
 Joel H. Tolentino
Joel H. Tolentino
- Molecular Oncology Program, H. Lee Moffitt Cancer Center, Tampa, FL, USA
Chronic myeloid leukemia (CML) is initially driven by the bcr–abl fusion oncoprotein. The identification of bcr–abl led to the discovery and rapid translation into the clinic of bcr–abl kinase inhibitors. Although, bcr–abl inhibitors are efficacious, experimental evidence indicates that targeting bcr–abl is not sufficient for elimination of minimal residual disease found within the bone marrow (BM). Experimental evidence indicates that the failure to eliminate the leukemic stem cell contributes to persistent minimal residual disease. Thus curative strategies will likely need to focus on strategies where bcr–abl inhibitors are given in combination with agents that specifically target the leukemic stem cell or the leukemic stem cell niche. One potential target to be exploited is the Janus kinase (JAK)/signal transducers and activators of transcription 3 (STAT3) pathway. Recently using STAT3 conditional knock-out mice it was shown that STAT3 is critical for initiating the disease. Interestingly, in the absence of treatment, STAT3 was not shown to be required for maintenance of the disease, suggesting that STAT3 is required only in the tumor initiating stem cell population (Hoelbl et al., 2010). In the context of the BM microenvironment, STAT3 is activated in a bcr–abl independent manner by the cytokine milieu. Activation of JAK/STAT3 was shown to contribute to cell survival even in the event of complete inhibition of bcr–abl activity within the BM compartment. Taken together, these studies suggest that JAK/STAT3 is an attractive therapeutic target for developing strategies for targeting the JAK–STAT3 pathway in combination with bcr–abl kinase inhibitors and may represent a viable strategy for eliminating or reducing minimal residual disease located in the BM in CML.
Introduction
The signal transducers and activators of transcription 3 (STAT3) was identified as a DNA-binding protein capable of transducing signals from cytokine-stimulated cell surface receptors to the nucleus, where it was capable of selectively binding to a palindromic Interferon (IFN)-γ-activated sequence element and induce transcription of acute-phase genes (Akira et al., 1994; Lutticken et al., 1994; Zhong et al., 1994). Coincidentally, at the time of STAT3 discovery, investigators in the chronic myeloid leukemia (CML) field were trying to identify the major downstream substrate of bcr–abl causative for CML transformation. In fact, when researchers were just beginning to understand the biological properties and functions of STAT3, more than 30 years of research work had already been conducted on CML. For example, it was already established that; (a) CML could be diagnosed by the presence of Philadelphia chromosome (Nowell and Hungerford, 1964), (b) the Philadelphia chromosome was created by the reciprocal translocation between chromosomes 9 and 22 and contained a gene called BCR–ABL which encoded for a fusion protein called bcr–abl (Lugo et al., 1990; Gishizky et al., 1993), (c) tyrosine kinase activity of the bcr–abl proteins is required for the transformation into CML (Carlesso et al., 1994), (d) herbimycin A, an inhibitor of tyrosine kinase, showed inhibition of growth in cells expressing bcr–abl (Okabe et al., 1992, 1994a,b). This final observation would ultimately lead to the discovery and use of the present-day clinical bcr–abl kinase inhibitors, Imatinib (IM), Nilotinib (NI), and Dasatinib (DA; Buchdunger et al., 1996; Druker et al., 1996; Lombardo et al., 2004; Weisberg et al., 2005; Kantarjian et al., 2006, 2009, 2010, 2011; Hochhaus et al., 2009).
Signal transducers and activators of transcription 3 is a member of a family of seven proteins, namely, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6, all of whom relay signals from the plasma membrane to the nucleus, where they regulate gene expression (for review see, Darnell, 1997; Levy and Darnell, 2002). STAT3 gene is located on chromosome 17q21 and was first independently cloned by Zhong et al. (1994). The STAT3 gene codes for a 92-kDa protein spanning 770 amino acids long with structural similarity to all other STAT member of proteins. Briefly, the STAT3 proteins structurally comprises of an N-terminal coiled–coiled domain involved in protein–protein interactions, a DNA-binding domain, a linker, a Src homology-2 (SH-2) domain, and a C-terminal transactivation domain (for review see, Groner et al., 2008; Aggarwal et al., 2009). In order to delineate the distinctive functions of each STAT family member, gene targeting approaches proved very fruitful, except for STAT3. Unlike all other STAT family members, targeted disruption of STAT3 gene, lead to early embryonic lethality (Takeda et al., 1997). Because of this, normal physiological functions of STAT3 had to be defined by tissue specific deletions of STAT3 (Akira, 2000).
Signal transducers and activators of transcription 3 plays a pivotal role in the hematopoietic system. The role of STAT3 in the hematopoietic development has been studied by the use of floxed-STAT3 mice that were crossed with transgenic mice expressing Cre protein in specific hematopoietic cell type. For example, (a) Takeda et al. (1998) showed that T-cell specific deletion of STAT3 resulted in normal development of T-cells, however these cells showed impaired IL-6 dependent suppression of apoptosis and the resulting T-cell proliferation. Furthermore, these T-cells show a defective IL-2-induced IL-2α expression leading to a partial defect in IL-2 dependent T-cell proliferation (Akaishi et al., 1998). (b) Mice showing STAT3 deficiency in macrophages and neutrophils developed chronic enterocolitis (Takeda et al., 1999). STAT3 depleted mice macrophages and neutrophil showed complete inhibition of IL-10 mediated anti-inflammatory response leading to excessive Th1 response in vivo. Additional phenotypic analysis showed that these mice where susceptible to endotoxin shock and demonstrated excessive secretions of cytokines like TNFα, IL-1, and IFNγ (aiding the Th1 type immune response; Takeda et al., 1999). (c) Studies performed on a subset of T helper cell, called Th17, showed that this cells require the activity of STAT3 for its development and in its ability to mount a Th17-dependent autoimmunity (Harris et al., 2007). (d) In addition to T-cells, STAT3 also regulates the early steps of B cell development, since deletion of STAT3 in the B cell compartment leads to increase in pre–pro-B cells along with a decrease in the pre-B and pro-B cell compartment (Chou et al., 2006). STAT3 is also very critical in the T-cell-dependent terminal differentiation of B cells (Fornek et al., 2006). (e) In the dendritic cell compartment deletion of STAT3 abrogates the effects of Flt3L on dendritic cell development and results in absence of dendritic cell progenitors and thus results in a decrease in the dendritic cell compartment (Laouar et al., 2003). (f) Finally, various studies have shown that STAT3 may aid hematopoietic stem cell renewal in the very early stages of stem cell regeneration, however in adult homeostatic conditions STAT3 activity did not alter self-renewal or differentiation (Oh and Eaves, 2002; Takizawa et al., 2003; Kato et al., 2005; Chung et al., 2006). Even though, the above mentioned studies identify STAT3 as an essential player in the hematopoietic compartment, the phenotype arising from the complete depletion of STAT3 in the all of the components of the hematopoietic compartment remains to be seen.
Experimental evidence indicates that STAT3 plays a very crucial role in hematopoiesis, in mediating immune response and in regulation of differentiation, proliferation, angiogenesis, metastasis, and apoptosis through its modulation of STAT3 target genes (Seita et al., 2008; Hankey, 2009). Inhibition of STAT3 activity by either STAT3 knock-out or through inhibition of STAT3 function, blocked v-Src-induced transformation, indicating the important role of STAT3 in malignant transformation (Bromberg et al., 1998; Turkson et al., 1998). The oncogenic potential of STAT3 was demonstrated by induction of malignant transformation and tumor formation in mice by over-expressing a constitutive form of STAT3 (STAT3C; Bromberg et al., 1999). Constitutively active STAT3 induces the expression of anti-apoptotic genes including Bcl-2, Bcl-xl, Mcl-1, and also up-regulates the expression of inhibitors of apoptotic machinery like survivin and c-IAP2 (Epling-Burnette et al., 2001; Bhattacharya et al., 2005; Cuevas et al., 2006; Gritsko et al., 2006). STAT3 activation can promote cell cycle progression by inducing expression of cyclin D1 mRNA and hence protein levels (Sinibaldi et al., 2000; Cuevas et al., 2006). The cumulative effect of persistent STAT3 activity in cells is the promotion of cell growth and survival, leading to transformation, and tumorigenesis. Thus, it is not surprising that STAT3 is a critical player for development, progression, and maintenance of many human tumors (for a list of STAT3-mediated tumors see, Johnston and Grandis, 2011). In the following sections we briefly review the activation, regulation, and targeting of STAT3 in CML.
STAT3 Activation Pathway
In the canonical STAT3 activation pathway, STAT3 lies latent in the cytoplasm in a resting cell (see Figure 1). STAT3 can be activated by a variety of diverse agents ranging from cytokines, growth factors, oncogenes, non-receptor tyrosine kinases to even peptides like angiotensin II (Marrero et al., 1995; Herrington et al., 2000, Reddy et al., 2000; Ihle, 2001). The first step in the activation of STAT3 involves a tyrosine phosphorylation activation event-mediated creation of a docking site for recruitment of the dormant cytoplasmic STAT3 (Zhang et al., 2000). This docking is mediated through the SH-2 domain of STAT3. Once docked the latent STAT3 is targeted for phosphorylation, either by the intrinsic kinase activity of the activated receptor tyrosine kinase or through the aid of the tyrosine kinase receptor-associated Janus kinase (JAK; Heinrich et al., 1998, 2003; Quesnelle et al., 2007). Irrespective of the activating kinase protein, STAT3 gets phosphorylated at the Tyr705 residue in the C-terminal domain and this phosphorylation leads to its activation (Heinrich et al., 1998, 2003; Quesnelle et al., 2007). The activated phosphorylated STAT3 disengages from its activating kinases and undergoes homodimerization by reciprocal interaction between the SH-2 domain of one monomer and the phosphorylated Tyr705 residue of its dimerizing partner (see Figure 1). STAT3 dimers translocate to the nucleus and bind specific DNA sequence and regulate the transcription of the responsive gene (Shuai et al., 1993, 1994). In addition, STAT3 can also be phosphorylated on the Ser727 residue, located in the transactivation domain, which allows for the maximal activation of transcription of genes (Zhang et al., 1995; Wen et al., 1995). It should be noted that in a normal cell, STAT3 activation sequence is very rapid and transient and is very tightly regulated as will be discussed in the following sections.
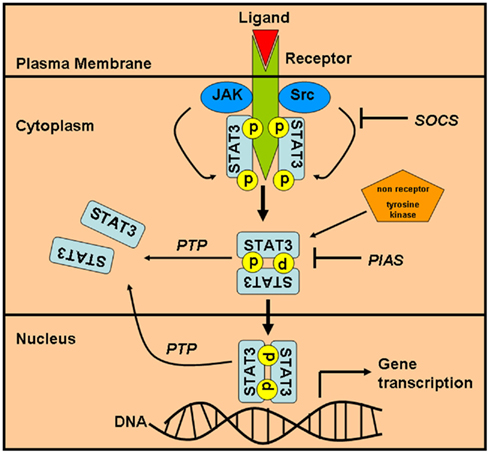
Figure 1. Activation and regulation of the STAT3 signaling pathway. The STAT3 signaling pathway is turned on by the activation of cell surface receptor (growth factor or cytokine receptors) leading to recruitment and activation of JAK-family of proteins, which in turn recruits and phosphorylates STAT3. STAT3 can also be directly phosphorylated by non-tyrosine kinase receptors (Src or c-abl). Phosphorylated STAT3 homodimerizes and translocates to the nucleus where it regulates gene expression. The signaling pathway can be switched off by the actions of a phosphatase which dephosphorylates STAT3 and prevents dimer formation. Also, PIAS and SOCS family of protein can directly compete with STAT3 for either binding opportunities with the activating receptor or for dimerization and translocation into the nucleus. STAT3, signal transducers and activators of transcription 3; JAK, Janus kinase; SOCS, suppressors of cytokine signaling; PTP, protein-tyrosine phosphatase; PIAS, protein inhibitor of activated STATs.
Bcr–abl Induced Activation of STAT3 in CML
Both bcr–abl and STAT3 protein share the ability to induce transformation in cells when they are over-expressed or activated. Thus it was speculated that one of the likely downstream signaling events that leads to CML transformation is the activation of the STAT3 pathway by oncogenic bcr–abl protein. To test this hypothesis, Ilaria and Van Etten (1996) transfected BaF/3 cells with p210 and p190 forms of BCR–ABL cDNA. BaF/3 cells are IL-3 dependent and non-leukemogenic, however these cells are transformed into IL-3 independence and are leukemogenic when expressing BCR–ABL cDNA (Daley and Baltimore, 1988; Ilaria and Van Etten, 1995). Thus, any differences in STAT activity between the parental and bcr–abl transformed BaF/3 cells could be directly attributed to the action of bcr–abl. Ilaria and Van Etten (1996) demonstrated that STAT1, STAT3, and STAT5 are constitutively tyrosine phosphorylated in bcr–abl transformed BaF/3 cells, with the intensity of the phosphorylation being STAT5 ≫ STAT3 ∼ STAT1. Moreover, the intensity of phosphorylation matched the DNA-binding activity of STAT5, STAT3, and STAT1 in the BCR–ABL transformed BaF/3 cells. Surprisingly, STAT6 was only phosphorylated in the p190 BCR–ABL cDNA expressing BaF/3 cells and STAT2 and STAT4 were not phosphorylated in any of the bcr–abl transformed BaF/3 cells (Ilaria and Van Etten, 1996). Furthermore, the activation of STATs by bcr–abl was found to be JAK-independent thereby indicating a mechanism of direct activation of STATs by bcr–abl. On the other hand, Carlesso et al. (1996) in their study, only detected constitutive STAT5 and STAT1 activity in bcr–abl positive CML cell lines, but like the previous study they also confirmed the observation that STAT activation in bcr–abl positive cells was not consistent with JAK kinase activity, suggesting the possibility of direct STAT activation by bcr–abl.
The role of STAT5 as the direct downstream target of bcr–abl was further confirmed by (de Groot et al., 1999) and Sillaber et al. (2000) by over-expressing a dominant negative isoform of STAT5. In both of these studies, the over-expression of the negative isoform of STAT5 led to inhibition of STAT5 phosphorylation and subsequent inhibition in cell growth in CML cell lines (Sillaber et al., 2000; de Groot et al., 1999). Further a direct interaction between bcr–abl and STAT5 was demonstrated by Nieborowska-Skorska et al. (1999) by showing that the SH-2 and SH-3 domain of bcr–abl protein is required for STAT5 activation, since mutations in these domains completely abolished STAT5 activation by bcr–abl. More recently, Hantschel et al. (2012) confirmed the finding that STAT5 was directly phosphorylated by bcr–abl and this activation was required for the maintenance of bcr–abl driven CML disease.
On the other hand, Coppo et al. (2006) have shown that in bcr–abl expressing cell lines (UT7-210, MO7E-p210, and K562) and in primary CD34+ CML progenitor cells, bcr–abl greatly phosphorylated STAT3 Ser727 residue and, to a lesser extent STAT3 Tyr705 residue. The tyrosine kinase activity of bcr–abl and the Tyr177 residue of the bcr–abl oncoprotein was required for this activation, since the Δ1172 tyrosine kinase mutant and the Y177F mutant bcr–abl constructs expressed in UT7 cell lines led to not only a reduction in phosphorylation of the STAT3 Ser727 residue but also to the reduction in the total STAT3 protein level. Also, they reported JAK1 and JAK2 were constitutively phosphorylated in UT7/9 and K562 cells and this was required for specifically phosphorylating the STAT3 Tyr705 residue but not the STAT5 Tyrosine residue (Coppo et al., 2006). Furthermore, by independently utilizing a MEK inhibitor and a MEK1 construct with a dominant negative activity, these same investigators were able to show that phosphorylation of the STAT3 Ser727 residue, but not STAT3 Tyr705 residue, required the activity of MEK in CML cells and in CD34+ CML progenitor cells from two patients (Coppo et al., 2006). Finally, in the same study inhibition of bcr–abl activity by the use of IM led to a dose-dependent down-regulation of STAT3 protein and mRNA, suggesting that bcr–abl transcriptionally regulates STAT3 gene.
In summary, there are many conflicting reports published concerning the status of STAT3 activity in CML cells. Our own observations in CML cells (K562 and KU812), show that these cells have very high phosphorylation of the STAT5 Ser694 residue and barely detectable levels of phosphorylated STAT3 Try705 residue as seen by western blotting (Bewry et al., 2008). However, when cultured with bone marrow (BM) stromal cells, these cells showed increased STAT3 phosphorylation. In addition, the constitutive STAT5 phosphorylation, but not the STAT3 phosphorylation, was inhibited when these co-cultured CML cells were treated with bcr–abl kinase inhibitors (Bewry et al., 2008; Nair et al., 2011).
STAT3 Activity Requirement for bcr–abl Induced Transformation
In the studies enumerated above, only a correlative link is provided for STAT3 activation by bcr–abl to cellular transformation. In order to provide more direct evidence, Spiekermann et al. (2002) used a different molecular methodology to address whether STAT3 and STAT5 are required for the transformation of hematopoietic cells. In their study, they utilized constitutively active STAT5 (STAT5A1*6) and STAT3 (STAT3C) mutants to stably transfect BaF/3 cells. Not surprisingly, both mutants induced long-term cytokine-independent growth in BaF/3 cells, a finding supporting a fundamental role for STAT3 and STAT5 in the malignant transformation of hematopoietic cells (Spiekermann et al., 2002). A more conclusive role for STAT3 and STAT5 in driving Bcr–abl dependent transformation of myeloid cells was recently provided by Hoelbl et al. (2010). In the study, they first reported that p210-BCR–ABL cDNA expressed in STAT5null/null fetal liver hematopoietic stem cells resulted in complete absence of myeloid transformation, indicating the critical dependence of STAT5 for the bcr–abl induced transformation event. Next they utilized BM cells derived from STAT3fl/flMx1Cre mice that were treated with polyinosinic: polycytidylic acid to induce STAT3 deletion in vivo. Similar to STAT5, significant reduction in colony numbers were observed for STAT3Δ/Δ cells upon transduction with p210-BCR–ABL cDNA. Taken together, the study indicate that STAT3 and STAT5 were absolutely required for the initial Bcr–abl dependent transformation event (Hoelbl et al., 2010). Further experiments in the same study also confirmed that once the cells are transformed, STAT5 but not STAT3 is required for bcr–abl induced myeloid leukemia maintenance in vivo. However, the role of STAT3 in maintaining minimal residual disease following bcr–abl inhibitor treatment was not addressed in this disease model.
STAT3 Activation by bcr–abl in Embryonic Stem Cells
Chronic myeloid leukemia is a stem cell disease as demonstrated by the presence of minimal residual disease in the form of a consistently detectable population of quiescent bcr–abl positive CD34+ progenitor cells that possess the ability for self-renewal and the capability to differentiate (Holyoake et al., 1999). Murine embryonic stem cells proved to be a very suitable system to study the effects of bcr–abl expression in an in vitro self-renewal and differentiation clonal model. For example, the direct effect of bcr–abl expression on murine embryonic stem cell differentiation was demonstrated in an in vitro embryonic stem cell differentiation system that utilized stromal cell co-culture to aid the differentiation of the stem cells into distinct hematopoietic lineages (Era and Witte, 2000). These stem cells where then transduced with a tetracycline regulated expression of bcr–abl vector employing a promoter that ensures 100% expression of bcr–abl in all the differentiated cells (Gossen and Bujard, 1992; Nakano et al., 1994). The results of their study indicated a bcr–abl dependent increase in the number of multipotent and myeloid lineage committed progenitor cells and at the same time a suppression of the development of committed erythroid progenitors (Era and Witte, 2000). This study established that bcr–abl was sufficient to establish the chronic phase of CML without the need for any additional genetic changes.
Even though the above murine embryonic stem cell model described the ability of bcr–abl to differentiate and propagate into CML chronic phase like disease, the molecular mechanism of the bcr–abl dependent self-renewal of murine embryonic stem cell was unknown. Previous studies showed that self-renewal of murine embryonic stem cell required the activation of STAT3 by leukemia inhibitory factor (LIF) acting through the gp130–JAK activation pathway (Niwa et al., 1998; Ernst et al., 1999). To investigate if bcr–abl can substitute for the action of LIF by maintenance of a self renewing capacity in cells despite LIF withdrawal, Coppo et al. (2003) utilized 129/SV-derived CCE embryonic stem cell. They demonstrated that bcr–abl expressing embryonic stem cells retained their “stemness” in the absence of LIF, confirming the absence of differentiation. The persistence of the embryonic stem cell morphology was directly attributed to specific STAT3 but not STAT5 activation and was independently verified by EMSA and over-expression of a dominant negative STAT3 mutant in bcr–abl expressing embryonic stem cells (Coppo et al., 2003). Next, to address whether bcr–abl directly phosphorylate STAT3 or did it require a messenger as an intermediary signaling protein, Nakamura et al. (2005) stably transfected a wild type murine embryonic stem cell line and a MEKK1−/− embryonic stem cell line with p210-bcr–abl called WTp210 and MEKK1−/−p210, respectively. They reported that, on LIF withdrawal, MEKK1−/−p210 showed less STAT3 activity than WTp210 and formed large flattened colonies having weak alkaline phosphatase activity, indicating a phenotype of differentiated embryonic stem cells (Nakamura et al., 2005). These results indicate that MEKK1 plays a role in bcr–abl induced activation of STAT3 (see Figure 2). Finally, in order to explain how bcr–abl driven activation of STAT3 can cue stem cells to differentiate and also self-renew, Coppo et al. (2009) proposed that bcr–abl induced these opposite phenotypes based on STAT3 activity in murine embryonic stem cells (Coppo et al., 2009). They reported in their study that embryonic stem cells expressing bcr–abl maintained an undifferentiated phenotype through activation of a STAT3-mediated self-renewal program. However, decreased STAT3 levels led to a rapid commitment of the bcr–abl expressing cells to differentiate, through the activation of the MAPK–ERK pathway (see Figure 2; Coppo et al., 2009). At present, the reason for decreased STAT3 activity within the bcr–abl expressing murine embryonic cells is not well understood and remains to be investigated. Taken together, all this studies confirm the role of STAT3 as a self-renewal factor operating downstream of bcr–abl in murine embryonic stem cells.
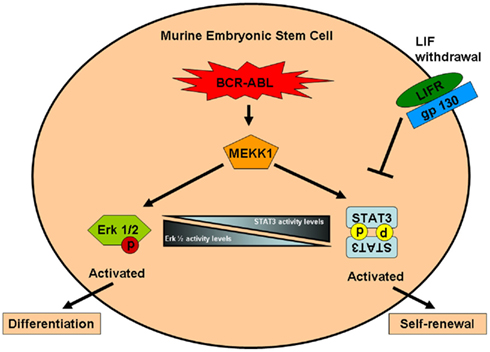
Figure 2. Regulation of self-renewal and differentiation in bcr–abl transformed murine embryonic stem cell. In bcr–abl transformed murine embryonic stem cell, STAT3 is activated in the LIF-independent MEKK1-dependent manner. Activation of MEKK1 by bcr–abl also leads to activation of Erk 1/2. It has been proposed that increased STAT3 activity within the stem cell promotes self-renewal and inhibition of differentiation, however decreased STAT3 activity leads to increased Erk 1/2 activity which in turn promotes rapid differentiation. This model has to be tested in bcr–abl transformed human stem cells. STAT3, signal transducers and activators of transcription 3; LIFR, leukemia inhibitory factor; Erk 1/2, extracellular signal-regulated kinases 1/2; MEKK1, mitogen-activated protein kinase kinase kinase.
All the studies enumerated above where carried out in murine embryonic stem cell model, wherein LIF–gp130–STAT3 activity is very much essential for maintenance of self-renewal of the stem cells. However, it should be noted that in non-human primate embryonic stem cells like in cynomolgus monkey embryonic stem cells, the maintenance of the undifferentiated embryonic state is maintained through a LIF/gp130/STAT3-independent pathway (Sumi et al., 2004). Similarly, Daheron et al. (2004) reported that in human embryonic stem cells, human LIF can induce STAT3 activation and nuclear translocation. However, the LIF–gp130–STAT3 pathway is unable to maintain pluripotency in human embryonic stem cells. Also, undifferentiated human embryonic stem cells did not show activated STAT3, indicating that in these embryonic stem cells STAT3-independent mechanism govern the self-renewal machinery (Daheron et al., 2004). In light of these reports, transformation of human embryonic cells with bcr–abl and the resultant dependence on STAT3 for self-renewal and differentiation in these transformed embryonic cells remain to be tested.
Regulation of the STAT3 Activation Pathway
Since STAT3 plays a very important role in a myriad of cellular functions, the activation of STAT3 is tightly regulated in normal cells (see Figure 1). This regulatory control is exerted at multiple levels of the STAT3 activation pathway and begins with the negative regulation of the activating receptor. Either after ligand binding or after receptor phosphorylation, the activated receptor gets tagged for endocytosis, thus preventing any further activation of receptor and thereby attenuating the signaling to STAT3 (Bild et al., 2002). The next level of regulation comes from protein-tyrosine phosphatase (PTP), which can dephosphorylate the receptor tyrosine kinases, JAKs or STAT3 directly thus ensuring the termination of STAT3 signaling (see Figure 1). PTPs implicated in regulating STAT3 signaling include SHP-1, SHP-2, PTP1B, and PTPεC (for review see, Xu and Qu, 2008). A unique PTP is the nuclear PTP TC45 which dephosphorylates STAT3 dimers that are present within the nucleus and thereby terminates STAT3-mediated transcription (Herrmann et al., 2007).
The family of suppressors of cytokine signaling (SOCS1–SOCS7) can antagonize STAT3 signaling by; (a) by binding and inhibiting JAKs, (b) by competing with STAT3 for docking sites on the activated tyrosine kinase receptor, and (c) by binding to JAKs and kinase receptors and targeting them for ubiquitin proteosomal degradation (for review see, Croker et al., 2008). Interestingly, SOCS is a part of the negative feedback loop, wherein activation of STAT3 increases the expression of SOCS, which then terminates the STAT3 activation by one of the before mentioned mechanisms (Yoshimura et al., 2003). Finally, the last level of regulation is exerted by the constitutively expressed protein inhibitor of activated STATs (PIAS; for review see, Shuai, 2006). PIAS, specifically PIAS3, can bind the dimeric activated STAT3 and block the DNA-binding ability of STAT3 (Chung et al., 1997). In addition to preventing binding of activated STAT3 to the promoter region of its regulatory genes, PIAS can utilize its SUMO ligase activity to recruit co-repressors at the STAT3 transcriptional complex and thus modulate STAT3-mediated transcription (Yamashina et al., 2006; Lee et al., 2007).
Expression of the Regulators of the STAT3 Activity in CML
Inactivation or reduced expression of SOCS, PIAS, or PTP proteins may lead to sustained activation of STAT3. Deregulation of the JAK–STAT3 pathway due to variations in the expression of the SOCS and PTP have been reported in CML cells. Not surprisingly, in most of the cases decrease in the proteins involved in the negative feedback mechanism, results in sustained activation of JAK–STAT3 pathway leading to increased progression of CML or to development of resistance toward anti-leukemic drugs. For example, SHP-1, the non-receptor type PTP, expressed primarily in hematopoietic cells has been shown to be silenced in various highly aggressive lymphoma and leukemia cell lines (Oka et al., 2001). Oka et al. (2002) demonstrated that this gene silencing is due a aberrant promoter methylation of the SHP-1 gene in leukemias and lymphomas. Specifically, Oka et al. (2002) showed that 73% of the CML patients tested showed very strong CpG island methylation of the SHP-1 gene and remaining 27% showed very weak methylation of the same gene. Similarly, Amin et al. (2007) in their study reported that SHP-1 levels were markedly decreased in advanced stage CML patients compared to patients in chronic phase and this correlated with IM resistance seen in advanced stage CML patients. However, in contrast to the study published by Oka et al. this paper did not find any DNA methylation in the promoter region nor did they find any mutations in the SHP-1 protein rather they attributed the decreased expression of SHP-1 to an as-yet undefined post-transcriptional modification (Amin et al., 2007). Finally, SHP-1 and SHP-2 have antagonistic biological functions in cells, in that, SHP-1 is a negative transducer while SHP-2 is a positive transducer of growth factor signaling pathway (Qu et al., 1999; You et al., 1999; Wang et al., 2006; Chong and Maiese, 2007). Indeed, in CML cells, SHP-1 physically associates with bcr–abl and blocks bcr–abl dependent transformation and in addition mediates PP2A-induced bcr–abl proteosome degradation (Bruecher-Encke et al., 2001; Neviani et al., 2005). Esposito et al. (2011) showed that in CML cell lines there is a physical interaction between SHP-1 protein with SHP-2 such that SHP-1 modulates the activation signals that SHP-2 receives from both bcr–abl and membrane receptor tyrosine kinase. Moreover, decreased SHP-1 expression is a determinant of IM sensitivity in CML patients, with patients failing IM treatment showing significantly lower levels of SHP-1 protein. Interestingly, knocking down SHP-2 levels in IM-resistant KCL22-R CML cells reduced STAT3 activation and cell survival in IM treated cells (Esposito et al., 2011).
Various studies have been reported showing that variations in expression, due to single nucleotide polymorphisms (SNPs,) within the STAT3 regulatory protein genes can have an impact on disease prognosis. For example, Guillem et al. (2012) showed that SNPs in SOCS1 (rs243327) and PTPN22 (rs2476601) genes correlated with the risk of primary resistance to IM were adverse prognostic factors for failure-free survival in CML patients. Polymorphism in the STAT3 gene has already been shown to co-relate with higher proclivity in patients for asthma, inflammation, and autoimmune diseases, so it is attractive to speculate that SNPs affecting the expression levels of STAT3 protein may also predict the treatment outcome when utilizing immune therapy like IFN-α in CML (Litonjua et al., 2005; Ferguson et al., 2010; Jabalameli, 2011). For example, IFN-α therapy is very effective in inducing hematological remission and a sustained cytogenetic response in CML (Talpaz et al., 1986, 1991). However, the effect of the therapy is very limited since only 8–30% of the treated patients achieve complete cytogenetic remission (Allan et al., 1995; Bonifazi et al., 2001). The molecular basis for the resistance or the heterogeneous response among patients is poorly understood. However, it was hypothesized that SNPs encoded within the STAT3 gene regulating its expression could be responsible for the observed heterogeneity. Indeed, Kreil et al. (2010) demonstrated that a polymorphism associated with STAT3 expression in CML could explain the difference in treatment response of CML patients to IFN. Specifically, the levels of STAT3 mRNA, but not STAT5a or STAT5b, correlated with an rs6503691 genotype and response of CML patients to INF-α (Kreil et al., 2010). In light of this studies, it is interesting to speculate if determination of individual status of STAT3 SNPs would help lead to a personalized treatment regimen for clinical decisions for initiating IFN-α treatment in CML patients.
Targeting STAT3 for Inhibition in CML
In the preceding sections, we have enumerated how the dysfunction of STAT3 regulatory pathway leads to a drug resistant phenotype in CML. Hence it is attractive to speculate that inhibition of STAT3 activity would lead to drug sensitization and cell death in CML cells. Evidence for direct inhibition of STAT3 leading to cell death in CML cells was provided by a study published by Ma et al. (2010), wherein they showed that CML K562 cell line transduced with STAT3 siRNA lead to inhibition of growth and proliferation, cell cycle blockade, and induction of apoptosis. These observations were further strengthened in the study conducted by Mencalha et al. (2010), who utilized a STAT3 specific inhibitor, LLL-3. LLL-3 reduced cell viability and induced cell death through the activation of Caspases-3/7 pathway in K562 cells. Additionally, when used in combination with IM, LLL-3 showed an additive effect on cell death thereby suggesting a potential therapeutic value of combining these two drug regimens for the treatment of CML (Mencalha et al., 2010).
Various studies have also looked at treatment regimens that indirectly decreased STAT3 activity and have reported its effect on CML cell survival. Wetzler et al. (2006) studied the effect of arsenic trioxide on JAKs and bcr–abl, both of which has been shown to activate STAT3. In their study, arsenic trioxide not only reduced STAT3 activation by JAKs but also reduced the phosphorylation of bcr–abl kinase. Not surprisingly, in vitro study has shown additive to synergistic cytotoxic activity of IM (depending on CML cell line and drug concentrations) when used in conjunction with arsenic trioxide in CML cells (Wetzler et al., 2006). Expression of Src family kinase in CML cells has been shown to induce resistance to IM in a kinase-dependent manner and inhibition of Src activity restored IM sensitivity (Pene-Dumitrescu and Smithgall, 2010). However, in solid tumors, the requirement for simultaneous inhibition of Src and STAT3 activity to induce cell death has been observed. In these tumors, cancer cell survival after Src kinase inhibition has been demonstrated to be due to reactivation of STAT3 as a compensatory pathway in surviving cells. Abrogation of STAT3 activity, by use of JAK inhibitors, along with Src inhibition was necessary to result in synergistic cytotoxicity (Johnson et al., 2007). Interestingly, Jia et al. (2009) have corroborated this finding in resistant K562 CML cells (IM-resistant (K562/G) and Adriamycin-resistant-P gp-expressing K562 CML cells (K562/A)), in which Src family kinases and p-STAT3 activity was increased and consequently the Bax/Bcl-2 ratio was decreased, when compared to the parental K562 cells. Use of ZD6474, an orally available, small-molecule tyrosine kinase inhibitor resulted in an in vitro and in vivo growth arrest of the parental and the resistant cell lines. The mechanism of action of ZD6474 was reported to be simultaneous inhibition of Src kinase and STAT3 activity and the resultant increase in Bax/Bcl-2 ratio (Jia et al., 2009). Zhu et al. (2011) in their study utilized Icaritin, a hydrolytic product of Icariin which is a constituent of Chinese herbal medicine Epimedium, to show potent anti-leukemic activity on CML in vitro and in vivo. Icaritin was shown to inhibit proliferation of CML cell line K562 and primary CD34+ CML cells and induce apoptosis through a bcr–abl independent manner. Instead the cause of death was shown to correlate with down-regulation of phosho-STAT3, -JAK2, -ERK, -AKT, and -p38 in a dose and time-dependent manner (Zhu et al., 2011).
Combination drug therapy have also been studied with the purpose of circumventing the problem of bcr–abl kinase inhibitor resistance in CML. Dai et al. (2004), have shown that combination therapy of sub-toxic concentrations of a proteosome inhibitor, Bortezomib with a broad spectrum inhibitor of cyclin-dependent kinases, Flavopiridol resulted in a synergistic increase in mitochondrial dysfunction, and apoptosis in CML cells. In this study, cell death observed in both IM-sensitive cell lines and IM-resistant cell lines co-related with, among other events, marked reduction in bcr–abl, STAT3 and STAT5 activity. In another study, Dasmahapatra et al. (2006) used BaF/3 cells expressing wild type bcr–abl that is sensitive to IM and BaF/3 cells expressing three clinically relevant bcr–abl mutations, namely, E225K, M351T, and T351I, that are known to cause resistance toward IM. They reported that the use of adaphostin, an adamantly ester derivative of the bcr–abl kinase inhibitor tryphostin AG957, induced cell death in these cell lines via caspase-3 activation and PARP degradation. The cell death was also accompanied by an attenuation of STAT3 and STAT5 phosphorylation. More importantly, in combination with proteosome inhibitor, Bortezomib, adaphostin potently induced reactive oxygen species and cell death in the resistant cell lines. These results indicate that adaphostin plus Bortezomib circumvent IM-resistance due to point mutations and thus offer a treatment strategy to treat resistant CML (Dasmahapatra et al., 2006).
In addition to the direct or indirect inhibition of STAT3 phosphorylation, the effects of STAT3 activity could also be nullified by inhibiting the expression/activity of its regulated gene. Not surprisingly the expression of a large number of genes with overlapping functions are regulated by both STAT3 and STAT5, and thus for the above mentioned strategy to work, choosing the right target is critical. One such candidate protein is Mcl-1, a well characterized member of the Bcl-2 family with a strong anti-apoptotic activity that is up-regulated by both STAT3 and STAT5 (Kozopas et al., 1993, Fukuchi et al., 2001; Allen et al., 2011). More importantly, Mcl-1 was shown to be expressed in primary CML cells in a constitutive manner (Aichberger et al., 2005). As proof of principle, to demonstrate that Mcl-1 is a viable target for inhibition in CML, Aichberger et al. (2005), utilized a Mcl-1 antisense oligonucleotide and showed co-operative anti-leukemic effects when used in combination with IM.
Taken together the reported studies indicate that STAT3 is an important target in CML and inhibition of STAT3 activity may provide an important strategy to induce cell death in CML cells that are resistant to the currently available therapy of bcr–abl kinase inhibitors.
Targeting STAT3 for Inhibition in CML within the Context of the Bone Marrow Microenvironment
It has become very clear that minimal residual disease (MRD) in CML is found predominantly in the BM, thus giving rise to the hypothesis that eradication of the disease would require not only shutting down the oncogenic signals arising from within the CML cells (the seed), but also from disrupting the interactions between the cells and the BM microenvironment (the soil; Paget, 1889). For example, our laboratory has demonstrated that when CML cells were cultured in conditioned media (CM) derived from human BM stromal cells, the CML cells were resistant to multiple bcr–abl kinase inhibitors including imatinib, nilotinib, and dasatinib mediated cell death (Bewry et al., 2008). This resistance was attributed to BM stromal-derived cytokine and growth factor milieu-mediated activation of STAT3 in the CML cells. The sustained and persistent activation of STAT3, was not abolished by the presence of bcr–abl kinase inhibitors and resulted in the increased expression of anti-apoptotic genes Mcl-1, Bcl-xl, and survivin in CML cells (Bewry et al., 2008). Indeed, our study found that knockdown of STAT3 by use of siRNA technology could sensitize CML cells, cultured in stromal cell derived CM, toward IM-mediated cell death.
In our follow-up study, we showed that activation of STAT3 is important not only for protection against bcr–abl inhibitors but also for the survival of CML cells within the BM microenvironment (Nair et al., 2011). Indeed, we established that in CML cells, the BM stromal-derived cytokine and growth factor milieu induced phosphorylation of STAT1, STAT3-S727, and STAT5 but not of STAT3-Y705, was abolished in the presence of NI, indicating that phosphorylated STAT1, STAT3-S727, and STAT5 were not important players in mediating BM milieu-induced NI resistance (Nair et al., 2011). Finally, reducing the expression of both JAK2 and TYK2 or utilizing a pan-JAK inhibitor blocked CM-mediated STAT3 activation and sensitized CML cells to NI-mediated cell death. The hypothesis that the BM microenvironment contributes to drug resistance by providing survival cues through the JAK–STAT3 pathway has also be explored by other laboratories. Traer et al. (2011) used CYT387, a JAK1/JAK2 inhibitor, and TG101209, a relatively specific JAK2 inhibitor and demonstrated that the inclusion of these inhibitors abolished the anti-apoptotic effect of BM stromal HS-5 CM on CML cell lines. Interestingly, in their study IM treatment alone increased STAT3 phosphorylation which was further enhanced in the presence of CM. They concluded from their observations that inhibition of bcr–abl induces a shift to an adaptive JAK2–STAT3 survival pathway that gets substantially reinforced within the BM microenvironment (Traer et al., 2011). Furthermore, in vivo studies utilizing a high-dose combination of TG101209 and NI while effective in eliminating CML cells also lead to toxicity in the non-leukemic hematopoietic cells, suggesting that such combination should be used with caution (Traer et al., 2011).
Similarly, Hiwase et al. (2010) have showed that when CML-CD34+ cells were treated with DA and then exposed to a mixture of cytokines [six growth factors cocktail or granulocyte-macrophage colony stimulating factor (GM-CSF) or granulocyte-colony stimulating factor (G-CSF)], the cells remained viable, suggesting that the bcr–abl dependence for survival could be overcome by exposure to cytokines. Like previous studies, re-establishment of the sensitivity to bcr–abl inhibitors in this progenitor cells required inhibition of JAK-family of kinases (Hiwase et al., 2010). However, amidst these reports suggesting that cytokine and growth factor exposure can lead to drug resistance in CML cells, Jorgensen et al. (2006) have shown that intermittent exposure to growth factor, G-CSF, together with IM leads to significant reduction in leukemic stem cells in vitro (Jorgensen et al., 2006). The hypothesis put forward to explain this reduction was that leukemic stem cells are quiescent and IM-resistant, however, G-CSF exposure pushes these cells into cell cycle and thus restores IM sensitivity. Indeed, this treatment regimen was further tested in 11 CML patients with favorable results with the authors recommending further evaluation of this therapy for newly diagnosed CML patients (Fang et al., 2011). At the same time small pilot studies looking at eradication of CML stem cells with the use of combination of C-CSF and IM have concluded that there is no therapeutic benefit of adding G-CSF to IM (Drummond et al., 2009; Foo et al., 2009). At present, more studies have to be performed to delineate the exact effects of G-CSF with bcr–abl inhibitors in CML patients.
In addition to exposure to the cytokine milieu within the BM, CML cells also physically interact with the stromal cells utilizing adhesion molecules like β1-integrins (Damiano et al., 2001). Such interaction give rise to cell-adhesion mediated drug resistance (CAM-DR) and has been demonstrated in multiple myeloma to occur through a β1-integrins/STAT3/IL-6 autocrine activation loop (Shain et al., 2009). Interestingly, inhibition of JAK–STAT3 pathway either by use of a pan-JAK inhibitor or a specific JAK2 inhibitor abolished the CAM-DR phenotype when primary CML progenitor cells are co-cultured with HS-5 BM stromal cells (Nair et al., 2011; Traer et al., 2011). Also, Guo-Bao et al. (2010) have demonstrated a CAM-DR phenotype in K562 when this cells were co-cultured with BM stromal cells derived from patients with CML. In their study, sensitization of these K562 co-cultured cells required treatment with arsenic trioxide, which was shown to down-regulate the expression of β1-integrins in K562 cells and thus cause a dose-dependent reduction in adhesion ability of K562 cells to the BM stromal cells. However, none of the studies above delineated the role of cytokine milieu versus the adhesion component as the critical contributing factor essential for development of drug resistant phenotype.
There is accumulating evidence showing that in human CML stem cells, enhanced bcr–abl kinase inhibition does not result in decreased cell survival of these cells (Konig et al., 2008). Furthermore, within the context of the BM microenvironment, cytokine support along with adhesive interactions with the stromal cells permitted CML stem cells to grow and survive even in the absence of bcr–abl activity (see Figure 3; Corbin et al., 2011). In fact, Reynaud et al. (2011) in their study have shown that proinflammatory tumor microenvironment affects leukemic progenitor cells and contributes to CML pathogenesis (Reynaud et al., 2011). Specifically, they showed that IL-6 expression is induced by bcr–abl resulting in a paracrine feedback loop that sustained CML development. Indeed, their study demonstrated that strategies utilized to block IL-6 signaling significantly delayed the onset of CML development even when the bcr–abl activity was maintained in cells (Reynaud et al., 2011). Since IL-6 is a very potent activator of STAT3, it does remains to be seen if targeting STAT3 for inhibition within the context of the BM also delays the onset of CML.
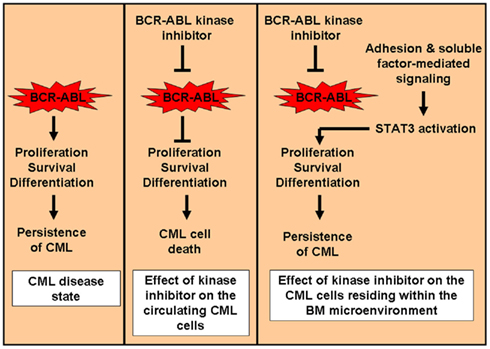
Figure 3. Mechanism for persistence of CML cells in the BM microenvironment. After the acquisition of the oncogene BCR–ABL, cells are dependent on bcr–abl for proliferation, survival, and differentiation (oncogenic addiction). In circulating CML cells, shut down of the kinase activity by use of a bcr–abl kinase inhibitor results in cell death and decrease in circulating CML cell burden. However, within the BM microenvironment, CML cells do not dependent upon bcr–abl activity for survival. These cells depend on the adhesion and soluble factor-mediated signaling for survival within the BM. In this scenario, inhibition of the bcr–abl kinase activity has no effect on the survival of the CML cells leading to the persistence of disease within the BM (minimal residual disease).
Conclusion
In light of reported data on the role of STAT3 in CML, it still remains to be seen, (i) if STAT3 is required for CML stem cell regeneration and CML development; (ii) does STAT3 polymorphism have an effect on CML disease progression, development of drug resistance and in maintenance of minimal residual disease; (iii) is there a functional overlap between STAT3 and STAT5 in CML cells so that JAK inhibitors might be a better option to specific inhibition of STAT3 activity. And lastly, (iv) what is the role of STAT3 in maintaining the minimal residual disease during bcr–abl kinase inhibitor treatment. Taken together, it would be interesting to test STAT3 as a very important target for inhibition in CML that could complement the existing bcr–abl kinase inhibitors in combating and reversing drug resistant phenotype and bring us closer to a cure for CML.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aggarwal, B. B., Kunnumakkara, A. B., Harikumar, K. B., Gupta, S. R., Tharakan, S. T., Koca, C., Dey, S., and Sung, B. (2009). Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann. N. Y. Acad. Sci. 1171, 59–76.
Aichberger, K. J., Mayerhofer, M., Krauth, M. T., Skvara, H., Florian, S., Sonneck, K., Akgul, C., Derdak, S., Pickl, W. F., Wacheck, V., Selzer, E., Monia, B. P., Moriggl, R., Valent, P., and Sillaber, C. (2005). Identification of Mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and Mcl-1 antisense oligonucleotides. Blood 105, 3303–3311.
Akaishi, H., Takeda, K., Kaisho, T., Shineha, R., Satomi, S., Takeda, J., and Akira, S. (1998). Defective IL-2-mediated IL-2 receptor alpha chain expression in Stat3-deficient T lymphocytes. Int. Immunol. 10, 1747–1751.
Akira, S., Nishio, Y., Inoue, M., Wang, X. J., Wei, S., Matsusaka, T., Yoshida, K., Sudo, T., Naruto, M., and Kishimoto, T. (1994). Molecular cloning of Aprf, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71.
Allan, N. C., Richards, S. M., and Shepherd, P. C. (1995). UK Medical Research Council randomised, multicentre trial of interferon-alpha n1 for chronic myeloid leukaemia: improved survival irrespective of cytogenetic response. The UK Medical Research Council’s Working Parties for Therapeutic Trials in Adult Leukaemia. Lancet 345, 1392–1397.
Allen, J. C., Talab, F., Zuzel, M., Lin, K., and Slupsky, J. R. (2011). c-Abl regulates Mcl-1 gene expression in chronic lymphocytic leukemia cells. Blood 117, 2414–2422.
Amin, H. M., Hoshino, K., Yang, H., Lin, Q., Lai, R., and Garcia-Manero, G. (2007). Decreased expression level of SH2 domain-containing protein tyrosine phosphatase-1 (Shp1) is associated with progression of chronic myeloid leukaemia. J. Pathol. 212, 402–410.
Bewry, N. N., Nair, R. R., Emmons, M. F., Boulware, D., Pinilla-Ibarz, J., and Hazlehurst, L. A. (2008). Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol. Cancer Ther. 7, 3169–3175.
Bhattacharya, S., Ray, R. M., and Johnson, L. R. (2005). STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 392, 335–344.
Bild, A. H., Turkson, J., and Jove, R. (2002). Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J. 21, 3255–3263.
Bonifazi, F., DE Vivo, A., Rosti, G., Guilhot, F., Guilhot, J., Trabacchi, E., Hehlmann, R., Hochhaus, A., Shepherd, P. C., Steegmann, J. L., Kluin-Nelemans, H. C., Thaler, J., Simonsson, B., Louwagie, A., Reiffers, J., Mahon, F. X., Montefusco, E., Alimena, G., Hasford, J., Richards, S., Saglio, G., Testoni, N., Martinelli, G., Tura, S., and Baccarani, M. (2001). Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood 98, 3074–3081.
Bromberg, J. F., Horvath, C. M., Besser, D., Lathem, W. W., and Darnell, J. E. Jr. (1998). Stat3 activation is required for cellular transformation by v-Src. Mol. Cell Biol. 18, 2553–2558.
Bromberg, J. F., Wrzeszczynska, M. H., Devgan, G., Zhao, Y., Pestell, R. G., Albanese, C., and Darnell, J. E. Jr. (1999). Stat3 as an oncogene. Cell 98, 295–303.
Bruecher-Encke, B., Griffin, J. D., Neel, B. G., and Lorenz, U. (2001). Role of the tyrosine phosphatase SHP-1 in K562 cell differentiation. Leukemia 15, 1424–1432.
Buchdunger, E., Zimmermann, J., Mett, H., Meyer, T., Muller, M., Druker, B. J., and Lydon, N. B. (1996). Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 56, 100–104.
Carlesso, N., Frank, D. A., and Griffin, J. D. (1996). Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med. 183, 811–820.
Carlesso, N., Griffin, J. D., and Druker, B. J. (1994). Use of a temperature-sensitive mutant to define the biological effects of the p210BCR-ABL tyrosine kinase on proliferation of a factor-dependent murine myeloid cell line. Oncogene 9, 149–156.
Chong, Z. Z., and Maiese, K. (2007). The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol. Histopathol. 22, 1251–1267.
Chou, W. C., Levy, D. E., and Lee, C. K. (2006). STAT3 positively regulates an early step in B-cell development. Blood 108, 3005–3011.
Chung, C. D., Liao, J., Liu, B., Rao, X., Jay, P., Berta, P., and Shuai, K. (1997). Specific inhibition of Stat3 signal transduction by PIAS3. Science 278, 1803–1805.
Chung, Y. J., Park, B. B., Kang, Y. J., Kim, T. M., Eaves, C. J., and Oh, I. H. (2006). Unique effects of Stat3 on the early phase of hematopoietic stem cell regeneration. Blood 108, 1208–1215.
Coppo, P., Dusanter-Fourt, I., Millot, G., Nogueira, M. M., Dugray, A., Bonnet, M. L., Mitjavila-Garcia, M. T., Le Pesteur, D., Guilhot, F., Vainchenker, W., Sainteny, F., and Turhan, A. G. (2003). Constitutive and specific activation of STAT3 by BCR-ABL in embryonic stem cells. Oncogene 22, 4102–4110.
Coppo, P., Dusanter-Fourt, I., Vainchenker, W., and Turhan, A. G. (2009). BCR-ABL induces opposite phenotypes in murine ES cells according to STAT3 activation levels. Cell. Signal. 21, 52–60.
Coppo, P., Flamant, S., De Mas, V., Jarrier, P., Guillier, M., Bonnet, M. L., Lacout, C., Guilhot, F., Vainchenker, W., and Turhan, A. G. (2006). BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 134, 171–179.
Corbin, A. S., Agarwal, A., Loriaux, M., Cortes, J., Deininger, M. W., and Druker, B. J. (2011). Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 121, 396–409.
Croker, B. A., Kiu, H., and Nicholson, S. E. (2008). SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 19, 414–422.
Cuevas, P., Diaz-Gonzalez, D., Sanchez, I., Lozano, R. M., Gimenez-Gallego, G., and Dujovny, M. (2006). Dobesilate inhibits the activation of signal transducer and activator of transcription 3, and the expression of cyclin D1 and bcl-XL in glioma cells. Neurol. Res. 28, 127–130.
Daheron, L., Opitz, S. L., Zaehres, H., Lensch, M. W., Andrews, P. W., Itskovitz-Eldor, J., and Daley, G. Q. (2004). LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells 22, 770–778.
Dai, Y., Rahmani, M., Pei, X. Y., Dent, P., and Grant, S. (2004). Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood 104, 509–518.
Daley, G. Q., and Baltimore, D. (1988). Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc. Natl. Acad. Sci. U.S.A. 85, 9312–9316.
Damiano, J. S., Hazlehurst, L. A., and Dalton, W. S. (2001). Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 15, 1232–1239.
Dasmahapatra, G., Nguyen, T. K., Dent, P., and Grant, S. (2006). Adaphostin and bortezomib induce oxidative injury and apoptosis in imatinib mesylate-resistant hematopoietic cells expressing mutant forms of Bcr/Abl. Leuk. Res. 30, 1263–1272.
de Groot, R. P., Raaijmakers, J. A., Lammers, J. W., Jove, R., and Koenderman, L. (1999). STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood 94, 1108–1112.
Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., Zimmermann, J., and Lydon, N. B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 2, 561–566.
Drummond, M. W., Heaney, N., Kaeda, J., Nicolini, F. E., Clark, R. E., Wilson, G., Shepherd, P., Tighe, J., Mclintock, L., Hughes, T., and Holyoake, T. L. (2009). A pilot study of continuous imatinib vs pulsed imatinib with or without G-CSF in CML patients who have achieved a complete cytogenetic response. Leukemia 23, 1199–1201.
Epling-Burnette, P. K., Liu, J. H., Catlett-Falcone, R., Turkson, J., Oshiro, M., Kothapalli, R., Li, Y., Wang, J. M., Yang-Yen, H. F., Karras, J., Jove, R., and Loughran, T. P. Jr. (2001). Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Invest. 107, 351–362.
Era, T., and Witte, O. N. (2000). Regulated expression of P210 Bcr-Abl during embryonic stem cell differentiation stimulates multipotential progenitor expansion and myeloid cell fate. Proc. Natl. Acad. Sci. U.S.A. 97, 1737–1742.
Ernst, M., Novak, U., Nicholson, S. E., Layton, J. E., and Dunn, A. R. (1999). The carboxyl-terminal domains of gp130-related cytokine receptors are necessary for suppressing embryonic stem cell differentiation. Involvement of STAT3. J. Biol. Chem. 274, 9729–9737.
Esposito, N., Colavita, I., Quintarelli, C., Sica, A. R., Peluso, A. L., Luciano, L., Picardi, M., Del Vecchio, L., Buonomo, T., Hughes, T. P., White, D., Radich, J. P., Russo, D., Branford, S., Saglio, G., Melo, J. V., Martinelli, R., Ruoppolo, M., Kalebic, T., Martinelli, G., and Pane, F. (2011). SHP1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood 118, 3634–3644.
Fang, B., Mai, L., Li, N., Song, Y., and Chunhua Zhao, R. (2011). Imatinib plus granulocyte colony-stimulating factor in chronic myeloid leukemia patients who have achieved partial or complete cytogenetic response while on imatinib. Case. Rep. Oncol. 4, 192–197.
Ferguson, L. R., Han, D. Y., Fraser, A. G., Huebner, C., Lam, W. J., Morgan, A. R., Duan, H., and Karunasinghe, N. (2010). Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat. Res. 690, 108–115.
Foo, J., Drummond, M. W., Clarkson, B., Holyoake, T., and Michor, F. (2009). Eradication of chronic myeloid leukemia stem cells: a novel mathematical model predicts no therapeutic benefit of adding G-CSF to imatinib. PLoS Comput. Biol. 5, e1000503. doi:10.1371/journal.pcbi.1000503
Fornek, J. L., Tygrett, L. T., Waldschmidt, T. J., Poli, V., Rickert, R. C., and Kansas, G. S. (2006). Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood 107, 1085–1091.
Fukuchi, Y., Kizaki, M., Yamato, K., Kawamura, C., Umezawa, A., Hata, J., Nishihara, T., and Ikeda, Y. (2001). Mcl-1, an early-induction molecule, modulates activin A-induced apoptosis and differentiation of CML cells. Oncogene 20, 704–713.
Gishizky, M. L., Johnson-White, J., and Witte, O. N. (1993). Evaluating the effect of P210 BCR/ABL on growth of hematopoietic progenitor cells and its role in the pathogenesis of human chronic myelogenous leukemia. Semin. Hematol. 30, 6–8.
Gossen, M., and Bujard, H. (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551.
Gritsko, T., Williams, A., Turkson, J., Kaneko, S., Bowman, T., Huang, M., Nam, S., Eweis, I., Diaz, N., Sullivan, D., Yoder, S., Enkemann, S., Eschrich, S., Lee, J. H., Beam, C. A., Cheng, J., Minton, S., Muro-Cacho, C. A., and Jove, R. (2006). Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 12, 11–19.
Groner, B., Lucks, P., and Borghouts, C. (2008). The function of Stat3 in tumor cells and their microenvironment. Semin. Cell Dev. Biol. 19, 341–350.
Guillem, V., Amat, P., Cervantes, F., Alvarez-Larran, A., Cervera, J., Maffioli, M., Bellosillo, B., Collado, M., Marugan, I., Martinez-Ruiz, F., and Hernandez-Boluda, J. C. (2012). Functional polymorphisms in SOCS1 and PTPN22 genes correlate with the response to imatinib treatment in newly diagnosed chronic-phase chronic myeloid leukemia. Leuk. Res. 36, 174–181.
Guo-Bao, W., Xiao-Qin, C., Qi-Rong, G., Jie, L., Gui-Nan, L., and Yue, L. (2010). Arsenic Trioxide overcomes cell adhesion-mediated drug resistance through down-regulating the expression of beta(1)-integrin in K562 chronic myelogenous leukemia cell line. Leuk. Lymphoma 51, 1090–1097.
Hankey, P. A. (2009). Regulation of hematopoietic cell development and function by Stat3. Front. Biosci. 14, 5273–5290.
Hantschel, O., Warsch, W., Eckelhart, E., Kaupe, I., Grebien, F., Wagner, K. U., Superti-Furga, G., and Sexl, V. (2012). BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 8, 285–293.
Harris, T. J., Grosso, J. F., Yen, H. R., Xin, H., Kortylewski, M., Albesiano, E., Hipkiss, E. L., Getnet, D., Goldberg, M. V., Maris, C. H., Housseau, F., Yu, H., Pardoll, D. M., and Drake, C. G. (2007). Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 179, 4313–4317.
Heinrich, P. C., Behrmann, I., Haan, S., Hermanns, H. M., Muller-Newen, G., and Schaper, F. (2003). Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20.
Heinrich, P. C., Behrmann, I., Muller-Newen, G., Schaper, F., and Graeve, L. (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 334(Pt 2), 297–314.
Herrington, J., Smit, L. S., Schwartz, J., and Carter-Su, C. (2000). The role of STAT proteins in growth hormone signaling. Oncogene 19, 2585–2597.
Herrmann, A., Vogt, M., Monnigmann, M., Clahsen, T., Sommer, U., Haan, S., Poli, V., Heinrich, P. C., and Muller-Newen, G. (2007). Nucleocytoplasmic shuttling of persistently activated STAT3. J. Cell Sci. 120, 3249–3261.
Hiwase, D. K., White, D. L., Powell, J. A., Saunders, V. A., Zrim, S. A., Frede, A. K., Guthridge, M. A., Lopez, A. F., D’Andrea, R. J., To, L. B., Melo, J. V., Kumar, S., and Hughes, T. P. (2010). Blocking cytokine signaling along with intense Bcr-Abl kinase inhibition induces apoptosis in primary CML progenitors. Leukemia 24, 771–778.
Hochhaus, A., O’Brien, S. G., Guilhot, F., Druker, B. J., Branford, S., Foroni, L., Goldman, J. M., Muller, M. C., Radich, J. P., Rudoltz, M., Mone, M., Gathmann, I., Hughes, T. P., and Larson, R. A. (2009). Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 23, 1054–1061.
Hoelbl, A., Schuster, C., Kovacic, B., Zhu, B., Wickre, M., Hoelzl, M. A., Fajmann, S., Grebien, F., Warsch, W., Stengl, G., Hennighausen, L., Poli, V., Beug, H., Moriggl, R., and Sexl, V. (2010). Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2, 98–110.
Holyoake, T., Jiang, X., Eaves, C., and Eaves, A. (1999). Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 94, 2056–2064.
Ilaria, R. L. Jr., and Van Etten, R. A. (1995). The SH2 domain of P210BCR/ABL is not required for the transformation of hematopoietic factor-dependent cells. Blood 86, 3897–3904.
Ilaria, R. L. Jr., and Van Etten, R. A. (1996). P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 271, 31704–31710.
Jabalameli, M. R. (2011). Role of STAT3 Locus in autoimmune diseases. Genes Immun. 12, 155 [author reply 156].
Jia, H. Y., Wu, J. X., Zhu, X. F., Chen, J. M., Yang, S. P., Yan, H. J., Tan, L., Zeng, Y. X., and Huang, W. (2009). ZD6474 inhibits Src kinase leading to apoptosis of imatinib-resistant K562 cells. Leuk. Res. 33, 1512–1519.
Johnson, F. M., Saigal, B., Tran, H., and Donato, N. J. (2007). Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin. Cancer Res. 13, 4233–4244.
Johnston, P. A., and Grandis, J. R. (2011). STAT3 signaling: anticancer strategies and challenges. Mol. Interv. 11, 18–26.
Jorgensen, H. G., Copland, M., Allan, E. K., Jiang, X., Eaves, A., Eaves, C., and Holyoake, T. L. (2006). Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin. Cancer Res. 12, 626–633.
Kantarjian, H., Giles, F., Wunderle, L., Bhalla, K., O’Brien, S., Wassmann, B., Tanaka, C., Manley, P., Rae, P., Mietlowski, W., Bochinski, K., Hochhaus, A., Griffin, J. D., Hoelzer, D., Albitar, M., Dugan, M., Cortes, J., Alland, L., and Ottmann, O. G. (2006). Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 354, 2542–2551.
Kantarjian, H., Pasquini, R., Levy, V., Jootar, S., Holowiecki, J., Hamerschlak, N., Hughes, T., Bleickardt, E., Dejardin, D., Cortes, J., and Shah, N. P. (2009). Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia resistant to imatinib at a dose of 400 to 600 milligrams daily: two-year follow-up of a randomized phase 2 study (START-R). Cancer 115, 4136–4147.
Kantarjian, H., Shah, N. P., Hochhaus, A., Cortes, J., Shah, S., Ayala, M., Moiraghi, B., Shen, Z., Mayer, J., Pasquini, R., Nakamae, H., Huguet, F., Boque, C., Chuah, C., Bleickardt, E., Bradley-Garelik, M. B., Zhu, C., Szatrowski, T., Shapiro, D., and Baccarani, M. (2010). Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 362, 2260–2270.
Kantarjian, H. M., Hochhaus, A., Saglio, G., Souza, C. D., Flinn, I. W., Stenke, L., Goh, Y. T., Rosti, G., Nakamae, H., Gallagher, N. J., Hoenekopp, A., Blakesley, R. E., Larson, R. A., and Hughes, T. P. (2011). Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 12, 841–851.
Kato, Y., Iwama, A., Tadokoro, Y., Shimoda, K., Minoguchi, M., Akira, S., Tanaka, M., Miyajima, A., Kitamura, T., and Nakauchi, H. (2005). Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J. Exp. Med. 202, 169–179.
Konig, H., Holtz, M., Modi, H., Manley, P., Holyoake, T. L., Forman, S. J., and Bhatia, R. (2008). Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 22, 748–755.
Kozopas, K. M., Yang, T., Buchan, H. L., Zhou, P., and Craig, R. W. (1993). MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. U.S.A. 90, 3516–3520.
Kreil, S., Waghorn, K., Ernst, T., Chase, A., White, H., Hehlmann, R., Reiter, A., Hochhaus, A., and Cross, N. C. (2010). A polymorphism associated with STAT3 expression and response of chronic myeloid leukemia to interferon alpha. Haematologica 95, 148–152.
Laouar, Y., Welte, T., Fu, X. Y., and Flavell, R. A. (2003). STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 19, 903–912.
Lee, S. J., Lee, J. R., Hahn, H. S., Kim, Y. H., Ahn, J. H., Bae, C. D., Yang, J. M., and Hahn, M. J. (2007). PIAS1 interacts with the KRAB zinc finger protein, ZNF133, via zinc finger motifs and regulates its transcriptional activity. Exp. Mol. Med. 39, 450–457.
Levy, D. E., and Darnell, J. E. Jr. (2002). Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662.
Litonjua, A. A., Tantisira, K. G., Lake, S., Lazarus, R., Richter, B. G., Gabriel, S., Silverman, E. S., and Weiss, S. T. (2005). Polymorphisms in signal transducer and activator of transcription 3 and lung function in asthma. Respir. Res. 6, 52.
Lombardo, L. J., Lee, F. Y., Chen, P., Norris, D., Barrish, J. C., Behnia, K., Castaneda, S., Cornelius, L. A., Das, J., Doweyko, A. M., Fairchild, C., Hunt, J. T., Inigo, I., Johnston, K., Kamath, A., Kan, D., Klei, H., Marathe, P., Pang, S., Peterson, R., Pitt, S., Schieven, G. L., Schmidt, R. J., Tokarski, J., Wen, M. L., Wityak, J., and Borzilleri, R. M. (2004). Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 47, 6658–6661.
Lugo, T. G., Pendergast, A. M., Muller, A. J., and Witte, O. N. (1990). Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247, 1079–1082.
Lutticken, C., Wegenka, U. M., Yuan, J., Buschmann, J., Schindler, C., Ziemiecki, A., Harpur, A. G., Wilks, A. F., Yasukawa, K., Taga, T., Kishimoto, T., Barbieri, G., Pellegrini, S., Sendtner, M., Heinrich, P. C., and Horn, F. (1994). Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 263, 89–92.
Ma, L. D., Zhou, M., Wen, S. H., Ni, C., Jiang, L. J., Fan, J., and Xia, L. (2010). Effects of STAT3 silencing on fate of chronic myelogenous leukemia K562 cells. Leuk. Lymphoma 51, 1326–1336.
Marrero, M. B., Schieffer, B., Paxton, W. G., Heerdt, L., Berk, B. C., Delafontaine, P., and Bernstein, K. E. (1995). Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 375, 247–250.
Mencalha, A. L., Du Rocher, B., Salles, D., Binato, R., and Abdelhay, E. (2010). LLL-3, a STAT3 inhibitor, represses BCR-ABL-positive cell proliferation, activates apoptosis and improves the effects of Imatinib mesylate. Cancer Chemother. Pharmacol. 65, 1039–1046.
Nair, R. R., Tolentino, J. H., Argilagos, R. F., Zhang, L., Pinilla-Ibarz, J., and Hazlehurst, L. A. (2011). Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk. Res. doi: 10.1016/j.leukres.2011.12.002. [Epub ahead of print].
Nakamura, Y., Yujiri, T., Nawata, R., Tagami, K., and Tanizawa, Y. (2005). MEK kinase 1 is essential for Bcr-Abl-induced STAT3 and self-renewal activity in embryonic stem cells. Oncogene 24, 7592–7598.
Nakano, T., Kodama, H., and Honjo, T. (1994). Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science 265, 1098–1101.
Neviani, P., Santhanam, R., Trotta, R., Notari, M., Blaser, B. W., Liu, S., Mao, H., Chang, J. S., Galietta, A., Uttam, A., Roy, D. C., Valtieri, M., Bruner-Klisovic, R., Caligiuri, M. A., Bloomfield, C. D., Marcucci, G., and Perrotti, D. (2005). The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 8, 355–368.
Nieborowska-Skorska, M., Wasik, M. A., Slupianek, A., Salomoni, P., Kitamura, T., Calabretta, B., and Skorski, T. (1999). Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 189, 1229–1242.
Niwa, H., Burdon, T., Chambers, I., and Smith, A. (1998). Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–2060.
Nowell, P. C., and Hungerford, D. A. (1964). Chromosome changes in human leukemia and a tentative assessment of their significance. Ann. N. Y. Acad. Sci. 113, 654–662.
Oh, I. H., and Eaves, C. J. (2002). Overexpression of a dominant negative form of STAT3 selectively impairs hematopoietic stem cell activity. Oncogene 21, 4778–4787.
Oka, T., Ouchida, M., Koyama, M., Ogama, Y., Takada, S., Nakatani, Y., Tanaka, T., Yoshino, T., Hayashi, K., Ohara, N., Kondo, E., Takahashi, K., Tsuchiyama, J., Tanimoto, M., Shimizu, K., and Akagi, T. (2002). Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 62, 6390–6394.
Oka, T., Yoshino, T., Hayashi, K., Ohara, N., Nakanishi, T., Yamaai, Y., Hiraki, A., Sogawa, C. A., Kondo, E., Teramoto, N., Takahashi, K., Tsuchiyama, J., and Akagi, T. (2001). Reduction of hematopoietic cell-specific tyrosine phosphatase SHP-1 gene expression in natural killer cell lymphoma and various types of lymphomas/leukemias: combination analysis with cDNA expression array and tissue microarray. Am. J. Pathol. 159, 1495–1505.
Okabe, M., Kawamura, K., Miyagishima, T., Itaya, T., Goodwyn, D., Shoji, M., Vogler, W. R., Sakurada, K., Uehara, M., and Miyazaki, T. (1994a). Effect of herbimycin A, an inhibitor of tyrosine kinase, on protein tyrosine kinase activity and phosphotyrosyl proteins of Ph1-positive leukemia cells. Leuk. Res. 18, 213–220.
Okabe, M., Uehara, Y., Noshima, T., Itaya, T., Kunieda, Y., and Kurosawa, M. (1994b). In vivo antitumor activity of herbimycin A, a tyrosine kinase inhibitor, targeted against BCR/ABL oncoprotein in mice bearing BCR/ABL-transfected cells. Leuk. Res. 18, 867–873.
Okabe, M., Uehara, Y., Miyagishima, T., Itaya, T., Tanaka, M., Kuni-Eda, Y., Kurosawa, M., and Miyazaki, T. (1992). Effect of herbimycin A, an antagonist of tyrosine kinase, on bcr/abl oncoprotein-associated cell proliferations: abrogative effect on the transformation of murine hematopoietic cells by transfection of a retroviral vector expressing oncoprotein P210bcr/abl and preferential inhibition on Ph1-positive leukemia cell growth. Blood 80, 1330–1338.
Paget, S. (1889). The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573.
Pene-Dumitrescu, T., and Smithgall, T. E. (2010). Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner. J. Biol. Chem. 285, 21446–21457.
Qu, C. K., Yu, W. M., Azzarelli, B., and Feng, G. S. (1999). Genetic evidence that Shp-2 tyrosine phosphatase is a signal enhancer of the epidermal growth factor receptor in mammals. Proc. Natl. Acad. Sci. U.S.A. 96, 8528–8533.
Quesnelle, K. M., Boehm, A. L., and Grandis, J. R. (2007). STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 102, 311–319.
Reddy, E. P., Korapati, A., Chaturvedi, P., and Rane, S. (2000). IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 19, 2532–2547.
Reynaud, D., Pietras, E., Barry-Holson, K., Mir, A., Binnewies, M., Jeanne, M., Sala-Torra, O., Radich, J. P., and Passegue, E. (2011). IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 20, 661–673.
Seita, J., Asakawa, M., Ooehara, J., Takayanagi, S., Morita, Y., Watanabe, N., Fujita, K., Kudo, M., Mizuguchi, J., Ema, H., Nakauchi, H., and Yoshimoto, T. (2008). Interleukin-27 directly induces differentiation in hematopoietic stem cells. Blood 111, 1903–1912.
Shain, K. H., Yarde, D. N., Meads, M. B., Huang, M., Jove, R., Hazlehurst, L. A., and Dalton, W. S. (2009). Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 69, 1009–1015.
Shuai, K. (2006). Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 16, 196–202.
Shuai, K., Horvath, C. M., Huang, L. H., Qureshi, S. A., Cowburn, D., and Darnell, J. E. Jr. (1994). Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 76, 821–828.
Shuai, K., Stark, G. R., Kerr, I. M., and Darnell, J. E. Jr. (1993). A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 261, 1744–1746.
Sillaber, C., Gesbert, F., Frank, D. A., Sattler, M., and Griffin, J. D. (2000). STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood 95, 2118–2125.
Sinibaldi, D., Wharton, W., Turkson, J., Bowman, T., Pledger, W. J., and Jove, R. (2000). Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene 19, 5419–5427.
Spiekermann, K., Pau, M., Schwab, R., Schmieja, K., Franzrahe, S., and Hiddemann, W. (2002). Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp. Hematol. 30, 262–271.
Sumi, T., Fujimoto, Y., Nakatsuji, N., and Suemori, H. (2004). STAT3 is dispensable for maintenance of self-renewal in nonhuman primate embryonic stem cells. Stem Cells 22, 861–872.
Takeda, K., Clausen, B. E., Kaisho, T., Tsujimura, T., Terada, N., Forster, I., and Akira, S. (1999). Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10, 39–49.
Takeda, K., Kaisho, T., Yoshida, N., Takeda, J., Kishimoto, T., and Akira, S. (1998). Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161, 4652–4660.
Takeda, K., Noguchi, K., Shi, W., Tanaka, T., Matsumoto, M., Yoshida, N., Kishimoto, T., and Akira, S. (1997). Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 94, 3801–3804.
Takizawa, M., Nobuhisa, I., Igarashi, K., Ueno, M., Nakashima, K., Kitamura, T., and Taga, T. (2003). Requirement of gp130 signaling for the AGM hematopoiesis. Exp. Hematol. 31, 283–289.
Talpaz, M., Kantarjian, H., Kurzrock, R., Trujillo, J. M., and Gutterman, J. U. (1991). Interferon-alpha produces sustained cytogenetic responses in chronic myelogenous leukemia. Philadelphia chromosome-positive patients. Ann. Intern. Med. 114, 532–538.
Talpaz, M., Kantarjian, H. M., Mccredie, K., Trujillo, J. M., Keating, M. J., and Gutterman, J. U. (1986). Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N. Engl. J. Med. 314, 1065–1069.
Traer, E., Mackenzie, R., Snead, J., Agarwal, A., Eiring, A. M., O’Hare, T., Druker, B. J., and Deininger, M. W. (2011). Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. doi:10.1038/leu.2011.325. [Epub ahead of print].
Turkson, J., Bowman, T., Garcia, R., Caldenhoven, E., De Groot, R. P., and Jove, R. (1998). Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 18, 2545–2552.
Wang, N., Li, Z., Ding, R., Frank, G. D., Senbonmatsu, T., Landon, E. J., Inagami, T., and Zhao, Z. J. (2006). Antagonism or synergism. Role of tyrosine phosphatases SHP-1 and SHP-2 in growth factor signaling. J. Biol. Chem. 281, 21878–21883.
Weisberg, E., Manley, P. W., Breitenstein, W., Bruggen, J., Cowan-Jacob, S. W., Ray, A., Huntly, B., Fabbro, D., Fendrich, G., Hall-Meyers, E., Kung, A. L., Mestan, J., Daley, G. Q., Callahan, L., Catley, L., Cavazza, C., Azam, M., Neuberg, D., Wright, R. D., Gilliland, D. G., and Griffin, J. D. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7, 129–141.
Wen, Z., Zhong, Z., and Darnell, J. E. Jr. (1995). Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250.
Wetzler, M., Brady, M. T., Tracy, E., Li, Z. R., Donohue, K. A., O’Loughlin, K. L., Cheng, Y., Mortazavi, A., Mcdonald, A. A., Kunapuli, P., Wallace, P. K., Baer, M. R., Cowell, J. K., and Baumann, H. (2006). Arsenic trioxide affects signal transducer and activator of transcription proteins through alteration of protein tyrosine kinase phosphorylation. Clin. Cancer Res. 12, 6817–6825.
Xu, D., and Qu, C. K. (2008). Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 13, 4925–4932.
Yamashina, K., Yamamoto, H., Chayama, K., Nakajima, K., and Kikuchi, A. (2006). Suppression of STAT3 activity by Duplin, which is a negative regulator of the Wnt signal. J. Biochem. 139, 305–314.
Yoshimura, A., Mori, H., Ohishi, M., Aki, D., and Hanada, T. (2003). Negative regulation of cytokine signaling influences inflammation. Curr. Opin. Immunol. 15, 704–708.
You, M., Yu, D. H., and Feng, G. S. (1999). Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 19, 2416–2424.
Zhang, T., Kee, W. H., Seow, K. T., Fung, W., and Cao, X. (2000). The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell. Biol. 20, 7132–7139.
Zhang, X., Blenis, J., Li, H. C., Schindler, C., and Chen-Kiang, S. (1995). Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science 267, 1990–1994.
Zhong, Z., Wen, Z., and Darnell, J. E. Jr. (1994). Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98.
Zhu, J. F., Li, Z. J., Zhang, G. S., Meng, K., Kuang, W. Y., Li, J., Zhou, X. F., Li, R. J., Peng, H. L., Dai, C. W., Shen, J. K., Gong, F. J., Xu, Y. X., and Liu, S. F. (2011). Icaritin shows potent anti-leukemia activity on chronic myeloid leukemia in vitro and in vivo by regulating MAPK/ERK/JNK and JAK2/STAT3/AKT signalings. PLoS ONE 6, e23720. doi:10.1371/journal.pone.0023720
Keywords: STAT3, bone marrow microenvironment, drug resistance, transformation, chronic myeloid leukemia
Citation: Nair RR, Tolentino JH and Hazlehurst LA (2012) Role of STAT3 in transformation and drug resistance in CML. Front. Oncol. 2:30. doi: 10.3389/fonc.2012.00030
Received: 01 December 2011; Accepted: 15 March 2012;
Published online: 10 April 2012.
Edited by:
Gautam Borthakur, MD Anderson Cancer Center, USAReviewed by:
Parameswaran N. Hari, Medical College of Wisconsin, USADean Anthony Lee, University of Texas MD Anderson Cancer Center, USA
Gautam Borthakur, MD Anderson Cancer Center, USA
Copyright: © 2012 Nair, Tolentino and Hazlehurst. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lori A. Hazlehurst, Molecular Oncology Program, H. Lee Moffitt Cancer Center, Tampa, FL 33612 USA. e-mail:bG9yaS5oYXpsZWh1cnN0QG1vZmZpdHQub3Jn