
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nucl. Med., 04 April 2024
Sec. Radionuclide Therapy
Volume 4 - 2024 | https://doi.org/10.3389/fnume.2024.1331364
This article is part of the Research TopicEvolving Targeted Radiotherapies to Treat CancerView all 4 articles
 Amanda G. Shea1,†
Amanda G. Shea1,† Malick Bio Idrissou2,†
Malick Bio Idrissou2,† Ana Isabel Torres1Tessa Chen1
Ana Isabel Torres1Tessa Chen1 Reiner Hernandez2,3
Reiner Hernandez2,3 Zachary S. Morris1,3
Zachary S. Morris1,3 Quaovi H. Sodji1,3*
Quaovi H. Sodji1,3*
Radiation therapy (RT) is a pillar of cancer therapy used by more than half of all cancer patients. Clinically, RT is mostly delivered as external beam radiation therapy (EBRT). However, the scope of EBRT is limited in the metastatic setting, where all sites of disease need to be irradiated. Such a limitation is attributed to radiation-induced toxicities, for example on bone marrow and hematologic toxicities, resulting from a large EBRT field. Radiopharmaceutical therapy (RPT) has emerged as an alternative to EBRT for the irradiation of all sites of metastatic disease. While RPT can reduce tumor burden, it can also impact the immune system and anti-tumor immunity. Understanding these effects is crucial for predicting and managing treatment-related hematological toxicities and optimizing their integration with other therapeutic modalities, such as immunotherapies. Here, we review the immunomodulatory effects of α- and β-particle emitter-based RPT on various immune cell lines, such as CD8+ and CD4+ T cells, natural killer (NK) cells, and regulatory T (Treg) cells. We briefly discuss Auger electron-emitter (AEE)-based RPT, and finally, we highlight the combination of RPT with immune checkpoint inhibitors, which may offer potential therapeutic synergies for patients with metastatic cancers.
The intricate interplay between radiation-induced DNA damage and immune response underscores the evolving understanding of the impact of RT on tumor control. Historically, radiation-induced DNA damage has been regarded as the primary mechanism by which most solid tumors respond to ionizing radiation. This damage leads to various cellular responses, such as apoptosis, senescence, and autophagy, ultimately resulting in tumor control (1–3). Since these cytotoxic effects can also have deleterious effects on bone marrow and systemic immune cell populations, RT has been considered immunosuppressive (4, 5). Yet, emerging data suggests that RT has the potential to elicit a favorable immune response by stimulating the immune system, which in turn contributes to tumor eradication (6–8). However, harnessing these therapeutic benefits hinges on preserving the function of effector immune cells amidst the deleterious effects of radiation (8–10). Recent advances in diagnostic imaging, tumor delineation, and motion management have resulted in the accurate delivery of radiation to the tumor while sparing healthy organs and minimizing hematological toxicities, including immune cell depletion (11). Despite these advances, EBRT cannot effectively be used to irradiate all disease sites in patients with widespread metastatic disease due to radiation-induced toxicity, including bone marrow and hematologic toxicities (12). In contrast to EBRT, RPT combines a radionuclide (radioactive isotope) with a tumor-targeting agent (e.g., antibody, peptide, small molecule) that can potentially deliver systemic radiation to all tumor sites. Although RPT shows considerable potential in the oncology field, it faces numerous limitations, including the restricted availability of certain radionuclides and biodistribution (13–15). Additionally, RPT can also have dose-limiting toxicities. In this review, we will cover the two most common groups of therapeutic radionuclides: alpha (α) and beta (β−) emitters. While the effects of EBRT on the immune system and healthy tissues are beginning to be understood and are still being explored, we are only starting to understand the effects of RPT on the various immune cell lineages. Here, we summarize existing pre-clinical and clinical data describing the impact of RPT on CD8+ T cells, CD4+ T cells, NK cells, and Treg cells.
Clinically, RT is delivered by EBRT or brachytherapy, with EBRT representing the more commonly used treatment modality (16–18). EBRT is a mainstay of oncological care and has been a pillar of cancer therapy for more than a century (19). As such, the majority of the described immunomodulatory effects of RT have been gleaned from radiation delivered by EBRT (6, 20, 21). In addition to inducing lethal DNA damage resulting in cell death, RT can induce immunogenic cell death (ICD) of tumor cells, which is characterized by the extracellular release of adenosine triphosphate (ATP) and high-mobility group box 1 protein (HMGB1) (22–25). RT can also induce calreticulin translocation from the endoplasmic reticulum onto the cell surface, which can result in immune cell recognition, infiltration in the tumor microenvironment (TME), and activation (26–30). RT-induced ICD can stimulate antigen presentation by dendritic cells (DCs), activating cytotoxic effector cells like CD8+ T cells (31). Additionally, RT sensitizes tumor cells to immune-cell-mediated killing by upregulating MHC-1 expression and inducing the release of pro-inflammatory cytokines via a type 1 interferon (IFN) response, which further stimulates CD8+ T cells (7, 32–36). While the immunomodulatory effects of RT delivered by EBRT are being elucidated, RT can also be detrimental to healthy cells, including immune cells (12, 37, 38). These effects are of increased concern with large radiation fields, such as those that would be needed to treat all lesions in patients with widely metastatic cancer.
EBRT can cause potentially harmful side effects on the healthy tissue surrounding the tumor (11, 18, 39). In addition, lymphopenia is a concerning side effect of radiation observed in several solid tumors, such as high-grade glioma, head and neck, lung, esophageal, pancreatic, and cervical cancers treated with EBRT (40–48). The observed lymphopenia appears to correlate with not only the size of the radiation field, but also the radiation dose delivered to lymphoid organs such as the spleen and lymph nodes (49, 50). Moreover, exposure to therapeutic radiation can lead to the development of secondary malignancies (51). The risk of such secondary malignancies is also associated with the size of the radiation field (52). Furthermore, EBRT cannot effectively target radiographically occult lesions. Thus, it is generally not used when there is a need to irradiate all tumor sites in patients with metastatic cancers (Figure 1). In these instances, RPT is an attractive therapeutic option that can deliver radiation systematically to all metastatic lesions (15, 53).
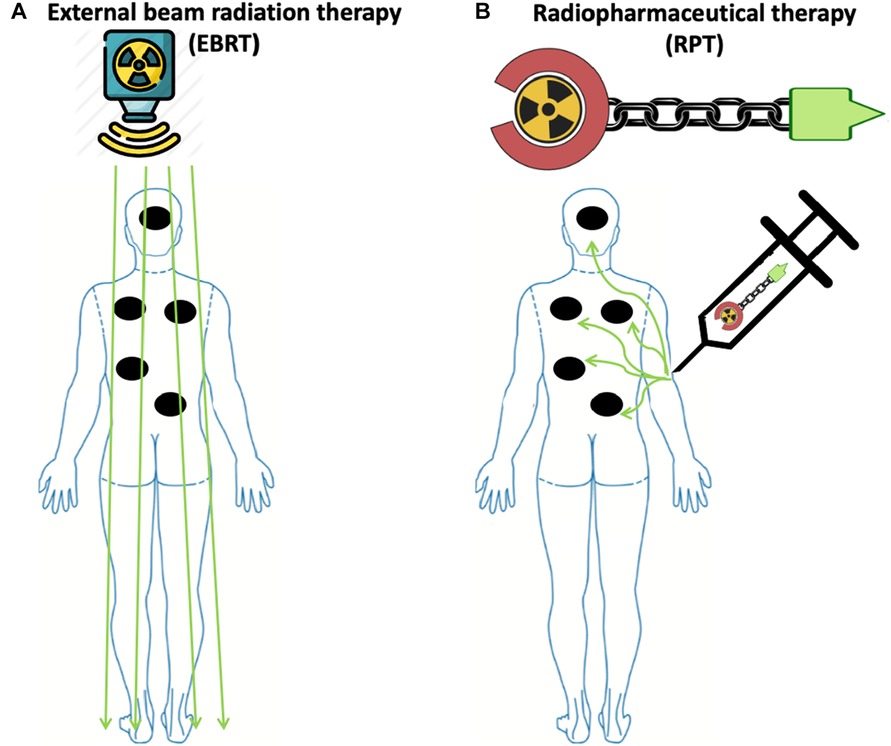
Figure 1. Radiopharmaceutical therapy delivers radiation to all sites of disease with potentially less toxicity compared to external beam radiation therapy in widely metastatic disease. (A) To irradiate all sites of disease in patients with widely metastatic disease, including microscopic disease using EBRT, a large radiation field is needed, thus increasing the risk of toxicity. (B) In contrast to EBRT, due to the molecular targeting in RPT, radiation is delivered to malignant cells expressing the target, while irradiation to healthy tissue is minimized due to the differential expression of the target. Made with Biorender.com.
RPT is a growing class of cancer therapeutics in which a radionuclide is linked to a ligand such as a small molecule, peptide, or antibody directed toward a cell surface antigen upregulated on malignant cells (15, 54) (Figure 2). Following the intravenous administration of a radiopharmaceutical agent, it selectively accumulates in the tumor and the TME, thus sparing healthy tissues that do not express the targeted antigens and allowing the targeted delivery of radiation to malignant cells. This makes RPT particularly attractive for the treatment of metastatic and microscopic tumors (15, 55–57). Indeed, several radiopharmaceutical agents have been shown to increase the survival of patients with metastatic diseases, for example prostate cancer and neuroendocrine cancer (58–60).
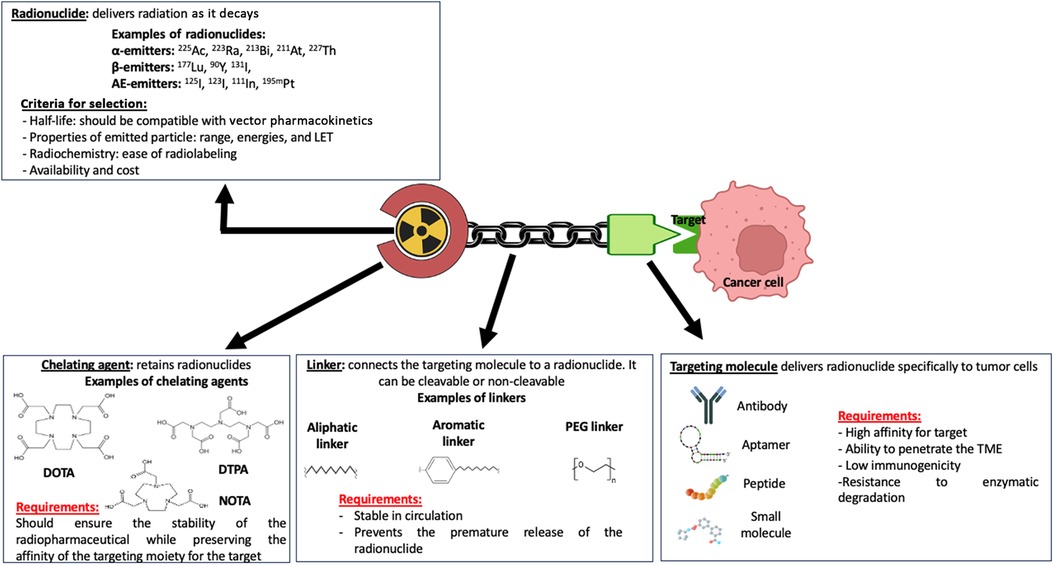
Figure 2. Pharmacophoric model of a radiopharmaceutical agent. The therapeutic effect of a radiopharmaceutical is impacted by the properties of the various domains of the pharmacophoric model, including the radionuclide, chelating agent, linker, and targeting molecule or moiety. Made with Biorender.com.
The recent approvals by the FDA of 223Ra-dichloride (223RaCl2; Xofigo®) for the treatment of castration-resistant prostate cancer with symptomatic bone metastases, 177Lu-DOTATATE (Lutathera®) for gastroenteropancreatic neuroendocrine tumors (GEP-NET), and 177Lu-PSMA-167 (Pluvicto®) for metastatic castration-resistant prostate cancer (mCRPC) have resulted in a renewed enthusiasm for the development of novel RPT agents (61–63).
Radionuclides used for RPT can emit different forms of therapeutic radiation, for example α-particles, β-particles, γ-rays, and Auger electrons. Often, a given radionuclide will emit multiple forms of radiation before decaying to a non-radioactive element. Each form of emitted radiation is characterized by unique properties, including range in biological tissues, relative biological effectiveness (RBE), physical half-life, and linear energy transfer (LET), which is the energy released per unit of distance (54, 55, 64). While RPT offers the advantage of targeted radiation delivery to tumor cells, it can induce dose-limiting toxicities including bone marrow and hematologic toxicity, nephrotoxicity, neurotoxicity, and hepatotoxicity (65–69).
β-particle emitters, which include Lutetium-177 (177Lu), Yttrium-90 (90Y), and Iodine-131 (131I) are currently the most commonly used radionuclides employed in RPT. β-particles have a low LET (∼0.2 keV/µm), and their range in biological tissue is several millimeters (up to 12 mm) (54, 70), making them well-suited for the irradiation of large tumors and tumors with heterogeneous expression of the RPT antigen target. Due to their low LET, β-emitters induce single-stranded DNA breaks, DNA base modifications, and DNA-protein crosslinks, which are more readily repairable compared to double-stranded DNA breaks (71, 72).
α-particle emitters include radionuclides such as Radium-223 (223Ra), Actinium-225 (225Ac), and Astatine-211 (211At). They have a short tissue range (50–100 µm), making them suitable for the treatment of micrometastases, and a high LET (50–230 keV/µm), thus making them highly cytotoxic (54, 71, 73). α-particles mostly induce clusters of DNA damage, including double-stranded DNA breaks, which are difficult to repair (54). The relative biological effectiveness (RBE) for α-particles is 5-fold that of β particles, highlighting the higher therapeutic potential of α-emitters compared to β-emitters (74).
Auger electron emitters include radionuclides such as Iodine-123 (123I), Iodine-125 (125I), and Indium-111 (111In). While they have a very short range in tissue (< 1 µm), they possess a medium to high LET (4–26 keV/µm) (54, 57, 73). Because of the short range, therapeutic benefits may be achieved with AEEs when they are in proximity of a sensitive cellular target such as nuclear DNA.
The majority of β-particle emitters and AEEs can also emit γ rays as they decay, which can be used for imaging purposes (75). The utilization of γ rays for imaging to assess the distribution of the uptake RPT is crucial for verifying that the uptake pattern aligns with the intended therapeutic target and for estimating the absorbed doses in both the target tissue and organs at risk. For example, γ rays emitted by 177Lu and 131I can be used to monitor in vivo biodistribution (76, 77). In some instances, data subsequently gleaned from such biodistribution can be used to increase the radiation dose and enhance therapeutic benefits (78, 79).
Similar to EBRT, RPT may also influence the immune system's response to tumors (7, 8, 80–86). This can be achieved through the direct or indirect irradiation of immune cells in the TME (87) (Figure 3). The latter, known as the “bystander effect”, refers to the phenomenon where cells that are not directly exposed to radiation exhibit biological responses as a result of signals emitted by neighboring irradiated cells (88, 89). However, in the context of RPT, this classic definition has been challenged. Emerging data, such as those by Leung et al., suggest that bystander cells include irradiated and non-irradiated cells, while the bystander effect emanates from irradiated cells, and this contributes to the mechanism by which 223Ra is able to delay the development of metastatic lesions (90–92). Indeed, signaling molecules released from irradiated cells may not only affect non-irradiated adjacent cells, but may also affect irradiated cells in the vicinity. Moreover, the abscopal effect, defined as tumor regression outside the irradiated region, is also thought to be involved in the biological effects of RPT (87). However, because of the different physical properties of the radionuclides, the effects of RPT on immune cells may differ. Here, we provide a summary of relevant studies evaluating the immunomodulatory effects of α-emitter, β-emitter, and AEE-based RPT.
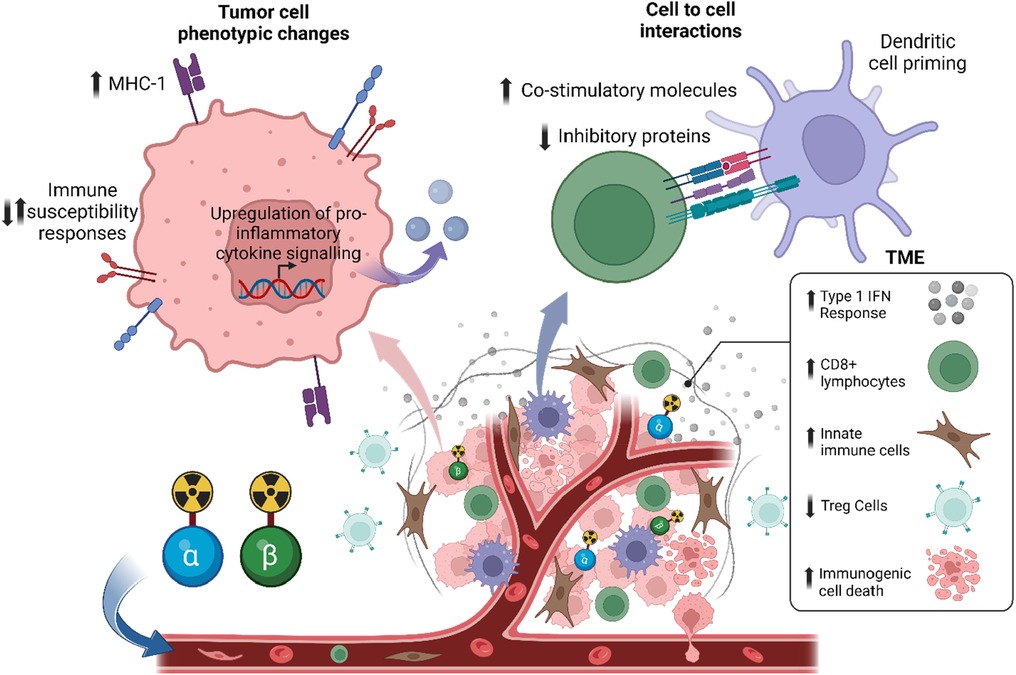
Figure 3. Systemic radiopharmaceutical treatment may have direct and indirect effects on the tumor microenvironment and immune cells. Preclinical data suggests that RPT with α-particles or β-particles has an immunostimulatory effect, such as an increase in type 1 interferon response, a decrease in regulatory T cells (Tregs), and an increase in immunogenic cell death.
177Lu and 131I are examples of radionuclides that emit β-particles and γ rays, while 90Y is an example of an almost pure β-emitting radionuclides. With the recent FDA approval of 177Lu-based RPTs Lutathera® and Pluvicto®, there has been an increased and renewed interest in not only understanding the mechanism of action on the TME but also the hematological side effects of RPT and dose-dependent toxicities in the clinic (59, 60, 62, 63). Several studies have evaluated the safety and immunomodulatory effects of β-particle RPT in the preclinical setting (7, 8, 81, 93, 94). β-particle RPT can indirectly regulate the function of immune cells in the TME through anti-tumor response. For example, 90Y-NM600 (a radiolabeled tumor-targeting alkylphosphocholine analog) triggers a type 1 interferon response in irradiated malignant cells (7, 8). Furthermore, β-particle RPT can also induce ICD in tumor cells (15, 64). Rouanet and colleagues demonstrated that 131ICF01012 induced the release of damage-associated molecular patterns (DAMPs) such as HMGB1 and ATP into the extracellular milieu. 131ICF01012 also induced cell surface translocation of calreticulin, along with the secretion of type I IFN (80). Here, we will discuss the direct effects of β-particle-emitter RPT on specific immune cell populations.
Following administration of 90Y-based RPT, FOXP3+ immunohistochemistry (IHC) staining revealed a notable reduction of Treg cells in the TME of tumors generated by the EL4 lymphoma cell line in murine syngeneic models (81).
In murine models of medullary breast adenocarcinoma, Vito and colleagues observed that while the 177Lu-BSA-tetrazine RPT did not significantly impact the level of CD4+ T cells in the peripheral blood after 4 days, an analysis of the TME by IHC demonstrated a significant decrease in intratumoral CD4+ T cells (95). Notably, Vito et al. did not analyze the CD4+ T cell subset to determine if they were specifically Tregs, however, Tregs are defined as CD4+/Foxp3+, indicating that this treatment combination effectively diminished nearly all CD4+ T cells. Similar to the observations of Hernandez et al., the findings by Vito et al. suggest that this treatment reduces the immunosuppressive tumor environment. In addition to this direct effect of 177Lu RPT reported by Vito et al., another study highlighted the indirect effect of 90Y-NM600 RPT on CD4+ T cells. Jagodinsky and colleagues co-cultured murine B16 melanoma or MOC2 head and neck squamous cell carcinoma cells in 90Y-NM600 until the tumor cells received a cumulative radiation dose of 6 Gy. They found that the viability of CD4+ T cells in the presence of 90Y-NM600 was enhanced when co-cultured with previously irradiated MOC2 and B16 tumor cells. Whereas in the absence of tumor cells, irradiation of CD4+ T cells with 90Y-NM600 significantly reduced their viability. They also investigated the expression of the immune checkpoint marker CTLA-4 and the immune activation signal IFN-γ on the T cell surface. They found that CTLA-4 was significantly upregulated on the CD4+ T cells in the presence of 90Y-NM600-treated MOC2 or 90Y-NM600-treated B16 cells, compared to the untreated controls. Additionally, they found that in the same experiment, IFN-γ was not significantly changed (7). This suggests that RPT to tumor cells induces signaling to CD4+ T cells that increases viability, but also increases immune inhibitory signals, potentially indicating a need for immunotherapy to block the upregulated immune checkpoint pathways.
Following treatment with 90Y-NM600 RPT, mice bearing EL4 lymphoma tumors revealed not only an increased infiltration of CD8+ T cells in the TME but also a significant activation of these infiltrated CD8+ T cells (81). Conversely, in a murine model of medullary breast adenocarcinoma, intratumorally delivered 177Lu-BSA-tetrazine RPT significantly decreased CD8+ T cells in the TME; however, this decrease was not seen in the CD8+ T cells in the peripheral blood (95). This was likely due to the targeting agent that was used, as in another experiment they found that intravenous administration of 177Lu-BSA-tetrazine RPT had poor tumor retention and high uptake in other tissues. Intratumoral delivery of 177Lu-BSA-tetrazine RPT allowed for local treatment while minimizing the systemic effect on peripheral CD8+ T cells. As described for CD4+ T cells, co-culture of CD8+ T cells with MOC2 or B16 tumor cells previously irradiated with 90Y-NM600 RPT indirectly enhanced the proliferation and activation of CD8+ T cells as demonstrated by increased viability and an increased secretion of IFN-γ. Moreover, CTLA-4 was similarly upregulated on the CD8+ T cells (7). In a murine model of prostate cancer, Potluri et al. observed that 90Y-NM600 RPT increased the infiltration of CD8+ T cells in the TME. However, similar to the observations by Jagodinsky et al., immune checkpoints including PD-1, CTLA-4, LAG-3, and VISTA were upregulated on the CD8+ T cells. They also revealed that the CD8+ T cell compartment showed an early increase in effector memory CD8+ T cells, with no increase in either the central memory, resident memory, or short-lived effector cells (93). While Hernandez et al. did not quantify specifically the memory populations following 90Y-NM600 treatment of EL4 tumors, they found in a rechallenge experiment that previously treated complete responder mice inoculated again with EL4 did not grow tumors, suggesting that RPT can induce memory formation (81).
Patel et al. found that NK cell numbers were significantly increased in the TME of B78 tumors, a murine melanoma model, following 90Y-NM600 (8). A similar effect on NK cells was noted when a murine model of human neuroendocrine tumors was treated with 177Lu-based RPT. Additionally, such treatment enhances the activation of the status of infiltrated NK cells, as demonstrated by their expression of Fas ligand (FasL) (96).
Several preclinical studies have reported a decrease in macrophages in the TME following 177Lu-based RPT (81, 95). By staining for the macrophage F4/80 antigen, it was found that 177Lu-BSA-tetrazine RPT moderately decreased the infiltration of macrophages in the TME of medullary breast adenocarcinoma (95). Potluri et al. showed that 3 days after 90Y-based RPT, dendritic cells (DCs) from a murine prostate cancer model showed a non-significant increase in PD-L1 expression (93). Wu et al. demonstrated that 177Lu-DOTATATE increased the infiltration of antigen-presenting cells expressing CD86+ within the TME of a murine model of a human neuroendocrine tumor (96).
Through these preclinical studies, it has become clear that systemically delivered RPT has dynamic effects on the immune cells present in the TME. In several of the aforementioned studies, RPT monotherapy did not elicit a complete tumor response. As such, many included a combination with immune checkpoint inhibition (ICI) to synergize the immunomodulatory effects of the RPT (7, 8, 81, 93). This indicates a need for careful study of the immune cell types present in the TME, along with the activation and exhaustion status, to determine which, if any, tumor models will benefit from RPT monotherapy or require combinations with other therapies, such as immunotherapy, to increase the effectiveness of the RPT.
Peptide receptor radionuclide therapy (PRRT) has been clinically employed for nearly two decades for the management of selected malignancies (97). PRRT is safe and considered to be an effective treatment for two types of solid tumors: neuroendocrine tumors (NET) and gastroenteropancreatic neuroendocrine neoplasms (NENs). 90Y-octreotide and 177Lu-ocreotate are the two most common PRRTs used for NETs and NENs (98, 99). Clinically, these β-particle-emitter-based PRRT agents show that less than 13% of patients exhibit grade 3 or higher hematological toxicities. However, in rare cases, myelodysplastic syndromes or acute leukemia have been reported. Although the expected absorbed doses were below the conventional toxicity threshold, the persistence of acute and long-lasting bone marrow toxicities still raises concerns, especially with repeated administration of PRRT (97). Phase 1 studies have suggested that the upper limit of total activity per cycle of 90Y to minimize hematological toxicity is 5.18 GBq (98). The rate of hematological toxicity is further increased in patients who received cytotoxic chemotherapy prior to PRRT agents. To this end, Kwekkeboom et al. showed that the rate of grade 2 or 3 leukocytopenia or thrombocytopenia was 67% in patients with a history of chemotherapy prior to 177Lu-based PRRT compared to 22% in patients without prior chemotherapy (100).
Two seminal phase 3 trials led to the FDA approval of 177Lu-DOTATATE and 177Lu-PSMA-617. In the NETTER-1 trial, 116 patients were administered 7.4 GBq 177Lu-DOTATATE every 8 weeks for 4 cycles. In total, 9% of patients had a grade 3 or higher lymphopenia (60). In the phase 3 VISION trial, which enrolled 831 patients with mCRPC, patients received either standard of care only or standard of care plus 177Lu-PSMA-617. Compared to the standard of care-only group, there was a 38% reduction in the risk of death and a 60% decrease in disease progression with the addition of 177Lu-PSMA-617 to the standard of care. Although side effects were reported, these did not impact the quality of life of the patients (59). Further analysis of the VISION trial indicated that the average total absorbed dose for 6 × 7.4 GBq (44.4 GBq) was 1.5 Gy (±0.9) in the bone marrow (101). Moreover, in the cohort receiving 177Lu-PSMA-617, the rate of hematological toxicity correlated with the extent of bony metastatic disease burden, as the absorption in these metastatic sites resulted in the irradiation of adjacent bone marrow (102).
Clinically, the β-particle emitter radioisotopes have been shown to be well tolerated and safe. The renewed interest in the field has inspired new clinical trials and unique approaches to better target tumors. However, many of the clinical trials’ primary outcomes is overall survival or disease-free progression, with few analyzing the effects on the systemic immune system, only on the hematologic toxicities of the radionuclides. Understanding the effects and limits of each of these RPTs in the TME and on the immune cells present will be critical to determining optimal combinations of RPTs and immunotherapy, including ICI, in the future.
Because damage induced by β-particle emitters may be easily repaired, higher doses may be necessary to achieve a therapeutic response, which can paradoxically lead to more treatment-related toxicities. Bone marrow and hematologic toxicity, such as anemia, leukopenia, and thrombocytopenia, are the frequent side effects of β-particle emitter-based RPT (103). Furthermore, some β-emitting radiopharmaceuticals are excreted through the kidneys, posing a risk of nephrotoxicity (104). We have summarized the major clinical toxicities observed during the phase 3 trials that led to FDA approval of 177Lu-PSMA-617 in mCRPC (Table 1) and of 177Lu-DOTATATE for metastatic midgut neuroendocrine tumors (Table 2).
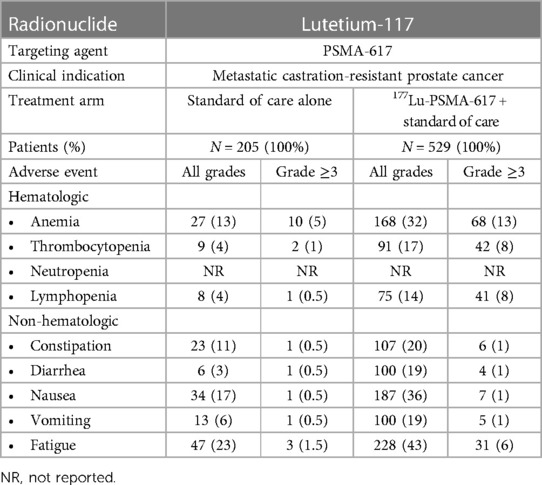
Table 1. Summary of clinical toxicities of β-particle-emitter-based RPT: lutetium-117-PSMA-617 (59).
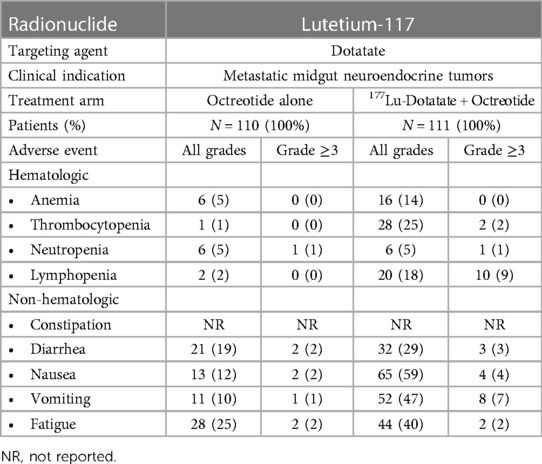
Table 2. Summary of clinical toxicities of β-particle-emitter-based RPT: lutetium-117-dotatate (60).
The advantages of α-particles resulting from their physical properties (short tissue range and high LET) and the regulatory approval of 223RaCl2 for patients with mCRPC have stimulated the development of several α-particle-emitter-based RPTs. Several reports indicate that α-particle irradiation elicits immune activation (82, 84, 105). Here, we will discuss some preclinical studies that have evaluated the effects of α-particles on specific immune cell populations.
Ferreira et al. showed that 225Ac-NM600 abrogated the infiltration of Treg cells in the TME of murine prostate cancer (105). In this study, they compared the anti-tumor and immune modulatory effects of 225Ac-NM600 and 177Lu-NM600 in two different immunocompetent prostate cancer models: MyC-Cap in FVB/NJ mice and Tramp-C1 in C57BL/6 mice. They injected these mice intravenously with either 7.4 or 18.5 kBq of 225Ac-NM600, or 5.5 or 18.5 MBq of 177Lu-NM600. After demonstrating the higher anti-tumor effect of 225Ac-NM600 compared to 177Lu-NM600, the immune response was compared. Immunophenotyping of the TME revealed that 7.4 or 18.5 kBq of 225Ac-NM600 resulted in a significant decrease of Tregs at 28 days post-injection in the Tramp-C1 TME and at 7-, 14-, and 28-days post-injection in the MyC-Cap TME, whereas 177Lu-NM600 had no effect. As Tregs are immunosuppressive cells, their decrease in the TME is beneficial for the anti-tumor response and could explain the superior anti-tumor effect observed with 225Ac-NM600 compared to 177Lu-NM600.
Malamas et al. reported the immunomodulation in tumor cells exposed to 223Ra and demonstrated that 223Ra enhanced T cell-mediated lysis of tumor cells by CD8+ T cells (82). In this study, breast (MDA-MB-231, ZR75-1), prostate (LNCaP, PC3), and lung (H1703, H441) carcinoma cells were first exposed for 96 h to 223Ra, delivering a radiation dose of 4 or 10 Gy. After establishing that there was no substantial tumor cell death, treated cells were co-cultured with CD8+ T cells specific for CEA, MUC, and brachyury epitopes. Their result showed a significant increase in T cell-mediated killing of all the cancer cells tested after 4 or 10 Gy of 223Ra. They concluded that sublethal doses of 223Ra enhance HLA-restricted, antigen-specific, CTL-mediated lysis of various human carcinomas and that such killing can be achieved by targeting a broad repertoire of tumor-associated antigens (82).
Leung et al. reported that 223RaCl2 decreased the splenic CD8+ T cells in Swiss mice (106). Their study aimed to determine the effect of 223RaCl2 on splenic immune cell population size and function. Three groups of mice received intravenously 0, 50, or 600 kBq/kg of 223RaCl2, and the spleens were harvested at days 5, 12, and 19 post-injections for analysis. Compared with the 0 kBq/kg group, there was no significant difference in the total number of harvested splenocytes in the 50 and 600 kBq/kg treated groups at all time points. However, treatment with 600 kBq/kg of 223RaCl2 was found to decrease splenic CD8+ T cells (p = 0.043) significantly and the 50 kBq/kg-treated group approached significance (p = 0.059) at day 19 post-treatment. In contrast, a very slight decrease in CD8+ T cells was observed before 19 days. Based on these results, they concluded that the effect of 223Ra on splenic cells is time- and dose-dependent.
Ferreira et al. observed a dose- and time-dependent decrease in the frequency of CD8+ T cells in the MyC-Cap TME of mice injected with 7.4 (∼300 kBq/kg) or 18.5 kBq (∼700 kBq/kg) of 25Ac-NM600, with the highest decrease in CD8+ T cells at 28 days post-injection with approximately 700 kBq/kg (69). They observed the highest decrease in CD8+ T cells at 19 days post-injection with 600 kBq/kg. However, no significant decrease in CD8+ T cells was observed in the Tramp-C1 TME after injection with 7.4 (∼300 kBq/kg) or 18.5 kBq (∼700 kBq/kg) of 225Ac-NM600 (105). Moreover, 225Ac-NM600 promoted a more active CD8+ T cell repertoire with increased expression of activation and proliferation markers such as CD44+, CD69+, and Ki67+ on CD8+ T cells, suggesting the induction of an immunogenic TME by 225Ac-NM600, which could be responsible for the anti-tumor response. However, no difference in tumor growth delay was observed after treatment with 18.5 kBq of 225Ac-NM600 + anti-CD8 antibody injected twice a week (for depletion of CD8 + cells). This indicated that the anti-tumor effect observed with 225Ac-NM600 did not depend on CD8+ T cell infiltration or stimulation but instead on Treg cell decrease in the TME (105).
Leung et al., after investigating the effect of 223RaCl2 on splenic immune cell population size and function in Swiss Webster mice (106), reported a constant decrease in NK cell population at 5, 12, and 19 days post-injection of 600 kBq/kg of 223RaCl2, while 50 kBq/kg had no effect on NK cell population. As NK cells are a vital part of the immune cells to eliminate cancer cells, a decreased level of NK cells is counter-beneficial for anti-tumor immunity. Furthermore, the cytotoxic activity of NK cells in mice treated with 50 kBq/kg was significantly increased on day 12, while only a marginal increase in cytotoxicity was noted in the group treated with 600 kBq/kg. Based on these results, Leung et al. suggested that low activity of 223Ra would be beneficial to avoid a steady decrease in the NK cell population while potentiating their immune response (106).
Gorin et al. studied the immunogenicity of bismuth-213 (213Bi) in mice with MC-38 adenocarcinoma using a vaccination approach (84). Mice were vaccinated 7 days before MC38 engraftment by subcutaneous injection of 213Bi-irradiated MC-38 cells (6-h incubation with 2.22 MBq/mL). Only 12% (3 of 25) of vaccinated mice developed tumors, compared to 84% (21 of 25) in the control group (non-vaccinated mice), suggesting that 213Bi-treated MC-38 cells are highly immunogenic and can elicit a robust anti-tumor response in vivo. Therefore, they explored in vitro the mechanisms supporting the α-particle-induced anti-tumor immune response by analyzing the immature bone marrow-derived dendritic cell (BMDC) phenotype after 48 h of incubation with conditioned medium from control or 213Bi-treated MC-38. Their results revealed that conditioned medium from 213Bi-treated MC-38 elicited a significant increase of 32% in CD40 expression, 44.8% in CD86 expression, and a non-significant upward trend of 4.4% in CD80 expression on BMDCs. The increased expression of these costimulatory molecules (CD40, CD80, and CD86 on the surface of BMDCs) is a characteristic of DC activation. However, no activation was observed when immature BMDCs were co-cultured with control media. These results suggest that 213Bi induces the release of soluble agents from MC-38 that are capable of activating DCs in vitro.
Furthermore, 213Bi increased the release of DAMPs such as HMGB1 and Hsp70 in the conditioned medium from treated MC-38 cells, which may contribute to the anti-tumor response by activating DCs (84). Similarly, Hagemann et al. demonstrated that the thorium-227 (227Th)-conjugate BAY2287411 targeting mesothelin induced an upregulation of DAMPs (83). After the exposure of OVCAR-3 cells to BAY2287411, upregulation of the DAMPs, calreticulin, HSP70, HSP90, and HMGB1 was observed. Another study also confirmed the upregulation of DAMPs after exposure of MC-38 hMSLN cells to thorium-227 delivered by a mesothelin-targeting compound, resulting in the subsequent activation of dendritic cells (107).
Only a few clinical studies have assessed the immunomodulatory effects of α-particle emitter-based RPT. Some are translational observational studies, and others investigate the clinical benefits of treatment combinations. Here, we will discuss the two primary studies that have evaluated the immunological changes in 223RaCl2-treated mCRPC patients by phenotyping the peripheral blood mononuclear cells (PBMCs) during 223Ra-treatment.
In a study reported in 2017, Kim et al. collected PBMCs before and 3–4 weeks after treatment with 223RaCl2 in 15 men with mCRPC and analyzed the CD8+ T cell population along with their subsets (naive, central memory, and effector memory) by flow cytometry (108). After IV administration of 50 kBq/kg, there was no change in the CD8+ T cell population. However, the proportion of effector memory CD8+ T cells expressing PD-1 was significantly reduced from 20.6% to 14.6% (108).
To better understand the immunological effects of 223Ra, Creemers et al. investigated the composition and abundance of circulating PBMCs in mCRPC patients before, during, and after treatment with 223RaCl2 (109). A total of 30 patients with mCRPC had their PBMCs collected, and longitudinal alterations in circulating immune cell populations were examined through immunophenotyping analysis. Patients received 6 monthly injections of 55 kBq/kg of 223RaCl2. They reported an increase in the proportion of CD8+ T cells expressing the immune checkpoint molecules PD-L1, ICOS, PD-1, or TIM-3, while the percentage of T cells within the PBMC population decreased during 223RaCl2 treatment. Their study was not limited to CD8+ T cells. They also reported a decrease in the total lymphocyte counts by approximately a factor of 2 during treatment, while monocyte counts remained relatively stable during this time. In addition to an increase in the proportion of T cells expressing inhibitory checkpoint molecules (PD-L1, PD-1, and TIM-3), an increase in the proportion of Treg and myeloid-derived suppressor cells (MDSC), two immunosuppressive subsets, was also observed during 223RaCl2 treatment. While no previous data on the effect of 223RaCl2 on Tregs or MDSCs are available, these findings are supported by several other studies reporting that ionizing radiation can lead to the accumulation of circulating and tumor-infiltrating Tregs (110–112) and MDSCs (113). A mechanistic understanding of the cause of this observation might be helpful to stimulate the immune system and optimize combined treatment approaches with RPT and immunotherapies.
α-particle emitters have diverse, profound, and unique immunostimulatory effects. The use of α-particle emitters as radiopharmaceuticals offers several advantages. α-particles have high linear energy transfer and a short path length, making them effective in killing cancer cells while minimizing damage to surrounding healthy tissue. α-particles may also be more effective in treating radioresistant tumors, providing an alternative option for patients who may not respond well to other forms of radiation therapy. This may be due to some of the immunomodulation occurring with the use of α-particle RPT. As interest increases in the field, it will be just as critical to determine the timing and dosing of α-particle RPT administration as it will be to investigate which immune populations are critical to achieving a complete response. Understanding the impact of α-RPT on immune cell populations is essential to elucidating the mechanisms behind its immunostimulatory effects. Such insights could pave the way for the development of combination therapies that act synergistically with α-RPT, potentially promoting tumor elimination and improving treatment outcomes for cancer patients.
α-particle-emitting RPT can lead to hematologic toxicity, including anemia, lymphocytopenia, leukopenia, thrombocytopenia, and neutropenia. However, due to the shorter path length of α-particles, these toxicities tend to be less severe compared to those induced by β-particle emitters (115, 115). Table 3 summarizes the key hematological and non-hematological toxicities observed during the phase 3 trial evaluating the safety and efficacy of 223RaCl2 vs. placebo in patients with mCRPC and bone metastases.

Table 3. Summary of clinical toxicities of α-particle-emitter-based RPT (58).
The probability of AE hitting immune cells present in the TME is very low because of their short range (< 1 µm). Even at the cellular level, AE ionization is so localized that AEEs must be delivered close to a sensitive cellular target (i.e., nuclear DNA, mitochondria, or cell membrane) to maximize their efficacy. For this reason, the immunomodulatory effect of AEE-RPT is the least explored among the forms of emitted radiation from RPT. Only two studies have investigated the effectiveness of AEE radioimmunoconjugates in eradicating the leukemic stem cell population in acute myeloid leukemia or in eliminating myeloid leukemia cells (116, 117). However, these studies are outside the scope of the present report. On the other hand, AEEs are also responsible for non-targeted bystander effects occurring in non-irradiated cells (57, 118). As more efforts are needed to elucidate the immune effects of AEE-based RPT, understanding the AE-induced bystander effects will be critical. This bystander effect involves communication between irradiated and non-irradiated cells through gap junctions or releasing cytokine signals to the extracellular matrix (119, 120). Signals mediated by the bystander effect include cell death, genomic instability, cell cycle, proliferation, and responses to radiation, including radionuclides (56). Both in vitro and in vivo models have been used to experimentally demonstrate the bystander effect with AEE. Using deoxyuridine labeled with the AEE 125I (125IUdR), Howell et al. labeled 50% and 10% of V79 cells, and after replating the cells for clonogenic survival, the observed cell viability in both groups was lower than predicted, implying the existence of intercellular communication between irradiated and non-irradiated cells (121). Using a xenograft model of human colon adenocarcinoma in nude mice and the AEE (125IUdR), Xue et al. demonstrated the bystander response in an in vivo model (122). While the bystander effect occurs with various radionuclides, it is crucial for the therapeutic effect of AEE due to their very short range in tissue. The bystander effect may also be mediated by exosomes, which have emerged as one of the most attractive and promising candidates to initiate bystander effects (123, 124). Double-stranded DNA contained in the exosomes is increasingly recognized for triggering immune responses by acting as DAMP signals (125–129). Taken together, these findings suggest the need for additional studies exploring AE-RPT-induced immune response.
AEEs are often considered to be less toxic compared to α- and β- emitters due to their short range in tissues, which limits their ability to traverse cellular membranes and cause widespread damage (130, 131). However, it is important to note that the emission of other radiation types, such as gamma rays or conversion electrons, in non-pure AEEs like 111In and 195mPt could contribute to toxicity and should be investigated.
Given the abundance of preclinical evidence suggesting the immunostimulatory effects of RPT, the potential benefits of combining RPT with ICI was investigated. Whether using β- or α-particle-emitter-based-RPT in combination with ICI, preclinical studies have reported mixed results, with various studies reporting the therapeutic benefit of such combination therapy while others have reported no benefit over RPT or ICI monotherapy (133). While some of the results have been mixed, there has been a promising positive trend with these combinations. Several studies have shown the efficacy and safety of RPT and ICI combination therapy, along with publishing the immunostimulatory effects of the combinations (7, 8, 93). For example, a lower dose outside the typical therapeutic range of 90Y was used to improve tumor response, reduce spontaneously arising metastatic tumor burden, increase complete response rates, and prolong overall survival compared to ICIs or RPT alone (8). This study also focused on the inherent characteristics of 90Y for dosimetric guidance during treatment. Indeed, other groups have also begun to investigate the therapeutic benefits of combining RPT and ICI while simultaneously using RPT for imaging and dosimetry. Kleinendorst et al. suggested the incorporation of dosimetric tools to correlate the absorbed radiation dose with the immunological effects (133). Other parameters such as the characteristics of the TME, such as tumor metabolism, hypoxia, and immunogenicity, may impact the immunomodulatory effects of RPT agents and the response to ICI. Thus, a better understanding of these parameters in the context of a therapeutic combination with RPT will be critical to harnessing a potential clinical therapeutic benefit. The selection of the optimal pairing of RPT (β- or α-particle emitters) and ICI (PD-1/PD-L1 inhibitors or CTLA-4 inhibitors) will also be crucial in implementing an effective therapeutic combination, as several studies have shown the upregulation of immune susceptibility markers following RPT (7, 93). As with any combination therapy, determining the optimal timing of the combination will also be critical.
RPT is revolutionizing cancer management, especially for patients with metastatic disease. In addition to its direct cytotoxic effects on tumor viability through DNA damage, RPT has the ability to indirectly affect tumor burden by altering the immune system response. This modulation involves various mechanisms including, but not limited to, induction of ICD, alteration of the TME, and enhancement of antigen presentation. These immune-mediated effects contribute to a comprehensive response against cancer, highlighting the multifaceted nature of RPT's therapeutic impact. Understanding these dual mechanisms of action is essential for harnessing the full potential of RPT in cancer treatment strategies. In this review, we have covered several avenues that can be addressed, including a better understanding of the immune cells that play a role in the anti-tumor activity following RPT. Elucidating the mechanisms of RPT-mediated immune response, incorporating RPT dosimetry, selecting the optimal ICI and radionuclide for RPT, and determining the optimal timing, dose, and fractionation of RPT for the therapeutic combination with ICI will be critical to fully harnessing the therapeutic benefits of RPT while minimizing the toxicities, especially in patients with metastatic disease. Through dedicated exploration of these research paths, we can unveil the full therapeutic potential of RPT and pave the way for tailored, life-prolonging treatments for patients battling metastatic cancer. This proactive approach not only fosters a deeper understanding of RPT's mechanisms of action but also enables the development of novel strategies for optimizing treatment efficacy and minimizing adverse effects. Ultimately, these endeavors hold the promise of revolutionizing cancer care by offering personalized therapeutic solutions that address the unique needs of each patient.
AS: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. MI: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. AT: Writing – review & editing, Writing – original draft. TC: Writing – review & editing, Writing – original draft. RH: Writing – review & editing, Resources. ZM: Writing – review & editing, Writing – original draft, Resources. QS: Writing – review & editing, Writing – original draft, Resources, Data curation, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
We acknowledge generous support from the National Cancer Institute (NCI)/National Institute of Health (NIH) P01 CA250972 (RH, ZSM), the University of Wisconsin (UW)−Madison Office of the Vice Chancellor for Research and Graduate Education, and funding from the Wisconsin Alumni Research Foundation (RH, ZSM). The contents of this article do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US government.
The authors would like to thank Tracy Berg for the editorial review of this manuscript.
RH received patent royalties from the Wisconsin Alumni Research Foundation and consulting fees from Archeus Technologies Inc. and Monopar Therapeutics. ZSM is a member of the Scientific Advisory Board for Archeus Technologies, Seneca Therapeutics, and NorthStar Medical Isotopes; received royalties from patents held by the Wisconsin Alumni Research Foundation; received stock/stock options from the Archeus Technologies Scientific Advisory Board and the Seneca Therapeutics Scientific Advisory Board; received research support from Point Biopharma, Telix Pharmaceuticals, and XRD Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
The author(s) declared that they were editorial board members of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors or the reviewers. Any product that may be evaluated in this article, or any claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rupnow BA, Murtha AD, Alarcon RM, Giaccia AJ, Knox SJ. Direct evidence that apoptosis enhances tumor responses to fractionated radiotherapy. Cancer Res. (1998) 58:1779–84.9581811
2. Dewey WC, Ling CC, Meyn RE. Radiation-induced apoptosis: relevance to radiotherapy. Int J Radiat Oncol Biol Phys. (1995) 33:781–96. doi: 10.1016/0360-3016(95)00214-8
3. Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. (2010) 31:363–72. doi: 10.1007/s13277-010-0042-8
4. Merrick A, Errington F, Milward K, O’Donnell D, Harrington K, Bateman A, et al. Immunosuppressive effects of radiation on human dendritic cells: reduced IL-12 production on activation and impairment of naive T-cell priming. Br J Cancer. (2005) 92(8):1450–8. doi: 10.1038/sj.bjc.6602518
5. McMahon RA, D’Souza C, Neeson PJ, Siva S. Innate immunity: looking beyond T-cells in radiation and immunotherapy combinations. Neoplasia. (2023) 46:100940. doi: 10.1016/j.neo.2023.100940
6. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. (2018) 24(12):1845–51. doi: 10.1038/s41591-018-0232-2
7. Jagodinsky JC, Jin WJ, Bates AM, Hernandez R, Grudzinski JJ, Marsh IR, et al. Temporal analysis of type 1 interferon activation in tumor cells following external beam radiotherapy or targeted radionuclide therapy. Theranostics. (2021) 11(13):6120–37. doi: 10.7150/thno.54881
8. Patel RB, Hernandez R, Carlson P, Grudzinski J, Bates AM, Jagodinsky JC, et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci Transl Med. (2021) 13(602):eabb3631. doi: 10.1126/scitranslmed.abb3631
9. Darragh LB, Gadwa J, Pham TT, Van Court B, Neupert B, Olimpo NA, et al. Elective nodal irradiation mitigates local and systemic immunity generated by combination radiation and immunotherapy in head and neck tumors. Nat Commun. (2022) 13(1):7015. doi: 10.1038/s41467-022-34676-w
10. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. (2009) 114(3):589–95. doi: 10.1182/blood-2009-02-206870
11. Bhide S, Nutting C. Recent advances in radiotherapy. BMC Med. (2010) 8:25. doi: 10.1186/1741-7015-8-25
12. Quast U. Whole body radiotherapy: a TBI-guideline. J Med Phys. (2006) 31:5. doi: 10.4103/0971-6203.25664
13. Volkert WA, Goeckeler WF, Ehrhardt GJ, Ketring AR. Therapeutic radionuclides: production and decay property considerations. J Nucl Med. (1991) 32:174–85.1988628
14. Zimmermann RG. Why are investors not interested in my radiotracer? The industrial and regulatory constraints in the development of radiopharmaceuticals. Nucl Med Biol. (2013) 40:155–66. doi: 10.1016/j.nucmedbio.2012.10.012
15. Sgouros G, Bodei L, McDevitt MR, Nedrow JR. Radiopharmaceutical therapy in cancer: clinical advances and challenges. Nat Rev Drug Discov. (2020) 19:589–608. doi: 10.1038/s41573-020-0073-9
16. Chargari C, Deutsch E, Blanchard P, Gouy S, Martelli H, Guérin F, et al. Brachytherapy: an overview for clinicians. CA Cancer J Clin. (2019) 69(5):386–401. doi: 10.3322/caac.21578
17. Delaney GP, Barton MB. Evidence-based estimates of the demand for radiotherapy. Clin Oncol. (2015) 27:70–6. doi: 10.1016/j.clon.2014.10.005
18. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. (2005) 104:1129–37. doi: 10.1002/cncr.21324
19. Connell PP, Hellman S. Advances in radiotherapy and implications for the next century: a historical perspective. Cancer Res. (2009) 69:383–92. doi: 10.1158/0008-5472.CAN-07-6871
20. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. OncoImmunology. (2014) 3(4):e28518. doi: 10.4161/onci.28518
21. Morris ZS, Guy EI, Francis DM, Gressett MM, Werner LR, Carmichael LL, et al. In situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Research. (2016) 76(13):3929–41. doi: 10.1158/0008-5472.CAN-15-2644
22. Yamazaki T, Vanpouille-Box C, Demaria S, Galluzzi L. Immunogenic cell death driven by radiation—impact on the tumor microenvironment. In: Lee PP, Marincola FM, editors. Tumor Microenvironment. Cham: Springer International Publishing (2020). p. 281–96.
23. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
24. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. (2009) 16(1):3–11. doi: 10.1038/cdd.2008.150
25. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. (2020) 8(1):e000337. doi: 10.1136/jitc-2019-000337
26. Sia J, Szmyd R, Hau E, Gee HE. Molecular mechanisms of radiation-induced cancer cell death: a primer. Front Cell Dev Biol. (2020) 8:41. doi: 10.3389/fcell.2020.00041
27. Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. (2020) 21:120–34. doi: 10.1038/s41590-019-0561-4
28. Feng M, Marjon KD, Zhu F, Weissman-Tsukamoto R, Levett A, Sullivan K, et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun. (2018) 9(1):3194. doi: 10.1038/s41467-018-05211-7
29. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13(1):54–61. doi: 10.1038/nm1523
30. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
31. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13(9):1050–9. doi: 10.1038/nm1622
32. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. (2005) 174(12):7516–23. doi: 10.4049/jimmunol.174.12.7516
33. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. (2015) 15:405–14. doi: 10.1038/nri3845
34. Guillot B, Portales P, Thanh AD, Merlet S, Dereure O, Clot J, et al. The expression of cytotoxic mediators is altered in mononuclear cells of patients with melanoma and increased by interferonalpha treatment. Br J Dermatol. (2005) 152(4):690–6. doi: 10.1111/j.1365-2133.2005.06512.x
35. Jin WJ, Zangl LM, Hyun M, Massoud E, Schroeder K, Alexandridis RA, et al. ATM inhibition augments type I interferon response and antitumor T-cell immunity when combined with radiation therapy in murine tumor models. J Immunother Cancer. (2023) 11(9):e007474. doi: 10.1136/jitc-2023-007474
36. Lim JYH, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol Immunother. (2014) 63:259–71. doi: 10.1007/s00262-013-1506-7
37. Cole LJ, Fishler MC, Ellis ME, Bond VP. Protection of mice against X-irradiation by spleen homogenates administered after exposure. Exp Biol Med. (1952) 80:112–7. doi: 10.3181/00379727-80-19540
38. Thomas E, Buckner C, Banaji M, Clift R, Fefer A, Flournoy N, et al. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. Blood. (1977) 49(4):511–33. doi: 10.1182/blood.V49.4.511.511
39. Mattiuzzi C, Lippi G. Current cancer epidemiology. J Epidemiol Glob Health. (2019) 9:217–22. doi: 10.2991/jegh.k.191008.001
40. Liu LT, Chen QY, Tang LQ, Guo SS, Guo L, Mo HY, et al. The prognostic value of treatment-related lymphopenia in nasopharyngeal carcinoma patients. Cancer Res Treat. (2018) 50(1):19–29. doi: 10.4143/crt.2016.595
41. Mendez JS, Govindan A, Leong J, Gao F, Huang J, Campian JL. Association between treatmentrelated lymphopenia and overall survival in elderly patients with newly diagnosed glioblastoma. J Neurooncol. (2016) 127(2):329–35. doi: 10.1007/s11060-015-2037-1
42. Rudra S, Hui C, Rao YJ, Samson P, Lin AJ, Chang X, et al. Effect of radiation treatment volume reduction on lymphopenia in patients receiving chemoradiotherapy for glioblastoma. Int J Radiat Oncol Biol Phys. (2018) 101(1):217–25. doi: 10.1016/j.ijrobp.2018.01.069
43. Tang C, Liao Z, Gomez D, Levy L, Zhuang Y, Gebremichael RA, et al. Lymphopenia association with gross tumor volume and lung V5 and its effects on non-small cell lung cancer patient outcomes. Int J Radiat Oncol Biol Phys. (2014) 89(5):1084–91. doi: 10.1016/j.ijrobp.2014.04.025
44. Cho O, Chun M, Chang S-J, Oh Y-T, Noh OK. Prognostic value of severe lymphopenia during pelvic concurrent chemoradiotherapy in cervical cancer. Anticancer Res. (2016) 36:3541–7.27354621
45. Cho O, Oh Y-T, Chun M, Noh OK, Lee H-W. Radiation-related lymphopenia as a new prognostic factor in limited-stage small cell lung cancer. Tumour Biol. (2016) 37:971–8. doi: 10.1007/s13277-015-3888-y
46. Davuluri R, Jiang W, Fang P, Xu C, Komaki R, Gomez DR, et al. Lymphocyte nadir and esophageal cancer survival outcomes after chemoradiation therapy. Int J Radiat Oncol Biol Phys. (2017) 99(1):128–35. doi: 10.1016/j.ijrobp.2017.05.037
47. Balmanoukian A, Ye X, Herman J, Laheru D, Grossman SA. The association between treatment-related lymphopenia and survival in newly diagnosed patients with resected adenocarcinoma of the pancreas. Cancer Invest. (2012) 30:571–6. doi: 10.3109/07357907.2012.700987
48. Wild AT, Ye X, Ellsworth SG, Smith JA, Narang AK, Garg T, et al. The association between chemoradiation-related lymphopenia and clinical outcomes in patients with locally advanced pancreatic adenocarcinoma. Am J Clin Oncol. (2015) 38(3):259–65. doi: 10.1097/COC.0b013e3182940ff9
49. Chadha AS, Liu G, Chen HC, Das P, Minsky BD, Mahmood U, et al. Does unintentional splenic radiation predict outcomes after pancreatic cancer radiation therapy? Int J Radiat Oncol Biol Phys. (2017) 97(2):323–32. doi: 10.1016/j.ijrobp.2016.10.046
50. Liu J, Zhao Q, Deng W, Lu J, Xu X, Wang R, et al. Radiation-related lymphopenia is associated with spleen irradiation dose during radiotherapy in patients with hepatocellular carcinoma. Radiat Oncol. (2017) 12(1):90. doi: 10.1186/s13014-017-0824-x
51. Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans V, et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. (2005) 366(9503):2087–106. doi: 10.1016/S0140-6736(05)67887-7
52. Demoor-Goldschmidt C, de Vathaire F. Review of risk factors of secondary cancers among cancer survivors. Br J Radiol. (2019) 92:20180390. doi: 10.1259/bjr.20180390
53. Dumortier J, Decullier E, Hilleret MN, Bin-Dorel S, Valette PJ, Boillot O, et al. Adjuvant intraarterial lipiodol or 131 I-lipiodol after curative treatment of hepatocellular carcinoma: a prospective randomized trial. J Nucl Med. (2014) 55(6):877–83. doi: 10.2967/jnumed.113.131367
54. Pouget JP, Navarro-Teulon I, Bardiès M, Chouin N, Cartron G, Pèlegrin A, et al. Clinical radioimmunotherapy—the role of radiobiology. Nat Rev Clin Oncol. (2011) 8(12):720–34. doi: 10.1038/nrclinonc.2011.160
55. Pouget J-P, Lozza C, Deshayes E, Boudousq V, Navarro-Teulon I. Introduction to radiobiology of targeted radionuclide therapy. Front Med (Lausanne). (2015) 2:12. doi: 10.3389/fmed.2015.00012
56. Brady D, O’Sullivan JM, Prise KM. What is the role of the bystander response in radionuclide therapies? Front Oncol. (2013) 3:215. doi: 10.3389/fonc.2013.00215
57. Idrissou MB, Pichard A, Tee B, Kibedi T, Poty S, Pouget JP. Targeted radionuclide therapy using auger electron emitters: the quest for the right vector and the right radionuclide. Pharmaceutics. (2021) 13(7):980. doi: 10.3390/pharmaceutics13070980
58. Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Fosså SD, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. (2013) 369(3):213–23. doi: 10.1056/NEJMoa1213755
59. Sartor O, De Bono J, Chi KN, Fizazi K, Herrmann K, Rahbar K, et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. (2021) 385(12):1091–103. doi: 10.1056/NEJMoa2107322
60. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 Trial of 177 Lu-dotatate for midgut neuroendocrine tumors. N Engl J Med. (2017) 376(2):125–35. doi: 10.1056/NEJMoa1607427
61. Poeppel TD, Handkiewicz-Junak D, Andreeff M, Becherer A, Bockisch A, Fricke E, et al. EANM guideline for radionuclide therapy with radium-223 of metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. (2018) 45(5):824–45. doi: 10.1007/s00259-017-3900-4
62. Hennrich U, Eder M. [177lu]lu-PSMA-617 (PluvictoTM): the first FDA-approved radiotherapeutical for treatment of prostate cancer. Pharmaceuticals. (2022) 15:1292. doi: 10.3390/ph15101292
63. Hennrich U, Kopka K. Lutathera®: the first FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals. (2019) 12:114. doi: 10.3390/ph12030114
64. Gill MR, Falzone N, Du Y, Vallis KA. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. (2017) 18:e414–23. doi: 10.1016/S1470-2045(17)30379-0
65. Valkema R, De Jong M, Bakker WH, Breeman WA, Kooij PP, Lugtenburg PJ, et al. Phase I study of peptide receptor radionuclide therapy with [In-DTPA]octreotide: the Rotterdam experience. Semin Nucl Med. (2002) 32(2):110–22. doi: 10.1053/snuc/2002.31025
66. Anthony LB, Woltering EA, Espenan GD, Cronin MD, Maloney TJ, McCarthy KE. Indium-111-pentetreotide prolongs survival in gastroenteropancreatic malignancies. Semin Nucl Med. (2002) 32(2):123–32. doi: 10.1053/snuc.2002.31769
67. Balster DA, O’Dorisio MS, Summers MA, Turman MA. Segmental expression of somatostatin receptor subtypes sst(1) and sst(2) in tubules and glomeruli of human kidney. Am J Physiol Renal Physiol. (2001) 280:F457–465. doi: 10.1152/ajprenal.2001.280.3.F457
68. Amro H, Wilderman SJ, Dewaraja YK, Roberson PL. Methodology to incorporate biologically effective dose and equivalent uniform dose in patient-specific 3-dimensional dosimetry for non-hodgkin lymphoma patients targeted with 131I-tositumomab therapy. J Nucl Med. (2010) 51:654–9. doi: 10.2967/jnumed.109.067298
69. Fowler JF. Radiobiological aspects of low dose rates in radioimmunotherapy. Int J Radiat Oncol Biol Phys. (1990) 18:1261–9. doi: 10.1016/0360-3016(90)90467-X
70. Marcu L, Bezak E, Allen BJ. Global comparison of targeted alpha vs targeted beta therapy for cancer: in vitro, in vivo and clinical trials. Crit Rev Oncol Hematol. (2018) 123:7–20. doi: 10.1016/j.critrevonc.2018.01.001
71. Dekempeneer Y, Keyaerts M, Krasniqi A, Puttemans J, Muyldermans S, Lahoutte T, et al. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin Biol Ther. (2016) 16(8):1035–47. doi: 10.1080/14712598.2016.1185412
72. Czerwińska M, Bilewicz A, Kruszewski M, Wegierek-Ciuk A, Lankoff A. Targeted radionuclide therapy of prostate cancer—from basic research to clinical perspectives. Molecules. (2020) 25:1743. doi: 10.3390/molecules25071743
73. Poty S, Francesconi LC, McDevitt MR, Morris MJ, Lewis JS. α-emitters for radiotherapy: from basic radiochemistry to clinical studies-part 1. J Nucl Med. (2018) 59:878–84. doi: 10.2967/jnumed.116.186338
74. Sgouros G, Roeske JC, McDevitt MR, Palm S, Allen BJ, Fisher DR, et al. MIRD pamphlet no. 22 (abridged): radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J Nucl Med. (2010) 51(2):311–28. doi: 10.2967/jnumed.108.058651
75. Stokke C, Kvassheim M, Blakkisrud J. Radionuclides for targeted therapy: physical properties. Molecules. (2022) 27:5429. doi: 10.3390/molecules27175429
76. Ljungberg M, Celler A, Konijnenberg MW, Eckerman KF, Dewaraja YK, Sjögreen-Gleisner K. MIRD pamphlet no. 26: joint EANM/MIRD guidelines for quantitative 177 Lu SPECT applied for dosimetry of radiopharmaceutical therapy. J Nucl Med. (2016) 57(1):151–62. doi: 10.2967/jnumed.115.159012
77. Dewaraja YK, Ljungberg M, Green AJ, Zanzonico PB, Frey EC, SNMMI MIRD Committee, et al. MIRD pamphlet no. 24: guidelines for quantitative 131 I SPECT in dosimetry applications. J Nucl Med. (2013) 54(12):2182–8. doi: 10.2967/jnumed.113.122390
78. Othman MFB, Mitry NR, Lewington VJ, Blower PJ, Terry SYA. Re-assessing gallium-67 as a therapeutic radionuclide. Nucl Med Biol. (2017) 46:12–8. doi: 10.1016/j.nucmedbio.2016.10.008
79. Pirovano G, Jannetti SA, Carter LM, Sadique A, Kossatz S, Guru N, et al. Targeted brain tumor radiotherapy using an auger emitter. Clin Cancer Res. (2020) 26(12):2871–81. doi: 10.1158/1078-0432.CCR-19-2440
80. Rouanet J, Benboubker V, Akil H, Hennino A, Auzeloux P, Besse S, et al. Immune checkpoint inhibitors reverse tolerogenic mechanisms induced by melanoma targeted radionuclide therapy. Cancer Immunol Immunother. (2020) 69(10):2075–88. doi: 10.1007/s00262-020-02606-8
81. Hernandez R, Walker KL, Grudzinski JJ, Aluicio-Sarduy E, Patel R, Zahm CD, et al. 90YNM600 targeted radionuclide therapy induces immunologic memory in syngeneic models of Tcell non-Hodgkin’s lymphoma. Commun Biol. (2019) 2(1):79. doi: 10.1038/s42003-019-0327-4
82. Malamas AS, Gameiro SR, Knudson KM, Hodge JW. Sublethal exposure to alpha radiation (223Ra dichloride) enhances various carcinomas’ sensitivity to lysis by antigen-specific cytotoxic T lymphocytes through calreticulin-mediated immunogenic modulation. Oncotarget. (2016) 7:86937–47. doi: 10.18632/oncotarget.13520
83. Hagemann UB, Ellingsen C, Schuhmacher J, Kristian A, Mobergslien A, Cruciani V, et al. Mesothelin-targeted thorium-227 conjugate (MSLN-TTC): preclinical evaluation of a new targeted alpha therapy for mesothelin-positive cancers. Clin Cancer Res. (2019) 25(15):4723–34. doi: 10.1158/1078-0432.CCR-18-3476
84. Gorin JB, Ménager J, Gouard S, Maurel C, Guilloux Y, Faivre-Chauvet A, et al. Antitumor immunity induced after α irradiation. Neoplasia. (2014) 16(4):319–28. doi: 10.1016/j.neo.2014.04.002
85. Gorin JB, Guilloux Y, Morgenstern A, Chérel M, Davodeau F, Gaschet J. Using α radiation to boost cancer immunity? Oncoimmunology. (2014) 3(9):e954925. doi: 10.4161/21624011.2014.954925
86. Constanzo J, Bouden Y, Godry L, Kotzki PO, Deshayes E, Pouget JP. Immunomodulatory effects of targeted radionuclide therapy. Int Rev Cell Mol Biol. (2023):105–36. doi: 10.1016/bs.ircmb.2023.02.001
87. Morris ZS, Wang AZ, Knox SJ. The radiobiology of radiopharmaceuticals. Semin Radiat Oncol. (2021) 31:20–7. doi: 10.1016/j.semradonc.2020.07.002
88. Nagasawa H, Little JB. Induction of sister chromatid exchanges by extremely low doses of α-Particles1. Cancer Res. (1992) 52:6394–6.1423287
89. Murphy JB, Morton JJ. The effect of roentgen rays on the rate of growth of spontaneous tumors in micE. J Exp Med. (1915) 22:800–3. doi: 10.1084/jem.22.6.800
90. Leung CN, Canter BS, Rajon D, Bäck TA, Fritton JC, Azzam EI, et al. Dose-dependent growth delay of breast cancer xenografts in the bone marrow of mice treated with 223 Ra: the role of bystander effects and their potential for therapy. J Nucl Med. (2020) 61(1):89–95. doi: 10.2967/jnumed.119.227835
91. Rajon DA, Canter BS, Leung CN, Bäck TA, Fritton JC, Azzam EI, et al. Modeling bystander effects that cause growth delay of breast cancer xenografts in bone marrow of mice treated with radium-223. Int J Radiat Biol. (2021) 97(9):1217–28. doi: 10.1080/09553002.2021.1951392
92. Canter BS, Leung CN, Fritton JC, Bäck T, Rajon D, Azzam EI, et al. Radium-223–induced bystander effects cause DNA damage and apoptosis in disseminated tumor cells in bone marrow. Mol Cancer Res. (2021) 19(10):1739–50. doi: 10.1158/1541-7786.MCR-21-0005
93. Potluri HK, Ferreira CA, Grudzinski J, Massey C, Aluicio-Sarduy E, Engle JW, et al. Antitumor efficacy of 90 Y-NM600 targeted radionuclide therapy and PD-1 blockade is limited by regulatory T cells in murine prostate tumors. J Immunother Cancer. (2022) 10(8):e005060. doi: 10.1136/jitc-2022-005060
94. Hernandez R, Grudzinski JJ, Aluicio-Sarduy E, Massey CF, Pinchuk AN, Bitton AN, et al. 177Lu-NM600 targeted radionuclide therapy extends survival in syngeneic murine models of triple-negative breast cancer. J Nucl Med. (2020) 61(8):1187–94. doi: 10.2967/jnumed.119.236265
95. Vito A, Rathmann S, Mercanti N, El-Sayes N, Mossman K, Valliant J. Combined radionuclide therapy and immunotherapy for treatment of triple negative breast cancer. IJMS. (2021) 22(9):4843. doi: 10.3390/ijms22094843
96. Wu Y, Pfeifer A, Myschetzky R, Garbyal R, Rasmussen P, Knigge U, et al. Induction of anti-tumor immune responses by peptide receptor radionuclide therapy with 177Lu-DOTATATE in a murine model of a human neuroendocrine tumor. Diagnostics. (2013) 3(4):344–55. doi: 10.3390/diagnostics3040344
97. Bodei L, Cremonesi M, Grana C, Rocca P, Bartolomei M, Chinol M, et al. Receptor radionuclide therapy with 90Y-[DOTA]0-Tyr3-octreotide (90Y-DOTATOC) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. (2004) 31(7):1038–46. doi: 10.1007/s00259-004-1571-4
98. Bodei L, Cremonesi M, Zoboli S, Grana C, Bartolomei M, Rocca P, et al. Receptor-mediated radionuclide therapy with 90Y-DOTATOC in association with amino acid infusion: a phase I study. Eur J Nucl Med Mol Imaging. (2003) 30(2):207–16. doi: 10.1007/s00259-002-1023-y
99. Gabriel M, Nilica B, Kaiser B, Virgolini IJ. Twelve-year follow-up after peptide receptor radionuclide therapy. J Nucl Med. (2019) 60:524–9. doi: 10.2967/jnumed.118.215376
100. Kwekkeboom DJ, Bakker WH, Kam BL, Teunissen JJM, Kooij PPM, Herder WW, et al. Treatment of patients with gastro-entero-pancreatic (GEP) tumours with the novel radiolabelled somatostatin analogue [177Lu-DOTA0,Tyr3]octreotate. Eur J Nucl Med Mol Imaging. (2003) 30(3):417–22. doi: 10.1007/s00259-002-1050-8
101. Herrmann K, Rahbar K, Eiber M, Krause BJ, Lassmann M, Jentzen W, et al. Dosimetry of 177 Lu-PSMA-617 for the treatment of metastatic castration-resistant prostate cancer: results from the VISION trial sub-study. JCO. (2022) 40(6 Suppl):97. doi: 10.1200/JCO.2022.40.6_suppl.097
102. Emmett L, Willowson K, Violet J, Shin J, Blanksby A, Lee J. Lutetium 177 PSMA radionuclide therapy for men with prostate cancer: a review of the current literature and discussion of practical aspects of therapy. J Med Radiat Sci. (2017) 64(1):52–60. doi: 10.1002/jmrs.227
103. Sharkey RM, Brenner A, Burton J, Hajjar G, Toder SP, Alavi A, et al. Radioimmunotherapy of non-Hodgkin’s lymphoma with 90Y-DOTA humanized anti-CD22 IgG (90Y-Epratuzumab): do tumor targeting and dosimetry predict therapeutic response? J Nucl Med. (2003) 44(12):2000–18.14660727
104. Bergsma H, Konijnenberg MW, van der Zwan WA, Kam BLR, Teunissen JJM, Kooij PP, et al. Nephrotoxicity after PRRT with (177)Lu-DOTA-octreotate. Eur J Nucl Med Mol Imaging. (2016) 43(10):1802–11. doi: 10.1007/s00259-016-3382-9
105. Ferreira CA, Potluri HK, Massey C, Grudzinski JJ, Carston A, Clemons N, et al. Profound immunomodulatory effects of 225 Ac-NM600 drive enhanced anti-tumor response in prostate cancer. Immunology. (2022). doi: 10.1101/2022.09.26.509374
106. Leung CN, Howell DM, Howell RW. Radium-223 dichloride causes transient changes in natural killer cell population and cytotoxic function. Int J Radiat Biol. (2021) 97:1417–24. doi: 10.1080/09553002.2021.1956002
107. Lejeune P, Cruciani V, Berg-Larsen A, Schlicker A, Mobergslien A, Bartnitzky L, et al. Immunostimulatory effects of targeted thorium-227 conjugates as single agent and in combination with anti-PD-L1 therapy. J Immunother Cancer. (2021) 9(10):e002387. doi: 10.1136/jitc-2021-002387
108. Kim JW, Shin MS, Kang Y, Kang I, Petrylak DP. Immune analysis of radium-223 in patients with metastatic prostate cancer. Clin Genitourin Cancer. (2018) 16:e469–76. doi: 10.1016/j.clgc.2017.10.010
109. Creemers JHA, Van Der Doelen MJ, Van Wilpe S, Hermsen R, Duiveman-de Boer T, Somford DM, et al. Immunophenotyping reveals longitudinal changes in circulating immune cells during radium-223 therapy in patients with metastatic castration-resistant prostate cancer. Front Oncol. (2021) 11:667658. doi: 10.3389/fonc.2021.667658
110. Balázs K, Kis E, Badie C, Bogdándi EN, Candéias S, Garcia LC, et al. Radiotherapy-induced changes in the systemic immune and inflammation parameters of head and neck cancer patients. Cancers (Basel). (2019) 11(9):1324. doi: 10.3390/cancers11091324
111. Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, et al. Stereotactic radiotherapy increases functionally suppressive regulatory T cells in the tumor microenvironment. Cancer Immunol Res. (2017) 5(11):992–1004. doi: 10.1158/2326-6066.CIR-17-0040
112. Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. (2011) 81(4):1128–35. doi: 10.1016/j.ijrobp.2010.09.034
113. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. (2013 1) 73(9):2782–94. doi: 10.1158/0008-5472.CAN-12-3981
114. Nguyen NC, Shah M, Appleman LJ, Parikh R, Mountz JM. Radium-223 therapy for patients with metastatic castrate-resistant prostate cancer: an update on literature with case presentation. Int J Mol Imaging. (2016) 2016:2568031. doi: 10.1155/2016/2568031
115. Tranel J, Feng FY, James SS, Hope TA. Effect of microdistribution of alpha and beta-emitters in targeted radionuclide therapies on delivered absorbed dose in a GATE model of bone marrow. Phys Med Biol. (2021) 66:035016. doi: 10.1088/1361-6560/abd3ef
116. Leyton JV, Hu M, Gao C, Turner PV, Dick JE, Minden M, et al. Auger electron radioimmunotherapeutic agent specific for the CD123+/CD131− phenotype of the leukemia stem cell population. J Nucl Med. (2011) 52(9):1465–73. doi: 10.2967/jnumed.111.087668
117. Chen P, Wang J, Hope K, Jin L, Dick J, Cameron R, et al. Nuclear localizing sequences promote nuclear translocation and enhance the radiotoxicity of the anti-CD33 monoclonal antibody HuM195 labeled with 111In in human myeloid leukemia cells. J Nucl Med. (2006) 47(5):827–36.16644753
118. Pouget J-P, Georgakilas AG, Ravanat J-L. Targeted and off-target (bystander and abscopal) effects of radiation therapy: redox mechanisms and risk/benefit analysis. Antioxid Redox Signaling. (2018) 29:1447–87. doi: 10.1089/ars.2017.7267
119. Ku A, Facca VJ, Cai Z, Reilly RM. Auger electrons for cancer therapy – a review. EJNMMI Radiopharm Chem. (2019) 4:27. doi: 10.1186/s41181-019-0075-2
120. Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. (2009) 9:351–60. doi: 10.1038/nrc2603
121. Howell RW, Bishayee A. Bystander effects caused by nonuniform distributions of DNA-incorporated (125)I. Micron. (2002) 33:127–32. doi: 10.1016/S0968-4328(01)00007-5
122. Xue LY, Butler NJ, Makrigiorgos GM, Adelstein SJ, Kassis AI. Bystander effect produced by radiolabeled tumor cells in vivo. Proc Natl Acad Sci U S A. (2002) 99:13765–70. doi: 10.1073/pnas.182209699
123. Bewicke-Copley F, Mulcahy LA, Jacobs LA, Samuel P, Akbar N, Pink RC, et al. Extracellular vesicles released following heat stress induce bystander effect in unstressed populations. J Extracell Vesicles. (2017) 6(1):1340746. doi: 10.1080/20013078.2017.1340746
124. Karam J, Constanzo J, Pichard A, Gros L, Chopineau J, Morille M, et al. Rapid communication: insights into the role of extracellular vesicles during Auger radioimmunotherapy. Int J Radiat Biol. (2023) 99(1):109–18. doi: 10.1080/09553002.2021.1955999
125. Al-Mayah A, Bright S, Chapman K, Irons S, Luo P, Carter D, et al. The non-targeted effects of radiation are perpetuated by exosomes. Mutat Res/Fundam Mol Mech Mutagen. (2015) 772:38–45. doi: 10.1016/j.mrfmmm.2014.12.007
126. Jelonek K, Widlak P, Pietrowska M. The influence of ionizing radiation on exosome composition, secretion and intercellular communication. Protein Pept Lett. (2016) 23:656–63. doi: 10.2174/0929866523666160427105138
127. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res. (2018) 6(8):910–20. doi: 10.1158/2326-6066.CIR-17-0581
128. Buzas EI. The roles of extracellular vesicles in the immune system. Nat Rev Immunol. (2023) 23:236–50. doi: 10.1038/s41577-022-00763-8
129. Zhou X, Xie F, Wang L, Zhang L, Zhang S, Fang M, et al. The function and clinical application of extracellular vesicles in innate immune regulation. Cell Mol Immunol. (2020) 17(4):323–34. doi: 10.1038/s41423-020-0391-1
130. Costantini DL, McLarty K, Lee H, Done SJ, Vallis KA, Reilly RM. Antitumor effects and normal-tissue toxicity of 111 in-nuclear localization sequence-trastuzumab in athymic mice bearing HER-positive human breast cancer xenografts. J Nucl Med. (2010) 51(7):1084–91. doi: 10.2967/jnumed.109.072389
131. Chen P, Cameron R, Wang J, Vallis KA, Reilly RM. Antitumor effects and normal tissue toxicity of 111In-labeled epidermal growth factor administered to athymic mice bearing epidermal growth factor receptor-positive human breast cancer xenografts. J Nucl Med. (2003) 44:1469–78.12960194
Keywords: radiopharmaceutical therapy, radiation therapy, radionuclide, alpha-particle emitter, beta-particle emitter, auger electron emitter, metastatic cancer, immune system
Citation: Shea AG, Idrissou MB, Torres AI, Chen T, Hernandez R, Morris ZS and Sodji QH (2024) Immunological effects of radiopharmaceutical therapy. Front. Nucl. Med. 4:1331364. doi: 10.3389/fnume.2024.1331364
Received: 1 November 2023; Accepted: 14 March 2024;
Published: 4 April 2024.
Edited by:
Roger W. Howell, The State University of New Jersey, United StatesReviewed by:
Nahum Puebla-Osorio, University of Texas MD Anderson Cancer Center, United States© 2024 Shea, Idrissou, Torres, Chen, Hernandez, Morris and Sodji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quaovi H. Sodji, cXNvZGppQGh1bW9uYy53aXNjLmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.