 Stellan Holgersson
Stellan Holgersson Pawan Kumar
Pawan Kumar- Chalmers University of Technology, Department of Chemistry and Chemical Engineering, Göteborg, Sweden
The literature for radionuclide sorption on four common granitic minerals have been surveyed. Mainly, such studies were modelling using Thermodynamic Sorption Models were investigated. Although the studies give a far from concerted results, they agree on the necessity to model radionuclide uptake by granitic minerals with a combination of ion exchange and surface complexation reactions. For the sheet-silicates biotite and chlorite alkaline and alkaline earth mainly bind by ion exchange but there is also a clear pH effect for this, which shows the importance of protons competing with metal cations for the exchange sites. For multivalent metal cations, surface complexation is the model of choice since the binding to mineral surfaces seems to be strongly dependent on pH and to be little affected by an increase in ion strength. Anion sorption seems to be taking place also by surface complexation, where the sorption mainly takes place at low pH. For the feldspar minerals K-feldspar and plagioclase the sorption is also modelled by the two reaction mechanisms ion exchange and surface complexation. Surface complexation seems to be especially prevalent for the M(III) and M(VI) state, while ion exchange probably dominates M (II) uptake. Although the literature on these minerals is sparse, the studies show that also these minerals have considerable sorption capacity and must be considered if sorption onto granite is to be modelled from single mineral data. What is usually missing from these studies are more systematic variations in pH, ion strength and temperature. Instead, there is a certain overemphasis on the establishment of sorption isotherms.
1 Introduction
In countries like Sweden, nuclear energy has been playing a large role in providing stable and emission-free energy for the last 50 years and will undoubtedly do so also in the near future, where global energy demand, especially in the form of electricity, is foreseen to double or triple compared with today. This expected increase is of course due to the enforced phasing out of fossil fuels, which still today constitutes about 83% (in 2021) of the world energy mix (OurWorldinData, 2023). Apart from the research on “Generation IV” nuclear technology, which mainly is a future concept for closing the loop of nuclear fuel and thereby minimizing waste and more importantly storage time, the research on final repositories have been going on for more than 40 years. The research has now reached a state of maturity, where actual spent fuel repositories are under construction. Today, both Sweden and Finland have advanced construction plans for such repositories. The disposal method will be a geological repository for radioactive waste, according to the KBS-3 concept (SKBF/KBS, 1983). The concept includes the water-tight and corrosion-resistant copper canister with spent nuclear fuel to be further protected by a placement in bentonite clay in the geological formation of granitic rock 500 m below the surface. Based on extensive geological surveys the Swedish Nuclear Fuel and Waste handling Company (SKB AB) has selected the Forsmark area in the municipality of Östhammar, about 150 km north of Stockholm as the most promising site for such a repository.
Deep geological repository designs like the KBS-3 concept, are constructed with multiple and consecutive barriers of which the host rock itself is considered to be the penultimate barrier to radionuclide transport, should all the other barriers fail. In such scenarios, which must be considered in safety analysis, the uptake of radionuclides by host rock, with constituent minerals, plays a major role for minimizing release to the biosphere. Studies on the sorption of radionuclides on rock and its constituent minerals do not only provide data for safety analyses but can also potentially give a more fundamental understanding of the sorption process, provided that concepts of chemical reactivity are applied for minerals surfaces and that these are coupled with reactions in aqueous solution. Such understanding will give increased predictive capability in modelling future scenarios, where groundwater conditions change.
Radionuclide sorption data is usually collected as empirical Rd values (distribution coefficients) without providing any details about the underlying mechanistic chemical reactions that govern these values. The Rd values are only valid for exactly the same conditions for which they are measured and thereby of limited applicability. Thermodynamic Sorption Modeling (TSM) is a collective term for modelling the Rd values from assigning detailed chemical reactions between mineral surfaces and solution. The assignment of these reactions is usually based on a combination of knowledge of the active surface groups of mineral surfaces and by measurements of certain mineral surface characteristics.
The present review is intended to provide literature data that potentially can be used for employing TSM of radionuclide sorption on four particular minerals that are judged to be important for the general radionuclide sorption properties of granite. The minerals are biotite, chlorite, K-feldspar and labradorite (a mineral in the plagioclase feldspar series) and these can be judged as representative constituent minerals for the Forsmark site type of rock. This selection of these four minerals was based on three presumptions: 1) a known relatively large radionuclide sorption capability but often a minor constituent (biotite and chlorite), 2) a known relatively less sorption capability but a major constituent (K-feldspar and labradorite) and 3) an appreciable porosity. The latter condition excludes minerals like quartz.
The present literature review is therefore made for scientific papers that present work regarding radionuclide sorption onto the four selected granitic minerals for this study: biotite, chlorite, labradorite(plagioclase) and K-feldspar, and more specifically, such studies where TSM have been used for evaluating the experimental data. It should be emphasized that this review does not cover all papers with experimental data of radionuclide sorption for the four minerals, but preferentially those where some sort of TSM have been used for the interpretation of the data.
2 Some characteristics of the host rock
According to the surveys made in the Forsmark area, and based on dominant rock type characteristics, there are four major groups (Group A to D) of rocks present. Each rock group is further subdivided into domains based on the grain size, composition, and relative age of the rock type present. The rock domain (SKB code RFM029) in the target volume that hosts the proposed repository is dominated by a rock of the metamorphic medium-grained meta-granite to granodiorite type (SKB rock type code 101057), which relates to major group B and which covers 84% of the targeted rock domain RFM029. Other rock types in this domain are fine-to medium-grained granodiorite, granite and tonalite (10%), amphibolite (3%) and subordinate pegmatite (2%) (Byegård, et al., 2006).
A detailed mineralogical composition analysis of this metamorphic medium-grained meta-granite to granodiorite by thin section point-counting of six samples gave the following results: quartz (26%–39%), biotite (3%–12%), K-feldspar (14%–29%), plagioclase (27%–41%), chlorite (<0–0.4%), muscovite (0%–1%), epidote (0%–1%), titanite (0%–1%), zircon (0%–0.2%) and opaque (0%–0.8%), the latter is probably iron oxide i.e., magnetite (Selnert, et al., 2008). The porosity of the rock type (101,057) has also been measured by the water-saturation method and the average value for 105 crushed samples was found to be 0.24 ± 0.12 (Byegård, et al., 2006). In the cited work, it was also noted that measurements with the same method on intact drill-core samples instead of crushed samples usually gave a slightly smaller porosity, for example, 0.16 ± 0.03 for three 5 cm long samples. The BET specific surface area of the rock type (101,057) has been measured for one intact drill-core sample and the results was 0.024 ± 0.012 m2/g (Selnert, et al., 2008). The cation-exchange capacity (CEC) of the rock type (101,057) for a 0.063–0.125 mm fraction of crushed rock has been measured with a standard method and the results was about 0.6–0.9 cmoles/g (Selnert, et al., 2008).
3 Theory of thermodynamic sorption modelling
Thermodynamic sorption modelling (TSM), or sometimes, surface complexation, is a research field that is under development and only a limited number of databases are available for inclusion in, for example, geochemical speciation codes such as PHREEQC (Parkhurst and Appelo, 2013). The research area has been developed since the 1970′s from a combination of a physical-chemical description of surface phenomena that appears between a solid-water interface in combination with classical water-solution chemistry (Dzombak and Morel, 1990). The latter topic describes reactions for dissolved elements (“species”) which forms compounds, aqueous complexes, which often have a net charge.
In order to perform TSM, three fundamental assumptions are usually made: 1) chemical reactions on surfaces can be described as a binding of dissolved species to specific functional groups (“sites”) on these surfaces, 2) these surface reactions can be described, just as reactions in solution, with equations for equilibrium and mass balances, 3) electrostatic effects may be taken into account by employing a coulombic term, taken from the theory about the electrical multi-layer of surfaces in aqueous solutions. Since the sorption of charged species potentially also involves a build-up of a surface charge with same sign as these species, further sorption may be obstructed by electrostatic repulsion. The columbic term can therefore be regarded as an activity coefficient for charged surface complexes. A provision for employing these steps is that the surface is well-defined in its composition, which usually limits TSM studies to single-solid phases, such as pure minerals. The three most common electrostatic interface models employed in TSM are the Diffuse Layer Model (DLM), The Constant Capacitance Model (CCM) and the Triple Layer Model (TLM), and each model describes the solid-solution interface slightly different with a different number of parameters (Hayes, et al., 1990). To this must be added the most common approach, the Non-electrostatic Model (NEM), which neglects any electrostatic effects between surface and solution and simply describes sorption with reactions and mass-balances only.
Today, there is no general agreement on how sorption data should be collected or interpreted, contrary to reactions in aqueous solution or precipitation reactions. This have led to several theories for the application of the electrical multi-layer which are often incompatible with each other. In their classical monography of TSM with hydrous ferric oxide (Dzombak and Morel, 1990) did an effort to clear up the among the different models and therefore selected the so-called Diffuse Layer Model (DLM, although they call it the generalized Double Layer Model) as the most promising candidate for describing the sorption of solution species to different surface sites.
For example, the DLM describes the deprotonation of a surface site ≡XOH2+ as
This is the “internal” equilibrium constant, valid for the inner plane or surface; the proton has not yet been transported through the outer diffuse layer with counterions, attracted by the surface charge. This is annotated with an index “s” on the proton symbol. For the outer plane, outside the diffuse layer, the corresponding equilibrium coefficient is
This is an apparent equilibrium constant or rather a coefficient which is dependent on the momentarily charge of the surface. The relation between the two equilibrium equations is
The exponential term includes variables of surface charge ΔZ (in this case, for a proton =1) and the surface potential Ψ (V), the latter assures that the equilibrium for the inner plane is constant. The surface potential can be deduced from the equation for the specific surface charge σ(C/m2)
F(C/mol) is the Faraday constant, SSA(m2/m3) the specific surface area and ΓH (mol/m2) is the momentarily proton surface density. According to deGouy-Chapman theory for electrical double-layers at the surface-water interface, the surface charge can be related to the surface potential with
On the right side of this equation everything, except ΔZ, Ψ and T are physical constants. If the surface site density ΓH is known, the surface potential Ψ can be calculated. An amphoteric surface site requires a second protonation constant or surface pKa value for the formation also of negatively charged sites ≡XO− in addition to the first one described with Eqs1, 2. With this and other additional charged surface species (for example, those formed by adsorbed metal cations and metal complexes) additional equilibria must be added and the expression for the surface charge must be expanded with additional terms.
To be able to numerically solve the complete non-linear equation system, the equations shown here must be complemented with the usual mass balances, for example, [M]tot, [H]tot and [≡X]tot. The former two mass balances are usually given by the initial experimental conditions, while the latter must be experimentally determined, preferentially in a separate solid phase characterization experiment or, and often with less success, by having this entity as a parameter to be optimized in the modelling. The reason why the latter method is less successful is that this parameter tends to serve as collector of model errors (that is, wrong assumptions about surface reactions) like a dust bin and this, in turn, will often give non-consistent results between different experimental conditions (for example, different pH, ionic strength, metal concentration).
To be able to model the variability of measured distribution coefficients certain surface reactions must be assumed. A model usually consists of a number of these surface reactions, for example, protonation of acidic/amphoteric surface sites, the uptake of cations and anions on these sites or a cation exchange (=electrostatic, or physical bonding) to other type of sites. In most cases, the sorption can be formulated to also depend on the variable surface charge, at least for chemical bonding sites according to the electrostatic interface models mentioned above. The most basic electrostatic interface model consists of a non-specific attraction between the charged surface and dissolved ions consisting of a “diffuse layer”.
Electrostatic interface models for the solid-solution interface can be classified as Single, Double and Triple-layer Plane models. The different planes normally consider, respectively, non-specific electrostatic attraction in a diffuse layer, chemical sorption on specific surface sites and finally specific physical sorption sites that takes place outside the diffuse layer. The three different interface models are illustrated in Figure 1 below.
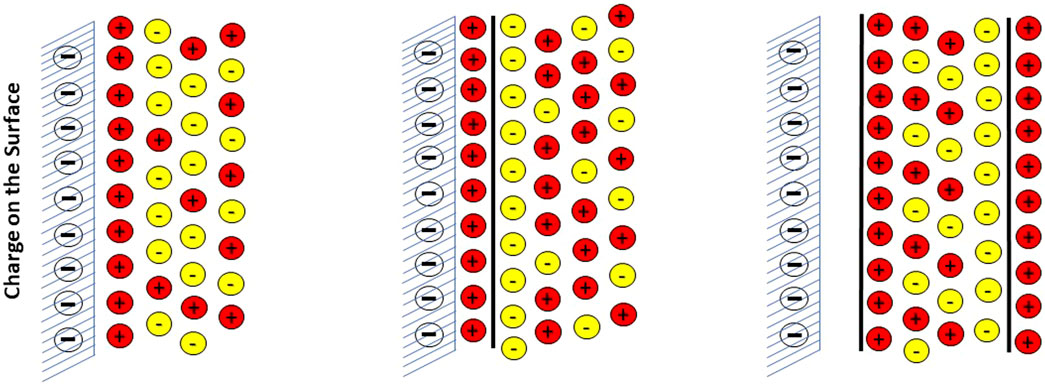
FIGURE 1. Illustration of the three commonly used electrostatic surface-water interface models: Single (with diffuse Layer), Double and Triple Plane models.
Thus, the DLM is a Double plane model and the TLM is a Triple plane model, while the CCM is essentially a single plane model, but instead of a diffuse layer it describes the potential transfer from surface to solution with a constant capacitance. In the literature, there is often a confusion in terminology between the type of electrostatic interface model (i.e., DLM, CCM, TLM) and the classification of a model (if they employ single, double or triple planes).
In a recent review of the status of TSM (Payne, et al., 2013) some recommendations on their usage were given.
1) The models should be calibrated within a wide range of conditions
2) Pure solid phases must be well defined and characterized
3) If complex phase is to be modelled, all the constituent phases must be characterized
This review identified two different approaches for modelling complex solid phases: the Component Additive (CA) method and the Generalized Composite (GC) method.
The CA method utilizes linear combinations of TSM made for the individual minerals, which have been corrected for their abundance in the rock and their specific surface area. The scaling factor for the minerals should then be the available surface area of that mineral in the rock. These scaling factors can be difficult to access for the rock, but microscopic investigation of thin slices of rock may be a method. The GC method instead utilizes a parameter optimization directly on sorption data for the rock. This leads to at least two problems: 1) the number of parameters to be fitted are large compared to experimental data and 2) there is no reference material to validate the model fitting with, because it is used in the fitting itself. Also, the GC model has limited applicability to other rock types, while a CA model may be attempted with several rock types, if the requisite pure mineral data are available. The two methods have been described in detail elsewhere (Davis, et al., 1998; Davis, et al., 2004).
4 Experimental methods
Several methods have been used for collecting sorption data. The use of pure minerals is preferred if the results should be interpreted using a set of specific surface reactions, like in TSM. Proton uptake and release of a mineral surface is usually investigated with titrations on very fine particle size fractions of the minerals. If the particle size is large, it will take exceedingly long time to reach an equilibrium, due to the in-diffusion of protons. The drawback with using fine size fraction is that mineral dissolution reaction may also increase which can muddle the results of the titrations of surface groups. Exceedingly acidic or basic conditions should be avoided, or the data should be collected comparatively fast so that any dissolution reactions can be ignored.
Coarser fractions can be used in batch experiments, where the experiments proceed over a long time, sometimes several months to reach equilibrium. The batch experiment consists of a crushed solid phase in contact with a water phase, normally at a fixed pH, ionic strength and the initial concentration of the sorbing metal in question. Because of the pH buffering property of most minerals, it is often necessary to use a buffer to fix the pH. The extent of mineral dissolution during long time sorption experiments should be checked with elemental analyses.
Sometimes the course fractions are used in column experiments, with a steady flow of water phase and where a “spike” of radioactive metal solution is added to inlet solution, followed by sampling of the outlet solution. The retention of metal on the column material due to in-diffusion and sorption gives characteristic properties of the break-through curves of the measured radioactivity in outlet water samples.
A further increase in the size of the solid is the diffusion experiment where complete pieces of minerals in a definite geometric shape (usually cylindrical) are used. The pieces of minerals are mounted in diffusion cells where the solution is in contact with some, or all of the surfaces. Depending on the thickness of the sample, an equilibrium may eventually be reached in several months up to years. Generally, the larger the mineral sample the longer the diffusion equilibrium will take.
If the solid material is initially tracer-free and the tracer uptake has attained equilibrium all the mentioned methods will result in a distribution coefficient Rd of the measured specie A
Note that the total concentration of adsorbed A on the solid, [A]ads, is usually not directly measurable but have to be deduced from the concentration of A in water phase, [A]aq and from initially added tracer using the mass balance. Some method for estimating the sorption on vessel walls should also be used, at least to investigate if this will affect the measured Rd or not. Another condition to be noted is if the uptake has not reached equilibrium after a certain time (which is very often the case) the measured Rd values are only apparent (temporal) and will change with time. In this case, a transport equation (usually, uptake by diffusion) is used for evaluation of the final Rd value.
If one assumes that an equilibrium reaction between a solution specie A and a surface site ≡X takes place,
This equilibrium is governed by the equilibrium constant K. Eq. 7, together with the mass balances for ≡X and A, are known as the Langmuir isotherm.
The isotherm Eq. 8 can be regarded as the most basic form of TSM. For the certain case where two conditions apply: 1) the concentration of A is relatively low compared with concentration of ≡X, one can assume that there is no risk of saturation of sites, and 2) there are just one type of A species in solution, then from the mass balance and the equilibrium constant it is then possible to assign a Kd value:
This is also called a linear sorption isotherm which is characterized by the Kd value only, the actual distribution constant for A between solid and liquid phase. The incorrect use of the designation Kd instead of Rd is almost always made in the literature. Kd values are, in fact, rarely directly measurable but have usually to be modelled.
Kd values (or the Langmuir reaction constants K when working near saturation conditions), should therefore be treated as fitting parameters in TSM and not as measurable experimental values.
Finally, a commonly used empirical isotherm is the Freundlich isotherm
The constant n is usually less than 1. The Freundlich isotherm cannot be used for TSM since it lacks the reaction-mechanistic approach, but it has nevertheless found wide use, especially for heterogenous compounds. It has been considered as an integrated version of a set of Langmuir isotherms with normal distributed ln K values (Sposito, 1980).
Measured Rd values are dependent on water composition variables (pH, ionic strength, metal concentration, temperature, type of background electrolyte etc.). This means that to model Rd values the conditions must be varied to investigate the effects on Rd. It is possible to use complementary techniques such as spectroscopic methods for trying to establish the nature of the bound surface complex. Methods that have been used for this purpose includes EXAFS (Extended X-ray Absorption Fine Structure spectroscopy), ATR-FTIR (Attenuated Total Reflection Fourier Transform InfraRed spectroscopy), XANES (X-ray Adsorption Near Edge Structure spectroscopy), XPS (X-ray Photoelectron Spectroscopy), TRLFS (Time-Resolved Laser-induced Fluorescence Spectroscopy) and AFM (Atomic Force Microscopy). However, these methods are mostly qualitative, they do not say anything about the strength of sorption.
Apart from measuring the distribution coefficients variation with water composition, such as pH, ionic strength and metal concentration, other parameters included in the model may have to be measured. The total surface site density is such a quantity, which is preferentially measured in separate experiments rather than used as a fitting parameter. Methods that can be used for acidic/amphoteric sites are gas adsorption measurements of the specific surface area SSA (m2/kg) according to the BET method and the reactive surface area (mol/kg) according to the tritium uptake method (Berubé, et al., 1967), or similar methods. These two combined can give the surface site density (mol/m2).
For ion-exchange sites, the total cation-exchange capacity (CEC) can usually be measured with standardized methods and this in combination with BET, can give a corresponding ion-exchange surface site density. It has also been reported that the uptake of ethyleneglycol monoethylether (EGME) may be used for measuring interlayer surface site density in sheet minerals, which is otherwise non-accessible with gas adsorption methods (Yukselen-Aksoy and Kaya, 2006). Other methods to access the total surface site density includes potentiometric titrations, isotope exchange methods, anion saturation methods and reactions with Grignard reagents. Experimental values of surface site density may be compared with theoretical calculations like those made with DFT (Density Functional Theory). Of the different parameters that is included in TSM, it seems that the models are especially sensitive to the surface site density (Goldberg, 1991), methods for determining this parameter independently of other parameters (surface pKa, Kd values etc) are therefore preferred.
5 Literature review
5.1 Biotite
Biotite, also called black mica, is a solid solution whose compositional range lies between the end members phlogopite KMg3AlSi3O10(OH)2 and annite KFe3AlSi3O10(OH)2.
The mineral is a member of the phyllosilicate mineral group and the mica sub-group or family. It has a trioctahedral structure with formula K(Mg0.6–1.8, Fe2.4–1.2)Si3AlO10(F,OH)2. Biotite is a layered silicate that follows the TOT (T: tetrahedral, O: Octahedral) + C(cation) structural pattern where the thickness of one TOT + C layer varies within 9.5–10Å. The two tetrahedral sheets consist of Si4+ tetrahedrally coordinated to oxygen and hydroxyl ions. Si4+ can be substituted with Al3+, or less commonly Fe3+. These two sheets are bound together with one octahedral sheet of cations C(oct), mostly Fe2+ or Mg2+. In trioctahedral sheets all the three sites are occupied by divalent cations. Ideally, one can write a trioctahedral TOT structure formula as
A net negative charge is created in the TOT unit structure when Si4+ is partially replaced by Al3+ in the tetrahedral sites. This negative charge is balanced by additional cations C(int) in an interlayer space between two TOT sheets. This cation is usually K+ for biotite (Nesse, 2000). A very rigid structure is formed due to the existence of K+ in the nonahydrate form K+(H2O)9 in the interlayer. The bonding between different TOT layers is purely electrostatic. Ion-exchange of K+ in biotite is usually the first step in the transformation of biotite to other secondary minerals like vermiculite.
5.2 Overview of biotite sorption models
In this section the main findings about tracer sorption onto biotite are collected and commented upon. In order to make the comparisons between different data more relevant, the section is divided into parts corresponding to tracer oxidation state for cationic tracers. Anionic tracers are treated separately. The individual references (B1-B14) are presented in more detail in Supplementary Appendix S1A.
In Table 1 below, all the literature data collected for tracer sorption onto biotite is briefly presented in alphabetical order of tracer element. Note that references in this section are abbreviated B1-B14, according to Table 1.
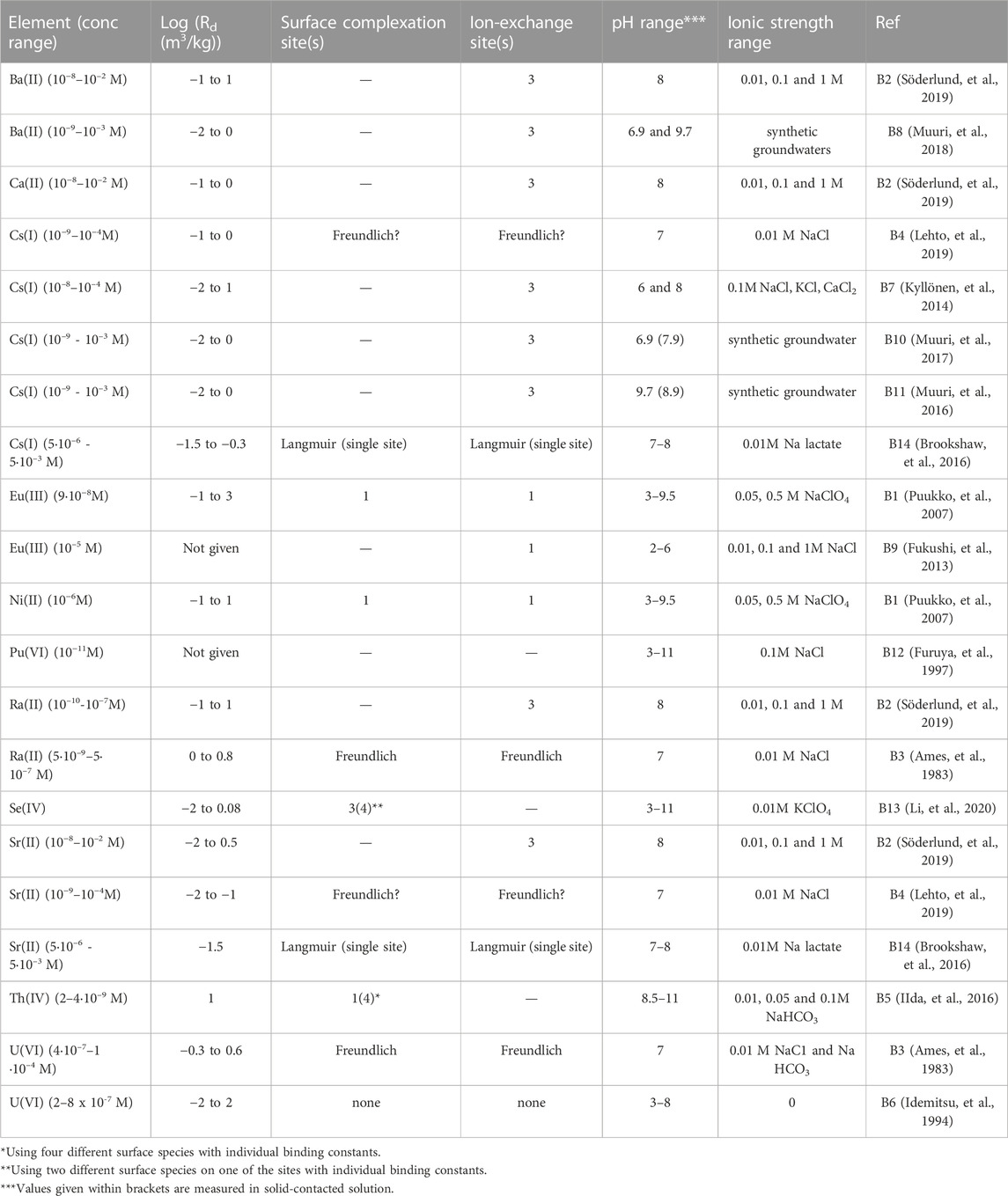
TABLE 1. Overview of radionuclide sorption experiments with biotite.
5.2.1 Cationic elements of oxidation state I
The only element of this type used in the references is Cs, but there are several studies available (refs. B4, B7, B10, B11, B14). The common experimental configuration for all the five studies is the sorption isotherm, where the pH is held fixed and about neutral while the tracer concentration is varied. There are no studies about any effects from ion strength, but in one paper (ref B7) the effect of the main electrolyte cation is studied.
A common finding seems to be a strong dependency of sorption of the tracer concentration, which would indicate a continuous filling up of multiple sorption sites. The most commonly applied model is the one suggested for the mica mineral illite (Bradbury and Baeyens, 2000), a model which employs no less than three different cation-exchange reactions, each site apparently dominating sorption within a specific Cs concentration range. In contrast, (ref B14) used a single site Langmuir isotherm, although the single site was not identified as either surface complexation or ion-exchange site. Only reference B4 instead used the empirical Freundlich isotherm. For Cs, it seems that at least one ion exchange site is needed to explain the uptake on biotite. The most likely site would then be the basal plane sites, normally occupied by K+ ions. Strangely, the effect of ionic strength on Cs sorption seems to be lacking in literature data but in case of an ion exchange mechanism, the uptake can be expected to be highly dependent on background electrolyte strength.
5.2.2 Cationic elements of oxidation state II
The most used elements in this group of cations are alkaline earths Ca (ref B2), Sr (refs B2, B14), Ba (refs B2, B8) and Ra (refs B2, B3). The sorption isotherm experiment is exclusively used, although the effect of electrolyte ion strength has also been studied (ref B2) or the salinity of the simulated groundwater (ref B8). The main findings are that cation sorption is fairly constant at low concentration but at about what can be considered to be the upper limit for tracer concentration (10−6M), the sorption decreased, indicating the saturation of a certain type of sorption sites. Increasing ionic strength or salinity decreased sorption, as can be expected from the additional competition for sorption sites of the ion-exchange type.
Modelling was usually made with the three-site ion exchange model based on the model for illite (Bradbury and Baeyens, 2000). However, (ref B14) used a single site Langmuir isotherm, although the single site was not identified as either surface complexation or ion-exchange site. Reference B3 instead used the empirical Freundlich isotherm to account for the decreasing sorption at higher concentration.
Only one reference studied a transition metal, Ni(II) by (ref B1). Here the employed experimental method was a pH variation with fixed tracer concentration together with a variation of electrolyte strength. The uptake of Ni was found to have a distinct sorption “edge” at pH about 7, possibly indicating the presence of surface sites with acidic behavior and which are deprotonated at a surface pKa. The ionic strength had little effect on Ni sorption which makes the case stronger for the presence of an acidic surface site. A model with one pKa surface complexation site (apparently without inclusion of surface charge) together with a one ion exchange site was fitted to the data (ref B1).
Since there are no systematic comparison of the effect of tracer concentrations and the pH for the different M(II) tracers, it is impossible to say 1) if the three models used (the three-site ion-exchange model, the single-site Langmuir model and the one pKa plus one ion exchange site model) are compatible with each other and 2) if they describe the sorption behavior completely for all type of M(II) elements.
5.2.3 Cationic elements of oxidation state III
In this group of elements only Eu(III) has been studied (refs. B1, B9). The studies are of the pH variation type and with metal concentration fixed. The ionic strength has been varied in 2-3 steps. Both of the references found a sharp sorption “edge” around neutral pH, which is typical for an amphoteric (that is, a charge dependency on pH) surface complexation site which can be activated by stepwise de-protonation at certain fixed pH values that is governed by one or two surface pKa values.
It is interesting that the two references found the location of the “edge” at the slightly different pH values: pH 3–4 (ref B9) and pH 5–6 (ref B1). The ionic strength had a profound effect on reducing Eu sorption above the pH 3–4 “edge” according to (ref B9), while the other work found the ionic strength only affecting sorption below the pH 5-6 edge.
Modelling of Eu(III) was made using a combination of one pKa value surface complexation site together with one ion-exchange site (ref B1). In the other work (ref B9) only one ion-exchange site was used for modelling the sorption. Apparently, the activation of such site with pH is then accomplished by exchanging protons with Eu, a process that is similar but not exactly the same as the de-protonation of a surface-complexation site.
It seems that a model with one surface complexation site plus one ion exchange site is enough to explain Eu (III) sorption at low metal concentration when pH is varied over a fairly large range (pH4-10).
Here it should be mentioned that varying the metal concentration behavior of sorption at a fixed pH (= the sorption isotherm) is much more difficult to investigate for elements with oxidation state (III) and higher, compared with the lower oxidation states, since the corresponding cations of type M(III), M(IV), M(V)O2 and M(VI)O2 will readily form hydroxide precipitations as soon as the pH of solution is anything but acidic unless total metal concentration is kept well below tracer level (<<1 10-6M).
5.2.4 Cationic elements of oxidation state IV
Only one reference was found for the (IV) state, the work (ref B5) which used Th(IV) at fixed low concentration, varying pH and with a non-inert electrolyte (NaHCO3). The latter means that carbonate complexation can be expected to interfere with Th sorption onto biotite and this in turn will presumably make the results harder to interpret with a model since the carbonate species must also be taken into the account. The sorption was found to be more or less constant in the limited pH range of the investigation (pH 8.5–10.5) and no effects were seen from the variation of ionic strength. A two pKa surface complexation model was applied with no less than four different species binding to the same site, each with a different binding constant.
To this can be commented that 1) it is highly questionable if such an ambitious model for the pH-dependent sorption can be used considering the limited pH range investigated, and 2) for explaining the sorption data in the actual pH range investigated, the binding of no less than four different Th species to the same site can probably be reduced to the binding of one surface specie only but instead with a fairly strong binding constant, coupled with the appropriate Th complexation constants in solution. It seems unlikely that Th(IV) sorption should be that weak compared with its speciation in solution, so that hydrolysis species is reflected in a corresponding set of similar surface complexes.
5.2.5 Cationic elements of oxidation state VI
For the oxidation state (VI) three references were found: two for U(VI) (refs B3 and B6) and one for Pu(VI) (ref B12). Although none of these works did include any TSM modelling they were included in this review because M(IV-VI) sorption studies are not that common.
In (ref B3) the sorption isotherm of U(VI) was investigated at fixed pH and electrolyte strength and temperature variated. The sorption was found to decrease for increasing values of the temperature and tracer concentration. The empirical Freundlich isotherm was used to explain the variability of sorption on metal concentration. In both (ref B6) and (ref B12) the pH variability of sorption was investigated at a fixed metal concentration. Both works show comparable results in that sorption was highly dependent on pH, peaking at about pH 6.5, above this pH the sorption decreased again most likely due to the formation of carbonate complexes (all studies were made at ambient conditions).
These few studies on M(VI) sorption onto biotite indicates that both pH and metal concentration can have an influence. The inverse temperature dependency of sorption shown in (ref B3) is also interesting since the effect of temperature on sorption can possible be used to identify which type of surface reaction (a chemical binding or an ion-exchange) that prevail by utilizing the van´t Hoff equation for determining ΔH and ΔS for the surface reaction.
5.2.6 Anionic elements
For anion sorption onto biotite, only Se(IV) have been studied (ref B13). This element exists in water solutions as the species SeO32-, HSeO3− and H2SeO3 with respective pKa values about 8 and 2.
The sorption was investigated by, in turn, varying pH and metal concentration at a fixed ionic strength. The pH variation results show a “reversed” sorption edge, where sorption is stronger for lower pH, while neutral and alkaline region gives lower sorption. This is in accordance with anionic sorption onto positively charged surface complexation sites, activated by proton uptake at a certain pH corresponding to a pKa value for biotite surface. Interestingly the “edge” was not very sharp, showing a gradual decrease in sorption in a broad pH range. This could be modelled as a 2-pKa, one surface complexation site model, where the surface charge de-protonated from ≡SOH2+ to neutral ≡SOH groups at about pH 5. Then the latter sde-protonated to ≡SO− groups at about pH 9.
Two different surface complexes on this single “strong” site were used in the model: one for SeO32- and one for HSeO3−.
For the sorption isotherm, the pH was fixed at pH 7.7 and showed the usual decrease at higher metal concentration. Interestingly, this behaviour was modelled as an additional two “weak” sites, also of the surface complexation type where only HSeO3− sorption was considered, although little evidence for a pH influence on these was presented.
Altogether this leads to no less than three distinct surface complexation sites for anions on biotite surface, where one is particularly strong and exhibits two pKa values. If these anionic sorption sites are in any way related to the cationic sorption sites described above, remains to be established.
At least the “strong” sorption site for Se could be identical with the surface complexation site that was used to model Ni and Eu sorption (ref B1).
5.2.7 Conclusions for sorption onto biotite
From the collected data on biotite sorption and modelling one can conclude that there should exist at least one surface complexation site for cations, with either one or two pKa values. There have been no attempts to include electrostatic effects in that model.
In addition, it seems that one has to assume a number of ion-exchange sites for explaining the sorption isotherms of alkaline and alkaline earth metals, which have very little tendency to bind with a chemical bond by surface complexation. For tracer concentrations, however, one can expect that only one such ion-exchange site should suffice to explain sorption of these type of cations. Whether these exchange sites are basal surface sites or edge sites is an open question. Cation-exchange may also be valid for other cations, especially transition metals, below their sorption “edge” of the surface complexation site.
The aforementioned amphoteric surface complexation site should (ideally) be the same for explaining the relatively high sorption at low pH of anions, at least the anionic element Se, while neutral to alkaline pH show low anionic sorption.
Additional “weak” surface complexation sites for the Se anion sorption have been suggested to prevail in this, neutral to high pH region, but more investigations on this matter is recommended, not least considering the importance such sorption may have for repository retention performance of anionic components, which are usually those found most problematic to retain.
5.3 Chlorite
Chlorite is, like biotite, a sheet-mineral or phyllosilicate mineral but does not belong to the mica sub-group but instead constitutes its own sub-group of chlorite minerals. The most common chlorite minerals are clinochlore ((Mg,Fe)5Al) AlSi3O10(OH)8, pennantite (Mn5Al) AlSi3O10(OH)8 and chamosite ((Fe,Mg)5Al) AlSi3O10(OH)8. The chlorites are solid solutions of theoretical end members, containing either Mg, Fe, Mn or Ni, although other substituents are also found. The ideal formula for the Mg end member can be written as Mg3AlSi3O10(OH)2⋅(Mg, Al)3(OH)6 (Nesse, 2000). The chlorites are considered to be alteration minerals of parent minerals like amphibole, pyroxene and biotite. The name was given by the commonly found green colors of the chlorite minerals.
Like biotite, the chlorites consist of TOT layers, with the middle octahedral sheet partially substituted with two divalent cations (a so called dioctahedral layer) with the ideal formula (C(oct)2+)2(Si2O5(OH)3-)2 and giving the structure a net negative charge. Like biotite, a partial replacement of Si4+ with Al3+ in the tetrahedral layers gives an additional net negative charge.
Unlike biotite, however, this negative charge is not balanced by additional interlayer cations but instead by a positively charged extra octahedral layer between the different TOT layers with the ideal formula (Mg0-3, Al3-0) (OH)6. Due to the similar formula of brucite, this extra octahedral layer is called a brucite layer and the sheet structure of chlorites is often called TOT + O. Like the micas, the bonding between different TOT layers is purely electrostatic and the hardness of the biotite minerals are also similar to the micas (Nesse, 2000).
5.4 Overview of chlorite sorption models
In this section the main findings about tracer sorption onto chlorite are collected and commented upon. In order to make the comparisons between different data more relevant, the section is divided into parts corresponding to tracer oxidation state for cationic tracers. Anionic tracers are treated separately. The individual references (C1-C8) are presented in more detail in Supplementary Appendix S1B.
In Table 2 below, all the literature data collected for tracer sorption onto chlorite is briefly presented in alphabetical order of tracer element. Note that references in this section are abbreviated C1-C8, according to Table 2.

TABLE 2. Overview of radionuclide sorption experiments with chlorite.
5.4.1 Cationic elements of oxidation state I
The only element of this type used in the references is Cs with two studies (refs. C5 and C8).
The experimental configuration in both the references is the sorption isotherm, where the pH is held fixed and about neutral, while the tracer concentration is varied. There are no studies about any effects from ion strength or pH variations for Cs sorption on chlorite. A common finding seems to be a strong dependency of sorption of the tracer concentration, which would indicate a continuous filling up of sorption sites.
A multiple-site sorption is indicated by a successful fitting of a Freundlich isotherm to the data (ref C5), while a single-site Langmuir isotherm was fitted to data in (ref C8). Taken together, this means that a multi-site sorption of Cs on chlorite is not unambiguously proved.
Interestingly, the enthalpy of reaction was determined in (ref C5), and its relatively small value of -8 kJ/mol gives further proof that the sorption reaction should be an ion-exchange one and not involving a stronger chemical bonding to the surface.
5.4.2 Cationic elements of oxidation state II
Several references were found for the M(II) state and these are discussed in two groups: one with the alkaline earths: Ba (ref C3), Sr (ref C8), and one with the transition metals: Co (ref C6) and Ni (ref C1) and (ref C4).
For the alkaline earth group, only sorption isotherm experiments were reported. It is the same situation as for the alkali metal Cs: one work (ref C3) uses the Freundlich isotherm, indicating multi-site sorption and one work (ref C8) uses a single-site Langmuir isotherm. Consequently, the multi-site ion-exchange of chlorite is not unambiguously proved also for this metal group.
The microbial reduction of Fe(III) surface groups was studied in (ref C8) but it had no effect on Sr sorption, which may confirm that the sorption is rather an ion-exchange reaction than a chemical bonding reaction, at least not to any ferric (hydro-)oxide groups, which seems logical for this group of metals.
For the transition metals, the sorption modeling that was found was more of a mixed bag.
In (ref C6) the sorption isotherm for Co(II) was modelled with the Freundlich isotherm, again indicating a multi-site sorption process, where multiple sites are sequentially filled up (Bradbury and Baeyens, 2000). The enthalpy and entropy of the sorption reaction was also determined, which gave the particularly interesting results that the sorption was entropy driven. The enthalpy change was found to be typical in value for an ion-exchange reaction but unfavorable, this was, however, compensated for by a favorable entropy change. The latter indicates at least a partial removal of the hydrous shell around the Co cation upon binding to the surface, which is somewhat surprising since ion-exchange reaction are usually considered to take place with the hydrous shells around the cations intact.
Two papers reports sorption with Ni(II), (ref C1) and (ref C4), and these are the only work found that have varied pH and ionic strength for a M(II) sorption study on chlorite. Both works agree so far as no effect was found from varying the ionic strength and this is usually what is found for a surface complexation reaction. Upon varying the pH, a sorption edge was evident in the sorption behavior found in both studies and its location was similar: (ref C1) show the edge stretching from pH 5 to pH 8, while in (ref C4) it was from pH 4 to pH 7.
This fairly extended sorption edge suggests either 1) a two-pKa single surface site or 2) two different surface sites that are “activated” at slightly different pH, and these two cases were actually used for models in (ref C1) and (ref C4), respectively. Another notable difference is that a Non-Electrostatic Model was used in (ref C1), while a Double Diffuse Layer model was used in (ref C4).
To conclude for M(II) sorption on chlorite, it seems that ion-exchange reaction(s) on the surface is what can be predicted for the alkaline earths. For the transition metals, the models are more inconsistent. For Co an ion-exchange is suspected to happen when varying metal concentration (foremost considering the fairly low enthalpy of reaction measured), while for Ni a very pronounced and extended sorption edge instead suggests that a surface complexation reaction is activated by increasing the pH.
It can therefore be worthwhile to do a more systematic mapping of the variable space regarding pH, ionic strength and maybe also temperature for transition metal M(II) sorption on chlorite. It is also important that the metal concentration variable should not be extended too far (not over 10–7 M) since hydroxide precipitations are likely to occur at non-acidic conditions. Metal hydroxide precipitations have often been wrongly interpreted as strong sorption.
5.4.3 Cationic elements of oxidation state VI
For the metal oxidation state M(VI) there are two studies of U(VI) on chlorite, (ref C2) and (ref C7).
In both (ref C2) and (ref C7) the pH was varied, while the metal concentration and ion strength were held constant. The pH variation results from these two works were fairly consistent in so far that (ref C2) measured a sorption “edge” starting about pH 5 to reach a maximum at pH 8 to then “reverse” back to lower sorption at higher pH. Although (ref C7) only measured sorption at three pH values, the varying degree of sorption with pH at least showed the same up and down trend as in (ref C2).
(Ref C7) also investigated the influence of ionic strength but effects were negligible. In the same work the metal concentration was varied from 10–5 to 10–4 M and here the results indicated an onset of sorption saturation. However, the latter concentration is relatively high for U(VI), especially in an alkaline solution (pH 10) and from an outside view, one may suspect that precipitations can be responsible for the perceived surface saturation. The sorption was nevertheless successfully modelled (see below).
Both works successfully used the same type of surface complexation model with a Diffuse Double electrostatic Layer interface model, utilizing one (ref C2) or two (ref C7) pKa values.
5.4.4 Conclusions for sorption onto chlorite
From the collected data on chlorite sorption and modelling, one can conclude that there should exist at least one surface complexation site for cations with two pKa values. However, it seems like models with two separate surface complexation sites, each with one pKa value also works. An electrostatic DDL interface model has been included in some of these models, while other models seem to do without any consideration of electrostatic surface effects.
In addition, it seems that one has to assume at least one ion-exchange site for explaining the sorption isotherms of alkaline and alkaline earth metals, which have very little tendency to bind with a chemical bond by surface complexation. It may also be the case, at least for transition metals M(II), that both type of sorption sites has to be considered.
5.5 K-feldspar
The feldspars are the most frequent mineral of the earth’s crust, with a total content of about 60%. Feldspars have three main compositional end members: K-feldspar (KAlSi3O8), albite (NaAlSi3O8), and anorthite (CaAl2Si2O8). A continuous series of solid solutions are mainly found only between K-feldspar and albite (alkali feldspars) and between albite and anorthite (plagioclases) with very little inclusion of Ca2+ and K+ in the respective series. Orthoclase is an additional designation for a certain structural variant of K-feldspar. The feldspar is made up of SiO4 tetrahedrons with corner-shared oxygen atoms, which gives a ring-like structure comprising of four tetrahedrons. Further corner-sharing gives a network of such four-tetrahedron rings. This also allows the formation of open sites in the structure that can easily accommodate cations such as Ca2+, Na+ and K+. In such site, the cation is co-ordinated to nine oxygen atoms in the immediate vicinity. When Ca2+ is replacing Na+ in the plagioclase series of solid solutions, charge neutrality is maintained by replacing Si4+ with Al3+ in the main structure (Nesse, 2000).
Feldspars can have a very complex macrostructure and therefore occur in polymorphs. The lattice stability of feldspars is dependent on the specific temperature cooling evolution at their formation. Moreover, during the cooling even more complicated textures can be formed due to exsolution (exclusion from the solid solution) giving streaks of a minor mineral variety in the major mineral. Also due to their relatively open structure, feldspars are prone to undergo weathering reactions, where in the first place the cations in the structure are replaced by protons. This opens up the structure further, leading to proton interaction with oxygens and structure dissolution (Bahlburg and Breitkreuz, 2008).
Considering K-feldspar, the Al:Si ratio in K-feldspar is 1:3 and it is found in three polymorphs: orthoclase, sanidine, and microcline. The main difference is the distribution of Al and Si in the tetrahedron structure. Rapid cooling leads to completely random distribution of Al and Si in sanidine, while slow cooling leads to the most ordered distribution of Al and Si in microcline. The orthoclase polymorphic structure can be considered as an intermediate between these two. The different polymorphs can only be reliably distinguished by their microscope-optical and cleavage properties. To the naked eye, sanidine is a white mineral, while microcline and orthoclase are usually colored pink, gray or yellow (Nesse, 2000).
5.6 Overview of K-feldspar sorption models
In this section the main findings about tracer sorption onto K-feldspar are collected and commented upon. In order to make the comparisons between different data more relevant, the section is divided into parts corresponding to tracer oxidation state for cationic tracers. No data were found for anionic elements. However, one may note that in the alkaline range some cationic elements may hydrolyze to a neutral or anionic specie. The individual references (K1-K9) are presented in more detail in Supplementary Appendix S1C.
In Table 3 below, the literature data collected for tracer sorption onto K-feldspar is briefly presented in alphabetical order of tracer element. In Table 3 below, all the literature data collected for tracer sorption onto chlorite is briefly presented in alphabetical order of tracer element. Note that references in this section are abbreviated K1-K9, according to Table 3.

TABLE 3. Overview of radionuclide sorption experiments with K-feldspar.
5.6.1 Cationic elements of oxidation state I
Only one reference was found for this oxidation state, the Cs(I) in (ref K2).
Results for both sorption isotherms and pH variation experiments are available. Since Cs is known to preferentially interact with surfaces by ion-exchange reaction rather than a surface complexation reaction, one may assume that the uptake is governed by a relatively low-affinity ion-exchange site but also with a fairly high site density, since there was no evidence of surface saturation: the measured isotherms were linear and were not levelling-off.
Despite that there was no apparent evidence for multiple-site sorption, typically shown as different linear slopes in different concentration regions, other than a suggested mechanism that Ca could possibly interfere with Cs sorption for one type of sites, the authors stated that a multiple-site Freundlich isotherm fitted the data better than a single-site Langmuir isotherm. Interestingly, the Cs sorption on K-feldspar in (ref K2) was found to be highly pH dependent, showing less sorption at higher pH, which is usually not the case for ion-exchange reactions. Normally, one would expect less sorption at low pH, where protons may compete with exchange sites. In this case one can therefore suspect that the Na+ ions from NaOH added to adjust pH may had an effect, especially since no background electrolyte was used to give an overall constant ionic strength.
5.6.2 Cationic elements of oxidation state II
Sorption of M(II) ions onto K-feldspar was studied in three works: (ref K4) using Pb, (ref K5) using Cd and (ref K9) using Sr. The two first studies investigated isotherms and the pH behavior of sorption, the last study only the ionic strength behavior.
The results for Pb(II) show linear isotherms with no sign of multiple sites or sorption saturation. Only at very low pH 2 the sorption was found to be reduced, which can be expected due to competition from protons. The results for Cd(II) also show linear sorption isotherms with no influence on sorption with the variation with pH. Both Langmuir and Freundlich isotherm were fitted successfully, however, since the isotherm slope is constant, that is, no indication of site saturation, it is doubtful if the use of a Freundlich isotherm can be justified since it implies presence of multi-site sorption.
The results for Sr(II) show a dependency of ionic strength, indicating a ion-exchange reaction.
To conclude, it seems that M(II) bind to K-feldspar by ion-exchange only, since sorption is more or less pH independent, except at very low pH 2-3, normally not encountered with natural groundwaters. Here one can expect proton competition for ion-exchange sites. Only one ion-exchange binding site seems to be validated.
5.6.3 Cationic elements of oxidation state III
Sorption of M(III) ions onto K-feldspar was studied in two works: (ref K7) using several different lanthanides and also Am, and (ref K8) using Nd. Both studies mainly investigated the pH behavior of sorption, although at 2-3 different metal concentrations.
All results show a common behavior: a sharp rise of a sorption “edge” in the pH range 5-6, especially at lowest tracer concentrations. Rising tracer concentration seems to make the “edge” region fuzzy, probably indicating problems with lanthanide/actinide precipitations at the highest concentration of 10−5M, since hydroxides are likely to precipitate already at 10−7M. Also, below the sorption “edge”, in the acidic range, a certain sorption capacity seems to persist. In both work, sorption was modelled as one-site surface-complexation reaction, although several different surface species were considered: two in (ref K8) and no less than four in (ref K7), including one carbonato surface complex.
(Ref K7) considered only hydroxide complexes, justified by spectrometric studies. Also, by support of spectroscopic studies, the model used a multi-dentate binding to the surface, which is a seldom used approach to formulate the bonding of a surface complex. A multi-site bonding has more commonly been assumed for ion-exchange reactions where several exchange sites bind to one multi-valent cation. In both studies the ionic strength had no significant influence on sorption, except that in (Ref K8) a reduced sorption effect was seen only at the highest metal concentration 10−5M. One can speculate if this may be due to metal colloid formation. In (Ref K7) the is a suggestion of an ion-exchange reaction to govern the residual weak sorption found below pH 5.
To conclude, M(III) elements seems to bind by surface complexation reactions, possibly assisted by ion exchange reaction at low pH, below the main sorption “edge”. A metal coordination-favorable, multi-site bonding involving several oxygen atoms on surface may explain the particularly strong sorption for the M(III) elements.
5.6.4 Cationic elements of oxidation state V
Sorption of M(V) ions onto K-feldspar was studied in two works: (ref K4) and (ref K8), both using Np.
In (ref K4) both isotherms and pH dependency of sorption were studied. The isotherms, where the Np concentration was varied in a fairly wide range (4⋅10−8–4⋅10−5M), were linear with a constant slope, thereby they suggest only one binding site. The sorption at pH6 and 10 were quite similar, while at pH2 sorption was considerably reduced. There was no study of the effect of ion strength, which may had given further clues about the type of surface reaction.
In (ref K8) the sorption of Np was measured at only one tracer concentration, but pH was varied in a broad range, pH 5–12. Initially sorption was comparatively weak (metal uptake below 20%), showing a small but constant increase above pH 7. However, this excluded the data points at the upper alkaline range, which showed increased sorption (up to 60%). Ion strength did not seem to have any effect on sorption. Sorption was modeled with a one-site surface complexation site.
To conclude, Np sorption onto K-feldspar seems to be fairly weak compared with U (which has typically 80%–90% uptake in ref K8) and also the lanthanides (100% uptake, ibid.) at least below pH 11. A surface complex reaction was used in (ref K8) with partial success (the upper alkaline range was not included). Despite the rather low sorption, usually associated with an ion-exchange reaction, there is no support in the literature for such a binding mechanism for NpO2+ ions.
It should be noted that Np is probably present in solution only partially as NpO2+ and partially as a hydrolyzed neutral or even anionic specie.
5.6.5 Cationic elements of oxidation state VI
Sorption of U(VI) ions onto K-feldspar was studied in four different works: (refs K1, K3, K4 and K6).
(Ref K1) is a very comprehensive work, where the effects of ion strength, tracer metal concentration (two steps), pH, temperature and CO2(g) on the sorption of U(VI) onto K-feldspar were studied. (Ref K3) mainly studied the pH effect, trace metal concentration was varied in two steps. (Ref K4) is mainly a spectroscopic study but also presents sorption isotherms in a wide concentration range. (Ref K6) finally, also studied the effect of temperature, pH, and metal concentration on sorption. The temperature had the unanimous effect of increasing sorption (refs. K1 and K6) and the van´t Hoff plots gave evidence for an entropy driven surface reaction, probably by surface-complexation thereby releasing most of the hydration shell of the uranyl ion. The pH variation generally shows a typical sorption “edge” of increasing sorption between pH 5-7, giving constant sorption above this pH if CO2(g) is not present (ref K1). If CO2 is present, a decreased sorption above this pH is measured due to the formation of uranyl-carbonate complexes (refs. K1, K3, K4 and K6).
In general, both tracer metal concentration and the ionic strength have little effect on U(VI) sorption on K-feldspar. The very wide range of tracer concentrations used in (ref K4) showed essentially linear behavior, however, at pH 6, two slopes were present. An increased slope with metal concentration rather than decreased slope with concentration indicate tracer precipitation, which was also confirmed with spectroscopic studies. The models used were one site surface complexation (refs. K1 and K3) with a varying number of surface complexes formed. It seems that the models used are either somewhat vaguely defined (ref K1) or are based on previous data for the analogue mineral albite (ref K3). The latter work, however, also presents spectroscopic data for the support of the selected surface species.
To conclude, the temperature dependency of U(VI) sorption, and the absence of ion strength dependency, together provide clear evidence for a surface complexation reaction. There is no evidence for multi-site sorption. The presence of CO2(g) has a strong influence on sorption in the alkaline pH range, probably by the formation of aqueous uranyl-carbonate complexes.
5.6.6 Conclusions for sorption onto K-feldspar
From the collected data on K-feldspar sorption and modelling one can conclude that there should exist at least one surface complexation site with either one or two pKa values, bonding to M(III), M(VI) and possibly also M(V). It is particularly interesting that a multi-dentate bonding model (a chelate surface complex) have been applied to M(III) sorption, which is an indication of a particular strong sorption mechanism.
An electrostatic DDL interface model has been included in some of these models, while other models seem to do without any consideration of electrostatic surface effects. In addition, it seems that one has to assume at least one ion-exchange site for explaining the sorption isotherms of alkaline, alkaline-earths and also divalent transition metals. This type of bonding may also be valid for the low sorption of M(III) and M(VI) in the acidic region, below surface complexation “edges”.
5.7 Labradorite (plagioclase)
The plagioclases also belong to the feldspar group of minerals, see the section about K-feldspar above for a general overview of the feldspars. The compositional range of plagioclase feldspars shows the continuous distribution of a solid solution between end members albite (NaAlSi3O8), and anorthite (CaAl2Si2O8). Ca2+ replaces Na+ due to their same size, charge neutrality in the mineral is maintained by replacing Si4+ with Al3+ in the tetrahedral structure. The plagioclase feldspars can be classified into six separate compositional ranges, based on their percentage of anorthite: oligoclase (10%–30%), andesine (30%–50%), labradorite (50%–70%) and bytownite (70%–90%).
Labradorite is the plagioclase mineral of main interest here since it is a constituent of the rock type at the Forsmark-site in Sweden.
5.8 Overview of labradorite(plagioclase) sorption models
In this section the main findings about tracer sorption onto labradorite/plagioclase are collected and commented upon. No references on labradorite were found, therefore all plagioclases were included in the review. In order to make the comparisons between different data more relevant, the section is divided into parts corresponding to tracer oxidation state for cationic tracers. No data were found for anionic elements. However, one may note that in the alkaline range some cationic elements may hydrolyze to a neutral or anionic specie. The individual references (P1-P5) are presented in more detail in Supplementary Appendix S1D.
In Table 4 below, all the literature data collected for tracer sorption onto plagioclases is briefly presented in alphabetical order of tracer element. Note that references in this section are abbreviated P1-P5, according to Table 4.
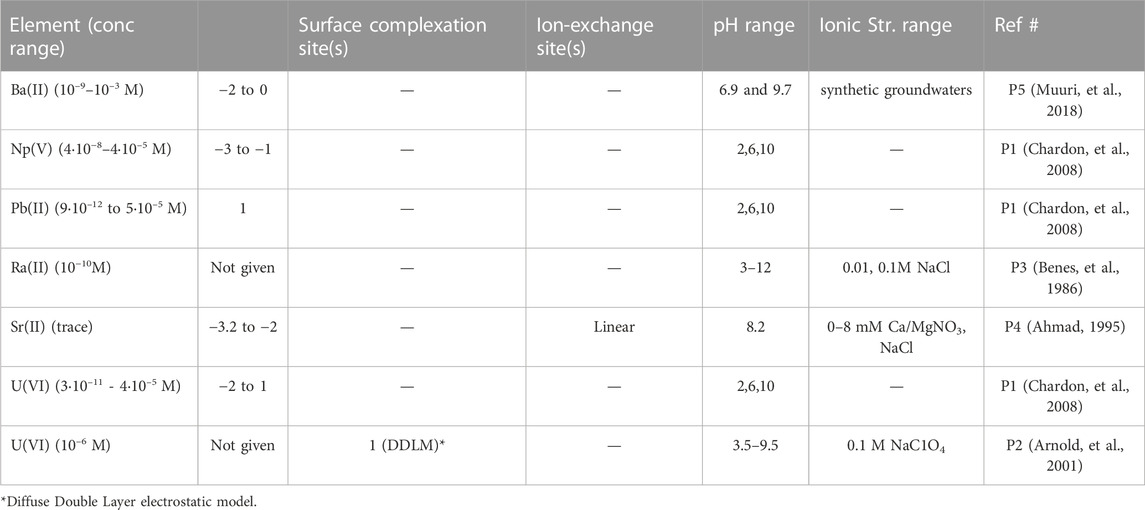
TABLE 4. Overview of plagioclase sorption experiments.
5.8.1 Cationic elements of oxidation state II
Sorption of M(II) ions onto plagioclases was studied in four works: (ref P1) using Pb, (ref P5) using Ba, (ref P3) using Ra and (ref P4) using Sr. The three first studies investigated variation of sorption with pH, the last two studies the variation with ionic strength and only the first study measured sorption isotherms. Of these four studies, only (ref P4) and (ref P5) did a theoretical modelling of results.
The results for Pb(II) show linear isotherms with no sign of multiple sites or sorption saturation. Only at very low pH 2 the sorption was found to be reduced, which can be expected due to competition from protons for the ion-exchange sites at this low pH value. The results for Ra(II) also show a reduced sorption with low pH, but also an increased ionic strength had a clear reduction on the sorption of Ra. The results for Sr(II)sorption show a dependency of ionic strength, indicating ion-exchange. The results for Ba(II) show a dependency of the synthetic groundwater used, where the more alkaline water (pH 9.7 vs. 6.9) gave stronger sorption of Ba on the plagioclase mineral. However, since the latter groundwater contained much more Na+ than the former, this is probably only an effect of the increased ionic strength on an ion exchange sorption and not an effect from a lower pH value.
To conclude, just as for K-feldspar, it seems that M(II) binds to the plagioclase feldspars exclusively by ion-exchange since sorption is more or less pH independent, except at very low pH 2-3, due to proton competition for ion-exchange sites. Only one ion-exchange binding site seems to be validated.
5.8.2 Cationic elements of oxidation state V
Only one work was found for Np(V) sorption onto plagioclase: (ref P1) which studied isotherms and the effect of pH variation. The sorption of Np(V) was fairly weak compared with both Pb(II) and U(VI) and also further reduced at low pH. Interestingly, the sorption at pH6 and 10 were found to be equally strong, a feature typical for metal uptake by ion-exchange, which is weakened only by increased proton concentrations at low pH. There was no modeling made of sorption although it was interpreted as a “weak” surface complexation reaction. These results for Np(V) are thus similar to K-feldspar.
5.8.3 Cationic elements of oxidation state VI
Two studies for U(VI) sorption onto plagioclase were found: (ref P1) which studied isotherms and the effect of pH variation at zero ionic strength and (ref P2) which studied the effect of pH variation at fixed tracer concentration and ionic strength. Both works show a clear pH dependency of U(VI) sorption, although (ref P1) indicated problems with precipitations at pH6. At higher pH carbonate complexes reduces sorption of U(VI) and also mitigates precipitations to form.
In (ref P2) the sorption was modeled with a single-site, two pKa Diffuse Double Layer electrostatic surface complexation model.
5.8.4 Conclusions for sorption onto plagioclases
From the few collected data of plagioclase sorption and modelling one can conclude that these confirms the findings that were made for sorption on K-feldspar: firstly, that one surface complexation site, (here with two pKa values), is responsible for bonding to M(VI) and possibly also M(V). Secondly, it seems that one has to assume at least one ion-exchange site for explaining the sorption isotherms of alkaline, alkaline-earths and also divalent transition metals. Contrary to K-feldspar however, any sorption of M(VI) on plagioclase in the acidic region was not indicated.
6 Discussion
The literature for sorption of radionuclides on four common granitic minerals have been investigated. Most of the work found concerns the sheet-silicates biotite and chlorite, both which are known to exhibit relatively strong metal-binding capability. The proposed binding mechanisms include at least one ion-exchange mechanism, which accounts for uptake of alkaline and alkaline earths. If metal concentration is increased, multiple ion-exchange sites that are consecutively saturated have to be assumed. For multi-valent metals, however, a chemical bonding surface complexation site is preferred to explain metal uptake. This type of site is probably located at mineral edges and have amphoteric properties. Both 1-pKa and 2-pKa surface site acidity models have been suggested.
For the feldspars K-feldspar and plagioclase, the picture is mostly the same: a combination of one ion-exchange sites and one surface complexation seems to be mostly employed in the models. Especially M (III) seems to have strong sorption on feldspars. But also alkaline earth metals like Ba (II) were found to exhibit the same sorption strength on plagioclase as biotite (Muuri, et al., 2018), 2018), which suggests that the feldspar components should be considered in modelling the sorption of the composite granitic rock, especially since feldspars are dominating the rock mass. A reliance on the low-abundance sheet-silicates only for modelling radionuclide sorption onto granite will probably fail.
The studies found are usually over-emphasizing the sorption isotherm study, the metal concentration variation. However, trace releases of radionuclides are usually what can be expected from scenarios were the barrier system has failed. Instead, a more systematic investigation on effects of pH, ionic strength and, not at least, the temperature would therefore be desirable. Van’t Hoff diagrams on the variation of sorption constants with temperature can give values for both enthalpy and entropy, this in turn may indicate if the sorption mechanism is due to surface complexation (entropy driven) or ion-exchange (enthalpy driven) (Rotenberg, et al., 2009).
Most studies found here in this review are of limited scope. It has been emphasized that for a reasonably successful modeling of sorption, a wide range of parameters have to be investigated, including pH and ionic strength (Payne, et al., 2013). Obviously, the limited scope of the investigations hinders the improvement in knowledge.
Another deficiency that can be found in some studies are that data for crucial solid characteristics, like the surface site density and acidity, are taken from studies of other minerals. This will most likely give unsatisfactory results for the modelling.
A very interesting question is also the dependency of sorption on the specific surface area (SSA). For TSM to work, the measured Rd (m3/kg) values must be converted to SSA-compensated Ra (m) values. But there are very few systematic studies of the effect of SSA on sorption. Especially in anisotropic materials like the sheet silicates biotite and chlorite, the SSA can be suspected to have little relation to the most important sorption sites, which are usually located only on edge surfaces.
Finally, one should not underestimate the importance of using a sound methodology for sorption studies. Equilibrium must be allowed to be reached, or if not, it must be modelled by an in-diffusion transport process. Also, all too often, too large metal concentration has been used, that may provoke precipitations of metal hydroxides, thereby masking sorption. Especially for weakly sorbing radionuclides, any wall sorption must be compensated for. Another pitfall is the use of filters for phase-separation, where the sorption onto another solid phase must be corrected for.
Author contributions
SH: manuscript writing PK: literature search. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Strålsäkerhetsmyndigheten, the Swedish Radiation Safety Authority.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnuen.2023.1227170/full#supplementary-material
References
Ahmad, S. H. S. S. (1995). Competitive adsorption of 90Sr on soil sediments, pure clay phases and feldspar minerals. Appl. Radiat. Isotopes 46, 287–292. doi:10.1016/0969-8043(95)00001-t
Ames, L. L., McGarrah, J. E., and Walker, B. A. (1983). Sorption of uranium and radium by biotite, muscovite, and phlogopite. Clays Clay Minerals 31, 343–351. doi:10.1346/ccmn.1983.0310503
Arnold, T., Zorn, T., Zänker, H., Berhard, G., and Nitsche, H. (2001). Sorption behavior of U(VI) on phyllite: experiments and modeling. J. Contam. Hydrology 47, 219–231. doi:10.1016/s0169-7722(00)00151-0
Asci, Y., Nurbas, M., and Acikel, S. (2008). A comparative study for the sorption of Cd(II) by K-feldspar and sepiolite as soil components and the recovery of Cd(II) using rhamnolipid biosurfactant. J. Environ. Manag. 88, 383–392. doi:10.1016/j.jenvman.2007.03.006
Bahlburg, H., and Breitkreuz, C. (2008). Grundlagen der Geologie. Heidelberg, Germany: Spektrum Akademischer Verlag.
Beneš, P., Borovec, Z., and Strejc, P. (1986). Interaction of radium with freshwater sediments and their mineral components. J. Radioanalytical Nucl. Chem. 98, 91–103. doi:10.1007/bf02060436
Berubé, Y. G., Onoda, G. Y., and De Bruyn, P. L. (1967). Proton adsorption at the ferric oxide/aqueous solution interface. Surf. Sci. 8, 448–461. doi:10.1016/0039-6028(67)90032-5
Bradbury, M. H., and Baeyens, B. (2000). A generalised sorption model for the concentration dependent uptake of caesium by argillaceous rocks. J. Contam. Hydrology 42, 141–163. doi:10.1016/s0169-7722(99)00094-7
Brookshaw, D. R., Lloyd, J. R., Vaughan, D. J., and Pattrick, R. A. D. (2016). Effects of microbial Fe(III) reduction on the sorption of Cs and Sr on biotite and chlorite. Geomicrobiol. J. 33, 206–215. doi:10.1080/01490451.2015.1076543
Byegård, J., Gustavsson, E., Tullborg, E-L., and Selroos, J-O. (2006). Bedrock transport properties, Preliminary site description Forsmark area -version 1.2 SKB Report R-05-86. Solna, Sweden: SKB.
Chardon, E. S., Bosbach, D., Bryan, N. D., Lyon, I. C., Marquardt, C., Römer, J., et al. (2008). Reactions of the feldspar surface with metal ions: sorption of Pb(II), U(VI) and Np(V) and surface analytical studies of reaction with Pb(II) and U(VI). Geochimica Cosmochimica Acta 72, 288–297. doi:10.1016/j.gca.2007.10.026
Davis, J. A., Coston, J. A., Kent, D. B., and Fuller, C. C. (1998). Application of the surface complexation concept to complex mineral assemblages. Environ. Sci. Technol. 32, 2820–2828. doi:10.1021/es980312q
Davis, J. A., Meece, D. E., Kohler, M., and Curtis, G. P. (2004). Approaches to surface complexation modeling of uranium(VI) adsorption on aquifer sediments. Geochem. Cosmochimica Acta 68, 3621–3641. doi:10.1016/j.gca.2004.03.003
Ding, D., Fu, P., Li, L., Xin, X., Hu, N., and Li, G. (2014). U (VI) ion adsorption thermodynamics and kinetics from aqueous solution onto raw sodium feldspar and acid-activated sodium feldspar. J. Radioanalytical Nucl. Chem. 299, 1903–1909. doi:10.1007/s10967-013-2903-2
Dzombak, D. A., and Morel, F. M. M. (1990). Surface complexation modelling – hydrous ferric oxide. New York, NY, USA: Wiley.
Eylem, C., Erten, H. N., and Göktürk, H. (1989). Sorption of barium on kaolinite, montmorillonite and chlorite. Analyst 114, 351–353. doi:10.1039/an9891400351
Fukushi, K., Hasegawa, Y., Maeda, K., Aoi, Y., Tamura, A., Arai, S., et al. (2013). Sorption of Eu (III) on granite: ePMA, LA–ICP–MS, batch and modeling studies. Environ. Sci. Technol. 47, 12811–12818. doi:10.1021/es402676n
Furuya, H., Idemitsu, K., Inagaki, Y., Arima, T., Sasaki, T., Kuroda, Y., et al. (1997). Sorption of plutonium on a biotite mineral. Sci. Rep. Res. Institutes Tohoku Univ. Ser. A-Physics 45, 89–91.
Gao, X., Bi, M., Shi, K., Chai, Z., and Wu, W. (2017). Sorption characteristic of uranium (VI) ion onto K-feldspar. Appl. Radiat. Isotopes 128, 311–317. doi:10.1016/j.apradiso.2017.07.041
Goldberg, S. (1991). Sensitivity of surface complexation modeling to the surface site density parameter. J. Colloids Interface Sci. 145, 1–9. doi:10.1016/0021-9797(91)90095-p
Gustafsson, Å., Molera, M., and Puigdomenech, I. (2004). Study of Ni(II) sorption on chlorite – A fracture filling mineral in granites. Mater. Res. Soc. Symposium Proc. 824, 373–378. doi:10.1557/proc-824-cc7.3
Hayes, K. F., Redden, G., Wendell, E., and Leckie, J. O. (1990). Surface complexation models: an evaluation of model parameter estimation using FITEQL and oxide mineral titration data. J. Colloid Interface Sci. 142, 448–469. doi:10.1016/0021-9797(91)90075-j
Idemitsu, K., Obata, K., Furuya, H., and Inagaki, Y. (1994). Sorption behavior of uranium (VI) on a biotite mineral. Mater. Res. Soc. Symposium Proc. 353, 981–988. doi:10.1557/proc-353-981
Iida, Y., Yamaguchi, T., Tanaka, T., and Hemmi, K. (2016). Sorption behavior of thorium onto granite and its constituent minerals. J. Nucl. Sci. Technol. 53, 1573–1584. doi:10.1080/00223131.2016.1138901
Ishikawa, N. K., Kuwata, M., Ito, A., and Umita, T. (2017). Effect of pH and chemical composition of solution on sorption and retention of cesium by feldspar, illite, and zeolite as cesium sorbent from landfill leachate. Soil Sci. 182, 63–68. doi:10.1097/ss.0000000000000194
Kyllönen, J., Hakanen, M., Lindberg, A., Harjula, R., Vehkamäki, M., and Lehto, J. (2014). Modeling of cesium sorption on biotite using cation exchange selectivity coefficients. Radiochim. Acta 102, 919–929. doi:10.1515/ract-2013-2180
Lehto, J., Puukko, E., Lindberg, A., and Voutilainen, M. (2019). Batch sorption experiments of cesium and strontium on crushed rock and biotite for the estimation of distribution coefficients on intact crystalline rock. Heliyon 5, e02296. doi:10.1016/j.heliyon.2019.e02296
Li, X., Puhakka, E., Liu, L., Zhang, W., Ikonen, J., Lindberg, A., et al. (2020). Multi-site surface complexation modelling of Se(IV) sorption on biotite. Chem. Geol. 533, 119433. doi:10.1016/j.chemgeo.2019.119433
Muuri, E., Ikonen, J., Matara-aho, M., Lindberg, A., Holgersson, S., Voutilainen, M., et al. (2016). Behavior of Cs in grimsel granodiorite: sorption on main minerals and crushed rock. Radiochim. Acta 104, 575–582. doi:10.1515/ract-2016-2574
Muuri, E., Matara-aho, M., Puhakka, E., Ikonen, J., Martin, A., Koskinen, L., et al. (2018). The sorption and diffusion of 133Ba in crushed and intact granitic rocks from the Olkiluoto and Grimsel in-situ test sites. Appl. Geochem. 89, 138–149. doi:10.1016/j.apgeochem.2017.12.004
Muuri, E., Siitari-Kauppi, M., Matara-aho, M., Ikonen, J., Lindberg, A., Qian, L., et al. (2017). Cesium sorption and diffusion on crystalline rock: Olkiluoto case study. J. Radioanalytical Nucl. Chem. 311, 439–446. doi:10.1007/s10967-016-5087-8
Neumann, J., Brinkmann, H., Britz, S., Lützenkirchen, J., Bok, F., Stockmann, M., et al. (2021). A comprehensive study of the sorption mechanism and thermodynamics of f-element sorption onto K-feldspar. J. Colloid Interface Sci. 591, 490–499. doi:10.1016/j.jcis.2020.11.041
Our World in Data, (2023). Our world in data. https://ourworldindata.org/sources-global-energy.
Parkhurst, D. L., and Appelo, C. A. J. (2013). PHREEQC version 3 – a computer program for speciation, batch reaction, one-dimensional transport and inverse geochemical calculations. U.S. Geol. Surv.
Payne, T. E., Brendler, V., Ochs, M., Bayens, B., Brown, P. L., Davis, J. A., et al. (2013). Guidelines for thermodynamic sorption modelling in the context of radioactive waste disposal. Environ. Model. Softw. 42, 143–156. doi:10.1016/j.envsoft.2013.01.002
Puukko, E., Olin, M., Puhakka, E., Hakanen, M., Lindberg, A., and Lehikoinen, J. “Sorption of nickel and europium on biotite,” in Proceedings of the FUNMIG 3rd Annual Meeting, Edinburgh, United Kingdom, November 2007, 265–272.
Richter, C., Müller, K., Drobot, B., Steudtner, R., Großmann, K., Stockmann, M., et al. (2016). Macroscopic and spectroscopic characterization of uranium (VI) sorption onto orthoclase and muscovite and the influence of competing Ca2+. Geochimica Cosmochimica Acta 189, 143–157. doi:10.1016/j.gca.2016.05.045
Richter, C. (2015). Sorption of environmentally relevant radionuclides (U(VI), Np(V)) and lanthanides (Nd(III)) on feldspar and mica Thesis. Dresden, Saxony, Germany: Technischen Universität.
Rotenberg, B., Morel, J-P., Marry, V., Turq, P., and Morel-Desrosiers, N. (2009). On the driving force of cation exchange in clays: insights from combined microcalorimetry experiments and molecular simulation. Geochimica Cosmochimica Acta 73, 4034–4044. doi:10.1016/j.gca.2009.04.012
Selnert, E., Byegård, J., and Widestrand, H. (2008).Forsmark site investigation- Laboratory measurements within the site investigation programme for the transport properties of the rock SKB Report P-07-139. Solna, Sweden: SKB.
Shahwan, T., and Erten, H. N. (1999). Radiochemical study of Co2+ sorption on chlorite and kaolinite. J. Radioanalytical Nucl. Chem. 241, 151–155. doi:10.1007/bf02347303
Shahwan, T., and Erten, H. N. (2002). Thermodynamic parameters of Cs+ sorption on natural clays. J. Radioanalytical Nucl. Chem. 253, 115–120. doi:10.1023/a:1015824819940
Singer, D. M., Maher, K., and Brown, G. E. (2009). Uranyl-chlorite sorption/desorption: evaluation of different U(VI) sequestration processes. Geochimica Cosmochimica Acta 73, 5989–6007. doi:10.1016/j.gca.2009.07.002
SKBF/KBS (1983). Kärnbränslecykelns slutsteg, Använt kärnbränsle -KBS-3 (Del I-IV). Stockholm, Sweden: SKBF/KBS.
Söderlund, M., Ervanne, H., Muuri, E., and Lehto, J. (2019). The sorption of alkaline earth metals on biotite. Geochem. J. 53, 223–234. doi:10.2343/geochemj.2.0561
Sposito, G. (1980). Derivation of the Freundlich equation for ion exchange reactions in soils. Soil Sci. Soc. Am. J. 44, 652–654. doi:10.2136/sssaj1980.03615995004400030045x
Yukselen-Aksoy, Y., and Kaya, A. (2006). Comparison of methods for determining specific surface area of soils. J. Geotechnical Geo-environmental Eng. 132, 931–936. doi:10.1061/(asce)1090-0241(2006)132:7(931)
Keywords: radionuclide sorption, granite, biotite, chlorite, plagioclase, K feldspar, thermodynamic sorption model
Citation: Holgersson S and Kumar P (2023) A literature review on thermodynamic sorption models of radionuclides with some selected granitic minerals. Front. Nucl. Eng. 2:1227170. doi: 10.3389/fnuen.2023.1227170
Received: 22 May 2023; Accepted: 27 July 2023;
Published: 17 August 2023.
Edited by:
Delphine Durce, Belgian Nuclear Research Centre (SCK CEN), BelgiumReviewed by:
Tiziana MISSANA, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas, SpainNathalie Wall, University of Florida, United States
Copyright © 2023 Holgersson and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stellan Holgersson, c3RlaG9sQGNoYWxtZXJzLnNl