham, S. A., Lee, J. H., Chang, K. C., Kim, J. H., Han, C. W., and Seo, H. G. (2008). Ran suppresses paclitaxel-induced apoptosis in human glioblastoma cells. Apoptosis 13, 1223–1231.
Woo, I. S., Jin, H., Kang, E. S., Kim, H. J., Lee, J. H., Chang, K. C., Park, J. Y., Choi, W. S., and Seo, H. G. (2011). TMEM14A inhibits N-(4-hydroxyphenyl)retinamide-induced apoptosis through the stabilization of mitochondrial membrane potential. Cancer Lett. 309, 190–198.
Wu, W., Chaudhuri, S., Brickley, D. R., Pang, D., Karrison, T., and Conzen, S. D. (2004). Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 64, 1757–1764.
Wu, W., Liu, P., and Li, J. (2012). Necroptosis: an emerging form of programmed cell death. Crit. Rev. Oncol. Hematol. 82, 249–259.
Wyllie, A. H. (2010). “Where, o death, is thy sting?” a brief review of apoptosis biology. Mol. Neurobiol. 42, 4–9.
Yacoubian, T. A., and Standaert, D. G. (2009). Targets for neuroprotection in Parkinson’s disease. Biochim. Biophys. Acta 1792, 676–687.
Yang, Z., Khoury, C., Jean-Baptiste, G., and Greenwood, M. T. (2006). Identification of mouse sphingomyelin synthase 1 as a suppressor of Bax-mediated cell death in yeast. FEMS Yeast Res. 6, 751–762.
Yenari, M. A., Liu, J., Zheng, Z., Vexler, Z. S., Lee, J. E., and Giffard, R. G. (2005). Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann. N. Y. Acad. Sci. 1053, 74–83.
Zdralevic, M., Guaragnella, N., Antonacci, L., Marra, E., and Giannattasio, S. (2012). Yeast as a tool to study signaling pathways in mitochondrial stress response and cytoprotection. ScientificWorldJournal 2012, 912147.
Keywords: heart failure, apoptosis, programmed cell death, anti-apoptotic genes, Bax, genetic screen, pre-condition, hormesis
Citation: Clapp C, Portt L, Khoury C, Sheibani S, Eid R, Greenwood M, Vali H, Mandato CA and Greenwood MT (2012) Untangling the roles of anti-apoptosis in regulating programmed cell death using humanized yeast cells. Front. Oncol. 2:59. doi: 10.3389/fonc.2012.00059
Received: 24 April 2012; Accepted: 24 May 2012;
Published online: 13 June 2012.
Edited by:
Frank Madeo, Karl-Franzens-Universität, AustriaReviewed by:
Gavin McStay, Columbia University, USACopyright: © 2012 Clapp, Portt, Khoury, Sheibani, Eid, Greenwood, Vali, Mandato and Greenwood. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael T. Greenwood, Department of Chemistry and Chemical Engineering, Royal Military College, P.O. Box 17000, Station Forces, Kingston, ON, Canada K7K 7B4. e-mail:bWljaGFlbC5ncmVlbndvb2RAcm1jLmNh
†Caitlin Clapp and Liam Portt have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.